Embed Size (px)
Citation preview

Kidney International, Vol. 32 (1987), pp. 1—12
EDITORIAL REVIEW
The clinical significance of inhibition of renal prostaglandinsynthesis
Clinical interest in renal prostaglandins has been stimulatedby the repeated observation that nonsteroidal anti-inflammatorydrugs (NSAD) can decrease renal function [1—4]. The purposeof this editorial review is to analyze the physiologic significanceof renal prostaglandin synthesis and the pathophysiologic con-sequences of inhibition of prostaglandin production through theuse of NSAD. In order to adequately develop this theme, it isnecessary to review some biochemical principles of eicosanoidsynthesis.
Eicosanoid is the generic term which refers to lipoxygenaseand cyclooxygenase products of arachidonic acid metabolism.Figure 1 outlines some of these pathways. Prostaglandins andthe other eicosanoids are autacoids which do not have acirculating, endocrine function but rather act in a paracrine orautocrine manner affecting cells close to or at the site ofprostaglandin synthesis [5]. NSAD inhibit either irreversibly(aspirin) or reversibly the cyclooxygenase enzymes whichconvert arachidonic acid to the prostaglandin endoperoxides[61. Presently, no similar inhibitory agents are clinically avail-able which inhibit the lipoxygenase enzymes converting arachi-donic acid to leukotrienes and monohydroxy fatty acids. Al-though glucocorticoids can limit substrate availability throughthe induction of a recently characterized phospholipase A2-inhibitory protein named lipocortin [7], their in vivo effects onendogenous prostaglandin production are rather inconsistent [8,9]. Recent in vitro studies support the concept that corticoster-oids inhibit mitogen—induced T-cell proliferation by suppressingendogenous LTB4 production [10].
Localization of renal prostaglandin synthesisEicosanoid synthesis in the kidney is localized to specific
sites and is not uniformly present throughout the nephron [5].Since the initial discovery of prostaglandin synthesis in therenal medulla, it has been recognized that medullary tissuesynthesizes greater amounts of prostaglandins than the cortex[11]. It is generally accepted that the regional heterogeneity ofarachidonate metabolism, as well as the lack of vascular com-munications between the medulla and cortex, dictate thatprostaglandins synthesized in the cortex (glomeruli and vascu-lature) regulate cortical function and prostaglandins synthe-sized in the medulla (collecting tubule and medullary interstitialcells) regulate medullary function [5]. Table I summarizes theprincipal sites of prostaglandin synthesis in the kidney, themajor prostaglandins produced at each site, and their principalactions. Assessment of prostaglandin synthesis by different
Received for publication December 23, 1985and in revised form June 24, 1986
© 1987 by the International Society of Nephrology
1
components of the nephron is based on immunofluorescentmicroscopy [12—14], separation of glomeruli and nephron seg-ments with measurement of prostaglandin synthesis [15—17] orby cell cultures of specific components of the nephron withmeasurement of eicosanoid turnover [18—22]. Some speciesvariation exists and the majority of data are based on studies ofrat, rabbit, and human kidney. In the cortex, the major sites ofprostaglandin synthesis include arteries, arterioles and theglomerulus. The proximal tubule and the loop of Henle showlittle cyclooxygenase activity, but may convert arachidonate toseveral epoxygenase derivatives [reviewed in 23]. The largeamounts of PGE2 produced by the renal medulla primarilyderive from the substantial biosynthetic activity of collectingtubule and interstitial cells. Eicosanoids synthesized within thekidney are either degraded in the kidney, by enzymes acting onthe M3,14 double bond and on the hydroxyl group at C-iS, orare removed from the kidney in the lymphatic and venousdrainage or by excretion into the urine [5]. As discussed below,measurement of urinary unmetabolized prostaglandins or theirstable hydrolysis products provides the best clinical assessmentof the state of renal prostaglandin production.
Does urinary prostaglandin excretion reflect intrarenalprostaglandin synthesis?
In vivo studies of renal prostaglandin synthesis, based on theassessment of renal secretory and/or excretory rates of PGE2and PGF2a, have been reported in several species including therat, rabbit, dog and humans [reviewed in 24]. Renal venousprostaglandins are presumably derived from direct venousdrainage of cellular sites of synthesis and also from venousremoval of reabsorbed prostaglandins. Measurement of unme-tabolized prostaglandins in renal venous plasma, however,suffers from major limitations, that is, very low levels, plateletsynthesis during and after sampling (only partially prevented bycyclooxygenase inhibitors), and invasive procedure. As a con-sequence, measurement of unmetabolized prostaglandins inurine has gained general acceptance following the originalreport by Frolich et al [25] suggesting their renal origin inhealthy women. Besides having a two orders of magnitudehigher prostaglandin concentration, urine has the additionaladvantage over renal venous plasma of being free of plateleteicosanoids and of providing a noninvasive, continuous ratherthan episodic measure of renal prostaglandin synthesis. Anumber of methodologic as well as biologic variables can,however, affect urinary prostaglandin measurements in humanstudies. These include storage conditions (due to chemicalinstability of some eicosanoids), purification and assay methods(due to the presence in urine of a large array of structurallyrelated eicosanoids), and possible contribution from extrarenalsources [26]. Thus, trace amounts of seminal fluid, having l0 to

Fig. 1. Summary of prostaglandin andthromboxane formation from arachidonicacid. The site of action of nonsteroidalantiinflammatory drugs is indicated on thecyclooxygenase enzyme which convertsarachidonic acid to PG endoperoxides.
Site Eicosanoid Action
Vasculature PGI. VasodilationGlomerulus PGI2, PGE�
TxA2Maintain GFRReduce GFR°
Collecting tubule PGE2, PGF2,. Enhance excretion of NaCland water
Medullary interstitial PGE2 Vasodilation and natriure-cells sis—diuresis
106 higher PGE2 concentration than urine, can contribute ahighly variable though substantial fraction of the measuredurinary PGE2 in man, making the overall interpretation of suchmeasurements highly questionable [27]. In addition to the"classical" primary prostaglandins, that is, PGE2 and PGF2a,human urine contains measurable amounts of 6-keto-PGF1a [28]and TXB2 [29], chemically stable hydration products of PGI2and TXA2, respectively. Drugs that selectively spare renalcyclooxygenase activity, such as sulindac or low—dose aspirin[30], do not reduce the urinary excretion of 6-keto-PGF1a andTXB2 in health [31—33] and disease [31, 34]. Human diseaseshave been characterized in which markedly—elevated urinaryexcretion of 6-keto-PGF1a (Bartter's syndrome) or TXB2 (sys-temic lupus erythematosus) were proven to reflect enhancedintrarenal production of PG!2 [35] or TXA2 [36], respectively,on the basis of unchanged excretion of the major urinarymetabolites reflecting an extra—renal origin [371, that is, 2,3-dinor-6-keto-PGFia and 2,3-dinor-TXB2.
Radioimmunoassay (RIA) as well as gas chromatography—mass spectrometry have been employed to quantitate urinary
prostaglandin and thromboxane excretion [25, 26, 38, 39].While the latter can provide definitive identification of a giveneicosanoid, with adequate sensitivity particularly in the nega-tive ion, chemical ionization mode [39], the former represents ahighly sophisticated form of bioassay substituting the classicalsmooth—muscle strip with a soluble antibody as the biologicalreactant. Consequently, the biological read—out, that is variablyreduced binding of the labeled eicosanoid to a presumably"specific" anti-eicosanoid serum as a function of increasingconcentrations of the unlabeled homologous ligand, is liable toall forms of artifacts possibly affecting bioassay. In particular,the presence in urine of a large number of potentially cross—reacting enzymatic derivatives of prostaglandins and thrombox-ane B2 excreted in a 10- to 100-fold excess vis-a-vis unmetab-olized eicosanoids represents a serious potential limitation tothe specificity of such measurements. Unfortunately, immuno-logic characterization of anti-eicosanoid sera is often limited toprimary prostaglandins and TXB2 with limited information, ifany, on the cross—reactivity of major urinary metabolites suchas the 2,3-dinor—derivatives of PGI2 and TXB2. Moreover,reduction of the measured urinary prostaglandin—like immuno-reactivity by cyclooxygenase inhibitors is often presented asevidence of the "specificity" of such measurements - This maybe misleading, inasmuch as the renal and extra—renal synthesisof all eicosanoids will be suppressed by non-selective cyclooxy-genase inhibitors (such as indomethacin) thereby causing asimultaneous reduction in the urinary excretion of both primaryunmetabolized prostaglandins (presumably reflecting a renalorigin) and potentially cross—reacting systemic metabolites. Onthe other hand, such methodologic problems will confound theeffects of selective cyclooxygenase inhibitors on the kidney(such as sulindac, low—dose aspirin, sulfinpyrazone) by virtueof the reduced excretion of cross—reacting systemic metabolites
2 Patrono and Dunn
Table 1. Principal renal sites of prostaglandin synthesis and majoractions
a TxA2 reduces GFR in pathophysiologic situations such as glomeru-lonephritis, transplant rejection, and ureteral obstruction.
Adapted from [5].

Inhibition of renal prostaglandin synthesis 3
masking the continued synthesis and excretion of renal prosta-glandins.
Reliable etiteria allowing the assessment of the specificity ofurinary PG measurements by RIA include: a) characterizationof the thin—layer or high—pressure liquid chromatographic pat-tern of distribution of the extracted prostaglandin—like immuno-reactivity; b) use of multiple antisera; and c) comparison withmass spectrometric determinations [28, 36]. Such an integratedapproach can provide meaningful information on renal prosta-glandin and thromboxane synthesis in health and disease.
Finally, it should be pointed out that urinary prostaglandinsonly represent a fraction of total renal synthesis, with substan-tial amounts of prostaglandins degraded by or secreted into therenal venous blood. Furthermore, renal venous blood as well asurinary unmetabolized prostaglandins probably manifest anheterogeneous origin within the kidney, since no data areavailable to indicate a preferential secretion or excretion ofprostaglandins by any particular component of the nephron.Purely on the basis of differential eicosanoid production inisolated human glomeruli and papillae [40] it could be specu-lated that urinary 6-keto-PGF1a and TXB2 primarily reflect theglomerular synthesis of PG!2 and TXA2, respectively. How-ever, human medullary synthesis of PGI2 has been shown invitro [411. Urinary PGE2 may be largely a result of medullaryPGE2 production. The medullary origin of urinary PGE2 issupported by experiments showing 75% reductions of urinePGE2 after chemical inedullectomy induced by bromethylamine[42].
Renal prostaglandin—synthesis inhibition by NSAD in man
Drug induced inhibition of renal prostaglandin synthesis isthought to be reflected by a variably reduced excretion ofprimary unmetabolized prostaglandins [24—26]. The specificdetection of such pharmacologic changes is severely hamperedby a) the virtually ubiquitous distribution of the enzyme cyclo-oxygenase and b) the ratio of urinary eicosanoid metabolites toprimary prostaglandins being 10—100 to 1. Particularly, in thecase of a selective sparing of renal cyclooxygenase activity [30],the continued excretion of renal prostaglandins can be largelyor totally obscured in the face of a concomitant reduction in theelimination of extrarenal eicosanoid metabolites. Inasmuch asbiochemical measurements, putatively related to renal cyclo-oxygenase activity, are used to interpret drug—induced changesin renal function and to project clinical implications, it is ofutmost importance to establish some criteria for the validity ofsuch measurements. Short of providing definitive proof ofidentification by obtaining a full mass—spectrum of the urinaryeicosanoid of interest [25], validation criteria of radioim-munologic measurements should include the following:
1) a prerequisite is that such studies be conducted in femalesubjects inasmuch as primary prostaglandins may largelyderive from extra—renal (seminal) sources in male urine[27];
2) evidence that urinary prostaglandin—like immunoreactiv-ity is reduced by >50% following the acute and/or chronicadministration of a nonselective cyclooxygenase inhibitor(such as indomethacin) at antiinflammatory dosage;
3) evidence that extra-renal eicosanoid metabolites (such as,PGE-M, PGF-M, 2,3-dinor-6-keto-PGF1a, 2,3-dinor-TXB2) do not contribute to the apparent prostaglandin—
like immunoreactivity, through cross—reactivity studiesand/or comparison with an independent method of analy-sis [28, 36];
4) under appropriate pathophysiologic conditions, evidencefor a functional correlate (that is, changes in renal bloodflow, glomerular filtration rate, renin secretion and/orsodium chloride excretion) of reduced renal prostaglan-din—synthesis.
Most, if not all, widely used NSAD have been tested for theiracute and/or chronic effects on urinary prostaglandin excretion.These include indomethacin [43—46], ibuprofen [34, 43, 44, 47,48], naproxen [47—50], aspirin [33, 51], fenoprofen [47],diclofenac [49, 52], sulindac [32, 34, 45, 46, 48, 49, 50, 52—63],piroxicam [49], and flurbiprofen [52]. Their effects have beencharacterized in healthy subjects [32, 33, 45, 46, 48, 53, 63] aswell as in patients with renal [34, 43, 47, 51, 52, 55, 58], hepatic[44, 56, 57, 61, 62], rheumatic [49, 50] and cardiovascular [50,54, 59, 60] disease. Some generalizations can be drawn from theresults of such studies: a) with the possible exception ofsulindac (see below), all of these NSAD have been shown toreduce urinary prostaglandin excretion by at least 50% whenused at full antiinflammatory dosage; a maximal reduction in therange of 60 to 80% has been described with the vast majority ofthese agents, although no detailed dose—response studies havebeen carried out to allow a quantitative assessment of relativepotencies; b) such a maximal suppression of renal prostaglandinsynthesis can be shown to occur within 24 to 48 hours oftreatment, and is fully reversible within 48 to 72 hours after drugwithdrawal, depending upon the pharmacokinetics of the indi-vidual agent; c) the apparent reduction in urinary prostaglandinexcretion following a given cyclooxygenase inhibitor is notsubstantially modified by disease processes affecting renal orhepatic function, although the latter can obviously alter thepharmacokinetics of some NSAD; d) despite early reports ofpartial attenuation of NSAD-induced biochemical and func-tional changes during continued drug administration [47], sucha phenomenon has not been confirmed in controlled studies[34].
In 1980, Ciabattoni et al [32] reported that renal prostaglandinexcretion in healthy women and patients with Bartter's syn-drome was not decreased by sulindac, possibly because of itsredox pro-drug nature confining formation and/or access of itsactive sulfide metabolite to extra—renal sites of cyclooxygenaseactivity [65]. These preliminary observations were later ex-tended and confirmed by ourselves [34, 45, 49, 63] as well as byother investigators [46, 52, 54, 58—62]. However, several studiesin health [48, 53] as well as in disease [50, 55—57] have reportedconflicting evidence regarding the biochemical selectivity ofsulindac.
As clarification of this issue is relevant to the interpretation ofchanges in renal function and cardiovascular homeostasis asso-ciated with NSAD administration, we shall attempt to brieflyoutline the different views and factors possibly responsible forthe conflicting findings. In doing so, we shall review separatelythose studies that have measured urinary eicosanoid excretionin the basal state from those studies that have quantitated theeffect of sulindac during i.v. furosemide—enhanced diuresis. Anoverview of 18 studies (123 patients) falling in the formercategory, is presented in Table 2. Obviously, two differentviews have been expressed by the investigators involved in
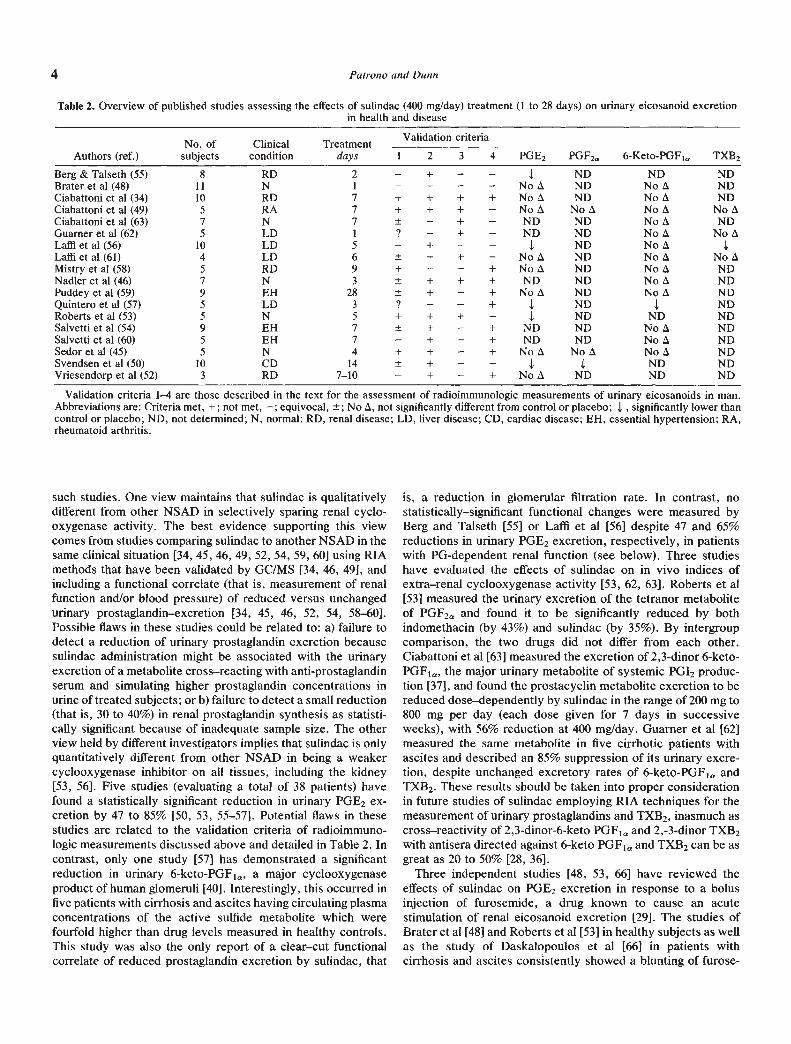
4 Patrono and Dunn
Table 2. Overview of published studies assessing the effects of sulindac (400 mg/day) treatment (ito 28 days) on urinary eicosanoid excretionin health and disease
Authors (ref.)No. ofsubjects
.Clinical
conditionTreatment
days
Validation criteria
PGE2 PGF2. 6-Keto-PGF1, TXB21 2 3 4
Berg & Talseth (55) 8 RD 2 — + — — ND ND NDBrater et al (48) 11 N I — — — — No A ND No A NDCiabattoni et al (34) 10 RD 7 + + + + No A ND No A NDCiabattoni et al (49) 5 RA 7 + + + No A No A No A No ACiabattoni et al (63) 7 N 7 ND ND No A NDGuarner et al (62) 5 LD I ? — + — ND ND No A No ALaffi et a! (56) 10 LD 5 — + — — ND No ALaffi et a! (61) 4 LD 6 — + — No A ND No A No AMistry et al (58) 5 RD 9 + — — + No A ND No A NDNadler et al (46) 7 N 3 ND ND No A NDPuddey et a! (59) 9 EH 28 — + No A ND No A NDQuintero et al (57) 5 LD 3 ? — — + I ND I NDRoberts et al (53) 5 N 5 + + + — I ND ND NDSalvetti et a! (54) 9 EH 7 — + ND ND No A NDSalvetti et al (60) 5 EH 7 — + — -I- ND ND No A NDSedor et a! (45) 5 N 4 + + — + No A No A No A NDSvendsen et al (50) 10 CD 14 — — ND NDVriesendorp et al (52) 3 RD 7—10 — + — + No A ND ND ND
Validation criteria 1—4 are those described in the text for the assessment of radioimmunologic measurements of urinary eicosanoids in man.Abbreviations are: Criteria met, +; not met, equivocal, No A, not significantly different from control or placebo; I , significantly lower thancontrol or placebo; ND, not determined; N, normal; RD. renal disease; LD, liver disease; CD, cardiac disease; EH, essential hypertension; RA,rheumatoid arthritis.
such studies. One view maintains that sulindac is qualitativelydifferent from other NSAD in selectively sparing renal cyclo-oxygenase activity. The best evidence supporting this viewcomes from studies comparing sulindac to another NSAD in thesame clinical situation [34, 45, 46, 49, 52, 54, 59, 60] using RIAmethods that have been validated by GC/MS [34, 46, 49], andincluding a functional correlate (that is, measurement of renalfunction and/or blood pressure) of reduced versus unchangedurinary prostaglandin—excretion [34, 45, 46, 52, 54, 58—60].Possible flaws in these studies could be related to: a) failure todetect a reduction of urinary prostaglandin excretion becausesulindac administration might be associated with the urinaryexcretion of a metabolite cross—reacting with anti-prostaglandinserum and simulating higher prostaglandin concentrations inurine of treated subjects; or b) failure to detect a small reduction(that is, 30 to 40%) in renal prostaglandin synthesis as statisti-cally significant because of inadequate sample size. The otherview held by different investigators implies that sulindac is onlyquantitatively different from other NSAD in being a weakercyclooxygenase inhibitor on all tissues, including the kidney[53, 56]. Five studies (evaluating a total of 38 patients) havefound a statistically significant reduction in urinary PGE2 ex-cretion by 47 to 85% [50, 53, 55—57]. Potential flaws in thesestudies are related to the validation criteria of radioimmuno-logic measurements discussed above and detailed in Table 2. Incontrast, only one study [57] has demonstrated a significantreduction in urinary 6-keto-PGF1a, a major cyclooxygenaseproduct of human glomeruli [40]. Interestingly, this occurred infive patients with cirrhosis and ascites having circulating plasmaconcentrations of the active sulfide metabolite which werefourfold higher than drug levels measured in healthy controls.This study was also the only report of a clear—cut functionalcorrelate of reduced prostaglandin excretion by sulindac, that
is, a reduction in glomerular filtration rate. In contrast, nostatistically—significant functional changes were measured byBerg and Talseth [55] or Laffi et al [56] despite 47 and 65%reductions in urinary PGE2 excretion, respectively, in patientswith PG-dependent renal function (see below). Three studieshave evaluated the effects of sulindac on in vivo indices ofextra—renal cyclooxygenase activity [53, 62, 63]. Roberts et al[53] measured the urinary excretion of the tetranor metaboliteof PGF2,, and found it to be significantly reduced by bothindomethacin (by 43%) and sulindac (by 35%). By intergroupcomparison, the two drugs did not differ from each other.Ciabattoni et al [63] measured the excretion of 2,3-dinor 6-keto-PGF1, the major urinary metabolite of systemic PG!2 produc-tion [37], and found the prostacyclin metabolite excretion to bereduced dose—dependently by sulindac in the range of 200 ing to800 mg per day (each dose given for 7 days in successiveweeks), with 56% reduction at 400 mg/day. Guarner et al [62]measured the same metabolite in five cirrhotic patients withascites and described an 85% suppression of its urinary excre-tion, despite unchanged excretory rates of 6-keto-PGF1,,. andTXB2. These results should be taken into proper considerationin future studies of sulindac employing RIA techniques for themeasurement of urinary prostaglandins and TXB2, inasmuch ascross—reactivity of 2,3-dinor-6-keto PGF1a and 2,-3-dinor TXB2with antisera directed against 6-keto PGF1,. and TXB2 can be asgreat as 20 to 50% [28, 36].
Three independent studies [48, 53, 66] have reviewed theeffects of sulindac on PGE2 excretion in response to a bolusinjection of furosemide, a drug known to cause an acutestimulation of renal eicosanoid excretion [291. The studies ofBrater et al [48] and Roberts et a! [53] in healthy subjects as wellas the study of Daskalopoulos et al [66] in patients withcirrhosis and ascites consistently showed a blunting of furose-

inhibition of renal prostaglandin synthesis 5
mide—enhanced POE2 excretion after sulindac. Althoughmerely a theoretical speculation, the above findings could beconsistent with a differential oxidation of the sulfide metaboliteof sulindac in the medulla as compared to the cortex, therebyexposing medullary sites (primarily synthesizing PGE2) but notcortical sites (primarily synthesizing PGI2) of cyclooxygenaseactivity to effective concentrations of the biologically activeform of the drug. The mechanism(s) responsible for the allegedbiochemical selectivity of sulindac is still poorly understood. Itis not related to a differential sensitivity of the renal cyclooxy-genase to the drug, since in vitro comparison of sulindac sulfideand indomethacin using renal medullary interstitial cells andpapillary collecting tubule cells in culture showed sulindacsulfide to be equipotent to indomethacin as an inhibitor of POE2synthesis [64]. Differential inhibition of renal and extra—renalprostaglandin synthesis might be related to the redox pro-drugnature of sulindac [65], as suggested by similar observationsmade with sulfinpyrazone [67]. Animal studies [68—73] haveyielded similarly conflicting results, depending upon the animalspecies, experimental design, dosage and route of administra-tion of sulindac. The study by Miller, Bednar and McGiff [74] inthe isolated perfused kidney of the rabbit and rat has estab-lished that: a) the kidney possesses the metabolic pathways forthe interconversion of sulindac sulfoxide and sulfide as well assulfone formation; b) whereas all three forms exist in renaltissue, the sulfide is inactivated before its release into venous orurinary effluents; c) administration of either sulindac sulfide orsulfoxide can result in effective cyclooxygenase inhibition.
Thus, to summarize our own experience and personal readingof the current literature on this rather controversial issue: 1)when given to human subjects at the recommended therapeuticdose of 400 mg/day, sulindac does not appear to influencecortical sites of renal cyclooxygenase activity, as reflected byurinary 6-keto-PGF,a, with the possible exception of patientswith cirrhosis and ascites; 2) under the same circumstances, thedrug may affect medullary sites of cyclooxygenase activity, asreflected by urinary POE2, in approximately one third of thecases thus far evaluated; 3) the drug does appear to blunt i.v.furosemide—induced PGE2 release; 4) the mechanism(s) under-lying these peculiar features of sulindac is largely unknown,although likely related to its redox pro-drug nature.
A different mechanism is responsible for the selective sparingof renal cyclooxygenase activity by low—dose aspirin [33]. Themechanism underlying this selectivity is probably related to adifferent rate of recovery of cyclooxygenase activity in glomer-uli versus platelets, following irreversible acetylation of theenzyme, and possibly to a different aspirin "sensitivity" ofglomerular cyclooxygenase. The finding of a normal pattern offurosemide—induced renin release and urinary 6-keto-PGF1,excretion after three to four weeks of low—dose (0.45 mg/kg/day) aspirin treatment in healthy subjects [33] indicates thatrenal PGI2-producing cells are readily activatable at a time ofvirtually complete suppression of platelet cyclooxygenase ac-tivity. Thus, low-dose aspirin represents an ideal pharmacolog-ic tool to investigate the role of intraglomerular platelet activa-tion in the progression of chronic glomerular disease [75]without the risk inherent to higher conventional dosage whichmight compromise prostaglandin—dependent renal function.
The effects of prostaglandins and nonsteroidal antiinflammatorydrugs on renal cortical and medullary functions
Renal function is not dependent upon the integrity ofprostaglandin synthesis, at least under ordinary circumstances.Inhibition of prostaglandin synthesis in normal animals andhumans does not induce a significant decline of renal function[1]. It is probable that basal, endogenous prostaglandin synthe-sis does contribute to control of renal vascular resistance,glomerular filtration and salt excretion in healthy subjects, butadministration of NSAD does not alter renal function sinceother regulatory mechanisms (adrenergic tone, renin secretion,dopamine, adenosine) are unstressed and can compensate forthe inhibition of prostaglandins [5]. In animals under anesthe-sia, especially after laparotomy, acute inhibition of prostaglan-din synthesis reduces renal blood flow substantially, althoughglomerular filtration rate is largely unaffected [76]. Differentresponses of anesthetized surgically—stressed dogs and con-scious animals to the acute inhibition of prostaglandin synthesisare partially attributable to the effects of anesthesia and surgeryon major vasoconstrictor hormones, especially angiotensin II,vasopressin, and catecholamines. In experimental and clinicalcircumstances, in which these vasoconstrictor hormones areincreased, renal prostaglandin synthesis is augmented and renalfunction becomes "prostaglandin dependent" [5]. Infusion ofangiotensin II, vasopressin, or norepinephrine into the renalartery stimulates renal prostaglandin synthesis measured eitheras secretion into renal venous blood or excretion into the urine[77—81]. In vitro studies have confirmed that angiotensin andvasopressin stimulate vasodilatory prostaglandin release inglomerular mesangial and epithelial cells, and renal medullaryinterstitial cells [19—21, 82, 83]. Pretreatment with nonsteroidalantiinflammatory agents potentiated the renal vasoconstrictioninduced by angiotensin II, alpha-adrenergic nerve stimulation,and norepinephrine or alpha adrenergic drugs [S4—89]. Theseexperiments reinforce the belief that the vasoconstrictor sub-stances stimulated renal cortical and medullary POE2 and P012which modulated the constrictor action of the angiotensin ornorepinephrine. The major clinical circumstances in whichvasoconstrictor hormones increase to maintain cardiovascularhomeostasis include extracellular volume depletion, congestiveheart failure, nephrotic syndrome and hepatic disease.
Diseases such as arteriosclerotic cardiovascular disease andchronic glomerular disease (including lupus nephritis) mayreduce renal POE2 or P012 [34, 36] and thereby enhance thesusceptibility to NSAD despite normal levels of constrictorhormones. Figure 2 summarizes our hypothesis about themodulating effect of renal POE2 and P012 on the intrarenalconstrictor action of angiotensin II, vasopressin, and norepi-nephrine. According to this hypothesis, drug— and/or disease—induced suppression of cortical prostaglandin synthesis is asso-ciated with a lower threshold for the contractile response of theglomerular mesangium and arterioles to a variety of agonists.This simplified scheme does not include the important possibil-ity that glomerular and vascular PGE2 and P012 may antagonizethe renal effects of diverse contractile compounds, such asplatelet activating factor, thromboxane A2, leukotriene C4-D4,and other vasoconstrictor agents. Perhaps more prevalent thanthe acute changes in glomerular filtration rate and renal bloodflow observed above is NSAD-induced sodium retention.

6 Patrono and Dunn
Constrictor hormone
Fig. 2. Scheme depicting the balance between vasoconstriction andvasodilatation existing in the pre- and post-glomerular circulation, andthe glonierular Inesangium. Abbreviations include: arterioscleroticcardiovascular disease (ASCVD); chronic glomerulonephritis (CGN);systemic lupus erythematosus (SLE); congestive heart failure (CHF);angiotensin II (ANG LI); nonsteroidal antiinflammatory drugs (NSAD).The graph schematically depicts the enhanced arteriolar and mesangialcontraction to constrictor hormones after inhibition of prostaglandinsynthesis.
Edema has been reported to occur in approximately 25% ofpatients with arthritis who were receiving either aspirin orfenoprofen and in about 10% of patients who were givenibuprofen [2]. The mechanisms underlying the retention ofsodium associated with NSAD administration are probablymultifactorial and have been discussed previously [1—5]. Waterretention that is disproportionate to sodium retention may leadto the development of hyponatremia in some patients, particu-larly as a result of the combined use of NSAD and thiazidescausing an additive effect on free—water excretion, as empha-sized by Clive and Stoff [2]. In addition, NSAD may producehyperkalemia by inducing hyporeninemic hypoaldosteronism asfirst shown by Tan et a! [90]. In a prospective study of 50patients requiring indomethacin treatment for various medicalreasons, 13 patients developed an increase in serum potassiumconcentration equal to or greater than 1.0 mM/liter within threeto seven days of treatment [91]. These patients were character-ized as being elderly (68 12 yrs) and with a relatively high(6 1%) prevalence of pre-existing mild to moderate azotemia.The vast majority of clinical studies published prior to 1985have been discussed extensively in previous reviews of thesubject [1—4]. It is important to realize that NSAD-inducedchanges in cortical and/or medullary functions have been doc-umented in short—term studies, that is, one day to four weeks oftreatment (5 to 7 days in most studies). The long—term conse-quences of chronic administration of renal prostaglandin syn-thesis inhibitors are largely unknown. In the next section, wewill review clinical and experimental studies published duringthe last two years, which have primarily focused on thecomparative evaluation of sulindac versus other NSAD inhealth and disease.
The effects of sulindac on renal function: Experimental andclinical studies
Interest in a possible renal—sparing effect of sulindac can beattributed to the clinical prevalence of acute renal failure as acomplication of nonsteroidal antiinflammatory drug therapy. Amajority of studies, summarized below, show negligible deteri-oration of renal function when sulindac is administered orally inconventional doses. In normal volunteers, glomerular filtrationrate, measured as either creatinine clearance or inulin clear-ance, was unaffected by sulindac [32, 45, 53, 92]. These resultsin normal volunteers are not surprising as other nonsteroidalantiinflammatory drugs, such as indomethacin and ibuprofen,also have no effect on GFR in normal volunteers, despitesubstantial inhibition of renal prostaglandin synthesis [1]. Sedoret al also did not observe any reductions of urine Na or Cl aftersulindac administration, whereas indomethacin substantiallyreduced salt excretion in normal volunteers [45].
A possible renal—sparing action of sulindac has been bestdemonstrated in patients with liver disease or chronic renalfailure. Patients with alcoholic cirrhosis and ascites treated witheither indomethacin or sulindac have very different renal func-tional responses to these NSAD. Daskalopoulous et al [66]administered indomethacin, 50 mg two hours and eight hoursprior to study and found reduced glomerular filtration rate byapproximately 50% accompanied by substantial decrements ofurinary PGE2. In similar cirrhotic patients, sulindac, 150 mgtwo and eight hours prior to evaluation, reduced glomerularfiltration rate insignificantly (15%) with no changes in urinaryprostaglandin excretion. Despite the absence of any sulin-dac—induced decrement of renal function in these experiments,sulindac as well as indomethacin antagonized furosemide—stimulated prostaglandin excretion and furosemide—inducednatriuresis and diuresis [66]. Laffi and coworkers studied tenpatients with alcoholic cirrhosis and ascites in a comparison ofsulindac 200 mg twice daily for five days, followed by ibuprofen400 mg every eight hours for one day [56]. Sulindac did notreduce glomerular filtration rate or renal plasma flow whereasibuprofen induced substantial decrements of these renal hemo-dynamic parameters in five of the ten cases. Quintero et al [57]have obtained contrasting results as they observed reductionsof glomerular filtration rate and prostaglandin excretion of 39%and 78% in five patients with cirrhosis and ascites who receivedsulindac 400 mg a day for three days. These workers noted, asdid Laffi et al, that patients with liver disease have significantlyincreased plasma levels of the active component of sulindac,sulindac sulfide. These observations of 2.5 [56] to fourfold [57]increases in plasma sulindac sulfide levels, after conventionaloral doses to cirrhotic patients, confirm the earlier work of Juhlet al [93] who found that the area under the curve for plasmasulindac sulfide increased fourfold in patients with poor hepaticfunction. These results raise a note of caution about theadministration of sulindac, particularly in conventional doses,to patients with severe hepatic dysfunction. Zambraski andcoworkers, studying dogs with biliary cirrhosis and portalhypertension, also documented high plasma sulindac sulfidelevels for prolonged periods of time after intravenous adminis-tration of sulindac [68]. In these animals, who had chronic bileduct ligation, sulindac or sulindac sulfide acutely decreasedGFR and RPF.
Afferentarteriole
Vasopressin
Ang IICatecholeminesVasopressin
C0U
C00

Inhibition of renal prostaglandin synthesis 7
Patients with mild to moderate chronic renal failure (GFR of90 21 mI/mm) have been compared for their responses toNSAD by Ciabattoni and coworkers [34]. Twenty women withchronic glomerular disease were treated with either ibuprofenor sulindac for seven days. Whereas ibuprofen reduced glomer-ular filtration rate and renal plasma flow by 35%, sulindac hadno significant effect on renal function or prostaglandin excretion[34]. These results have been confirmed by others. In patientswith greater reductions of GFR (37 to 41 ml/min) indomethacinbut not sulindac reduced filtration 25% and decreased PGE2excretion to a greater extent [55]. Swainson and Griflithsevaluated six patients with chronic renal disease and basalglomerular filtration rates of 18 to 48 ml/min [92]. Twenty—eightdays of treatment with sulindac, 600 mg per day, inducednegligible (12%) or no changes in renal plasma flow and glomer-ular filtration rate, respectively. As one patient did have greaterdecrements of renal plasma flow and glomerular filtration rate,the authors conclude that although sulindac is relatively safe, anoccasional patient may develop acute renal failure [92]. Acutereversible decline of renal function, attributable to sulindac, hasbeen reported in only two other cases. Blackshear et al [3, 94]described an 81 year—old woman with hypertension and atrialfibrillation, receiving diuretic therapy, who was treated withsulindac 800 mg over 24 hours. She suffered reversible in-creases in blood urea nitrogen and serum creatinine. Corwinand Bonventre reported the case of a 79 year—old woman,treated with sulindac 150 mg twice daily, with concomitantproblems of congestive heart failure, hypertension andadult—onset diabetes mellitus [95]. Renal function declinedsubstantially and, with cessation of sulindac therapy, the pa-tient recovered over 10 days. These cases highlight the possi-bility that elderly patients with cardiovascular and renal diseasemay develop acute renal failure despite any relative, renal—spar-ing effect of sulindac. Despite pretreatment GFR of 37 mllmin,nine patients with chronic renal failure studied by Mistry et al[58] had only 10% decrements of GFR and no changes of RPFafter nine days therapy with sulindac. Consistent with humanstudies, the administration of sulindac (30 mg/kg over 48 hours)to 5/6 nephrectomized rats had no effect on renal function,electrolyte excretion or renal PGE2 production [691. In con-trast, indomethacin dramatically reduced the urinary excretionof Na and the fractional excretion of Na in these animals,concomitantly with a marked suppression of renal PGE2 pro-duction [69]. Other reports do not confirm that sulindac has noeffects on renal prostaglandin synthesis or GFR in experimentalanimals. Sulindac, compared to indomethacin and naproxen,reduced urine PGE2, free water clearance, and GFR to a similarextent in sodium—depleted rats [71]. Measurements of renalvenous 6-keto-PGF1 in rats after endotoxin [70] or dogs afterhemorrhagic hypotension [72] also demonstrated inhibition ofPG!2 synthesis after parenteral administration of sulindac.Henrich, Brater and Campbell, using the model of hemorrhagichypotension in the dog, measured comparable reductions ofGFR, RBF and renal venous 6-keto-PGF10, in animals receivingsulindac, indomethacin or benoxaprofen intravenously [72].These results are in agreement with the earlier work byZambraski, Chremos and Dunn showing that intravenoussulindac (sulfide or sulfoxide) acutely reduced renal functionand prostaglandin synthesis in chronic bile duct—ligated dogs[681. It is noteworthy that all of the aforementioned experimen-
tal studies utilized parenteral rather than oral routes of therapy.Additional prospective clinical studies using a double—blindcrossover design in patients with significant renal insufficiencyare needed to unequivocally answer the question of renalsparing by sulindac.
It is well established that indomethacin reduces proteinuriaand enhances the selectivity of the proteinuria in patients withthe nephrotic syndrome [96]. These effects of indomethacinmay be secondary to inhibition of renal cortical prostaglandinsynthesis, accompanied by reductions of glomerular hydro-static pressure. De Zeeuw and coworkers have compared fourdifferent NSAD, including sulindac, for their therapeutic effi-cacy to decrease proteinuria in nephrotic patients [52]. Consist-ent with a lack of prostaglandin inhibition, sulindac did notreduce proteinuria whereas indomethacin, diclofenac andflurbiprofen reduced proteinuria and PGE2 excretion 50 to 75%.Reductions of proteinuria and glomerular filtration rate aftertreatment with these three nonsteroidal agents correlated highlywith reduced POE2 excretion [52]. These results reinforce thebelief that sulindac has minimal capacity to inhibit renal glomer-ular and cortical vascular eicosanoid—synthesis.
Does inhibition of renal prostaglandin synthesis blunt thenatriuretic response to furosemide?
When injected intravenously into healthy subjects, furose-mide acutely enhances the urinary excretion of unmetabolizedprostaglandins and TXB2 [29] with no change in urinary 2,3-dinor-6-keto-PGF1a [97], consistent with a localized intrarenalenhancement of arachidonate availability. However, whengiven orally the same drug produces a marked increase ofsodium excretion without increasing urinary prostaglandin ex-cretion both in female volunteers and patients with essentialhypertension [98]. Reports of the effects of prostaglan-din—synthesis inhibition on furosemide—induced natriuresis arealso conflicting, with some studies demonstrating up to 30%blunting of the natriuretic response whereas in others therewere no significant effects [99]. As salt balance is an importantvariable influencing the overall effects of cyclooxygenase inhib-itors [100], it is not surprising to find varying results in theliterature in which salt balance has not been controlled. In dogs,the effects of furosemide on "cortical events," that is, renalvasodilation and renin release can be dissociated from thenatriuretic effect on the basis of a different time—course anddose—requirement [101]. Only renal blood flow and renin secre-tion correlate with enhanced renal PGI2 production, while thenatriuretic response has a different time course and can bedemonstrated at lower doses of the drug which do not alterrenal PG!2 synthesis [101]. Thus, furosemide—induced natriure-sis is probably not prostaglandin—mediated, in contrast toincreases in renin release and renal blood flow. Riley et alreached a similar conclusion by showing that, in healthy men ona 100 mEq Na diet, the natriuretic response to intravenousfurosemide was not inhibited by either ibuprofen or sulindac,whereas both drugs reduced the stimulation of renin release[102]. These authors also concluded, since renin secretion wassuppressed by sulindac, that this NSAD was not "renal spar-ing." Further animal work suggests that indomethacin reducesthe diuretic and natriuretic responses to furosemide because ofan effect on furosemide—induced renal vasodilation rather than

8 Patrono and Dunn
an effect on tubular function or on delivery of the drug to theascending limb of Henle [100]. A similar conclusion has beenreached on the basis of human studies [103]. In the recent studyby Brater et al [48], however, naproxen, sulindac and ibuprofenall significantly decreased by 14 to 34% the maximal natriureticresponse to an intravenous furosemide injection in healthy men,despite no significant changes in urinary 6-keto-PGF1 excre-tion and renal plasma flow. Although neither total amounts offurosemide nor the time course of its delivery into urine wereaffected by these NSAD, an alteration of early (first 10 to 15mm) delivery of the drug to its tubular site of action could notbe excluded. In fact, only a very small change in the initialdelivery of furosemide to the ascending limb of Henle couldprobably explain the observed effects on sodium excretion. Theclinical relevance of such transient acute effects of NSAD to themaintenance of sodium excretion during chronic diuretic ther-apy with oral furosemide has probably been overemphasized.
Does inhibition of renal prostaglandin synthesis attenuate theantihypertensive effect of' concomitant antihypertensive
therapy?
The action of prostaglandins on blood vessels and the role ofprostaglandins in arterial hypertension have been reviewedrecently [1041. Probably renal as well as extra—renal vascularsites of cyclooxygenase activity participate in the regulation ofblood pressure, through the release of various eicosanoidsdirectly acting on vascular smooth—muscle function and/orindirectly affecting its response to vasopressor substances[104].
No consistent effects of orally administered indomethacin onblood pressure of healthy subjects have been described [104].However, when given by i.v., indomethacin resulted in aprompt increase in both systolic and diastolic blood pressure[105], an effect possibly related to a more rapid and profoundsuppression of cyclooxygenase activity and/or to its transientnature being closely monitored. As no prostaglandin measure-ments were reported in the vast majority of studies, theapparent discrepancies among different investigations are diffi-cult to reconcile. In three different studies, indomethacin wasconsistently shown to enhance the pressor response to angio-tensin II in normal man [106—108]. Most likely, the integrity ofother cardiovascular regulatory mechanisms largely accountsfor the lack of measurable changes in blood pressure associatedwith prostaglandin synthesis inhibition in healthy subjects.
When administered to patients with essential hypertensionbeing treated with various antihypertensive agents, indometh-acm has been shown consistently to increase both systolic anddiastolic blood pressure by 5 to 15 mm Hg [104]. The extent andduration of such an effect varied in different investigations, anda few studies have described an attenuation of the hypertensiveeffect of indomethacin upon continued treatment [59, 109].However, no study had assessed the long—term consequences(>6 weeks) of such a pharmacologic interaction. Drugs whoseantihypertensive effect appeared to be attenuated by concomi-tant administration of indomethacin include beta—adrenergicblocking agents, thiazide diuretics, converting enzyme inhibi-tors and various combinations of the above, thus making itunlikely that such phenomenon reflects a specific pharmacoki-netic interaction.
Several distinct mechanisms might account for the observedeffects of indomethacin: a) enhanced sodium retention causingan increase of plasma volume; b) enhanced renal vascularresistance; c) altered pressor responsiveness to angiotensin IIand/or noradrenaline; d) an inherent vasoconstrictor effect ofthe drug. Although the first three mechanisms provide plausibleexplanations and are compatible with reduced renal and/orextra—renal PGI2 production, the fourth explanation should bealso considered in the light of recently—demonstrated vasocon-strictor effects of indomethacin in the human cerebral [110] andcoronary [111] circulation, which are not shared by othercyclooxygenase inhibitors. In comparative studies with indo-methacin, neither aspirin (1,950 mg/day) nor naproxen (500mg/day) significantly affected blood pressure control in treatedhypertensives [109, 112]. In contrast, piroxicam (20 mg/day)was recently shown to blunt the antihypertensive effect ofpropranolol in mild to moderate hypertensives [1131. Inasmuchas no information on the effects of the different drugs onprostaglandin synthesis was provided in these studies, no firmconclusion can be reached on the involvement of renal and/orsystemic prostaglandin—synthesis inhibition in the observedchanges.
Several recent studies have demonstrated that sulindac (400mg/day) does not attenuate the hypotensive effects of thiazides[109, 114], beta—adrenergic blocking agents [54, 113], captopril[601 or various combination regimens [59, 114—116]. In the studyby Puddey et al [59] and in the studies by Salvetti et al [54, 60]indomethacin significantly reduced urinary PGE2 and/or 6-keto-PGF1, excretion while sulindac had no effect. Wong et alcompared placebo, sulindac, naproxen and piroxicam in 20treated hypertensive patients and found significantly higherdiastolic blood pressure and lower urine 6-keto-PGF1a afternaproxen or piroxicam therapy for four weeks, compared tosulindac [115]. Inasmuch as sulindac does inhibit extra—renalPGI2 production, as assessed by a dose—dependent reduction ofurinary 2,3-dinor-6-keto-PGF1a excretion during chronic dosing[631, its lack of hypertensive effect vis-a-vis other cyclooxygen-ase inhibitors (that is, indomethacin and piroxicam) wouldargue in favor of the importance of intrarenal mechanisms a)and b), outlined above, in mediating the hemodynamic effects ofnonselective inhibitors. This conclusion is supported by therecent observations of Trimarco et al [117] that indomethacin aswell as ibuprofen, but not sulindac, increase blood pressure inuntreated hypertensive patients in response to a salt load. Thisstudy suggests that renal prostaglandins participate in the renaldisposal of a chronic salt load in hypertensive patients to agreater extent than in healthy subjects.
Clearly, further investigations are needed in order to clarifysome of the unresolved issues raised by these studies. Incor-poration of biochemical as well as functional measurements intofuture experiments and use of different cyclooxygenase inhibi-tors should help to better define the role of renal prostaglandinsin blood pressure regulation.
Conclusions
Integrity of renal prostaglandin synthesis is necessary tomaintain renal cortical and medullary function in patients withdiverse types of disease. A comparison of the biochemicaleffects of various nonsteroidal antiinflammatory drugs necessi-tates that renal prostaglandin synthesis, as reflected by urinary

Inhibition of renal prostaglandin synthesis 9
immunoreactive prostaglandin excretion, be assessed withproper attention to problems created by seminal fluid contam-ination, inadequate chromatographic separation of samples,and largely unknown cross reactivities of urinary eicosanoidmetabolites with most available antibodies. Most, but not all,clinical studies support the observation that conventional dosesof sulindac, administered orally, do not inhibit renal prostaglan-din synthesis or alter renal function. Caution is in order,however, about the use of any nonsteroidal antiinflammatorydrug, including sulindac at conventional dosage, in patientswith substantial hepatic disease as the plasma levels of sulindacsulfide are increased and prolonged compared to patients withnormal liver function. Some NSAD attenuate the efficacy ofantihypertensive therapy whereas sulindac, despite inhibitionof systemic vascular PGI2 production, does not antagonize theantihypertensive drugs. These observations point to intrarenal,PG-dependent mechanisms, such as NaC1 excretion, in medi-ating the hemodynamic, that is, prohypertensive effects ofnon-selective NSAD.
CARLO PATRONO AND MICHAEL J. DUNNCatholic University of Rome, Rome italy
andUniversity Hospitals of Cleveland, Cleveland, Ohio, USA
AcknowledgmentsWe wish to thank Ms. Norma Minear for help in preparing the
manuscript.
Reprint requests to Dr. Carlo Patrono, Department of Pharmacol-ogy, Catholic University School of Medicine, Largo Francesco Vito 1,00168 Rome, Italy.
References
I. DUNN MJ: Nonsteroidal antiinflammatory drugs and renal func-tion. Ann Rev Med 35:411—428, 1984
2. CLIVE DM, STOFF JS: Renal syndromes associated withnonsteroidal antiinflammatory drugs. N EngI J Med 310:563—572,1984
3. BLACKSHEAR JL, NAPIER JS, DAVIDMAN M, STILLMAN MT:Renal complications of nonsteroidal antiinflammatory drugs: iden-tification and monitoring of those at risk. Sem Arth Rheum14:163—175, 1985
4. CARMICHAEL J, SFIANKEL SW: Effect of nonsteroidal antiinflam-matory drugs on prostaglandins and renal function. Am J Med78:992—1000, 1985
5. DUNN MJ: Renal prostaglandins, in Renal Endocrinology, editedby DUNN MJ. Baltimore, Williams and Wilkins, 1983, pp. 1—74
6. METZ SA: Antiinflammatory agents as inhibitors of prostaglandinsynthesis in man. Med Clin N Am 65:713—756, 1981
7. FLOWER RJ: The mediators of steroid action. (abstract) Nature320:20, 1986
8. BOMBARDIERI 5, CATTANI P, CIABATTONI G, DI MUNNO 0,PASERO GP, PATRONO C, PINCA E, PUGLIESE F: The synovialprostaglandin system in chronic inflammatory arthritis. Differen-tial effects of steroidal and nonsteroidal anti-inflammatory drugs.Br J Pharmacol 73:893—897, 1981
9. NARAY—FEJES—TOTH A, FEJES—TOTH G, FISCHER C, FROLICH JC:Effect of dexamethasone on in vivo prostanoid production in therabbit. J Clin Invest 74:120—123, 1984
10. GOODWIN JS, ATIURU D, SIERAKOwSKL 5, Lftos EA: Mecha-nism of action of glucocorticosteroids. Inhibition of T cell prolif-eration and interleukin 2 production by hydrocortisone is reversedby leukotriene B4. J Clin Invest 77:1244—1250, 1986
11. LARSSON C, ANGGARD E: Regional differences in the formationand metabolism of prostaglandins in the rabbit kidney. Eur JPharmacol 21:30—36, 1973
12. SMITH WL, WILKIN GP: Immunochemistry of prostaglandinendoperoxide—forming cyclooxygenases: The detection of thecyclooxygenases in rat, rabbit and guinea pig kidneys by im-munofluorescence. Prostaglandins 13:873—892, 1977
13. SMITH WL, BELL TG: Immunohistochemical localization of theprostaglandin—forming cyclooxygenase in renal cortex. Am JPhysiol 235:F451—F457, 1978
14. M0RI Y, MINE M: The localization of prostaglandins in the rabbitkidneys demonstrated with indirect immunofluorescence. Biomed-ical Res 2:281—284, 1981
15. HASSID A, KoNIEczKowsKI M, DUNN Mi: Prostaglandin synthe-sis in isolated rat kidney glomeruli. Proc Nail Acad Sci USA76:1155—1159, 1979
16. SRAER J, SRAER JD, CIIANSEL D, Russo MARIE F, KOUZNETZOvAB, ARDAILLOU R: Prostaglandin synthesis by isolated rat renalglomeruli. Mol Cell Endocrinol 16:29—37, 1979
17. FOLKERT VW, SCHLONDORFF D: Prostaglandin synthesis in iso-lated glomeruli. Prostaglandins 17:79—86, 1979
18. SRAER i, FOIDART J, CHANSEL D, MAHIEU P. KOUZNETZ0VA B,ARDAILLOU R: Prostaglandin synthesis by mesangial and epithe-hal glomerular cultured cells. FEBS Let! 104:420—424, 1979
19. PETRULUS AS, AIKAWA M, DUNN MJ: Prostaglandin and throm-boxane synthesis by rat glomerular epithehial cells. Kidney In!20:469—474, 1981
20. SCHARSCHMIDT L, DUNN MJ: Prostaglandin synthesis by ratglomerular mesangial cells in culture. The effects of angiotensin IIand arginine vasopressin. J Clin Invest 71:1756—1764, 1983
21. BECKTR, HASSID A, DUNN Mi: The effect of arginine vasopressinand its analogs on the synthesis of prostaglandin £2 by rat renalmedullary interstitial cells in culture. J Pharmacol Exp Ther215:15—19, 1980
22. GRENIER FC, ROLLINS TE, SMITH WL: Kinin—induced prosta-glandin synthesis by renal papillary collecting tubule cells inculture. Am J Physiol 241:F94—F104, 1981
23. SCHLONDORFF D: Renal prostaglandin synthesis: Sites of produc-tion and specific actions of prostaglandins. Am J Med 81:1—11, 1986
24. PATRONO C, PUGLIESE F: The involvement of arachidonic acidmetabolism in the control of renin release. J Endocrinol In vest3:193—201, 1980
25. FROLICH JC, WILSON TW, SWEETMAN BJ, SMIGEL M, NIEs AS,CARR K, WATSON iT, OATES JA: Urinary prostaglandins; identi-fication and origin. J Clin Invest 55:763—770, 1975
26. PUGLIESE F, CIABATTONI G: Investigations of renal arachidonicacid metabolites by radioimmunoassay, Prostaglandins and theKidney: Biochemistry, Physiology, Pharmacology and ClinicalApplications, edited by MJ DUNN, C PATRONO, GA CINOTrI.New York, Plenum Press, 1983, pp. 83—98
27. PATRONO C, WENNMALM A, CIABATTONI G, NOWAK J, PUGLIESEF, CIN0rTI GA: Evidence for an extrarenal origin of urinaryprostaglandin E2 in healthy men. Prostaglandins 18:623—629, 1979
28. PATRONO C, PUGLIESE F, CIABATTONI G, PATRIGNANI P, MASERIA, CHIERCHIA 5, PESKAR BA, CIN0TTI GA, SIMONETTI BM,PIERUCCI A: Evidence for a direct stimulatory effect of prosta-cycline on renin release in man. J Clin Invest 69:231—239, 1982
29. CIABATTONI G, PUGLIESE F, CINOTTI GA, STIRATI G, R0NcI R,CASTRUCCI G, PIERUCCI A, PATRONO C: Characterization offurosemide—induced activation of the renal prostaglandin system.EurJ Pharmacol 60:181—187, 1979
30. PATRONO C: Tissue—selective inhibition of prostaglandin andthromboxane synthesis in man: Investigative and therapeuticimplications. Clin Physiol 4:443—447, 1984
31. PATRONO C, CIABATTONI G, PATRIGNANI P, FILABOZZI P. PINCAE, SATTA MA, VAN DORNE D, CIN0rTI GA, PUGLIESE F,PIERUCCI A, SIMONETTI BM: Evidence for a renal origin ofurinary thromboxane B2 in health and disease, in Advances inProstaglandin, Thromboxane and Leukotriene Research, editedby B SAMUELSSON, R PAOLETTI, P RAMWELL, (vol. 11). NewYork, Raven Press, 1983, pp. 493—498
32. CIABATTONI G, PUGLIESE F, CINorri GA, PATRONO C: Renaleffects of anti-inflammatory drugs. Eur J Rheumatol 3:210—221,1980
33. PATRIGNANI P, FILABOZZI P, PATRONO C: Selective cumulative

10 Patrono and Dunn
inhibition of platelet thromboxane production by low—dose aspirinin healthy subjects. J Clin Invest 69:1366—1372, 1982
34. CIABATTONI G, CINOTTI GA, PIERuccI A, SIMONETTI BM,MANZI M, PUGLIESE F, BARSOTTI P, PECCI G, TAGGI F, PATRONOC: Effects of sulindac and ibuprofen in patients with chronicglomerular disease. Evidence for the dependence of renal functionon prostacyclin. N Engl J Med 310:279—283, 1984
35. WATSON ML, GILL JR. BRANCH RA, OATES JA, BRAsI-I AR:Systemic prostaglandin 12 synthesis is normal in patients withBartter's syndrome. Lancet 2:368—370, 1983
36. PATRONO C, CIABArI-ONI G, REMuzzi G, GOTTI E, BOMBARDIER!S, D MUNNO 0, TARTARELLI G, CINOTTI GA, SIMONETTI BM,PIERuCcI A: Functional significance of renal prostacyclin andthromboxane A2 production in patients with systemic lupuserythematosus. J Cliii Invest 76: 101 1—1018, 1985
37. FITZGERALD GA, PEDERSEN AK, PATRONO C: Analysis ofprostacyclin and thromboxane biosynthesis in cardiovascular dis-ease. Circulation 67:1174—1177, 1983
38. ZIPSER RD, MARTIN K: Urinary excretion of arterial bloodprostaglandins and thromboxane in man. Am J Physiol 242:El7l—E177, 1982
39. LAWSON J, BRASH AR, DORAN J, FITZGERALD GA: Measurementof urinary 2,3-dinor-thromboxane B2 and thromboxane B2 usingbonded—phase thenyl-boronic acid columns and capillary gaschromatography—negative—ion chemical ionization mass spectom-etry. Anal Biochem 150:463—470, 1985
40. Ss.AER J, ARDAILLOU N, SRAER J-D, ARDAILLOU R: in vitroprostaglandin synthesis by human glomeruli and papillae. Pro sta-
glanduns 23:855—864, 198241. HASSID A, DUNN M: Biosynthesis and metabolism of prostaglan-
dins in human kidney in vitro, in Prostaglandins and The Kidney.Biochemistry, Physiology, Pharmacology and Clinical Applica-tions, edited by MJ DUNN, C PATRONO, GA CIN0TTI. New York,Plenum Press, 1983, pp. 3—16
42. BING RF, RUSSEL GI, THURSTON H, SWALES JD, GODFREY N,LAZARUS Y, JACKSON J: Chemical renal medullectomy. Effect onurinary PGE2 and plasma renin in response to variations in sodiumintake and in relation to blood pressure. Hypertension 5:951—957,1983
43. GILL JR JR, FROLICH JC, BOWDEN RE, TAYLOR AA, KEISER HR,SEYBERTH HW, OATES JA, BARTTER FC: Bartter's syndrome: Adisorder characterized by high urinary prostaglandins and a de-pendence of hyperreninemia on prostaglandin synthesis. Am JMed 61:43—51, 1976
44. ZIPSER RD. HOEFS JC, SPECKART PF, ZIA PK, HORTON R:Prostaglandins: Modulators of renal function and pressor resist-ance in chronic liver disease. J Clin Endocrinol Metabol 48:895—900, 1979
45. SEDOR JR. WILLIAMS SL, CHREMOS AN, JOHNSON CL, DUNNMJ: Effects of sulindac and indomethacin on renal prostaglandinsynthesis. Clin Pharmacol Ther 36:85—91, 1984
46. NADLER JL, MCKAY M, CAMPESE V, VRBANAC J, HORTON R:Evidence that prostacyclin modulates the vascular actions ofcalcium in man. J Clin Invest 77:1278—1284, 1986
47. KIMBERLY RP, BOWDEN RE, KEISER HR, PLOTZ PH: Reductionof renal function by newer nonsteroidal antiinflammatory drugs.Am J Med 64:804—807, 1978
48. BRATER DC, ANDERSON S. BAIRD B, CAMPBELL WB: Effects ofibuprofen, naproxen, and sulindac on prostaglandins in men.Kidney mt 27:66—73, 1985
49. CIABATTONI G, BIANCHI—PORRO G, CARUSO I, FUMAGALLI M,PUGLIESE F, PATRONO C: Differential inhibition of prostaglandinand thromboxane synthesis in human tissues by nonsteroidalantiinflammatory drugs. (abstract) Clin Res 32:462A, 1984
50. SVENDSEN UG, GERSTOFT J. HANSEN TM, CHRISTENSEN P,LORENZEN Ill: The renal excretion of prostaglandins and changesin plasma renin during treatment with either sulindac or naproxenin patients with rheumatoid arthritis and thiazide—treated heartfailure. JRheumatol 11:779—782, 1984
51. KIMBERLY RP, GILL JR JR, BOWDEN RE, KEISER HR, PLOTZ PH:Elevated urinary prostaglandins and the effects of aspirin on renalfunction in lupus erythematosus. Ann mt Med 89:336—341, 1978
52. VRIESENDORP R, DE ZEEUW D, DE JONG PE, DONKER AJM,
PRATT JJ, VAN DER HEM GK: Reduction of urinary protein andprostaglandin E2 excretion in the nephrotic syndrome bynonsteroidal anti-inflammatory drugs. Clin Nephrol 25:105—110,1986
53. ROBERTS DG, GERBER JG, BARNES JS, ZEE GO, NIEs AS:Sulindac is not renal—sparing in man. Clin Pharmacol Ther38:258—265, 1985
54. SALVETTI A, PEDRINELLI R, ALBERICI P, MAGAGNA A,ABDEL—HAQ B: The influence of indomethacin and sulindac onsome pharmacological actions of atenolol in hypertensive patients.BrJ Cliii Pharmacol 17:lO8S—llIS, 1984
55. BERG KJ, TALSETH T: Acute renal effects of sulindac and indo-methacin in chronic renal failure. Clin Pharmacol Ther 37:447—452, 1985
56. LAFFI G, DASKALOPOULOS G, KRONBORG I, HsUEH W,GENTILINI P. ZIPSER RD: Effects of sulindac and ibuprofen inpatients with cirrhosis and ascites: An explanation for therenal—sparing effect of sulindac. Gastroenterology 90:182—187,1986
57. QUINTERO E, GINES P, ARROYO V, RIMOLA A, CAMPS J, GAYA J,GUEVARA A, RODAMILANS M, RODES J: Sulindac reduces theurinary excretion of prostaglandins and impairs renal function incirrhosis with ascites. Nephron 42:298—303, 1986
58. MISTRY CD, LOTE CJ, GOKAL R, CURRIE WJ, VANDENBURG M,MALLICK MP: Effects of sulindac on renal function andprostaglandin synthesis in patients with moderate chronic renalinsufficiency. Clin Sci70:501—505, 1986
59. PUDDEY lB. BEILIN U, VANDONGEN R, BANKS R, RousE 1Differential effects of sulindac and indomethacin on blood pres-sure in treated essential hypertensive subjects. Clin Sci 69:327—336, 1985
60. SALVETTI A, PEDRINELLI R, MAGAGNA A, UGENTI P: Differentialeffects of selective and non-selective prostaglandin—synthesis in-hibition on the pharmacological responses to captopril in patientswith essential hypertension. Gun Sd 63:261S—263S, 1982
61. LAFFI G, LA VILLA G, PINzANI M, CIABATTONI G, PATRIGNANIP. MANNELLI M, COMINELLI F, GENTILINI P: Altered and renaland platelet arachidonic acid metabolism in cirrhosis. Gastroen-terology 90:274—282, 1986
62. GUARNER F, GUARNER C, PRIETO J, COLINA I, QUIROGA J, CASASJ, FREIXA R, ROSELLO J, GELPI E, BALANZO J: Increased synthe-sis of systemic prostacyclin in cirrhotic patients. Gastroenterology90:687—694, 1986
63. CIABATTONI 0, Boss AH, PATRIGNANI P. CATELLA F,SIMONETTI BM, PIERUCCI A, FILABOZZI P, PATRONO C: Dose—dependent inhibition of extra—renal prostacyclin production bysulindac in man. Clin Pharmacol Pher (in press)
64. SEDOR JR, DAVIDSON E, DUNN MJ: Effects of nonsteroidalanti-inflammatory drugs in healthy subjects. Am J Med 81:58-70,1986
65. DUGGAN DE, HARE LE, DITZLER CA, LEI BW, KWAN KC: Thedisposition of sulindac. Cliii Pharmacol Ther 21:326—335, 1977
66. DASKALOPOULOS G, KRONBORG I, KATKOV W, GONZALEZ M,LAFFI 0, ZIPSER R: Sulindac and indomethacin suppress thediuretic action of furosemide in patients with cirrhosis and ascites:Evidence that sulindac affects renal prostaglandins. Am J KidneyDis 6:217—221, 1985
67. CATELLA F, PUGLIESE F, PATRIGNANI P, FILABOZZI P, CIABAT-TONI G, PATRONO C: Differential platelet and renal effects ofsulfinpyrazone in man. (abstract) Cliii Res 32:239A, 1984
68. ZAMBRASKI EJ, CHREMOS AN, DUNN MJ: Comparison of theeffects of sutindac with other cyclooxygenase inhibitors onprostaglandin excretion and renal function in normal and chronicbite duct—ligated dogs and swine. J Pharmacol Exp Ther 228:560—566, 184
69. Rubinger D, Frishberg Y, Eldor A, Popovtzer MM: The effect ofsuppression of prostaglandin synthesis on renal function in ratswith intact and reduced renal mass. Prostaglanduns 30:651—668,1985
70. Olanoff LS, Cook JA, Halushka PV: Sulindac does not preserverenal prostacyclin synthesis during endotoxemia. Prostaglandins29:1031—1037, 1985
71. IZUMI Y, FRANCO—SAENZ R, MULROW P1: Effects of prostaglan-

Inhibition of renal prostaglandin synthesis 11
din synthesis inhibition on the renin-angiotensin system and renalfunction. Hypertension 7:791—796, 1985
72. HENRICH WL, BEATER DC, CAMPBELL WB: Renal hemodynamiceffects of therapeutic plasma levels of sulindac sulfide duringhemorrhage. Kidney mt 29:484—489, 1986
73. BERTANI T, BENIGN! A, CUTILLO F, RoccHI G, MORELLI C,CARMINATI C, VERROUST P, REMUZZI G: Effect of aspirin andsulindac in rabbit nephrotoxic nephritis. J Lab C/in Med 107:
261—268, 198674. MILLER Mi, BEDNAR MM, MCGIFF JC: Renal metabolism of
sulindac: Functional implications. J Pharmacol Exp Ther 231:449—456, 1984
75. CAMERON JS: Platelets in glomerular disease. Ann Rev Med35: 175—180, 1984
76. TERRAGNO NA, TERRAGNO DA, MCGIFF JC: Contribution ofprostaglandins to the renal circulation in conscious, anesthetizedand laparotomized dogs. Circ Res 40:590—595, 1977
77. MCGIFF JC, CROWSHAW K, TERRAGNO NA, LONIGRO AJ: Re-lease of a prostaglandin—like substance into renal venous blood inresponse to angiotensin II. Circ Res 26 and 27 (Suppl. I):1121—1130, 1970
78. MCGIFF JC, CROWSHAW K, TERRAGNO NA, L0NIGR0 AJ: Renalprostaglandins: Possible regulators of the renal actions of pressorhormones. Nature 227:1255—1257, 1970
79. MCGIFF JC, CROWSHAW K, TERRAGNO NA, MALIK KU,LONIGRO AJ: Differential effect of noradrenaline and renal nervestimulation on vascular resistance in the dog kidney and therelease of a prostaglandin E-like substance. C/in Sci 42:223—233,1972
80. NEEDLEMAN P, DOUGLAS JR JR, JAKSCHIK B, STOECKLEIN PB,JOHNSON EM JR: Release of renal prostaglandin by catechola-mines: Relationship to renal endocrine function. J Pharmacol ExpTher 188:453—460, 1974
81. ZIPSER RD, MYERS SI, NEEDLEMAN P: Stimulation of renalprostaglandin synthesis by the pressor activity of vasopressin.Endocrinology 108:495—499, 1981
82. ZUSMAN RM, KEISER HR: Prostaglandin biosynthesis by rabbit
renomedullary interstitial cells in tissue culture: Stimulation byangiotensin II, bradykinin, and arginine vasopressin. J Gun Invest60:215—223, 1977
83. DUNN Mi, LIARD JF, DRAY F: Basal and stimulated rates of renalsecretion and excretion of prostaglandins E2, Fa, and 13,14-dihydro-l5-Fa in the dog. Kidney mt 13:136—143, 1978
84. AIKEN JW, VANE JR: Intrarenal prostaglandin release attenuatesthe renal vasoconstrictor activity of angiotensin. J PharmacolExpTher 184:678—687, 1973
85. SATOH 5, ZIMMERMANBG: Influence of the renin—angiotensinsystem on the effect of prostaglandin synthesis inhibitors in therenal vasculature. Circ Res 36 and 37 (Suppl. I):189—196, 1975
86. SWAIN iA, HEYNDRICKX GR, BOETTCHER DH, VATNER SF:Prostaglandin control of renal circulation in the unanesthetizeddog and baboon. Am J Physiol 229:826—830, 1975
87. FINN WF, ARENDSHORST WJ: Effect of prostaglandin synthetaseinhibitors on renal blood flow in the rat. Am J Physiol 231:1541—1545, 1976
88. NEEDLEMAN P, MARSHALL GR, JOHNSON EM JR: Determinantsand modification of adrenergic and vascular resistance in thekidney. Am J Physiol 227:665—669, 1974
89. MALIK KU, MCGIFF JC: Modulation of prostaglandins of adren-
ergic transmission in the isolated perfused rabbit and rat kidney.Circ Res 36:599—609, 1975
90. TAN SY, SHAPIRO R, FRANCO R, STOCKARD H, MULROW Pi:Indomethacin—induced prostaglandin inhibition with hyperkale-mia: A reversible cause of hyporeninemic hypoaldosteronism.Ann mt Med 90:783—785, 1979
91. ZIMRAN A, KRAMER M, PLASKIN M, HERSHKO C: Incidence ofhyperkalemia induced by indomethacin in a hospital population.
BrMed J 291:107—108, 1985
92. SWAINSON CP, GRIFFITHS P: Acute and chronic effects of
sulindac on renal function in chronic renal disease. Clin
Pharmacol Ther 37:298—300, 198593. JUHL RP, VANTHIEL DH, DITTERT LW, ALBERT KS, SMITH RB:
Ibuprofen and sulindac kinetics in alcoholic liver disease. C/in
Pharmacol Ther 34:164—109, 198394. BLACKSHEAR JL, DAVIDMAN M, STILLMAN MT: Identification of
risk for renal insufficiency from nonsteroidal antiinflammatorydrugs. Arch mt Med 143:1130—1134, 1983
95. Coiwi HL, BONvENTRE iV: Renal insufficiency associated withnonsteroidal antiinflammatory agents. Am J Kidney Dis 4:147—152,
1984
96. TIGGELERRG, HULME B, WIJDEVELD PG: Effect of indomethacinon glomerular permeability in the nephrotic syndrome. Kidney mt16:312—321, 1979
97. MACKAY 1G. MUIRAL, WATSON ML: Contribution of prostaglan-dins to the systemic and renal vascular response to furosemide innormal man. Br J C/in Pharmacol 17:513—519, 1984
98. HALUSHKA PV, MARGOLIUS HS, ALLENH, CONRAD EC: Urinaryexcretion of prostaglandin E-like material and kallikrein: Effectsoffurosemide. Prostaglandins 18:359—364, 1979
99. OLSEN UB: Diuretics and Kidney Prostaglandins, in Prostaglan-dins and the Kidney: Biochemistry, Physiology, Pharmacology,and Clinical Applications, edited by DUNN Mi, PATRONO C,CINOTTI GA. New York, Plenum Publishing Co., 1983, pp.205—212.
100. NIE5 AS, GAL J, FADUL 5, GERBER iG: Indomethacin—furosemideinteraction: The importance of renal blood flow. J Pharmacol ExpTher 226:27—32, 1983
101. WILSON TW, LOADHOLT CB, PRIVITERA PJ, HALUSHKA PVFurosemide increases urine 6-keto-prostaglandin F1,—Relation tonatriuresis, vasodilation, and renin release. Hypertension 4:634—641, 1982
102. RILEY Li, VLASSES PH, ROTMENSCH HH, SWANSON BN,CHREMOS AN, ioHNsoN CL, FERGUSON RK: Sulindac andibuprofen inhibit furosemide—stimulated renin release but notnatriuresis in men on a normal sodium diet. Nephron 41:283—288,1985
103. CHENNAVASIN P, SEIWELL R, BEATER DC: Pharmacoki-
netic—dynamic analysis of the indomethacin—furosemide interac-tion in man. J Pharmacol Exp Ther 215:77—81, 1980
104. GROENE H-i, DUNN Mi: The role of prostaglandins in arterialhypertension: A critical review. Adv Nephrol 14:241—272, 1985
105. WENNMALM A: Influence of indomethacin on the systemic andpulmonary vascular resistance in man. C/in Sci Mol Med54:141—145, 1978
106. NEGUS P, TANNEN RL, DUNN Mi: Indomethacin potentiates thevasoconstrictor actions of angiotensin II in normal man. Prosta-glandins 12:175—180, 1976
107. SPECKART P, ZIA P, ZIP5ER R, HORTON R: The effect of sodium
restriction and prostaglandin inhibition on the renin—angiotensinsystem in man. J Endocrinol Metab 44:832—837, 1977
108. VIERHAPPER H, WALDHAUSL W, NOWOTNYP: Effect of indo-
methacin upon angiotensin—induced changes in blood pressure andplasma aldosterone in normal man. Eur J C/in Invest 11:85—89,1981
109. KOOPMANS PP, THIEN TH, GRIBNAU FWJ: Influence of
nonsteroidal antiinflammatory drugs on diuretic treatment of mildto moderate essential hypertension. Br Med J 289:1492—1494, 1984
110. ERIK550N 5, HAGENFELDT L, LAW D, PATRONO C, PINCA E,WENNMALM A: Effect of prostaglandin synthesis inhibitors onbasal and carbon dioxide stimulated cerebral blood flow in man.Acta Physiol Scand 117:203—211, 1983
111. EDLUNDA, BERGLUND B, VAN DORNE D, KAUSER L, NOWAKi,PATRONO C, SOLLEVI A, WENNMALM A: Coronary flow regula-tion in patients with ischemic heart disease: Release of purines andprostacyclin and the effect of inhibitors of prostaglandin forma-tion. Circulation 71:1113—1120, 1985
112. MILLS EH, WHITWORTH JA, ANDREWS J, KINCAID—SMITH P:Nonsteroidal antiinflammatory drugs and blood pressure. Aust NZJMed 12:478—482, 1982
113. EBEL DL, RHYMER AR, STAHL E: Effects of sulindac, piroxicamand placebo on the hypotensive effect of propranolol in patientswith mild to moderate essential hypertension. Adv Ther 2:131—142,1985
114. STEINESS E, WALDORFF 5: Different interaction of indomethacin

12 Patrono and Dunn
and sulindac with thiazides in hypertension. Br Med J 285:1702—1703, 1982
115, WONG DO, SPENCE JD, LAMKI L, FREEMAN D, MCDONALD JW:Effect of nonsteroidal anti-inflammatory drugs on control of hy-pertension by beta blockers and diuretics. Lancet i:997—1001, 1986
116. LEWIS RV, TONER JM, JACKSON PR, RAMSAY LE: Effects of
indomethacin and sulindac on blood pressure of hypertensivepatients. Br Med J 292:934—935, 1986
117. TRIMARCO B, DE SIMONE A, CuocoLo A, RICCIARDELLI B,VOLPE M, PATRIGNANI P. SACCA L, CONDORELLI M: Role ofprostaglandins in the renal handling of a salt load in essentialhypertension. Am J Cardiol 55:116—121, 1985





























