Embed Size (px)
Citation preview

Parkin Facilitates the Elimination of Expanded PolyglutamineProteins and Leads to Preservation of Proteasome Function*
Received for publication, December 2, 2002, and in revised form, March 26, 2003Published, JBC Papers in Press, April 3, 2003, DOI 10.1074/jbc.M212235200
Yien Che Tsai‡, Paul S. Fishman§¶, Nitish V. Thakor‡, and George A. Oyler§¶�
From the ‡Department of Biomedical Engineering, The Johns Hopkins University, Baltimore, Maryland 21205, the§Department of Neurology, University of Maryland, Baltimore, Maryland 21201, and ¶Research Services,Baltimore Veterans Affairs Medical Center, Baltimore, Maryland 21201
Parkin, the most commonly mutated gene in familialParkinson’s disease, encodes an E3 ubiquitin ligase. Anumber of candidate substrates have been identified forparkin ubiquitin ligase action including CDCrel-1, o-glycosylated �-synuclein, Pael-R, and synphilin-1. Wenow show that parkin promotes the ubiquitination anddegradation of an expanded polyglutamine protein.Overexpression of parkin reduces aggregation and cy-totoxicity of an expanded polyglutamine ataxin-3 frag-ment. Using a cellular proteasome indicator systembased on a destabilized form of green fluorescent pro-tein, we demonstrate that parkin reduces proteasomeimpairment and caspase-12 activation induced by anexpanded polyglutamine protein. Parkin forms a com-plex with the expanded polyglutamine protein, heatshock protein 70 (Hsp70) and the proteasome, whichmay be important for the elimination of the expandedpolyglutamine protein. Hsp70 enhances parkin bindingand ubiquitination of expanded polyglutamine proteinin vitro suggesting that Hsp70 may help to recruit mis-folded proteins as substrates for parkin E3 ubiquitinligase activity. We speculate that parkin may function torelieve endoplasmic reticulum stress by preserving pro-teasome activity in the presence of misfolded proteins.Loss of parkin function and the resulting proteasomalimpairment may contribute to the accumulation of toxicaberrant proteins in neurodegenerative diseases includ-ing Parkinson’s disease.
Parkin, the most commonly mutated gene known to resultin familial Parkinson’s disease (PD),1 encodes an E3 ubiq-uitin ligase (1). Several substrates for parkin have been iden-tified including CDCrel-1, an o-glycosylated form of�-synuclein �Sp22, Pael-R (2), and synphilin-1 (3–5). These
parkin substrates have little sequence or functional similar-ities; however, Pael-R and �-synuclein have a propensity tomisfold and aggregate (2, 4). This common property of knownparkin substrates suggests that parkin may play a generalrole in the degradation of misfolded proteins, which mightotherwise overwhelm the ubiquitin-proteasome system(UPS).
Parkin has been demonstrated to function in the endoplas-mic reticulum-associated degradation (ERAD) of misfolded ERproteins (2, 6). Parkin is up-regulated during the unfoldedprotein response (6). Pael-R overexpression results in ER ac-cumulation of the protein, causing ER stress-induced celldeath; parkin overexpression ameliorates these effects (2). Pro-teasome function is also important for normal ERAD and pro-teasomal dysfunction can cause ER stress (7, 8). WhereasERAD is an important pathway for eliminating misfolded pro-teins in the ER, there are many misfolded aggregation-proneproteins that are translated in the cytosol including most of thepolyglutamine (poly(Q)) containing proteins. Accumulation ofmisfolded cytosolic and ER-translated proteins can ultimatelyinhibit proteasomal activity (9–11).
Whereas the cytotoxicity of expanded poly(Q) proteins maybe because of a variety of mechanisms (12–16), expandedpoly(Q) proteins impair proteasome function (9–11). Proteaso-mal dysfunction has also been demonstrated in PD brain andmay play a role in the accumulation of aberrant proteins andneuronal loss that characterize several of the adult neurode-generative diseases (17). Accumulation of aberrant proteins isa hallmark of both PD and the poly(Q) expansion diseases,which include Huntington’s disease (HD) and several spino-cerebellar ataxias. Overexpression of aberrant proteins hasbeen very useful for identifying genes and proteins capable ofmodifying their accumulation or toxicity. Molecular chaperonessuch as Hsp70 improve cell viability (18–20) and facilitate theelimination of poly(Q) proteins in cellular models and amelio-rate disease phenotype in transgenic Drosophila models (18,21–25). Hsp70 also improves the phenotype in a Drosophila PDmodel in which human �-synuclein is overexpressed (26). Par-kin appears to interact with Hsp70 along with the ubiquitinat-ing factor CHIP (27). The N terminus of parkin contains adomain homologous to ubiquitin called the ubiquitin-like (Ubl)domain. A similar domain in the human homologue of the yeastDNA repair factor (hhRad23) has been shown to interact withexpanded poly(Q) proteins (28) and bind the proteasome (29,30). Other Ubl domain containing proteins such as Dsk2 (31,32) and Ubp6 (33) also bind the proteasome. These findingssuggest that parkin may also bind expanded poly(Q) proteinsand proteasomes via its Ubl domain.
In the current investigations we have chosen to use an ex-panded poly(Q) protein as a model for cellular pathology medi-ated by misfolded proteins more generally. The relationship
* This work was supported by National Institutes of Health GrantNS24282 (to N. V. T.), a Veterans Affairs Merit and REAP Awards (toP. S. F.), National Institutes of Health Grant NS43658-01 and a Bur-roughs Welcome Fund Career Award (to G. A. O.), and the Society forProgressive Supranuclear Palsy (to P. S. F. and G. A. O.). The costs ofpublication of this article were defrayed in part by the payment of pagecharges. This article must therefore be hereby marked “advertisement”in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
� To whom correspondence should be addressed: Dept. of Neurology,10 North Greene St., Baltimore MD 21201. Tel.: 410-605-7000 (ext.6623); Fax: 410-605-7906; E-mail: [email protected].
1 The abbreviations used are: PD, Parkinson’s disease; E1, ubiquitin-activating enzyme; E2, ubiquitin carrier protein; E3, ubiquitin-proteinisopeptide ligase; UPS, ubiquitin-proteasome system; ERAD, endoplas-mic reticulum-associated degradation; ER, endoplasmic reticulum; HD,Huntington’s disease; Ubl, ubiquitin-like; poly(Q), polyglutamine; GST,glutathione S-transferase; GFP, green fluorescent protein; PBS, phos-phate-buffered saline; HA, hemagglutinin; HEK293, human embryonickidney-derived 293; htt, Huntingtin.
THE JOURNAL OF BIOLOGICAL CHEMISTRY Vol. 278, No. 24, Issue of June 13, pp. 22044–22055, 2003Printed in U.S.A.
This paper is available on line at http://www.jbc.org22044
by guest on May 14, 2018
http://ww
w.jbc.org/
Dow
nloaded from

between genetically determined neurodegeneration and abnor-mal misfolding of a disease causing protein is well establishedin poly(Q) diseases (34). Poly(Q)-mediated neurodegenerationis likely to serve as a model for a number of neurodegenerativedisorders in which genetic mutations of the disease-relatedproteins causes misfolding and aggregation such as in�-synuclein, SOD1, and Tau mutations (35–37). Investigationsof the role of parkin in facilitating the degradation of misfoldedproteins may also be relevant to sporadic PD and several of theneurodegenerative conditions (38, 39).
In this study, we address whether parkin promotes theubiquitination and degradation of expanded poly(Q) proteins,thus reducing impairment of the UPS. We also examine theinteraction of parkin with expanded poly(Q) proteins, Hsp70,and the proteasome. Our goal was to further understand therole of Hsp70 binding in parkin function and the role ofparkin in preventing cell death induced by misfolded pro-teins. Proteasomal dysfunction can result in ER stress-associated cell death because retrotranslocation of misfoldedproteins from the ER requires ongoing ubiquitination andproteasome function in the cytosol (8, 40). For this reason, weassess the effects of parkin overexpression not only on UPSfunction but also on the activation of pathways involved inER stress-induced cell death.
MATERIALS AND METHODS
Plasmid Construction and Protein Expression
Total RNA was extracted from cultures of human embryonic kidney-derived 293 (HEK293) cells using TRIzol reagent (Invitrogen). Oli-go(dT)-primed first strand cDNAs were generated using Thermo-ScriptTM reverse transcriptase-PCR system (Invitrogen). Polymerasechain reaction (PCR) amplification of parkin cDNA was performedusing parkin-specific primers. Parkin mutants were generated by PCRwith wild-type parkin cDNA as template and cloned into the mamma-lian expression vectors pcDNA3.1(�) (Invitrogen) and CMV-FLAG 7.1(Sigma). Wild type and mutant parkin were cloned into pGex-2T(Amersham Biosciences) and pRSETA vector (Invitrogen). GST-E6AP,GST-parkin, and mutants were produced in the BL21 strain of Esche-richia coli and purified with glutathione-Sepharose 4B (AmershamBiosciences). His6-parkin was expressed in BL21(DE3) bacteria andpurified with TALON resin (Clontech). Poly(Q) proteins (Gln26 andGln79) fused to green fluorescent protein (GFP) were generated using afragment of ataxin-3 with 26 and 79 glutamine repeats, respectively.The ataxin-3 fragments were generated by PCR and subcloned intoEGFP-C1 vector (Clontech) leaving 44 amino acids of ataxin-3 N-ter-minal to the poly(Q) tract and 26 amino acids at the C terminus.GST-E6AP (originally from A. Weissman) and His6-ubiquitin (original-ly from D. Bohman (41)) cDNAs were generous gifts from Cecile Pickart(Johns Hopkins). GFPu plasmid was kindly provided by Ron Kopito(Stanford); myc-E6AP cDNA was kindly provided by Allan Weissman(National Institutes of Health, NCI); mouse procaspase-12 cDNA waskindly provided by Junying Yuan (Harvard); cDNA for Hsp70 andataxin-3 Gln28 and Gln84 were kindly provided by Henry Paulson (Uni-versity of Iowa).
Transfection
N18 and HEK293 cells were transfected with various expressionvectors using FuGENE 6 (Roche Diagnostics) according to the manu-facturer’s recommendations. Total amounts of plasmid DNA in individ-ual transfections were adjusted to be equivalent in all transfectionswith empty vector. Transfected cells were cultured for 48–72 h andharvested for immunoprecipitation, Western blotting, or cell deathassay.
Generation of GFPu Cell Lines
HEK293 cells were transfected with GFPu plasmid and cultured inmedium containing G418. Isolated foci were selected for expansion andthe cell lines were screened for an increase in GFPu fluorescence upontreatment with the proteasome inhibitor MG132.
Immunofluorescence
The YAC72 (42) transgenic mice, which express human Huntingtinwith 72 glutamine repeats as a model for Huntington’s disease, were
used in this study. Brains from 12–15-month-old YAC72 transgenicmice were removed after perfusion with 4% formaldehyde and post-fixed overnight in 2% paraformaldehyde and 30% sucrose in phosphate-buffered saline (PBS). Brains were sectioned in series of 10 20-�mcoronal sections on a cryostat and collected in PBS (pH 7.5). Immuno-fluorescence detection in cultured cells was performed as described inRef. 43. The following antibodies were used: anti-FLAG M2 (Sigma),anti-HA (Clontech), anti-parkin (Cell Signaling Technologies), anti-Huntingtin (Chemicon), and anti-ubiquitin (generous gift from CecilePickart). Cy5-conjugated donkey anti-rabbit and fluorescein isothiocya-nate or Cy3-conjugated donkey anti-mouse were used as secondaryantibodies (Chemicon). Controls included omission of primary antibodyor primary antibody alone. As additional controls of parkin immunoflu-orescent colocalization with poly(Q)-containing proteins, pre-adsorptionof the parkin antibody was performed using purified parkin expressedin bacteria (5 mg of parkin protein/ml of parkin antibody).
All tissue was collected in accordance with the institutional reviewboard-approved guidelines. Human HD brains were obtained anony-mously from the Brain and Tissue Bank for Developmental Disorders atthe University of Maryland. Frontal cortex and caudate tissue sampleswere taken from two different patients. The brains were from malepatients with genetically confirmed HD. The patients died at ages 47and 51 years old, respectively. The brains were preserved in 10%formalin for 5 to 6 years prior to immunofluorescence observations.Tissue from the selected regions were dissected into 1-cm cubes andimmersed in 30% sucrose in PBS for 72 h at 4 °C. The tissue sampleswere sectioned on a cryostat in 10-�m sections and collected in PBS.Immunofluorescence was performed using anti-Huntingtin (Chemicon)and anti-parkin antibodies HP2A recognizing amino acids 342–353 ofparkin (a generous gift of Michael Schlossmacher). The HP2A antibodyhas been successfully used on human brain autopsy samples to demon-strate localization of parkin to Lewy bodies (44). Fluorescein isothio-cyanate-conjugated donkey anti-mouse and Cy5-conjugated donkeyanti-rabbit were used as secondary antibodies (both from Chemicon).
Immunoprecipitation and Immunoblot Analysis
Transfected cells were harvested, washed in PBS, and lysed in lysisbuffer (50 mM Tris, pH 7.5, 150 mM NaCl, 1 mM phenylmethylsulfonylfluoride, 2 �g/ml aprotinin, 0.5 �g/ml leupeptin, 0.5–1.0% TritonX-100). Lysates were centrifuged at 15,000 � g for 10 min, and thesupernatant was precleared before immunoprecipitation. Samples (300�g) were incubated with 40 �l of anti-FLAG M2 affinity gel (Sigma) at4 °C for 2 h with constant mixing. The immobilized immunocomplexeswere collected by centrifugation, washed 3� with lysis buffer, andboiled in SDS sample buffer for SDS-PAGE. For brain homogenates,mouse brains were removed and homogenized in lysis buffer containing2 mM ATP and 1% Nonidet P-40 with a Dounce homogenizer. Brainhomogenates were centrifuged at 15,000 � g for 10 min at 4 °C and usedfor immunoprecipitation (1 mg/ml) as described above using anti-Hun-tingtin (Chemicon) or anti-parkin antibodies (Cell Signaling Technolo-gies or HP2A kindly provided by Michael Schlossmacher). Rabbit anti-mouse was used as control IgG for nonspecific co-immunoprecipitation.The antigen complex was eluted and processed for SDS-PAGE. Forimmunoblots, cells were lysed in lysis buffer and briefly sonicated.Equal amounts (50 �g) of cell lysates were separated by SDS-PAGE,transferred to membranes, probed with the appropriate antibody, andvisualized using chemiluminescence. GFP-Gln79, GFP-Gln26, and GFPwere detected with rabbit polyclonal anti-GFP (Clontech). FLAG-tagged wild-type and mutant parkin were detected using anti-FLAGM2 (Sigma). HA-tagged poly(Q) proteins were detected with anti-HA(Clontech). Caspase-12 was detected by rabbit polyclonal anti-caspase-12 (Cell Signaling Technologies). The Rpt6/S8 and HC3 sub-units of the proteasome were detected with rabbit polyclonal antibodiesagainst p45 and HC3, respectively (Affiniti).
Confocal Microscopy and Quantification of Aggregatesand Cell Viability
All images were acquired on a Zeiss LSM 510 confocal microscope.Images were minimally processed for presentation. In all experimentsin which aggregates and cell viability were quantified, the same ob-server, blinded to the transfection, scored the cells. For GFP-Gln79
aggregates, cells with large visible inclusions were counted (see Fig.4A). Cell viability was assayed by propidium iodide exclusion under afluorescence microscope (Zeiss Axiovert). Only cells expressing GFP-poly(Q) proteins were scored for propidium iodide exclusion. Transfec-tion efficiency is about 50%. For each experiment, 200-500 cells werecounted for each treatment.
Parkin Preserves Proteasome Function 22045
by guest on May 14, 2018
http://ww
w.jbc.org/
Dow
nloaded from
Ubiquitination Assays
Auto-ubiquitination—FLAG-tagged wild-type and mutant parkin ex-pressed in HEK293 cells were immunoprecipitated using anti-FLAGM2 antibody (300 �g, described above). The ubiquitination reactioncontained the immunocomplexes, mammalian ubiquitin-activating en-zyme E1 (70 nM), the E2 ubiquitin-conjugating enzyme UbcH7 (100 nM),and 125I-labeled ubiquitin (5 �M) in a reaction buffer of 50 mM Tris (pH7.6), 5 mM MgCl2, 2 mM ATP with an ATP regenerating system. Thereaction (50 �l) was incubated at 37 °C with gentle agitation for theindicated period of time and quenched with 2� SDS sample buffer (2).
In Vitro Ubiquitination of Expanded Poly(Q) Proteins—35S-Labeledataxin-3 Gln79 was translated in vitro using either a rabbit reticulocytelysate or S30 T7 bacteria lysate system (Promega). Ubiquitinationreactions contained 1 �l of translation mixture, mammalian E1 (100nM), UbcH7 (300 nM), bovine ubiquitin (5 �M, Sigma), E3 (GST-E6AP,GST-parkin or mutants, 500 nM) in 50 �l of reaction buffer (above).Hsp70 (1 �g, Sigma) was added where indicated. The reaction wasincubated for 2 h at 37 °C and quenched with 2� SDS sample buffer.The reaction mixtures were separated by SDS-PAGE and processed forvisualization on a Storm PhosphorImager (Amersham Biosciences).
Pulse-Chase Experiments
HEK293 cells were transfected with the indicated expression plas-mids and cultured for 30 h. Cells were washed and starved in Met/Cys-free medium for 1 h before labeling with 50 �Ci/ml [35S]Met and 35S-Cysfor 1 h (Promix, Amersham Biosciences). After labeling, cells werewashed three times and chased in normal medium supplemented withunlabeled Met and Cys. Where indicated, 50 �M MG132 was added tothe labeling and chase medium to inhibit proteasome activity. At theindicated times, cells were washed twice with PBS, lysed in RIPAbuffer, and briefly sonicated before immunoprecipitation with anti-GFPantibody. Immunocomplexes were washed, boiled in SDS sample buffer,and separated by SDS-PAGE. Radiolabeled proteins were visualized byexposure to phosphorimage screens and analyzed with a Storm Phos-phorImager (Amersham Biosciences).
Assay for the Ubiquitin-Proteasome System in GFPu Cells
HEK293 cells stably expressing GFPu were transiently transfectedwith HA-Gln79 and FLAG-tagged wild-type or mutant parkin. After72 h, cells were imaged for GFPu fluorescence and HA-Gln79 andFLAG-parkin expression (immunofluorescence). For cells transfectedwith HA-Gln79, GFPu fluorescence of individual cells expressing HA-
Gln79 was measured. To evaluate the effect of overexpressing parkin onGFPu fluorescence, GFPu fluorescence of cells expressing FLAG-parkinwere measured.
In Vitro Binding Assays
An aliquot containing 10 �g of GST, GST-parkin, or GST-parkinmutants immobilized on glutathione-Sepharose 4B was incubated with0.1 �M purified bovine 26 S proteasomes (gift of Y. Lam, Johns Hopkins(45)) in 20 �l of binding buffer (50 mM Tris, pH 7.6, 1 mM dithiothreitol,1 mM ATP) for 2 h at 4 °C with constant mixing. The immobilizedproteins were collected by centrifugation, washed 3� with bindingbuffer, and boiled in SDS sample buffer for SDS-PAGE (2). HA-Gln84
was translated in vitro using S30 T7 bacteria lysate. An aliquot of thetranslation mixture was incubated with His6-parkin (1 �g) in 25 �l ofbuffer containing 50 mM Tris (pH 7.6), 1 mg/ml ovalbumin for 2 h atroom temperature with constant mixing. HA-Gln84 was immunoprecipi-tated with anti-HA antibody and His6-parkin immunoprecipitated withanti-His6 antibody (Clontech).
Statistical Analysis
Non-parametric statistics were used in this study to avoid assump-tions about the underlying distributions of the data. The Kruskal-Wallis test was used for multiple comparisons and p � 0.05 consideredstatistically significant. For two-sample comparison, treatment wascompared with control (transfection with empty vector) using two-tailedWilcoxon signed-rank test. Data were analyzed with S-Plus and pre-sented as mean � S.D.
RESULTS
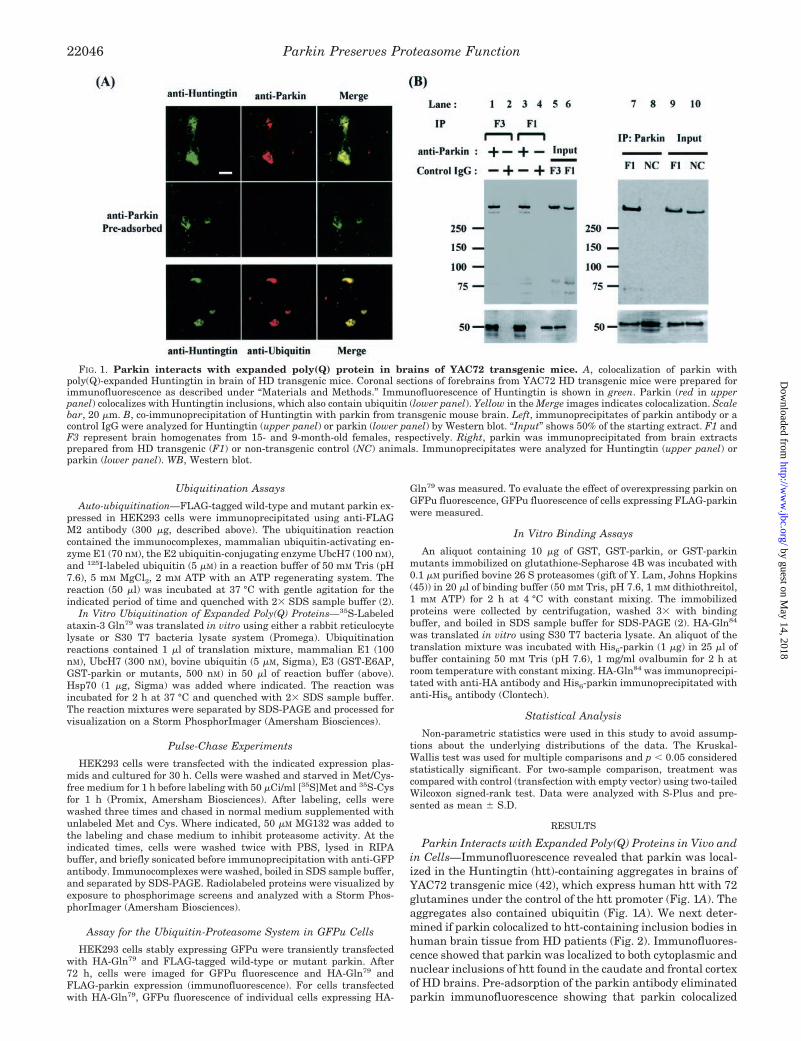
Parkin Interacts with Expanded Poly(Q) Proteins in Vivo andin Cells—Immunofluorescence revealed that parkin was local-ized in the Huntingtin (htt)-containing aggregates in brains ofYAC72 transgenic mice (42), which express human htt with 72glutamines under the control of the htt promoter (Fig. 1A). Theaggregates also contained ubiquitin (Fig. 1A). We next deter-mined if parkin colocalized to htt-containing inclusion bodies inhuman brain tissue from HD patients (Fig. 2). Immunofluores-cence showed that parkin was localized to both cytoplasmic andnuclear inclusions of htt found in the caudate and frontal cortexof HD brains. Pre-adsorption of the parkin antibody eliminatedparkin immunofluorescence showing that parkin colocalized
FIG. 1. Parkin interacts with expanded poly(Q) protein in brains of YAC72 transgenic mice. A, colocalization of parkin withpoly(Q)-expanded Huntingtin in brain of HD transgenic mice. Coronal sections of forebrains from YAC72 HD transgenic mice were prepared forimmunofluorescence as described under “Materials and Methods.” Immunofluorescence of Huntingtin is shown in green. Parkin (red in upperpanel) colocalizes with Huntingtin inclusions, which also contain ubiquitin (lower panel). Yellow in the Merge images indicates colocalization. Scalebar, 20 �m. B, co-immunoprecipitation of Huntingtin with parkin from transgenic mouse brain. Left, immunoprecipitates of parkin antibody or acontrol IgG were analyzed for Huntingtin (upper panel) or parkin (lower panel) by Western blot. “Input” shows 50% of the starting extract. F1 andF3 represent brain homogenates from 15- and 9-month-old females, respectively. Right, parkin was immunoprecipitated from brain extractsprepared from HD transgenic (F1) or non-transgenic control (NC) animals. Immunoprecipitates were analyzed for Huntingtin (upper panel) orparkin (lower panel). WB, Western blot.
Parkin Preserves Proteasome Function22046
by guest on May 14, 2018
http://ww
w.jbc.org/
Dow
nloaded from

with inclusions containing poly(Q)-expanded htt in HD brainand in transgenic mouse models of HD.
We next examined if parkin interacts with the poly(Q)-ex-panded htt in the soluble fraction by co-immunoprecipitation(Fig. 1B). Parkin immunoprecipitates from YAC72 brain homo-genates contain htt, showing that parkin interacts with htt invivo. To determine whether the state of htt aggregation effectedits interaction with parkin, immunoprecipitation of parkin wasperformed from brain homogenates of 9-month-old (before ag-gregation occurs) and 15-month-old YAC72 transgenic mice,which contain htt inclusions (42). The age of the animal had noeffect on the co-immunoprecipitation of parkin with htt, dem-onstrating that the state of aggregation of htt does not effectthe interaction of parkin and htt (Fig. 1B, left). Htt was absentfrom parkin immunoprecipitates from non-transgenic controlbrain (Fig. 1B, right). Because the YAC72 mice express humanhtt at only one-third to one-half the level of endogenous htt(42), the interaction of parkin with poly(Q)-expanded htt inthese animals is unlikely to be an artifact of overexpression ofthe human htt. The htt antibody used in this experiment rec-ognizes both mouse and human htt. These results suggest thatparkin interacts preferentially with poly(Q)-expanded htt.
To determine whether parkin interacts with poly(Q)-ex-panded proteins in cells, we used a fragment of ataxin-3 con-taining Gln79 fused to GFP (GFP-Gln79). Human embryonickidney-derived 293 (HEK293) and mouse neuroblastoma N18cells overexpressing GFP-Gln79 develop fluorescent aggregateslocated in the cytoplasmic, perinuclear, or intranuclear regions(see Figs. 3 and 5). Immunofluorescence analysis in N18 cellsshowed that endogenous parkin colocalized with GFP-Gln79 inthese aggregates (Fig. 3A), even though it is normally ex-pressed in the cytoplasm and excluded from the nucleus (Fig.
3B). We next examined if parkin interacts GFP-Gln79 in thesoluble fraction by co-immunoprecipitation. HEK293 cells weretransfected with FLAG-tagged parkin and GFP-Gln79. Immu-noprecipitates of FLAG-tagged parkin contain GFP-Gln79
showing that parkin interacts with GFP-Gln79 in cells (Fig. 3C,left). We next determined if parkin interacts preferentially withexpanded poly(Q) proteins in our model. HEK293 cells weretransfected with FLAG-tagged parkin together with GFP,GFP-Gln26, or GFP-Gln79. Immunoprecipitation of FLAG-tagged parkin from cell lysates preferentially co-precipitatedGFP-Gln79 in comparison with GFP-Gln26, showing that par-kin preferentially interacts with Gln79 in these cells (Fig. 3C,right, compare lanes 1 and 2). Immunoprecipitation of FLAG-parkin did not co-precipitate GFP indicating that the interac-tion was specific for the poly(Q) region.
Parkin Promotes the Ubiquitination and Degradation of anExpanded Poly(Q) Protein—To establish a functional role forFLAG-tagged parkin, we immunoprecipitated FLAG-taggedparkin from HEK293 cell lysates and examined its E3 ubiq-uitin ligase activity in vitro (Fig. 4A). ImmunoprecipitatedFLAG-parkin showed autoubiquitination in vitro in the pres-ence of an E1 and UbcH7 (Fig. 4A). The disease-associatedparkin truncation mutant, Q311X, on the other hand, did notshow any E3 activity in this assay (lane 4), consistent withprevious reports (1, 3, 6). Interestingly, a single mutation of acritical cysteine residue of the distal RING (C418R) completelyabolished parkin E3 activity (Fig. 4A, lane 5), suggesting thetwo RING fingers may function as a single motif (46).
Because parkin exhibits ubiquitin ligase activity and inter-acts with GFP-Gln79, it is natural to ask if parkin can ubiquiti-nate the expanded poly(Q) protein. To address this question,we prepared 35S-labeled ataxin-3 Gln79 by in vitro translationand tested if bacterially produced GST-parkin promoted itsubiquitination (Fig. 4B). As shown in Fig. 4B, wild-type parkinbut not the Q311X or C418R parkin mutants, promoted theubiquitination of Gln79. Although monoubiquitinated Gln79
was the predominant product, more polyubiquitinated forms ofGln79 were also produced (for example, lane 8). Previous ge-netic studies suggested that E6AP promotes the ubiquitinationof a different poly(Q)-expanded protein, ataxin-1 (47). As acontrol, we determined if E6AP could promote the ubiquitina-tion of Gln79 in vitro. Contrary to our expectation, E6AP did notpromote ataxin-3 Gln79 ubiquitination in our in vitro assay(Fig. 4B, right panel).
The best known function of protein ubiquitination is to targetproteins for degradation by the 26 S proteasome. We thereforeassessed if parkin facilitated the degradation of GFP-Gln79 incultured cells. Pulse-chase experiments showed that co-expres-sion of parkin accelerated the degradation of GFP-Gln79 (Fig.4C, ● versus Œ) and the effect could be blocked with a protea-some inhibitor MG132 (● versus �). Co-expression of E6AP orthe parkin mutant Q311X had no detectable effect on the rateof turnover of GFP-Gln79 (Fig. 4C), in agreement with thefailure of these proteins to promote GFP-Gln79 ubiquitinationin vitro (Fig. 4B). These data show that parkin promotes thedegradation of GFP-poly(Q) proteins via the ubiquitin-protea-some pathway.
Parkin Suppresses Aggregation and Toxicity of GFP-Gln79—Based on the results presented above, we considered it possiblethat parkin could protect cells from deleterious effects of over-expressing expanded poly(Q) proteins. N18 cells transientlytransfected with GFP-Gln79 developed fluorescent aggregates(Fig. 5A), as previously seen for this and other expandedpoly(Q) proteins (9, 11, 13, 15, 18–20, 48–50). Using confocalmicroscopy, we assigned transfected N18 cells to one of threegroups: cells with 1) large fluorescent inclusions; 2) small mul-
FIG. 2. Colocalization of parkin with poly(Q)-expanded Hun-tingtin in human HD brains. Fixed sections of caudate (upper panel)or frontal cortex (lower panel) from two separate HD brains wereprepared for immunofluorescence as described under “Materials andMethods.” Immunofluorescence of Huntingtin is shown in green. Parkin(red) colocalizes with both cytoplasmic and nuclear inclusions in boththe caudate and frontal cortex. Yellow in the Merge images indicatescolocalization. Scale bar, 5 �m.
Parkin Preserves Proteasome Function 22047
by guest on May 14, 2018
http://ww
w.jbc.org/
Dow
nloaded from

tiple inclusions; or 3) diffuse fluorescence (Fig. 5A). Co-trans-fection of FLAG-parkin with GFP-Gln79 significantly reducedthe percentage of cells showing large inclusions (Fig. 5B), in amanner that was dependent on the amount of parkin cDNAtransfected (Fig. 5B, left). The parkin-dependent reduction inaggregate size was greatly diminished in the presence of theproteasome inhibitor MG132, whereas MG132 only minimallyincreased aggregate size in the absence of co-expressed parkin(Fig. 5B, right). No parkin-dependent reduction in GFP-Gln79
aggregation occurred upon co-transfection of the parkin mu-tants Q311X and C418R (Fig. 4C); nor did co-expression of twoother E3s, E6AP and XIAP, mimic the effect of wild-type par-kin. These results are consistent with the failure of the parkinmutants and E6AP to promote GFP-Gln79 ubiquitination (Fig.4B) or degradation (Fig. 4C). These results thus suggest thatwild-type parkin inhibits the formation of GFP-Gln79 inclu-sions by promoting the ubiquitination and degradation of thepoly(Q) protein. Consistent with this interpretation, immuno-blot analysis showed that overexpression of wild-type parkin,but not mutant Q311X, reduced the steady-state level of GFP-Gln79 (Fig. 5D, lanes 2–4 versus 5) and this effect was blockedby MG132 (lane 6). The failure of parkin mutant Q311X toreduce GFP-Gln79 levels was not because of poor expression(Fig. 5D, lower panel).
To investigate if parkin protects against toxicity of expandedpoly(Q) proteins, we co-expressed FLAG-tagged parkin withGFP-Gln79 in N18 cells. The ratio of parkin to GFP-Gln79
plasmid DNA was varied (while keeping the total amount ofDNA constant with empty vector) to investigate the dose effect
of parkin overexpression. Co-expression of wild-type parkinsignificantly enhanced cell viability as assayed by propidiumiodide exclusion (Fig. 6A). Adding MG132 greatly reduced theprotective effect of parkin indicating that its effects on proteinaggregation and cell survival both depend on the ubiquitin-proteasome pathway. Co-expression of parkin mutants Q311Xor C418R showed no protective effect (Fig. 6B). Co-expressionof two other E3s, E6AP and XIAP, offered no protection againstGFP-Gln79 toxicity (Fig. 6B), indicating that protection in thismodel system is specific to enzymatically active parkin ratherthan a general property of overexpression of any ubiquitinligase. It is reasonable to propose that this protection relies onthe parkin-dependent ubiquitination and degradation of GFP-Gln79 documented in Figs. 4 and 5 above.
Parkin Preserves the Ubiquitin-Proteasome System from Im-pairment by an Expanded Poly(Q) Protein—Expanded poly(Q)proteins have been shown to inhibit the proteasome that leadsto ER stress contributing to expanded poly(Q)-mediated celldeath (9–11). To study the effect of parkin overexpression onproteasome function, we generated HEK293 cell lines stablyexpressing GFPu, a form of GFP that is selectively targeted toproteasomes, as shown by a 6–7-fold increase in GFPu fluores-cence following treatment with 10 �M MG132 for 6 h (Fig. 7A,panel 6; see also Ref. 9). Transient transfection of HA-Gln79 inGFPu cells increased GFPu fluorescence by 7–8-fold (Fig. 7, A,second panel, and B) confirming a previous demonstration ofproteasome inhibition by overexpression of a poly(Q)-expandedhtt exon 1 (9). Co-expression of wild-type parkin reduced theincrease in GFPu fluorescence caused by HA-Gln79 overexpres-
FIG. 3. Interaction of parkin withGFP-poly(Q) proteins in culturedcells. A, colocalization of parkin withGFP-Gln79 in N18 cells. N18 rat neuro-blastoma cells were transfected withGFP-Gln79. GFP-Gln79 was visualized byGFP fluorescence (green) and endogenousparkin by anti-parkin immunofluores-cence (red). A fraction of the cells devel-oped cytoplasmic and nuclear aggregates,which contained both GFP-Gln79 and par-kin. Scale bar, 10 �m. B, distribution ofendogenous parkin in untransfected N18neuroblastoma cells by immunofluores-cence. C, co-immunoprecipitation of GFP-Gln79 with parkin. Left, HEK293 cellswere transfected with FLAG-parkin andGFP-Gln79. Cell lysates were processedfor immunoprecipitation with anti-FLAGantibody. Pre-adsorption of anti-FLAGantibody with 3� FLAG peptide pre-vented immunoprecipitation of FLAG-parkin. Right, HEK293 cells were trans-fected with FLAG-parkin and GFP-Gln79
(lanes 1 and 4), GFP-Gln26 (lanes 2 and 5),or GFP (lanes 3 and 6). Cell lysates wereprocessed for immunoprecipitation withanti-FLAG antibody and immunoblottedfor GFP (upper panel) or Fll (lower panel).WB, Western blot.
Parkin Preserves Proteasome Function22048
by guest on May 14, 2018
http://ww
w.jbc.org/
Dow
nloaded from

sion, resulting in a distribution that was closer to the controllacking HA-Gln79 expression (Fig. 7, A, third versus fourthpanels; see also Fig. 7C). Co-expression of the inactive C418R-parkin mutant failed to ameliorate the increase in GFPu fluo-rescence caused by HA-Gln79 overexpression (Fig. 7B, fourthpanel; see also Fig. 7C). Importantly, overexpression of parkinalone did not significantly decrease GFPu fluorescence showingthat parkin did not directly target GFPu for degradation (Fig.7C). The reduction in GFPu fluorescence observed when parkinwas co-expressed with HA-Gln79 therefore indicated improvedUPS function in these cells. This improvement in UPS functionresults from parkin-facilitated degradation of expandedpoly(Q) proteins
Parkin Reduces Caspase-12 Activation—Proteasome func-tion is essential for normal ERAD and relief of ER stress (7, 8).Overexpression of poly(Q) containing proteins has been shownto induce ER stress by inhibiting proteasome activity (12, 51).Blocking ER stress-mediated apoptosis via the ASK pathwayprevented the neuronal death induced by poly(Q) overexpres-sion (51), suggesting that ER stress resulting from poly(Q)inhibition of the proteasome may be important in poly(Q)-mediated cell death. To determine whether the ability of parkinto preserve proteasome function in the setting of poly(Q) over-
expression translates into reduced ER stress, we monitoredcaspase-12 activation as a marker for ER stress. Activation ofcaspase-12 in murine neurons is an important effector pathwayfor ER stress-induced neuronal cell death (52). HEK293 cellsco-transfected with procaspase-12 and GFP-Gln79 plasmidsshowed increased activation of caspase-12 (Fig. 8, lanes 1 and2) and cell death (data not shown). Co-expression of wild-typeparkin, but not the inactive mutant parkin-Q311X, reduced theactivation of caspase-12 (Fig. 8, lanes 3 and 4). These resultssuggest that parkin can protect cells against ER stress-inducedcell death by improving proteasome function and consequentlymaintaining ERAD, which reduces ER stress.
Parkin Interacts with Hsp70 and the Proteasome—The mo-lecular chaperone Hsp70 has been previously shown to reduceaggregation of expanded poly(Q) proteins (18, 19, 22, 23, 25,53). Hsp70 is also involved in the degradation of certain mis-folded proteins (21, 54–56). We have shown that parkin inter-acts with, and promotes the degradation of, misfolded poly(Q)proteins. We therefore tested if parkin interacts with Hsp70.We found that antibody against FLAG-parkin co-precipitatedHsp70 (Fig. 9A, top panel), confirming a previous report byImat et al. (27) that parkin forms a complex with Hsp70 in cells.In contrast, Hsp70 did not co-precipitate with a different E3
FIG. 4. Parkin promotes the ubiquitination and degradation of an expanded poly(Q) protein. A, parkin possesses E3 activity.FLAG-tagged wild-type and mutant parkin were immunoprecipitated from lysates of HEK293 cells transfected with the indicated expressionplasmids and used for in vitro ubiquitination assays with 125I-labeled ubiquitin (see “Materials and Methods”). Anti-FLAG M2 affinity gelpreincubated with 3� FLAG peptide was used for immunoprecipitation in the control reaction in lane 3 (without E3). B, left, parkin promotes theubiquitination of Gln79 in vitro. 35S-Labeled ataxin-3 Gln79 was prepared by in vitro translation and used for ubiquitination assay with GST-fusedwild-type or mutant parkin proteins expressed in E. coli. The asterisk (*) highlights the product formed from the addition of a single ubiquitin toGln79. Right, E6AP, an E3 implicated in ataxin-1 ubiquitination, did not promote ubiquitination of Gln79 in this assay. C, parkin accelerates theturnover of GFP-Gln79 in cultured cells. HEK293 cells were co-transfected with GFP-Gln79 and the indicated plasmids. Thirty hours post-transfection, cells were pulse-labeled for 1 h and the degradation of 35S-labeled GFP-Gln79 was monitored by immunoprecipitation with anti-GFPduring a chase with unlabeled amino acids. In one experiment with wild-type parkin, MG132 was added at 50 �M. Top, autoradiograph ofimmunoprecipitates. Bottom, quantitation of data (n � 3, mean � S.D.). Symbols: co-transfection of GFP-Gln79 with empty vector (Œ); wild-typeparkin (●); wild-type parkin with 50 �M MG132 (�); parkin mutant Q311X (�); or E6AP (�).
Parkin Preserves Proteasome Function 22049
by guest on May 14, 2018
http://ww
w.jbc.org/
Dow
nloaded from

(E6AP, bottom panel). To determine the region of parkin in-volved in this interaction, we constructed FLAG-tagged dele-tion mutants of parkin and examined their interaction withHsp70 by co-immunoprecipitation from lysates of cells trans-fected with FLAG-parkin and GFP-Gln79 (Fig. 9B). The resultsrevealed that an intact RING-IBR-RING region was essentialfor Hsp70 interaction, consistent with a previous report ofHsp70 binding to parkin�Pael-R complex (27). Because Hsp70has been previously shown to interact with misfolded proteinsincluding expanded poly(Q) proteins (11, 18–21, 23, 53, 57),co-precipitation of Hsp70 and parkin could reflect either adirect interaction between parkin and the expanded repeatprotein, or an indirect interaction with a complex containingHsp70 together with the expanded repeat protein.
The interaction of parkin with Hsp70 may be functionallyimportant in at least two ways. Hsp70 may enhance the bind-ing and ubiquitination of the substrate by parkin similar toRefs. 58 and 59. Alternatively, Hsp70 may inhibit the E3 ac-tivity of parkin to promote refolding of the substrate suggestedin Ref. 27. To better understand the role of parkin interactionwith Hsp70, we examined the interaction of parkin and HA-tagged poly(Q) protein in vitro. HA-Gln84 was in vitro trans-lated, in the presence or absence of Hsp70, using a bacterialsystem, and incubated with bacterially expressed His6-parkin.Hsp70 enhanced the binding of HA-Gln84 to His6-parkin (Fig.9C, right versus left, top panels). In a parallel experiment,Hsp70 similarly enhanced the co-immunoprecipitation of His6-parkin with HA-Gln84 (Fig. 9C, bottom panels). We next exam-ined the effect of Hsp70 on the ability of parkin to ubiquitinatein vitro translated poly(Q)-expanded proteins. 35S-Labeled
ataxin-3 Gln79 was translated in vitro, in the absence of Hsp70,using a bacterial system and then used as a substrate in aubiquitination assay with or without added Hsp70 (Fig. 9D).The addition of Hsp70 enhanced parkin-dependent ubiquitina-tion of Gln79, possibly by enhancing the binding of parkin toGln79 (cf. panel C). We did not observe this requirement inHsp70 for in vitro ubiquitination of Gln79 translated using areticulocyte lysate system (Fig. 4). This observation may reflectthe differences between bacteria lysate and reticulocyte lysatesystems; reticulocyte lysate already contains substantialamounts of many molecular chaperones (estimated at 2 �M
functional Hsp70 and Hsp90 (60)). Imai et al. (27) have alsoobserved that larger amounts of Hsp70 can inhibit parkin-de-pendent ubiquitination of the substrate Pael-R in vitro. Takentogether, our results suggest that parkin preferentially bindsand ubiquitinates misfolded poly(Q) proteins via its interactionwith Hsp70.
The efficient degradation of misfolded proteins requires theefficient presentation of these substrates to the proteasome.Substrate recognition is generally dependent on polyubiquiti-nation, but additional factors may be necessary for efficientrecognition or processing in some cases. The Ubl domains ofcertain other Ubl proteins have been shown to bind the protea-some, an event that may recruit additional factors to the deg-radation machinery (29, 61–63). We therefore investigated thepossibility that parkin interacts with the proteasome. We ob-served that Rpt6, a proteasome ATPase subunit, co-precipi-tates with parkin from lysates of cells co-transfected withFLAG-parkin and GFP-Gln79 (Fig. 10A). Because proteasomesubunits have been found in inclusions containing expanded
FIG. 5. Parkin reduces aggregationof GFP-poly(Q) proteins. A, GFP-Gln79
inclusion body formation in N18 cells.GFP-Gln79 was expressed by transfection(see “Materials and Methods”). Confocalimages representative of the three typesof inclusions (see text) are shown. B, par-kin reduces GFP-Gln79 aggregation. Left,N18 cells were co-transfected with GFP-Gln79 and empty vector (control) or vary-ing quantities (0.5, 1.0, and 2.0 �g) ofFLAG-parkin. Right, cells co-transfectedwith empty vector and 2.0 �g of FLAG-parkin were cultured in the presence of aproteasome inhibitor MG132 (added 48 hpost-transfection). These experimentswere performed together but presented ontwo panels for clarity. C, specificity of par-kin-dependent reduction in aggregatesize. N18 cells were co-transfected withGFP-Gln79 and empty vector, FLAG-par-kin, the indicated parkin mutants, E6APand XIAP, and assayed for GFP-Gln79 ag-gregation. Data shown are means witherror bars indicating S.D. (n � 9; *, p �0.01; **, p � 0.005 compared with controltransfection with empty vector). D, par-kin reduces steady state level of GFP-Gln79. HEK293 cells were transfectedwith GFP-Gln79 and varying quantities(0, 0.5, 1.0, and 2.0 �g) of parkin or 2.0 �gof mutant Q311X. 50 �g of total cell lysatewere analyzed by Western blot for GFP(top panel) or FLAG epitope (bottom).Cells were cultured in 50 �M MG132 (add-ed 48 h post-transfection) overnight inlane 6.
Parkin Preserves Proteasome Function22050
by guest on May 14, 2018
http://ww
w.jbc.org/
Dow
nloaded from

poly(Q) proteins, this interaction could be indirect (10, 18, 64).To determine whether parkin interacts directly with the pro-teasome, we examined binding of GST-parkin to purified 26 Sproteasomes in vitro. We found that GST-parkin indeed bounddirectly to 26 S proteasomes as indicated by pull-down of Rpt6(Fig. 10B, left panel). Similar assays with parkin variants iden-tified the Ubl domain as being critical for this interaction (Fig.10C, right panel).
DISCUSSION
Parkin has previously been demonstrated to function inERAD for degrading unfolded proteins from the ER (2, 6).However, many misfolded proteins are translated in the cytosolincluding �-synuclein and several expanded poly(Q) proteins.In this study, we show that parkin also promotes the ubiquiti-nation and degradation of a misfolded protein translated in thecytosol. The model misfolded protein used in this study, apoly(Q) expanded ataxin-3 fragment, misfolds in the cytosoland inhibits the proteasome. Parkin preferentially binds andfacilitates the degradation of the misfolded poly(Q) proteinthereby reducing the impairment of proteasome function. On-going proteasome activity is required for the retrotranslocationof misfolded ER proteins to the cytosol for degradation. Whenproteasome activity is inhibited, retrotranslocation of mis-folded ER proteins fails and the ERAD mechanism for thereduction of ER stress is blocked. ER stress can then lead to theactivation of apoptosis pathways including the activation ofcaspase-12 and ASK. Thus ER stress-mediated cell death canarise from either overexpression of a misfolded ER protein suchas Pael-R or by proteasome inhibition by cytosolic translatedmisfolded proteins such as the poly(Q) containing proteins.
Parkin appears to be capable of protecting the proteasome frominhibition by expanded poly(Q) proteins. Our results thereforeextend the observations of Imai et al. (2, 6) and suggest thatparkin targets the degradation of several misfolded substratesin multiple cellular compartments.
This study also demonstrates that parkin is an E3 ubiquitinligase for poly(Q) expanded ataxin-3. The interaction of parkinwith expanded poly(Q) proteins is not simply a result of over-expression, as evidenced by the colocalization and co-immuno-precipitation of parkin with poly(Q) expanded htt in the brainsof YAC72 HD transgenic mice and colocalization with htt in-clusions in HD brains. These findings suggest a role for parkinfunction in neurodegenerative diseases such as HD. Previousgenetic studies have implicated E6AP, a HECT domain E3ubiquitin ligase, in the ubiquitination of poly(Q) expandedataxin-1. Mutation of E6AP results in a reduction in the for-mation of visible inclusions but enhanced cytotoxicity in trans-genic mice overexpressing expanded poly(Q) ataxin-1 (47). Al-though E6AP did not show E3 activity for poly(Q)-expandedataxin-3 protein in our in vitro assay, it is possible that E6APmight specifically ubiquitinate ataxin-1 but not ataxin-3. Theseproteins are substantially different with respect to protein size,sequence, protein interacting partners, and subcellular local-ization. It is also conceivable that another factor is required foreffective ubiquitination of ataxin-3 by E6AP. Alternatively,E6AP may ubiquitinate a poly(Q) interacting protein importantfor inclusion body formation without directly ubiquitinatingthe poly(Q) protein.
Several other candidate substrates for parkin have beenpreviously identified (2–5). These candidate substrates havelittle sequence homology. However, the substrates Pael-R and�Sp22 share with the expanded poly(Q) proteins a commonpropensity to misfold and aggregate (2, 4). Our results indicatethat parkin preferentially ubiquitinates expanded poly(Q) con-taining proteins. It is not known how parkin recognizes itsapparently diverse protein substrates. We show that Hsp70associates with parkin and expanded poly(Q) proteins in acomplex similar to the reported complex of Hsp70, parkin, andPael-R (27). Hsp70 binding appears to be mediated primarilyby the RING-IBR-RING domain. Hsp70 enhances the bindingand ubiquitination of expanded poly(Q) proteins by parkin invitro where the expanded poly(Q) proteins are translated in abacteria lysate system. Hsp70 is important in the recognitionand unfolding of misfolded proteins and degradation of certainmisfolded proteins (21, 54, 59). Hsp70 has also been shown topreferentially bind htt with expanded poly(Q) repeats com-pared with normal repeats (19) and promote the degradation ofpoly(Q)-expanded androgen receptor (21). Our results suggestthat parkin may preferentially recognize misfolded poly(Q) pro-teins via interaction with Hsp70, which may partially accountfor its apparent broad substrate specificity. This result differsfrom that of Imai et al. (27) where adding larger amounts ofHsp70 (3–4 �g) inhibited parkin-dependent ubiquitination ofPael-R in vitro. In their assay, Pael-R was translated in rabbitreticulocyte lysate, which contains a substantial amount ofHsp70 and many other components of the UPS. The differenteffects of Hsp70 on parkin E3 activity may reflect differencesbetween an ER versus cytosolic protein substrate or differencesbetween bacteria and rabbit reticulocyte lysates or the quantityof Hsp70 added. In our model, parkin is able to recognizemisfolded poly(Q) proteins by its interaction with Hsp70. Thissuggests that parkin may recognize a wide variety of misfoldedproteins by forming a complex with Hsp70 and thus parkinmay function as an E3 for misfolded proteins more broadly.
The efficient degradation of misfolded proteins may requireeffective presentation of these substrates to the proteasome.
FIG. 6. Parkin reduces toxicity of GFP-poly(Q) proteins. A,wild-type parkin reduced GFP-Gln79 toxicity in a concentration- andproteasome-dependent manner. N18 cells were co-transfected withGFP-Gln79 and empty vector (control) or increasing quantities (0.5, 1.0,and 2.0 �g) of FLAG-parkin. Where indicated, 50 �M MG132 was addedovernight. These experiments were performed together but presentedon two panels for clarity. B, specificity of parkin amelioration of GFP-Gln79 toxicity. N18 cells were transfected with GFP-Gln79 and 2.0 �g ofparkin, and the indicated parkin mutants, E6AP or XIAP. Cells wereassayed for propidium iodide exclusion 60 h post-transfection. Datashown are means with error bars indicating S.D. (n � 9; *, p � 0.01; **,p � 0.005 compared with control transfection with empty vector).
Parkin Preserves Proteasome Function 22051
by guest on May 14, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Substrate recognition is generally dependent on polyubiquiti-nation, but additional factors may be necessary for efficientrecognition or processing in some cases. It has also been sug-gested that some E3 ubiquitin ligases directly binds the pro-teasome, which may help to present the substrate to the pro-teasome (65) or recruit additional factors to the degradationmachinery (62, 63). We found that parkin binds the proteasomeand the Ubl domain is critical for this interaction. Recent NMRstudies show that the Ubl domain of parkin interacts with theRpn10 subunit of the proteasome in vitro and the interactionregion includes Arg42 of the Ubl domain of parkin (66). A singlepoint mutation R42P in the Ubl domain has been linked toearly-onset Parkinsonism (67), suggesting that proteasomebinding may be important for parkin function. The recent work
of Imai et al. (27) demonstrates that parkin forms a complexthat contains not only the substrate Pael-R but also Hsp70 andthe ubiquitinating factor CHIP, which appears to enhance theE3 activity of parkin (27). CHIP-related polyubiquitination ofsubstrates appears to be important for degradation of ER mis-folded proteins (58, 59, 68, 69). Hsp70 has been shown to beinvolved in the degradation of certain misfolded proteins (21,54, 56, 70). We speculate that parkin may interact with Hsp70and the proteasome to facilitate direct presentation of the ubiq-uitinated substrate in an unfolded state more suitable for entryinto the proteasomal pore for successful degradation.
Parkin improves proteasome function in the presence of mis-folded cytosolic poly(Q) proteins, which can otherwise impairthe UPS and activate cell death (9, 12, 51). Proteasome dys-function occurs in the presence of misfolded ER proteins suchas mutant cystic fibrosis transmembrane conductance regula-tor or misfolded cytosolic proteins such as expanded poly(Q)proteins and �-synuclein (9, 10, 12, 71). Whereas protein ag-gregation, proteasome dysfunction, and cell death appear to beclosely related in cell culture models, the relationship betweeninclusion body formation and cell death in vivo is less clear. Intransgenic mice that overexpress poly(Q)-expanded ataxin-1,mutation in the E3 ubiquitin ligase E6AP results in enhancedcytotoxicity despite a reduction in the formation of visible in-clusions (47). Nevertheless, there is a growing amount of recentevidence that proteasome dysfunction and accumulation ofmisfolded proteins may play a role in the pathogenesis of sev-eral neurodegenerative diseases. Proteasomal subunits arefound in poly(Q) protein aggregates (10, 11, 64) as well as inLewy bodies, the hallmark intracellular inclusion of PD (72).
FIG. 8. Parkin reduces activation of caspase-12 induced byGFP-Gln79 overexpression. HEK293 cells were co-transfected withprocaspase-12, GFP-Gln79, and wild-type (WT) or mutant parkin asindicated. 50 �g of cell lysates were monitored for activation ofcaspase-12 by Western blot.
FIG. 7. Parkin improves proteasome function in the presence of expanded poly(Q) protein. A, HEK293 cells stably expressing GFPuwere transfected with the indicated plasmids and GFPu fluorescence was quantitated on a fluorescence microscope (“Materials and Methods”). B,co-expression of HA-Gln79 and GFPu stabilizes GFPu. HEK293 cell lines stably expressing GFPu were transfected with HA-Gln79 and monitoredfor GFPu fluorescence and expression of HA-Gln79 (HA immunofluorescence). Arrows indicate HA-Gln79 expressing cells. C, smoothed densityestimates of GFPu fluorescence for selected histograms in panel A. The curves are normalized to a maximum of 1 for easy visualization. AFU,arbitrary fluorescence units.
Parkin Preserves Proteasome Function22052
by guest on May 14, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Autopsy-derived brain tissue from PD patients show reducedproteasomal activity (17). Overexpression of expanded poly(Q)proteins or mutant �-synuclein inhibits proteasome activityand pharmacological inhibition of the proteasome enhancestoxicity and accumulation of these proteins (9–12, 71, 73–75).
The impairment of proteasome function by misfolded cytoso-lic proteins such as expanded poly(Q) proteins can result infailure of ERAD and consequently ER stress (9, 10, 12). ERstress in turn activates apoptosis pathways involving ASK andcaspase-12 (12, 51, 76–78). Blocking these pathways for ERstress-mediated apoptosis attenuates expanded poly(Q)-in-duced cell death. Our results demonstrate that by promotingdegradation of misfolded poly(Q) proteins and preserving pro-teasome function, parkin is able to reduce the activation ofcaspase-12, an important effector for ER stress-induced cell
death thereby improving cell viability. Thus parkin may beimportant in stress response pathways for eliminating mis-folded proteins and preserving proteasome function. ER stress-induced cell death has been implicated in several neurodegen-erative diseases (2, 12, 51, 52). Our results raise the possibilitythat parkin may play a role in modulating some of these neu-rodegenerative conditions.
Several adult neurodegenerative diseases including both thepoly(Q) expansion diseases and PD are associated with thepresence of intracellular protein inclusions. The relationshipbetween Lewy bodies and parkin is currently unclear. Parkin-linked PD is commonly cited to lack Lewy bodies; however, theactual number of autopsies is quite small (fewer than 10 total)and one actually contained Lewy bodies (79–83). Despite theabsence of Lewy bodies, accumulation of the candidate sub-
FIG. 9. Parkin interacts with Hsp70. A, co-immunoprecipitation of Hsp70 with parkin. HEK293 cells were transfected with GFP-Gln79 andFLAG-tagged parkin or myc-tagged E6AP. Cell lysates were processed for immunoprecipitation with anti-FLAG or anti-myc antibodies andimmunoblotted for Hsp70. B, parkin structure-function relationship in Hsp70 interaction. The indicated variants were tested for the ability toco-precipitate Hsp70. Top, Hsp70 and FLAG blots; bottom, schematic of the constructs used and summary of the co-immunoprecipitation results.Symbols: Ubl, f; proximal RING, wide spaced lines in oval; distal RING, thin spaced lines in oval; IBR, �. C, Hsp70 enhances interaction of parkinand poly(Q) protein in vitro. HA-tagged Gln84 was translated in vitro using a bacterial system in the presence or absence of Hsp70. His6-parkinwas expressed in E. coli, purified with TALON resin, and incubated with the translation products. His6-parkin was then immunoprecipitated andHA-Gln84 detected with anti-HA antibody. In a reciprocal experiment, HA-Gln84 was immunoprecipitated and His6-parkin detected with ananti-parkin antibody. D, Hsp70 enhances ubiquitination of Gln79 by parkin. 35S-Labeled ataxin-3 Gln79 was prepared by in vitro translation inbacteria lysate and used for ubiquitination assay with GST-parkin with or without Hsp70 added; asterisk indicates mono-ubiquitinated Gln79.
Parkin Preserves Proteasome Function 22053
by guest on May 14, 2018
http://ww
w.jbc.org/
Dow
nloaded from

strates of parkin (�Sp22 and a detergent-insoluble form ofPael-R) has been demonstrated in autosomal recessive juvenileParkinsonism brains, consistent with impaired protein degra-dation (2, 4). These observations show that accumulation ofparkin candidate substrates does not always lead to Lewy bodyformation. It has been proposed that Lewy body formation mayrequire specific ubiquitination of �Sp22 and the �-synuclein-associated protein synphilin-1 (4, 5). Thus the reported absenceof Lewy bodies in autosomal recessive juvenile onset Parkin-sonism does not contradict our hypothesis that parkin is in-volved in the degradation of misfolded cytosolic and ER-resi-dent proteins.
It is worth emphasizing that expanded poly(Q) proteins areused as a model for studying parkin degradation of cytosolicmisfolded proteins. Overexpression of expanded poly(Q) pro-teins has been a useful tool for the study of cellular handling ofaberrant proteins. Cellular models and transgenic Drosophilaoverexpressing poly(Q)-expanded disease proteins have identi-fied molecular chaperones and other novel genes that modulatepoly(Q) expansion diseases (18, 20, 23, 25, 49, 53, 57). Resultsfrom these model systems may be relevant to other neurode-generative diseases, such as PD as evidenced by a recent dem-onstration that the molecular chaperone Hsp70 also protectsagainst overexpression of human �-synuclein in a transgenicDrosophila model of PD (26). Obviously one does not expect tofind poly(Q) aggregates in autosomal recessive juvenile Parkin-sonism brains unless the patient also has a genetic mutationinvolving poly(Q) expansion. Rather we have used poly(Q) pro-teins as a model to explore mechanisms by which parkin pro-motes degration of cytosolic misfolded proteins and reduces celldeath. It is not yet known if parkin is a modifier for poly(Q)expansion diseases. Our results suggest that parkin may be adisease modifier for poly(Q) disorders; it would be interesting totest this prediction in transgenic animal models. Although wepresent results with expanded poly(Q) proteins, we are cur-rently testing if parkin can promote the ubiquitination anddegradation of other misfolded proteins involved in neurode-
generative diseases. We have also observed that parkin pro-motes the degradation of another unrelated misfolded mutantof the coral red fluorescent protein DsRed.2 Based on our re-sults and the apparent broad substrate specificity of parkin, itis reasonable to believe that parkin is involved in the degrada-tion of misfolded proteins in multiple cellular compartments.
In conclusion, we have demonstrated that parkin promotesthe ubiquitination and degradation of expanded poly(Q) pro-teins. The resulting preservation of proteasome function re-duces activation of caspase-12-mediated cell death associatedwith ER stress. We have also shown that parkin binds Hsp70primarily through its C-terminal RING-IBR-RING domain.Hsp70 enhances both binding and ubiquitination of the ex-panded poly(Q) proteins by parkin and may serve to recruitmisfolded proteins as substrates for parkin ubiquitin ligaseactivity. We propose that parkin recognizes misfolded proteinsand promotes the degradation of these aberrant proteins fromboth the cytosolic and ER compartments. We speculate thatloss of parkin function increases cell susceptibility to protea-some dysfunction caused by misfolded proteins and ER stress-induced cell death.
Acknowledgments—We thank Cecile Pickart for helpful advice andcritical review of the manuscript, and Kenneth Fischbeck andHenry Paulson for critical review of the manuscript. We have benefitedgreatly from discussion with Jianxin You and Amy Lam on proteasomebinding assays. J. Shi, D. Yarnell, M. Remington, and T. Bowen pro-vided technical assistance for this study.
REFERENCES
1. Shimura, H., Hattori, N., Kubo, S., Mizuno, Y., Asakawa, S., Minoshima, S.,Shimizu, N., Iwai, K., Chiba, T., Tanaka, K., and Suzuki, T. (2000) Nat.Genet. 25, 302–305
2. Imai, Y., Soda, M., Inoue, H., Hattori, N., Mizuno, Y., and Takahashi, R. (2001)Cell 105, 891–902
3. Zhang, Y., Gao, J., Chung, K. K., Huang, H., Dawson, V. L., and Dawson, T. M.(2000) Proc. Natl. Acad. Sci. U. S. A. 97, 13354–13359
2 Y. C. Tsai, P. S. Fishman, N. V. Thakor, and G. A. Oyler, unpub-lished data.
FIG. 10. Parkin interacts with theproteasome. A, co-immunoprecipitationof proteasome subunit with parkin.HEK293 cells were transfected with GFP-Gln79 and FLAG-parkin. FLAG-parkinwas immunoprecipitated from the cell ly-sates and proteasome subunit Rpt6/S8was detected by immunoblotting. For thecontrol, FLAG beads were preincubatedwith 3� FLAG peptide. B, parkin bindsproteasomes directly via its Ubl domain.Left, immobilized bacterially expressedGST-parkin (or GST control) was incu-bated with purified 26 S proteasome. Af-ter washing, bound subunit Rpt6/S8 wasdetected by immunoblotting. Right pan-els, pull-down assays were performed us-ing purified 26 S proteasomes and differ-ent regions of parkin fused to GST. Boundproteasomes were detected by immuno-blotting against Rpt6/S8 (19 S subunit) orHC3 (20 S subunit). C, Coomassie stain-ing of the GST fusion constructs used inpanel B.
Parkin Preserves Proteasome Function22054
by guest on May 14, 2018
http://ww
w.jbc.org/
Dow
nloaded from

4. Shimura, H., Schlossmacher, M. G., Hattori, N., Frosch, M. P., Trockenbacher,A., Schneider, R., Mizuno, Y., Kosik, K. S., and Selkoe, D. J. (2001) Science293, 263–269
5. Chung, K. K., Zhang, Y., Lim, K. L., Tanaka, Y., Huang, H., Gao, J., Ross,C. A., Dawson, V. L., and Dawson, T. M. (2001) Nat. Med. 7, 1144–1150
6. Imai, Y., Soda, M., and Takahashi, R. (2000) J. Biol. Chem. 275, 35661–356647. Werner, E. D., Brodsky, J. L., and McCracken, A. A. (1996) Proc. Natl. Acad.
Sci. U. S. A. 93, 13797–138018. Chillaron, J., and Haas, I. G. (2000) Mol. Biol. Cell 11, 217–2269. Bence, N. F., Sampat, R. M., and Kopito, R. R. (2001) Science 292, 1552–1555
10. Jana, N. R., Zemskov, E. A., Wang, G., and Nukina, N. (2001) Hum. Mol.Genet. 10, 1049–1059
11. Waelter, S., Boeddrich, A., Lurz, R., Scherzinger, E., Lueder, G., Lehrach, H.,and Wanker, E. E. (2001) Mol. Biol. Cell 12, 1393–1407
12. Kouroku, Y., Fujita, E., Jimbo, A., Kikuchi, T., Yamagata, T., Momoi, M. Y.,Kominami, E., Kuida, K., Sakamaki, K., Yonehara, S., and Momoi, T. (2002)Hum. Mol. Genet. 11, 1505–1515
13. McCampbell, A., Taylor, J. P., Taye, A. A., Robitschek, J., Li, M., Walcott, J.,Merry, D., Chai, Y., Paulson, H., Sobue, G., and Fischbeck, K. H. (2000)Hum. Mol. Genet. 9, 2197–2202
14. Monoi, H., Futaki, S., Kugimiya, S., Minakata, H., and Yoshihara, K. (2000)Biophys. J. 78, 2892–2899
15. Ross, C. A., Margolis, R. L., Becher, M. W., Wood, J. D., Engelender, S., Cooper,J. K., and Sharp, A. H. (1998) Prog. Brain Res. 117, 397–419
16. Luthi-Carter, R., Strand, A. D., Hanson, S. A., Kooperberg, C., Schilling, G., LaSpada, A. R., Merry, D. E., Young, A. B., Ross, C. A., Borchelt, D. R., andOlson, J. M. (2002) Hum. Mol. Genet. 11, 1927–1937
17. McNaught, K. S., and Jenner, P. (2001) Neurosci. Lett. 297, 191–19418. Cummings, C. J., Mancini, M. A., Antalffy, B., DeFranco, D. B., Orr, H. T., and
Zoghbi, H. Y. (1998) Nat. Genet. 19, 148–15419. Jana, N. R., Tanaka, M., Wang, G., and Nukina, N. (2000) Hum. Mol. Genet. 9,
2009–201820. Kobayashi, Y., Kume, A., Li, M., Doyu, M., Hata, M., Ohtsuka, K., and Sobue,
G. (2000) J. Biol. Chem. 275, 8772–877821. Bailey, C. K., Andriola, I. F., Kampinga, H. H., and Merry, D. E. (2002) Hum.
Mol. Genet. 11, 515–52322. Fernandez-Funez, P., Nino-Rosales, M. L., de Gouyon, B., She, W. C., Luchak,
J. M., Martinez, P., Turiegano, E., Benito, J., Capovilla, M., Skinner, P. J.,McCall, A., Canal, I., Orr, H. T., Zoghbi, H. Y., and Botas, J. (2000) Nature408, 101–106
23. Warrick, J. M., Chan, H. Y., Gray-Board, G. L., Chai, Y., Paulson, H. L., andBonini, N. M. (1999) Nat. Genet. 23, 425–428
24. Marsh, J. L., Walker, H., Theisen, H., Zhu, Y. Z., Fielder, T., Purcell, J., andThompson, L. M. (2000) Hum. Mol. Genet. 9, 13–25
25. Kazemi-Esfarjani, P., and Benzer, S. (2000) Science 287, 1837–184026. Auluck, P. K., Chan, H. Y., Trojanowski, J. Q., Lee, V. M., and Bonini, N. M.
(2002) Science 295, 865–86827. Imai, Y., Soda, M., Hatakeyama, S., Akagi, T., Hashikawa, T., Nakayama,
K. I., and Takahashi, R. (2002) Mol. Cell 10, 55–6728. Wang, G., Sawai, N., Kotliarova, S., Kanazawa, I., and Nukina, N. (2000)
Hum. Mol. Genet. 9, 1795–180329. Hiyama, H., Yokoi, M., Masutani, C., Sugasawa, K., Maekawa, T., Tanaka, K.,
Hoeijmakers, J. H., and Hanaoka, F. (1999) J. Biol. Chem. 274,28019–28025
30. Elsasser, S., Gali, R. R., Schwickart, M., Larsen, C. N., Leggett, D. S., Muller,B., Feng, M. T., Tubing, F., Dittmar, G. A., and Finley, D. (2002) Nat. CellBiol. 4, 725–730
31. Kaye, F. J., Modi, S., Ivanovska, I., Koonin, E. V., Thress, K., Kubo, A.,Kornbluth, S., and Rose, M. D. (2000) FEBS Lett. 467, 348–355
32. Saeki, Y., Sone, T., Toh-e, A., and Yokosawa, H. (2002) Biochem. Biophys. Res.Commun. 296, 813–819
33. Leggett, D. S., Hanna, J., Borodovsky, A., Crosas, B., Schmidt, M., Baker,R. T., Walz, T., Ploegh, H., and Finley, D. (2002) Mol Cell 10, 495–507
34. Sherman, M. Y., and Goldberg, A. L. (2001) Neuron 29, 15–3235. Goedert, M. (2001) Nat. Rev. Neurosci. 2, 492–50136. Johnston, J. A., Dalton, M. J., Gurney, M. E., and Kopito, R. R. (2000) Proc.
Natl. Acad. Sci. U. S. A. 97, 12571–1257637. DeTure, M., Ko, L. W., Easson, C., and Yen, S. H. (2002) Am. J. Pathol. 161,
1711–172238. Souza, J. M., Giasson, B. I., Chen, Q., Lee, V. M., and Ischiropoulos, H. (2000)
J. Biol. Chem. 275, 18344–1834939. Giasson, B. I., Duda, J. E., Murray, I. V., Chen, Q., Souza, J. M., Hurtig, H. I.,
Ischiropoulos, H., Trojanowski, J. Q., and Lee, V. M. (2000) Science 290,985–989
40. Mancini, R., Fagioli, C., Fra, A. M., Maggioni, C., and Sitia, R. (2000) FASEBJ. 14, 769–778
41. Treier, M., Staszewski, L. M., and Bohmann, D. (1994) Cell 78, 787–79842. Hodgson, J. G., Agopyan, N., Gutekunst, C. A., Leavitt, B. R., LePiane, F.,
Singaraja, R., Smith, D. J., Bissada, N., McCutcheon, K., Nasir, J., Jamot,L., Li, X. J., Stevens, M. E., Rosemond, E., Roder, J. C., Phillips, A. G.,Rubin, E. M., Hersch, S. M., and Hayden, M. R. (1999) Neuron 23, 181–192
43. Harlow, E., and Lane, D. (1990) Antibodies: A Laboratory Manual, Cold SpringHarbor Laboratory Press, Cold Spring Harbor, NY
44. Schlossmacher, M. G., Frosch, M. P., Gai, W. P., Medina, M., Sharma, N.,Forno, L., Ochiishi, T., Shimura, H., Sharon, R., Hattori, N., Langston,J. W., Mizuno, Y., Hyman, B. T., Selkoe, D. J., and Kosik, K. S. (2002) Am. J.
Pathol. 160, 1655–166745. Lam, Y. A., Pickart, C. M., Alban, A., Landon, M., Jamieson, C., Ramage, R.,
Mayer, R. J., and Layfield, R. (2000) Proc. Natl. Acad. Sci. U. S. A. 97,9902–9906
46. Rankin, C. A., Joazeiro, C. A., Floor, E., and Hunter, T. (2001) J. Biomed. Sci.8, 421–429
47. Cummings, C. J., Reinstein, E., Sun, Y., Antalffy, B., Jiang, Y., Ciechanover,A., Orr, H. T., Beaudet, A. L., and Zoghbi, H. Y. (1999) Neuron 24, 879–892
48. de Cristofaro, T., Affaitati, A., Feliciello, A., Avvedimento, E. V., and Varrone,S. (2000) Biochem. Biophys. Res. Commun. 272, 816–821
49. Wyttenbach, A., Carmichael, J., Swartz, J., Furlong, R. A., Narain, Y., Rankin,J., and Rubinstein, D. C. (2000) Proc. Natl. Acad. Sci. U. S. A. 97,2898–2903
50. Warrick, J. M., Paulson, H. L., Gray-Board, G. L., Bui, Q. T., Fischbeck, K. H.,Pittman, R. N., and Bonini, N. M. (1998) Cell 93, 939–949
51. Nishitoh, H., Matsuzawa, A., Tobiume, K., Saegusa, K., Takeda, K., Inoue, K.,Hori, S., Kakizuka, A., and Ichijo, H. (2002) Genes Dev. 16, 1345–1355
52. Nakagawa, T., Zhu, H., Morishima, N., Li, E., Xu, J., Yankner, B. A., andYuan, J. (2000) Nature 403, 98–103
53. Muchowski, P. J., Schaffar, G., Sittler, A., Wanker, E. E., Hayer-Hartl, M. K.,and Hartl, F. U. (2000) Proc. Natl. Acad. Sci. U. S. A. 97, 7841–7846
54. Zhang, Y., Nijbroek, G., Sullivan, M. L., McCracken, A. A., Watkins, S. C.,Michaelis, S., and Brodsky, J. L. (2001) Mol. Biol. Cell 12, 1303–1314
55. Nishikawa, S. I., Fewell, S. W., Kato, Y., Brodsky, J. L., and Endo, T. (2001)J. Cell Biol. 153, 1061–1070
56. Wagner, I., Arlt, H., van Dyck, L., Langer, T., and Neupert, W. (1994) EMBOJ. 13, 5135–5145
57. Chan, H. Y., Warrick, J. M., Gray-Board, G. L., Paulson, H. L., and Bonini,N. M. (2000) Hum. Mol. Genet. 9, 2811–2820
58. Murata, S., Minami, Y., Minami, M., Chiba, T., and Tanaka, K. (2001) EMBORep. 2, 1133–1138
59. Meacham, G. C., Patterson, C., Zhang, W., Younger, J. M., and Cyr, D. M.(2001) Nat. Cell Biol. 3, 100–105
60. Frydman, J., Nimmesgern, E., Ohtsuka, K., and Hartl, F. U. (1994) Nature370, 111–117
61. Luders, J., Demand, J., and Hohfeld, J. (2000) J. Biol. Chem. 275, 4613–461762. Suzuki, T., Park, H., Kwofie, M. A., and Lennarz, W. J. (2001) J. Biol. Chem.
276, 21601–2160763. Demand, J., Alberti, S., Patterson, C., and Hohfeld, J. (2001) Curr. Biol. 11,
1569–157764. Chai, Y., Koppenhafer, S. L., Shoesmith, S. J., Perez, M. K., and Paulson, H. L.
(1999) Hum. Mol. Genet. 8, 673–68265. Xie, Y., and Varshavsky, A. (2000) Proc. Natl. Acad. Sci. U. S. A. 97,
2497–250266. Sakata, E., Yamaguchi, Y., Kurimoto, E., Kikuchi, J., Yokoyama, S., Yamada,
S., Kawahara, H., Yokosawa, H., Hattori, N., Mizuno, Y., Tanaka, K., andKato, K. (2003) EMBO Rep. 4, 301–306
67. Terreni, L., Calabrese, E., Calella, A. M., Forloni, G., and Mariani, C. (2001)Neurology 56, 463–466
68. Connell, P., Ballinger, C. A., Jiang, J., Wu, Y., Thompson, L. J., Hohfeld, J.,and Patterson, C. (2001) Nat. Cell Biol. 3, 93–96
69. Jiang, J., Ballinger, C. A., Wu, Y., Dai, Q., Cyr, D. M., Hohfeld, J., andPatterson, C. (2001) J. Biol. Chem. 276, 42938–42944
70. Bercovich, B., Stancovski, I., Mayer, A., Blumenfeld, N., Laszlo, A., Schwartz,A. L., and Ciechanover, A. (1997) J. Biol. Chem. 272, 9002–9010
71. Petrucelli, L., O’Farrell, C., Lockhart, P. J., Baptista, M., Kehoe, K., Vink, L.,Choi, P., Wolozin, B., Farrer, M., Hardy, J., and Cookson, M. R. (2002)Neuron 36, 1007–1019
72. Ii, K., Ito, H., Tanaka, K., and Hirano, A. (1997) J. Neuropathol. Exp. Neurol.56, 125–131
73. Tanaka, Y., Engelender, S., Igarashi, S., Rao, R. K., Wanner, T., Tanzi, R. E.,Sawa, A., Dawson, V. L., Dawson, T. M., and Ross, C. A. (2001) Hum. Mol.Genet. 10, 919–926L. D.
74. Rideout, H. J., and Stefanis, L. (2002) Mol. Cell Neurosci. 21, 223–23875. McNaught, K. S., Mytilineou, C., Jnobaptiste, R., Yabut, J., Shashidharan, P.,
Jennert, P., and Olanow, C. W. (2002) J. Neurochem. 81, 301–30676. Rao, R. V., Castro-Obregon, S., Frankowski, H., Schuler, M., Stoka, V., del Rio,
G., Bredesen, D. E., and Ellerby, H. M. (2002) J. Biol. Chem. 277,21836–21842
77. Fujita, E., Kouroku, Y., Jimbo, A., Isoai, A., Maruyama, K., and Momoi, T.(2002) Cell Death Differ. 9, 1108–1114
78. Morishima, N., Nakanishi, K., Takenouchi, H., Shibata, T., and Yasuhiko, Y.(2002) J. Biol. Chem. 277, 34287–34294
79. Mori, H., Kondo, T., Yokochi, M., Matsumine, H., Nakagawa-Hattori, Y.,Miyake, T., Suda, K., and Mizuno, Y. (1998) Neurology 51, 890–892
80. Takahashi, H., Ohama, E., Suzuki, S., Horikawa, Y., Ishikawa, A., Morita, T.,Tsuji, S., and Ikuta, F. (1994) Neurology 44, 437–441
81. Hayashi, S., Wakabayashi, K., Ishikawa, A., Nagai, H., Saito, M., Maruyama,M., Takahashi, T., Ozawa, T., Tsuji, S., and Takahashi, H. (2000) Mov.Disord. 15, 884–888
82. Portman, A. T., Giladi, N., Leenders, K. L., Maguire, P., Veenma-van der Duin,L., Swart, J., Pruim, J., Simon, E. S., Hassin-Baer, S., and Korczyn, A. D.(2001) Neurology 56, 1759–1762
83. Farrer, M., Chan, P., Chen, R., Tan, L., Lincoln, S., Hernandez, D., Forno, L.,Gwinn-Hardy, K., Petrucelli, L., Hussey, J., Singleton, A., Tanner, C.,Hardy, J., and Langston, J. W. (2001) Ann. Neurol. 50, 293–300
Parkin Preserves Proteasome Function 22055
by guest on May 14, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Yien Che Tsai, Paul S. Fishman, Nitish V. Thakor and George A. OylerPreservation of Proteasome Function
Parkin Facilitates the Elimination of Expanded Polyglutamine Proteins and Leads to
doi: 10.1074/jbc.M212235200 originally published online April 3, 20032003, 278:22044-22055.J. Biol. Chem.
10.1074/jbc.M212235200Access the most updated version of this article at doi:
Alerts:
When a correction for this article is posted•
When this article is cited•
to choose from all of JBC's e-mail alertsClick here
http://www.jbc.org/content/278/24/22044.full.html#ref-list-1
This article cites 82 references, 34 of which can be accessed free at
by guest on May 14, 2018
http://ww
w.jbc.org/
Dow
nloaded from


![Universal Business Language Version 2docs.oasis-open.org/ubl/cs1-UBL-2.1/UBL-2.1.pdf · OASIS Universal Business Language TC Chairs: Jon Bosak (bosak@pinax.com), Individual ... [UBL-2.1]](https://img.pdfslide.us/doc/110x75/5e7065bd965725432c6cc8bd/universal-business-language-version-2docsoasis-openorgublcs1-ubl-21ubl-21pdf.jpg)


























