Embed Size (px)
Citation preview

С U о
The chemistry of the nitro and nitroso
groups Edited by
H E N R Y F E U E R
Purdue University Lafayette, Indiana
Part 2
1970 I N T E R S C I E N C E P U B L I S H E R S
a division of John Wiley & Sons
N E W Y O R K L O N D O N S Y D N E Y T O R O N T O

CHAPTER 2
Directing effects of the nitro group in electro philic and radical aro matic substitutions
T A D E U S Z U R B A Ń S K I ч
Warsaw Institute of Technology (Politechnika), Warszawa, Poland
I. ELECTROPHILIC SUBSTITUTION . . . . . . . 49 A . Introduction . . . . . . . . . . 49 B. Directing Effect of the Nitro Group 50
1. Historical review . . . . . . . . 50 2. Modern theories . . . . . . . . 54 3. me/a-Directing effect of the nitro group . . . . . 5 5 4. orMo-/wr«-Dirccting effect of the nitro group . . . . 5 7 5. Directing effect of a nitro group placed in the side chain or ring 59
G. Indirect Substitution . . . . . . . . 60 II. FREE-RADICAL SUBSTITUTION. . . . . . . . 64
A . Directing Effect of the Nitro Group . . . . . . 6 4 B. Activating Effect of the Nitro Group. . . . . . 6 9
III. REFERENCES . . . . . . . . . . 7 1
I. ELECTROPHILIC SUBSTITUTION
A. Introduction
It is now accepted that ni trat ion is due to the action of a positively charged ion (i.e., a cation) N 0 2 + . Therefore a ni trat ion by ionic substitution (or displacement) is an electrophilic substitution, according to the well-known nomenclature introduced by Ingold 1 ( 'Ka t iono id ' substitution according to L a p w o r t h 2 and Robinson 3 ) . In electrophi l ic substitution the two electrons which form the new covalent
49

50 Tadeusz Urbański
bond (i.e., the bond between the aromatic compound and the electrophilic reagent) are both supplied by the aromatic compound (equation 1).
(l)
(1) (slow) (2) (fast) (3)
In 1 two 'free' electrons are supplied by the aromatic system (77-electron sextet). The addi t ion of the electrophilic agent N 0 2 + results i n the formation of the intermediate cr-complex 2 ( 'Wheland intermediate ') 4 . O n the basis of experiments, part icularly those of Me lande r 5 , the formation of 2 constitutes the rate-determining step. T h e overall reaction is of second order and follows the S E 2 mechanism, i.e., is a bimolecular electrophilic substitution.
B. Directing Effect of the Nitro Group
I. Historical review"
The systematic study of aromatic substitution became possible only after the correct structural relationship was established between the ortho, meta, and para isomers of benzene. The first attempts to formulate the orientation rules were made as early as 1875 by H u b n e r 7 and i n the following year by Noe l t ing 8 .
They found that the substitution of benzene derivatives i n the ortho and para positions occurred without simultaneous substitution i n the meta position and that the mode of substitution depended largely on the substituent group already present in the benzene ring. Further, Noel t ing tried to establish a relation between the directing effect of a substituent and its chemical character. H e pointed out that m^a-directing groups such as N 0 2 , S 0 3 H , and C O O H are acidic, whereas basic (e.g., N H 2 ) or neutral groups (e.g., C H 3 and/or CI) are ortho and para directing.
The rule could not explain why the phenolic O H group is ortho and para directing, although it should be considered as acidic.
Later , Arms t rong 9 drew attention to the fact that simple substituents containing double or triple bonds, e.g.,
О о / /
С N , or C s N , \ \
ОН о

Directing Effects of the Nitro Group 51
are meta directing. This was supported by V o r l â n d e r 1 0 . T h e rule was accepted for some time, but eventually a number of exceptions were found and this reduced its value.
C r u m Brown and Gibson 1 1 advanced an original concept of the substitution rule. Regarding substituent X in C e H 6 X as a derivative of H X , they stated that X will be a meta-divecting group if H X can be oxidized to H O X in a one-step process. Thus N 0 2 should be meta directing, for H N 0 2 can be oxidized to H O N 0 2 .
T h e most important systematic collection of experimental facts related to substitution was given by H o l l e m a n n 1 2 1 3 . H e also examined substitution reactions of disubstituted benzene derivatives, C 6 H 4 X Y , and was able to show which of the two groups had the stronger orienting effect. Further a considerable number of experiments were carried out by Hol lemann using kinetic measurements. O n the basis of the relative speed and the yield of substitution, Hollemann classified substituents not only according to their directing effect, but also their 'directing powers' (Table 1).
TAIÎLE 1. 'Directing powers' of substituent directing groups.
ortho-para O H > N H 2 > N R 2 > NHAcyl > Cl > lir > C H 3 > higher alkyls > I
meta С О О Н > S O s H > N 0 2
Although the rule is empirical, it did help to predict the nature of the product obtained on substitution of most aromatic compounds. Thus , if OT-nitrotolucne is further nitrated, the nitro group enters the ortho and para positions relative to the methyl group. It follows from this experimental fact that the directive effect of the nitro group is less than that of the methyl group.
Hol lemann also pointed out that ortho-para-d\rcctu\g groups increase the rate of aromatic (electrophilic in present day terminology) substitution, whereas OT«/a-orienting groups greatly decrease it. Thus phenol, containing the ог/Ло-^ara-direct ing O H group, can readily be nitrated even with dilute nitric acid, but nitrobenzene with its ?wto-directing nitro group requires a mixture of concentrated nitric and sulfuric acids. Nitrobenzene also requires vigorous conditions (e.g., high temperature) in order to be chlorinated or sulfonated. Generally speaking, the rate of the reactions of nitrobenzene is about 10~7 lower than that of benzene.
Addit ional attempts to establish general rules for the directing effect in substitution were due to H a m m i c k and Ill ingworth 1 4 , and to Mason and coworkers1 5 .

52 Tadeusz U r b a ń s k i
Several important attempts to explain the substitution rule in more modern terms were initiated in 1902 by Flurscheim 1 6 who introduced a concept of alternately strong and weak distribution of 'chemical affinity' around an aromatic ring. T h e difference of reactivity distribution due to the influence of meta- ( N 0 2 ) and ortho-para- (CI) directing groups is shown in formulas 4 and 5, respectively. T h e thick and thin lines indicated large and small
(4) (5)
quantities of 'chemical affinity,' respectively, and arrows indicated 'residual' or 'free affinity.' Although the term 'affinity' is rather meaningless in the present state of chemical theories, the Flurscheim bonds of strong and weak 'affinity' may be compared with bonds of different orders between positions 1 and 2, and the arrows should indicate 'affinity forces' or 'free affinity'6 available at reactive carbon atoms1 7 .
A n important feature of Flurshcim theory was an introduction of the concept that alternations in degree of chemical reactivity can be transmitted from a substituent group situated in a relatively distant part of a molecule.
T h e alternation in chemical character of the atoms of any substituted benzene derivative was also considered later by F r y 1 8 . H e suggested that positive and negative charges resided upon the atoms constituting the aromatic molecule and he suggested formula 6 for benzene. Thus the nitro group would induce a positive charge at
H +
I H
(6)
the hydrogen atoms in the meta positions (structure 7) , whereas chlorine, being essentially electronegative, would induce the positive
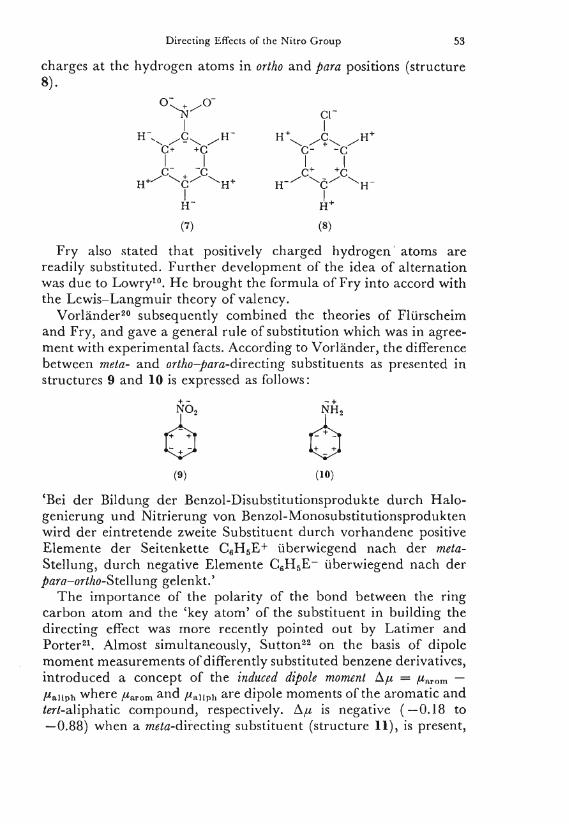
Directing Effects of the Nitro Group 53
charges at the hydrogen atoms i n ortho and para positions (structure 8). '
(7) (8)
F r y also stated that positively charged hydrogen atoms are readily substituted. Further development of the idea of alternation was due to L o w r y 1 0 . H e brought the formula of F ry into accord with the L e w i s - L a n g m u i r theory of valency.
V o r l â n d e r 2 0 subsequently combined the theories of Flurscheim and Fry , and gave a general rule of substitution which was in agreement wi th experimental facts. Accord ing to V o r l â n d e r , the difference between meta- and or//w-/>ara-directing substituents as presented i n structures 9 and 10 is expressed as follows:
NÓ-2 N H ,
(9) (10)
'Be i der Bi ldung der Benzol-Disubstitutionsprodukte durch H a l o -genierung und Ni t r ie rung von Benzol-Monosubstitutionsprodukten w i r d der eintretende zweite Substituent durch vorhandene positive Elemente der Seitenkette C 6 H 5 E + uberwiegend nach der meta-Stellung, durch negative Elemente C 6 H 5 E ~ uberwiegend nach dcr para-ort/w-StcWung gelenkt.'
The importance of the polarity of the bond between the r ing carbon atom and the 'key atom' of the substituent in bui ld ing the directing effect was more recently pointed out by Lat imer and Porter 2 1 . Almost simultaneously, Su t ton 2 2 on the basis of dipole moment measurements of differently substituted benzene derivatives, introduced a concept of the induced dipole moment Д/г = fi&TOm — caliph where /чатот and u i l U v h are dipole moments of the aromatic and ter/-aliphatic compound, respectively. A/u is negative (—0.18 to — 0.88) when a me/a-directing substituent (structure 11), is present,

54 Tadeusz Urbański
and positive (0.21-0.88) when an or/Ao-jfxzra-directing substituent is present (structure 12).
R R
(11) (12)
T h e induced moment for the nitro group is —0.88 (determined from / < a r o m = - 3 . 9 3 and / < a l i p h = - 3 . 0 5 ) .
M o r e recent data on dipole moments 2 3 give a value of —0.52 for the nitro group. This value was obtained from measurements of nitrobenzene and ter/-nitrobutane i n the gas phase.
Sv i rbe ly 2 4 further developed this idea, and stated that i f the dipole moment of a monosubstituted benzene derivative were larger than 2.07 D , further substitution would occur i n the meta position. T h e dipole moment o f nitrobenzene is 4.08 D and hence is meta directing.
Subsequently this line of thought led E y r i n g and R i 2 5 to calculate dipole moments and charge distribution from rates of ni trat ion o f substituted benzene derivatives. T i t o v 9 1 has given an or iginal approach to the problem of the directing effect of substituents i n electrophilic substitution. H e divided al l substituents into two classes: (1) those facilitating the oxidation of the benzene r ing to the quinonoid one, and (2) those inhibi t ing the formation of the quinonoid system.
Electron donating groups which are ortho-para directing belong to class (1) and electron attracting, meta directing (hence the nitro group) belong to class (2).
2. Modern theories
The F ry and V o r l â n d e r concepts can be regarded as precursors o f the more modern electronic theory which was developed by L a p w o r t h 2 6 and R o b i n s o n 2 7 .
Lapwor th applied his earlier theories2 8 of polar i ty and chemical changes to aromatic substitutions. H e explained the reactivity at particular points in a benzene r ing by the action of substituents of polar character which induce the electrical polarity. A n electrical polarizat ion can be transmitted wi th in an aromatic ring at the moment of reaction, just as the alternate polarity was induced i n conjugated systems2 8.

Directing Effects of the Nitro Group 55
A n important feature of Lapworth 's v iews 2 6 was that he stressed the importance of the presence of a key atom of a definite polar character i n a directing group. T h e more pronounced the polar character of the key atom, the more the substitution is restricted to one type. Thus , the methyl group i n toluene favors ortho-para substitution, but meta substitution occurs to the extent of 4 % . This is due to a very weak electronegativity of the carbon atom and very weak electropositivity of hydrogen atoms i n the methyl group. O n the other hand, phenol is substituted exclusively i n the ortho and para positions due to a very strongly negative oxygen atom i n phenol .
R o b i n s o n 2 7 amplified and developed Lapworth 's views, and I n g o l d 2 9 , 3 0 , together with other British authors introduced a special terminology and symbolism.
Accord ing to this terminology an electrophilic group, like the nitro group produces a negative inductive effect ' — L ' This is i n accordance wi th the presentation of V ic to r M e y e r 3 1 who described the nitro group as being a negative substituent.
Conversely, nucleophil ic substituents are represented by the symbol ' + Г (a positive inductive effect).
Bri t ish workers also introduced the concept of the mesomeric effect ' M ' which can be of a different sign than the inductive effect, but wh ich is negative for the nitro group. T h e effect can be considered as a form of a permanent displacement of the charge. It can be related to the concept of induced dipole Afi mentioned previously.
T h e H a m m c t t substituent constant, a, of the nitro group is positive i n both meta and para positions, and its value is relatively high. Th is is typical of electron-attracting substituents.
3. meta-Directing effect of the nitro group
The nitro group is a substituent wi th a dipolar structure i n which the positive end of the formal dipole is attached to the nucleus.
o <5 +^ +/
— N < у — N \ 4
о - О
Other wzete-dirccting groups have similar features, e.g. o-
• * - \ ± nitrile - C = N , sulfone R—S++—, and carbonyl С — O -
o-

56 Tadeusz Urbański
N o r m a n and T a y l o r 3 2 pointed out recently that substituents wi th dipolar double or triple bonds, and among them the nitro group, are electron wi thdrawing through both their —I effect (which reduces the electron density at each nuclear carbon) and their — M effect which results i n further electron wi thdrawal from ortho and para carbons. Both effects act i n the same sense, giving rise to deactivation at a l l nuclear positions and part icularly the ortho and para positions.
Consequently the isolated molecule wi th a nitro group (or any other m^a-directing group) should be polarized as shown i n structure 13; structure 14 shows the polarization by the action of an ortho-/>ara-directing group.
T h e mechanism of electrophilic substitution of monosubstituted. benzene derivatives i n which the meta position is least deactivated, is shown in equation 2.
(15) (16) (17)
W i t h molecular orbi ta l calculations as a basis, 'Whe land intermediates,' such as 16 are sometimes represented by formula 18, where the positive charge is equally shared between three carbon atoms. However , nuclear magnetic resonance measurements of the proton shifts i n pentamethylcyclohexadienyl cat ion do not agree wi th an equal distr ibution of the positive charge 3 3 .

Directing Effects of the Nitro Group 57
Structures 13 and 14 give a qualitative estimate of the electron density distr ibution around the aromatic r ing.
A more modern quantitative representation of charge distribution i n aromatic rings is based on the theory of molecular orbital and simplified wave mechanical ca lcu la t ions 3 4 , 3 5 .
T h e results of 77-elcctron density calculations 3 6 for nitrobenzene are presented i n diagram 19 (for comparison, the 77-electron distribution i n an i l i ne 3 5 is shown i n diagram 20).
Substitution of 19 i n the meta position by positively charged species (elcctrophiles) is thus substantiated.
Also, the calculation of localization energy introduced by W h e l a n d 4 can be helpful i n establishing the directing effect of substituents.
4 ortho-poro-Directing effect of the nitro group Al though the nitro group is meta directing, a considerable amount
o f 0- and a much smaller amount of/>-dinitrobenzenes are formed i n the nitrat ion of nitrobenzene. Accord ing to H o l l e m a n 1 3 , the nitration of nitrobenzene yields те-dinitrobcnzene (93.2%), o-dinitrobenzene (6.4%), and />-dinitrobenzene (0.3%). T h e ratio of 1/2 ortlw.para is 11:0. ortho Substitution is even more pronounced i n the chlorina-tion of nitrobenzene (by G1+) leading to m-chloronitrobenzene (80.9%), o-chloronitrobenzene (17.6%), and jfr-chloronitrobenzene (1 .5%). Here the ratio of 1/2 ortho :para is lower (5.9), but the absolute values are much higher than i n the previous example;
There has been some controversy over the reasons why the nitro group leads to such a high 1 /2 ortho :para ratio. Accord ing to De la M a r e and R i d d 3 7 the three ma in views on the subject are:
(a) that the nitro group and other отг/a-directing groups specifically deactivate the para pos i t ion 3 8 ;
(b) that те^а-directing groups provide an addit ional reaction path for ortho substitution by the prior addit ion of the reagent to

58 Tadeusz Urbański
the group, followed by rearrangement to the ortho position, i.e., nitration occurs indirect ly 3 9 ; and
(c) that the lower 1/2 ortho :para ratios are a consequence of steric hindrance at the ortho pos i t ion 4 0 , 4 1 .
However, since correlation of oricntational data for unsaturated substituents conjugated with the aromatic ring have shown that the 1 /2 ortho :para and 1 /2 meta :para decrease together in the same order, D e la M a r e and R i d d 3 7 suggest that electronic rather than steric factors determine the ratios of substitution.
It should be added to all these views that the calculated values of ^--electron density shown in diagram 19 predict a high ratio of 1/2 ortho :para in the electrophilic substitution of nitrobenzene. Baciocchi and I l luminat i 4 2 examined the rates of bromination and chlorination of 3-nitrodurene (21) into the p o s i t i o n n a i to the nitro
C H 3 y x O j
C H 3
(21)
group. T h e y found a strong deactivating action of the nitro group, of the order of 106-107, and this was also the case with 2-nitro-mesitylene and 3-nitroisodurene. However, the reactivity of 21 with molecular halogen was found to be higher than that predicted from the electrical effects in electrophilic substitution. T h e authors ascribed the higher rate to a steric inhibition of resonance of the nitro group. O n the other hand, with 2-nitromesitylene and 3-nitroisodurene, where meta substitution occurred, a slight decrease of deactivation was observed. Here the expected minor effect of steric inhibition of resonance might be overshadowed by increased hindrance to the approach of the reagent at the reaction center.
It should also be mentioned that in the homologs of benzene, the influence of the alkyl groups should not be neglected. Thus N o r m a n and R a d d a 4 3 have pointed out that when nitrotolucnes are subjected to electrophilic substitution, the electron-donating methyl group acts contrary to the nitro group: the nitro group destabilizes intermediate 16 in the order meta < ortho < para, whereas the methyl group stabilizes it in the same order.

Directing Effects of the Nitro Group 59
5. Directing effect of a nitro group placed in the side chain or ring
The mete-directing effect of the nitro group placed i n a side chain is reduced. It decreases wi th an increase o f the distance between the nitro group and the aromatic r ing subjected to substitution. T h e corresponding data are collected i n Table 2.
TABLE 2. Effect of a nitro group in the side chain on electrophilic substitution (nitration).
m-Nitro derivative,
Compound % yield Ref
Nitrobenzene 93 44 Phenylnitromethane 67
48 45 Phenyl-co-nitroethane 13 44 co-Nitrostyrene 2 44, 46
T h e low yie ld of meta substitution i n ro-nitrostyrene shows that the nitro group on the v iny l group, which is known to be ortho-para directing, has practically no directing effect i n electrophilic substitutions.
The influence of the side-chain nitro group upon meta substitution (e.g., i n nitration) can also be altered by other side-chain substituents, as indicated i n structures 22-244 6.
N 0 2 N O , N 0 2
(22) (23) (24)
67% 29% 84%
A n interesting problem of the directing effect of o-, m-, and />-nitrophenyl groups upon the electrophilic substitution has been investigated by M i z u n o and S i m a m u r a 4 7 . They examined the nitration of mononitrodiphenyl with nitric acid and acetic anhydride at 0° and found that the nitrophenyl substituent is deactivating but
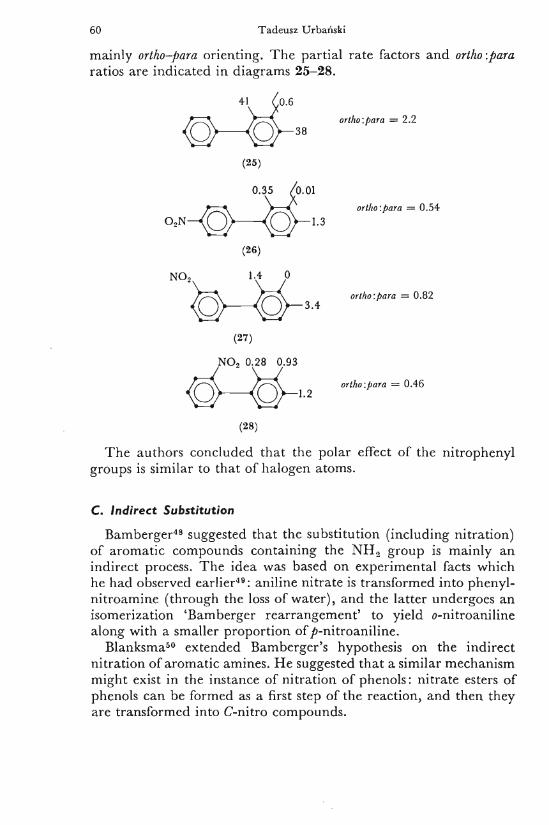
60 Tadeusz Urbański
mainly ortho-para orienting. T h e part ial rate factors and ortiw.para ratios are indicated i n diagrams 25-28.
ortho:para = 2.2
ortiw.para — 0.54
ortho-.para — 0.82
(27)
ortho-.para = 0.46
(28)
T h e authors concluded that the polar effect of the nitrophenyl groups is similar to that of halogen atoms.
C. Indirect Substitution
Bamberger 4 8 suggested that the substitution ( including nitration) of aromatic compounds containing the N H , group is mainly an indirect process. T h e idea was based on experimental facts wh ich he had observed ear l ie r 4 9 : aniline nitrate is transformed into phcnyl -nitroamine (through the loss of water), and the latter undergoes an isomcrization 'Bamberger rearrangement' to yie ld o-nitroaniline along with a smaller proportion of/>-nitroaniline.
B l a n k s m a 5 0 extended Bamberger's hypothesis on the indirect nitration of aromatic amines. H e suggested that a similar mechanism might exist i n the instance of nitrat ion of phenols: nitrate esters o f phenols can be formed as a first step of the reaction, and then they are transformed into C-nitro compounds.

Directing Effects of the Nitro Group Gl
Further experiments on Bamberger's rearrangement were carried out by O r t o n and coworkers5 1 . They came to the conclusion that the rearrangement is an acid-catalyzed reaction.
Ho l l eman and coworkers 5 2 attempted to test Bamberger's hypothesis by comparing the orientation when aniline was nitrated (1) by a direct substitution and (2) by a rearrangement.
T h e y found that the orientation through a rearrangement may differ considerably from the orientation through direct substitution. Thus the action of sulfuric acid upon an i l in ium nitrate yielded mainly p- and те-nitroaniline wi th a very small proport ion of the ortho derivative, whereas the rearrangement of phenylnitroamine yielded (as i n Bamberger's experiments) o-nitroaniline as the main product.
M o r e recently a number of papers were published on Bamberger's rearrangement.
Thus Hughes and Jones 5 3 came to the following conclusions : ( 1 ) the acid-catalyzed rearrangement of phenylnitromethane and
the nitration of aniline, under comparable conditions, involve two very different types of orientations ;
(2) nitr ic acid or any other nitrat ing agent are not intermediates o f any importance i n the Bamberger rearrangement and ;
(3) the acid-catalyzed rearrangement i n polar solvents is essential ly intramolecular.
T h e rearrangement is slow, and of first order 5 4 . Isotopic t e s t s 5 5 - 5 7
seem to confirm an intramolecular mechanism. For no ionic or radical fission was noticed which would be followed by intermolecular recombination of the counterfragments. However , more recently, split t ing off of nitrite ions from nitramines was noticed by Whi te and coworkers 5 8 and confirmed by Banthorpe, et Й / . 5 6 5 9
W h i t e 5 8 used the isotope di lu t ion method. H e found that N-nit ro- /Y-methylani l ine- 1 4 C in 0 . 1 7 V H C 1 at 40° produced: 5 2 . 1 % o-nitro-A^methylaniline, 30.9 % />-nitro-/V-methylaniline, 9 . 9 % N-methylanil ine, and no ?n-nitro-/V-methylaniline.
Banthorpe, et я / . 5 6 , 5 9 , found much evidence against a 7r-complex and a radical cage mechanism, and explained the rearrangement as shown i n equation 3. They also suggested that C-nitrites can be
+ +
(3)
(29) (30) (31)
formed as intermediates of the rearrangement.
62 Tadeusz Urbański
Banthorpe and T h o m a s 6 0 also found that iV-methyl-vV-nitro-l-naphthylamine, in solvents such as toluene, rapidly rearranges to form 2- and 4-nitro isomers on heating to 100°, and also on exposure to ultraviolet i r radiat ion at room temperature. T h e reaction appears to be more complex than the acid-catalyzed process, but no evidence was found for a mechanism involving homolytic or heterolytic fission. Consequently, as before, an intramolecular migration is suggested as i n the acid-catalyzed rearrangement.
The Bamberger rearrangement was found to be responsible for the migrat ion of the nitro group in C-nitro derivatives of aromatic amines described by Pausacker and Scroggie 6 1 . They found that heating of 2,3-dinitroacetanilide with sulfuric acid yielded 2,5-dinitroanil ine (46 % ) , 3,4-dinitroaniline (23 % ) , and a small quantity of 2,3-dinitroaniline (5%) .
It was originally suggested that the nitration reaction may be reversible. However, more recent studies of these workers 6 2 showed that the mechanism of the reaction involves a reversed Bamberger rearrangement and the mechanism can be expressed as shown i n equation 4.
/ N 0 2
N N H R N H R
(33) (34) N O ,
(36)
R = H or C O C H 3
It was found by the same authors that heating 2,3-dinitrophenol with sulfuric acid leads to partial isomerization to 2,5-dinitrophenol. Th is probably could be explained in terms of the B l a n k s m a 5 0
hypothesis, i.e., through the formation of an intermediate nitrate ester of phenol (38) (equation 5).

Directing Effects of the Nitro Group 63
Other dinitro compounds, v iz . , those substituted i n the 2,5 and 3,4 positions do not undergo such rearrangement. Th is fact is evidence that only that group can migrate which is subjected to steric hindrance, i.e., the nitro group i n ortho position to the adjacent groups.
O H O H
Й)
(38) (39)
The reversibility of C-nitrat ion seems to be l imi ted to the above-mentioned cases. In general the reversibility is possible when a group such as N H R or O H is present, which can form an intermediate wi th the mobile nitro group.
T h e mobi l i ty of a те-nitro group i n nitro derivatives of toluene was recently verified by U r b a ń s k i and Ost rowski 6 3 . These workers kept solutions of various nitro derivatives of toluene in concentrated sulfuric acid at 9 0 - 9 5 ° for ca. 60 hours. o-Nitrotolucne (41), m-nitrotoluenc (42), /;-nitrotoluene (43), 2,4,6-trinitrotoluene (44), and 2,4,5-trinitrotoluenc (45) were examined.
N o change was found i n the boi l ing points of 41 and 42, and i n the melt ing points of 43-45. It was, however, found that solutions containing 42 or 45 eventually produced a slight blue color wi th diphenylamine.
However, G o r e 6 4 reported that on heating 9-nitroanthracene i n a mixture of sulfuric and trichloroacetic acids at 6 5 - 9 5 ° for 25 minutes 'the odor of nitrous fumes was noticeable.' Work-up of the reaction mixture gave nitr ic acid (81 %) and anthraquinone (21 % ) , the latter probably formed by oxidative action of nitr ic acid. T h e hydrolysis of the nitro group in 9-nitroanthracene on acid treatment is perhaps not so surprising i f one considers the high reactivity o f this pos i t ion 3 6 .

64 Tadeusz U r b a ń s k i
II FREE-RADICAL SUBSTITUTION
A. Directing Effect of the Nitro Group
W h e n a 'meta-directing group' such as the nitro group is present i n the aromatic r ing and the r ing is attacked by a free radical , the homolytic substitution does not occur i n the meta but i n ortho and para positions, i.e., i n a way similar to nucleophil ic substitutions.
F r o m theoretical considerations W h e l a n d 4 has pointed out that any radical reagent should attack preferentially the ortho and para positions.
D a t a for the energy distribution i n nitrobenzene when subjected to electrophilic and radical substitutions are shown i n structures 46 and 47.
N O , N O
electrophilic radical substitution substitution
(46) (47)
T h e relative reaction rates for both, electrophilic and radical substitutions as calculated from atom localizat ion energies at 18° and 80° , respectively4, are given i n structures 48 and 49.
N O , N O ,
0.454
electrophilic radical substitution substitution
(48) (49)
T h e differences between the directing effects in the two types o f substitution can be clearly seen (at least qualitatively) although the experimental values may be somewhat different.
W h e l a n d 6 5 also drew attention to the possible formation of quinonoid-type intermediates 50-52. Th is view was later emphasized B y Weiss and coworkers 6 6 . T h e y concluded from hydroxylat ion experiments of nitrobenzene under free-radical conditions {vide infra) that the influence o f the nitro group is due (1) to the greater
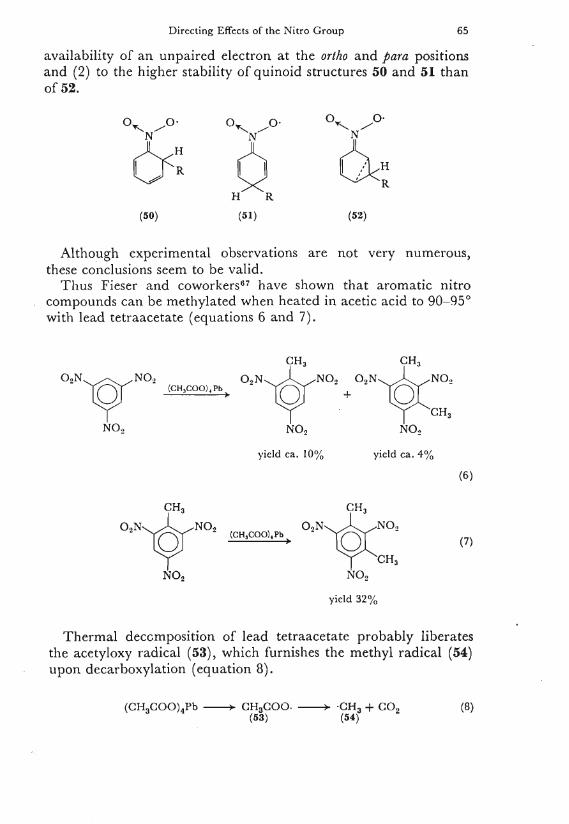
Directing Effects of the Nitro Group 65
availability of an unpaired electron at the ortho and para positions and (2) to the higher stability of quinoid structures 50 and 51 than of 52.
< 4 / ° -N
(50)
N
H R
(51)
О о-N
R
(52)
Although experimental observations are not very numerous, these conclusions seem to be valid.
Thus Fieser and coworkers6 7 have shown that aromatic nitro compounds can be methylated when heated in acetic acid to 9 0 - 9 5 ° with lead tetraacetate (equations 6 and 7).
C H ,
O j N ^ ^ \ „ N 0 2 0 2 N (СН,СОО)4РЬ ¥
N O ,
C H
(6)
0 2 N N O , (CH,COO),Pb
0 „ N
N O ,
(7)
T h e r m a l decomposition of lead tetraacetate probably liberates the acetyloxy radical (53), which furnishes the methyl radical (54) upon decarboxylation (equation 8).
(GH,GOO) 4 Pb > C H 3 G O C > C H 3 + C O , (8) (53) (54)
66 Tadeusz Urbański
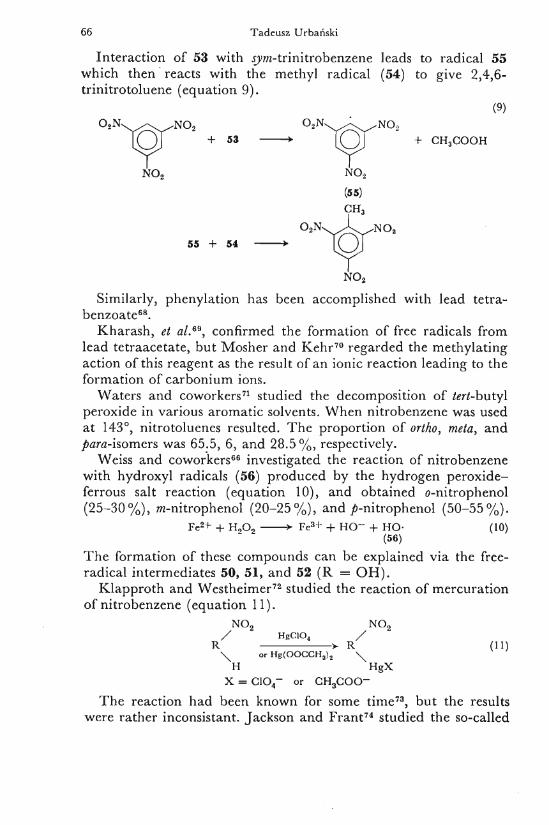
Interaction of 53 with yym-trinitrobenzene leads to radical 55 which then reacts with the methyl radical (54) to give 2,4,6-trinitrotoluene (equation 9) .
(9)
[Cjj + 53 • iÇjj - c:ii : ,C()<)ii
55 + 54 N O ,
Similarly, phenylation has been accomplished with lead tetra-benzoate6 8 .
Kharash , et a/ . 6 9 , confirmed the formation of free radicals from lead tetraacetate, but Mosher and K e h r 7 0 regarded the methylating action of this reagent as the result of an ionic reaction leading to the formation of carbonium ions.
Waters and coworkers7 1 studied the decomposition of ter/-butyl peroxide in various aromatic solvents. W h e n nitrobenzene was used at 143°, nitrotoluenes resulted. T h e proportion of ortho, meta, and />ara-isomers was 65.5, 6, and 28.5 % , respectively.
Weiss and coworkers6 6 investigated the reaction of nitrobenzene with hydroxyl radicals (56) produced by the hydrogen peroxide-ferrous salt reaction (equation 10), and obtained o-nitrophenol (25-30%), m-nitrophenol (20-25%), and />-nitrophenol (50-55%) .
Fe2+ + H „ 0 , > Fe3+ + H O " + H O - (10) (56)
T h e formation of these compounds can be explained via the free-radical intermediates 50, 51, and 52 (R = O H ) .
Klapproth and Westheimer 7 2 studied the reaction of mercuration of nitrobenzene (equation 11).
N 0 2 N 0 2
/ HgC10 4 / R >- R (11)
V or H i î ( O O C C H : j ) 2 V H H g X
X = C 1 0 4 - or сн 3 соо -T h e reaction had been known for some t ime 7 3 , but the results
were rather inconsistant. Jackson and F r a n t 7 4 studied the so-called

Directing Effects of the Nitro Group 67
'classical' mercuration which consists o f heating aromatic compounds wi th mercuric acetate i n a non-polar medium. 'Classical ' mercuration of nitrobenzene at 150° gave 53, 32, and 1 5 % of ortho, meta, and para isomers, respectively, suggesting that a homolytic substitution may be involved.
K l a p p r o t h and Westheimer have shown that many of the apparent anomalies in the orientation i n aromatic mercuration can be better understood i f attention is pa id to mercuration either wi th ionized mercuric salts i n strong acid solution or wi th largely undissociated mercuric acetate i n non-polar solvents.
W i t h mercuric perchlorate i n aqueous perchloric acid and part icularly at low temperature, the orientation is typical of electrophi l ic substitution (meta in the case of nitrobenzene). In the reaction wi th mercuric acetate, considered to be partly radical substitution, the orientation is largely ortho-para (see Table 3).
T A B L E 3. Grientation effects in the mercuration of nitrobenzene72.
Isomer, %
Exptl conditions ortho + para meta
Hg(C10 4 ) 2 in 60% H C 1 0 4 at 25° 11 89 Hg(G10 4 ) 2 in 40% H C 1 0 4 at 95° 37 63 H g ( O O C G H 3 ) 2 in nitrobenzene at 95° 52 48 H g ( O O C C H 3 ) 2 in nitrobenzene at 150° 57 43
Similar results were obtained by Oga ta and T s u c h i d a 7 5 . Thus , the proportions of o-, p-, and ти-mercurated nitrobenzene were up to 37.5, 6.5, and 57.0 % , respectively, when mercuration was carried out by mercuric nitrate in nitr ic acid at 90° .
The large proportion of wz^a-substitution product is probably due to the lack of a sharp demarcation line between the electrophilic and the radical substitution i n the discussed reactions.
A part ia l radical substitution may be responsible for the nitrat ion of nitrobenzene to dinitrobenzencs by nitr ic acid i n the presence o f mercuric oxide, as reported by Ogata and T s u c h i d a 7 6 . T h e y found as much as 26 % 0- and only 24 % m-dinitrobenzenes. These authors d id not report the formation of the para isomer.
H e y and coworkers 7 7 - ' 8 2 studied the arylat ion of nitrobenzene through the action of various sources of the aryl radicals / > - R C 6 H 4 - , generated from such sources as diazotates, j ( > - R C 6 H 4 N 2 O N a , nitroso-acylarylamincs, / ; - R C 6 H 4 N ( N O ) C O C H 3 , and acyl peroxides, (p-R C e H 4 C 0 2 ) 2 .
(iii Tadeusz Urbański
The average substitution i n the meta position for R = Br and C H 3 was only 12.1 and 8.6%, respectively, and was essentially independent of the source of the aryl radical .
The phenylation of nitrobenzene gave the figures collected i n Tab le 4.
TABLE 4. Substitution of nitrobenzene with phenyl radical.
Nitrophenyls, % Source of phenyl
radical orllio meta para
Sodium benzenediazoate 54 ± 4 9 ± 2 37 ± 4.4 Benzoyl peroxide 59.5 ± 4 8.5 ± 2 32 ± 4
The high proportion of ortho and para substituents could be explained i n terms of quinonoid structures 50 and 51. It should, however, be born in mind that an aromatic radical of the type X C 6 H 4 - may acquire a polar character owing to the electron-attracting or electron-repelling properties of the substituent X .
TAULE 5. Arylation of benzotrichloride (80 ) M .
Isomer, %
Radical ortho meta para
G 6 H 5 ' 12 49 39 / - - N 0 2 C 6 H 4 - 0 73 27
Thus the radical / > - N 0 2 C e H 4 - , i f considered as somewhat electrophi l ic i n character, might be expected to react most readily at nucleophil ic sites. O n the other hand, the jfr-tolyl radical should be considered as somewhat nucleophilic in character.
T h e electrophilic character of the />-nitrophenyl radical was demonstrated by H e y , el al.7S, Dann lcy and Sternfeld 8 3 , and more recently by Saunders 8 4 (Table 5).
Chang , Hey , and W i l l i a m s 8 0 studied the /;-nitropbenylation of chloro- and bromobenzencs, and compared the results wi th the phenylation of chloro-, bromo-, and nitrobenzencs (Table 6). T h e products were analyzed by infrared spectroscopy and isotope d i lu t ion methods.
'The electrophilic character of the />-halogcnophenyl radicals is demonstrated by the increased proportion of the substitution at the meta position, i n agreement with the directing influence of the nitro group in electrophilic substitution.

Directing Effects of the Nitro Group 69
TABLE 6. />-Halogenophenylation of nitrobenzene ( 8 0 ° ) 8 0 .
Isomer, %
Compound Radical ortho meta para
G e H 5 N O „ G e H 5 " 62.5 9.8 27.7 G 6 H 6 N 0 2 * - C l C e H 4 - 59.0 13.8 27.2 C e H 5 N 0 2 ^ - B r C e H 4 - 57.7 13.2 29.1
B. Activating Effect of the Nitro Group
H e y and G r i e v e 8 5 found already i n 1934 that the nitro group activates the aromatic r ing toward homolytic substitution. For instance the competitive phenylation of toluene and nitrobenzene by phenyl radicals showed that the yie ld of nitrodiphenyls was about four times greater than the yield of methyldiphenyls. Th is is the result of the general character of free-radical reactions wh ich are free of the powerful electrostatic forces which dominate heterolytic reactions.
H e y and his c o w o r k e r s 7 7 , 7 9 ' 8 1 gave a quantitative analysis of the rate of homolytic attack on nitrobenzene in terms of part ia l rate factors8 6 . The rates of the substitution by the phenyl radical of nitrobenzene as compared with chlorobenzene, and the part ial rate factors are given i n Tables 7 and 8, respectively.
T A B L E 7. Rate ratios and isomer distributions for phenylation of nitrobenzenes and chlorobenzenes.
Isomer, % 7 7
Rate ratio -Compound X P h X / P h H 8 1 ortho meta para
Nitrobenzene N O , 2.94 58 10 32 Chlorobenzene CI 1.06 62 24 14
TABLE 8. Partial rate factors for phenylation of nitrobenzenes and chlorobenzenes81.
Partial rate factors F. Fm Fv
P h N 0 2 / P h H 5.5 0.86 4.9 PhCl/PhH 1.6 1.0 1.2
H e y 7 7 also calculated the part ial rate factors for homolytic substitution in nitrobenzene from data given by W h e l a n d 4 . H e obtained for P h N O 2 / P h H : F 0 = 2.25, Fn = 0.85, and Fv = 8.7.

70 Tadeusz U r b a ń s k i
There are, however, exceptions to this rule. Thus H e y and coworkers 8 2 found that the />-chlorophenyl radical attacks nitrobenzene less rapidly (rate ratio 0.53) than benzene (rate ratio 2.94) (Table 7). Moreover , o- and/>-nitrophenyl radicals are less reactive toward nitrobenzene than toward benzene. Also wi th these negatively substituted radicals the proportion of meta substitution i n nitrobenzene is increased, because of the electrophilic character of the substituent. In other words, the radicals wi th a nitro group (and possibly also wi th chlorine) acquire an electrophilic character.
Accord ing to Dannley and G i p p i n 8 8 the thermal decomposition of benzoyl peroxide i n a-nitronaphthaline (and also i n a-chloro-and a-bromonaphthalines) leads to monosubstitution by a benzoyl-oxy group i n the 2, 4, and 5 positions (equation 12). I n addit ion,
O C O C e H 5 O C O C 6 H 5
18% 10%
benzoic acid, carbon dioxide, benzene, and esters are also formed. T h e substituents have the following relative activating influence
toward attack of the benzoyloxy radical :
N O . , > Br > C l > H
19 2.0 1.2 1.0
O n the other hand, the attack of t r iphenylmethyl radical on aromatic substrates i n the presence of benzoyl peroxide indicates the following order of reactivity according to Benkcser and Schroeder 8 0 : C 6 H 5 O C H 3 > C 6 H 5 C 1 > C 6 H 5 H > C 6 H 6 C O O C H 3 > C 6 H 5 C F 3 > C G H 5 N 0 2 . Nitrobenzene failed to react i n this system, and the experimental results indicate the electrophilic nature of the t r iphenylmethyl radical .
A n excellent review of homolytic aromatic substitution has been given by W i l l i a m s 9 0 . It includes a description of some unpublished work.

Directing Effects of the Nitro Group 71
III. REFERENCES
1. G. K . Ingold, Chem. Rev., 15, 265 (1934). 2. A . Lapworth, Nature, 115, 625 (1925). 3. R. Robinson, Solway Reports, 1931, 434. 4. G . W. Wheland, J. Am. Chem. Soc, 64, 900 (1942). 5. L . Melander, Nature, 163, 599 (1949); Acta Chem. Scand., 3, 95 (1949). 6. A more detailed historical review is presented by: F. Henrich in Theorien der Organi-
schen Chemie, Vieweg and Sohn, Braunschweig, 1921 ; W. A . Waters in Physical Aspects of Organic Chemistry, 5th ed., Routledge and Kegan Paul, London, 1953; and P. H . Hermans in Introduction to Theoretical Organic Chemistry (Ed. R. E . Reeves), Elsevier Publishing Co., Amsterdam, 1954.
7. H . Hubner, Chem. Ber., 8, 873 (1875). 8. E . Noelting, Chem. Ber., 9, 1797 (1876). 9. H . E . Armstrong, J. Chem. Soc, 51, 258 (1887).
10. D. Vorlânder, Ann. Chem., 320, 122 (1902). 11. A. Crum Brown and J . Gibson, J. Chem. Soc, 61, 367 (1892). 12. A . F. Hollemann, Die direkte EinfUlmmg von Subslituenten in den Benzolkern, Veit and
Co., Leipzig, 1910. 13. A . F. Hollemann, Chem. Rev., 1, 187 (1925). 14. D. L . Hammiclc and W. S. Illingworth, J. Chem. Soc, 1930, 2358. 15. S. F. Mason, E . Race, and F. E . Pounder, J. Chem. Soc, 1935, 1673. 16. B. Flurscheim, J. Prakt. Chem., 66, 321 (1902); 71, 497 (1905); Chem. Ber., 39, 2015
(1906). 17. С. A . Coulson and G . S. Rushbrooke, Proc. Cambridge Phil. Soc, 36, 193 (1940). 18. H . S. Fry, Z. Phys. Chem., 76, 385, 396, 591 (1911) ; J. Am. Chem. Soc, 34, 664 (1912);
36, 248, 1035 (1914). 19. T . M . Lowry, J. Chem. Soc, 123, 826 (1923). 20. D. Vorlânder, Chem. Ber., 52, 263 (1919); 58, 1893 (1925). 21. W. M . Latimer and С. W. Porter, J. Am. Chem. Soc, 52, 206 (1930). 22. L . E . Sutton, Proc Roy. Soc, A133, 668 (1931). 23. 'Landolt-Bórnstein Zahlenwcrte und Funktionen aus Physik, Chemie, Astronomie,
Geophysik und Technik,' 6 Aufl., Band I, Teil 3, Springer Vcrlag, Berlin, 1951, pp. 460-463; О. A . Osipov, V . I. Minkin, and Y u . B. Kletnik, Spravochnikpo Dipolnym Momenlam (reference book on dipole moments). University Press, Rostov, 1961.
24. W. J . Svirbcly and L . C. Warner, ./. Am. Chem. Soc, 57, 655 (1935) : W. J . Svirbely, J. Am. Chem. Soc, 61, 2555 (1939).
25. H . Eyring and T . Ri , J. Chem. Phys., 8, 433 (1940). 26. A. Lapworth, J. Chem. Soc, 121, 416 (1922). 27. W. O . Kcrmack and R. Robinson, J. Chem. Soc, 121, 427 (1922); J . Allan, A . E .
Oxford, and R. Robinson, J. Chem. Soc, 1926, 401 ; R. Robinson, Two Lectures on an Outline of an Electrochemical (Electronic) Theory of the Course of Organic Reactions, Institute of Chemistry of Great Britain and Ireland, London, 1932; R. Robinson, J. Soc. of Dyers and Colourists, 50, 65 (1934).
28. A. Lapworth, J. Chem. Soc, 73, 445 (1898). 29. С. K . Ingold and E . H . Ingold, J. Chem. Soc, 1926, 1310. 30. С. K . Ingold, Structure and .Mechanism in Organic Chemistry, Cornell University Press,
Ithaca, N.Y. , 1953. 31. V . Meyer, Chem. Ber.. 20, 534, 2994 (1887); 21, 1295, 1306, 1331, 1334 (1888).

72 Tadeusz Urbański
32. R . O. C. Norman and R. Taylor, Electrophilic Substitution in Benzenoid Compounds, Elsevier Publishing Co., Amsterdam, 1965, p. 71.
33. L . P. Colpa, G. MacLean, and E . L . Mackor, Tetrahedron, 19, Suppl. 2, 65 (1963). 34. J.D.Roberts, Notes on Molecular Orbital Calculations, W. A . Benjamin, New York, 1961. 35. A . Streitwieser, Jr., Molecular Orbital Theory for Organic Chemistry, John Wiley and
Sons, New York, 1961. 36. K . Higasi, H . Baba, and A . Rembaum, Quantum Organic Chemistry, Interscience
Publishers, New York, 1965. 37. P. B. D. de la Mare, and J . H . Ridd, Aromatic Substitution, Butterworths Publication
Ltd., London, 1959, p. 82. 38. G. K . Ingold, Ann. Rept. Progr. Chem., 23, 140 (1926). 39. A . Lapworth and R. Robinson, Memoirs Proc. Manchester Lit. Phil. Soc, 72, 43 (1928). 40. J . D. Roberts and A . Streitwieser, Jr., J. Am. Chem. Soc, 74, 4723 (1952). 41. R . D. Brown, J. Am. Chem. Soc, 75, 4077 (1953). 42. E . Baciocchi and G. Illuminati, J. Am. Chem. Soc, 86, 2677 (1964). 43. R . O. G. Norman and G . K . Radda, J. Chem. Soc, 3610 (1961). 44. J . W. Baker, J. Chem. Soc, 1929, 2225; J . W. Baker and I. S. Wilson, J. Chem. Soc,
1927, 872. 45. T. Urbański, Compt. Rend., 206, 122 (1938); T. Urbański and Gedroyć, Rocz. Chem.,
18, 125 (1938). 46. J . W. Baker and G. K . Ingold, J. Chem. Soc, 1926, 2462; J . W. Baker, J. Chem. Soc,
1929, 2257. 47. Y . Mizuno and O . Simamura, J. Chem. Soc, 1958, 3875. 48. E . Bamberger, Chem. Ber., 27, 584 (1894); 28, 399 (1895); 30, 1248 (1897); E .
Bamberger and E . Hoff, Ann. Chem., 311, 91 (1900). 49. E . Bamberger, Chem. Ber., 26, 471, 485 (1893); 27, 359 (1894). 50. J . J . Blanksma, Rec Trav. Chim., 21, 281 (1902); 23, 202 (1904). 51. K . J . P. Orton, J. Chem. Soc, 81, 490, 806 (1902) ; K . J . P. Orton and A . E . Smith,
J. Chem. Soc, 87, 389 (1905); 91, 146 (1907); K . J . P. Orton and G. Pearson, J. Chem. Soc, 93, 725 (1908); A . E . Bradfield and K . J . P. Orton, J. Chem. Soc, 1929, 915.
52. A . F. Hollemann, J . G. Hartogs, and T. van der Linden, Chem. Ber., 44, 704 (1911). 53. E . D. Hughes and G . T. Jones, J. Chem. Soc, 1950, 2678. 54. E . D. Hughes, С. K . Ingold, and R. B. Pearson, J. Chem. Soc, 1958, 435. 55. S. Brownstein, G. A . Burton, and E. D. Hughes, J. Chem. Soc, 1958, 4354. 56. D . V . Banthorpe, E . D. Hughes, and D. L . H . Williams, J. Chem. Soc, 1964, 5349. 57. D. V . Banthorpe, J . A . Thomas, and D. L . H . Williams, J. Chem. Soc, 1965, 6135. 58. W. N . White, J . R . Klink, D. Lazdins, G. Hathaway, J . T. Golden, and H . S. White,
J. Am. Chem. Soc, 83, 2024 (1961); 86, 1517 (1964). 59. D . V . Banthorpe and J . A . Thomas, J. Chem. Soc, 1965, 7149. 60. D . V . Banthorpe and J . A . Thomas, J. Chem. Soc, 1965, 7158. 61. К . H . Pausacker and J . G . Scroggie, Chem. Ind. (London), 1954, 1290. 62. К . H . Pausacker and J . G . Scroggie, J. Chem. Soc, 1955, 1897. 63. T . Urbański and T. Ostrowski, according to T. Urbański, Chemistry and Technology
of Explosives, Vol . I, Pergamon Press, Oxford-PWN, Warszawa, 1964, p. 41. 64. P. H . Gore, J. Chem. Soc, 1957, 1437. 65. G . W. Wheland, The Theory of Resonance, John Wiley and Sons, New York, 1945. 66. H . Loebl, G . Stein, and J . Weiss, J. Chem. Soc, 1949, 2074. 67. L . F. Fieser, R. G. Klapp, and W. H . Daudt, J. Am. Chem. Soc, 64, 2052 (1942). 68. D . H . Hey, G. J . N . Stirling, and G . H . Williams, J. Chem. Soc, 1954, 2747.

Directing Effects of the Nitro Group 73
69. M . S. Kharash, H . N . Fricdlander, and W. H . Urry, J. Org. Chem., 16, 553 (1951). 70. W. A . Mosher and G. L . Kchr, J. Am. Chem. Soc, 75, 3172 (1953). 71. W. A . Waters, B. R. Cowley, and R. O . G. Norman, according to G . H . Williams,
Ref. 90. 72. W. J . Klapproth and F. H . Westheimer, J. Am. Chem. Soc, 72, 4461 (1950). 73. J . Jurgcns, Rec. Trav. Chim., 45, 61 (1926); S. Coffey, J. Chem. Soc, 1926, 3215. 74. G . R. Jackson and M . S. Frant, J. Am. Chem. Soc, 77, 5625 (1955). 75. Y. Ogata and M . Tsuchida, J. Org. Chem., 20, 1637 (1955). 76. Y . Ogata and M . Tsuchida, J. Org. Chem., 21, 1065 (1956). 77. D. H . Hey, J. Chem. Soc, 1952, 1974. 78. D . H . Hey, A . Nechvatal, and T . S. Robinson, J. Chem. Soc, 1951, 2892. 79. D . R. Augood, D . H . Hey, and G . H . Williams, J. Chem. Soc, 1952, 2094. 80. Chang Shih, D . H . Hey, and G . H . Williams, J. Chem. Soc, 1958, 1885. 81. D . H . Hey, S. Orman, and G . H . Williams, J. Chem. Soc, 1961, 565. 82. J . I. G . Gadogan, D. H . Hey, and G . H . Williams, J. Chem. Soc, 1955, 1425. 83. R. L . Dannley and M . Stcrnfeld, J. Am. Chem. Soc, 76, 4543 (1954). 84. F. C . Saunders, Ph.D. Thesis, London, 1958, according to G . H . Williams, Ref. 90. 85. W. S. M . Grieve and D. H . Hey, J. Chem. Soc, 1934, 1797. 86. G. K . Ingold, A . Lapworth, E . Rothstein, and D . Ward, J. Chem. Soc, 1931, 1959;
G. K . Ingold and M . S. Smith, J. Chem. Soc, 1938, 905; M . L . Bird and C . K . Ingold, J. Chem. Soc, 1938, 918.
87. D . H . Hey, H . N . Moulden, and G . H . Williams, J. Chem. Soc, 1960, 3769. 88. R. L . Dannley and M . Gippin, J. Am. Chem. Soc, 74, 332 (1952). 89. R. A. Benkcser and W. Schroeder, J. Am. Chem. Soc, 80, 3314 (1958). 90. G . W. Williams, Hemolytic Aromatic Substitution, International Monographs Vol. 4,
Pcrgamon Press, Oxford, 1960. 91. A . I. Titov, Voprosy Reaktsionnoi Sposobnosti i Orientatsii v Teorii Nitrovania
Aromaticheskikh Soedinenii po Ionno Kompleksnomu Tipu (Problems of reactivity and orientation in theory of nitration of aromatic compounds according to an ionic complex type) in Organicheskie Poluprodukty i Krasiteli (Organic intermediates and dyes), Vol . 2, ed. A . I. Korolev, p. 46, Goskhimizdat, Moskva, 1961.



![N-Nitroso-bis(2-acetoxypropyl)amine as a Further Pancreatic … · [CANCERRESEARCH36, 2877-2884,August1976] SUMMARY N-Nitroso-bis(2-acetoxypropyl)amine , a possible f3 me tabolite](https://img.pdfslide.us/doc/110x75/60cc9fbec22aeb481e38fedc/n-nitroso-bis2-acetoxypropylamine-as-a-further-pancreatic-cancerresearch36-2877-2884august1976.jpg)

























