Embed Size (px)
Citation preview

Eur. J. Biochem. 45,623-627 (1974)
The N-Terminal Amino- Acid Sequences of DNA Polymerase I from Escherichia coZi and of the Large and the Small Fragments Obtained by a Limited Proteolysis
Henning JACOBSEN Institut for Riokemisk Genetik, K~benhavns Universitet
Hans KLENOW and Kay OVEROAARD-HANSEN
Biokemisk Institut B, K~lbenhavns Universitet
(Received March 4, 1974)
The two fragmcnts obtained by proteolytic cleavage of DNA polymerase I from Escherichia coli have been isolated by chromatography on hydroxyapatite. The amino-acid composition and the N-terminal amino-acid sequences of the native enzyme and of the two fragments have been determined. The sequences for the native enzyme and the small fragment were identical while the large fragment had a different N-terminal sequence. This shows unambiguously that the small fragment is placed in the N-terminal end of the native enzyme.
DNA polymerase I from Escherichia coli consists of a single polypeptide chain with a molecular weight of 109000 [l]. By gel filtration the enzyme is eluted in a volume larger than that expected for a globular protein of that molecular weight. Because of this observation the DNA polymerase is considered as an enzyme with an unusual conformation [Z].
By limited proteolysis with subtilisin the native enzynie is split into two fragments [Z-41. Both fragments have enzyme activity and their molecular weights are about 75000 and 35000, respectively. The large fragment retains the polymerase and 3' + A ' exonuclease activity which is specific for single-stranded DNA, while the small fragment retains only the 5' + 3' exonuclease activity of the native DNA polymerase. The latter activity is spe- cific for double-stranded DNA. On basis of this the DNA polymerase may be regarded as a bifunctional enzyme and from the available data we may surmise that it is folded into two compact tertiary structures connected with a more open hinge consisting of a peptide chain [ 5 ] .
Abbreviations. Dansyl, l-dimethylaminonaphthalene-5- sulphonyl. Abbreviations for polynnclootides follow CBN rules see Eur. J . Biochenz. 15, 206 (1970).
Enzymes (CBN Recommandations 1972). DNA poly- merase I (EC 2.7.7.7), subtilisin (EC 3.4.21.14); DNase (EC 3.1.4.5); catalase (EC 1.11.1.6); chymotrypsinogen (EC 3.4.21.1).
In the present work the amino-acid compositions of the intact DNA polymerase and of the two frag- ments are reported. Determination of the N-terminal amino-acid sequences have been carried out by the dansyl-Edman technique. The N-terminal amino - acid sequence of the small fragment is found to be identical with that of the native enzyme while the N-terminal sequence of the large fragment is different. This suggests that the small fragment constitutes the N-terminal part of the native enzyme.
MATERIALS AND METHODS The DNA polymerase I was purified as reported
previously [5 ] . Cleavage of native DNA polymerase I by subtilisin and removal of DNA present in the reaction mixture was performed as already described [5 ] . The separation of the two fragments formed by the proteolytic reaction was obtained by chromatog- raphy on a column of hydroxyapatite (Bio-Rad Laboratories). The fragments were adsorbed to the column a t low phosphate buffer concentration (0.030 M and pH 6.3) and they were successively eluted with a concentration gradient of the same buffer.
DNA polymerase activity was determined as pre- viously described [5] with activated calf thymus DNA as primer and template. The exonuclease
Eur. J . Biochem. 45 (1974)

624 Structural Investigations on DNA IPolymerase I from Escherichia coli
activities were measured with the procedure previous- ly described [5]. The 3' + 5' exonuclease activity was determined in 80 mM glycine buffer pH 9.0 with 3H-labelled (dT),,, as substrate. The 5' + 3' cxonuclease activity was determined in 16 mM phosphate buffer pH 7.4 with DNase-treated 3H- labelled poly[d(A-T) * d(A-T)] as substrate. All mea- surements of enzyme activities were performed a t 37 "C.
Dodecylsulphate-polyacrylamide gel electropho- resis was performed according to Weber and Osborn [5 ,6] to test the purity and to estimate the molecular weight of the fragments. Staining was performed with Coomassie brilliant blue. In another series of experi- ments the proteins dissolved in 0.1 M NaHCO, pH 9.8 with 1 sodium dodecylsulphate were made fluorescent by labelling with dansyl chloride a t room temperature followed by reduction with 2-mercapto- ethanol before electrophoresis [7]. The dansyl hydroxide formed from the surplus reagent was not removed, but advantage was taken to use this com- pound as an internal marker for calculation of relative electrophoretic mobilities. The gels were made of 100/, acrylamide and 0.34O/, N,N'-methy- lenebisacrylamide and polymerized with 0.030/, N,N,N',N'-tetramethylethylenediamine and 0.05O/, ammonium persulphate. The buffer used was 0.1 M NH,HCO, containing 0.1 O/, sodium dodecylsulphate. Electrophoresis was performed at room temperature for 15 h a t 1.0 mA per gel (5x100 mm). Dansyl protein (0.2 pg) was applied to the gels for molecular weight determinations. To test the homogeneity 2 pg was used in each experiment. For determination of molecular weight the electrophoretic system was calibrated by use of DNA polymerase I, catalase, bovine serum albumin, ovalbumin and chymo- trypsinogen.
Amino-acid analyses were carried out on a Dur- rum analyzer, model D-500. Aliquots containing 0.1 nmol protein were hydrolyzed in 50 pl redistilled HCI a t 110 "C in vacuum-sealed tubes. The DNA polymerase and the large fragment were hydrolyzed for 24 and 72 h, while the small fragment was hydro- lyzed in duplicate for 24, 48 and 72 h.
The N-terminal amino acids were identified by labelling with dansyl chloride [8]. After hydrolysis the dansyl amino acids were identified by thin-layer chromatography on polyamide sheets (5 x 5 cm) [9,10]. The high content of dansyl-tyrosine in the unfractionated hydrolysate made the identification of the N-terminal amino acid by thin-layer chromatog- raphy unreliable. This prompted us to reduce the amount of dansyl-tyrosine before thin-layer chroma- tography by gel filtration on Sephadex G-25 (P. M. Andersen, personal communication). The dried hy- drolysate was redissolved in 5 pl 50°/, pyridine and applied onto the gel column ( 2 x 60 mm). The elution took place with 0.05 M ammonia a t a rate of 4 to 5
drops per minute and migration of the dansylated amino acids was visually observed under ultraviolet light. During gel filtratiion the original yellow fluores- cent sample was separated into a faster-moving greenish-yellow part and a slower-moving orange part. The two parts with different fluorescence were collect- ed separately, each in one to two fractions. All fluorescent fractions wax dried, redissolved each in 10 pl ethanol (93O/,, w/w) and applied onto poly- amide sheets. The dansylated amino acids except dansyl-tyrosine, bis(dansyl)tyrosine, and bis(dansy1)- lysine, may be found in the greenish-yellow fluores- cent fraction.
The technique applied to sequential Edman degradation was a modification of that described by Weiner et al. [8] using dodecylsulphate as detergent in an inorganic solvent An attempt to sequence the proteins following directly the procedure of Weiner et al. was not successful because the proteins were not solubilized after treatment with trifluoroacetic acid. The proteins formed gelly clumps which could not be divided into small particles by trituration as proposed. By combining the technique of Weiner et al. with the method described by Gray and Smith [12] the problem was overcome as follows. The protein solutions was divided into a series of aliquots, each containing 1 nmol protein. Several cycles of Edman degradation were performed on the aliquots. After each cycle one lube was removed from the series and the N-terminal amino acid was identified as described above.
RESULTS AND DISCUSSIONS By chromatography of the cleaved DNA polymer-
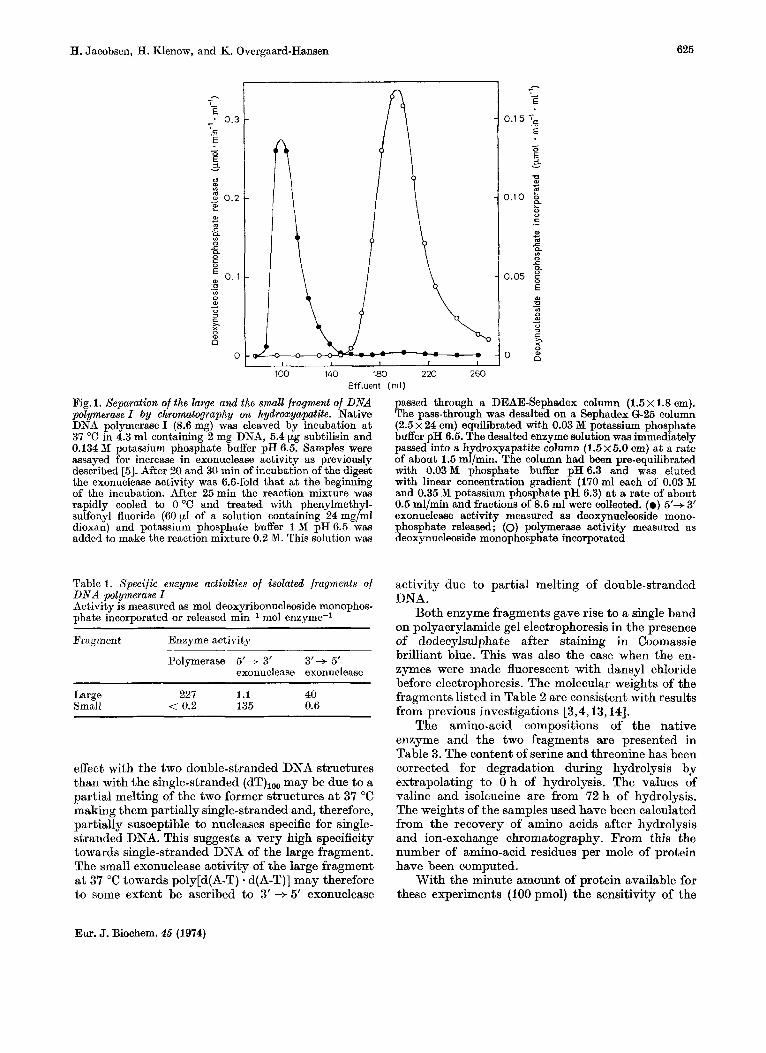
ase on hydroxyapatite the polymerase activity and the 5' --f 3' exonucleasle activity corresponding to the two fragments were (completely separated and the recovery of each activity was 60 -70°/, of that present a t the end of subtilisin treatment (Fig. 1). The specific activities of the two fragments are listed in Table 1. The figures show that t,he small fragment has high specific 5' + 3' exonuclease activity and that it is almost completely devoid of 3' --f 5' exonuclease activity [as measured with (dT),,, as substrate] and of polymerase activity. The large fragment has a high specific polymerase activity, a high 3' + 5' exo- nuclease activity and a very low 5' --f 3' exonuclease activity, as measured with poly[d(A-T) . d(A-T)] as substrate. The exonucllease activity of the large fragment has also been measured a t 37 "C with poly(dA) * oligo(dT) as substrate and this showed the same low activity as with poly[d(A-T) * d(A-T)]. When this exonuclease activity was measured a t 23 "C with (dT),,,, poly[d(A-T) * d(A-T)] or polg- (dA) - (dT),,, as substrates i t was found to be lower than that a t 37 "C by factors of3,10, and 40, respec- tively. The much more pronounced temperature
Eur. J. Biochem. 45 (1974)
H. Jacobsen, H. Klenow, and K. Overgaard-Hansen 625
I t I
I I I I I
100 140 180 220 260 Eff[uent ( m l )
- - - E
0.1 5 yc ._ E - 0
5
0.10 s .-, V c
5 0
c
II)
c
._
... 4
8 0.05 5 E
L
a, -0
m 0
0 3 c
.- - =.
o g
Fig. 1. Separation of the large and the small fragment of DNA polymerase I by chromatography on hydroxyapatite. Native DNA polymerase I (8.6 mg) was cleaved by incubation a t 37 "C in 4.3 ml containing 2 mg DNA, 5.4 pg subtilisin and 0.134 Df potassium phosphate buffer pH 6.5. Samples were assayed for increase in exonuclease activity as previously described 153. After 20 and 30 min of incubation of the digest the exonuclease activity was 6.6-fold that a t the beginning of the incubation. After 25min the reaction mixture was rapidly cooled to 0 "C and treated with phenylmethyl- sulfonyl fluoride (60 yl of a solution containing 24 mg/ml dioxan) and potassium phosphate buffer 1 M pH 6.5 was added t,o make the reaction mixture 0.2 M. This solution was
passed through a DEAE-Sephadex column (1.5 x 1.8 em). The pass-through was desalted on a Sephadex 6-25 column (2.5 x 24 em) equilibrated with 0.03 M potassium phosphate buffer pH 6.5. The desalted enzyme solution was immediately passed into a hydroxyapatite column (1 .5~5.0 em) a t a rate of about 1.5 ml/min. The column had been pre-equilibrated with 0.03M phosphate buffer pH6.3 and was eluted with linear concentration gradient (170 ml each of 0.03 M and 0.35 M potassium phosphate pH 6.3) a t a rate of about 0.5 ml/min and fractions of 8.6 ml were collected. (0 ) 5'+ 3' exonuclease activity measured as deoxynucleoside mono- phosphate released; (0) polymerase activity measured as deoxynucleoside monophosphate incorporated
Table 1. Specijic enzyme activities of isolated fragments of activity due to partial melting of double-stranded DNA. DNA polymerase I
Activity is measured as mol deoxyribonucleoside monophos- phate incorporated or released min-l mol enzyme-1 Both enzyme fragments gave rise to a single band
on polyacrylamide gel electrophoresis in the presence Fragment Enzyme activity of dodecylsulphate after staining in Coomassie
Polymerase 5'+ 3' 3'+ 5' exonuclease exonuclease
Large 227 1.1 40 Small < 0.2 135 0.6
effect with the two double-stranded DNA structures than with the single-stranded (dT),,, may be due to a partial melting of the two former structures a t 37 "C making them partially single-stranded and, therefore, partially susceptible to nucleases specific for single- stranded DNA. This suggests a very high specificity towards single-stranded DNA of the large fragment, The small exonuclease activity of the large fragment a t 37 "C towards poly[d(A-T) - d(A-T)] may therefore to some extent be ascribed to 3' + 5' exonuclease
brilliant blue.-This was also the case when the en- zymes were made fluorescent with dansyl chloride before electrophoresis. The molecular weights of the fragments listed in Table 2 are consistent with results from previous investigations [3,4,13,14].
The amino-acid compositions of the native enzyme and the two fragments are presented in Table 3. The content of serine and threonine has been corrected for degradation during hydrolysis by extrapolating to 0 h of hydrolysis. The values of valine and isoleucine are from 72 h of hydrolysis. The weights of the samples used have been calculated from the recovery of amino acids after hydrolysis and ion-exchange chromatography. From this the number of amino-acid residues per mole of protein have been computed.
With the minute amount of protein available for these experiments (100 pmol) the sensitivity of the
Eur. J. Biochem. 45 (1974)
626 Structural Investigations on DNA Polymerase I from Escherichia coli
methods has been pressed to the limit. However, in consideration of this there is a reasonable good agreement between the sum of amino acids in the two fragments and the amino acids in the native enzyme. One cannot exclude the possibility that a small peptide in the hinge region is cut out by the digestion with subtilisin, but it appears under all circumstances to be rather small.
The amino-acid analysis of the DNA polymerase and of the large fragment has previously been publish- ed by Jovin et a2. [I] and Setlow et al. [13] (Table 3). Setlow et al. found that the relative contents of aspartic acid and proline in the large fragment were respectively higher and lower than in the native
Table 2. The molecular weights and N-terminal sequences of the DNA polymerase I (E. coli) and of the two fragments obtained after limited protwlys/s with subtilisin The molecular weight determinations were performed by dodecylsulphate-polyacrylamide electrophoresis on the pro- teins which were made fluorescent by labelling with dansyl chloride
Enzyme fragment Molecular N-terminal amino-acid weight sequences
DNA polymerase I 109000 Met-Val-Glx-Ile-Pro- (whole) Glx-Leu-Asx-
Small fragment 34500 Met-Val-Glx-Ile- Large fragment 71 000 Val-Ile-Met-a
valyl-isoleucine bond quantitatively. a 72 h of hydrolysis are necessary to split the dansyl-
enzyme. However, in our experiments we did not find those differences significant.
The results of sequencing are shown in Table .2 The first three N-terminal residues were determined for both the intact DNA polymerase and for the small fragment. They were in both cases found to be Met- Val-Glx. This demonstrates the small fragment to be placed N-terminally in the intact enzyme. For the intact enzyme the following four residues were also determined. The first three residues in the large fragment were Val-Ile-Met.
Methionine was observed as the predominant amino acid in the determination of the N-terminal amino acid of both the native polymerase and of the small fragment. However, valine which is the next amino acid in both enzymes was also noticed in min- ute amount. Likewise after each step of Edman degra- dation the following amino acid was seen as trace. Aspartic acid which WELS detected in minute amount together with leucine a,fter the sixth step of Edman degradation of the native enzyme is therefore likely to represent the eight residue in DNA polymerase. The same reasonings place isoleucine as the fourth amino acid in the small fragment. Jovin et al. [l] determined the methionine to be the predominant N-terminal by the cyanate and iluorodinitrobenzene method in the amount of about 0.6mol per mol protein. Whether valine was found among the cont- aminating amino acids was not specified.
Methionine is found most often as the N-terminal amino acid in proteins from E. coli [I51 consistent with
Table 3. Camparison of amino-acid composition of DNA polymerase I , large fragment and small fragment
Amino acid Small Large Sum of DNA poly- Large DNA poly- fragment fragment amino acids merase I fragment merase I ( M , = 35000) ( M , = 75000) in fragments ( M , = 109000) (Setlow (Jovin
et al. [13]) et al. [l])
residues/ residues/ 680 residues 109000 g
residues/molecule
protein Lysine 21.7 45 67 69 42 61 Histidine 5.6 17 23 23 16 19 Arginine 8.3 268 348 45 37 48 Asparagine/aspar tate 28.0 69 97 81 82 88 Threonine 20.6 33 54 52 34 53 Serine 9.1 27 36 42 26 40 Glutamine/glutamate 43.6 102 146 125 87 126 Proline 24.6 35 60 56 22 53 Glycine 24.5 40 65 65 41 63 Alanine 34.2 71 105 113 72 102 Valine 22.5 41 64 69 39 61 Methionine 10.2 16 26 27 15 24 Isoleucine 14.5 41 56 55 42 55 Leucine 39.0 74 113 110 75 112 Tyrosine 9.8 21 31 32 27 32 Phenylalanine 6.9 17 24 29 19 25
Total 326 675 1001 993 676 962
a Too low due to interfering ammonia.
Eur. J. Biochem. 45 (1974)

H. Jacobsen, H. Klenow, and K. Overgaard-Hansen 627
the initiation mechanism of peptide chain synthesis in which methionine takes part as a formly derivative [16]. After deformylation the methionine may be removed by an aminopeptidase specific for hydrolyz- ing N-terminal methionyl bonds [17,18]. Our results suggest that in our preparation of DNA polymerase the N-terminal methionine has been partly removed, possibly catalysed by this aminopeptidase. Whether removal of the methionine changes the enzyme activity is uncertain.
The large fragment showed no trace of other N-terminal amino acids than valine. Similarly after the two cycles of Edman degradation and dansyla- tion only a single amino acid was detected. This indicates the action of subtilisin on the peptide chain between the large and the small fragment to be rather specific.
The results presented in Table 2 strongly indicate the small fragment to be placed in the N-terminal end of the native enzyme. The results of experiments by Lehman and Chien with the polA mutant P 3478 have led to the same conclusion [19]. They found that this mutant which contains very low DNA polymerase I activity contained a nearly normal level of 5' +3' exonuclease activity. The latter activity was found to be associated with a protein identical with the small fragment formed by proteo- lytic cleavage of the native wild-type enzyme DNA polymerase I. Since the polA mutant is an amber mutant [ 191 a reasonable explanation for the presence of the small fragment would be that it was part of the amber peptide. This peptide may be converted to the small fragment by digestion by proteolytic E. coli enzymes.
The authors are much indebted to Statens Naturviden- skabelige Forskningsr&d (Denmark) for support. The expert assistance by Mrs I. Henningsen and Mr E. A. Geertsen is acknowledged.
REFERENCES 1. Jovin T. M., Englund, P. T. & Bertsch, L. L. (1969) J .
2. Klenow. H. & Henninesen. I. (1970) Proc. NatZ. Acad. Biol. Chem. 244, 2996-3008.
\ I
Sci. U. S. A . 65, l68"-175. 3. Brutlae. D., Atkinson. M. R.. Setlow, P. & K0rnberg.A.
(1969) Biochem. Biophys. Res. Cmhun. 37, 982-589. 4. Klenow, H. & Overgaard-Hansen, K. (1970) BEBS Lett.
5. Klenow, H., Overgaard-Hansen, H. & Patkar, S. A.
6. Weber, K. & Osborn, N. (1969) J . Biol. Chem,. 244,
7. Talbot, D. N. & Yphantis, D. A. (1971) $rial. Biochem.
6, 25-27.
(1971) Eur. J . Biochem. 22, 371-381.
4406 - 4412.
44. 246-253 8. Weiner, A. M., Platt, T. & Weber, K. (1972) J . Biol.
9. Woods. K. R. & Wane. K. T. (1967) Biochim. BioDhhvs. Chem. 247, 3242-3251.
% , . " Act& 133, 369-370:'
10. Hartlev, B. S. (1970) Biochem. J . 119, 805-822. 11. Referekce deleted. '
12. Gray, W. R. & Smith, J. F. (1970) Anal. Biochem. 33,
13. Setlow, P., Brutlag, D. & Kornberg, A. (1972) J . Biol.
14. Setlow, P. & Kornberg, A. (1972) J . Biol. Chem. 247,
15. Waller, J.-P. (1963) J . Mol. Biol. 7 , 483-496. 16. Clark, B. C. F. & Marcker, K. A. (1966) J . Mol. Biol. 17,
17. Matheson,A.T. & Dich,A. J. (1970) FEBS Lett. 6,
18. Adams, J. M. (1968) J . MoZ. BWZ. 33, 571-589. 19. Lehman, J. R. & Chien, J. R. (1973) in D N A Synthesis
in vitro (Wells, R. D. & Inman, R., eds) p. 3, Uni- versity Park Press, Baltimore.
36-42.
Chem. 247, 224-231.
232-240.
394-405.
235 - 237.
H. Jacobsen, Genetisk Institut B, Kebenhavns Universitet, 0ster Farimagsgade 2A, DK-1353 Kebenhavn K, Denmark
H. Klenow and K. Overgaard-Hansen, Biokemisk Institut B, Kebenhavns Universitet, Juliane Maries Vej 30, DK-2100 Kebenhavn 0, Denmark
Correction
Volume 43, No. 3
The occurrence of multiple activities in the high-molecular-weight DNA polymerase fraction of mammalian tissues. A preliminary study of some of their properties, by A. M. Holmes, I. P. Hesslewood, and I. R. Johnston:
Page 492, Table 2, column 4, last-but-one line : for 7.6 f 1 (5 ) , read 7.6 f 0.1 (5).
Eur. J. Biochem. 45 (1974)





























