Embed Size (px)
Citation preview

The molecular spectrum of hemoglobinopathy; Thalassemia and Hb variants
Cornelis L. Harteveld
Dpt. Of Clinical Genetics, Hemoglobinopathy Reference Center
LEIDEN UNIVERSITY MEDICAL CENTER

I have no personal or financial interests to declare:
I have no financial support from an industry source at the
current presentation.
대한혈액학회 Korean Society of Hematology
COI disclosureName of author : 홍 길 동 or Gildong Hong

Epidemiology hemoglobinopathies
7% world population is carrier of hemoglobinopathy
~350,000 baby’s born each year with Sickle Cell Anemia (SCA) or Thalassemia Major (TM)

Wednesday, March 20, 2019C.L.Harteveld, LUMC 4
From Hemoglobin to Hemoglobinopathy
Hb molecule:
16p
11p

Wednesday, March 20, 2019C.L.Harteveld, LUMC 5
11p
16p ζ α2 α1
ε Gγ Aγ δ β
Alpha-globin gene cluster
Beta-globin gene cluster
Globin genes

Wednesday, March 20, 2019C.L.Harteveld, LUMC 6
Hemoglobine-switch
11p
16p ζ α2 α1
ε Gγ Aγ δ β
HbF HbA
Embryonisch HbHbA2
Hb switch: embryo (Hb Gower I,II, Hb Portland) > foetus (HbF) > adult (HbA)

C.L.Harteveld, LUMC
Sickle Cell Disease
Structural defect: sickle cell anemia (SCA)

C.L.Harteveld, LUMC
β-Thalassemia
Expression defect: Thalassemia
α-thalassemiaβ-thalassemia

Pathology determined by unbalance between α- and β-chains
20-Mar-199 Insert > Header & footer
Patient β-thal intermedia/major: αα/αα vs β+β+ , β+β0 or β0β0
Patient α-thal to HbH disease: -α/αα, --/αα , --/-α, --/-- vs β/β
βα

Prevention and management of Hemoglobinopathy
20-Mar-19Insert > Header & footer10
Implementation of basic health interventions1. Management of Sickle Cell Anemia (SCA)
-early diagnosis-penicillin prophylaxis-vaccination
To reduce excess mortality amongst children under 5 years
3. Prevention of SCA and TM-carrier detection -information and counseling-prenatal diagnosis
To reduce the number of affected births
2. Management of Thalassemia Major (TM)-early diagnosis-transfusion /chelation therapy
To prevent unnecessary complications of treatment

Carrier detection
through laboratory diagnosis
Diagnostics of hemoglobinopathy
20-Mar-19Insert > Header & footer12
HemoCytometry
Hbseparation
DNA ClinicalDiag-nosis

C.L.Harteveld, LUMC
Flow Chart for Diagnostics of HbP
Hematology HPLC and Hb electrophoresis
Gap-PCR for7 most commonα-thal deletions
normal
Sequencing α-genes
MicrocyticHypochromicNormal iron
MLPA
Hb variant
Sequencing β-gene
HbA2 HbF

RBC (x 1012 /l)
1716151413121110987
7
6
5
4
Hb(mmol/land g%)
normal βT/β βT/ βT
Hemocytometry thalassemia genotypes
normal βT/β βT/ βT
MCV (fl)100
90
80
70
60
50
MCH (fmoland pg)
323028262422201816
normal βT/β βT/ βT
normal βT/β βT/ βT
βT/β trait
normal
βT/ βT
Intermediamajor
10.5109.58.78.17.56.96.25.654.5
2.001.871.741.621.491.371.251.121.00
normal
α-thaltrait
HbH disease

C.L.Harteveld, LUMC
Flow Chart for Diagnostics of HbP
Hematology HPLC and Hb electrophoresis
Gap-PCR for7 most commonα-thal deletions
normal
Sequencing α-genes
MicrocyticHypochromicNormal iron
MLPA
Hb variant
Sequencing β-gene
HbA2 HbF

20-mrt-1916 Insert > Header & footer
Capillarys 2 FlexPiercing (Sebia)
Hb electrophoresis
Hb separation equipment: HPLC and CE
Premier Resolution (PR)Trinity Biotech(Menarini)
High Performance Liquid Chromatography (HPLC)
Together they make a strong combi….

C.L.Harteveld, Hemoglobinopathies Lab
HPLC and CE : Hb variants and quantitation
Normal HbS carrier
HbSHbAHbA97.5%
HbA22.5% HbA2
5.6%
β-thal carrier

C.L.Harteveld, LUMC
Flow Chart for Diagnostics of HbP
Hematology HPLC and Hb electrophoresis
Gap-PCR for7 most commonα-thal deletions
normal
Sequencing α-genes
MicrocyticHypochromicNormal iron
MLPA
Hb variant
Sequencing β-gene
HbA2 HbF

Fully Automated sample preparation and Sanger sequencing

• Approx. 10% of all alpha-thal defects• All Hb variants of the alpha-globin chain
S13F S6R S13F S8RF S18R
S3F R
2A
2BF S18R
S3F R
1A
1B
α2 α1
Direct sequencing for point mutation analysis
β
HBLBF1 HBLBR1
HBLBF2 HBLBR2
HBLBF3 HBLBR3
HBLBF4 HBLBR4
• Detection rate approx. 90% of all beta-thal defects• All Hb variants of the beta-globin chain
~10% deletions
~90% deletions

20-Mar-19Insert > Header & footer21
ε Gγ Aγ δ βNorm HetLW Het SEA Het MedI Het20.5 Het FILNorm HetRW HomRW α-tripl.
3000
20002500
150012001000
500
Multiplex gap-PCR for the most common thalassemia deletions

Other deletion types of α-thalassemia: MLPA
20-Mar-19Insert > Header & footer22

C.L.Harteveld, Hemoglobinopathies Lab
…. Sometimes patterns are more complex…
Can’t do without
DNA analysis
Wednesday, March 20, 2019C.L.Harteveld, LUMC 24
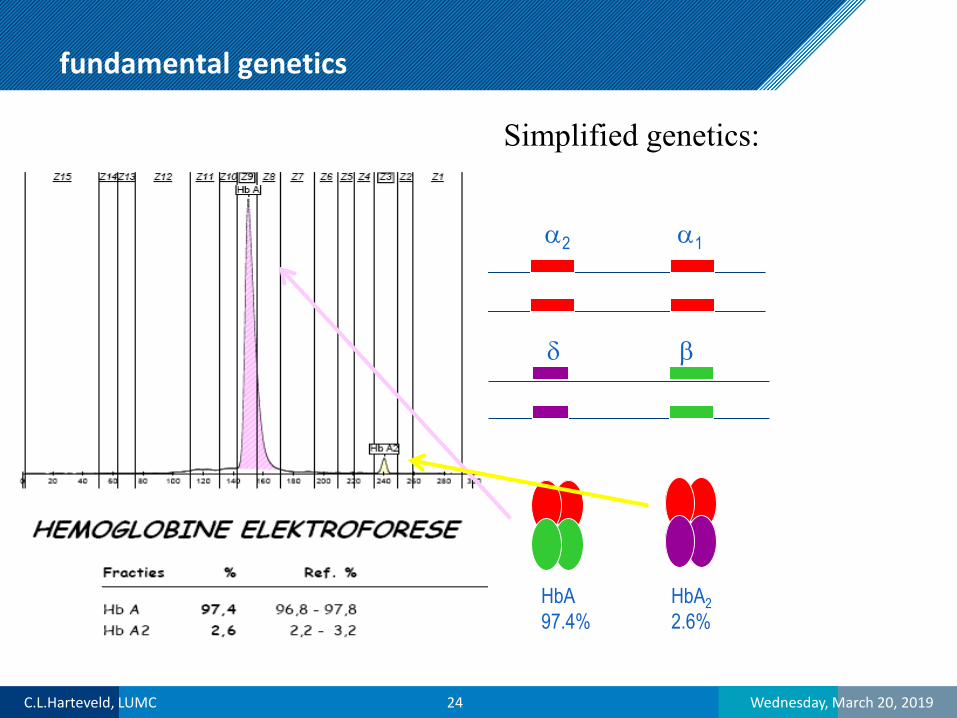
fundamental genetics
α2 α1
δ β
HbA22.6%
HbA97.4%
Simplified genetics:

Case 1: complex CE pattern
Hb Bart’s (γ4)
HbA (α2β2)
HbF (α2γ2)+ HbAQ (αQ2β2)
HbF Q-Thailand (αQ2γ2)
HbE (α2βE2)
HbE Q-Thailand (αQ2βE2)
Fraction % Hb type
Proband18 days
Child detected during neonatal screening:
Molecular analysis revealed 3 defects:
- Hb Q-Thailand (α1cd74 Asp>His)- -α4.2 deletion- HbE (βcd26Glu>Lys)
HbE HbA HbA2
α2 α1
γγ δ β
HbF
HbQE HbQ HbQA2 HbQF
HbBart’s

Fraction % Hb type
HbA (α2β2)HbF (α2γ2)+HbA Q-ThailandHbF Q-Thailand (αQ2γ2)HbE (α2βE2)HbA2 (α2δ2)HbE Q-Thailand (αQ2βE2)HbA2 Q-Thailand (αQ2δ2)
Proband 18 daysProband87 days
HbE HBA HbA2
α2 α1
γγ δ β
HbF
HbQE HBQ HbQA2 HbQF
HbBart’s
Case 1: complex CE pattern
Child detected during neonatal screening:
Molecular analysis revealed 3 defects:
- Hb Q-Thailand (α1cd74 Asp>His)- -α4.2 deletion- HbE (βcd26Glu>Lys)

HbBart’s
HbF+HbQ
HbA HbQFHbEHbQE
Hom HbE Het -α4.2HbQ-Thailand
Het -α4.2HbQ-Thailand /het HbE
2.5 wks
3 months2 yrs
Case 1: complex CE pattern

HbE particularly common in South-East Asia
20-Mar-19Insert > Header & footer28
Kaart Azië met verdeling hbE en beta-thal
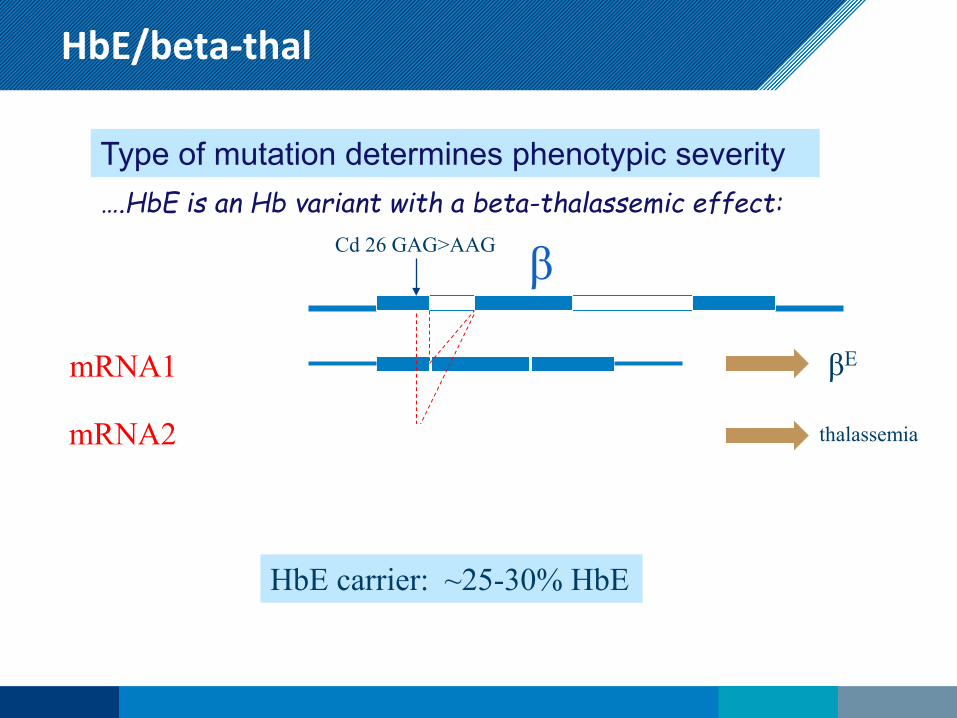
HbE/beta-thal
Insert > Header & footer29
Type of mutation determines phenotypic severity ….HbE is an Hb variant with a beta-thalassemic effect:
Cd 26 GAG>AAG β
mRNA1 βE
mRNA2 thalassemia
HbE carrier: ~25-30% HbE

Wednesday, March 20, 2019C.L.Harteveld, LUMC 30
This was reported asHbE carrier (!)
HbE coincides with HbA2 peak….but usually 30%, NOT 37.9%
Case 2: Pitfalls ….to (β)E or not to (β)E.
…but this is how a real HbE carrier looks like!
Hb D-IranSilent alpha-variantNormalhematology
Hb EMicrocytichypochromic
CE
CE

Unbalanced α / β synthesis
20-Mar-1931 Insert > Header & footer
Hemolytic anemia in beta-thalassemia intermedia/major
Due to unbalanced α- / β-chain synthesis
- alpha-thalassemia; excess beta-chains- beta-thalassemia; excess alpha-chains
Proteolytic processes degrade excess,
…..but if unbalance becomes too excessive this may result in
β-thal-intermedia
Everything depends on the balance in life

Case 3 Child is beta-thal carrier, but unexpected severely anemic
αααanti 3.7-α3.7 Cd 39 CAG>TAG
(HBB:c.118C>T)
1 2 3
Hb 15.5 11.6 7.2 g%
RBC 5.31 5.39 3.67 1012/l
Ht 0.46 0.37 0.25 l/l
MCV 86 69 67 fl
MCH 29.1 21.6 19.9 pg
ZPP 62 83 270 μmol/mol heme
Hp - - <24.3 Mg/100ml serum
HPLC A2 2.9 5.1 4.8 %
CE A2 2.8 5.2 4.4 %
1 2
3
Example : genetic consequences
1 2
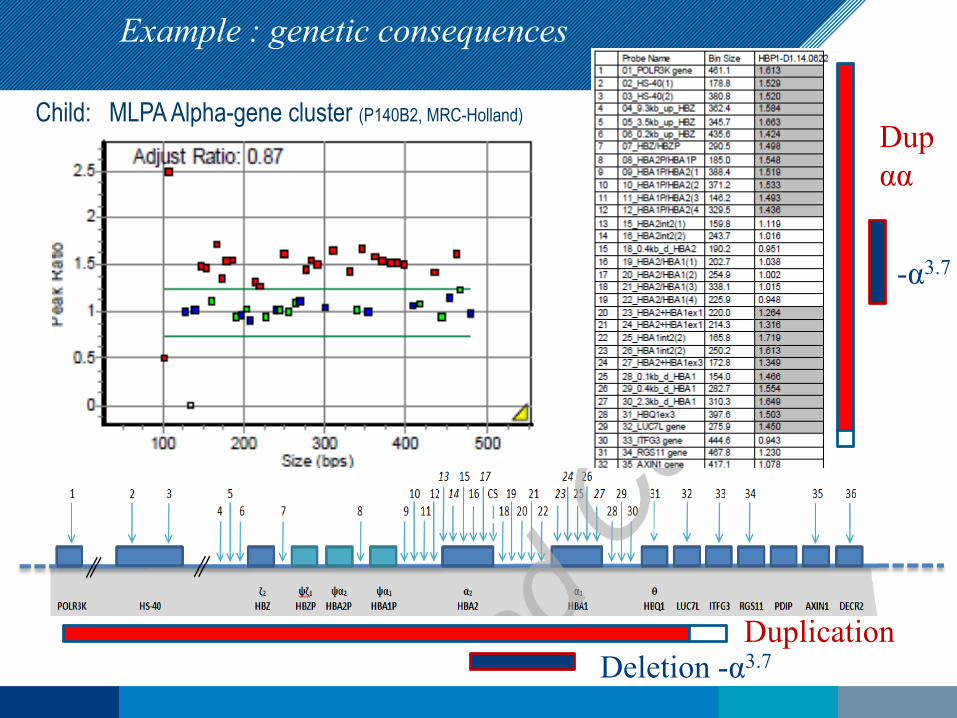
-α3.7
Dupαα
DuplicationDeletion -α3.7
Child: MLPA Alpha-gene cluster (P140B2, MRC-Holland)
Example : genetic consequences

αααanti 3.7-α3.7
1 2
3
ααTel MRE normal
Case 3 Child is beta-thal carrier, but unexpected severely anemic
Cd 39 CAG>TAG(HBB:c.118C>T)
αααα
ααTel MRE ααMRE-α3.7Tel MREβ0
β5 x α1 x β
Example : genetic consequences
Counseling:Triplication+ quadruplication+ beta-thal =Severe beta-thalintermedia

20-mrt-1935 Insert > Header & footer
A beta-thal carrier becoming transfusion dependent later in life: “late-onset TM”

Case 4: other case of ‘late-onset TM’ from Greece:
20-mrt-1936 Insert > Header & footer
Daughter (norm)
Hb (g/l) 75 140
RBC (x10 12/l)
- 4.8
MCV (fl) 79.7 87
MCH (pg) 27.4 29
HbF (%) 6.7 0
HbA2 3.2 2.5
treatment Transfusion dependent
HBB:c.315+1G>A (IVS2-1g>a)
5 Mb deletion

Two different mechanisms:Mosaicism due to ‘second’ (somatic) mutation!
20-mrt-1937 Insert > Header & footer
1. Uni Parental Iso Disomy (UPID) 11p
Greek patient2. Deletion 5Mb on 11p
Italian patient
*
* *
*

5Mb deletion takes away HBB and H19 + IGF2 :
38 Insert > Header & footer
CNV’s patient
CNV’s mother
SNPs patient
SNPs mother
Affymetrix CytoScan HD Array with Chromosome Analysis Suite (ChAS, version 3.0) (Thermo Fisher Scientific, CA, USA).
Chromosoom 11
IGF2 H19
IGF2 H19
M
Pmethylated
This might explain the late onset: growth advantage for cells deprived of beta-synthesis
IGF2 H19
IGF2 H19
P
Pmethylated
methylated

Conclusion
• More laboratory tools become available in time (Next Generation Sequencing)
• Clinical genetic lab is changing
• Whole Exome/Genome Sequencing is still expensive, but becomes cheaper
• More emphasis on functionality of Variants of Unknown Significance (VUS) found by WES/WGS
• Therefore RNA and protein technologies will become more important
• Hemoglobinopathy diagnosis relies on phenotype-genotype correlation:Hematology, Hb separation and DNA analysis

Acknowledgements
20-Mar-1940 Insert > Header & footer
LUMC/LDGA:Sandra ArkesteijnGreet BakkerJeanette ter HuurneSharda BisoenRianne SchaapMaaike VerschurenLinda VijfhuizenClaudia RuivenkampCathy Bosch
Frank BaasChristi van Asperen
LUMC/LGTC:Rolf VossenEmile de MeijerQuint HottentotStefan WhiteHenk Buermans
Human and Clinical Genetics:Johan den Dunnen
National and KapodistrianUniversity of Athens:
Joan Traeger-Synodinos





























