Embed Size (px)
Citation preview

Tissue Inhibitor of Metalloproteinases-2 (TIMP-2)Suppresses TKR-Growth Factor Signaling Independentof Metalloproteinase Inhibition*
Received for publication, September 6, 2000, and in revised form, October 16, 2000Published, JBC Papers in Press, October 19, 2000, DOI 10.1074/jbc.M008157200
Susan E. Hoegy, Hae-Ryong Oh, Marta L. Corcoran‡, and William G. Stetler-Stevenson§
From the Extracellular Matrix Pathology Section, Laboratory of Pathology, Division of Clinical Sciences,NCI, National Institutes of Health, Bethesda, Maryland 20892-1500
The tissue inhibitors of metalloproteinases (TIMPs)block matrix metalloproteinase (MMP)-mediated in-creases in cell proliferation, migration, and invasionthat are associated with extracellular matrix (ECM)turnover. Here we demonstrate a direct role for TIMP-2in regulating tyrosine kinase-type growth factor recep-tor activation. We show that TIMP-2 suppresses the mi-togenic response to tyrosine kinase-type receptorgrowth factors in a fashion that is independent of MMPinhibition. The TIMP-2 suppression of mitogenesis isreversed by the adenylate cyclase inhibitor SQ22536,and implicates cAMP as the second messenger in theseeffects. TIMP-2 neither altered the release of transform-ing growth factor a from the cell surface, nor epidermalgrowth factor (EGF) binding to the cognate receptor,EGFR. TIMP-2 binds to the surface of A549 cells in aspecific and saturable fashion (Kd 5 147 pM), that is notcompeted by the synthetic MMP inhibitor BB-94 and isindependent of MT-1-MMP. TIMP-2 induces a decreasein phosphorylation of EGFR and a concomitant reduc-tion in Grb-2 association. TIMP-2 prevents SH2-protein-tyrosine phosphatase-1 (SHP-1) dissociation from immu-noprecipitable EGFR complex and a selective increasein total SHP-1 activity. These studies represent a newfunctional paradigm for TIMP-2 in which TIMP sup-presses EGF-mediated mitogenic signaling by short-cir-cuiting EGFR activation.
In mature normal tissues, the structure and composition ofthe extracellular matrix (ECM)1 functions to maintain tissuehomeostasis and cellular quiescence. These anti-proliferativeand differentiation promoting effects of the ECM are attribut-able both to its composition and three-dimensional spatial or-
ganization, as well as the presence of soluble growth inhibitors,such as TGF-b (1–4). Compelling evidence for these effects alsocomes from transgenic animal studies in which altered ECMexpression or organization, disruption of ECM attachments, orproteolytic modification of ECM integrity results in altereddevelopmental and disease-related phenotypes (5–7). The ma-trix metalloproteinases (MMPs) are a major determinant ofECM turnover in tissue morphogenesis. Altered expression ofMMP activity is associated with a variety of pathologic condi-tions, including tumor progression and cancer invasion (5–8).
In addition to disrupting the structural organization of theECM, MMP proteolysis of ECM can result in release and/oractivation of sequestered growth factors (1, 3). In addition,MMP activity may expose cryptic sites in the ECM or directlymodify cell surface receptors or ligands involved in both cell-matrix, as well as cell-cell adhesion (1, 3, 9). The endogenousmetalloproteinase inhibitors, tissue inhibitors of MMPs(TIMPs), negatively regulate the proteolytic activity of MMPsduring ECM turnover. Reduction or ablation of TIMP geneexpression results in enhanced ECM proteolysis concomitantwith up-regulation of cell invasive activity of nontransformeddifferentiated cells (10, 11). In comparison, TIMP overexpres-sion results in decreased invasion of endothelial and tumorcells both in vitro and in vivo (12, 13). Recent transgenic animalstudies have demonstrated that alteration of the MMP/TIMPbalance in vivo in favor of TIMP-1 activity can block neoplasticproliferation in the SV40 T antigen-induced model of murinehepatocellular carcinoma (14). The mechanism of this TIMP-1effect was mediated by direct inhibition of MMP processingof insulin-like growth factor-binding protein-3 (IGFBP-3),thereby preventing the release of insulin-like growth factor IIand thus suppressing mitogenic activity. These and other stud-ies demonstrate that, through inhibition of MMP activity andprevention of ECM turnover, TIMPs can suppress cell prolifer-ation, invasion and reduce metastasis formation, i.e. TIMPs actas tumor suppressors. As a result of such studies, targetingMMP activity with synthetic MMP inhibitors has become anattractive strategy for therapeutic intervention in cancer pro-gression (15). However, recent studies suggest that TIMPs mayalso directly modulate cell growth in an MMP-independentfashion, although many of these studies lack detailed mecha-nistic insight (16–20). Thus, in addition to their action asinhibitors of metalloproteinases, it is important to investigatewhether TIMPs function to directly modulate cell growth andthe potential mechanisms for these effects.
Epidermal growth factor receptor (EGFR) is highly ex-pressed in human cancers and is detectable at low levels inmany normal tissues (21). Overexpression of EGFR has beenobserved in a variety of human tumors, and EGF-relatedgrowth factors play a role in human cancer growth through
* The costs of publication of this article were defrayed in part by thepayment of page charges. This article must therefore be hereby marked“advertisement” in accordance with 18 U.S.C. Section 1734 solely toindicate this fact.
‡ Current address: IGEN International Inc., 16020 Industrial Dr.,Gaithersburg, MD 20877.
§ To whom correspondence should be addressed: Laboratory of Pa-thology, Div. of Clinical Sciences, NCI, National Institutes of Health,Bldg. 10, Room 2A33, MSC 1500, Bethesda, MD 20892-1500. Tel.:301-496-2687; Fax: 301-402-2628; E-mail: [email protected].
1 The abbreviations used are: ECM, extracellular matrix; TIMP, tis-sue inhibitor of matrix metalloproteinases; MMP, matrix metallopro-teinase; Ala1TIMP-2, amino-terminal alanine appended TIMP-2; TKR,tyrosine kinase-type receptor; bFGF, basic fibroblast growth factor;EGF, epidermal growth factor; EGFR, epidermal growth factor recep-tor; TGF-a, transforming growth factor a; PDGF, platelet-derivedgrowth factor; ERK, extracellular regulated kinase; PTP, protein-ty-rosine phosphatase; DMEM, Dulbecco’s modified Eagle’s medium; PBS,phosphate-buffered saline solution; MT-1-MMP, membrane-type ma-trix metalloproteinase-1; MOPS, 4-morpholinepropanesulfonic acid.
THE JOURNAL OF BIOLOGICAL CHEMISTRY Vol. 276, No. 5, Issue of February 2, pp. 3203–3214, 2001Printed in U.S.A.
This paper is available on line at http://www.jbc.org 3203
by guest on August 9, 2019
http://ww
w.jbc.org/
Dow
nloaded from

autocrine and paracrine mechanisms (22). Overexpression ofEGFR (23) or structural alterations in the receptor protein,such as truncation of the cytoplasmic domain, may elicit li-gand-independent signaling and autonomous cell growth (24).Ligand binding to EGFR initiates receptor dimerization, auto-phosphorylation of tyrosyl residues on the cytoplasmic domainof EGFR, and subsequently Src-mediated activation of the ex-tracellular signal-regulated kinase mitogen-activated protein(MAP) kinase pathway (25). Mitogenic signaling of the EGFRseems to critically depend on activation of the extracellularsignal-regulated kinase/mitogen-activated kinase cascade.
Here we study the role of TIMP-2 in the regulation of cellgrowth in response to tyrosine kinase-type receptor (TKR)growth factor stimulation. In addition to soluble ligand bind-ing, membrane-anchored ligands can also stimulate TKR-me-diated mitogenic responses at high cell densities or followingproteolytic processing from the cell surface. Examples are themembrane-anchored EGFR ligands, which include heparin-bound epidermal growth factor, amphiregulin, transforminggrowth factor-a (TGF-a), and betacellulin, which are shed fromthe plasma membrane by proteolytic cleavage resulting in au-tocrine activation of the receptor (26, 27). Synthetic metallo-proteinase inhibitors, such as BB-94 (Batimastat), reduce cellproliferation in the human mammary epithelial cell line 184A1by blocking TGF-a release (26). BB94 also inhibits EGFRtrans-activation by G-protein-coupled receptors that occurs viaa metalloproteinase directed cleavage of pro-heparin-boundEGF (27). These findings suggest that the metalloproteinaseinhibitors prevent the release of membrane-anchored EGFRligands (e.g. TGF-a, pro-heparin-bound EGF), thereby inhibit-ing autocrine activation of the receptor protein (26). However,if soluble ligands that do not require metalloproteinase proc-essing (e.g. EGF) are present, BB-94 did not inhibit the mito-genic response in these experiments (26). Thus we have focusedour experiments on TIMP inhibition of cellular responses tosoluble mitogenic factors, in particular EGF.
We have examined the direct modulation of TKR growthfactor-stimulated proliferation of human, A549 lung carci-noma, MCF7 mammary carcinoma, HT1080 fibrosarcoma, andHs68 dermal fibroblast cells using both wild type TIMP-2 (wt-TIMP-2) and a null-inhibitor form of TIMP-2, Ala1TIMP-2.Both forms of TIMP-2 abrogate the TKR-mediated mitogenicresponses in these cells. We also investigated the mechanism ofthe diminished mitogenic response following TIMP-2 pretreat-ment prior to growth factor stimulation. The results demon-strate that these suppressive effects are mediated by disrup-tion of TKR activation proximal to the extracellular signal-regulated kinase pathway. To our knowledge this is the firstdemonstration that TIMP-2 can directly suppress activation ofa mitogenic response through suppression of TKR activation inan MMP-independent fashion.
EXPERIMENTAL PROCEDURES
Reagents—Recombinant human EGF, PDGF, and bFGF were ob-tained from R&D Systems, Minneapolis, MN. The following commer-cially available antibodies were obtained: human anti-EGFR, clone 528,mouse, monoclonal IgG2a (Santa Cruz Biotechnology, Santa Cruz, CA);human anti-phosphotyrosine, clone PY-20, mouse IgG2b, monoclonal,(Transduction Labs, Lexington, KY); human anti-MT-1-MMP, clone113-5B7 or 114-6G6 (catalytic), mouse, monoclonal (Chemicon Interna-tional, Temecula, CA), human anti-Grb2, clone C-23, rabbit, polyclonal,(Santa Cruz, Santa Cruz, CA); human anti-SH-PTP1, clone C-19, rab-bit, polyclonal (Santa Cruz Biotechnology); human anti-SH-PTP2, cloneC-18, rabbit, polyclonal (Santa Cruz Biotechnology); mouse and rabbitIgG-horseradish peroxidase conjugate, (Santa Cruz); and goat anti-mouse IgG (H1L) (Kirkegaard & Perry, Gaithersburg, MD). MMPsynthetic hydroxamate inhibitor, BB-94, was a gift from British Bio-technology, Ltd. (Oxford, United Kingdom). Adenylate cyclase inhibitor,SQ22536, and PKA inhibitor, H89 were purchased from Calbiochem (La
Jolla, CA). The recombinant MT-1-MMP catalytic domain (150 units/mg) was purchased from Chemicon International and metalloprotein-ase activity was determined by the thiopeptolide assay as describedpreviously (28). RIPA buffer consists of 50 mM Tris-HCl, pH 7.4, 150 mM
NaCl, 1% Nonidet P-40, 0.5% sodium deoxycholate, and 0.1% SDS.The rTIMP-2 protein was expressed using a vaccinia virus expression
system and purified as described (29). TIMP-2 was also expressed inEscherichia coli with the authentic sequence (wt) or with an alanineresidue appended to the amino-terminal cysteine (Ala1TIMP-2) asdescribed previously (28). TIMP-2 and Ala1TIMP-2 were purified bygel filtration in 4 M guanidine HCl, folded, and oxidized, and thenpurified by gel filtration under native conditions. Recombinant TIMP-1was isolated from the conditioned medium of EPA-transfected Chinesehamster ovary cells (8/8 2G EPA2) (Genetics Institute, Cambridge, MA)as described (30), and then purified by high performance liquid chro-matography gel permeation chromatography using 50 mM Tris-HCl,150 mM NaCl, pH 7.5. All TIMP preparations were endotoxin testedusing the Limulus amoebocyte lysis assay and found to contain less than2 EU/mg of protein.
Cell Culture Conditions—Human lung adenocarcinoma cells (A549;ATCC CCL 185), human fibrosarcoma cells (HT1080; ATCC CCL 121),human breast adenocarcinoma (MCF7; ATCC HTB 22), and humandermal fibroblasts (Hs68; ATCC CRL 1635) were obtained from Amer-ican Tissue Culture Collection (Manassas, VA). Cells were grown to80% confluence in Dulbecco’s modified Eagle’s media (DMEM; LifeTechnologies Inc.) containing 4500 mg/liter of D-glucose, D-glutamine,sodium pyruvate, 100 units/ml penicillin-G, 100 mg/ml streptomycinsulfate, and 10% heat-inactivated fetal bovine serum, unless otherwiseindicated. Cells were trypsinized using trypsin-EDTA (Life Technolo-gies, Inc., Bethesda, MD).
Cell Growth Assays—A549, Hs68, HT1080, and MCF7 cells wereplated at 5 3 105 cells/well on a 96-well Costar plate for 18 h in DMEMwith 10% fetal bovine serum. The cells were then starved for 18 h inDMEM without serum to synchronize cells in G1 (or G0) phase of the cellcycle. Fresh serum-free DMEM was added to the wells prior to treat-ment with TIMP-2. Cells were routinely incubated with TIMP-2 at theindicated concentrations for 30 min, followed by incubation in DMEMwith or without EGF (100 ng/ml, R & D Systems), bFGF (50 ng/ml, R &D Systems), or PDGF (50 ng/ml, R & D Systems) and incubated for 24 h.TIMP-2 pretreatment could be reduced to 1 min prior to addition ofgrowth factor without loss of an effect on growth factor stimulation.Following growth factor stimulation, the cells were incubated for 1 hwith the CellTiter 96TM AQueous One Solution reagent (Promega, Mad-ison, WI) containing 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxy-phenyl)-2-(4-sulfo-phenyl)-2H-tetrazolium, inner salt, and phenazineethosulfate. The quantity of formazan product was determined by the490 nm absorbance, and was directly proportional to the number ofliving cells in culture. The mean and S.D. for triplicate determinationswere recorded for all incubation conditions.
Alternatively, the mitogenic response to growth factor stimulationwith and without TIMP-2 pretreatment was quantitated by [3H]thymi-dine incorporation assays. Cells were synchronized in serum-free con-ditions and treated with TIMP-2 and growth factors as described above.Following growth factor treatment [3H]thymidine (0.1 mCi/ml; Amer-sham Pharmacia Biotech) was added and incubated for 2 h at 37 °C.The percentage of thymidine incorporated in a 2-h pulse correlated in alinear fashion with the cell number. The culture medium was subse-quently discarded, the wells were washed twice with phosphate-buff-ered saline (PBS), and the cells were fixed in methanol:glacial aceticacid (3:1). The incorporated [3H]thymidine was extracted as describedpreviously and quantitated by liquid scintillation counting (31). Themean and S.D. of triplicate assays were determined for all incubationconditions. SQ22536 or 9-(tetrahydro-2-furyl)adenine (Calbiochem/No-vabiochem) was solubilized in sterile deionized H2O and added to cellsat a final concentration of 100 mM (32). H-89 was dissolved in sterile,deionized H2O and added to give a final concentration of 0.1 mM (33).
The results of the growth assays are presented as the percentage ofmaximal growth factor response for the mitogen being tested aftercorrecting for nonstimulated growth in basal medium. This allowscomparison of the effects of TIMP-2 or Ala1TIMP-2 on the mitogenicresponse to various growth factors, as well as between cell lines.
Immunoprecipitation and Western Blotting—HT1080, Hs68, A549, orMCF7 cells were grown in a 6-well Costar plate, pretreated withTIMP-2 or Ala1TIMP, followed by growth factors (described above).Following incubation with growth factor for 5 min, 37 °C, cells werewashed with PBS and treated for 10 min at 4 °C with RIPA lysis buffercontaining freshly added protease inhibitors (10 mg/ml aprotinin, 30mg/ml 4-(2-aminoethyl)benzenesulfonyl fluoride, and 100 mM sodium
TIMP-2 Suppresses EGFR Activation3204
by guest on August 9, 2019
http://ww
w.jbc.org/
Dow
nloaded from

orthovanadate). Cells were disrupted by repeated aspiration through a21-gauge needle. Cell lysates were pre-cleared with normal mouse-IgGand Protein A/G (Pierce, Rockford, IL), and the supernatants were thenincubated with anti-EGFR monoclonal antibodies (clone LA22, UpstateBiotech, Lake Placid, New York) at 4 °C, 1 h. Immune complexes wereprecipitated with Protein A/G-agarose and washed extensively withRIPA buffer, 4 °C. Immunoprecipitated EGFR was resolved by poly-acrylamide gel electrophoresis and transferred to a polyvinylidene di-fluoride membrane (NOVEX, San Diego, CA) using a blotting apparatus(Bio-Rad). Tyrosine phosphorylation of the EGFR was visualized byincubating the membrane with anti-phosphotyrosine antibodies (pri-mary), followed by anti-mouse IgG horseradish peroxidase antibodies(secondary), and detection using the ECL system (DuPont Renaissance,PerkinElmer Life Science, Boston, MA). Total EGFR (loading control) orGrb2 was visualized by incubating the membrane with stripping buffer(2% SDS, 62.5 mM Tris-HCl, pH 7.4, 100 mM b-mercaptoethanol) for 2 h,23 °C, followed by extensive washing (0.05% Tween 20 in PBS) andblocking (0.25% nonfat milk, 0.12% Tween 20, SSC). The membranewas incubated with anti-EGFR (primary), or anti-GRB-2 (primary)antibody, followed by horseradish peroxidase-conjugated secondary an-tibody and ECL development, as above.
125I-EGF Binding Assay—A549 cell monolayers were washed withPBS, trypsinized off the tissue culture, and resuspended in DMEM togive a suspension of 1 3 106 cells/ml. Cells were equilibrated in DMEMcontaining 10% fetal calf serum for 1 h, 23 °C, followed by washing withPBS (3 times). TIMP-2, Ala1TIMP-2 6 EGF (unlabeled) was added tocell suspensions, in binding buffer (Amersham Pharmacia Biotech),followed by addition of 125I-EGF (100 mCi/ml, .75 Ci/mmol, AmershamPharmacia Biotech) and incubated for 3 h, 4 °C. The cells were dilutedin ice-cold binding buffer, collected by gentle centrifugation (,1000 3g), and washed three times with cold PBS. EGF bound to the A549 cellswas determined by g counting (cpm) on a Packard, Cobra auto-g-counter (Canberra Co., Downes Grove, IL).
Fluorescent Labeling and TIMP-2 Binding Assay—TIMP-2 andAla1TIMP-2 were labeled with BODIPY-Fl by addition of three molarequivalents (total) of BODIPY-Fl (Molecular Probes) in three batchesover 2 h, 23 °C. This labeling reaction was protected from light bycovering the reaction vessel with aluminum foil. The reaction wasquenched by addition of 1.5 M Tris-HCl, pH 7.5, to give a final concen-tration of 50 mM Tris-HCl. The crude reaction mixture was then passedover a Superose 6 (Amersham Pharmacia Biotech) gel filtration columnusing 50 mM Tris-HCl, pH 7.5, 100 mM NaCl as eluate. The BODIPY-labeled TIMP-2 or Ala1TIMP-2 containing peaks were collected andthe degree of labeling was calculated by determining the ratio of ab-sorbance at 450 nm/280 nm. An optimum value between 0.3 and 0.7 wasobtained for these labeling reactions.
Binding assays of BODIPY-labeled TIMP-2 to A549 and MCF7 cellswere performed in triplicate as follows. Cells were grown to 80% con-fluence in DMEM containing 10% fetal calf serum, in white-walled,clear-bottom 96-well Costar plates. Cells were washed with PBS andpretreated to dissociate preformed ligand-receptor complexes with 3 M
glycine buffer in 0.9% saline, pH 3.0, for 3 min. The cells were againwashed in PBS prior to addition of TIMP-2-BODIPY in PBS containing0.1% bovine serum albumin for 1 h, 37 °C. The supernatant was re-moved from cells and placed into empty wells for determination ofunbound TIMP-2-BODIPY. Cell monolayers were washed three timeswith ice-cold (4 °C) PBS containing 0.1% bovine serum albumin.Amounts of bound TIMP-2-BODIPY were analyzed using a plate reader(PerkinElmer Life Sciences HTS7000), and measuring fluorescencefrom each well (l excitation 5 494 nm and l emission 5 520 nm).Specific binding was calculated as the difference between boundBODIPY-TIMP-2 in the presence or absence of excess (100-fold) unla-beled ligand. Scatchard analysis of TIMP-2 binding was performed aspreviously reported (35).
Confocal Fluorescent Microscopy—A549 and MCF7 cells were platedat a range of 1–3 3 105 cells/ml in a Lab-Tek chambered glass slide(Nalgene). The growth medium was discarded and cells were washedwith PBS. Nonspecific binding was blocked using 1 mg/ml bovine serumalbumin (fatty acid-free), for 1 h. Cell were washed with PBS 3 timesand incubated with BODIPY-labeled TIMP-2 or Ala1TIMP-2, for 30min, 37 °C. Following incubation with BODIPY-labeled TIMP-2, thecells were washed with PBS 5 times and fixed in paraformaldehyde. Fordouble staining experiments cells were first incubated with BODIPY-labeled TIMP-2, washed, and fixed as described above. These cells werethen incubated with the appropriate primary antibody for 1 h, followedby washing with PBS, prior to incubation with secondary rhodamine-conjugated antibody for 45 min, 23 °C. Following this secondary anti-body incubation the cells were washed three times with PBS and fixed
as before. Two drops of Vectashield Mounting Medium (Vector Labora-tories, Burlingame, CA) containing DAPI (for nuclear counter staining)were added prior to coverslipping. Cell associated BODIPY-labeledTIMP-2 or Ala1TIMP-2, as well as anti-MT-1-MMP antibody stainingwas localized using confocal laser microscopy. All preparations wereexamined with a Leica confocal microscope, model TCS4D/DMIRBE,equipped with argon and argon-krypton lasers. Cells were originallyphotographed at 3 80 magnification.
Protein-tyrosine Phosphatase Assays—Protein-tyrosine phosphataseactivities were assayed following immunoprecipitation of either SHP-1(clone C-19, Santa Cruz Biotechnology) or SHP-2 (clone N-16, SantaCruz Biotechnology) utilizing selective antibodies. Phosphotyrosinephosphatase activity of the immunoprecipitates was measured as theamount of phosphotyrosine content remaining after addition of knownamounts of phosphotyrosine-containing peptide substrates, (Roche Mo-lecular Biochemicals). Hs68 or A549 cells were plated on 75-cm2 Nuncflasks (2 3 106/flask). Cells were preincubated with TIMP-2, followed bystimulation with growth factor (described above). Cells were harvestedin lysis buffer containing 10 mM MOPS, pH 6, 5 mM EDTA, 10%glycerol, 1% Triton X-100, 10 mM NaF, 25 mM glycerol phosphate, 10mg/ml 4-(2-aminoethyl)benzenesulfonyl fluoride, and 30 mg/ml aproti-nin at 4 °C for 10 min. The relative protein concentrations were deter-mined using the BCA assay (Pierce). Equal amount of protein (200 mg)were immunoprecipitated with anti-SHP-1 or SHP-2 antibodies (Trans-duction Labs) at 4 °C for 1 h, followed by incubation with anti-mouseagarose beads (Cappel) for 1 h. The beads were subsequently washed 3times with lysis buffer (without orthovanadate), and 3 times withphosphatase assay buffer. The immunoprecipitates were resuspendedin assay buffer and the reaction initiated by addition of phosphotyrosinepeptides (Roche Molecular Biochemicals). The optical densities weremeasured at 405 nm. Data was normalized to blank assay values andplotted as relative OD units of phosphotyrosine. Enhanced phosphataseactivity is represented by a decrease in phosphotyrosine levels.
RESULTS
TIMP-2 Suppresses Tyrosine Kinase Growth Factor-stimu-lated Cell Proliferation—Previous reports have shown thatTIMP-2 and Ala1TIMP-2 can stimulate the growth of quies-cent (serum starved) cells in culture (28, 34). However, thissystem does not represent a physiologic setting in which mul-tiple stimuli, both positive and negative, are integrated todetermine the cellular response. The effects of TIMP-2 on themitogenic response to TKR growth factors in several cells lines,including Hs68, HT1080, A549, and MCF7 cells were examinedin vitro. Treatment of these quiescent cells with a variety ofTKR growth factors, including EGF, bFGF, and PDGF, resultsin approximately a 2-fold stimulation of cell growth. A repre-sentative example of this growth stimulation is presented inthe inset in Fig. 1A. Preincubation of quiescent A549, HT1080,HS68, or MCF7 cells with increasing TIMP-2 concentrationsfollowed by addition of a TKR growth factor, such as EGF,results in dose-dependent inhibition of the mitogenic response(Fig. 1A). Preincubation of cells with TIMP-2, prior to additionof growth factor, was routinely performed for 30 min at 37 °C,but identical effects were obtained with preincubation periodsas short as 1 min, as previously reported (28). This suppressiveeffect on growth factor stimulation was not observed if TIMP-2was added concurrent with EGF stimulation or after treatmentof the cells with EGF (data not shown).
The observed effects of TIMP-2 on mitogenic response are notspecific for A549 cells. The effects of TIMP-2 on the EGF-stimulated responses of HT1080 human fibrosarcoma andMCF7 human mammary carcinoma cells are essentially iden-tical to those observed with the A549 cells (Fig. 1A). TIMP-2maximally inhibited (student t test, p , 0.01) the mitogenicresponse in all cell lines tested to 50–60% of the level achievedfollowing EGF stimulation alone (Fig. 1A). Similarly, TIMP-2reduced the mitogenic response to bFGF and PDGF in thesecell lines (data not shown). The effects of TIMP-2 on the mito-genic responses were observed at low nanomolar concentra-tions (,10 nM) with the maximal suppression of growth factor-mediated proliferation obtained at 20–50 nM TIMP-2.
TIMP-2 Suppresses EGFR Activation 3205
by guest on August 9, 2019
http://ww
w.jbc.org/
Dow
nloaded from

TIMP-2 Effect on Mitogenic Response Is Not Dependent onMMP Inhibition—To determine whether the effect of TIMP-2on growth factor mitogenic response in these cells was depend-ent on inhibition of MMP activity, we examined the effect ofother MMP inhibitors on the mitogenic response. We utilizedthe endogenous MMP-inhibitor, TIMP-1, a synthetic MMP in-hibitor (BB-94, Batimastat), and Ala1TIMP-2, a form ofTIMP-2 that lacks MMP inhibitor activity. Neither TIMP-1 northe synthetic hydroxamate inhibitor, BB-94, demonstrated anymodulating effects on mitogenic stimulation in any of thecell lines tested (data not shown, see below). However,Ala1TIMP-2 was effective at inhibiting the proliferative re-sponse stimulated by EGF, bFGF, and PDGF treatment of
A549 and Hs68 cells (Fig. 1). Pretreatment with 50 nM
Ala1TIMP-2 prior to exposure to bFGF or PDGF suppressedthe growth factor-mediated mitogenic response of Hs68 andA549 cells (Fig. 1B). Ala1TIMP-2 suppressed cell growth to thelevels observed with TIMP-2 stimulation alone, without addi-tion of growth factors (Fig. 1B). The suppressive effect ofAla1TIMP-2 also demonstrated a dose dependence (Fig. 1C),however, Ala1TIMP-2 suppressed the mitogenic response tolower levels than those achieved with TIMP-2.
TIMP-2 Inhibition of Growth Factor Response Requires Ad-enylate Cyclase Activity—We previously reported that the mi-togenic effects of TIMP-2 on quiescent Hs68 or HT1080 cellproliferation were dependent on activation of a heterotrimeric
FIG. 1. TIMP-2 and Ala1TIMP-2 in-hibit EGF, bFGF, and PDGF-inducedproliferation of A549, HT1080, Hs68,and MCF7 cells. A, A549, MCF7, orHT1080 cells were seeded onto gelatin (10mg/ml)-coated 96-well plates and were se-rum starved to quiescence, followed bytreatment with TIMP-2 (0–200 nM) for 30min, stimulated with EGF (200 ng/ml),and incubated for 24 h. Proliferation ofviable cells was determined by the changein absorbance at 490 nm on reduction of3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxy-methoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium salt or [3H]thymidine incor-poration. Results shown are thepercentage of maximum proliferation ob-tained by stimulation of cells with EGFalone (100%), after correction for basalrate of proliferation in serum-free condi-tions. Each data point represents the av-erage 6 S.D. of six determinations. B,A549 and Hs68 cells were treated with6Ala1TIMP-2 (50 nM) for 30 min, fol-lowed by stimulation with various growthfactors (*GF) (bFGF (50 ng/ml with 1.13units/mg of heparin), EGF (200 ng/ml),PDGF (50 ng/ml)), or with TIMP-2 (50 nM)alone (absence of growth factors or se-rum). C, A549 and Hs68 cells weretreated with Ala1TIMP-2 (0–50 nM), fol-lowed by stimulation with EGF (200ng/ml).
TIMP-2 Suppresses EGFR Activation3206
by guest on August 9, 2019
http://ww
w.jbc.org/
Dow
nloaded from
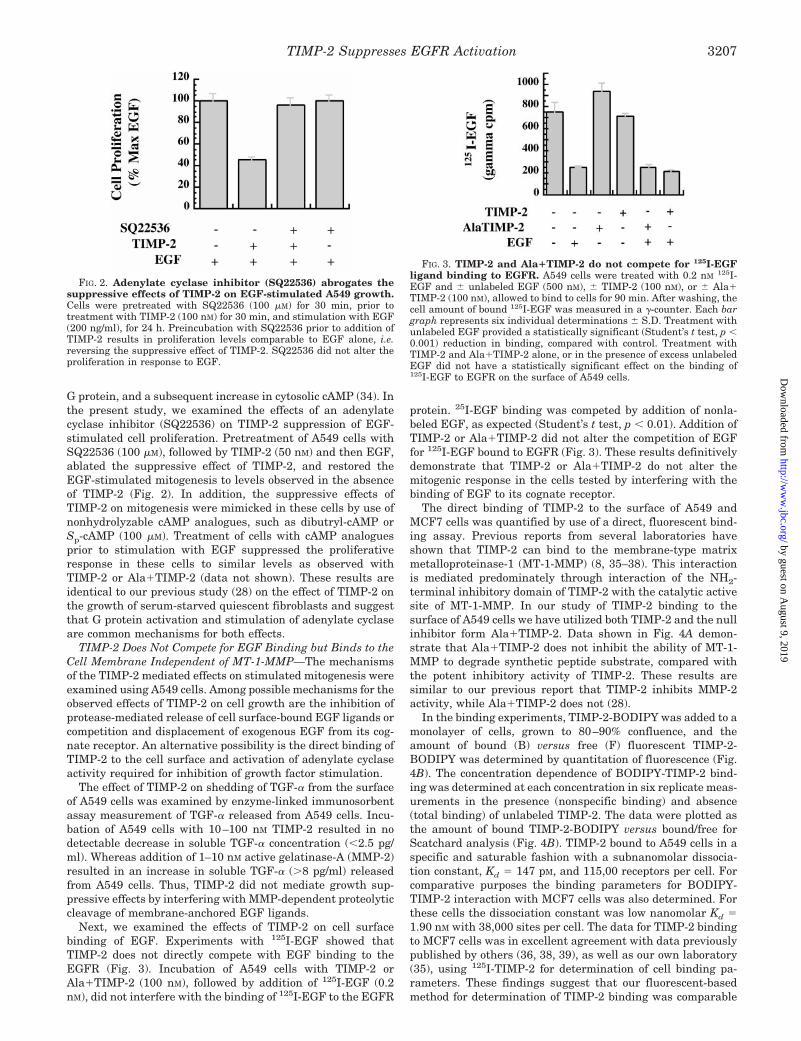
G protein, and a subsequent increase in cytosolic cAMP (34). Inthe present study, we examined the effects of an adenylatecyclase inhibitor (SQ22536) on TIMP-2 suppression of EGF-stimulated cell proliferation. Pretreatment of A549 cells withSQ22536 (100 mM), followed by TIMP-2 (50 nM) and then EGF,ablated the suppressive effect of TIMP-2, and restored theEGF-stimulated mitogenesis to levels observed in the absenceof TIMP-2 (Fig. 2). In addition, the suppressive effects ofTIMP-2 on mitogenesis were mimicked in these cells by use ofnonhydrolyzable cAMP analogues, such as dibutryl-cAMP orSp-cAMP (100 mM). Treatment of cells with cAMP analoguesprior to stimulation with EGF suppressed the proliferativeresponse in these cells to similar levels as observed withTIMP-2 or Ala1TIMP-2 (data not shown). These results areidentical to our previous study (28) on the effect of TIMP-2 onthe growth of serum-starved quiescent fibroblasts and suggestthat G protein activation and stimulation of adenylate cyclaseare common mechanisms for both effects.
TIMP-2 Does Not Compete for EGF Binding but Binds to theCell Membrane Independent of MT-1-MMP—The mechanismsof the TIMP-2 mediated effects on stimulated mitogenesis wereexamined using A549 cells. Among possible mechanisms for theobserved effects of TIMP-2 on cell growth are the inhibition ofprotease-mediated release of cell surface-bound EGF ligands orcompetition and displacement of exogenous EGF from its cog-nate receptor. An alternative possibility is the direct binding ofTIMP-2 to the cell surface and activation of adenylate cyclaseactivity required for inhibition of growth factor stimulation.
The effect of TIMP-2 on shedding of TGF-a from the surfaceof A549 cells was examined by enzyme-linked immunosorbentassay measurement of TGF-a released from A549 cells. Incu-bation of A549 cells with 10–100 nM TIMP-2 resulted in nodetectable decrease in soluble TGF-a concentration (,2.5 pg/ml). Whereas addition of 1–10 nM active gelatinase-A (MMP-2)resulted in an increase in soluble TGF-a (.8 pg/ml) releasedfrom A549 cells. Thus, TIMP-2 did not mediate growth sup-pressive effects by interfering with MMP-dependent proteolyticcleavage of membrane-anchored EGF ligands.
Next, we examined the effects of TIMP-2 on cell surfacebinding of EGF. Experiments with 125I-EGF showed thatTIMP-2 does not directly compete with EGF binding to theEGFR (Fig. 3). Incubation of A549 cells with TIMP-2 orAla1TIMP-2 (100 nM), followed by addition of 125I-EGF (0.2nM), did not interfere with the binding of 125I-EGF to the EGFR
protein. 25I-EGF binding was competed by addition of nonla-beled EGF, as expected (Student’s t test, p , 0.01). Addition ofTIMP-2 or Ala1TIMP-2 did not alter the competition of EGFfor 125I-EGF bound to EGFR (Fig. 3). These results definitivelydemonstrate that TIMP-2 or Ala1TIMP-2 do not alter themitogenic response in the cells tested by interfering with thebinding of EGF to its cognate receptor.
The direct binding of TIMP-2 to the surface of A549 andMCF7 cells was quantified by use of a direct, fluorescent bind-ing assay. Previous reports from several laboratories haveshown that TIMP-2 can bind to the membrane-type matrixmetalloproteinase-1 (MT-1-MMP) (8, 35–38). This interactionis mediated predominately through interaction of the NH2-terminal inhibitory domain of TIMP-2 with the catalytic activesite of MT-1-MMP. In our study of TIMP-2 binding to thesurface of A549 cells we have utilized both TIMP-2 and the nullinhibitor form Ala1TIMP-2. Data shown in Fig. 4A demon-strate that Ala1TIMP-2 does not inhibit the ability of MT-1-MMP to degrade synthetic peptide substrate, compared withthe potent inhibitory activity of TIMP-2. These results aresimilar to our previous report that TIMP-2 inhibits MMP-2activity, while Ala1TIMP-2 does not (28).
In the binding experiments, TIMP-2-BODIPY was added to amonolayer of cells, grown to 80–90% confluence, and theamount of bound (B) versus free (F) fluorescent TIMP-2-BODIPY was determined by quantitation of fluorescence (Fig.4B). The concentration dependence of BODIPY-TIMP-2 bind-ing was determined at each concentration in six replicate meas-urements in the presence (nonspecific binding) and absence(total binding) of unlabeled TIMP-2. The data were plotted asthe amount of bound TIMP-2-BODIPY versus bound/free forScatchard analysis (Fig. 4B). TIMP-2 bound to A549 cells in aspecific and saturable fashion with a subnanomolar dissocia-tion constant, Kd 5 147 pM, and 115,00 receptors per cell. Forcomparative purposes the binding parameters for BODIPY-TIMP-2 interaction with MCF7 cells was also determined. Forthese cells the dissociation constant was low nanomolar Kd 51.90 nM with 38,000 sites per cell. The data for TIMP-2 bindingto MCF7 cells was in excellent agreement with data previouslypublished by others (36, 38, 39), as well as our own laboratory(35), using 125I-TIMP-2 for determination of cell binding pa-rameters. These findings suggest that our fluorescent-basedmethod for determination of TIMP-2 binding was comparable
FIG. 2. Adenylate cyclase inhibitor (SQ22536) abrogates thesuppressive effects of TIMP-2 on EGF-stimulated A549 growth.Cells were pretreated with SQ22536 (100 mM) for 30 min, prior totreatment with TIMP-2 (100 nM) for 30 min, and stimulation with EGF(200 ng/ml), for 24 h. Preincubation with SQ22536 prior to addition ofTIMP-2 results in proliferation levels comparable to EGF alone, i.e.reversing the suppressive effect of TIMP-2. SQ22536 did not alter theproliferation in response to EGF.
FIG. 3. TIMP-2 and Ala1TIMP-2 do not compete for 125I-EGFligand binding to EGFR. A549 cells were treated with 0.2 nM 125I-EGF and 6 unlabeled EGF (500 nM), 6 TIMP-2 (100 nM), or 6 Ala1TIMP-2 (100 nM), allowed to bind to cells for 90 min. After washing, thecell amount of bound 125I-EGF was measured in a g-counter. Each bargraph represents six individual determinations 6 S.D. Treatment withunlabeled EGF provided a statistically significant (Student’s t test, p ,0.001) reduction in binding, compared with control. Treatment withTIMP-2 and Ala1TIMP-2 alone, or in the presence of excess unlabeledEGF did not have a statistically significant effect on the binding of125I-EGF to EGFR on the surface of A549 cells.
TIMP-2 Suppresses EGFR Activation 3207
by guest on August 9, 2019
http://ww
w.jbc.org/
Dow
nloaded from

in sensitivity and specificity to methods described previously.The addition of a 10-fold excess of unlabeled Ala1TIMP-2 to
A549 cells treated with TIMP-2-BODIPY leads to a statisticallysignificant (Student’s t test, p , 0.001) reduction in binding ofTIMP-2 to the cell surface. Ala1TIMP-2 competition results ina 65% decrease in BODIPY-TIMP-2 binding to A549 cells sug-gesting that it competes for binding to most, but not all, TIMP-2-binding sites (Fig. 4B). In contrast, addition of a broad spec-trum, hydroxamate MMP inhibitor, BB-94 (0.5 mM), did notsignificantly compete for binding of BODIPY-TIMP-2 to the cellsurface (Fig. 4B). This failure of BB-94 to compete with TIMP-2cell surface binding was in contrast to the effects of synthetichydroxamate MMP inhibitors which have been shown to in-hibit the binding of TIMP-2 to MT-1-MMP (36, 38). Westernblot analysis of A549 cell membranes demonstrated that thesecells have low but detectable levels of MT-1-MMP comparedwith well characterized cell lines such as HT1080 (data notshown). However, addition of an MT1-MMP antibody, specificfor the catalytic domain, maximally reduced the binding ofTIMP-2 to cells by 35% (Fig. 4B). Together these data suggestthat TIMP-2 binds to the cell surface and that this interactionmay consist of at least two binding sites, an interpretationconsistent with the Scatchard analysis shown in Fig. 4A, aswell as previous reports (36).
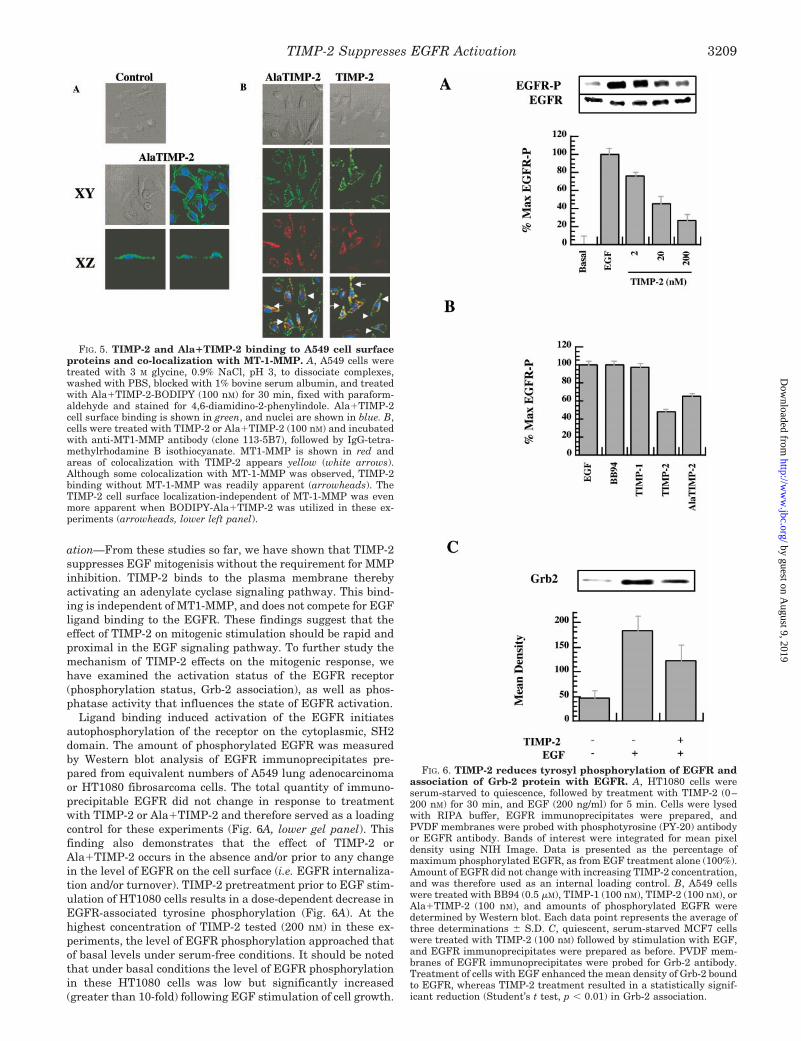
Confocal, laser fluorescent microscopy was utilized to exam-ine the localization of TIMP-2 on the cell surface and colo-calization with MT-1-MMP. Confocal fluorescent microscopydemonstrated fluorescent-labeled (BODIPY) TIMP-2 orAla1TIMP-2 bound to the surface of A549 cells (Fig. 5A). Anal-ysis of Nemarsky optic images of A549 cells with or withoutTIMP-2 or Ala1TIMP-2 treatment revealed no significant mor-phologic changes following short term (30 min) exposure toTIMP-2 (Fig. 5, A, top and middle left panels; B, top two panels).Fluorescence localization of BODIPY-TIMP-2 or Ala1TIMP-2demonstrates a linear, punctate pattern consistent with cellsurface localization (Fig. 5, A, lower panels, B, second panelsfrom top). xz-axis analysis of the confocal images (Fig. 5A,bottom panel) confirms that TIMP-2 binding occurs on thesurface of A549 cells. By fluorescent antibody staining the cellswith rhodamine anti-MT-1-MMP antibody complexes, the ma-jority of TIMP-2 binds to the surface of A549 cells independentof MT1-MMP localization (Fig. 5B, bottom panel right, andarrowheads). A minor component of the TIMP-2 on the cellsurface colocalized with MT-1-MMP (Fig. 5B, bottom panel, andarrows). The MT-1-MMP colocalization was markedly less ap-parent when the binding of Ala1TIMP-2 was examined (Fig.5B, bottom panel, left). Ala1TIMP-2 colocalization with MT1-MMP (Fig. 5, bottom panel, left, and arrows) was reducedcompared with that observed with wtTIMP-2 (Fig. 5, bottompanel, right, and arrows). These findings are consistent withour in vitro observations that the Ala1TIMP-2 mutant does notinhibit MT-1-MMP activity (Fig. 4A) (i.e. Ala1TIMP-2 does notbind to the MT-1-MMP active site).
TIMP-2 Disrupts EGFR Phosphorylation and Grb-2 Associ-FIG. 4. TIMP-2 and Ala1TIMP-2 activity against MT-1-MMP
catalytic domain and cell binding experiments. A, measurementof Ala1TIMP-2 suppressive activity against MT-1-MMP. TIMP-2 andAla1TIMP-2 inhibition of MT-1-MMP activity were determined usingthe thiopeptolide assays as described previously (28). These assayswere performed in 50 mM MOPS, 150 mM NaCl, 1 mM CaCl2, and 1 mM
5,59-dithiobis-(2-nitrobenzoic acid) at pH 7.0. Concentrations of thecatalytic domain of MT-1-MMP and thiopeptolide were 20 nM and 50mM, respectively. TIMP-2 effectively inhibits MT-1-MMP activity, butAla1TIMP-2 does not, as indicated by no decrease in Vi over the rangeof added Ala1TIMP-2 concentrations. B, measurement of Kd and num-ber of receptors per cell was determined for TIMP-2-BODIPY binding toA549 cells by a 96-well plate binding assay. A549 cells were seeded onto96-well plates and treated with TIMP-2-BODIPY (0–100 nM) for 30min, 37 °C. Supernatant was transferred to new wells and measured asthe amount of unbound TIMP-2-BODIPY. Cells were washed with
PBS and the amount of TIMP-2-BODIPY was measured as the amountof fluorescence remaining on the A549 cell surface. Concentration ofbound TIMP-2-BODIPY was determined with a standard curve (TIMP-2-BODIPY concentration versus fluorescence units) and plotted againstBound/Free, to give the resulting Scatchard plot. Scatchard analysiswas performed as described previously (35). Unlabeled TIMP-2 wasadded to cells to determine specific binding from total and nonspecificbinding. C, a 10-fold excess of Ala1TIMP-2 (unlabeled) was added tocells, resulting in a reduction of bound fluorescence (Student’s t test,p , 0.01), whereas addition of BB94 (0.5 mM) did not result in anysignificant change. However, addition of 10 mg/ml anti-MT1-MMP(clone 114–6G6) resulted in a reduction in bound TIMP-2-BODIPYfluorescence (Student’s t test, p , 0.05).
TIMP-2 Suppresses EGFR Activation3208
by guest on August 9, 2019
http://ww
w.jbc.org/
Dow
nloaded from
ation—From these studies so far, we have shown that TIMP-2suppresses EGF mitogenisis without the requirement for MMPinhibition. TIMP-2 binds to the plasma membrane therebyactivating an adenylate cyclase signaling pathway. This bind-ing is independent of MT1-MMP, and does not compete for EGFligand binding to the EGFR. These findings suggest that theeffect of TIMP-2 on mitogenic stimulation should be rapid andproximal in the EGF signaling pathway. To further study themechanism of TIMP-2 effects on the mitogenic response, wehave examined the activation status of the EGFR receptor(phosphorylation status, Grb-2 association), as well as phos-phatase activity that influences the state of EGFR activation.
Ligand binding induced activation of the EGFR initiatesautophosphorylation of the receptor on the cytoplasmic, SH2domain. The amount of phosphorylated EGFR was measuredby Western blot analysis of EGFR immunoprecipitates pre-pared from equivalent numbers of A549 lung adenocarcinomaor HT1080 fibrosarcoma cells. The total quantity of immuno-precipitable EGFR did not change in response to treatmentwith TIMP-2 or Ala1TIMP-2 and therefore served as a loadingcontrol for these experiments (Fig. 6A, lower gel panel). Thisfinding also demonstrates that the effect of TIMP-2 orAla1TIMP-2 occurs in the absence and/or prior to any changein the level of EGFR on the cell surface (i.e. EGFR internaliza-tion and/or turnover). TIMP-2 pretreatment prior to EGF stim-ulation of HT1080 cells results in a dose-dependent decrease inEGFR-associated tyrosine phosphorylation (Fig. 6A). At thehighest concentration of TIMP-2 tested (200 nM) in these ex-periments, the level of EGFR phosphorylation approached thatof basal levels under serum-free conditions. It should be notedthat under basal conditions the level of EGFR phosphorylationin these HT1080 cells was low but significantly increased(greater than 10-fold) following EGF stimulation of cell growth.
FIG. 5. TIMP-2 and Ala1TIMP-2 binding to A549 cell surfaceproteins and co-localization with MT-1-MMP. A, A549 cells weretreated with 3 M glycine, 0.9% NaCl, pH 3, to dissociate complexes,washed with PBS, blocked with 1% bovine serum albumin, and treatedwith Ala1TIMP-2-BODIPY (100 nM) for 30 min, fixed with paraform-aldehyde and stained for 4,6-diamidino-2-phenylindole. Ala1TIMP-2cell surface binding is shown in green, and nuclei are shown in blue. B,cells were treated with TIMP-2 or Ala1TIMP-2 (100 nM) and incubatedwith anti-MT1-MMP antibody (clone 113-5B7), followed by IgG-tetra-methylrhodamine B isothiocyanate. MT1-MMP is shown in red andareas of colocalization with TIMP-2 appears yellow (white arrows).Although some colocalization with MT-1-MMP was observed, TIMP-2binding without MT-1-MMP was readily apparent (arrowheads). TheTIMP-2 cell surface localization-independent of MT-1-MMP was evenmore apparent when BODIPY-Ala1TIMP-2 was utilized in these ex-periments (arrowheads, lower left panel).
FIG. 6. TIMP-2 reduces tyrosyl phosphorylation of EGFR andassociation of Grb-2 protein with EGFR. A, HT1080 cells wereserum-starved to quiescence, followed by treatment with TIMP-2 (0–200 nM) for 30 min, and EGF (200 ng/ml) for 5 min. Cells were lysedwith RIPA buffer, EGFR immunoprecipitates were prepared, andPVDF membranes were probed with phosphotyrosine (PY-20) antibodyor EGFR antibody. Bands of interest were integrated for mean pixeldensity using NIH Image. Data is presented as the percentage ofmaximum phosphorylated EGFR, as from EGF treatment alone (100%).Amount of EGFR did not change with increasing TIMP-2 concentration,and was therefore used as an internal loading control. B, A549 cellswere treated with BB94 (0.5 mM), TIMP-1 (100 nM), TIMP-2 (100 nM), orAla1TIMP-2 (100 nM), and amounts of phosphorylated EGFR weredetermined by Western blot. Each data point represents the average ofthree determinations 6 S.D. C, quiescent, serum-starved MCF7 cellswere treated with TIMP-2 (100 nM) followed by stimulation with EGF,and EGFR immunoprecipitates were prepared as before. PVDF mem-branes of EGFR immunoprecipitates were probed for Grb-2 antibody.Treatment of cells with EGF enhanced the mean density of Grb-2 boundto EGFR, whereas TIMP-2 treatment resulted in a statistically signif-icant reduction (Student’s t test, p , 0.01) in Grb-2 association.
TIMP-2 Suppresses EGFR Activation 3209
by guest on August 9, 2019
http://ww
w.jbc.org/
Dow
nloaded from
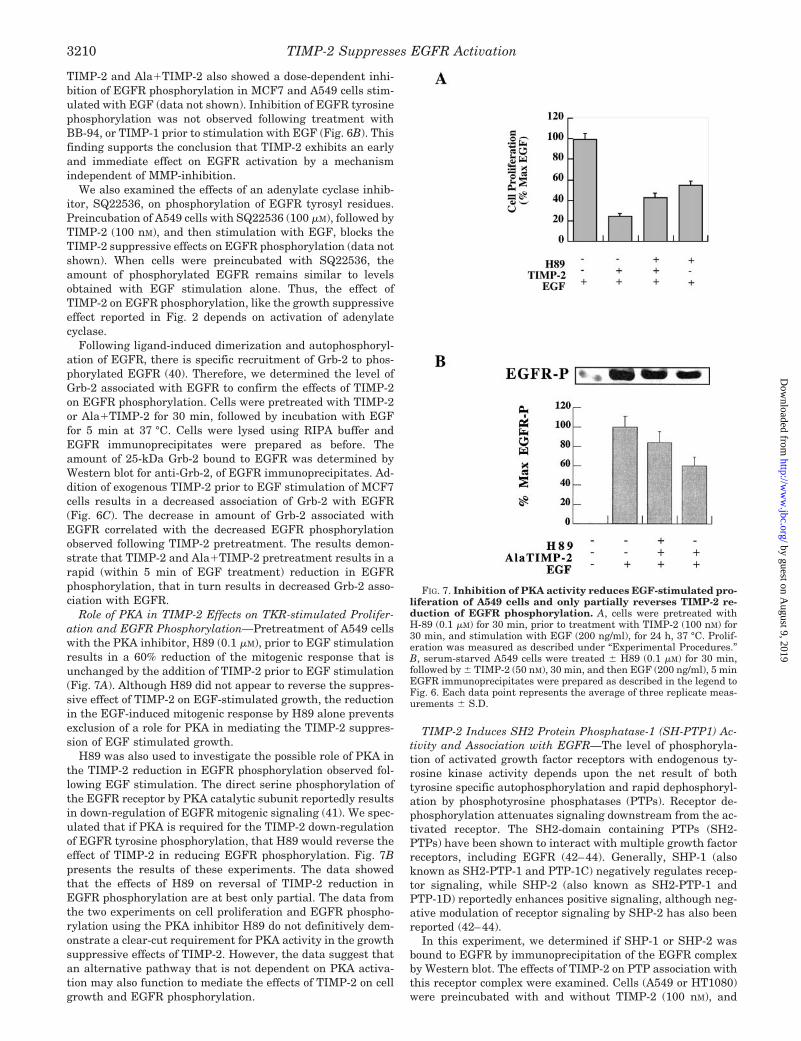
TIMP-2 and Ala1TIMP-2 also showed a dose-dependent inhi-bition of EGFR phosphorylation in MCF7 and A549 cells stim-ulated with EGF (data not shown). Inhibition of EGFR tyrosinephosphorylation was not observed following treatment withBB-94, or TIMP-1 prior to stimulation with EGF (Fig. 6B). Thisfinding supports the conclusion that TIMP-2 exhibits an earlyand immediate effect on EGFR activation by a mechanismindependent of MMP-inhibition.
We also examined the effects of an adenylate cyclase inhib-itor, SQ22536, on phosphorylation of EGFR tyrosyl residues.Preincubation of A549 cells with SQ22536 (100 mM), followed byTIMP-2 (100 nM), and then stimulation with EGF, blocks theTIMP-2 suppressive effects on EGFR phosphorylation (data notshown). When cells were preincubated with SQ22536, theamount of phosphorylated EGFR remains similar to levelsobtained with EGF stimulation alone. Thus, the effect ofTIMP-2 on EGFR phosphorylation, like the growth suppressiveeffect reported in Fig. 2 depends on activation of adenylatecyclase.
Following ligand-induced dimerization and autophosphoryl-ation of EGFR, there is specific recruitment of Grb-2 to phos-phorylated EGFR (40). Therefore, we determined the level ofGrb-2 associated with EGFR to confirm the effects of TIMP-2on EGFR phosphorylation. Cells were pretreated with TIMP-2or Ala1TIMP-2 for 30 min, followed by incubation with EGFfor 5 min at 37 °C. Cells were lysed using RIPA buffer andEGFR immunoprecipitates were prepared as before. Theamount of 25-kDa Grb-2 bound to EGFR was determined byWestern blot for anti-Grb-2, of EGFR immunoprecipitates. Ad-dition of exogenous TIMP-2 prior to EGF stimulation of MCF7cells results in a decreased association of Grb-2 with EGFR(Fig. 6C). The decrease in amount of Grb-2 associated withEGFR correlated with the decreased EGFR phosphorylationobserved following TIMP-2 pretreatment. The results demon-strate that TIMP-2 and Ala1TIMP-2 pretreatment results in arapid (within 5 min of EGF treatment) reduction in EGFRphosphorylation, that in turn results in decreased Grb-2 asso-ciation with EGFR.
Role of PKA in TIMP-2 Effects on TKR-stimulated Prolifer-ation and EGFR Phosphorylation—Pretreatment of A549 cellswith the PKA inhibitor, H89 (0.1 mM), prior to EGF stimulationresults in a 60% reduction of the mitogenic response that isunchanged by the addition of TIMP-2 prior to EGF stimulation(Fig. 7A). Although H89 did not appear to reverse the suppres-sive effect of TIMP-2 on EGF-stimulated growth, the reductionin the EGF-induced mitogenic response by H89 alone preventsexclusion of a role for PKA in mediating the TIMP-2 suppres-sion of EGF stimulated growth.
H89 was also used to investigate the possible role of PKA inthe TIMP-2 reduction in EGFR phosphorylation observed fol-lowing EGF stimulation. The direct serine phosphorylation ofthe EGFR receptor by PKA catalytic subunit reportedly resultsin down-regulation of EGFR mitogenic signaling (41). We spec-ulated that if PKA is required for the TIMP-2 down-regulationof EGFR tyrosine phosphorylation, that H89 would reverse theeffect of TIMP-2 in reducing EGFR phosphorylation. Fig. 7Bpresents the results of these experiments. The data showedthat the effects of H89 on reversal of TIMP-2 reduction inEGFR phosphorylation are at best only partial. The data fromthe two experiments on cell proliferation and EGFR phospho-rylation using the PKA inhibitor H89 do not definitively dem-onstrate a clear-cut requirement for PKA activity in the growthsuppressive effects of TIMP-2. However, the data suggest thatan alternative pathway that is not dependent on PKA activa-tion may also function to mediate the effects of TIMP-2 on cellgrowth and EGFR phosphorylation.
TIMP-2 Induces SH2 Protein Phosphatase-1 (SH-PTP1) Ac-tivity and Association with EGFR—The level of phosphoryla-tion of activated growth factor receptors with endogenous ty-rosine kinase activity depends upon the net result of bothtyrosine specific autophosphorylation and rapid dephosphoryl-ation by phosphotyrosine phosphatases (PTPs). Receptor de-phosphorylation attenuates signaling downstream from the ac-tivated receptor. The SH2-domain containing PTPs (SH2-PTPs) have been shown to interact with multiple growth factorreceptors, including EGFR (42–44). Generally, SHP-1 (alsoknown as SH2-PTP-1 and PTP-1C) negatively regulates recep-tor signaling, while SHP-2 (also known as SH2-PTP-1 andPTP-1D) reportedly enhances positive signaling, although neg-ative modulation of receptor signaling by SHP-2 has also beenreported (42–44).
In this experiment, we determined if SHP-1 or SHP-2 wasbound to EGFR by immunoprecipitation of the EGFR complexby Western blot. The effects of TIMP-2 on PTP association withthis receptor complex were examined. Cells (A549 or HT1080)were preincubated with and without TIMP-2 (100 nM), and
FIG. 7. Inhibition of PKA activity reduces EGF-stimulated pro-liferation of A549 cells and only partially reverses TIMP-2 re-duction of EGFR phosphorylation. A, cells were pretreated withH-89 (0.1 mM) for 30 min, prior to treatment with TIMP-2 (100 nM) for30 min, and stimulation with EGF (200 ng/ml), for 24 h, 37 °C. Prolif-eration was measured as described under “Experimental Procedures.”B, serum-starved A549 cells were treated 6 H89 (0.1 mM) for 30 min,followed by 6 TIMP-2 (50 nM), 30 min, and then EGF (200 ng/ml), 5 minEGFR immunoprecipitates were prepared as described in the legend toFig. 6. Each data point represents the average of three replicate meas-urements 6 S.D.
TIMP-2 Suppresses EGFR Activation3210
by guest on August 9, 2019
http://ww
w.jbc.org/
Dow
nloaded from

stimulated with EGF as above. Analysis of SH2-PTPs associ-ated with EGFR immunoprecipitates demonstrates that,compared with EGF treatment alone, TIMP-2 pretreatmentpreserves the association of SHP-1 with EGFR immunopre-cipitates to levels that are essentially identical with thoseobserved in the basal state. The levels of SHP-1 associated withEGFR complexes were inversely correlated with the level ofEGFR phosphorylation previously observed (Figs. 6A, and 8, Aand B). In contrast, the association of SHP-2 with EGFR com-plex remains unchanged following either TIMP-2 pretreatmentor EGF stimulation of cells (Fig. 8, A and B). We also assayedtotal cytoplasmic SHP-1 and SHP-2 activity utilizing an invitro tyrosine phosphatase assay following selective immuno-precipitation of total SHP-1 and SHP-2. The results of theseexperiments demonstrate a significant (Student’s t test, p ,
0.01) increase in total SHP-1 activity in cells pretreated withTIMP-2 prior to EGF stimulation (Fig. 8C, decrease in opticaldensity for phosphotyrosine staining reflects enhanced phos-photyrosine activity). No increase in SHP-2 activity was ob-served following TIMP-2 preincubation. In fact, EGF stimula-tion alone results in a significant induction of SHP-2 activityand a slight inhibition of SHP-2 activity was noted followingTIMP-2 pretreatment (Fig. 8C). These results demonstratethat TIMP-2 supression of EGFR activation was mediated, atleast in part, through persistent association of SHP-1 withEGFR and a selective increase total SHP-1 activity.
DISCUSSION
The tissue microenvironment is known to exert a profoundinfluence on cell proliferation and differentiation (45–47). Cell
FIG. 8. TIMP-2 enhances binding of SH2 protein-tyrosine phosphatase-1 (SH-PTP1), but not phosphatase-2 (SH-PTP2) to EGFR. A,A549 cells were serum starved to quiescence and treated with TIMP-2 (100 nM) followed by EGF (200 ng/ml) for 5 min. EGFR immunoprecipitateswere prepared, and polyvinylidene difluoride membranes were probed for SH-PTP1 or SH-PTP2. B, treatment of cells with EGF reduced SH-PTP1association, but TIMP-2 restored levels to that seen without EGF stimulation. Amounts of SH-PTP2 were not significantly altered on treatmentwith EGF or TIMP-2. C, cells were plated on 75-cm2 Nunc flasks 2 3 106 cells per flask and following attachment were incubated in serum-freeDMEM for 2 h then in serum-free DMEM for an additional 24 h. TIMP-2 (24 nM) was preincubated for 5 min prior to a 10-min stimulation withgrowth factor (EGF, 100 ng/ml). The cells were harvested in lysing buffer containing vanadate. Equal amounts of protein from the lysates (200 mg)were immunoprecipitated with anti-SHP-1 or SHP-2 antibodies (Transduction Labs) at 4 °C for 60 min followed by 60 min incubation withanti-mouse agarose beads (Cappel). The beads were subsequently washed three times in lysing buffer without orthovanadate and 3 times morewith phoshatase assay buffer. These immunoprecipitates were resuspended in assay buffer, transferred to 96-well plates, and the reaction initiatedby adding a phosphotyrosine peptide. After 10 min at 30 °C, the reaction was stopped by addition of 100 mM orthrovanadate. The relative levelsof phosphatase activity in these immunprecipitates was determined utilizing a phosphopeptide by enzyme-linked immunosorbent assay asdescribed by manufacturer’s instruction (Roche Molecular Biochemicals). Enzyme-linked immunosorbent assay results were determined in aMolecular Devices microplate reader. A decrease in optical density is indicative of enhanced phosphatase activity against the phosphopeptidecontaining substrate.
TIMP-2 Suppresses EGFR Activation 3211
by guest on August 9, 2019
http://ww
w.jbc.org/
Dow
nloaded from

fate is the net result of cellular integration of multiple signalsderived from soluble factors and adhesive interactions presentin the microenvironment (48–50). Evidence for the influence ofthis integrative process on cell behavior is derived from studiesusing reconstituted ECM, as well as alteration in the expres-sion of cell adhesion molecules, or the introduction of proteasesto disrupt the structure and/or composition of the microenvi-ronment (3, 6, 46, 49). ECM turnover is a critical event indevelopment, morphogenesis, and tissue remodeling (3, 6, 46,49). Enhanced MMP activity results in altered neonatal devel-opment and progression of pathologic conditions (6, 49). Vis avis their ability to inhibit MMP activity, TIMPs can suppressECM turnover associated with either embryonic developmentor pathologic conditions (2, 9, 51), resulting indirectly in sup-pression of cell growth and/or direct inhibition of cellular inva-sion initiated by ECM remodeling (12–14, 52).
However, studies demonstrate that TIMPs also directly alterin vitro cell growth and/or survival of a variety of cell types (16,17, 18, 19, 31, 34, 53–57). These effects are independent ofTIMP-mediated MMP inhibitory activity. TIMP-3 overexpres-sion induces apoptosis (58, 59), an effect variably reproduced bysynthetic MMP inhibitors and possibly related to stabilizationof TNF-a receptors or inhibition of TNF-a converting enzyme(60).
In the present report we examine how TIMP-2 induction ofintracellular signaling is integrated with TKR growth factorinduction of mitogenic signals. TIMP-2 abrogates the mitogenicresponse of a variety of cell types to several different TKR-typegrowth factors. The TIMP-2 inhibition of growth factor-stimu-lated mitogenesis occurs in a concentration range identical tothat observed for the maximal effect on stimulation of growthin quiescent, dermal fibroblasts (28, 34). These TIMP-2 concen-trations are only slightly lower than reported for the growthsuppressive effect of TIMP-2 on bFGF-stimulated endothelialcells (31). Mitogenic stimulation of various cell lines at sub-nanomolar concentrations of TIMPs has also been reported (19,53, 55). The differences in effective TIMP-2 concentrationsbetween these reports are possibly due to variation in theinnate sensitivity of the cell lines tested, or, alternatively,different TIMP preparations may have altered potency or con-taminants (e.g. endotoxin). All of the TIMP-2 preparations inthis study are known to be free of significant endotoxin con-tamination (see “Experimental Procedures”). It is also possiblethat differences in the levels of active MMPs produced by thecell lines may influence responsiveness to the growth modulat-ing activity of TIMP-2. This is because high concentrations ofavailable MMP active sites could sequester TIMP-2 and pre-vent it from binding to the cell surface.
The suppressive effect of TIMP-2 on growth factor-stimu-lated mitogenesis is specific and independent of MMP inhibi-tion. No effects on stimulated cell growth are observed withTIMP-1 or BB-94. Ala1TIMP-2, lacking MMP inhibitor activ-ity, remains active in abrogating the TKR-stimulated response.No evidence of cell death or apoptosis was observed in theseexperiments, and the cells remained viable following exposureto TIMP-2 or Ala1TIMP-2 alone. In fact, TIMP-2 orAla1TIMP-2 treatment without growth factor stimulation re-produces the modest mitogenic stimulation of quiescent cells aspreviously reported (28, 34). Ala1TIMP-2 shows a somewhatmore potent effect on inhibiting EGF-stimulated growth com-pared with the wild type protein. This is specific for EGF-stimulated growth and is not observed with the other growthfactors utilized in this study. Possible explanations for thisobservation include intrinsic differences in the growth factor-specific responses and/or greater availability of Ala1TIMP-2,
which did not bind effectively to the MT-1-MMP active sites(see below).
TIMP-2 regulation of TKR-induced mitogenesis occurs im-mediately downstream of receptor-ligand interaction duringreceptor activation. Previous reports demonstrate that metal-loproteinases contribute to shedding of EGFR ligands, such asTGF-a, as well as EGFR (61). TIMP-2 treatment did not alterEGFR levels, compete for EGF binding to EGFR, or alterTGF-a release. These findings are again consistent withTIMP-2 growth modulation that is independent of inhibition ofMMP activity required for either ligand or receptor processing.
TIMP-2 binds directly to the cell surface (19, 34, 36, 55).However, the identification of cell surface binding proteins forTIMP-2 is complicated by the presence of MT-MMPs. Theseintegral membrane proteins contain a transmembrane domainand catalytic site that is oriented toward the ECM (8, 37).Binding of TIMP-2 to MT-1-MMP involves interaction of theNH2-terminal of TIMP-2 with the catalytic site of MT-1-MMP(38), and is reportedly sensitive to synthetic, hydroxamateMMP inhibitor (36). Our in vitro analysis confirms thatAla1TIMP-2 does not inhibit MT-1-MMP activity, as we havepreviously reported for MMP-2 (28). This finding suggests thatAla1TIMP-2 will not bind to the MT-1-MMP catalytic site onthe cell surface.
We demonstrate specific and saturable binding of TIMP-2and Ala1TIMP-2 to the surface of A549 and MCF7 cells. Cellsurface binding of TIMP-2 to A549 cells was not competed byTIMP-1 or BB-94. Anti-MT-1-MMP catalytic domain antibodiesreduce TIMP-2 binding by only 35%, compared with the 65%reduction following addition of excess unlabeled Ala1TIMP-2.In fluorescence confocal microscopy experiments, minor colo-calization of TIMP-2 and MT-1-MMP in A549 cells is observed,but is essentially absent when Ala1TIMP-2 and MT-1-MMPare visualized. Collectively our studies on TIMP-2 binding andfluorescence co-localization confirm the presence of a high af-finity, TIMP-2-binding site on A549 cells that is independent ofMT-1-MMP. This is a principal binding site for TIMP-2 in A549cells that is specifically competed by Ala1TIMP-2, but is notblocked by synthetic hydroxamate MMP inhibitors nor anti-MT-1-MMP antibody. The presence of such sites has been sug-gested in previous studies that demonstrated TIMP-2 bindingin the presence of synthetic MMP inhibitor (36), but such siteshave remained poorly characterized. From these studies weconclude that TIMP-2 binds to the cell surface and this inter-action may consist of at least two binding sites, one MT-1-MMPindependent, as well as a MT-1-MMP site. This interpretationis consistent with Scatchard analysis, as well as previous re-ports of multiple TIMP-2-binding sites (36). TIMP-2 binding tothe MT-1-MMP-independent, high affinity site presumably re-sults in activation of adenylate cyclase that is required forTIMP-2 inhibition of TKR-stimulated cell growth. TIMP-2binding results in heterotrimeric G protein activation and anincrease in cytosolic cAMP level (34).
Activation of downstream signaling cascades by TKR(s) isdependent on the net level of receptor phosphorylation. Netphosphorylation is dependent on the level of autophosphoryla-tion induced by ligand binding and the level of associatedprotein-tyrosine phosphatase activity (62, 63). Cells treatedwith TIMP-2 or Ala1TIMP-2, prior to EGF, down-regulate thelevel of EGFR autophosphorylation in a dose-dependent fash-ion. This effect is again specific for TIMP-2 and is not observedwith synthetic, hydroxamate MMP inhibitor, BB94, or TIMP-1.Furthermore, the decrease in EGFR phosphorylation is con-firmed by a concomitant decrease in Grb-2 association withEGFR. This effect of TIMP-2 on EGFR autophosphorylation isdependent upon adenylate cyclase activity, but is not com-
TIMP-2 Suppresses EGFR Activation3212
by guest on August 9, 2019
http://ww
w.jbc.org/
Dow
nloaded from

pletely reversed by the PKA inhibitor, H89. The lack of PKAinhibitor to reverse the TIMP-2 effect on cell growth or EGFRphosphorylation is in contrast to the direct role of PKA inTIMP-2 mitogenic stimulation of quiescent cells previously re-ported (34). Also, the positive effect of TIMP-2 on the growth ofquiescent cells is reversed at higher TIMP-2 concentrations,this is not observed in the TIMP-2 suppression of stimulatedgrowth or EGFR down-regulation (34). These differences sug-gest that, although similar in requirement for adenylate cy-clase activity, these actions of TIMP-2 are mechanistically dis-tinct from one another. Direct phosphorylation of EGFR by theserine kinase activity of PKA is observed in vitro with purified,recombinant catalytic subunit of PKA and in cells followingadministration of cAMP analogues (41). The failure of H89 tocompletely reverse TIMP-2-mediated reduction in EGFR phos-phorylation is consistent with a TIMP-2 action that is imme-diately downstream to receptor-ligand interaction and TKRactivation. To our knowledge this is the first demonstrationthat TIMP-2 directly interferes with receptor signal transduc-tion at the level of TKR phosphorylation.
Activated, TKR(s) are rapidly dephosphorylated resulting indown-regulation of their signaling activity (62, 63). The SH2-PTPs are known to interact with multiple receptor systems,including the erythropoietin receptor and interleukin-3 recep-tor in hematopoietic cells, as well as the EGFR and vascularendothelial growth factor (VEGF) receptor (Flt, KDR) in epi-thelial and endothelial cells, respectively (42–44). The SH2-PTP, SHP-1, is involved in receptor dephosphorylation andnegative regulation of receptor signaling. SHP-2 has little ef-fect on receptor phosphorylation and positively mediates recep-tor signaling via mechanisms that are not well understood. Thedirect interaction of SHP-1 with the EGFR receptor has beendemonstrated in vitro (42–44). The catalytic domain of SHP-1is important for EGFR dephosphorylation and cannot be sub-stituted by the catalytic domain in SHP-2 (43). It is not knownif SHP-1 displays selectivity with respect to dephosphorylationof individual phosphotyrosine residues on EGFR (43). Also,EGFR dephosphorylation does not absolutely correlate withSHP-1 binding (43). This has been interpreted to suggest thatthe SHP-1 active site may be sterically hindered with respect tosome phosphotyrosine sites on EGFR, and/or not all SHP-1activity is directly bound to EGFR in vivo. SHP-1 may alsoassociate with the EGFR complex via an intermediary dockingprotein.
In our experiments TIMP-2 prevents dissociation and/or pro-motes association of SHP-1 with the EGFR complex in a fash-ion that correlates with the decrease in EGFR phosphorylation.These findings are confirmed by direct measurement of phos-phatase activity in SHP-1 and SHP-2 immunoprecipitates pre-pared from whole cell lysates. Comparison of the results ofSHP-1 levels associated with EGFR in the basal and TIMP-2-treated cells with the direct assay of SHP-1 activity is consist-ent with previous reports that suggest not all SHP-1 activity isbound to EGFR (43). Alternatively there may be a selectiveincrease in the specific activity of the SHP-1 associated withEGFR following TIMP-2 treatment. The observed increase inSHP-2 activity with EGF treatment is consistent with thereported positive modulation of EGFR signaling reported forthis PTP (44). Our findings are consistent with previous reportsdemonstrating that SHP-1 can negatively modulate EGFR ac-tivation and prevent downstream signal propagation from thisreceptor (43, 44). Also, TIMP-2 may induce a selective increasein SHP-1 activity that is not directly bound to EGFR, but mayrequire some auxiliary docking protein that is as yet unidenti-fied. Little is known about regulation of SHP-1 levels or activ-ity, but one report does suggest that a cAMP-regulated path-
way involving phosphorylation of SHP-1 may function toregulate this activity (64).
In summary, TIMP-2 suppresses the mitogenic response toTKR growth factor stimulation. TIMP-2 binds to the surface ofA549 cells independently of MT-1-MMP. TIMP-2 binding to thecell surface results in activation of adenylate cyclase and in-creased cAMP levels in the cytosol, presumably secondary toreceptor-mediated activation of the GTP-binding protein Gas.TIMP-2 modulates the phosphorylation of the EGFR followinggrowth factor stimulation and this effect is mediated by SHP-1.These effects of TIMP-2 on the mitogenic response are observedwith several different growth factors (EGF, bFGF, and PDGF)and in multiple cell types (including neoplastic cells as well asdermal fibroblasts). Based on these findings, we proposeTIMP-2 functions to suppress inappropriate growth factorstimulation of cells in G1/G0 phase of the cell cycle, as well as toinhibit MMP-mediated ECM turnover. Consistent with ourobservation that TIMP-2 needs to be available prior to growthfactor stimulation, and the proximal inhibition or down-regu-lation of EGFR activation (phosphorylation), TIMP-2 mustfunction during early G1 prior to entry in the restriction pointlate in G1. Both this G1 regulator-type and protease inhibitorfunctions of TIMP promote tissue homeostasis. The G1 regula-tor-type function may be a feedback mechanism that informscells that MMP-mediated remodeling of the ECM is complete.In this proposal TIMP-2 would not function to suppress mito-genesis until ECM remodeling is shutdown by inhibition ofMMP activity. Saturation of available MMP active sites in theremodeling matrix and/or on the cell surface would block ECMturnover, only then would excess free TIMP-2 begin to suppresscell responsiveness to residual growth factor stimulation. Ourproposal is consistent with the recent finding that timp-2-deficient mice display no abnormalities in fertility or develop-ment (65, 66). As a G1 cell cycle phase regulator, one wouldexpect that TIMP-2 would be a nonessential gene and thatelimination from the germ line would not necessarily result indisruption of fetal development (67). The results of the presentstudy demonstrate that TIMP-2 is multifunctional in suppress-ing growth factor stimulation, modulating MT-1-MMP activa-tion of pro-MMP-2, in addition to the well established role ofMMP inhibition, which prevents ECM remodeling. The growthsuppression and MMP inhibitor functions both act to preservetissue homeostasis. The control and integration of theseTIMP-2 functions is poorly understood and warrants furtherinvestigation.
Acknowledgments—We thank Dr. Larry Wahl for the Lumulus amoe-bocyte lysis (LAL) analysis, as well as Drs. Kazuyo Takeda and Zu-Xi Yufor technical support and assistance with the confocal mircoscopy. Wealso thank Drs. Liliana Gudez and David Roberts for helpfuldiscussions.
REFERENCES
1. Boudreau, N., Werb, Z., and Bissell, M. J. (1996) Proc. Natl. Acad. Sci. U. S. A.93, 3509–3513
2. Boudreau, N., and Bissell, M. J. (1998) Curr. Opin. Cell Biol. 10, 640–6463. Lukashev, M. E., and Werb, Z. (1998) Trends Cell Biol. 8, 437–4414. Hanahan, D., and Weinberg, R. A. (2000) Cell 100, 57–705. Vu, T. H., Shipley, J. M., Bergers, G., Berger, J. E., Helms, J. A., Hanahan, D.,
Shapiro, S. D., Senior, R. M., and Werb, Z. (1998) Cell 93, 411–4226. Sternlicht, M. D., Lochter, A., Sympson, C. J., Huey, B., Rougier, J. P., Gray,
J. W., Pinkel, D., Bissell, M. J., and Werb, Z. (1999) Cell 98, 137–1467. Holmbeck, K., Bianco, P., Caterina, J., Yamada, S., Kromer, M., Kuznetsov,
S. A., Mankani, M., Robey, P. G., Poole, A. R., Pidoux, I., Ward, J. M., andBirkedal-Hansen, H. (1999) Cell 99, 81–92
8. Werb, Z. (1997) Cell 91, 439–4429. Davis, G. E., Bayless, K. J., Davis, M. J., and Meininger, G. A. (2000) Am. J.
Pathol. 156, 1489–149810. Khokha, R., Waterhouse, P., Yagel, S., Lala, P. K., Overall, C. M., Norton, G.,
and Denhardt, D. T. (1989) Science 243, 947–95011. Alexander, C. M., and Werb, Z. (1992) J. Cell Biol. 118, 727–73912. Albini, A., Melchiori, A., Santi, L., Liotta, L. A., Brown, P. D., and Stetler-
Stevenson, W. G. (1991) J. Natl. Cancer Inst. 83, 775–77913. DeClerck, Y. A., Perez, N., Shimada, H., Boone, T. C., Langley, K. E., and
TIMP-2 Suppresses EGFR Activation 3213
by guest on August 9, 2019
http://ww
w.jbc.org/
Dow
nloaded from

Taylor, S. M. (1992) Cancer Res. 52, 701–70814. Martin, D. C., Sanchez-Sweatman, O. H., Ho, A. T. V., Inderdeo, D. S., Tsao,
M. S., and Khokha, R. (1999) Lab. Invest. 79, 225–23415. Gibbs, J. B. (2000) Science 287, 1969–197316. Guedez, L., Stetler-Stevenson, W. G., Wolff, L., Wang, J., Fukushima, P.,
Mansoor, A., and Stetler-Stevenson, M. (1998) J. Clin. Invest. 102,2002–2010
17. Li, G. Y., Fridman, R., and Kim, H. R. C. (1999) Cancer Res. 59, 6267–627518. Johnson, M. D., Choi, H.-R., Chesler, L., Tsao-Wu, G., Bouck, N., and
Polverini, P. J. (1994) J. Cell. Physiol. 160, 194–20219. Hayakawa, T., Yamashita, K., Ohuchi, E., and Shinagawa, A. (1994) J. Cell
Sci. 107, 2373–237920. Yamashita, K., Suzuki, M., Iwata, H., Koike, T., Hamaguchi, M., Shinagawa,
A., Noguchi, T., and Hayakawa, T. (1996) FEBS Lett. 396, 103–10721. Caraglia, M., Tagliaferri, P., Correale, P., Genua, G., Pinto, A., Del Vecchio, S.,
Esposito, G., and Bianco, A. R. (1993) Cancer Immunol. Immunother. 37,150–156
22. Aaronson, S. (1991) Science 254, 1146–115323. DiFiore, P. P., Pierce, J. H., Kraus, M. H., Segatto, O,., King, C. R., and
Aaronson, S. A. (1987) Science 237, 178–18224. Fedi, P., Tronick, S. R., Aaronson, S. A. (1997) in Cancer Medicine (Holland,
J. F., Bast, R. C., Morton, D. L., Frei, E., Kufe, D. W., and Weichselbaum,R. R., ed) pp. 41–64, Williams and Wilkins, Baltimore
25. Weiss, A., and Schlessinger, J. (1998) Cell 94, 277–28026. Dong, J., Opresko, L. K., Dempsey, P. J., Lauffenburger, D. A., Coffey, R. J.,
and Wiley, H. S. (1999) Proc. Natl. Acad. Sci. U. S. A. 96, 6235–624027. Prenzel, N., Zwick, E., Daub, H., Leserer, M., Abraham, R., Wallasch, C., and
Ullrich, A. (1999) Nature 402, 884–88828. Wingfield, P. T., Sax, J. K., Stahl, S. J., Kaufman, J., Palmer, I., Chung, V.,
Corcoran, M. L., Kleiner, D. E., and Stetler-Stevenson, W. G. (1999) J. Biol.Chem. 274, 21362–21368
29. Fridman, R., Fuerst, T. R., Bird, R. E., Hoyhtya, M., Oelkuct, M., Kraus, S.,Komarek, D., Liotta, L. A., Berman, M. L., and Stetler-Stevenson, W. G.(1992) J. Biol. Chem. 267, 15398–15405
30. Welgus, H. G., Jeffery, J. J., Stricklin, G. P., and Eisen, A. Z. (1982) J. Biol.Chem. 257, 11534–11539
31. Murphy, A. N., Unsworth, E. J., and Stetler-Stevenson, W. G. (1993) J. Cell.Physiol. 157, 351–358
32. Fabbri, E., Brighenti, L., and Ottolenghi, C. (1991) J. Enzym. Inhib. 5, 87–9833. Chijiwa, T., Mishima, A., Hagiwara, M., Sano, M., Hayashi, K., Inoue, T.,
Naito, K., Toshioka, T., and Hidaka, H. (1990) J. Biol. Chem. 265,5267–5272
34. Corcoran, M. L., and Stetler-Stevenson, W. G. (1995) J. Biol. Chem. 270,13453–13459
35. Emmert-Buck, M. R., Emonard, H. P., Corcoran, M. L., Krutzsch, H. C.,Foidart, J. M., and Stetler-Stevenson, W. G. (1995) FEBS Lett. 364, 28–32
36. Itoh, Y., Ito, A., Iwata, K., Tanzawa, K., Mori, Y., and Nagase, H. (1998) J. Biol.Chem. 273, 24360–24367
37. Nagase, H., and Woessner, J. F. (1999) J. Biol. Chem. 274, 21491–2149438. Zucker, S., Drews, M., Conner, C., Foda, H. D., DeClerck, Y., Langley, K. E.,
Bahou, W., Docherty, A. J. P., and Cao, J. (1998) J. Biol. Chem. 273,1216–1222
39. Cao, J., Drews, M., Lee, H. M., Conner, C., Bahou, W. F., and Zucker, S. (1998)J. Biol. Chem. 273, 34745–34752
40. Miloso, M., Mazzotti, M., Vass, W. C., and Beguinot, L. (1995) J. Biol. Chem.270, 19557–19562
41. Barbier, A. J., Poppleton, H. M., Yigzaw, Y., Mullenix, J. B., Wiepz, G. J.,Bertics, P. J., and Patel, T. B. (1999) J. Biol. Chem. 274, 14067–14073
42. Guo, D. Q., Wu, L. W., Dunbar, J. D., Ozes, O. N., Mayo, L. D., Kessler, K. M.,Gustin, J. A., Baerwald, M. R., Jaffe, E. A., Warren, R. S., and Donner, D. B.(2000) J. Biol. Chem. 275, 11216–11221
43. Keilhack, H., Tenev, T., Nyakatura, E., Godovac-Zimmermann, J., Nielsen, L.,Seedorf, K., and Bohmer, F. D. (1998) J. Biol. Chem. 273, 24839–24846
44. Tenev, T., Keilhack, H., Tomic, S., Stoyanov, B., Stein-Gerlach, M., Lammers,R., Krivtsov, A. V., Ullrich, A., and Bohmer, F. D. (1997) J. Biol. Chem. 272,5966–5973
45. Park, C. C., Bissell, M. J., and Barcellos-Hoff, M. H. (2000) Mol. Med. Today 6,324–329
46. Bissell, M. J., Weaver, V. M., Lelievre, S. A., Wang, F., Petersen, O. W., andSchmeichel, K. L. (1999) Cancer Res. 59, 1757s–1763s
47. Gumbiner, B. M. (1996) Cell 84, 345–35748. Sieg, D. J., Hauck, C. R., Ilic, D., Klingbeil, C. K., Schaefer, E., Damsky, C. H.,
and Schlaepfer, D. D. (2000) Nat. Cell Biol. 2, 249–25649. Streuli, C. (1999) Curr. Opin. Cell Biol. 11, 634–4050. Lee, Y. J., and Streuli, C. H. (1999) J. Biol. Chem. 274, 22401–2240851. Blavier, L., and DeClerck, Y. A. (1997) Mol. Biol. Cell 8, 1513–152752. Imren, S., Kohn, D. B., Shimada, H., Blavier, L., and DeClerck, Y. A. (1996)
Cancer Res. 56, 2891–285553. Stetler-Stevenson, W. G., Bersch, N., and Golde, D. W. (1992) FEBS Lett. 296,
231–23454. Bertaux, B., Hornebeck, W., Eisen, A. Z., and Dubertret, L. (1991) J. Invest.
Dermatol. 97, 679–68555. Hayakawa, T., Yamashita, K., Tanzawa, K., Uchijima, E., and Iwata, K. (1992)
FEBS Lett. 298, 29–3256. Nemeth, J. A., and Goolsby, C. L. (1993) Exp. Cell Res. 207, 376–38257. Nemeth, J. A., Rafe, A., Steiner, M., and Goolsby, C. L. (1996) Exp. Cell Res.
224, 110–11558. Baker, A. H., George, S. J., Zaltsman, A. B., Murphy, G., and Newby, A. C.
(1999) Br. J. Cancer 79, 1347–135559. Baker, A., Zaltsman, A., George, S., and Newby, A. (1998) J. Clin. Invest. 101,
1478–148760. Smith, M. R., Kung, H. F., Durum, S. K., Colburn, N. H., and Sun, Y. (1997)
Cytokine 9, 770–78061. Codony-Servat, J., Albanell, J., Lopez-Talavera, J. C., Arribas, J., and Baselga,
J. (1999) Cancer Res. 59, 1196–20162. Byon, J. C., Kenner, K. A., Kusari, A. B., and Kusari, J. (1997) Proc. Soc. Exp.
Biol. Med. 216, 1–2063. Hubbard, S. R., Mohammadi, M., and Schlessinger, J. (1998) J. Biol. Chem.
273, 11987–1199064. Catalan, R. E., Gargiulo, L., Martinez, A. M., Calcerrada, M. C., and Liras, A.
(1997) FEBS Lett. 400, 280–28465. Caterina, J. J., Yamada, S., Caterina, N. C. M., Longenecker, G., Holmback,
K., Shi, J., Yermovsky, A. E., Engler, J. A., and Birkedal-Hansen, H. (2000)J. Biol. Chem. 275, 26416–16422
66. Wang, Z., Juttermann, R., and Soloway, P. D. (2000) J. Biol. Chem. 275,26411–26415
67. Sherr, C. J. (2000) Cancer Res. 60, 3689–3695
TIMP-2 Suppresses EGFR Activation3214
by guest on August 9, 2019
http://ww
w.jbc.org/
Dow
nloaded from

Susan E. Hoegy, Hae-Ryong Oh, Marta L. Corcoran and William G. Stetler-StevensonSignaling Independent of Metalloproteinase Inhibition
Tissue Inhibitor of Metalloproteinases-2 (TIMP-2) Suppresses TKR-Growth Factor
doi: 10.1074/jbc.M008157200 originally published online October 19, 20002001, 276:3203-3214.J. Biol. Chem.
10.1074/jbc.M008157200Access the most updated version of this article at doi:
Alerts:
When a correction for this article is posted•
When this article is cited•
to choose from all of JBC's e-mail alertsClick here
http://www.jbc.org/content/276/5/3203.full.html#ref-list-1
This article cites 66 references, 33 of which can be accessed free at
by guest on August 9, 2019
http://ww
w.jbc.org/
Dow
nloaded from





























