Embed Size (px)
Citation preview

A R T I C L E
Journ
al of
Materials
Ch
emistry
ww
w.rsc.o
rg/m
aterials
The molecular structures and electrochemical response of
‘‘twisted’’ tetra(aryl)benzidenes
Paul J. Low,*a Michael A. J. Paterson,a Andres E. Goeta,a Dmitry S. Yufit,a
Judith A. K. Howard,a Julian C. Cherryman,b Daniel R. Tackleyb and Bev Brownb
aDepartment of Chemistry, University of Durham, South Road, Durham, UK DH1 3LE.
E-mail: [email protected] Ltd., PO Box 42, Hexagon House, Blackley, Manchester, UK M9 8ZS
Received 30th March 2004, Accepted 28th May 2004
First published as an Advance Article on the web 28th June 2004
The compounds N,N,N’,N’-tetra(4-methylphenyl)-(1,1’-biphenyl)-4,4’-diamine (4), N,N,N’,N’-tetra(4-
methylphenyl)-(2,2’-dimethyl)-(1,1’-biphenyl)-4,4’-diamine (5a) and N,N,N’,N’-tetra(4-methylphenyl)-
(2,2’,6,6’-tetramethyl)-(1,1’-biphenyl)-4,4’-diamine (6a) undergo two reversible one electron oxidations.
The first oxidation potential increases in the order 4 v 5a v 6a, while the separation between the first
and second oxidation decreases in the reverse order 4 (0.30 V) w 5a (0.16 V) w 6a (0.00 V), reflecting
the decreasing thermodynamic stability of the radical cations [41] w [51] w [6a1]. Electronic spectroscopy
and spectroelectrochemistry (UV-Vis-NIR) confirm expectations, and the introduction of methyl groups
at the 2,2’ and 6,6’ positions of the 1,1’-biphenyl moiety electronically decouple the arylamine moieties.
In contrast, N,N,N’,N’-tetra(phenyl)-(2,2’-dimethyl)-(1,1’-biphenyl)-4,4’-diamine (5b) and N,N,N’,N’-tetra(phenyl)-(2,2’,6,6’-tetramethyl)-(1,1’-biphenyl)-4,4’-diamine (6b) give much less kinetically stable
radical cations upon oxidation, which oligomerise/polymerise through the 4 positions of the
N-phenyl groups via a step-growth process. The molecular and crystal structures of 4 and 6b are
also reported.
Introduction
Tetra(aryl)benzidenes, such as TPD (1) represent a basicstructural motif often found in hole transport materials whichare used in a wide variety of applications, ranging fromphotocopiers to electroluminescent display devices.1 Thesesmall molecular materials are often incorporated into multi-layer devices as dispersions in an inert polymer matrix in orderto reduce problems associated with crystallisation duringdevice fabrication or device operation. Alternatively, linearor star shaped oligomers, dendrimers and polymeric polyaryl-amine derivatives may be used, as these relatively highmolecular weight materials are more resistant to crystallisationthan smaller molecular analogues, and have better electronicproperties which may be attributed to the modified interchaininteractions,2 yet typically remain sufficiently soluble for readyprocessing. Hence, there is considerable contemporary interestnot only in the synthesis of these polyarylamine based materialsbut also in the various molecular structure–property relation-ships which govern the oxidation (or ionisation) potentials ofthe resulting materials and the intermolecular interactionswhich occur in the solid state.
Polymeric polyarylamines are usually prepared from Ull-mann-style coupling reactions,3 or nickel catalysed amine–arylhalide cross coupling reactions.4 In either case, any metalcatalyst residue must be removed from the product polymerbefore it can be employed in electronics based applications. Theoxidative dimerisation of triphenylamine (2) has long beenknown,5 and Dunsch et al. have reported the polymerisation oftriphenylamine by anodic oxidation at high current densities innon-polar solvents.6 In addition, oxidation of a small numberof diphenylamines has been shown to afford oligomeric mate-rials in low yield,7 More recently, Lambert and Noll, as well asLeung’s team, have demonstrated that bis-triarylamines spannedby a variety of conjugated bridges can be polymerised electro-chemically to give conjugated poly{tetra(aryl)benzidene}D
OI:
10
.10
39
/b4
04
73
1a
2 5 1 6 J . M a t e r . C h e m . , 2 0 0 4 , 1 4 , 2 5 1 6 – 2 5 2 3 T h i s j o u r n a l i s � T h e R o y a l S o c i e t y o f C h e m i s t r y 2 0 0 4
Publ
ishe
d on
28
June
200
4. D
ownl
oade
d by
Uni
vers
ity o
f W
este
rn O
ntar
io o
n 28
/10/
2014
11:
41:3
8.
View Article Online / Journal Homepage / Table of Contents for this issue

polymers.8,9 These results have prompted us to disclose ourown work on the structural, electrochemical and spectroscopicproperties of tetra(aryl)benzidenes in which the conjugationbetween the two triarylamine moieties is disrupted throughthe introduction of methyl groups into the common biphenylmoiety.
Results and discussion
The distribution of the SOMO of triphenylamine radical cation[NPh3]1? ([2]1?), calculated using the BPW91 method10,11 inconjunction with the 6–31G(d,p) basis set in Gaussian 98,12,13
and visualised using the Molekel program,14 suggests that anappreciable amount of the unpaired electron character resideson the aromatic ring carbons para to the nitrogen centre(Fig. 1), and is clearly the underlying electronic basis for theobserved chemical reactivity of this species.5 In contrast, theSOMO of radical cations derived from tetra(aryl)benzidenesare delocalised over the benzidene-like central portion of themolecule, which lends significantly greater chemical stability tothese species.15,16 This orbital description demands a relativelyplanar structure to the benzidene core in these radical cations,and indeed this is observed in the solid state structure of[3]SbCl6.16 Given the wide range of molecular conformationslikely to be found in an amorphous film of device-grade TPD-style hole transporting material, we were interested in probing
more thoroughly the influence of molecular conformation onthe properties of these systems.
Polyarylamines and derivatives are most often prepared viaUllmann condensation protocols, which, in the most generalform, involves copper catalysed condensation of an unacti-vated aryl halide with a primary or secondary arylamine in thepresence of some additional base.3 The reference compound 4was readily prepared in good yield from the Ullmann-stylecondensation of 4,4’-diiodobenzene and di(p-tolyl)amine in thepresence of K2CO3. Commercially available 4,4’-diamino-2,2’-dimethylbiphenyl was diazotised, and converted to the corres-ponding diiodide by treatment with aqueous KI3, followed bycopper catalysed condensation with ditolylamine and diphenyl-amine to give 5a and 5b in good yield. A similar sequencebeginning with 4,4’-diamino-2,2’,6,6’-tetramethylbiphenyl gave6a and 6b.
Despite the great interest in tetra(aryl)benzidenes and relatedsystems as hole transporting materials, there is remarkablylittle solid state data relating to the molecular structures ofthese compounds.16,17 Single crystals of 4 (in form of dichlor-omethane solvate 1 : 1) and 6b suitable for X-ray diffractionwere obtained by slow evaporation of CH2Cl2 (4) or THF (6b)solutions. The molecular structure of 4 is illustrated in Fig. 2,and selected bond lengths and angles are summarised inTable 1. A C2-axis, which passes through the midpoint of thecentral biphenyl C–C single bond, relates the two halves ofthe molecule. There are no unusual bond lengths within themolecular structure, although it is worth noting that theshortest N–C bond length is associated with C(4) and reflectsthe predominant conjugation pathway in the molecule. Thebiphenyl moiety is characterised by a C(1)–C(1A) bond lengthof 1.480(2) A. Each nitrogen centre is approximately planar,
Fig. 1 A graphical representation of the DFT calculated Semi-Occupied Molecular Orbital (SOMO) of the triphenylamine radicalcation, 21.
Fig. 2 An ORTEP diagram (50%) illustrating the molecular structure of 4, and the atom labelling scheme. Hydrogen atoms have been omittedfor clarity.
Table 1 Selected bond lengths (A), bond angles and torsion angles (u)for compounds 4 and 6b
4 6b
N(1)–C(4) 1.410(1) 1.428(1)N(1)–C(7) 1.426(1) 1.411(2)N(1)–C(14) 1.429(1) 1.424(2)C(1)–C(2) 1.398(2) 1.404(2)C(2)–C(3) 1.391(2) 1.397(2)C(3)–C(4) 1.397(2) 1.390(2)C(4)–C(5) 1.401(2) 1.390(2)C(5)–C(6) 1.383(2) 1.395(2)C(6)–C(1) 1.405(2) 1.404(2)C(1)–C(1A) 1.480(2) 1.500(2)C(4)–N(1)–C(7) 119.52(9) 120.1(1)C(4)–N(1)–C(14) 120.03(9) 117.9(1)C(7)–N(1)–C(14) 116.51(9) 120.8(1)C(4)–N(1)–C(7)–C(12) 236.0(1) 35.6(2)C(4)–N(1)–C(14)–C(15) 268.2(1) 39.7(2)C(2)–C(1)–C(1A)–C(2A) 149.9(2) 298.3(2)
J . M a t e r . C h e m . , 2 0 0 4 , 1 4 , 2 5 1 6 – 2 5 2 3 2 5 1 7
Publ
ishe
d on
28
June
200
4. D
ownl
oade
d by
Uni
vers
ity o
f W
este
rn O
ntar
io o
n 28
/10/
2014
11:
41:3
8.
View Article Online

and the aryl groups are arranged in the usual propeller likefashion (Table 1). The biphenyl geometry is typical, with aninter-ring torsion angle C(2)–C(1)–C(1A)–C(6A) of 232.8u.The comparable biphenyl inter-ring angles in TPD (1), 3,and N,N’-di(p-tolyl)-N,N’-diphenyl-4,4’-diaminobiphenyl are234.7u, 237.1u and 244.5u,16,17 respectively. The molecularbackbone is distorted by intermolecular contacts, with H(16)making close contacts with both C(1) [2.878 A] and C(2)[2.851 A] on an adjacent molecule.
The molecular geometry of 6b (Fig. 3, Table 1) revealsclearly the influence of the methyl groups C(13) and C(20), withthe torsion angle C(6)–C(1)–C(1A)–C(6A) observed as 85.0u.The amine centres are approximately planar, with the aryl ringsystems defining the usual propeller geometry associated withtriarylamines. The N–Caryl bond lengths span a range identicalto that found in 4 [1.411(2)–1.428(2) A], although the shortestN–C bond in 6b is associated with C(7). The C(1)–C(1A) bondlength in 6b [1.500(2) A] is longer than the equivalent separa-tion in 4.
The packing of these molecules within the crystal lattice isalso of interest, given the scarcity of reports dealing with theintermolecular relationships of TPD-type molecules in the solidstate. Molecules 4 form a loose framework, with the solvent ofcrystallisation located in the cavities between molecules of thearylamine (Fig. 4). The framework is supported by a number ofweak intermolecular C–H…p interactions, the shortest beingC(16)–H(16)…C(2’)(0.5 2 x, 0.5 2 y, 2z) (H…C 2.85(1) A).Such weak C–H…p interactions are commonly observed in thesolid state structures of aromatic compounds, but it should benoted that there is no evidence for p…p stacking typeinteractions in 4. Not surprisingly, the solvent molecule ofCH2Cl2 also takes part in a number of weak CH…C andCH…Cl interactions. In contrast to 4, the molecules of 6bare found as layers perpendicular to the crystallographica-direction (Fig. 5). The individual molecules in each layer are
linked together by p…p interactions (the shortest C…Cdistance between parallel Ph-rings is C16…C16’ 3.282 A),while CH…p interactions (the shortest being C15–H15…C8,C8…H15 2.828 A) connect the molecules of the differentlayers.
While crystallographic studies reveal detail about solid statestructure in a crystalline environment, other methods canprovide indirect evidence about the physical and electronic
Fig. 3 An ORTEP plot (50%) showing the molecular structure of 6b and the atom labelling scheme, with hydrogen atoms omitted for clarity.
Fig. 4 Packing of the molecules of 4 within the crystal lattice. Viewedalong b-axis, H-atoms omitted for clarity.
Fig. 5 Crystal structure of 6b, viewed in the 011 direction, H-atoms omitted for clarity.
2 5 1 8 J . M a t e r . C h e m . , 2 0 0 4 , 1 4 , 2 5 1 6 – 2 5 2 3
Publ
ishe
d on
28
June
200
4. D
ownl
oade
d by
Uni
vers
ity o
f W
este
rn O
ntar
io o
n 28
/10/
2014
11:
41:3
8.
View Article Online

structure of these materials in solution. The electronic spectraof simple triarylamines, such as 2, typically feature a single UVp–p* absorption band (Fig. 6a). In contrast, tetra(aryl)benzi-denes such as 1 exhibit two absorption bands arising from p–p*transitions which may be more precisely defined as transitionsbetween the delocalised HOMO to unoccupied orbitals more orless localised on the biphenyl (lmax ca. 350 nm) and peripheralaryl (lmax ca. 300 nm) groups.16 This characteristic profile isalso observed for the conformationally unrestricted system 4(Fig. 6b). However, in the case of 5 (Fig. 6c) and 6 (Fig. 6d)only a single absorption band is observed near 300 nm indicat-ing that the triarylamine moieties in these compounds areelectronically decoupled.
The electrochemical response of 4, 5a, 5b, 6a and 6b wereexamined by cyclic and differential pulse voltammetry(Table 2). The unrestricted system 4 displayed two reversible,one electron oxidation waves, which were separated by 0.30 V.Introduction of methyl groups at the 2 and 6 positions in 5a, 5band 6a resulted in an increase in the first oxidation potential,and a decrease in the separation of the redox events, becominga single, apparently two electron, process in 6a. Therefore,despite the presence of the inductively electron donating methylgroups in the 2 and 6 positions in 5a, 5b and 6a, the oxidationof these compounds is thermodynamically less favourablethan 4 and probably reflects the greater reorganisation energy
required during the oxidation of these more sterically restrictedsystems. The electrochemical response of 6b was complicatedby the chemical instability of the redox product. Thus while theinitial oxidative sweep displayed an oxidation wave at 0.90 V,as the potential was cycled between 2.0 and 21.5 V this wavebecame broader, and a new wave at 0.58 V was detected. A thinfilm of material was clearly evident when the surface of theworking electrode was examined at the end of the experiment.
The spectroelectrochemical response of a small number oftetra(aryl)benzidenes have been reported recently, and notsurprisingly the spectral profiles obtained for 4n1 (n ~ 0–2)were very similar to those of 1n1, 3n1,16 and N,N,N’,N’-tetra(p-methoxyphenyl)-1,1’-diaminobiphenyl.18 Oxidation of 4 to 41
was characterised by collapse of the parent spectrum, and thegrowth of a lower energy p–p* transition at 490 nm and theappearance of an intense, asymmetric NIR band (lmax ~1470 nm), which arises from the (HOMO-1) A SOMOtransition (Fig. 7a). Further oxidation to 421 results in thecollapse of the spectral transitions characteristic of 41 and theshift of the p–p* band to 780 nm (Fig. 7b), which is entirely inkeeping with the spectral profiles observed for 121 and therelated systems mentioned above.
In general terms, the spectroelectrochemical response of 5awas similar to that of 4, but in keeping with the lower com-proportionation constant associated with 5a1 this species couldnot be generated free of 5a and 5a21. As the potential appliedto the working electrode increased, the p–p* bands observed inthe neutral species at 308 nm collapsed, with a concomitantincrease in the bands associated with the radical cation at 439and 1863 nm (Fig. 8a), while the p–p* bands from the dicationdominate at higher potentials still (Fig. 8b). The chemicalreversibility of the system was verified by the recovery of thespectrum of 5a after exhaustive reduction. Oxidation of 6aproceeded smoothly to the dication, with the observation ofa single isosbestic point at 430 nm (Fig. 9). The NIR regionremained transparent during this process, and 6a1 was notdetected.
While 5b1 was shown to be sufficiently stable kinetically togive a chemically reversible response under conditions of thecyclic voltammetry experiment (100–200 mV s21), the chemicalreactivity of this species was apparent on the longer timescaleof the OTTLE-based spectroelectrochemical experiments.
Fig. 6 The UV-Vis absorption spectra (l/e) of (a) 2 (302/23400), (b) 4 (307/29700; 355/36600), (c) 5a (309/38700) and (d) 6a (309/40300). The spectraof 5b and 6b are similar to those of their tolyl-substituted analogues 5a and 6a.
Table 2 The electrochemical response of 1, 4, 5a, 5b, 6a and 6b
CompoundE0’ vs. Fc/Fc1
CH2Cl2/VDE0’
CH2Cl2/VComproportionconstant Kc
a
1 E1 ~ 0.292 0.215 4.320E2 ~ 0.507
4 E1 ~ 0.251 0.303 133,140E2 ~ 0.554
5a E1 ~ 0.390 0.158 550E2 ~ 0.550
5b E1 ~ 0.442 0.146 295E2 ~ 0.588
6a E1 ~ 0.463 — —6b E1 ~ 0.90 (irrev) — —a Kc ~ exp(DE0RT/nF) b Data recorded in CH2Cl2 (0.1 M NBu4BF4)on a Pt working electrode. Potentials are referenced against Fc/Fc1.
J . M a t e r . C h e m . , 2 0 0 4 , 1 4 , 2 5 1 6 – 2 5 2 3 2 5 1 9
Publ
ishe
d on
28
June
200
4. D
ownl
oade
d by
Uni
vers
ity o
f W
este
rn O
ntar
io o
n 28
/10/
2014
11:
41:3
8.
View Article Online
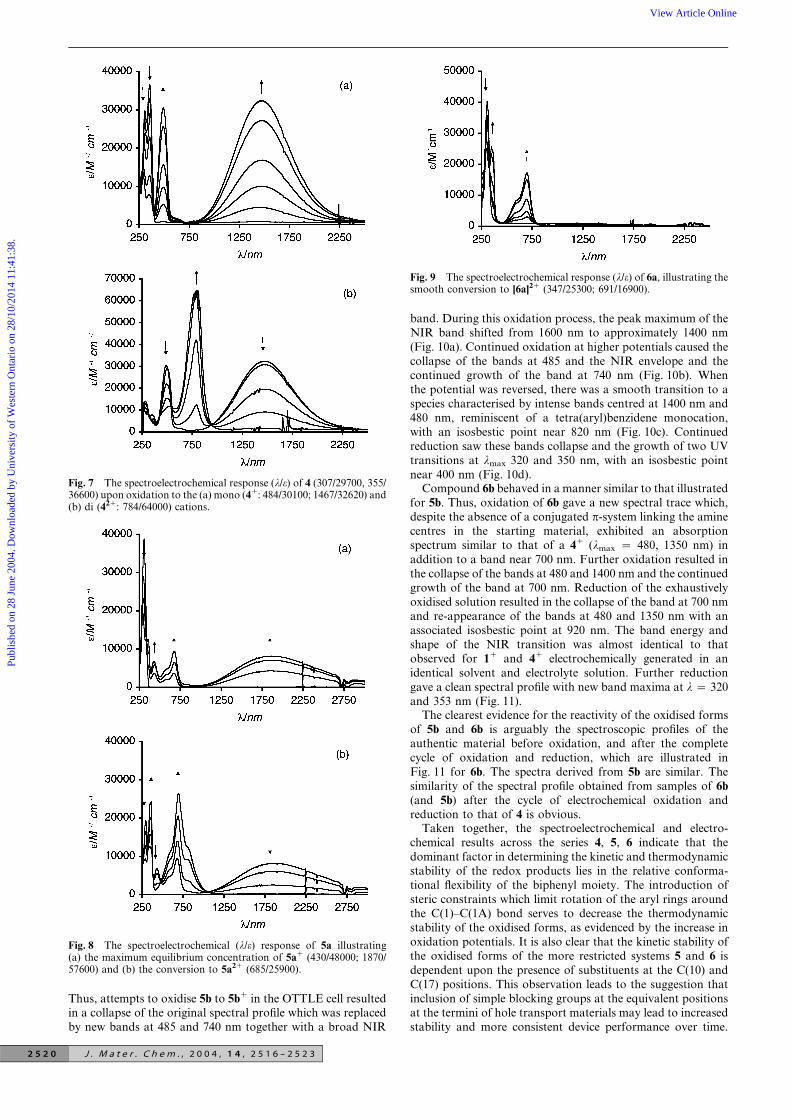
Thus, attempts to oxidise 5b to 5b1 in the OTTLE cell resultedin a collapse of the original spectral profile which was replacedby new bands at 485 and 740 nm together with a broad NIR
band. During this oxidation process, the peak maximum of theNIR band shifted from 1600 nm to approximately 1400 nm(Fig. 10a). Continued oxidation at higher potentials caused thecollapse of the bands at 485 and the NIR envelope and thecontinued growth of the band at 740 nm (Fig. 10b). Whenthe potential was reversed, there was a smooth transition to aspecies characterised by intense bands centred at 1400 nm and480 nm, reminiscent of a tetra(aryl)benzidene monocation,with an isosbestic point near 820 nm (Fig. 10c). Continuedreduction saw these bands collapse and the growth of two UVtransitions at lmax 320 and 350 nm, with an isosbestic pointnear 400 nm (Fig. 10d).
Compound 6b behaved in a manner similar to that illustratedfor 5b. Thus, oxidation of 6b gave a new spectral trace which,despite the absence of a conjugated p-system linking the aminecentres in the starting material, exhibited an absorptionspectrum similar to that of a 41 (lmax ~ 480, 1350 nm) inaddition to a band near 700 nm. Further oxidation resulted inthe collapse of the bands at 480 and 1400 nm and the continuedgrowth of the band at 700 nm. Reduction of the exhaustivelyoxidised solution resulted in the collapse of the band at 700 nmand re-appearance of the bands at 480 and 1350 nm with anassociated isosbestic point at 920 nm. The band energy andshape of the NIR transition was almost identical to thatobserved for 11 and 41 electrochemically generated in anidentical solvent and electrolyte solution. Further reductiongave a clean spectral profile with new band maxima at l ~ 320and 353 nm (Fig. 11).
The clearest evidence for the reactivity of the oxidised formsof 5b and 6b is arguably the spectroscopic profiles of theauthentic material before oxidation, and after the completecycle of oxidation and reduction, which are illustrated inFig. 11 for 6b. The spectra derived from 5b are similar. Thesimilarity of the spectral profile obtained from samples of 6b(and 5b) after the cycle of electrochemical oxidation andreduction to that of 4 is obvious.
Taken together, the spectroelectrochemical and electro-chemical results across the series 4, 5, 6 indicate that thedominant factor in determining the kinetic and thermodynamicstability of the redox products lies in the relative conforma-tional flexibility of the biphenyl moiety. The introduction ofsteric constraints which limit rotation of the aryl rings aroundthe C(1)–C(1A) bond serves to decrease the thermodynamicstability of the oxidised forms, as evidenced by the increase inoxidation potentials. It is also clear that the kinetic stability ofthe oxidised forms of the more restricted systems 5 and 6 isdependent upon the presence of substituents at the C(10) andC(17) positions. This observation leads to the suggestion thatinclusion of simple blocking groups at the equivalent positionsat the termini of hole transport materials may lead to increasedstability and more consistent device performance over time.
Fig. 7 The spectroelectrochemical response (l/e) of 4 (307/29700, 355/36600) upon oxidation to the (a) mono (41: 484/30100; 1467/32620) and(b) di (421: 784/64000) cations.
Fig. 8 The spectroelectrochemical (l/e) response of 5a illustrating(a) the maximum equilibrium concentration of 5a1 (430/48000; 1870/57600) and (b) the conversion to 5a21 (685/25900).
Fig. 9 The spectroelectrochemical response (l/e) of 6a, illustrating thesmooth conversion to [6a]21 (347/25300; 691/16900).
2 5 2 0 J . M a t e r . C h e m . , 2 0 0 4 , 1 4 , 2 5 1 6 – 2 5 2 3
Publ
ishe
d on
28
June
200
4. D
ownl
oade
d by
Uni
vers
ity o
f W
este
rn O
ntar
io o
n 28
/10/
2014
11:
41:3
8.
View Article Online

The spectroscopic profiles of the products derived from electro-chemical oxidation of 5b and 6b, which contain two UVabsorption bands, are indicative of an unrestricted tetraaryl-benzidene moiety. It seems likely that the increased barrier torotation caused by the methyl groups at the 2,2’ (and 6,6’)positions destabilises the planar arrangement of the biphenylmoiety, effectively decoupling the triarylamine moieties andrestoring triphenylamine-like chemical reactivity to the oxi-dised forms of these compounds. The chemical stability of thetolyl-based analogues 5a and 6a clearly indicates that the site ofreaction lies at the carbon centres on the peripheral rings trans
to the nitrogen centre [i.e. C(10) and C(17)].Guided by the electrochemical and spectroelectrochemical
results, preliminary preparative scale chemical oxidation of6b was undertaken using an equimolar quantity of SbCl5 inCH2Cl2. The addition of the oxidising agent was marked by aninstant deep blue colouration of the solution, which rapidlyfaded to orange. The progress of the reaction could be followedby UV-Vis spectroscopy, with two absorption bands at 480 and1400 nm observed from these orange solutions, consistent with
the spectroelectrochemically generated profiles. The electro-spray mass spectrum of the crude reaction mixture clearlyindicated the presence of dimeric (m/z 1086) and trimeric (m/z1628) products, although higher oligomers were not observed,possibly due to the lower solubility of the longer chain mate-rials. The reaction mixture was allowed to stir for 1 h atambient temperature, before being quenched by addition ofSnCl2. Filtration of the reaction mixture through a silica padto remove the tin and antimony by-products gave a colourlesssolution from which a white solid (7) was obtained afterconcentration and precipitation into ethanol.
In the 1H NMR spectrum of the resulting white solid, singletresonances at d 2.20 and d 6.91 ppm characteristic of the2,2’,6,6’-tetramethylbenzidene fragment were observed. In thearomatic region, an AB resonance centred at d 7.13 ppm indi-cated the presence of a 4,4’-disubstituted 1,1’-biphenyl moietyin the product, 7 as well. This suggestion is further supportedby the UV/Vis spectrum of 7 prepared in this manner, whichwas characterised by two absorption bands at 315 and 351 nm,similar to the spectral profile of 4, and other simple TPD-typemolecules. Estimates of the weight average of 7 obtained fromseveral reactions by GPC against polystyrene standards fell inthe range 1740 (3-mer) to 30500 (58-mer) depending upon therate of addition of the oxidising agent and the concentration ofthe reagents. While the conditions were not optimised, it is clearthat oxidation of 6b, either chemically or electrochemically,results in the production of oligo and polymeric derivatives.
Conclusion
The introduction of steric bulk at the 2 and 6 positions in4,4’-bis(diphenylamino)biphenyls can influence both the oxida-tion potential of the material and the thermodynamic stabilityof the oxidation products. The use of steric constraints tobreak the extended p-conjugated system in the benzidenemoiety results in materials which are susceptible to oxidativepolymerisation, either electrochemically or by chemical oxida-tion. The production of a polymeric polyarylamine in
Fig. 10 (a) The spectroelectrochemical response of 5b upon oxidation at lower potentials during the early stages. Note the shift in the low energyband maximum. (b) The spectroelectrochemical response during the latter stages of the complete oxidation of 5b. (c) The spectral profiles generatedduring early stages of the back-reduction. (d) The spectral profile after complete back-reduction.
Fig. 11 The UV-Vis spectra (l/e) of 6b before (solid line, 310/39400)and after (dashed line, 320/28700; 346/31900) the oxidation/reductioncycle.
J . M a t e r . C h e m . , 2 0 0 4 , 1 4 , 2 5 1 6 – 2 5 2 3 2 5 2 1
Publ
ishe
d on
28
June
200
4. D
ownl
oade
d by
Uni
vers
ity o
f W
este
rn O
ntar
io o
n 28
/10/
2014
11:
41:3
8.
View Article Online

electrochemical fashion, in which the only ‘‘reagent’’ is the solidelectrode, and the ‘‘by-products’’ are protons is an intriguingobservation. Studies aimed at optimisation of the benignelectrochemical synthesis, and investigation of the transportproperties of the resulting materials, are underway.
Experimental
All reactions were carried out under a nitrogen atmosphere inflame-dried glassware. Solvents were dried from an appropriateagent and distilled. Di(4-methylphenyl)amine, 4,4’-diamino-2,2’-dimethylbiphenyl dihydrochloride and ortho-dichloroben-zene were purchased from Aldrich; diphenylamine, 18-crown-6and K2CO3 were purchased from Avocado. All commercialreagents were used as received. The diiodides were prepared aspreviously described.19–21 The computational work was carriedout within the Gaussian 98 package.13 Molecular geometrieswere optimised for both the neutral and radical cation states of2 at a DFT level using the BPW91 functional (Becke’s 1998exchange functional, with Perdew and Wang’s 1991 gradient-corrected correlation functional) in conjunction with the6–31G(d,p) basis set.10–12 Post-processing for visualisation ofthe molecular orbitals generated by the calculations was carriedout using the Molekel programme.14
Instrumentation
Mass spectra were recorded on a Micromass Autospec instru-ment operating in EI mode. NMR spectra were recorded fromCDCl3 solutions on Varian XL-200 (1H), Varian Unity-300 (1Hand 13C{1H}) or Varian VXR-400 (1H, 13C, 1H–1H COSYand 1H–13C HETCOR). All chemical shifts are reported ind (ppm), referenced against solvent resonances. UV-Vis-NIRspectra were recorded on a Varian Cary-5 spectrophotometer,electrochemical measurements were made with an AutoLabPGSTAT30 from CH2Cl2 solutions containing 0.1 MNBu4BF4 as supporting electrolyte and potentials cited arereferenced against ferrocene such that the ferrocene couple fallsat 0 V. Spectroelectrochemical experiments were also carriedout in 0.1 M NBu4BF4/CH2Cl2 solutions using an OTTLEcell,22 fitted with a Pt gauze mesh semi-transparent workingelectrode, a Pt wire counter and pseudo-reference electrodes,and a home-built potentiostat.
Crystallographic details
Single-crystal X-ray data for both compounds 4 and 6b werecollected at 120(1) K on a Bruker SMART-CCD 6000diffractometer (v-scan, 0.3u/frame) using Mo-Ka- radiation(l ~ 0.71073 A). No absorption correction was applied. Thestructure was solved by direct methods and refined by full-matrix least squares on F2 for all data using SHELXTL soft-ware. All non-hydrogen atoms were refined with anisotropicdisplacement parameters, H-atoms were located on the diffe-rence map and refined isotropically.
Crystal data for 4. C40H36N2. CH2Cl2, M ~ 629.63, mono-clinic, space group C 2/c, a ~ 18.5659(3), b ~ 13.6399(2), c ~14.6862(2) A, b ~ 116.78(1)u, U ~ 3320.2(2) A3, F(000) ~1328, Z ~ 4, Dc ~ 1.260 mg m23, m ~ 0.228 mm21. 16149reflections (2.11 ¡ h ¡ 29.49u) were collected yielding 4596unique data (Rmerg ~ 0.041). Final wR2(F2) ~ 0.1190 for alldata (280 refined parameters), conventional R1(F) ~ 0.0426 for3703 reflections with I w 2s, GOF ~ 1.069. The largest peakon the residual map is 0.54 e A23. CCDC reference numberCCDC 235057. See http://www.rsc.org/suppdata/jm/b4/b404731a/ for crystallographic data in .cif or other electronicformat.
Crystal data for 6b. C40H36N2, M ~ 544.71, monoclinic,space group C 2/c, a ~ 9.7412(8), b ~ 16.670(1), c ~ 18.784(2)A, b ~ 97.068(4)u, U ~ 3027.1(4) A3, F(000) ~ 1160, Z ~ 4,Dc ~ 1.195 mg m23, m ~ 0.069 mm21. 20949 reflections(2.18 ¡ h ¡ 30.51u) were collected yielding 4619 unique data(Rmerg ~ 0.046). Final wR2(F2) ~ 0.1364 for all data (262refined parameters), conventional R1(F) ~ 0.0505 for 3278reflections with I w 2s, GOF ~ 1.021. The largest peak on theresidual map is 0.32 e A23. CCDC reference number CCDC235058. See http://www.rsc.org/suppdata/jm/b4/b404731a/ forcrystallographic data in .cif or other electronic format.
Synthesis and characterisation
N,N,N’,N’-tetra(4-methylphenyl)-(1,1’-biphenyl)-4,4’-diamine(4). A solution of diiodobiphenyl (2.0 g, 4.93 mmol), di(4-methylphenyl)amine (2.14 g, 10.9 mmol), K2CO3 (5.44 g,39.4 mmol), copper powder (0.67 g, 10.6 mmol) and 18-crown-6 (0.29 g, 0.99 mmol) in o-dichlorobenzene (10 ml) was heatedat reflux for 15 h. The reaction mixture was filtered and thesolvent volume reduced in vacuo. The crude mixture waspurified by column chromatography (silica, hexanes) (1.25 g,47%). 1H NMR: d 7.31 (d, JHH ~ 8 Hz, 4H, H2/H3), 6.97 (m,20H), 2.2 (s, 12H, CH3). 13C NMR: d 147.22, 145.54, 134.20,132.61, 130.08, 127.28, 124.74, 123.16, 21.07. EI MS m/z 544[M]1, 349 [M 2 NAr2]1. m.p. 203–205u. Calculated forC40H36N2: C 88.19%; H 7.66%; N 5.14%; found: C 88.18%; H7.74%; N 4.49%.
N,N,N’,N’-tetra(4-methylphenyl)-(2,2’-dimethyl)-(1,1’-biphenyl)-4,4’-diamine (5a). The compounds 2,2’-dimethyl-4,4’-diiodobi-phenyl (2.17 g, 5.0 mmol), di(4-methylphenyl)amine (2.36 g,12.0 mmol), K2CO3 (5.52 g, 40.0 mmol), copper powder (0.68 g,10.75 mmol) and 18-crown-6 (0.27 g, 1.0 mmol) were dissolvedin o-dichlorobenzene (10 ml) and heated at reflux for 45 h. Thereaction mixture was filtered and the solvent volume reducedin vacuo. The crude mixture was purified by column chromato-graphy (silica, hexanes) (2.35 g, 82%). 1H NMR d 7.05 (m,22H), 2.32 (s, 12H, H13), 1.99 (s, 6H, H20). 13C NMR d 145.79,144.54, 135.90, 134.08, 129.27, 128.76, 122.79, 118.96, 19.78,19.05. EI MS m/z 572 [M] 1, 377 [M 2 NAr2] 1, 286 [M]21. Mp248–250u.
N,N,N’,N’-tetraphenyl-(2,2’-dimethyl)-(1,1’-biphenyl)-4,4’-diamine (5b). A stirred solution of 2,2’-dimethyl-4,4’-diiodobi-phenyl (1.0 g, 2.3 mmol), diphenylamine (0.78 g, 4.6 mmol),K2CO3 (2.54 g, 18.4 mmol), copper powder (0.58 g, 9.2 mmol)and 18-crown-6 (0.12 g, 0.46 mmol) in o-dichlorobenzene(10 ml) was heated at reflux for 20 h. The reaction mixture wasfiltered and the solvent volume reduced in vacuo. The crudemixture was purified by column chromatography (silica,hexanes/CH2Cl2) to afford the title compound as a yellowpowder. (0.38 g, 35%). 1H NMR d 7.22 (s, 2H, H5), 7.19 (m,8H, H8), 7.06 (m, 8H, H9), 6.93 (m, 2H, H2/H3), 6.92 (m, 4H,H10), 6.85 (m, 2H, H2/H3), 1.94 (s, 6H, CH3). 13C NMR d147.95, 146.48, 137.10, 135.93, 130.40, 129.17, 125.10, 124.17,122.64, 121.23, 20.06. EI MS m/z 516 [M] 1, 349 [M 2 NPh2] 1.Mp 206–209u.
N,N,N’,N’-tetra(4-methylphenyl)-(2,2’,6,6’-tetramethyl)-(1,1’-biphenyl)-4,4’-diamine (6a). A stirred solution of 2,2’,6,6’-tetramethyl-4,4’-diiodobiphenyl (2.0 g, 4.33 mmol),di(4-methyl)phenylamine (1.87 g, 9.52 mmol), K2CO3 (4.78 g,34.63 mmol), copper powder (0.59 g, 9.3 mmol) and 18-crown-6 (0.27 g, 1.0 mmol) in o-dichlorobenzene (10 ml) was heated toreflux for 18 h. The reaction mixture was filtered and thesolvent volume reduced in vacuo. The crude mixture waspurified by column chromatography (silica, hexanes, CH2Cl2),(0.29 g, 11%). 1H NMR d 6.99 (d, JAB ~ 8 Hz, 8H, H8), 6.93(d, JAB ~ 8 Hz, 8H, H9), 6.73 (s, 4H, H3), 2.25 (s, 12H,
2 5 2 2 J . M a t e r . C h e m . , 2 0 0 4 , 1 4 , 2 5 1 6 – 2 5 2 3
Publ
ishe
d on
28
June
200
4. D
ownl
oade
d by
Uni
vers
ity o
f W
este
rn O
ntar
io o
n 28
/10/
2014
11:
41:3
8.
View Article Online

ArCH3), 1.76 (s, 12H, biphenyl CH3). 13C NMR d 146.88,146.10, 132.08, 130.89, 130.07, 128.06, 124.55, 122.56, 21.16,20.34. EI MS m/z 600 [M]1, 405 [M 2 NAr2]1.
N,N,N’,N’-tetra(phenyl)-(2,2’,6,6’-tetramethyl)-(1,1’-biphenyl)-4,4’-diamine (6b). A stirred solution of 2,2’,6,6’-tetramethyl-4,4’-diiodobiphenyl (2.0 g, 4.33 mmol), diphenylamine (1.61 g,9.5 mmol), K2CO3 (5.52 g, 40.0 mmol), copper powder (0.6 g,9.3 mmol) and 18-crown-6 (0.23 g, 0.86 mmol) in ortho-dichlorobenzene (10 ml) was heated at reflux for 19 h. Thereaction mixture was filtered and the solvent volume reducedin vacuo. The crude mixture was treated MeOH and placed inthe freezer, producing a pale precipitate, which was collectedand recrystallised from benzene/hexanes (1.64 g, 70%). 1HNMR d 7.26 (pseudo t, JHH ~ 16 Hz, 8H, H9), 7.12 (d, JHH ~7 Hz, 8H, H8), 7.00 (t, JHH ~ 14 Hz, 4H, H10), 6.86 (s, 4H,H3), 1.86 (s, 12H, CH3). 13C NMR d 148.34, 146.24, 137.14,135.07, 129.37, 124.19, 123.63, 122.48, 20.24. EI MS m/z 544[M]1, 377 [M 2 NAr2]1. Calculated for C40H36N2: C 88.19; H6.66%; N 5.14%; found: C 87.80%; H 6.95%; N 5.05%.
Acknowledgements
This work was supported by the University of Durham, theEPSRC, ONE-North East and Avecia Ltd. JAKH holds anEPSRC Senior Research Fellowship. MAJP held an EPSRC/Avecia CASE award. We are pleased to acknowledge helpfuldiscussions with Professor Dr C. Lambert (University ofWurzburg) and communication of results prior to publication.
References
1 (a) U. Mitschke and P. Bauerle, J. Mater. Chem., 2000, 10, 1471;(b) P. M. Borsenberger and D. S. Weiss, Organic Photoreceptorsfor Xerography, Marcel Dekker, New York, 1998; (c) D. M. Paiand B. E. Springett, Rev. Mod. Phys., 1993, 65, 163; (d) M. Strukelj,J. Am. Chem. Soc., 1996, 118, 1213; (e) J. Kalinaswki, Appl. Phys.Lett., 1996, 118, 2317.
2 J. Cornil, D. Beljonne, J. P. Calbert and J. L. Bredas, Adv. Mater.,2001, 13, 1053.
3 See, for example: (a) T. D. Selby, K.-Y. Kim and S. C. Blackstock,Chem. Mater., 2002, 14, 1685; (b) J. Pang, Y. Tao, S. Freiberg,X.-P. Yang, M. D’Iorio and S. Wang, J. Mater. Chem., 2002, 12,206; (c) H. B. Goodbrand and N.-X. Nu, J. Org. Chem., 1999, 64,670; (d) C. G. Frost and P. Mendonca, J. Chem. Soc., PerkinTrans. 1, 1998, 2615; (e) H. Tanaka, S. Tokito, Y. Taga andA. Okada, Chem. Commun., 1996, 2175; (f) M. Thelakkat, R. Fink,F. Haubner and H.-W. Scmidt, Macromol. Symp., 1997, 125, 157;(g) P. Strohriegl, G. Jesberger, J. Heinze and T. Moll, Makromol.Chem., 1992, 193, 909; (h) H. Weingarten, J. Org. Chem., 1964, 29,
3624; H. Weingarten, J. Org. Chem., 1964, 29, 977; (i) J. Lindley,Tetrahedron, 1984, 40, 1433.
4 (a) S. Tanaka, T. Iso and Y. Doke, Chem. Commun., 1997, 2063;(b) M. Ishikawa, M. Kawai and Y. Ohsawa, Synth. Met., 1991, 40,231.
5 E. T. Seo, R. F. Nelson, J. M. Fritsch, L. S. Marcoux, D. W. Leedyand R. N. Adams, J. Am. Chem. Soc., 1966, 88, 3498.
6 A. Petr, C. Kvarnstrom, L. Dunsch and A. Ivaska, Synth. Met.,2000, 108, 245.
7 H. Sato, A. Kanegae, R. Yamaguchi, K. Ogino and J. Kurjata,Chem. Lett., 1999, 79.
8 C. Lambert and G. Noll, 2003, personal communication.9 M. Leung, M.-Y. Chou, Y. O. Su, C. L. Chiang, H.-L. Chen,
C. F. Yang, C.-C. Yang, C.-C. Lin and H.-T. Chen, Org. Lett.,2003, 5, 839.
10 A. D. Becke, Phys. Rev. A, 1988, 38, 3098.11 J. P. Perdew, K. Burke and Y. Wang, Phys. Rev. B, 1996, 54,
16533; Erratum: Phys. Rev. B, 1998, 57, 14999.12 (a) R. Ditchfield, W. J. Hehre and J. A. Pople, J. Chem. Phys.,
1971, 54, 724; (b) W. J. Hehre, R. Ditchfield and J. A. Pople,J. Chem. Phys., 1972, 56, 2257; (c) P. C. Hariharan and J. A. Pople,Mol. Phys., 1974, 27, 209; (d) M. S. Gordon, Chem. Phys. Lett.,1980, 76, 163; (e) P. C. Hariharan and J. A. Pople, Theor. Chim.Acta., 1973, 28, 213.
13 M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria,M. A. Robb, J. R. Cheeseman, V. G. Zakrzewski,J. A. Montgomery, Jr., R. E. Stratmann, J. C. Burant,S. Dapprich, J. M. Millam, A. D. Daniels, K. N. Kudin,M. C. Strain, O. Farkas, J. Tomasi, V. Barone, M. Cossi,R. Cammi, B. Mennucci, C. Pomelli, C. Adamo, S. Clifford,J. Ochterski, G. A. Petersson, P. Y. Ayala, Q. Cui, K. Morokuma,P. Salvador, J. J. Dannenberg, D. K. Malick, A. D. Rabuck,K. Raghavachari, J. B. Foresman, J. Cioslowski, J. V. Ortiz,A. G. Baboul, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz,I. Komaromi, R. Gomperts, R. L. Martin, D. J. Fox, T. Keith,M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe,P. M. W. Gill, B. G. Johnson, W. Chen, M. W. Wong, J. L. Andres,C. Gonzalez, M. Head-Gordon, E. S. Replogle and J. A. Pople,GAUSSIAN 98 (Revision A.11), Gaussian, Inc., Pittsburgh, PA,2001.
14 S. Portmann and H. P. Luthi, Chimia, 2000, 54, 766.15 V. Coropceanu, M. Malagoli, J. M. Andre and J. L. Bredas, J. Am.
Chem. Soc., 2002, 124, 10519.16 P. J. Low, M. A. J. Paterson, H. Puschmann, A. E. Goeta,
J. A. K. Howard, C. Lambert, J. C. Cherryman, D. R. Tackley,S. Leeming and B. Brown, Chem. Eur. J., 2004, 10, 83.
17 A. R. Kennedy, W. E. Smith, D. R. Tackley, W. I. F. David,K. Shankland, B. Brown and S. J. Teat, J. Mater. Chem., 2002, 12,168.
18 C. Lambert and G. Noll, J. Am. Chem. Soc., 1999, 121, 8434.19 S. F. Nelsen, R. F. Ismagilov, K. E. Gentile and D. R. Powell,
J. Am. Chem. Soc., 1999, 121, 7108.20 A. Helms, D. Heiler and G. McLendon, J. Am. Chem. Soc., 1992,
114, 6227.21 R. B. Carlin, J. Am. Chem. Soc., 1945, 67, 928.22 C. M. Duff and G. A. Heath, Inorg. Chem., 1991, 30, 2528.
J . M a t e r . C h e m . , 2 0 0 4 , 1 4 , 2 5 1 6 – 2 5 2 3 2 5 2 3
Publ
ishe
d on
28
June
200
4. D
ownl
oade
d by
Uni
vers
ity o
f W
este
rn O
ntar
io o
n 28
/10/
2014
11:
41:3
8.
View Article Online





























