Embed Size (px)
Citation preview

0 1994 by The American Society for Biochemistry and Molecular Biology, Inc. THE JOURNAL OF BIOLCGICAL CHEMISTRY Vol. 269, No. 23, Issue of June 10, pp. 16034-16040, 1994
Printed in U.S.A.
Expression of a Dominant-negative Mutant Human Insulin Receptor in the Muscle of Transgenic Mice*
(Received for publication, March 9, 1994)
Pi-Yun ChangSO, Heike BeneckeSO, Yannick Le Marchand-Brusteln, Joel LawittsS, and David E. MollerSII From the $Charles A. Dana Research Institute and Harvard-Thorndike Laboratory of Beth Israel Hospital, Department of Medicine, Beth Israel Hospital and Harvard Medical School, Boston, Massachusetts 02215 and the lFaculte de Medicine, INSERM Unite 145, Nice, France
To examine the in vivo effects of a kinase-deficient mutant human insulin receptor, we used the muscle cre- atine kinase promoter to express a putative dominant- negative receptor: Ala”” -+ Thr (Moller, D. E., Yokota, A, White, M. F., Pazianos, A. G., and Flier, J. S. (1990) J. Biol. Chern. 265, 14979-14986) in transgenic mice. Two lines were generated, where receptor expression was re- stricted to striated muscle and was increased by 5-12- fold in skeletal muscle. Transgenic gluteal muscle insu- lin receptor kinase activity was reduced by 430% after maximal in vivo insulin stimulation. Glycogen content in this muscle was reduced by 46% in transgenic mice. In- sulin levels were 4-fold higher, and glucose concentra- tions were 12% higher in transgenics fed ad libitum. Transgenic mice exhibited reduced in vivo sensitivity to low dose (0.1 milliunitdg) intravenous insulin. In iso- lated soleus muscles from transgenics, where mutant receptors were expressed at lower levels, insulin -s t’ 1mu- lated receptor kinase activity was reduced by 42%, but insulin-stimulated 2-deoxyglucose uptake was unaf- fected. These results indicate that (i) overexpression of a kinase-deficient human insulin receptor in muscle causes dominant-negative effects at the level of receptor kinase activation, (ii) impairment of insulin-stimulated muscle receptor tyrosine kinase activity can cause de- creased insulin sensitivity in vivo, (iii) kinase-defective receptor mutants may be used to create novel animal models of tissue-specific insulin resistance.
The insulin receptor is a transmembrane oligomeric glyco- protein that binds insulin to the surface of target cells and mediates its subsequent actions. It is well established that the insulin receptor is a tyrosine-specific protein kinase (1 ,2) . Re- ceptor tyrosine kinase activity is stimulated by ligand binding and is further augmented by insulin-stimulated autophospho- rylation of tyrosine residues within the receptor j3-subunit (3, 4). Several lines of investigation have supported the hypothesis that activation of the tyrosine kinase is required for receptor- mediated signal transduction that results in most, if not all, of
* This work was supported by National Institutes of Health Grant DK45874 and a grant from the Harcourt General Charitable Founda- tion (both to D. E. M.) and NATO Grant CRG 931483 (to Y. M.-B.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “aduer- tisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
5 These two authors contributed equally to the studies described herein.
nology and Metabolism, Beth Israel Hospital, 330 Brookline Ave., Bos- 1 ) To whom correspondence should be addressed: Division of Endocri-
ton, MA 02215. Tel.: 617-735-2151; Fax: 617-735-2927.
the metabolic and growth promoting effects of insulin (4). Thus, with the possible exception of insulin-stimulated pyruvate de- hydrogenase activation (5), site-directed mutants of the recep- tor ATP-binding site fail to mediate any biological effects of insulin when expressed in transfected cells (6-8). With one exception (91, studies have reported that biological signaling also is (variably) impaired by site-directed mutagenesis of one or more tyrosyl autophosphorylation sites (residues 1158,1162, and 1163)’ present within the receptor kinase domain (10-13). Several studies also have suggested that marked overexpres- sion of site-directed kinase-deficient human receptor mutants can result in dominant-negative inhibition of insulin signaling via the residual population of endogenous rodent insulin recep- tors expressed by transfected cultured cells (6-8, 14).
The potential dominant-negative effects of kinase-defective insulin receptors are best illustrated by naturally occurring mutations of the insulin receptor gene, which we and others have identified in a number of human subjects with clinical syndromes of severe insulin resistance (reviewed in Ref. 15). Several of these patients are heterozygous for missense muta- tions in one allele which affect the insulin receptor tyrosine kinase domain (residues 1002-1257) (16-26). Patients with such mutations can inherit the phenotype of severe insulin resistance in a dominant fashion (18, 23-26), and the muta- tions have been shown by in vitro expression studies to be deficient in insulin-stimulated receptor kinase activity (18, 19, 22, 23, 27, 28). Since the clinical phenotype in such cases is more severe than would be expected to result from a 50% re- duction in the number of functional insulin receptors, domi- nant-negative inhibition of the product of the normal allele has been proposed as the explanation for in vivo insulin resistance.
Although it is clear that activation of the insulin receptor kinase plays an important role in mediating biological effects of insulin, better expression systems are needed to evaluate the relation between insulin receptor tyrosine kinase activity and the metabolic effects of insulin in vivo. Transgenic animals provide an important system for testing the physiologic role of specific proteins. Through the use of tissue-specific promoter- enhancer elements, the overexpression of a desired protein can be directed to selected cell types (29). In the present study we have succeeded in employing transgenic techniques to overex- press a putative dominant-negative human insulin receptor mutant in transgenic mice. The cDNA encoding a naturally occurring receptor mutant where Thr is substituted for Ala at codon 1134 was used (18). We previously demonstrated that the Thr1134 mutation was inherited as a dominant disease-causing allele in humans. Moreover, after expression in transfected
Amino acids are numbered according to Ebina et al. (33).
16034

Dominant-negative Insulin Receptors in Dansgenic Mice 16035
Chinese hamster ovary (CHO)' cells, Thr"34 receptors dis- played normal ligand binding but were devoid of detectable insulin-stimulated tyrosine kinase activity and failed to medi- ate several biological effects of insulin (18, 30).
Since skeletal muscle represents the predominant site of in vivo insulin-stimulated glucose disposal (31, 321, we elected to use muscle-specific DNA regulatory elements to drive expres- sion of mutant insulin receptors in mice. These mice have al- lowed us to directly test the dominant-negative hypothesis and, by perturbing muscle insulin receptor function in vivo, to as- sess the quantitative relationship between insulin receptor ty- rosine kinase activity and stimulation of metabolic effects of insulin in this tissue.
EXPERIMENTAL PROCEDURES Construction of Mutant Human Insulin Receptor Dansgenic
Vectors-A series of different vectors designed to express mutant human insulin receptors in the muscle of transgenic mice was constructed using standard recombinant DNA techniques (Ref. 34, Fig. 1). A construct (MLC/IR) containing mutant insulin receptor cDNAcoupled to promoter- enhancer elements from the rat myosin light chain 1/3 locus (35) and an 850-bp cassette (SVPA) containing the polyadenylation and splice site sequences of SV40 (provided by Nadia Rosenthal, Boston University Medical Center) was prepared as we described previously for construct pTMPE/HIR which contains the wild-type human insulin receptor cDNA (36). An alternative myosin light chain-based insulin receptor minigene construct (MLCMR) was prepared by subcloning intronic sequences from the human insulin receptor gene into the insulin receptor cDNA. Intron 8/9 and flanking sequences within exons 8 and 9 of the human insulin receptor gene (37) were obtained by polymerase chain reaction
TGTAAGGGTAACTG-3') and primer 2 (5"AGAGGTGAAGCAAAGTG- amplification (38) of human genomic DNA with primer 1 (5"CGGTCT-
CAT-3'). A 90-bp sequence of insulin receptor cDNA from BstEII to BamHI in MLC/IR was replaced by the 3.6-kb BstEII-BamHI-digested polymerase chain reaction product containing intron 8/9. Both MLC/IR and MLCMR constructs were excised from the parent plasmid (pGEM5- Zf-, Promega, Madison, WI) prior to microinjection (36).
Two additional insulin receptor transgenes were prepared using the 5' regulatory region (to -3,300 bp) of the rat muscle creatinine kinase (MCK) gene provided by Stephen D. Hauschka (University of Washing- ton, Seattle, WA). The 4.4-kb insulin receptor cDNA on HindIII linkers was subcloned into the HindIII site of PGEM3-Zf- (Promega) containing the 850-bp HindIII-BamHI SVPAcassette and modified by the presence of two MluI sites on either side of the polylinker to create pIWSVPA. After abolishing an MluI site within the promoter, a 3.3-kb HindIII- BstEII fragment was excised from an MCK promoter construct (-3300MCK-CAT) (39) and inserted by blunt end ligation at the 5' end of the insulin receptor cDNA to create MCK/IR. Alternatively, a 6.5-kb region of the MCK gene (provided by Jeff Chamberlain, University of Michigan) which contains the -3,300-bp 5"flanking region, exon 1 (non- coding), and the first intron into exon 2 was utilized. After abolishing the MluI site in the MCK promoter, the 6.5-kb XhoI fragment was
start codon in pIWSVPA to create MCK6.5/IR. The MCMR and subcloned into a Sal1 site located immediately 5' of the insulin receptor
MCKB.B/IR transgenes were excised from the parent plasmid using MluI. Certain constructs were tested by transient transfection into differentiating CzClz cultured myotubes as described previously (36).
Generation and Identification of Dansgenic Mice-Linearized trans- gene DNA was separated by electrophoresis in 1.0% agarose gels, fol- lowed by electroelution and purification using Elutip D columns (Schleicher and Schuell). Fertilized one-cell mouse zygotes were recov- ered from the oviducts of pregnant FVBNJ mice (40). Approximately 2 pl (300 copies) were microinjected into male pronuclei followed by a brief period of culture in vitro (40). Microinjected zygotes were then transferred to the oviducts of pseudopregnant recipient CD-1 mice and allowed to develop to term (40).
For Southern analysis, genomic DNA was prepared from tail biopsy material taken from 3-4-week old mouse pups (40). Tail DNA (10 pg) was digested with BamHI and subjected to agarose gel electrophoresis followed denaturation and subsequent transfer to nylon membranes (Micron Separations, Westborough, MA) (34). Transgene integration
The abbreviations used are: CHO, Chinese hamster ovary; bp, base pair%); kb, kilobase pair(s); MCK, muscle creatinine kinase; MLC, myo- sin light chain; ANOVA, analysis of variance; CAT, chloramphenicol acetyltransferase.
was assessed by probing nylon filters with a [CY-~~PI~CTP random- primed labeled BstEII fragment of the human insulin receptor cDNA (41). Membranes were hybridized, washed with high stringency, and subjected to autoradiography as described previously (26). Transgenic founder mice were mated with nontransgenic FVBNJ mice to establish hemizygous lines.
RNA Isolation and Northern Analysis-Total cellular RNA was ex- tracted using the method of Chomczynski and Sacchi (42). For Northern analysis, 20-pg samples of RNA were separated by electrophoresis on 1.2% formaldehyde-agarose gels followed by transfer to nylon mem- branes (34). Membranes were hybridized to a random-primed labeled human insulin receptor cDNA probe as described above. A [CY-~~PI~CTP random-primed labeled p-actin cDNA probe was also prepared as de- scribed (43). conditions for hybridization, washing, and exposure of membranes have been described previously (26, 43).
Immunodetection and Quantitation of Insulin Receptor Expression- Quantitation of total and human insulin receptor expression was per- formed using microtiter plates coated with either a nonspecies-specific anti-insulin receptor antibody (AB-3, Oncogene Science, Uniondale, N Y ) or a monoclonal human-specific anti-insulin receptor antibody (aIR1) (36). Samples (50 mg) of skeletal muscle or other tissues were snap-frozen in liquid nitrogen and then homogenized a t 4 "C in 500 pl of buffer A containing 100 mM HEPES, pH 7.6,3% Triton X-100,168 p~ diisopropylfluorophosphate, 0.1 mg/ml aprotinin, 1 mg/ml leupeptin, 0.1 mg/ml pepstatin. After incubation at 4 "C for 30 min and centrifugation for 10 min at 10,000 x g to remove insoluble material, duplicate 40-pl aliquots of each sample of lysate were incubated overnight a t 4 "C in microtiter wells coated with either antibody. After removal of the tissue lysate, wells were washed five times with ice-cold buffer B containing 50 mM NaCl, 1.2 mM KCl, 0.5 nm CaC1,.2Hz0, 10 mM HEPES, pH 7.6, 0.05% Triton X-100, 10% glycerol, 0.5% bovine serum albumin, 0.05% sodium azide. Forty pl of buffer B with the addition of 30,000 cpm of 1251-insulin (2200 Ci/mmol) was added to each well followed by incuba- tion at 20 "C for 2 h. Each well was then washed with buffer B as described above followed by determination of radioactivity after re- moval of bound insulin with 10% SDS. The protein concentration in each sample of solubilized tissue was measured using BCA Protein Assay Reagent (Pierce Chemical Co.). The relative expression of human or total insulin receptors (per mg of protein) was determined by com- parison with standard cuwes generated using variably diluted (over a 20-fold range) samples of solubilized transfected CHO cells which over- express wild-type human insulin receptors (44).
Measurement of Skeletal Muscle Insulin Receptor Tyrosine Kinase Activity-The tyrosine kinase activity of muscle insulin receptors was measured after immobilization on microtiter wells using a modification of methods reported by Klein et al. (45). For analysis of skeletal muscle insulin receptor tyrosine kinase activity following in vivo insulin ad- ministration, fasted (overnight) mice were anesthetized with Avertin (tribromoethanol, tertamyl alcohol, Aldrich) and placed on a warm sur- face. A sample of gluteal muscle (25-50 mg) was removed followed by intraperitoneal injection of the indicated insulin doses (in 100 p1 of 0.9% NaCI). Additional gluteal muscle biopsies were obtained 20 and 40 min after insulin administration. Muscle biopsies were snap-frozen in liquid nitrogen and homogenized as described above in 500 pl of buffer C (buffer A with the addition of 25 mM EDTA, 4.7 p~ NaF, 0.3 PM sodium pyrophosphate, 0.01% Na,VO,). After incubation and centrifugation as described above, duplicate 40-pl aliquots of each sample of lysate were incubated overnight at 4 "C in microtiter wells coated with either an- tibody (AB-3 or aIR1). Wells were washed five times with buffer B (see above). Tyrosine kinase activity of the bound insulin receptors was assayed by adding 20-pl poly-Glu-Tyr (4:l) (Sigma) (4 mg/ml in water) and 20 pl of buffer B with the addition of 10 mM M P , 5.0 mM Mn", 0.5 PM ATP (including 7.0 pCi of [Y-~~PIATP, 3000 CUmmol). After 20-min incubation at 20 "C, the reaction was stopped by spotting duplicate aliquots onto filter paper (Whatman 3"). Filter papers were washed extensively in ice-cold 10% trichloroacetic acid, 10 mM sodium pyrophos- phate. Radioactivity was determined by Cerenkov counting. Back- ground values obtained from reactions performed without the addition of solubilized muscle were subtracted; data was corrected for variation in protein concentrations as described above.
Measurement of Muscle Glycogen, Circulating Insulin, Glucose Tol- erance, and Insulin Tolerance-Muscle glycogen content was measured as follows: -100 mg samples of gluteal or gastrocnemius muscle were digested in 0.5 M KOH followed by precipitation of glycogen with 66% ethanol using Na,SO, as a carrier. Glycogen was digested with amylo- glucosidase, and the released glucose was measured by enzymatic de- termination as described previously (46).
Plasma insulin and blood glucose levels were measured with mice

16036 Dominant-negative Insulin Receptors in Dansgenic Mice
CONSTRUCT # O F #FOUNDERS FOUNDERS: EXPRESSING:
MLCilR ", l 1
0
MLCilllR 14 0
MCWlR - l 3 0
MCK6.5flR 11 7
FIG. 1. Muscle-specific transgene constructs. A series of con- structs designed to drive the expression of tyrosine kinase-deficient mutant human insulin receptors in the muscle of transgenic mice was generated. MLC/IR and MLCIIIIR constructs included 1.3 kb of the 5"flanking region of the rat myosin light chain (MLC) 113 locus and a 0.5-kb fragment of the 3' MLC gene-enhancer region. MCWIR and MCK6.5/IR constructs included a 3.3-kb fragment of the 5"flanking region of the rat muscle creatine kinase gene with (MCK6.5nR) or without (MCWIR) exon 1 and intron 1 of the MCK gene. Mutant human insulin receptor sequences consisted of a 4.4-kb cDNA containing the intact coding region with (MLCIIIIR) or without a 3.0-kb insulin recep- tor intron (8/9) cloned from human genomic DNA. A 0.85-kb SV40 polyadenylatiodsplice site sequence was placed at the 3' end of the insulin receptor cDNA. The number of founder transgenic mice gener- ated with each construct is noted. Individual founder mice were screened for the expression of human insulin receptor protein by im- munoassay of solubilized biopsied skeletal muscle using a human-spe- cific anti-insulin receptor antibody. Mutant human insulin receptor ex- pression was detected (7/11 founders) only in founder mice bearing the MCK6.5/IR transgene. 0, mutant IR cDNA, 0, intron; El, SV40 poly(A).
who were fasted overnight or fed ad libitum. Blood was collected from the retro-orbital sinus or tail vein with a heparinized capillary tube. Insulin was measured by radioimmunoassay using rat insulin stan- dards (Linco, St. Louis, MO). Blood glucose was measured by the glu- cose oxidase method as reported previously (36).
Glucose tolerance tests were performed with overnight fasted un- anesthetized mice as follows: small samples (10 pl) of blood were ob- tained from the tail vein at the indicated time points before and after the intraperitoneal administration of 2 mg of glucose (25% in 0.9% NaClUg of body weight.
For insulin tolerance tests, mice were fasted for 6 8 h followed by anesthesia with intraperitoneal pentobarbitol(O.1 mg/g of body weight). Animals were placed on a warm surface followed by intravenous (tail vein) administration of insulin (0.1 milliunitdg of body weight, Novo Nordisk, Princeton, NJ). Blood samples (retro-orbital sinus) were ob- tained at the indicated time points and assayed for glucose concentra- tion as noted above.
Measurement of Insulin Receptor orosine Kinase Activity and 2-De- oxyglucose Uptake by Isolated Mouse Soleus Muscles in Vitro-Mouse soleus muscles were obtained and incubated as described previously (47,48). Briefly, soleus muscles were rapidly removed and tied via their tendons to stainless steel clips. All incubations were conducted at 37 "C under 95% 02; 5% CO, in 1.0 ml of Krebs-Ringer bicarbonate buffer, pH 7.3 (471, which was supplemented with 1.0% bovine serum albumin and 2.0 mM pyruvate. Following a 15-min preincubation period, muscles were incubated for 30 min with or without insulin at the indicated concentrations before the addition of 0.1 mM 2-deoxyglucose with 0.5 pCi/ml 2-13Hldeoxyglucose (8.1 Ci/mmol) and 0.1 pCi/ml ['4Clsucrose (298 mCi/mmol) for a subsequent 10-min period.
Following the above incubations, muscles were snap-frozen in liquid nitrogen, homogenized by sonication in 500 pl of ice-cold buffer C (see above), and solubilization was achieved by agitating at 4 "C for 45 min. Following centrifugation a t 10,000 x g for 10 min a t 4 "C, aliquots (250 pl) of each sample (supernatant) were removed and analyzed for 'H and "C radioactivity. Duplicate 40-pl aliquots of each sample were assayed for insulin receptor tyrosine kinase activity as described above. Protein concentration was determined with 5.0-pl sample aliquots.
Statistical Analysis-Data are expressed as mean * S.E. of multiple determinations. Statistical significance was assessed using analysis of variance (ANOVA) with multiple sample comparison (Newman-Keul's test) and by Student's t test.
RESULTS
Generation of Dansgenic Mice Expressing Thr1134 Mutant Human Insulin Receptors in Muscle-As depicted in Fig. 1, several constructs designed to drive the expression of mutant
A.
z8sl 1 8 s
1 2 3 4
Frc. 2. Expression of mutant human insulin receptor mRNA in the skeletal muscle of transgenic mice. In this example, total RNA (20 pg) was extracted from gluteal muscle derived from two transgenic (lanes 1 and 2 ) and two nontransgenic (lanes 3 and 4 ) mice. This Northern blot was hybridized to random-primed [~r-~~PldCTP-labeled human insulin receptor (A) and p-actin ( B ) cDNA probes. The expected 4.4-kb insulin receptor transgene mRNA band is detected in muscle RNA from the two transgenic mice. A 1.8-kb actin mRNA band is de- tected in all four RNA samples. The location of 18 and 28 S ribosomal RNAs is noted.
human insulin receptors in the muscle of transgenic mice were prepared using DNA regulatory elements from the rat myosin light chain (MLC) 1/3 locus or the MCK gene. These four trans- gene constructs were microinjected into fertilized mouse zy- gotes and a total of 49 individual transgenic founder mice were subsequently identified by Southern blot analysis of genomic DNA obtained by tail biopsy. In order to screen for the expres- sion of mutant human insulin receptors, transgenic founder mice (or their transgenic offspring) underwent gluteal muscle biopsies. Insulin receptors were solubilized and immobilized on microtiter wells coated with a human-specific anti-insulin re- ceptor antibody (aIR1). The presence of human insulin recep- tors was detected by increased l2'1-insulin binding relative to low background countslmin of bound '2sI-insulin obtained with muscle from nontransgenic mice.
Analysis of 11 founder mice generated using the MLC/IR construct and 14 founder mice transgenic for the intron-con- taining MLC/I/IR construct failed to detect any with expression of mutant human receptors in muscle. In contrast, both MLC constructs were capable of directing the expression of human insulin receptors in transiently transfected C2C12 cells (not shown). Screening of 13 founder mice transgenic for the MCWIR founder also failed to demonstrate detectable levels of human insulin receptor expression in biopsied skeletal muscle. Importantly, use of the intron-containing MCK6.5/IR construct resulted in readily detectable gluteal muscle expression of mu- tant human insulin receptors in the majority (7/11) of trans- genic founders (Fig. 1). Two of the MCK6.5/IR founder mice with higher human receptor expression were selected and propagated by backcrossing to nontransgenic FVB mice to gen- erate two independent hemizygous lines (2-2 and 2-3) for fur- ther study. High level expression of the human insulin receptor transgene in these lines was confirmed by Northern analysis of skeletal muscle RNA probed with a human insulin receptor cDNA as shown in Fig. 2.
Quantitation of Thr1l3* Insulin Receptor Expression in Dans- genic Mice-Representative transgenic mice from lines 2-2 and 2-3 were sacrificed and several tissues analyzed for the poten- tial expression of human insulin receptors as described above. Analysis of intestine, kidney, brain, adipose tissue, spleen,

Dominant-negative Insulin Receptors in Dansgenic Mice 16037
1504 I .- 40 ,aa ,& Q 4 Q q 9 0 9 9
INSULIN DOSE (mulgrn) Time (min)
K w FIG. 4. Skeletal muscle insulin recentor tvrosine kinase activ- FIG. 3. Relative expression of mutated human insulin recep-
from gluteal (GLUT), heart, diaphragm (DZA), triceps brachii (TRZC), tors in different muscles of transgenic mice. Total protein extracts
erector spinae (EREC SP), gastrocnemius (GAST), and soleus (SOL) muscles were assayed to determine the level of mutant human insulin receptor expression by immobilization of insulin receptors on microtiter wells coated with human-specific anti-insulin receptor antibody fol- lowed by measurement of '251-insulin binding. After correction for pro- tein concentration and background values (55.0%) obtained using muscle derived from nontransgenic mice, results were expressed rela- tive to the level of expression in gluteal muscle (percent gluteal expres- sion). Each value shown represents the mean of muscles derived from five to nine mice (equal numbers of mice from lines 2-2 and 2-31,
liver, heart, and gluteal muscle revealed detectable levels of human receptor expression exclusively in skeletal muscle and heart as expected based on the tissue distribution of endog- enous MCK gene expression in rodents and the reported skel- etal and cardiac muscle-specificity of a -3,300 MCK promoter- chloramphenicol acetyltransferase fusion reporter gene in transgenic mice (39).
Transgenic mice from both lines were compared with non- transgenic littermates in order to establish the degree of mutant human insulin receptor overexpression in skeletal muscle. In- sulin receptors present in protein extracts of biopsied gluteal muscle were immobilized on microtiter wells coated with a non- species-specific antireceptor antibody (AB-3) and quantitated by '251-insulin binding. The abundance of 1251-insulin-labeled re- ceptors present in aliquots of protein extracts from both trans- genics and nontransgenics was within the linear range obtained with variable amounts of recombinant expressed insulin recep- tors (CHO cell lysate). Using this approach, total insulin recep- tor expression was determined to be increased by 8.3 z 0.5-fold in muscle from line 2-2 (n = 20) transgenic mice and increased by 11.5 2 0.7-fold in line 2-3 (n = 11).
In order to quantitate potential differences in the expression of mutant human insulin receptors in different striated muscles, several mice from each line were analyzed to deter- mine the relative abundance of human receptors in protein extracts from seven selected muscles. As shown in Fig. 3, higher levels of expression were evident in gluteal, erector spi- nae, gastrocnemius, and triceps muscles. In contrast, lower levels (23-35% of the level in gluteal muscle) of human receptor expression were evident in heart, diaphragm, and soleus muscle. The pattern of differential human receptor expression was similar for both lines of transgenic mice (Fig. 3).
In Vivo Activation of Muscle Insulin Receptor Tyrosine Kinase-An assay for measurement of muscle insulin receptor tyrosine kinase activation after i n vivo insulin stimulation was developed. Following intraperitoneal administration of insulin, samples of biopsied gluteal muscle were extracted under con- ditions designed to preserve the phosphorylation state of solu- bilized receptors. Tyrosine kinase activity was determined by phosphorylation of poly-Glu-Qr after immobilization of insulin receptors on antibody-coated microtiter wells.
Experiments performed with nontransgenic mice established
ity after in vivo insulin stimulation. Fasted anesthetized mice re- ceived an intraperitoneal injection of insulin followed by biopsy removal of gluteal muscle samples. After immobilization of solubilized insulin receptors on microtiter wells coated with a nonspecies-specific anti- insulin receptor antibody, tyrosine kinase activity toward poly-Glu-Tyr was measured. A shows a dose-response curve generated using increas- ing insulin doses administered to nontransgenic mice. Results are ex- pressed as fold stimulation of receptor kinase activity 30 min after insulin administration relative to kinase activity in muscle from mice who received no insulin. Each point represents mean of values obtained with two to four mice. B shows a time course of gluteal muscle insulin receptor kinase activation before and 20 or 40 min after nontransgenic (0, n = 6 ) or transgenic (0, n = 6 ) mice received insulin (50 milliunits/g of body weight). Kinase activity is expressed as nanomoles of [Y-~~PIATP transferred to the substrate per mg of muscle protein extract (trans- genic uersus control, p = 0.0001 by ANOVA).
that maximal stimulation of muscle receptor kinase activation occurred with insulin doses greater than 10 milliunits/g (Fig. 4A). Subsequently, we compared transgenic mice from lines 2-2 and 2-3 overexpressing mutant Thr"34 human insulin receptors in muscle with nontransgenic littermate control mice using a maximally stimulating insulin dose (50 milliunits/g). Isolation of human insulin receptors after immobilization on microtiter wells coated with the human-specific antibody demonstrated that the Thr"34 insulin receptors expressed in muscle were devoid of detectable insulin-stimulated kinase activity (not shown). When insulin receptors were isolated by immobiliza- tion with the nonspecies-specific antireceptor antibody, maxi- mal insulin-stimulation of total gluteal muscle receptor tyro- sine kinase activity (per mg of protein) was markedly impaired in transgenic mice. Although basal muscle receptor tyrosine activity (per mg of protein) did not differ significantly between the two groups of mice, insulin stimulated receptor kinase ac- tivity by 12.1 z 2.0-fold in nontransgenic muscle versus 2.2 5
0.3-fold in transgenic muscle (Fig. 4). In order to exclude the possibility that large numbers of
kinase-deficient human insulin receptors present in muscle protein extracts might have precluded the binding of normally activated endogenous insulin receptors to microtiter wells, the following control experiments were performed. 1) Parallel meas- urements of receptor abundance ('251-insulin binding) con- firmed that immunoadsorption of receptors in transgenic muscle extracts was within the linear range of standard curves generated with CHO cell extracts. 2) Kinase assays also were performed with aliquots of muscle protein extract which had been diluted by &fold. Absolute values of basal and insulin- stimulated kinase activity were proportionately lower with identical differences observed between transgenic and control mice. 3) No change in insulin-stimulated kinase activity was observed after muscle extracts from transgenic mice were first depleted of human receptor protein by immunoadsorption to wells coated with aIR; insulin-stimulated receptor kinase ac- tivity in nontransgenic muscle was preserved following pre- exposure to the human-specific antibody.
Altered in Vivo Physiologic Parameters in Dansgenic Mice- Having established that both lines of transgenic mice ex-

16038 Dominant-negative Insulin Receptors in Dansgenic Mice
" MALE FEMALE
control mice. A, intraperitoneal glucose tolerance (2.0 mglg of body FIG. 5. Glucose tolerance and insulinemia in transgenic uersus
weight) tests were performed with nine-week old overnight fasted transgenic (m) and littermate control (0) male mice and with transgenic (0) and littermate control (0) female mice. Blood samples were obtained at the indicated time points and analyzed for glucose concentration. Each point represents the mean of five to eight mice. B, plasma insulin levels (ad libitum fed) were measured in 9-week-old transgenic ( n = 13) and littermate control ( n = 9) male mice and in transgenic (n = 11) and littermate control ( n = 12) female mice (*, p < 0.05 uersus control).
pressed similar high levels of mutant insulin receptors in muscle and were characterized by impaired muscle insulin re- ceptor function, several parameters that relate to glucose ho- meostasis were assessed. All in vivo studies were conducted using similar numbers of 9-11-week-old transgenic mice from both lines (2-2,2-3) and equal numbers of age- and sex-matched nontransgenic littermate controls.
Intraperitoneal glucose tolerance tests performed with fasted mice revealed higher stimulated glucose levels in males versus females as other investigators have observed (491, but slightly higher mean glucose concentrations in transgenic mice did not achieve statistical significance (Fig. 5A). Thus, inter- animal variation in the glucose response to intraperitoneal glu- cose administration may possibly have prevented the detection of a small increase in glucose excursion in transgenic mice. In the fasted state, transgenic and nontransgenic mice exhibited similar levels of plasma insulin and blood glucose (Table I). In contrast, blood glucose concentrations in ad libitum fed trans- genic mice were 12% higher than in fed controls (Table I). Importantly, ad libitum fed transgenic mice also exhibited plasma insulin levels that were 2.0-2.4-fold higher than litter- mate controls (Table I, Fig. 5B).
Measurement of skeletal muscle glycogen content revealed a 45% decrease in mean gluteal muscle glycogen in transgenic mice (Table I). The glycogen content of transgenic gastrocne- mius muscle also tended to be lower than in controls, but did not achieve statistical significance.
As an independent and more direct assessment of in vivo insulin sensitivity, intravenous insulin tolerance tests were performed. As shown in Fig. 6, blood glucose concentrations remained higher in transgenic mice after administration of 0.1 milliunits/g of insulin. The calculated rate of decline in glucose concentration (0-10 min) was 88.3 i: 19 pmoHiter/min for transgenic mice versus 203 2 34 pmol/liter/min for littermate control mice (n = 12 each, p = 0.005).
Assessment of Insulin Receptor Qrosine Kinase Activity and Insulin-stimulated Glucose Uptake in Isolated Soleus MUS- cles-Although levels of mutant receptor expression in soleus muscles were only 27% of the level reached in gluteal muscle (Fig. 3), separate measurements established that total insulin receptor abundance in this muscle was increased by -3.5-fold relative to the level of endogenous receptor expression. We thus sought to determine the potential relationship between im- paired muscle receptor tyrosine kinase and insulin's effect on
TABLE I Physiologic parameters
numbers of 9-11-week old transgenic mice from lines 2-2 and 2-3 and The following physiologic parameters were measured using similar
equal numbers of age- and sex-matched nontransgenic littermate con- trol mice. Plasma immunoreactive insulin (IRI) and blood glucose con- centrations were measured in fasted and ad libitum fed mice. The glycogen content of gastrocnemius and gluteal muscles from ad libitum fed mice was also determined.
Control Transgenic
Fasting IRI (ng/ml) 0.41 f 0.1 (n = 7) 0.53 f 0.12 ( n = 7) Fed IRI (nglml) 0.63 f 0.09 ( n = 21) 1.42 f 0.3 ( n = 24)" Fasting glucose (mM) 8.49 f 0.32 ( n = 27) 8.66 f 0.35 ( n = 27) Fed glucose (mM) 8.47 2 0.25 (n = 31) 9.43 f 0.28 ( n = 26)" Glycogen content
(pg/mg protein) Gastrocnemius 2.20 f 0.25 ( n = 19) 1.75 -C 0.22 ( n = 19) Gluteal 1.33 f 0.18 ( n = 18) 0.73 f 0.07 ( n = 20)"
~
Indicates statistically different from control at p 5 0.01.
1°1
O \ Q ? p % P t P
TIME (mln)
FIG. 6. Intravenous insulin tolerance in transgenic uersus con- trol mice. 9-week-old transgenic (0, n = 12) or sex and age-matched littermate control mice (0, n = 12) were fasted for 6-8 h followed by administration of insulin (0.1 milliunits/g of body weight) via the tail vein. Blood samples were obtained at the indicated time points and assayed for glucose concentration. Glucose values in nontransgenic mice were lower than for transgenics ( p < 0.001 by ANOVA).
glucose uptake in this muscle which can be isolated and incu- bated in vitro (47,48). Muscles were incubated for 40 min with various insulin concentrations before measuring receptor tyro- sine kinase activity as described above. 2-[3H]Deoxyglucose up- take during the last 10 min of incubation with insulin was measured simultaneously. As shown in Fig. 7A, insulin receptor tyrosine kinase activity was decreased a t all insulin concentra- tions in transgenic muscles, including the maximally effective one (100 n M ) where a 40% reduction in kinase activity was observed. Despite impairment of insulin-stimulated receptor kinase activation in soleus muscle, insulin-stimulated 2-deoxy- glucose uptake was unaffected by the presence of mutant in- sulin receptors in this muscle, even at submaximally effective insulin concentrations (Fig. 7B 1.
DISCUSSION
Studies employing the euglycemic hyperinsulinemic clamp technique in man and rat have implicated muscle, which com- prises -40% of body weight, as the major site of insulin-medi- ated whole body glucose disposal (31,32). In the present study we utilized heterologous tissue-specific elements to drive the expression of mutant insulin receptors in the muscle of trans- genic mice in an attempt to test the hypothesis that kinase- deficient insulin receptors can cause dominant-negative impair- ment of in vivo insulin sensitivity. Four different transgenes that included promoter-enhancer domains from the rat MLC or MCK genes were used to generate a large number of founder mice. Use of the MLC promoter and the 3' MLC enhancer coupled to mutant human insulin receptor cDNAfailed to result in human receptor expression. In contrast, we demonstrated previously that an otherwise identical construct which con-
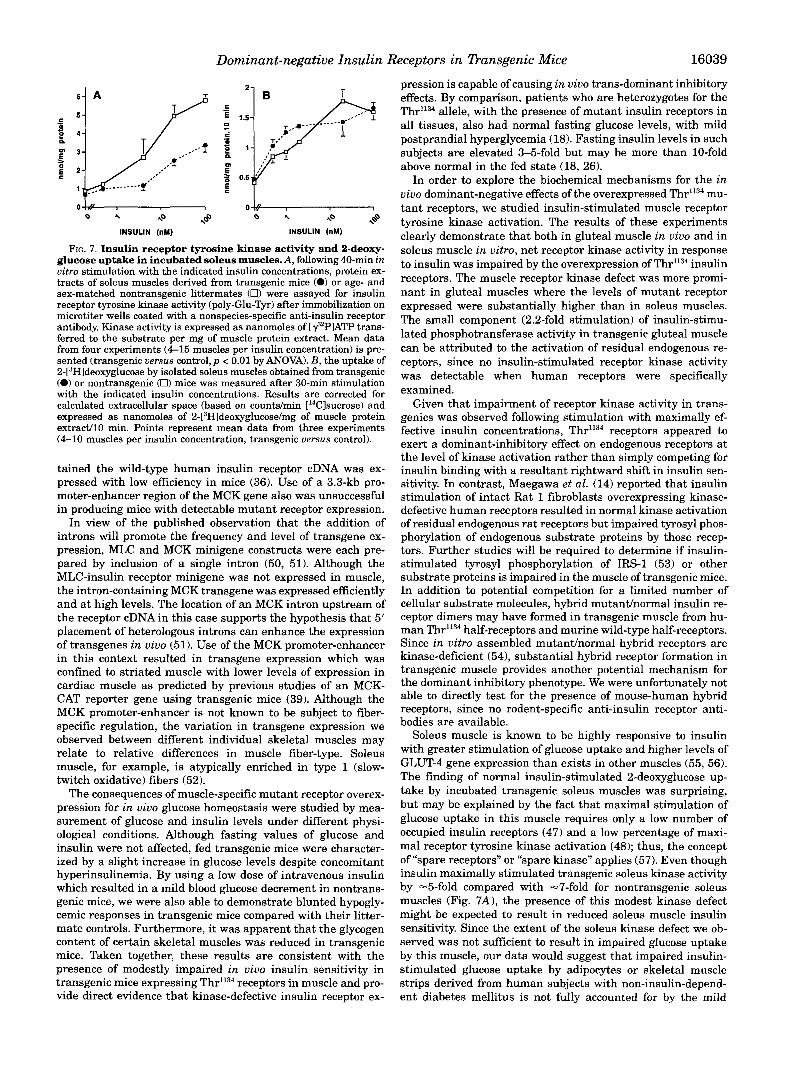
Dominant-negative Insulin Receptors in Dansgenic Mice 16039
a 4 40 ,@ a 4 40 \@ INSULIN (nM) INSULIN (nM)
FIG. 7. Insulin receptor tyrosine kinase activity and 2-deoxy- glucose uptake in incubated soleus muscles.A, following 40-min in vitro stimulation with the indicated insulin concentrations, protein ex- tracts of soleus muscles derived from transgenic mice (0) or age- and sex-matched nontransgenic littermates (0) were assayed for insulin receptor tyrosine kinase activity (poly-Glu-Tyr) after immobilization on microtiter wells coated with a nonspecies-specific anti-insulin receptor antibody. Kinase activity is expressed as nanomoles of [q2PlATP trans- ferred to the substrate per mg of muscle protein extract. Mean data from four experiments (4-15 muscles per insulin concentration) is pre- sented (transgenic uersus control, p < 0.01 by ANOVA). B, the uptake of 2-[3Hldeoxyglucose by isolated soleus muscles obtained from transgenic (0) or nontransgenic (0) mice was measured after 30-min stimulation with the indicated insulin concentrations. Results are corrected for calculated extracellular space (based on countdmin ['4Clsucrose) and expressed as nanomoles of 2-[3Hldeoxyglucose/mg of muscle protein extradl0 min. Points represent mean data from three experiments (4-10 muscles per insulin concentration, transgenic uersus control).
tained the wild-type human insulin receptor cDNA was ex- pressed with low efficiency in mice (36). Use of a 3.3-kb pro- moter-enhancer region of the MCK gene also was unsuccessful in producing mice with detectable mutant receptor expression.
In view of the published observation that the addition of introns will promote the frequency and level of transgene ex- pression, MLC and MCK minigene constructs were each pre- pared by inclusion of a single intron (50, 51). Although the MLC-insulin receptor minigene was not expressed in muscle, the intron-containing MCK transgene was expressed efficiently and at high levels. The location of an MCK intron upstream of the receptor cDNA in this case supports the hypothesis that 5' placement of heterologous introns can enhance the expression of transgenes in vivo (51). Use of the MCK promoter-enhancer in this context resulted in transgene expression which was confined to striated muscle with lower levels of expression in cardiac muscle as predicted by previous studies of an MCK- CAT reporter gene using transgenic mice (39). Although the MCK promoter-enhancer is not known to be subject to fiber- specific regulation, the variation in transgene expression we observed between different individual skeletal muscles may relate to relative differences in muscle fiber-type. Soleus muscle, for example, is atypically enriched in type 1 (slow- twitch oxidative) fibers (52).
The consequences of muscle-specific mutant receptor overex- pression for in vivo glucose homeostasis were studied by mea- surement of glucose and insulin levels under different physi- ological conditions. Although fasting values of glucose and insulin were not affected, fed transgenic mice were character- ized by a slight increase in glucose levels despite concomitant hyperinsulinemia. By using a low dose of intravenous insulin which resulted in a mild blood glucose decrement in nontrans- genic mice, we were also able to demonstrate blunted hypogly- cemic responses in transgenic mice compared with their litter- mate controls. Furthermore, it was apparent that the glycogen content of certain skeletal muscles was reduced in transgenic mice. Taken together, these results are consistent with the presence of modestly impaired in vivo insulin sensitivity in transgenic mice expressing Thr1134 receptors in muscle and pro- vide direct evidence that kinase-defective insulin receptor ex-
pression is capable of causing in vivo trans-dominant inhibitory effects. By comparison, patients who are heterozygotes for the Thr"34 allele, with the presence of mutant insulin receptors in all tissues, also had normal fasting glucose levels, with mild postprandial hyperglycemia (18). Fasting insulin levels in such subjects are elevated 3-5-fold but may be more than 10-fold above normal in the fed state (18, 26).
In order to explore the biochemical mechanisms for the in vivo dominant-negative effects of the overexpressed Thr1134 mu- tant receptors, we studied insulin-stimulated muscle receptor tyrosine kinase activation. The results of these experiments clearly demonstrate that both in gluteal muscle in vivo and in soleus muscle in vitro, net receptor kinase activity in response to insulin was impaired by the overexpression of Thr"34 insulin receptors. The muscle receptor kinase defect was more promi- nant in gluteal muscles where the levels of mutant receptor expressed were substantially higher than in soleus muscles. The small component (2.2-fold stimulation) of insulin-stimu- lated phosphotransferase activity in transgenic gluteal muscle can be attributed to the activation of residual endogenous re- ceptors, since no insulin-stimulated receptor kinase activity was detectable when human receptors were specifically examined.
Given that impairment of receptor kinase activity in trans- genics was observed following stimulation with maximally ef- fective insulin concentrations, Thr1'34 receptors appeared to exert a dominant-inhibitory effect on endogenous receptors at the level of kinase activation rather than simply competing for insulin binding with a resultant rightward shift in insulin sen- sitivity. In contrast, Maegawa et al. (14) reported that insulin stimulation of intact Rat 1 fibroblasts overexpressing kinase- defective human receptors resulted in normal kinase activation of residual endogenous rat receptors but impaired tyrosyl phos- phorylation of endogenous substrate proteins by those recep- tors. Further studies will be required to determine if insulin- stimulated tyrosyl phosphorylation of IRS-1 (53) or other substrate proteins is impaired in the muscle of transgenic mice. In addition to potential competition for a limited number of cellular substrate molecules, hybrid mutanthormal insulin re- ceptor dimers may have formed in transgenic muscle from hu- man Thr1134 half-receptors and murine wild-type half-receptors. Since in vitro assembled mutanthormal hybrid receptors are kinase-deficient (541, substantial hybrid receptor formation in transgenic muscle provides another potential mechanism for the dominant inhibitory phenotype. We were unfortunately not able to directly test for the presence of mouse-human hybrid receptors, since no rodent-specific anti-insulin receptor anti- bodies are available.
Soleus muscle is known to be highly responsive to insulin with greater stimulation of glucose uptake and higher levels of GLUT-4 gene expression than exists in other muscles (55, 56). The finding of normal insulin-stimulated 2-deoxyglucose up- take by incubated transgenic soleus muscles was surprising, but may be explained by the fact that maximal stimulation of glucose uptake in this muscle requires only a low number of occupied insulin receptors (47) and a low percentage of maxi- mal receptor tyrosine kinase activation (48); thus, the concept of "spare receptors" or "spare kinase" applies (57). Even though insulin maximally stimulated transgenic soleus kinase activity by -5-fold compared with -7-fold for nontransgenic soleus muscles (Fig. 7A), the presence of this modest kinase defect might be expected to result in reduced soleus muscle insulin sensitivity. Since the extent of the soleus kinase defect we ob- served was not sufficient to result in impaired glucose uptake by this muscle, our data would suggest that impaired insulin- stimulated glucose uptake by adipocytes or skeletal muscle strips derived from human subjects with non-insulin-depend- ent diabetes mellitus is not fully accounted for by the mild

16040 Dominant-negative Insulin Receptors in Transgenic Mice
receptor kinase defect seen in these same tissues (58-62). Simi- larly, impairment of insulin-stimulated glucose uptake by so- leus muscles derived from obese insulin resistant mice may result more from potentially independent observed defects in post-receptor signaling than from the mild insulin receptor kinase defect present in such mice (48).
In contrast to the modest degree of in vivo insulin resistance displayed by our transgenic mice, the expression of mutant receptors in all tissues results in a phenotype of severe insulin resistance in humans who are heterozygous for kinase-defi- cient insulin receptor alleles. Interestingly, the receptor kinase defect reportedly seen in cells derived from such subjects is typically modest, resulting in a -40% reduction in maximal receptor kinase activity that is similar to the kinase defect we observed in transgenic soleus muscles (63). An important as- pect of the in vivo phenotype of severe insulin resistance in affected patients may involve the role of mutant receptor ex- pression in the liver. We have recently determined that the Thr"34 receptor mutant is resistant to acute insulin-stimulated internalization and down-regulation (see Ref. 15). The presence of this defect in the liver would be predicted to impair in vivo insulin clearance leading to further degrees of hyperinsuline- mia. In this context, normally functioning receptors may be preferentially internalized and degraded resulting in dispro- portionate accumulation of kinase-defective receptors on the surface of cells in uivo. Marked degrees of in vivo hyperinsu- linemia may also result in further insulin resistance via desen- sitization of insulin signaling at the post-receptor level (64).
(INSERM, Nice, France) and Wei Tang and Steve Tollin (Beth Israel Acknowledgments-The technical assistance of Thierry Gremeaw
Hospital, Boston, MA) is greatly appreciated. Harald H. Klein (Univer-
sity, United Kingdom) provided valuable technical advice. We are grate- sity of Lubeck, Kubeck, Germany) and Ken Siddle (Cambridge Univer-
ful to Jeffrey S. Flier (Beth Israel Hospital) for valued advice and discussions, to Alan C. Moses (Beth Israel Hospital) for his careful review of the manuscript, and to Steve Hauschka (University of Wash- ington, Seattle, WA), Jeff Chamberlain (University of Michigan, Ann Arbor, MI) and Nadia Rosenthal (Massachusetts General Hospital, Bos- ton, MA) for providing important plasmid DNA samples.
REFERENCES
2. Rosen, 0. M., Herrera, R., Olowe, Y., Petruzzelli, L. M., and Cobb, M. H. (1983) 1. Kasuga, M., Karlsson, F. A,, and Kahn, C . R. (1982) Science 215, 185-187
3. White, M. F., Shoelson, S. E., Keutmann, H., and Kahn, C . R. (1988) J. Biol.
4. White, M. F., and Kahn, C. R. (1994) in Znsulin Resistance (Moller, D. E., ed)
6. Ebina, Y., Araki, E., Taira, M., Shimada, F., Mori, M., Craik, C . S., Siddle, K., 5. Gottschalk, W. K. (1992) J. B i d . Chem. 266, 8814-8819
Pierce, S. B., Roth, R. A,, and Rutter, W. J. (1987) Proc. Nutl. Acad. Sci.
7. Chou, C. K., Dull, T. J., Russell, D. S., Gherzi, R., Lebwohl, D., Ullrich,A., and U. S. A. 84, 704-708
Rosen, 0. R. (1987) J. Biol. Chem. 262, 1842-1847 8. McClain, D. A,, Maegawa, H., Lee, J., Dull, T. J., Ullrich, A., and Olefsky, J . M.
9. Rafaeloff, R., Maddw, B. A,, Brunetti, A., Sbraccia, P., Sung, C. K., Patel, R., (1987) J. Biol. Chem. 262, 14663-14671
Hawley, D. M., and Goldfine, I. D. (1991) Biochem. Biophys. Res. Commun. 179,912-918
10. Ellis, L., Clauser, E., Morgan, D. O., Edery, M., Roth, R. A.. and Rutter, W. J. (1986) Cell 45,721-732
11. Wilden, P. A., Backer, J. M., Kahn, C. R., Cahill, D. A,, Schroeder, G. J., and White, M. F. (1990) Proc. Natl. Acad. Sci. U. S. A. 87, 3358-3362
12. Debant, A,, Clauser, E., Ponzio, G., Filloux, C.,Auzan, C., Contreres, J. O., and Rossi, B. (1988) Proc. Nutl. Acad. Sci. U. S. A. 85, 8032-8036
13. Murakami, M. S., and Rosen, 0. M. (1991) J. Biol. Chem. 266,22653-22660 14. Maegawa, H., Olefsky, J. M., Thies, S., Boyd, D., Ullrich, A., and McClain, D.
15. Taylor, S. I., and Moller, D. E. (1993) in Znsulin Resistance (Moller, D. E., ed) A. (1988) J. B i d . Chem. 263,12629-12637
16. Odawara, M., Kadowaki, T., Yamamoto, R., Shibasaki, Y., Tobe, K., Accili, D., pp. 83-111, John Wiley and Sons, New York
Bevins, C . L., Mikami, Y., Matsuma, N., Akanuma,Y., Takaku, F., Taylor, S. I., and Kasuga, M. (1989) Science 245, 6 6 4 8
Proc. Natl. Acud. Sci. U. S. A. 80, 3237-3240
Chem. 263,2969-2980
pp. 9-24, John Wiley and Sons, New York
17. Moller, D. E., and Flier, J . S. (1988) N. Engl. J . Med. 319, 15261529 18. Moller, D. E., Yokota, A,, White, M. F., Pazianos, A. G., and Flier, J. S. (1990)
19. Cama, A,, Sierra, M. L., Ottini, L., Kadowaki, T., Gorden, P., Imperato-McGin-
20. Kim, H., Kadowaki, H., Sakura, H., Odawara, M., Momomura, K., Takahashi,
J. Biol. Chem. 265,14979-14985
ley, J., and Taylor, S. I. (1991) J. Clin. Endocrinol. Metab. 73, 894-901
Y., Miyazaki, Y, Ohtani, T., Akanuma, Y., Yazaki, Y., Kasuga, M., Taylor, S. I., and Kadowaki, T. (1992) Diabetologia 35, 261-266
21. Roach, P., Arakaki, R. F., Accili, D., Taylor, S. I., and Gorden, P. (1992) Clin. Res. 40, 311A
22. Cama, A,, Sierra, M. L., Quon, M., Ottini, L., Garden, P., and Taylor, S. I. (1993) J. B i d . Chem. 268,8060-8069
23. Haruta, T., Takata, Y., Iwanishi, M., Maegawa, H., Imamura, T., Egawa, K , Itazu, T., and Kobayashi, M. (1993) Diabetes 42, 1837-1844
24. Iwanishi, M., Haruta, T., Takata, Y., Ishibashi, O., Sasaoka, T., Egawa, K., Imamura, T., Naitou, K., Itazu, T., and Kobayashi, M. (1993) Diubetologia
25. Nozaki, O., Suzuki, Y., Shimada, F., Hashimoto, N., Taira, M., Hatanaka, Y., 36,414-422
Notoya, Y., Kanashiro, O., Makino, H., and Yoshida, S. (1993) J. Clin.
26. Moller, D. E., Cohen, 0.. Yamaguchi, Y., Azziz, R., Eberle, A,, and Flier, J. S. Endocrinol. Metab. 77, 169-172
(1994) Diabetes 43,247-255 27. Yamamoto-Honda, R., Koshio, O., Tobe, K., Shibasaki, Y., Momomura, K.,
Odawara, M., Kadowaki, T., Takaku, F., Akanuma, Y., and Kasuga, M. (1990) J. B i d . Chem. 265, 14777-14783
28. Moller, D. E., Yokota, A., Ginsburg-Fellner, F., and Flier, J. S. (1990) Mol.
29. Hanahan, D. (1989) Science 246, 1265-1275 Endocrinol. 4, 1183-1191
30. Moller, D. E., Benecke, H., and Flier, J. S. (1991) J. Biol. Chem. 266, 10995- 11001
31. DeFronzo, R. A,, Jacot, E., Jequier, E., Maeder, E., Wahren, J., and Felber, J. P. (1981) Diabetes 30, 100&1007
32. Kraegen, E. W., James, D. E., Jenkins, A. B., and Chisholm, D. J. (1985) Am. J. Physiol. 248, E353-E362
33. Ebina, Y., Ellis, L., Jarnagin, K., Edery, M., Graf, L., Clauser, E., Ou, J. H., Masiarz, F., Kan, Y. W., Goldfine, I. D., Roth, R. A,, and Rutter, W. J. (1985)
34. Ausubel, F. M., Brent, R., and Kingston, R. E. (1987) Current Protucols in Cell 40, 747-758
35. Rosenthal, N., Kornhauser, J. M., Donoghue, M., Rosen, K. M., and Merlie, J. Molecular Biology, Wiley and Sons, New York
36. Benecke, H., Flier, J. S., Rosenthal, N., Siddle, IC, Klein, H. H., and Moller, D. P. (1989) Proc. Natl. Acad. Sci. U. S. A. 86, 7780-7784
37. Seino, S., Seino. M., Nishi, S., and Bell, G. 1. (1989) Proc. Nutl. AcaJ .+i E. (1993) Diabetes 42, 206-212
_.
38. ORahilly, S., Choi, W. H., Patel, P., Turner, R. C., Flier, J. S., and Moller, D. E.
39. Johnson, J. E., Wold, B. J., and Hauschka, S. D. (1989) Mol. Cell. Bid . 9.
U. S. A. 86, 114-118
(1991) Diabetes 40, 777-782
33934399 40. Hogan, B., Constantini, F., and Lacy, E. (1986) Manipulating the Mouse Em-
bryo: A Laboratory Manual, Cold Spring Harbor Laboratory, Cold Spring
41. Whittaker, J., Okamoto, A. K., Thys, R., Bell, G. I., Steiner, D. F., and Hof- Harbor, NY
42. Chomczynski, P., and Sacchi, N. (1987)AnaL Biochem. 162,156-159 mann, C . A. (1987) Proc. Natl. Acad. Sci. U. S. A. 84,5237-5241
43. Mountjoy, K. G., Moller, D. E., Flier, J. S., and Holdaway, I. M. (199O)Biochem.
44. Benecke, H., Flier, J. S., and Moller, D. E. (1992) J. Clin. Inuest. 89,2066-2070 45. Klein, H. H., Kowalewski, B., Drenckhan, M., Neugebauer, S., Matthaei, S.,
46. Chan, T. M., and Exton, J. H. (1976) Anal. Biochem. 71, 96-105 47. Le Marchand-Brustel, Y., Jeanrenaud, B., and Freychet, P. (1978) Am. J.
Physiol. 234, E348-E358 48. Heydrick, S. J., Jullien, D., Tanti, J. F., Giorgetti, S., Van Obberghen, E., and
Le Marchand-Brustel, Y. (1993) J. Clin. Inuest. 91, 1358-1366 49. Shepherd, P. R., Gnudi, L., Tozzo, E., Yang, H., Leach, E, and Kahn, B. B.
(1993) J. B i d . Chem. 268, 22243-22246 50. Brinster, R. L., Allen, J. M., Behringer, R. R., Gelinas, R. E., and Palmiter, R.
D. (1988) Proc. Nutl. Acad. Sci. U. S. A. 85, 836-840 51. Palmiter, R. D., Sandgren, E. P., Avarbock, M. R., Allen, J. M., and Brinster, R.
L. (1991) Proc. Natl. Acad. Sci. U. S. A. 88, 478482 52. Ariano, M. A,, Armstrong, R. B., and Edgerton, V. R. (1973) J. Histochem.
Cytochem. 21 ,5145 53. Sun, X. J., Rothenberg, P., Kahn, C. R., Backer, J. M., Araki, E., Wilden, P. A.,
Cahill, D. A,, Goldstein, B. J., and White, M. F. (1991) Nature 352, 73-77 54. Treadway, J. L., Morrison, B. D., Soos, M. A,, Siddle, K., Olefsky, J., Ullrich,A.,
McClain, D. A,, and Pessin, J. E. (1991) Proc. Natl. Acud. Sci. U. S. A. 8 8 ,
55. James, D. E., Jenkins, A. B., and Kraegen, E. W. (1985) Am. J. Physiol. 248, 214-218
56. Henriksen, E. J., Bourey, R. E., Rodnick, K. J., Koranyi, L., Permutt, M. A., and E567-E574
57. Freidenberg, G. R., Suter, S. L., Henry, R. R., Reichart, D., and Olefsky, J. M. Holloszy, J. 0. (1990) Am. J. Physiol. 239, E593-E598
58. Caro, J. F., Sinha, M. K., Raju, S. M., Ittoop, O., Pones, W. J., Flickinger, E. G., (1991) J. Clin. Znuest. 87, 2222-2229
59. Dohm, G. L., Tapscott, E. B., Pones, W. J., Dabbs, D. J., Flickinger, E. G., Meelheim, D., and Dohm, G. L. (1987) J. Clin. Invest. 79, 1330-1337
Meelheim, D., Fushiki, T., Atkinson, S. M., Elton, C. W., and Caro, J. F. (1988) J. Clin. Znuest. 82,486-494
60. Garvey, W. T., Huecksteadt, T. P., Matthaei, S., and Olefsky, J. M. (1988) J. Clin. Inuest. 81, 1528-1536
61. Freidenherg, G. R., Henry, R. R., Klein, H. H., Reichart, D. R., and Olefsky, J. M. (1987) J. Clin. Invest. 79, 240-250
62. Sinha, M. K., Pones, W. J., Flickinger, E. G., Meelheim, D., and Caro. J. F.
Biophys. Res. Commun. 169,978-985
and Kotzke, G. (1993) Diabetes 42, 883-890
(1987) Diabetes 36, 620-625 63. Grigorescu, F., Flier, J. S., and Kahn, C. R. (1984) J. Biol. Chem. 259, 15003-
64. Marshall, S., and Olefsky, J . M. (1980) J. Clin. Inoest. 66, 763-772 15006






















