Embed Size (px)
Citation preview

Abstract In this study the activity of a number of novelimidazoline-based compounds (IMID series) was assessedby functional and binding studies to determine their ac-tions at KATP channels. The novel compounds, which wesynthesised, were methoxy-, methyl-, butyl- and fluoro-phenyl derivatives of clonidine.
In functional studies we determined the potency (bycalculating a pKB value) of the IMID compounds to an-tagonise levcromakalim responses in segments of isolatedpig coronary artery. The most potent compounds identi-fied (laboratory codes: IMID-1M, IMID-26F and IMID-4F) had apparent pKB values of approximately 7 which issimilar to that for the sulphonylurea, glibenclamide andthe lipophilic quaternary ion, tetraphenylphosphonium.This inhibitory action was specific for levcromakalimsince the imidazoline antagonist IMID-1M failed to effectvasorelaxation response-curves to the non-KATP channelopener, sodium nitroprusside. In the spontaneously beat-ing rat right atrium preparation the majority of the com-pounds were able to cause slowing of heart rate, but withlow EC50 values (approximately 10–30 µM). In bindingstudies, the compounds were unable to displace bindingof [3H]P1075 to bovine aortic smooth muscle preparationsnor [3H]glibenclamide binding to rat cerebral cortex mem-branes.
These studies show that some imidazoline-based com-pounds are potent antagonists of levcromakalim-mediatedvasorelaxation responses in the pig coronary artery. Thecompounds displayed only minimal bradycardic activity.The site of action of the imidazoline compounds does notappear to be the same as that used by KATP channel open-
ers or sulphonylurea-based antagonists. It is likely thatthese compounds interact with the KATP channel pore itself.
Key words Imidazolines · Levcromakalim · Smooth muscle relaxation · KATP channels · KATP channelantagonists · Rat right atria · Pig coronary artery
Introduction
In 1983, Noma (1983) identified specific K+ channels incardiac cells, which are depressed by intracellular ATP atlevels greater than 1 mM. Since then, ATP-sensitive K+
(KATP) channels have been identified in a wide variety oftissues including the pancreatic β-cell, skeletal muscle,the CNS and in vascular and non-vascular smooth muscle(see Yokoshiki et al. 1998). There has been a tremendousamount of interest in KATP channels due to the discoverythat some synthetic agents exert their effect by eitheropening or antagonising KATP channels in a variety of tis-sues. For example, the sulphonylurea compound gliben-clamide is a potent and selective KATP channel antagonistin the pancreas (see Ashford 1990).
There are also a number of groups of compounds,chemically unrelated to the sulphonylureas, which havealso been shown to antagonise the smooth muscle relaxantactions of KATP channel openers in vascular and non-vas-cular smooth muscle (see Challinor-Rogers and McPher-son 1994). These include imidazolines (e.g. alinidine) andchemically related compounds including phentolamine,which were shown to antagonise KATP channel openers,such as levcromakalim, in vascular and non-vascularsmooth muscle (McPherson and Angus 1989, 1990; Chal-linor-Rogers et al. 1994; see Challinor-Rogers and Mc-Pherson 1994). Other studies performed at the same timeby Schultz and Hasselblatt (1989) and Chan and Morgan(1990) also showed this to be true for phentolamine andother imidazolines against KATP channels in the pancreaticβ-cell. However, pA2 or pKB values calculated for theseimidazoline compounds have shown them to be some 30to 100 times less potent than glibenclamide itself. Thus, in
Karen Bell · Joanne Favaloro · Vivian Khalil ·Magdy M. Iskander · Grant A. McPherson
The identification of a potent imidazoline-based vascular KATP channel antagonist
Naunyn-Schmiedeberg’s Arch Pharmacol (2000) 362 :145–151Digital Object Identifier (DOI) 10.1007/s002100000261
Received: 22 November 1999 / Accepted: 28 March 2000 / Published online: 9 June 2000
ORIGINAL ARTICLE
K. Bell · J. Favaloro · G. A. McPherson (✉ )Department of Pharmacology, Monash University, Clayton 3800, Victoria, Australia e-mail: [email protected], Fax: +61-3-99055851
V. Khalil · M. M. IskanderDepartment of Medicinal Chemistry, Victorian College of Pharmacy, Monash University, 381 Royal Parade, Parkville 3052, Victoria, Australia
© Springer-Verlag 2000

vascular smooth muscle the pKB calculated for gliben-clamide antagonising the actions of levcromakalim is ap-proximately 7 while the pKB calculated for alinidine isapproximately 5.5 (Challinor-Rogers et al. 1994); i.e.alinidine is 50 times less potent than glibenclamide at an-tagonising the KATP channel-opening activity of levcro-makalim.
At present, there is a great amount of interest in com-pounds that antagonise KATP channels because they mayhave important therapeutic uses in the treatment of a numberof pathological conditions including cardiac arrhythmia,shock and diabetes mellitus (type II). In this paper we re-port the synthesis and testing of a group of imidazolineanalogues, related to alinidine, that are as potent as gliben-clamide in their ability to antagonise the KATP channel-open-ing activity of levcromakalim in vascular smooth muscle.
Materials and methods
Porcine coronary artery
Porcine hearts were obtained from freshly killed pigs at an abat-toir. The right circumflex artery was rapidly removed and placedin ice-cold physiological Krebs’ solution (composition in mM:NaCl 119, KCl 4.7, MgSO4 · 7H2O 1.17, KH2PO4 1.18, CaCl2 2.5,NaHCO3 25, and glucose 11). The artery was cut into 4-mm-longsegments and each segment was suspended on two stainless steelwire hooks, 400 µm in diameter, in 25-ml jacketed glass organbaths. The lower hook was fixed to a support leg attached to a mi-crometer, while the upper wire hook was suspended from a GrassFT03C force transducer. Isometric force was recorded with an on-line data acquisition system (CVMS Version 2.0; World PrecisionInstruments, USA). Vessels were left to equilibrate under zeroforce for 30 min at 37°C in Krebs’ solution gassed with 5% CO2 inO2 and an initial force of 5 g was then applied. After another 30min, the force was re-adjusted to 5 g and the tissues were left for afurther 30 min. Subsequently, a potassium depolarising solution(composition in mM: KCl 123, MgSO4 · 7H2O 1.17, KH2PO4 1.18,CaCl2 2.5, NaHCO3 25, and glucose 11) was added. This responsewas used to determine the maximum constrictor response of thetissue. After a plateau to the potassium depolarising solution wasreached, vessels were washed twice and left until the response returned to the initial baseline, before commencing the experi-ment.
After this period each coronary ring was submaximally (ap-proximately 50%) constricted with the thromboxane-mimeticU46619 (3–30 nM). Once a plateau response to this vasoconstric-tor was reached, a cumulative concentration-response curve to lev-cromakalim (0.1–30 µM; or in some cases sodium nitroprusside; 1 nM–30 µM) was then constructed (0.5 log increments). Concen-trations of levcromakalim were added when the response to theprevious concentration had reached a plateau. Only one concentra-tion-response curve was obtained on any one coronary artery ring.A single concentration of the imidazoline compound (IMID series)tested was added 20 min prior to submaximally constricting thetissue with U46619. Preliminary experiments indicated that this in-cubation period was sufficient for equilibrium antagonism to beproduced. In some cases levcromakalim did not cause full tissuerelaxation, hence sodium nitroprusside (10 µM) was added at theend of each curve to obtain maximal vessel relaxation.
Rat spontaneously beating right atria
In a separate series of experiments the bradycardic action of thenovel analogues was assessed on the spontaneously beating rat(Sprague-Dawley) right atrium as previously described (Challinor-
Rogers et al. 1994). Following removal of the heart, the rightatrium was dissected free, and placed vertically on stainless steelS-shaped hooks and suspended in Krebs’ buffer in a jacketed organbath maintained at 37°C. The atrium was washed and left to equi-librate for approximately half an hour until the resting atrial ratewas stable. Basal tension was maintained at approximately 0.5 gthroughout the experiments.
Cumulative concentration-response curves (0.5 log increments)were then constructed for each imidazoline analogue (0.1–30 µM).Concentrations of the compounds were added when the responseto the previous concentration had reached a plateau. Chronotropicresponses were recorded (as 5-s averages) with an online data ac-quisition system (CVMS Version 2.0; World Precision Instru-ments, USA).
[3H]P1075 binding
The ability of the novel imidazoline compounds to compete forsites labelled by the KATP channel opener [3H]P1075 was deter-mined. Bovine aorta smooth muscle cell membranes were pre-pared by adapting the method of Carman-Krzan et al. (1997).Bovine aortae were obtained from the local abattoir and stored inice-cold storage buffer [composition in mM: NaCl 139, KCl 5,MgCl2 1.2, HEPES (4-(2-hydroxyethyl)-1-piperazine-ethane-sul-phonic acid) 5, and EGTA (ethylene glycol-bis-(2-aminoethyl-ether)-N,N,N’,N’-tetra-acetic acid) 1, pH=7.4]. The aortae werethen cleared of fat and connective tissue and the smooth muscledissected from the inner elastic layer. The smooth muscle was thenminced in 3 vol/g (wet weight) of the mincing buffer [composition:HEPES 10 mM, EGTA 1 mM, phenylmethylsulphonyl fluoride(PMSF) 0.2 mM, pepstatin A 0.2 µM, leupeptin 10 µM, and soy-bean trypsin inhibitor 10 µg/ml] and homogenised. The homo-genate was then sieved through a fine nylon mesh, then cen-trifuged at 100 g for 10 min at 4°C. The supernatant was aspiratedand further centrifuged at 48,000 g for 15 min at 4°C. The result-ing pellet was then suspended in approximately 20 volumes ofHEPES buffer (composition in mM: NaCl 139, KCl 5, MgCl2 2,and HEPES 20, pH=7.4) and stored at –80°C.
Drug displacement experiments were carried out using themethod of Löffler-Walz and Quast (1998). Two hundred micro-litres of the membrane preparation was incubated with [3H]P1075(1 nM), the cold displacing drugs, levcromakalim (10 µM) andglibenclamide (10 µM) or the IMID compounds (10 µM) to give atotal volume of 1 ml in an incubation buffer [composition in mM:NaCl 139, KCl 5, MgCl2 25, CaCl2 1.25, HEPES 20, creatinephosphate 20, Na2ATP 3, and creatine phosphokinase (50 U/ml),pH=7.4] at 37°C for 30 min. The incubation was stopped byadding 4 ml of ice-cold quench solution (composition in mM:NaCl 154, Tris 50, pH=7.4). The bound and free ligand was thenseparated by filtration under vacuum over glass microfibre filters(Whatman GF/B). The filters were washed twice with 5 ml ofquench solution and counted for 3H in 2.5 ml scintillant (EcoLite;ICN Biomedicals, USA).
[3H]glibenclamide binding
The ability of the imidazoline compounds to interact with the sitelabeled by the KATP channel antagonist glibenclamide was as-sessed by using the technique of Challinor-Rogers et al. (1995).Male Sprague-Dawley rats (250–350 g) were sacrificed by 80%CO2/20% O2 narcosis and exsanguination. The cerebral cortex wasdissected free and placed in phosphate buffer (10 mM, pH=7.4)and maintained at 4°C. Membranes were prepared by homogenisa-tion of the cerebral cortex in 20 vol (w/v) of ice-cold phosphatebuffer. The homogenate was centrifuged (35,000 g for 15 min at4°C), the supernatant discarded and the resultant pellet washed andthe above procedure repeated. The final membrane suspension wasprepared by suspending the pellet in 50 vol (w/v) phosphate buffer.Each millilitre of homogenate therefore contained 20 mg wetweight of original tissue mass.
146
Drug displacement experiments were carried out by incubating500 µl of the membrane preparation with [3H]glibenclamide (0.3 nM), the cold displacing drug, glibenclamide (10 µM) or theIMID compounds listed in Fig.1 (10 µM) to give a total volume of1 ml in a phosphate buffer at room temperature for 1 h. The incu-bation was stopped by adding 4 ml of ice-cold phosphate buffer.The bound and free ligand were then separated by filtration undervacuum over glass microfibre filters (Whatman GF/B). The filterswere washed twice with 5 ml of phosphate buffer and counted for3H in 2.5 ml scintillant (EcoLite; ICN Biomedicals, USA).
Analysis of results
Porcine coronary artery. The contraction to U46619 (3–30 nM)was taken as 100% response and relaxation produced by differentconcentrations of levcromakalim expressed as a percentage of this
response. The percentages obtained were then represented graphi-cally and the pD2 (–log EC50) value calculated as the concentrationof relaxant required to cause 50% of the maximal relaxant re-sponse. To determine whether the antagonists directly affected thetissue’s ability to contract, the response to U46619 was expressedas a percentage of the maximal potassium depolarising solutioncontraction.
In this and previous studies (see Challinor-Rogers et al. 1994)it has been observed that, at high concentrations, some of the imi-dazoline analogues (particularly those with low potency) shift thelevcromakalim concentration-response curve in a non-parallelfashion. Thus the slope of the curve is often reduced and the max-imum response to levcromakalim depressed. This suggests that thetype of antagonism displayed by the compounds is non-competi-tive and prevented us from the use of Schild analysis (Jenkinson1991) as a means of determining the potency of the imidazolinecompounds. Instead, an ‘apparent pKB’ can be estimated, based on
147
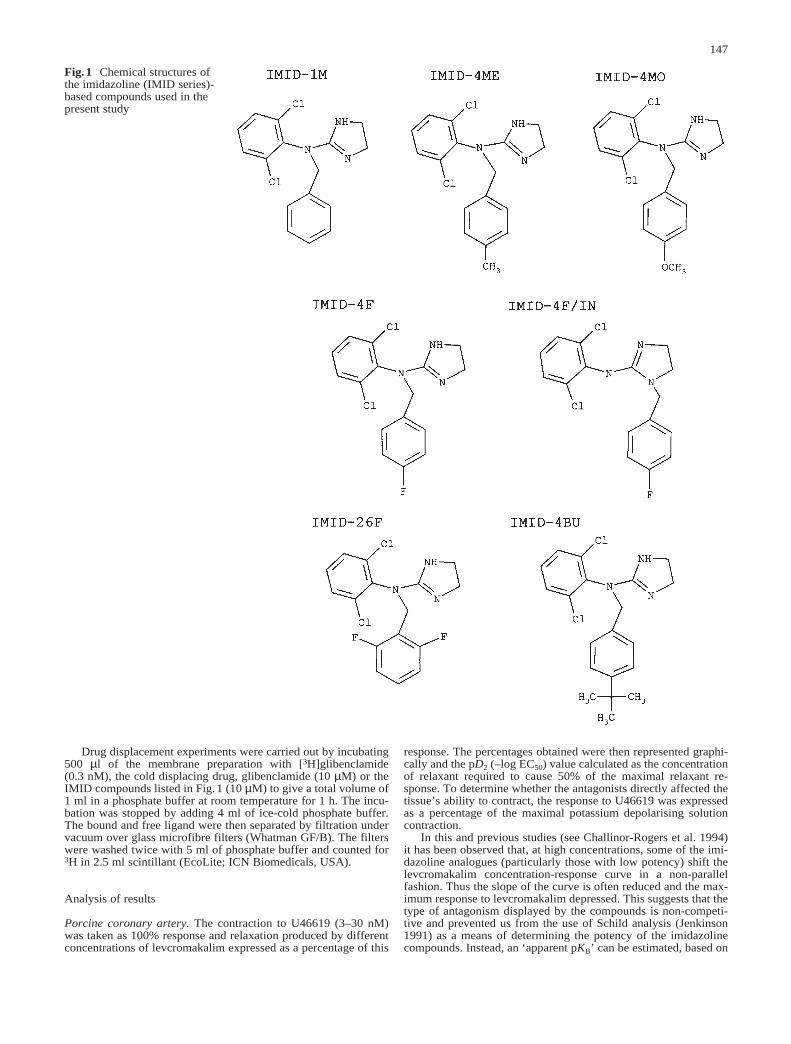
Fig.1 Chemical structures ofthe imidazoline (IMID series)-based compounds used in thepresent study
a single concentration of antagonist. The following equation wasused:
Apparent pKB = -Log([Antagonist Concentration, M]/ (1)[Concentration Ratio -1])
In the present study the single concentration of the antagonist usedto calculate the apparent pKB was selected on the basis that it pro-duced a parallel-like shift in the levcromakalim concentration-re-sponse curve without overtly affecting the maximal levcromakalimresponse.
Rat spontaneously beating right atria. Bradycardic responses wereexpressed as a percentage of the resting heart rate with 100% re-ferring to initial resting heart rate. The percentages obtained werethen represented graphically and the pD2 (–log EC50) calculated.
Statistics
One-way repeated measures analysis of variance (ANOVA) wasused to test several values that were dependent, Student-Newman-Keuls method for all pairwise multiple comparisons was used aspost ANOVA. All statistical calculations were performed usingSigmaStat (Jandal Scientific, USA). Results in the text are given asmeans ± SEM.
Drugs
Drugs used and their sources were: levcromakalim (Beecham,UK); sodium nitroprusside (David Bull, Australia); U46619 (Up-john, Kalamazoo, Mich., USA); the IMID series of imidazolinecompounds were synthesised by Vivian Khalil and Magdy Iskan-der at the Victorian College of Pharmacy and Eva Campi, Depart-ment of Chemistry, Monash University (Khalil 1995). Figure 1shows the structure of the derivatives. All novel imidazolines weresynthesized as HCl salts. Levcromakalim and the imidazoline ana-logues were made up in 100% methanol as stock solutions of 10mM. All dilutions were made in distilled water.
Results
Antagonism of levcromakalim responses by imidazoline analogues
The ability of seven novel imidazolines to antagonise lev-cromakalim-induced vasorelaxation was investigated inpig right circumflex coronary artery constricted withU46619 (3–30 nM). All compounds characterised in thisstudy were based on the compound IMID-1M and con-sisted of an n-benzyl-substituted derivative of clonidine(Fig.1). From IMID-1M several groups of compoundswere derived: methoxy-benzyl (IMID-4MO), fluoro-ben-zyl (IMID-4F, IMID-26F and IMID-4F/IN), butyl-benzyl(IMID-4BU) and methyl-benzyl derivatives (IMID-4ME).Table 1 summarizes the results obtained in this study.
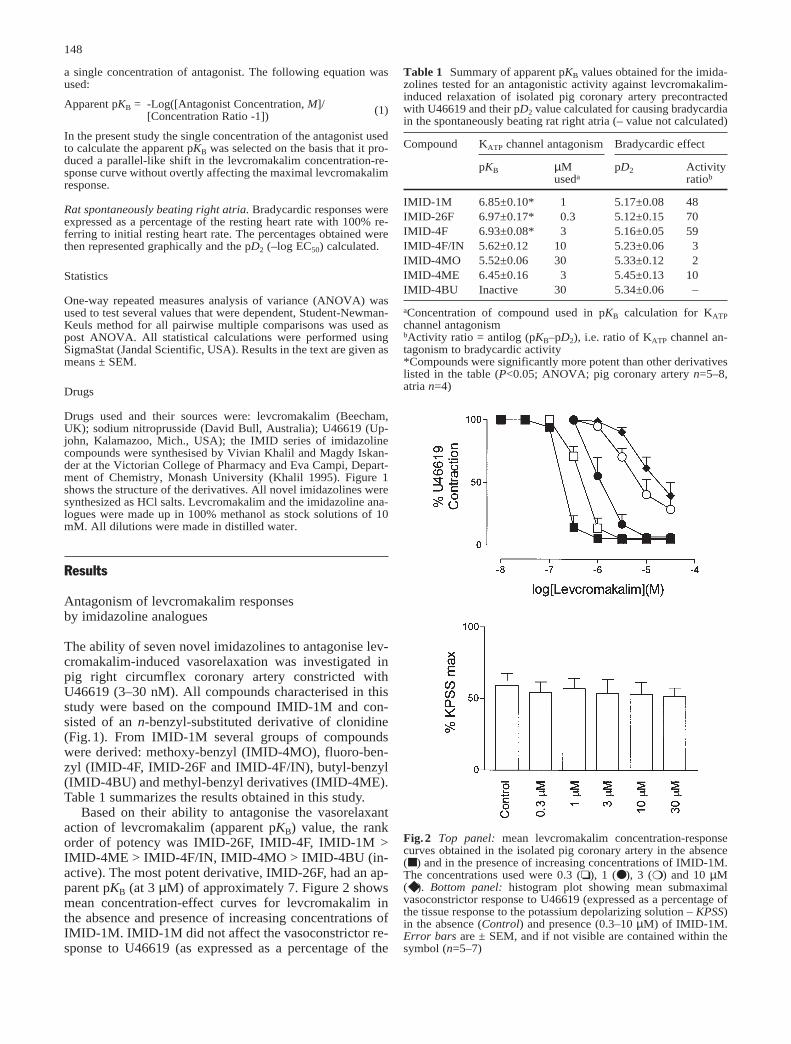
Based on their ability to antagonise the vasorelaxantaction of levcromakalim (apparent pKB) value, the rankorder of potency was IMID-26F, IMID-4F, IMID-1M >IMID-4ME > IMID-4F/IN, IMID-4MO > IMID-4BU (in-active). The most potent derivative, IMID-26F, had an ap-parent pKB (at 3 µM) of approximately 7. Figure 2 showsmean concentration-effect curves for levcromakalim inthe absence and presence of increasing concentrations ofIMID-1M. IMID-1M did not affect the vasoconstrictor re-sponse to U46619 (as expressed as a percentage of the
148
Table 1 Summary of apparent pKB values obtained for the imida-zolines tested for an antagonistic activity against levcromakalim-induced relaxation of isolated pig coronary artery precontractedwith U46619 and their pD2 value calculated for causing bradycardiain the spontaneously beating rat right atria (– value not calculated)
Compound KATP channel antagonism Bradycardic effect
pKB µM pD2 Activityuseda ratiob
IMID-1M 6.85±0.10* 1 5.17±0.08 48IMID-26F 6.97±0.17* 0.3 5.12±0.15 70IMID-4F 6.93±0.08* 3 5.16±0.05 59IMID-4F/IN 5.62±0.12 10 5.23±0.06 3IMID-4MO 5.52±0.06 30 5.33±0.12 2IMID-4ME 6.45±0.16 3 5.45±0.13 10IMID-4BU Inactive 30 5.34±0.06 –
aConcentration of compound used in pKB calculation for KATPchannel antagonismbActivity ratio = antilog (pKB–pD2), i.e. ratio of KATP channel an-tagonism to bradycardic activity*Compounds were significantly more potent than other derivativeslisted in the table (P<0.05; ANOVA; pig coronary artery n=5–8,atria n=4)
Fig.2 Top panel: mean levcromakalim concentration-responsecurves obtained in the isolated pig coronary artery in the absence(■ ) and in the presence of increasing concentrations of IMID-1M.The concentrations used were 0.3 (❏ ), 1 (● ), 3 (❍ ) and 10 µM(◆ ). Bottom panel: histogram plot showing mean submaximalvasoconstrictor response to U46619 (expressed as a percentage ofthe tissue response to the potassium depolarizing solution – KPSS)in the absence (Control) and presence (0.3–10 µM) of IMID-1M.Error bars are ± SEM, and if not visible are contained within thesymbol (n=5–7)
maximum tissue response to the potassium depolarizingsolution; Fig.2), at concentrations up to 30 µM. None ofthe other compounds tested significantly affected thevasoconstrictor response to U46619 (data not shown).
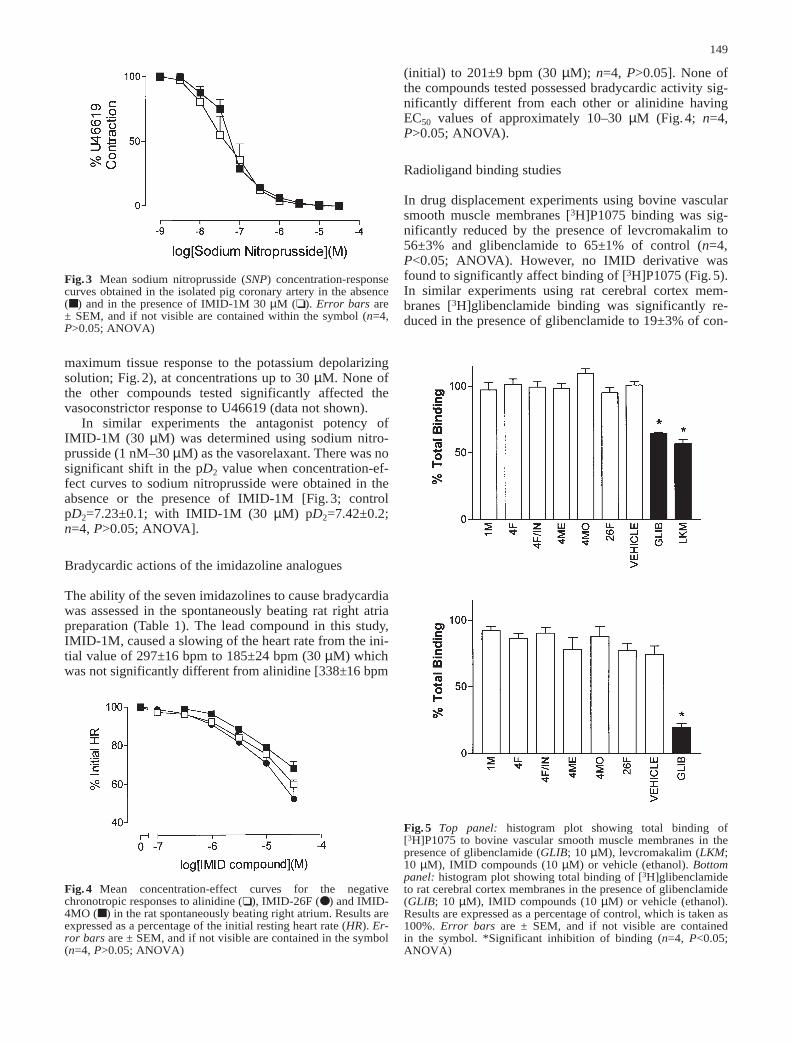
In similar experiments the antagonist potency ofIMID-1M (30 µM) was determined using sodium nitro-prusside (1 nM–30 µM) as the vasorelaxant. There was nosignificant shift in the pD2 value when concentration-ef-fect curves to sodium nitroprusside were obtained in theabsence or the presence of IMID-1M [Fig.3; controlpD2=7.23±0.1; with IMID-1M (30 µM) pD2=7.42±0.2;n=4, P>0.05; ANOVA].
Bradycardic actions of the imidazoline analogues
The ability of the seven imidazolines to cause bradycardiawas assessed in the spontaneously beating rat right atriapreparation (Table 1). The lead compound in this study,IMID-1M, caused a slowing of the heart rate from the ini-tial value of 297±16 bpm to 185±24 bpm (30 µM) whichwas not significantly different from alinidine [338±16 bpm
(initial) to 201±9 bpm (30 µM); n=4, P>0.05]. None ofthe compounds tested possessed bradycardic activity sig-nificantly different from each other or alinidine havingEC50 values of approximately 10–30 µM (Fig.4; n=4,P>0.05; ANOVA).
Radioligand binding studies
In drug displacement experiments using bovine vascularsmooth muscle membranes [3H]P1075 binding was sig-nificantly reduced by the presence of levcromakalim to56±3% and glibenclamide to 65±1% of control (n=4,P<0.05; ANOVA). However, no IMID derivative wasfound to significantly affect binding of [3H]P1075 (Fig. 5).In similar experiments using rat cerebral cortex mem-branes [3H]glibenclamide binding was significantly re-duced in the presence of glibenclamide to 19±3% of con-
149
Fig.3 Mean sodium nitroprusside (SNP) concentration-responsecurves obtained in the isolated pig coronary artery in the absence(■ ) and in the presence of IMID-1M 30 µM (❏ ). Error bars are ± SEM, and if not visible are contained within the symbol (n=4,P>0.05; ANOVA)
Fig.4 Mean concentration-effect curves for the negativechronotropic responses to alinidine (❏ ), IMID-26F (● ) and IMID-4MO (■ ) in the rat spontaneously beating right atrium. Results areexpressed as a percentage of the initial resting heart rate (HR). Er-ror bars are ± SEM, and if not visible are contained in the symbol(n=4, P>0.05; ANOVA)
Fig.5 Top panel: histogram plot showing total binding of[3H]P1075 to bovine vascular smooth muscle membranes in thepresence of glibenclamide (GLIB; 10 µM), levcromakalim (LKM;10 µM), IMID compounds (10 µM) or vehicle (ethanol). Bottompanel: histogram plot showing total binding of [3H]glibenclamideto rat cerebral cortex membranes in the presence of glibenclamide(GLIB; 10 µM), IMID compounds (10 µM) or vehicle (ethanol).Results are expressed as a percentage of control, which is taken as100%. Error bars are ± SEM, and if not visible are contained in the symbol. *Significant inhibition of binding (n=4, P<0.05;ANOVA)

trol (n=4, P>0.05; ANOVA), but not in the presence of theIMID compounds (Fig.5).
Discussion
The main finding from this study is that some but not alln-benzyl derivatives of clonidine are potent antagonists ofthe KATP channel opening effects of levcromakalim onvascular smooth muscle. We were the first to show thatimidazolines (e.g. alinidine and phentolamine) were an-tagonists of the vascular smooth muscle relaxant actionsof cromakalim (McPherson and Angus 1989, 1990). Us-ing identical techniques as those described in the presentexperiments, the apparent pKB for alinidine in rat thoracicaorta was calculated to be approximately 5.5 (Challinor-Rogers et al. 1994). Replacing the n-allyl substituent ofalinidine with either an unsubstituted or substituted n-ben-zyl derivative generated compounds (IMID-1M, IMID-26F, IMID-4F) that had apparent pKB values of approxi-mately 7, some 40 to 60 times more potent than alinidineitself. It has been known for some time that the substituenton the extra-cyclic nitrogen of alinidine was important indetermining KATP channel potency because the parentcompound of alinidine, clonidine (which lacks the n-allylsubstitution), is not active as a levcromakalim antagonist(McPherson and Angus 1989). Hence, in this study it wasapparent that the substitution of a benzene ring on the ex-tra-cyclic nitrogen caused a substantial increase in po-tency as a KATP channel antagonist. However, substitu-tions onto the benzene ring either had little affect on thepotency (IMID-4F and -26F) or actually decreased the po-tency (IMID-4F/IN, -4MO, -4ME and -4BU) of thesecompounds as levcromakalim antagonists. As noted ear-lier, previous work by us has shown that imidazolinesgenerally cause a shift to the right of the levcromakalimconcentration-effect curve associated with suppression ofthe maximum response (see Challinor-Rogers et al. 1994).With the more potent compounds (e.g. IMID-1M; Fig.4)that we have developed, this phenomenon is less obvious.However, we expect suppression of the maximum re-sponse would occur if concentrations of levcromakalimcould have been extended past our technical limit of 30 µM.
The antagonism of levcromakalim vasorelaxant re-sponses was also specific for this vasodilator since IMID-1M (30 µM) failed to affect the vasorelaxant responses tosodium nitroprusside, a nitro-vasorelaxant compound thatcauses its effect via cGMP generation. This is consistentwith previous findings which showed that imidazoline-based compounds specifically inhibited vasorelaxant re-sponses to KATP channel openers (McPherson and Angus1989). The compounds were also found to be very poorbradycardic agents with EC50 values between 10 µM and30 µM. Therefore, given the relatively high potency ofthese compounds as levcromakalim antagonists and theirpoor ability to cause bradycardia, many of the compoundswere found to be highly vascular-selective. In particularIMID-1M, IMID-4F and IMID-26F were the most selectiveagents with a calculated activity ratio between 50 and 70.
A number of chemically diverse compounds areknown to antagonise the KATP channel-opening effects oflevcromakalim. Most notable of these are the sulphonyl-ureas typified by glibenclamide. Previous studies (seeMcPherson et al. 1997) have shown that glibenclamidebehaves as a classical competitive antagonist of levcro-makalim-mediated vasorelaxant responses in a number ofvascular preparations, with a pA2/pKB of approximately 7and a slope value near unity. Thus when using sulphonyl-urea-based compounds, the levcromakalim concentration-effect curve is shifted to the right in a parallel mannerwith no obvious effect on the maximum response. How-ever, studies have shown that glibenclamide can antago-nise the effects of U46619 in some species by directly act-ing at the thromboxane A2 receptor (Cocks et al. 1990;Kemp and McPherson 1998). Hence, experimentally,compounds such as IMID-1M may be a useful adjunct inthe study of KATP channels since they appear to operate ata site within the KATP channel complex separate to that ofglibenclamide (i.e. at KIR rather than the sulphonylurea re-ceptor) and also they do not antagonise thromboxane A2receptors.
Another potent group of levcromakalim antagoniststhat have been identified are the lipophilic quaternary ionssuch as tetraphenylphosphonium (pKB=7.2; McPhersonand Piekarska 1994; Piekarska and McPherson 1997).While displaying the same potency as glibenclamide,tetraphenylphosphonium (and related compounds) show amarkedly different type of antagonism as that displayedby the sulphonylureas. In the case of the lipophilic quater-nary ion, these compounds cause a non-parallel shift tothe right of the levcromakalim concentration-effect curveand, at higher concentrations, suppress the maximum va-sorelaxant response that levcromakalim can elicit. At thistime it is uncertain where lipophilic quaternary ions inter-act with the KATP channel. However, previous studies withother quaternary ions have suggested that they interactwith the pore of the K+ channel to inhibit K+ efflux fromthe cell (see McPherson and Piekarska 1994). The po-tency and nature of the antagonism displayed by the imi-dazolines characterised in this study are similar to thatdisplayed by the lipophilic quaternary ions. Thus the imi-dazolines cause a non-competitive antagonism of the KATPchannel-opening effects of levcromakalim. Since imida-zolines can be positively charged at physiological pH (seeMcPherson and Piekarska 1994), it is possible that thesecompounds may affect KATP channel opening through amechanism similar to that utilized by the lipophilic qua-ternary ions.
The KATP channel is an octameric complex of two sub-units: a pore-forming inward rectifying K+ channel sub-unit, KIR6.x, and a regulatory subunit, the sulphonylureareceptor (SUR; Sakura et al. 1995; Inagaki et al. 1996).Different subunits are found in different tissues; for ex-ample, in vascular smooth muscle, the KATP channel iscomposed of SUR2B and KIR6.1, while in the pancreas itis SUR1 and KIR6.2 (see Babenko et al. 1998). Previousbinding studies have identified these subunits on thesulphonylurea receptor sites for both the KATP channel
150

openers (e.g. levcromakalim) and the sulphonylurea-based antagonists such as glibenclamide (Bray and Quast1992; Quast et al. 1992; Löffler and Quast 1997). As theIMID compounds failed to displace either the binding ofthe opener, [3H]P1075 in smooth muscle membranes, orthe antagonist, [3H]glibenclamide in rat cerebral cortexmembranes, it can be concluded they are acting neither atthe opener nor sulphonylurea binding site of the SUR. Webelieve that these compounds act at the pore of the KIRsubunit that constitutes the KATP channel. Indeed, this the-ory is aided by the recent finding by Proks and Ashcroft(1997) that the imidazoline, phentolamine, inhibited pan-creatic KATP channels by directly interacting with the KIRcomponent of the KATP channel albeit in this study it wasnot KIR6.1 as found in the vasculature.
In conclusion the results from this study have identi-fied potent imidazoline-based KATP channel antagonists.The compounds IMID-1M, -26F and -4F are as potent asthe sulphonylurea glibenclamide and the lipophilic qua-ternary ion tetraphenylphosphonium which have pKB val-ues of approximately 7 in vascular smooth muscle. Com-pounds that interact with KATP channels may have impor-tant therapeutic uses. For example, KATP channel antago-nists, of the type studied here, appear to have some anti-arrhythmic activity in a number of experimental situations(see Challinor-Rogers et al. 1997). It is currently uncer-tain whether the compounds described herein also interactwith pancreatic-type KATP channels to regulate insulin se-cretion and hence be useful in the treatment of diabetesmellitus. With further investigation these compounds maytherefore represent a new class of therapeutic agent. Inaddition, they represent a potential new experimental tooldue to their lack of activity at the thromboxane receptor invascular smooth muscle.
Acknowledgements. GAM is a Senior Research Fellow of theNH&MRC. A Project Grant from the NH&MRC of Australia sup-ported this project. We would like to thank Eva Campi and Profes-sor Jackson from the Department of Chemistry, for their input.These experiments complied with the NH&MRC (Australia) ani-mal ethics standards.
References
Ashford MLJ (1990) Potassium channels and modulation of in-sulin secretion. In: Cook NS (ed) Potassium channels: struc-ture, classification, function and therapeutic potential. EllisHorwood, Chichester, pp 300–325
Babenko AP, Aguilar-Bryan L, Bryan J (1998) A view of SUR/KIR6.x KATP channels. Annu Rev Physiol 60:667–687
Bray KM, Quast U (1992) A specific binding site for K+ channelopeners in rat aorta. J Biol Chem 267:11689–11692
Carman-Krzan M, Krzan M, Schunack W (1997) Pharmacolog-ical properties of cardiovascular histamine H1 receptor bind-ing sites: characterisation with 2-phenylhistamines. Naunyn-Schmeideberg’s Arch Pharmacol 355:431–437
Challinor-Rogers JL, McPherson GA (1994) Potassium channelopeners and other regulators of KATP channels. Clin Exp Phar-macol Physiol 21:583–597
Challinor-Rogers JL, Hay TK, McPherson GA (1994) Comparisonof the cromakalim antagonism and bradycardic actions of a se-ries of novel alinidine analogues in the rat. Naunyn-Schmeide-berg’s Arch Pharmacol 350:158–166
Challinor-Rogers JL, Kong DCM, Iskander MN, McPherson GA(1995) Structure-activity relationship of glibenclamide analogs:a comparison of potency as levcromakalim antagonists in rataorta vs. affinity for [3H]glibenclamide binding to membranesfrom rat cerebral cortex. J Pharmacol Exp Ther 273:778–786
Challinor-Rogers JL, Rosenfeldt FL, Du XJ, McPherson GA(1997) Antiischemic and antiarrhythmic activities of some novelalinidine analogues in the rat heart. J Cardiovasc Pharmacol29:499–507
Chan SL, Morgan NG (1990) Stimulation of insulin secretion byefaroxan may involve interaction with potassium channels. EurJ Pharmacol 176:97–101
Cocks TM, King SJ, Angus JA (1990) Glibenclamide is a competi-tive antagonist of the thromboxane A2 receptor in dog coronaryarteries in vitro. Br J Pharmacol 100:375–378
Inagaki I, Gonoi T, Clement JP IV, Wang C-Z, Aguilar-Bryan L,Bryan J, Seino S (1996) A family of sulphonylurea receptorsdetermines the pharmacological properties of ATP-sensitive K+
channels. Neuron 16:1011–1017Jenkinson DH (1991) How we describe competitive antagonists:
three questions of usage. Trends Pharmacol Sci 12:53–54Kemp BK, McPherson GA (1998) Interspecies differences in
thromboxane A2 receptors are distinguished by glibenclamide.Eur J Pharmacol 354:173–178
Khalil V (1995) Synthesis and testing of novel potassium channelligands. MSc thesis, Monash University, Clayton, Australia
Löffler C, Quast U (1997) Pharmacological characterisation of thesulphonylurea receptor in rat isolated aorta. Br J Pharmacol 120:476–480
Löffler-Walz C, Quast U (1998) Binding of KATP channel modula-tors in rat cardiac membranes. Br J Pharmacol 123:1395–1402
McPherson GA, Angus JA (1989) Phentolamine and structurallyrelated compounds selectively antagonize the vascular actionsof the K+ channel opener, cromakalim. Br J Pharmacol97:941–949
McPherson GA, Angus JA (1990) Characterization of responses tocromakalim and pinacidil in smooth and cardiac muscle by useof selective antagonists. Br J Pharmacol 100:201–206
McPherson GA, Piekarska AE (1994) Antagonism by lipophilicquaternary ions of the K+ channel opener, levcromakalim, invascular smooth muscle. Br J Pharmacol 112:1223–1229
McPherson GA, Choi RT, Kong DC, Iskander MN (1997) Thethromboxane-A2 and KATP channel antagonist actions of a se-ries of sulphonylurea derivatives in the pig coronary artery. EurJ Pharmacol 324:193–200
Noma A (1983) ATP-regulated K+ channels in cardiac muscle. Na-ture 305:147–148
Piekarska A, McPherson G (1997) Structure-activity relationshipof quaternary ion antagonism of levcromakalim-induced relax-ation in pig coronary artery. Eur J Pharmacol 322:37–44
Proks P, Ashcroft FM (1997) Phentolamine block of KATP chan-nels is mediated by Kir6.2. Proc Natl Acad Sci USA 94:11716–11720
Quast U, Bray KM, Andres H, Manley PW, Baumlin Y, DosogneJ (1992) Binding of the K+ channel opener [3H]P1075 in ratisolated aorta: relationships to functional effects of openers andblockers. Mol Pharmacol 43:474–481
Sakura H, Ämmälä C, Smith PA, Gribble FM, Ashcroft FM (1995)Cloning and functional expression of the cDNA encoding anovel ATP-sensitive potassium channel subunit expressed inpancreatic β-cells, brain, heart and skeletal muscle. FEBS Lett377:338–344
Schultz A, Hasselblatt A (1989) Dual action of clonidine on in-sulin release: suppression, but stimulation when alpha 2-adrenoceptors are blocked. Naunyn-Schmiedeberg’s Arch Phar-macol 340:321–327
Yokoshiki H, Sunagawa M, Seki T, Sperelakis N (1998) ATP-sen-sitive K+ channels in pancreatic, cardiac and vascular smoothmuscle cells. Am J Physiol 274:C25–C37
151





























