Embed Size (px)
Citation preview

The Autoxidation of TetralinAuthor(s): C. H. Bamford and M. J. S. DewarSource: Proceedings of the Royal Society of London. Series A, Mathematical and PhysicalSciences, Vol. 198, No. 1053 (Aug. 15, 1949), pp. 252-267Published by: The Royal SocietyStable URL: http://www.jstor.org/stable/98169 .
Accessed: 08/05/2014 03:34
Your use of the JSTOR archive indicates your acceptance of the Terms & Conditions of Use, available at .http://www.jstor.org/page/info/about/policies/terms.jsp
.JSTOR is a not-for-profit service that helps scholars, researchers, and students discover, use, and build upon a wide range ofcontent in a trusted digital archive. We use information technology and tools to increase productivity and facilitate new formsof scholarship. For more information about JSTOR, please contact [email protected].
.
The Royal Society is collaborating with JSTOR to digitize, preserve and extend access to Proceedings of theRoyal Society of London. Series A, Mathematical and Physical Sciences.
http://www.jstor.org
This content downloaded from 169.229.32.137 on Thu, 8 May 2014 03:34:34 AMAll use subject to JSTOR Terms and Conditions

The autoxidation of tetralin
BY C. H. BAMFORD AND M. J. S. DEWAR
Courtaulds Ltd., Maidenhead, Berks
(Communicated by A. H Wilson, F.R.S.-Received 28 December 1948)
The autoxidation of tetralin can be photosensitized by suitable vat dyes. This reaction can therefore be investigated by standard photochemical techniques. The kinetics have in this way been shown to conform to the scheme suggested by Bolland & Gee (1 946) for autoxidations. The absolute velocity constants for the elementary processes have been evaluated, and the activation energies and frequency factors estimated (table 4).
The frequency factors of free radical reactions in solution have been discussed. It is suggested that the 'normal' frequency factor for such a reaction is about 107.
It is pointed out that the techniques described in this paper should be generally applicable to the study of free radical reactions in solution.
INTRODUCTION
The autoxidation of tetralin has previously been studied by George, Rideal & Robert- son ( 1946), George & Robertson (1 946 a, b), George (1946 a, b) and Robertson & Waters
(1946). These authors now seem to agree that the oxidation is a radical chain process, following the general scheme of Bolland & Gee (1946), although the evidence that the reaction does follow these kinetics is somewhat scanty.
We have recently found that many vat dyes are able to photosensitize the autoxida- tion of tetralin (Bamford & Dewar 1948 a). By initiating the reaction in this way it is possible to make use of standard photochemical techniques, and we have thus been able to confirm the general mechanism, and also to determine the absolute values of the velocity constants of the individual radical reactions.
EXPERIMENTAL METHOD
The reactions were carried out in a Warburg type apparatus, and pressure changes were followed by means of a dibutyl phthalate manometer read to 0.01 mm. with a cathetometer. The consumption of oxygen could be calculated from the pressure change and the dimensions of the apparatus. The reaction vessel was a spherical bulb of approximately 20 ml. capacity, attached to the manometer by a glass spiral, and shaken mechanically at constant speed. The rate of shaking was normally about 7 times per sec., and was measured stroboscopically. Five ml. of tetralin were used in each run. The whole apparatus was immersed in a transparent thermostat (?+001?C). The system could be evacuated to 10-5mm.Hg by a three-stage mercury-diffusion pump backed by an oil pump and could be filled with oxygen (previously purified by fractional distillation) at pressures measured on mercury or Apiezon oil manometers.
In the photochemical experiments a IkW projection lamp was used as light source. A 21. flask filled with 10 % copper sulphate solution was used as a con- densing lens, and served also to remove infra-red radiation. The solution was
[ 252 ]
This content downloaded from 169.229.32.137 on Thu, 8 May 2014 03:34:34 AMAll use subject to JSTOR Terms and Conditions

The autoxidation of tetralin
circulated by a pump to avoid overheating. The rest of the system consisted of an iris
diaphragm, an electromagnetically operated Compur shutter, and two collimating lenses. Filters of Crookes A glass and saturated sodium nitrite solution could be inserted. Intensities were measured by a calibrated thermopile potentiometer system. Normally both filters were used, cutting off light with A < 4200 A.
Purification of materials
Tetralin was shaken with concentrated sulphuric acid, and after washing and
drying it was boiled under reflux for several hours with metallic sodium, in a nitrogen atmosphere, It was then distilled under nitrogen through a 15-plate column, and
preserved in evacuated ampoules. The dyes, specially pure specimens of which were kindly supplied by I.C.I., were
further purifiedby sublimation in vacuo. Solutions of the dyes in tetralin (0.05 mg./ml.) were made up in evacuated tubes.
Benzoyl peroxide was crystallized from chloroform and light petroleum, and dried in vacuo.
Hydroquinone, and ac- and f/-naphthols were pure commercial specimens.
KINETIC SCHEME
It is convenient to give the complete kinetic scheme at this stage. This differs from the scheme of Bolland & Gee only in the mode of initiation of the chains:
D+ hv-> D* (labs.)'
D* -> D
+
(kf) D*+02->D+02 (kl) D* + TH->DH + T (k2) T + 2 T02 (k3) (1) TO2 + TH-> TO2H + T (k4) 2T-> (k5) T+ T02- (k6) 2TO2-> (k7)
D and TH represent dye and tetralin respectively; T is a radical formed from tetralin
by loss of a hydrogen atom, and TO2 is a peroxide radical derived from T. For long chains, with the approximation ck = 4(k5k7) (Bolland & Gee 1946), this scheme leads to
d[02] k_ 2 -abs. [TH] k3kc4[02]1 [TH] dt k(f + k[O02]1 + k[TH] k 3 /k7[021 + k4 k,[TH] ' (2
where [0211 is the concentration of oxygen in the liquid. The term in the first bracket on the right of (2) gives the rate of chain starting I. These equations assume that the
light absorption is weak. It was established that this was so in the present experiments.
253
Vol. 198. A. I7
This content downloaded from 169.229.32.137 on Thu, 8 May 2014 03:34:34 AMAll use subject to JSTOR Terms and Conditions

C. H. Bamford and M. J. S. Dewar
RESULTS AT HIGH OXYGEN PRESSURES
At sufficiently high oxygen pressures the second term on the right of (2) becomes
effectively constant. If the rate of initiation is constant the rate of reaction should then be independent of the oxygen pressure. Initiation by benzoyl peroxide should fulfil this condition (cf. Bolland I948). Table 1 shows that in fact the rate of reaction remained sensibly constant down to pressures of about 50 mm. At low pressures the rate fell as equation (2) requires; the dependence of the rate on the pressure will be discussed later.
TABLE 1. AUTOXIDATION OF TETRALIN INITIATED BY BENZOYL PEROXIDE
T = 25? C, benzoyl peroxide, 1% f -,
k2
oxygen pressure - 107d[0]/dt (mm. Hg) (mol.l.-isec.-l) 600 3-09 400 3.04 300 3-04 200 3-01
50 2-98 10 2*91
1-93 2-72 1-55 2-64 1-16 2-53 0-772 2-30 0-618 2-07 0-463 1-85 0-386 1-68 0.309 1-48 0-231 1-21 0*154 0-86 0-116 0-66 0-077 047 0-039 0-25
T = 45? C, benzoyl peroxide, 0.1% frA ,
oxygen pressure - 107d[02]/dt (mm. Hg) (mol.l.'sec.-1) 600 5-98 400 5-97 200 5-95 100 5.98
5-42 564 2-89 5-30 1-45 4-57 1-30 4-31 0.99 4-02 0-685 3-37 0-640 3-03 0-535 2-77 0-383 214 0-283 1-73
When the reaction is photosensitized by dyes, the rate for a given dye and light intensity is not constant at high oxygen pressures, but falls off as the pressure increases. This indicates that the rate of initiation must depend on the pressure. It is known that excited dyes are deactivated by oxygen. The second and third equations in scheme (1) allow for spontaneous (or solvent) deactivation and deactivation by oxygen respectively. From equation (2) it follows that at high oxygen pressures
(dt k2kf + 1 [021 + k2 [TH]}. (3)
Assuming that solutions of oxygen in tetralin obey Henry's law a plot of (d[02]/dt)-2 against pressure should be linear, and the ratio of slope to intercept gives the 'relative
quenching coefficient' q for oxygen, defined by
k[ q = - (4)
kf + k2z [TH] '
254
This content downloaded from 169.229.32.137 on Thu, 8 May 2014 03:34:34 AMAll use subject to JSTOR Terms and Conditions
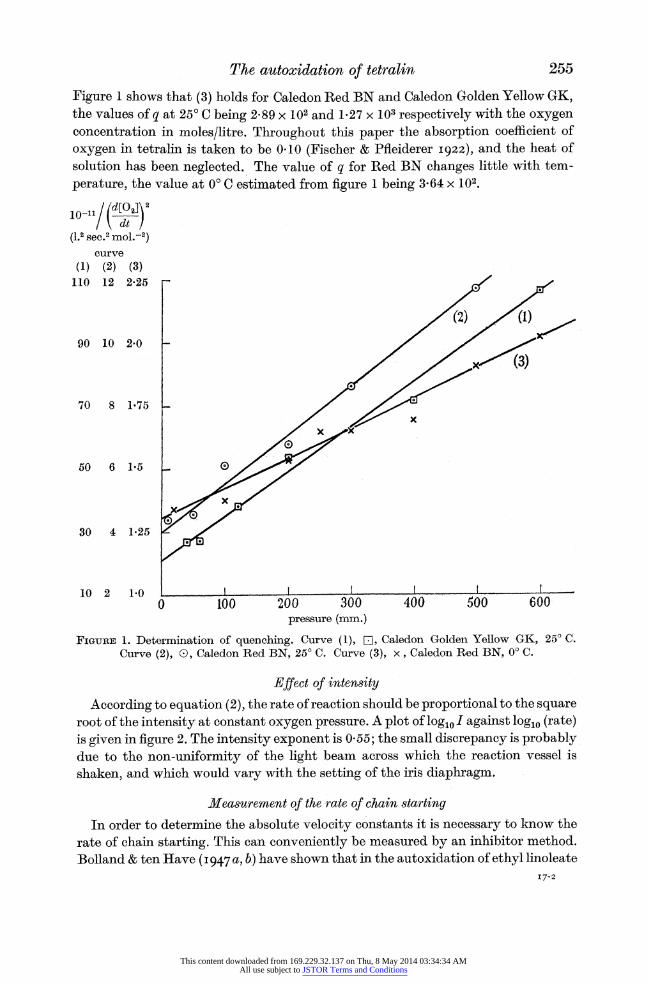
The autoxidation of tetralin
Figure 1 shows that (3) holds for Caledon Red BN and Caledon Golden Yellow GK, the values of q at 25? C being 2-89 x 102 and 1.27 x 103 respectively with the oxygen concentration in moles/litre. Throughout this paper the absorption coefficient of
oxygen in tetralin is taken to be 0-10 (Fischer & Pfleiderer 1922), and the heat of solution has been neglected. The value of q for Red BN changes little with tem-
perature, the value at 0? C estimated from figure 1 being 3-64 x 102.
0-11/ (d[02])2 \dt !
(1.2 sec.2 mol.-2) curve
(1) (2) (3) 110 12 2.25
(2) / )
90 10 20 -
(3)
70 8 1'75
50 6 1'5 _ /
30 4 1.25
10 2 1.0 ... I 1 t .... 0 100 200 300 400 500 600
pressure (mm.)
FIGURE 1. Determination of quenching. Curve (1), E[, Caledon Golden Yellow GK, 25? C. Curve (2), 0, Caledon Red BN, 25? C. Curve (3), x, Caledon Red BN, 0? C.
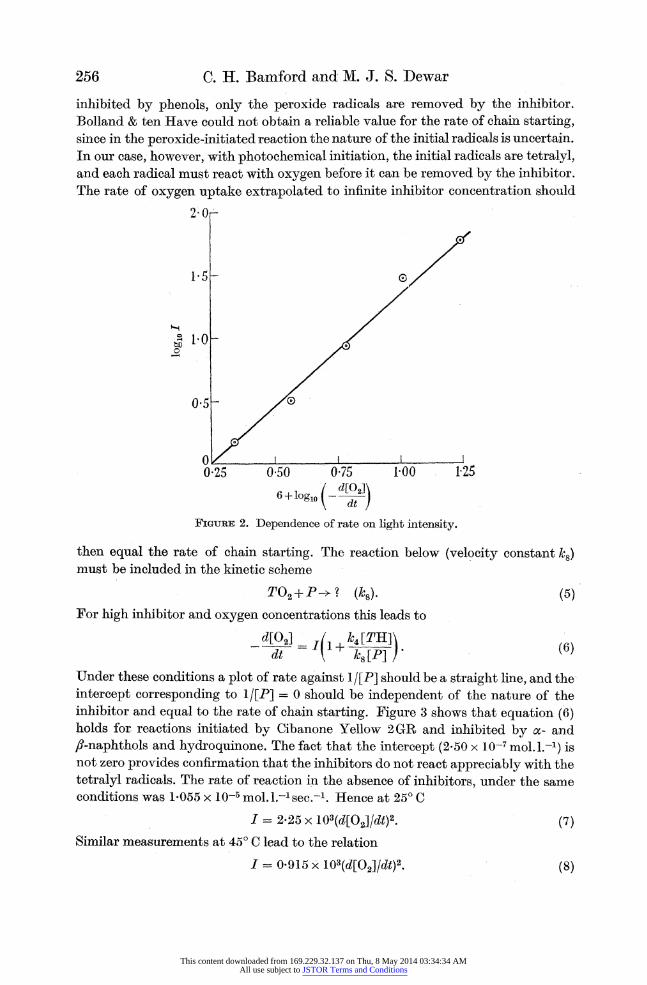
Effect of intensity
According to equation (2), the rate of reaction should be proportional to the square root of the intensity at constant oxygen pressure. A plot of log10 I against log1o (rate) is given in figure 2. The intensity exponent is 0-55; the small discrepancy is probably due to the non-uniformity of the light beam across which the reaction vessel is
shaken, and which would vary with the setting of the iris diaphragm.
Measurement of the rate of chain starting
In order to determine the absolute velocity constants it is necessary to know the rate of chain starting. This can conveniently be measured by an inhibitor method. Bolland & ten Have (1947 a, b) have shown that in the autoxidation of ethyl linoleate
17-2
255
This content downloaded from 169.229.32.137 on Thu, 8 May 2014 03:34:34 AMAll use subject to JSTOR Terms and Conditions
256 C. H. Bamford and M. J. S. Dewar
inhibited by phenols, only the peroxide radicals are removed by the inhibitor. Bolland & ten Have could not obtain a reliable value for the rate of chain starting, since in the peroxide-initiated reaction the nature of the initial radicals is uncertain. In our case, however, with photochemical initiation, the initial radicals are tetralyl, and each radical must react with oxygen before it can be removed by the inhibitor. The rate of oxygen uptake extrapolated to infinite inhibitor concentration should
2-0-
1.5- e/
0.5 /-
0 .. .. .. A'.. 0'25 0.50 0'75 1-00 125
6 + log (d2)
FIGUuE 2. Dependence of rate on light intensity.
then equal the rate of chain starting. The reaction below (velocity constant k8)
must be included in the kinetic scheme
TO2+P - (ksg). (5)
For high inhibitor and oxygen concentrations this leads to
d[021TH]) (6) dt I1+ k8 E[PI (6
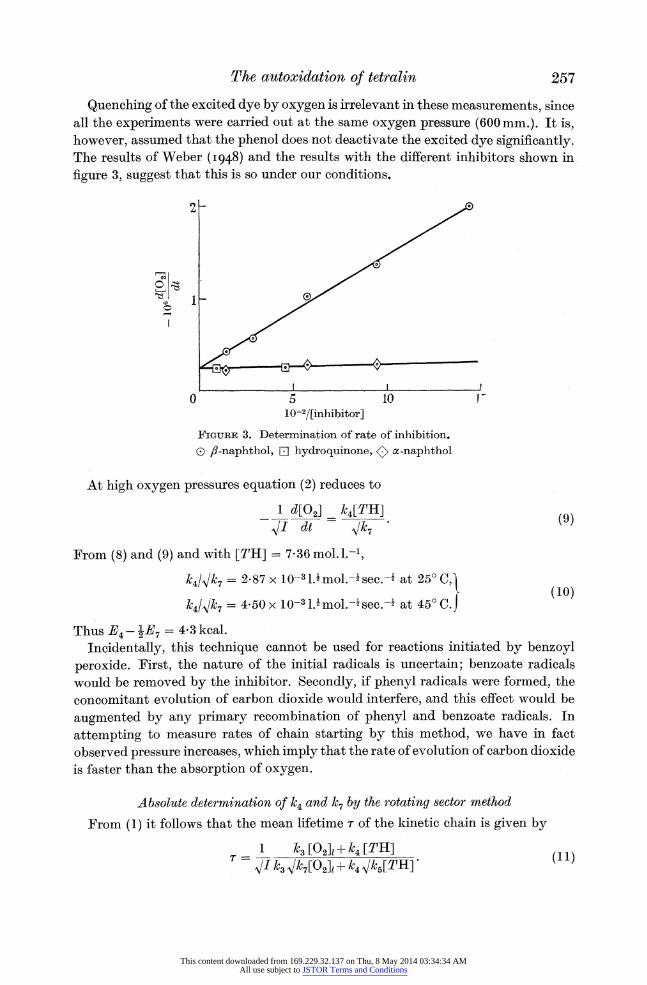
Under these conditions a plot of rate against 1/[P] should be a straight line, and the intercept corresponding to 1/[P] 0= should be independent of the nature of the inhibitor and equal to the rate of chain starting. Figure 3 shows that equation (6) holds for reactions initiated by Cibanone Yellow 2GGR and inhibited by a- and Pf-naphthols and hydroquinone. The fact that the intercept (2-50 x 10-7 mol.l .-) is not zero provides confirmation that the inhibitors do not react appreciably with the tetralyl radicals. The rate of reaction in the absence of inhibitors, under the same conditions was 1-055 x 10-5 mol 1.- see-1. Hence at 25? C
I = 2-25 x l03(d02]ldt)2. (7)
Similar measurements at 45? C lead to the relation
I = 0-915 x 103(d[O0]/dt)2. (8)
This content downloaded from 169.229.32.137 on Thu, 8 May 2014 03:34:34 AMAll use subject to JSTOR Terms and Conditions
The autoxidation of tetralin
Quenching of the excited dye by oxygen is irrelevant in these measurements, since all the experiments were carried out at the same oxygen pressure (600mm.). It is, however, assumed that the phenol does not deactivate the excited dye significantly. The results of Weber (I948) and the results with the different inhibitors shown in
figure 3, suggest that this is so under our conditions.
2-
I't, 1
0 5 10 I 10-2/[inhibitor]
FIGURE 3. Determination of rate of inhibition. 0 fl-naphthol, W hydroquinone, <) a-naphthol
At high oxygen pressures equation (2) reduces to
1 d[02 _ k4[TH]
VI dt Vk7
From (8) and (9) and with [TH] = 7-36 mol. 1.-1,
k4d/k7 = 2.87 x 10- l.mol.-isec.-' at 25? C, (10)
k4/k7 = 450 x 10-3 l.mol.-isec.-l at 45? C.J
Thus E4- E7 = 4-3 kcal.
Incidentally, this technique cannot be used for reactions initiated by benzoyl peroxide. First, the nature of the initial radicals is uncertain; benzoate radicals would be removed by the inhibitor. Secondly, if phenyl radicals were formed, the concomitant evolution of carbon dioxide would interfere, and this effect would be
augmented by any primary recombination of phenyl and benzoate radicals. In
attempting to measure rates of chain starting by this method, we have in fact observed pressure increases, which imply that the rate of evolution of carbon dioxide is faster than the absorption of oxygen.
Absolute determination of k4 and k7 by the rotating sector method
From (1) it follows that the mean lifetime T of the kinetic chain is given by
1 Ik3 [021 + k4 [TH] T =I k3 /k70 + k4k s[TH] 1)
257
This content downloaded from 169.229.32.137 on Thu, 8 May 2014 03:34:34 AMAll use subject to JSTOR Terms and Conditions
C. H. Bamford and M. J. S. Dewar
At high oxygen pressures this reduces to
T = 1/V(k7I). (12)
Thus the determination of at high oxygen pressures enables k4 and k7 to be calculated
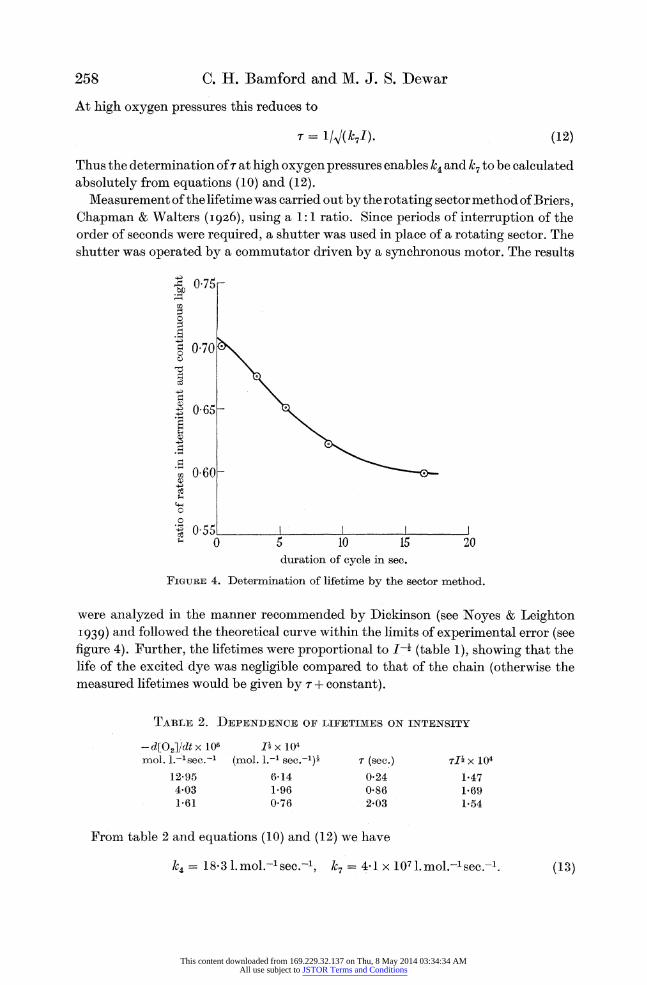
absolutely from equations (10) and (12). Measurement of the lifetime was carried out by the rotating sector method of Briers,
Chapman & Walters (I926), using a 1: 1 ratio. Since periods of interruption of the order of seconds were required, a shutter was used in place of a rotating sector. The shutter was operated by a commutator driven by a synchronous motor. The results
) 0.75-
0
.*. 0-70
4-\
0 0-65- E
* 060- a)
0 X 0-55 1 I
0 5 10 15 20 duration of cycle in sec.
FIGURE 4. Determination of lifetime by the sector method.
were analyzed in the manner recommended by Dickinson (see Noyes & Leighton I939) and followed the theoretical curve within the limits of experimental error (see figure 4). Further, the lifetimes were proportional to I-i (table 1), showing that the life of the excited dye was negligible compared to that of the chain (otherwise the measured lifetimes would be given by - + constant).
TABLE 2. DEPENDENCE OF LIFETIMES ON INTENSITY
-d[O2]/dt x 106 I_ x 104 mol. l.-lsec.-' (mol. 1.-i sec.-l)
12.95 6-14 4-03 1-96 1-61 0-76
r (sec.) 0-24 0-86 2-03
7li x 104
1-47 1-69 1-54
From table 2 and equations (10) and (12) we have
= 18'3 1. mol.-lsec.-l, 7 = 4-1 x 107 . mol.-lsec.-I.
258
(13)
This content downloaded from 169.229.32.137 on Thu, 8 May 2014 03:34:34 AMAll use subject to JSTOR Terms and Conditions
The autoxidation of tetralin
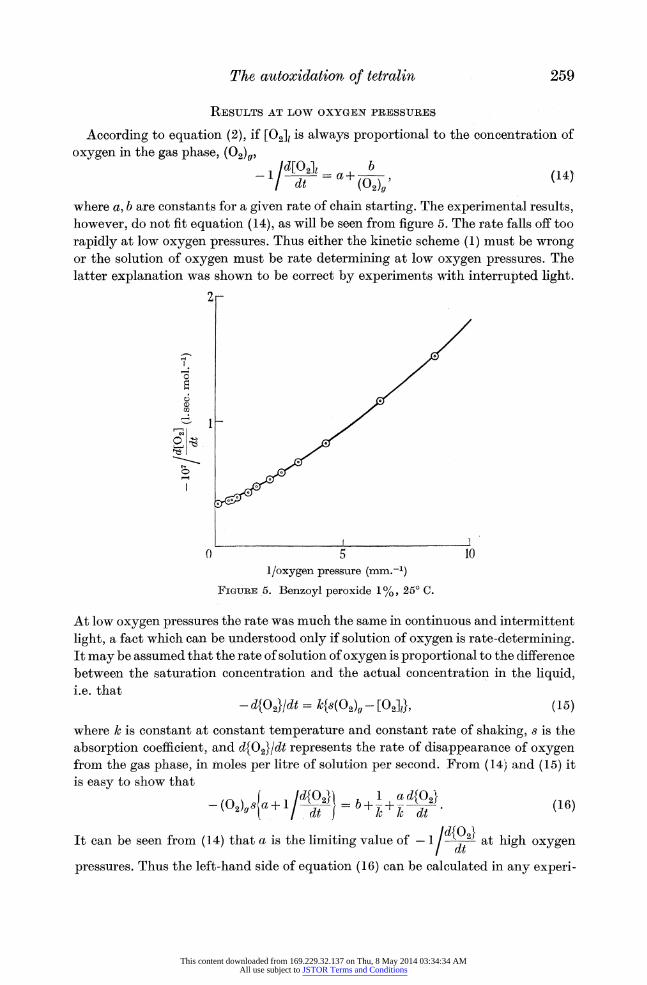
RESULTS AT LOW OXYGEN PRESSURES
According to equation (2), if [O2]1 is always proportional to the concentration of
oxygen in the gas phase, (02)O,
- 1/d[2= a+(0b - (14- dt (02)g'
where a, b are constants for a given rate of chain starting. The experimental results, however, do not fit equation (14), as will be seen from figure 5. The rate falls off too
rapidly at low oxygen pressures. Thus either the kinetic scheme (1) must be wrong or the solution of oxygen must be rate determining at low oxygen pressures. The latter explanation was shown to be correct by experiments with interrupted light.
2-
0
1
1 p
II-1 o 5 10
1/oxygen pressure (mm.-x)
FIGURE 5. Benzoyl peroxide 1%, 25? C.
At low oxygen pressures the rate was much the same in continuous and intermittent
light, a fact which can be understood only if solution of oxygen is rate-determining. It may be assumed that the rate of solution of oxygen is proportional to the difference between the saturation concentration and the actual concentration in the liquid, i.e. that
-d{02}/dt = k{8(02)- [02]1}, (15)
where k is constant at constant temperature and constant rate of shaking, s is the
absorption coefficient, and d{02}/dt represents the rate of disappearance of oxygen from the gas phase, in moles per litre of solution per second. From (14) and (15) it is easy to show that
{ /d{dot 2 1- ad d(0(16) c e i iii dt
2}} b? Id02} at It can be seen from (14) that a is the limiting value of - 1 / at high oxygen
pressures. Thus the left-hand side of equation (16) can be calculated in any experi-
259
This content downloaded from 169.229.32.137 on Thu, 8 May 2014 03:34:34 AMAll use subject to JSTOR Terms and Conditions
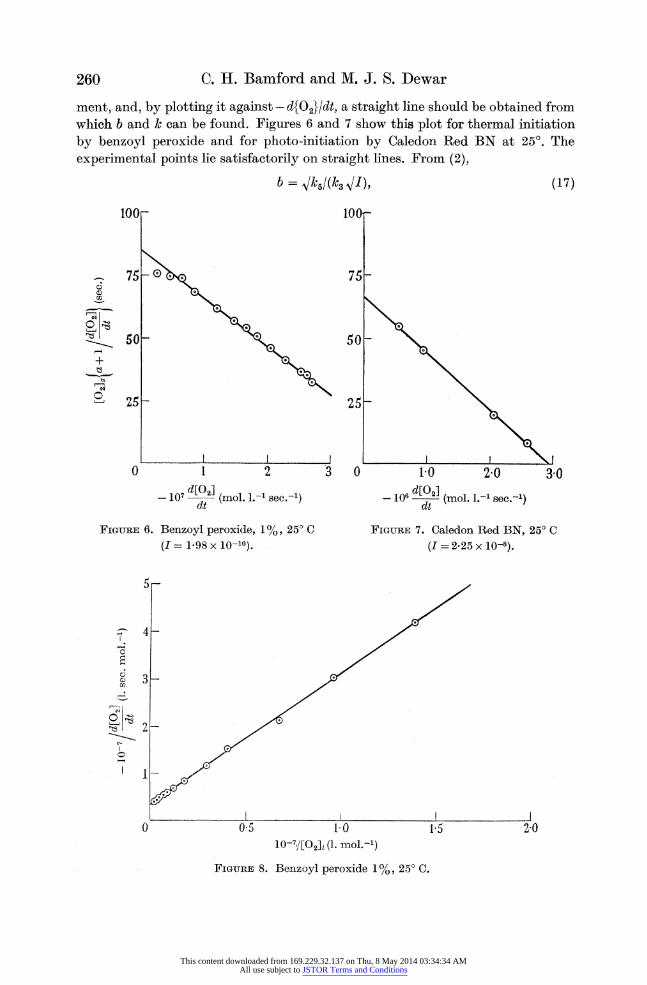
C. H. Bamford and M. J. S. Dewar
ment, and, by plotting it against - d{0}/dt, a straight line should be obtained from which b and k can be found. Figures 6 and 7 show this plot for thermal initiation
by benzoyl peroxide and for photo-initiation by Caledon Red BN at 25?. The
experimental points lie satisfactorily on straight lines. From (2),
6b = V5(k3VI),
(a m r-l
r2sI
r-"
aq
C)
1 2 3 0 10 2-0
- d107 d2] (mol. 1.-1 sec.-) -106 d 2] (mol. 1.-1 sec.-) dt- 10 dt
(17)
FIGURE 6. Benzoyl peroxide, 1%, 25? C
(I = 1-98 x 10-10).
5-
4-
2- -
I 1-
o) yf^ T -
0.
0 1.0
10-7,/[O2l (1. mol.-
FIGURE 7. Caledon Red BN, 25? C
(I = 2-25 x 10-8).
15 2-0 -1)
FIGURE 8. Benzoyl peroxide 1%, 25? C.
260
This content downloaded from 169.229.32.137 on Thu, 8 May 2014 03:34:34 AMAll use subject to JSTOR Terms and Conditions
The autoxidation of tetralin
and b /I should be constant. The values obtained for this quantity are 3-94 x 10-5
(figure 6) and 3-90 x 10-5 (figure 7). We may take the value of Vk5/k3 to be 3-94 x 10-5 at 25? C, since figure 6 represents the more accurate series of experiments. Using the value of k from figure 6, values of [O2]1 may be calculated from (15). According
to equation (14) a plot of - 1I dt against l/[O2]1 should be a straight line. Figure 8
shows that this is so. Although the straight lines in figures 6 and 8 are consequences of the same equations, we consider that b can be found more conveniently from
figure 8. In this case the value agrees almost exactly with that obtained from
figure 6. It will be noticed that the magnitudes of k found from figures 6 and 7 are not quite the same. The value of k should be sensitive to the experimental conditions, e.g. the exact position of the reaction bulb, and much greater variations were obtained by altering the rate of shaking.
100-
75
50-
25-
I I I 0 2 4 6
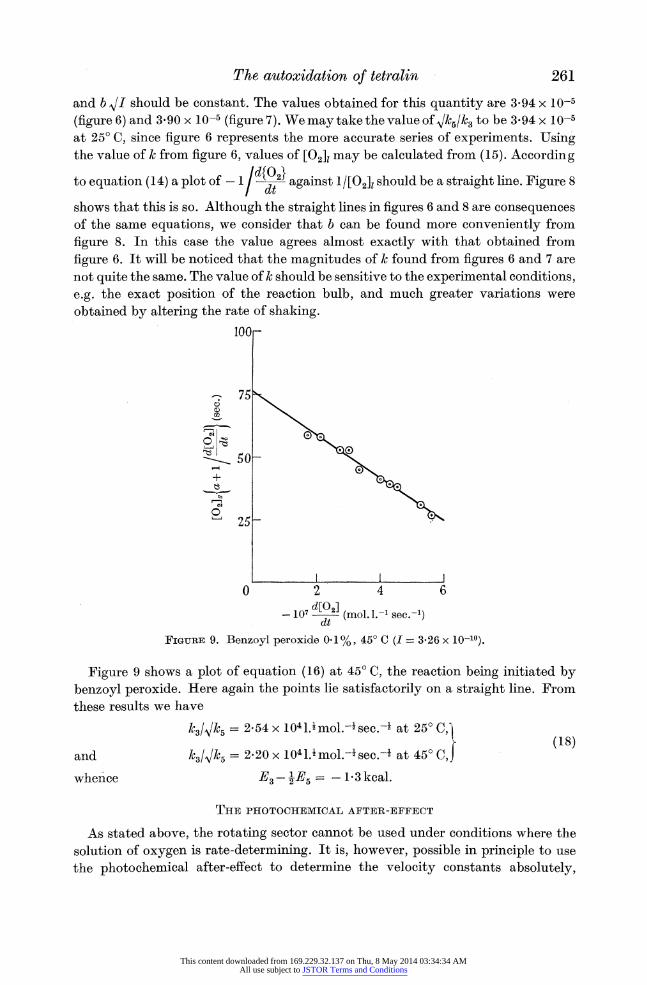
d[02] - 107 ] (mol. 1.-1 sec.-1) dt FIGURE 9. Benzoyl peroxide 0-1%, 45? C (I = 3-26 x 10-10).
Figure 9 shows a plot of equation (16) at 45? C, the reaction being initiated by
benzoyl peroxide. Here again the points lie satisfactorily on a straight line. From these results we have
Al3/1k5 = 2-54 x 104 l.Imol.-I sec.-i at 25? C, r (18)
and k3//k = 2-20 x 1041.mol.-sec.- at 45? C,
whence E3 - E = - 1 3 kcal.
THE PHOTOCHEMICAL AFTER-EFFECT
As stated above, the rotating sector cannot be used under conditions where the solution of oxygen is rate-determining. It is, however, possible in principle to use the photochemical after-effect to determine the velocity constants absolutely,
261
This content downloaded from 169.229.32.137 on Thu, 8 May 2014 03:34:34 AMAll use subject to JSTOR Terms and Conditions

C. H. Bamford and M. J. S. Dewar
provided that suitable corrections can be applied. It may readily be shown from
equation (2) that the photochemical after-effect AO2 is given by
_ _ _ _ 1 1 AG dt k3[2]+ k4[TH[]i (19)
in which - d{02}/dt is the rate in the light, and 6 is the ratio of the rates in the light and in the dark. (If the rate in the dark is not sufficiently large, it is convenient to
supply continuous weak illumination.) The total after-effect measured when the
light is cut off is A'02 AO2 + [02]1 (dark)
- [021 (light)
A- 2 + s8(2) - [02]1 (light)
A= 02 o, (20)
from equation (15). In deriving (19) it has been assumed that the concentration of
oxygen in solution during the time of the after-effect is constant, and equal to its value in the light. This is an approximation which will be justified below.
It was found possible to measure A'02 directly over the whole range of oxygen pressures. Values of AO2 could be calculated from (20), since k was known from
previous measurements. From (19) it follows that a plot of
IA02/[L{d{02} } log ? (1 +
against 1/[02] should be a straight line. In place of l/[02] it is more convenient to
plot the equivalent expression (see equation (2)), (a + 1 , where a and b have
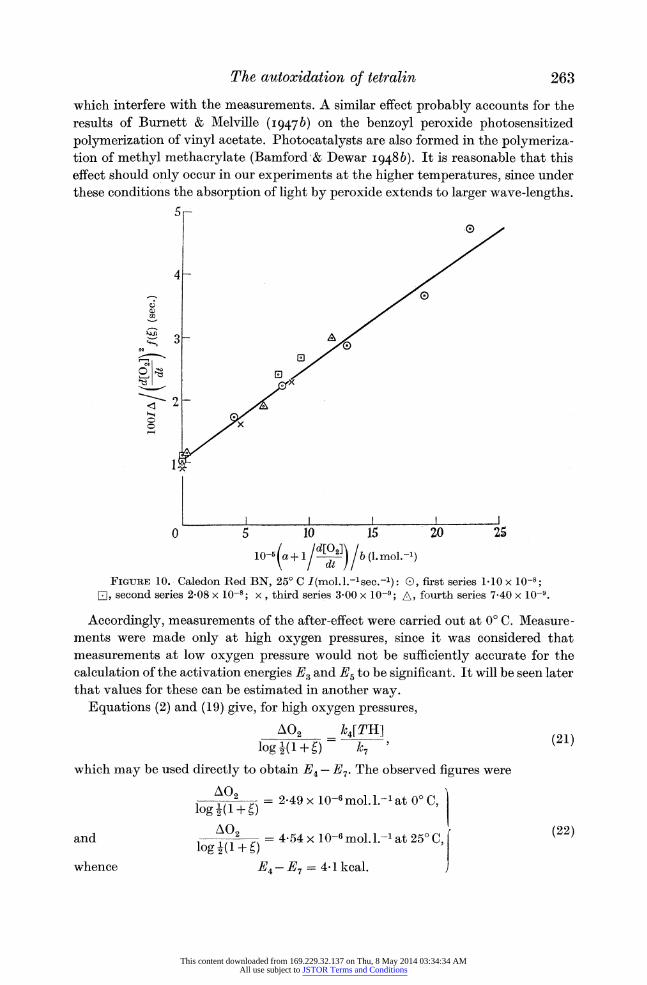
their previous significance. Figure 10 shows this plot for several series of experiments at 25? C, in which the rate of initiation was varied by a factor of about 10. All the
points lie satisfactorily on the same straight line. This indicates that the change in
[021 during the after-effect is not serious under our conditions, for the error so introduced would be very sensitive to the rate of initiation.
From the slope and intercept of the line in figure 10, k3 and k4 may be found
absolutely (equation (19)) and hence the other velocity constants. Values are given in table 3. The values of k4 and k7 agree quite satisfactorily with those obtained by the rotating sector method (equation (13)). There are a number of uncertainties in the latter method, and we therefore consider that the values in table 3 are the more reliable.
TABLE 3. VELOCITY CONSTANTS AT 25? C (l. mol.1sec.-1)
k3 6-76 x 107 k5 7-10 x 106
k4 13.3 k7 2-15 x 107
The after-effects could not be measured satisfactorily at 45? C, but were always unreasonably large. The cause is not clear, but there is some evidence that photo- decomposition of tetralin hydroperoxide at 45? C produces very active catalysts
262
This content downloaded from 169.229.32.137 on Thu, 8 May 2014 03:34:34 AMAll use subject to JSTOR Terms and Conditions
The autoxidation of tetralin
which interfere with the measurements. A similar effect probably accounts for the results of Burnett & Melville (i947b) on the benzoyl peroxide photosensitized polymerization of vinyl acetate. Photocatalysts are also formed in the polymeriza- tion of methyl methacrylate (Bamford & Dewar 9I48b). It is reasonable that this effect should only occur in our experiments at the higher temperatures, since under these conditions the absorption of light by peroxide extends to larger wave-lengths.
5
"r
4-
3s
-~2-8 o ,x
I I . I 0 5 10 15 20 25
10-5 (a+l/ d[O]t b (L.mol.-1)
FIGURE 10. Caledon Red BN, 25? C I(mol.l.-1sec.-l): 0, first series 1 10 x 10-8; E1, second series 2-08 x 10-8; x, third series 3-00 x 10-9; A, fourth series 7-40 x 10-9.
Accordingly, measurements of the after-effect were carried out at 0? C. Measure- ments were made only at high oxygen pressures, since it was considered that measurements at low oxygen pressure would not be sufficiently accurate for the calculation of the activation energies E3 and E5 to be significant. It will be seen later that values for these can be estimated in another way.
Equations (2) and (19) give, for high oxygen pressures,
AO2 k4 TH]
log (1+) k ( )
which may be used directly to obtain E4 - E. The observed figures were
log(1 = 2- 49 x 0-6 mol. 1.-1 at 0? C,
AG2 10-6 (22) and l 2 'l= 4-54 x 10-6 mol. 1.-1 at 25? C,
whence E4 - E = 4-1 kcal.
263
This content downloaded from 169.229.32.137 on Thu, 8 May 2014 03:34:34 AMAll use subject to JSTOR Terms and Conditions

C. H. Bamford and M. J. S. Dewar
ACTIVATION ENERGIES AND FREQUENCY FACTORS
Equations (10) and (22) give the values for E4 and E7 shown in table 4. Estimates of E, and E5 may be made as follows. From (18), E5> 26 kcal., since E3 cannot be
negative. E; corresponds to the reaction between two tetralyl radicals. The activa- tion energy for the reaction between two polystyryl radicals is 2.8 kcal. (Bamford & Dewar 1948c). In both cases the radicals are substituted benzyl radicals, and in the case of tetralin the activation energy should if anything be less, since any contribution due to diffusion would be small. Thus E5 28 kcal. It is therefore reasonable to take E5 = 2*6 and E3 = 00 kcal.
TABLE 4. ACTIVATION ENERGIES AND FREQUENCY FACTORS
E3 = 0-0 kcal. A3 = 6-8 x 107 E4 = 4-5 kcal. A4 = 2.5 x 104 E5 = 2-6 kcal. A5 = 5-5 x 10s E7 = 0-4 kcal. A7 = 42 x 107
DISCUSSION
(i) Three papers by Robertson & Waters (I948a,b,c) on the autoxidation of tetralin have recently appeared. As their conclusions differ from ours in certain
respects, some comment seems to be required. The main difference lies in the mode of termination postulated. Robertson & Waters
conclude that in the initial stage of the oxidation termination occurs by the reaction of tetralyl radicals and later by the reaction
TO2 + OH-TOH + 02. (23)
The main argument appears to be the improbability of termination mechanisms of other types.
In the first place, equation (2) implies that reaction between tetralyl radicals can
only be important at very low oxygen pressures, for under other conditions the concentration of tetralyl is extremely low. This might have been anticipated from the work of Bolland (I946). Secondly, the analytical results of Robertson & Waters not only can be interpreted in terms of our three termination reactions, but also throw some light on their mechanisms, which are probably as follows:
2 I \\ }\J+ I (a) \/\/ %/~/ \/\/
(T) 0--0 OOH
/) +11 }/ ) 1~\+ /\/1(b) >(24) ^\/\ ^\/\ or + I 1 +02
0 -0' OOH
2 1 1 - | l I + II , (C) %/\/ %/x/~ 1%/\/)
264
This content downloaded from 169.229.32.137 on Thu, 8 May 2014 03:34:34 AMAll use subject to JSTOR Terms and Conditions

The autoxidation of tetralin
The formation of dihydronaphthalene in the autoxidation at high oxygen pressures and the evolution of oxygen in the decomposition of tetralin hydroperoxide (Robert- son & Waters 1948a,b) provide strong evidence for reaction (24c). Robertson & Waters overlooked this possibility and regarded the presence of dihydronaph- thalene as evidence for the disproportionation of tetralyl radicals. At very high rates, when the chains are short, allowance would have to be made for this evolution of
oxygen. In our experiments the correction would never exceed about 3 %. It is also evident that the decomposition of tetralin hydroperoxide (Robertson
& Waters 1948b) can be interpreted in a similar way. (ii) Although absolute velocity constants have now been determined for a number
of radical reactions in solution (Swain & Bartlett I946; Burnett & Melville I947a; Bamford & Dewar I947a, 1948a,b,c, and other papers in the press from this
laboratory), all these have involved large radicals from polymerization processes. The measurements described in this paper seem to be the first on reactions of small radicals. It is now possible to make a preliminary survey of the field, and table 5
summarizes the data at present available for the activation energies and frequency factors of radical reactions in solution. The values for the propagation and transfer
reactions in the polymerizations of styrene have been calculated from the data of
Gregg & Mayo (1947), and Lewis, Walling, Cummings, Briggs & Mayo (1948) using our value (1948 a) for the propagation constant in the polymerization. The values for
vinyl acetate were obtained by Dixon-Lewis in these laboratories. In general the frequency factors are much lower than the 'normal' value for
bimolecular reactions in solution. This can hardly be a size effect, since the termina-
tion reactions which involve two large radicals have in fact the highest frequency factors, and since the frequency factors in the tetralin reactions are also low. Attention
was drawn to these points in a previous communication (Bamford & Dewar 1949), where it was suggested that the normal frequency factor for radical reactions in
solution is probably about 107.
(iii) The chain lengths were in the range 25 to 600 depending on the rate of initiation. The lifetime of a TO2 radical at 25? C is 1-02 x 10-2 sec.; this is of the same
order as the life of a polystyryl radical in the polymerization of styrene at 25? C
(6-2 x 10-3 sec., Bamford & Dewar 1948a). The lifetime of a T radical depends on the
oxygen pressure. At 100 mm. and 25? C it is 2-75 x 10-5 sec. It is interesting that the
life of a kinetic chain is of the order of seconds under our conditions, since it has
generally been assumed that the growth of radical chains is always extremely rapid. In the thermal polymerization of styrene at 0? C the mean lifetime of the kinetic
chain is 11 hr. (iv) It is interesting to compare the rate of initiation by benzoyl peroxide with the
published rate of decomposition of the peroxide. From the values quoted by Swain & Bartlett (I 946) the rate of decomposition of benzoyl peroxide in 1 % solution is about 10-9 mol. 1.-lsec.-1. The observed rate of initiation was 2-09 x 10-10mol. 1.-i sec.-1.
Since each molecule of peroxide could theoretically initiate two chains, there is
clearly a discrepancy of a factor of 10. This might be explained in several ways
(e.g. primary recombination, non-radical decomposition of the peroxide, or induced
decomposition of the peroxide by radicals), but in any case it throws doubt on the
265
This content downloaded from 169.229.32.137 on Thu, 8 May 2014 03:34:34 AMAll use subject to JSTOR Terms and Conditions

266 C. H. Bamford and M. J. S. Dewar
TABLE 5
reaction E1 kcal. log,o A
polymerization of styrene: propagation 6-5 6.01 transfer 14-2 7-18 termination 2-8 8-49
polymerization of vinyl acetate:
propagation 3.0 5.84 transfer 5-9 3-86 termination 0.0 8-30
co-polymerization, reactions of
polystyryl radicals with:
methyl methacrylate 6-0 6-01 methyl acrylate 6-1 5.90 diethyl maleate 7*2 5.60 diethyl fumarate 5-4 5 83 p-chlorostyrene 6*1 5-91
chain transfer reactions of styrene with solvents:
benzene 21-3 15-07 toluene 16-6 10-04 ethylbenzene 12-0 4-74 isopropylbenzene 12-0 4-92 tertbutylbenzene 20-2 14-68 diphenylmethane 10-2 3-25 triphenylmethane 11-6 6-25 fluorene 9-6 5-82 cyclohexane 19-9 13'22 n-heptane 11-5 3-57 ethylene dichloride 21-2 11.10* ethylene dibromide 16-2 8-48* carbon tetrachloride 10-7 6-88* carbon tetrabromide 6-7 6-59*
autoxidation of tetralin:
k3 0 7-82 k4 4-5 4-40 k5 2-6 8-74 k7 0-4 7-62
* Bamford & Dewar (1947b).
validity of work in which the rate of chains starting has been equated to the rate of decomposition of the peroxide (cf. Swain & Bartlett 1946; Bolland 1948. The work of the former authors will be discussed in this connexion elsewhere).
(v) The techniques described in this paper can clearly be extended to other free radical reactions, e.g. the addition of halogen compounds to olefines, and work along such lines is in progress here.
REFERENCES
Bamford, C. H. & Dewar, M. J. S. I947a Disc. Faraday Soc. 2, 310. Bamford, C. H. & Dewar, M. J. S. I947b Disc. Faraday Soc. 2, 314. Bamford, C. H. & Dewar, M. J. S. 1948a Proc. Roy. Soc. A, 192, 309. Bamford, C. H. & Dewar, M. J. S. I948b Proc. Roy. Soc. A, 192, 329. Bamford, C. H. & Dewar, M. J. S. 1948c In course of publication.
This content downloaded from 169.229.32.137 on Thu, 8 May 2014 03:34:34 AMAll use subject to JSTOR Terms and Conditions

The autoxidation of tetraltn 267
Bamford, C. H. & Dewar, M. J. S. I949 Nature, 163, 256.
Bolland, J. L. 1946 Proc. Roy. Soc. A, 186, 218.
Bolland, J. L. 1948 Trans. Faraday Soc. 44, 669.
Bolland, J. L. & Gee, G. I946 Trans. Faraday Soc. 42, 236, 244. Bolland, J. L. & ten Have, P. 1947a Trans. Faraday Soc. 43, 201. Bolland, J. L. & ten Have, P. I947b Disc. Faraday Soc. 2, 252. Briers, F., Chapman, D. L. & Walters, E. 1926 J. Chem. Soc. p. 562. Burnett, G. M. & Melville, H. W. 1947a Proc. Roy. Soc. A, 189, 456. Burnett, G. M. & Melville, H. W. I947b Proc. Roy. Soc. A, 189, 474. Fischer, F. & Pfleiderer, G. 1922 Z. anorg. Chem. 124, 61.
George, P. 1946a Proc. Roy. Soc. A, 185, 337.
George, P. I946b Trans. Faraday Soc. 42, 210.
George, P., Rideal, E. K. & Robertson, A. 1946 Proc. Roy. Soc. A, 185, 288. George, P. & Robertson, A. x946a Proc. Roy. Soc. A, 185, 309.
George, P. & Robertson, A. 9I46b Trans. Faraday Soc. 42, 217.
Gregg, R. A. & Mayo, F. R. 1947 Disc. Faraday Soc. 2, 328. Lewis, F. M., Walling, C., Cummings, W., Briggs, E. R. & Mayo, F. R. I948 J. Amer. Chem.
Soc. 70, 1519.
Noyes, W. A. & Leighton, P. A. 194I Photochemistry of gases. New York: Reinhold. Robertson, A. & Waters, W. A. 1946 Trans. Faraday Soc. 42, 201. Robertson, A. & Waters, W. A. 1948a J. Chem. Soc. p. 1574. Robertson, A. & Waters, W. A. x948b J. Chem. Soc. p. 1578. Robertson, A. & Waters, W. A. I948c J. Chem. Soc. p. 1585. Swain, C. G. & Bartlett, P. D. I946 J. Amer. Chem. Soc. 68, 2381. Weber, G. 1948 Trans. Faraday Soc. 44, 185.
The dissociation energy of the N-N bond in hydrazine BY M. SZWARC, Chemistry Department, University of Manchester
(Communicated by M. G. Evans, F.R.S.-Received 19 January 1949)
The thermal decomposition of hydrazine was investigated over a temperature range of 630 to 780? C and at pressures of a few mm. Hg. The experiments were carried out in a flow system, toluene being used as a carrier gas. This technique makes it possible to discriminate between the heterogeneous decomposition of hydrazine
3N2H4- N2 + 4NH,, (1)
and 2N2H4- H2 + N2 + 2NH (2)
and the homogeneous decomposition to
N2H4-> 2NH2.. (3)
The NH2 radicals produced by the latter process were removed, in the presence of excess toluene, by the rapid reaction (4)
C6H5. CH, + NH2,- C6H5. CH2. + NH3. (4) Thus the rate of formation of dibenzyl measures the rate of reaction (3). The study of the stoichiometry of the overall process, of the kinetics of various steps and of the effect of packing the reaction vessel led to the conclusion that (3) is a homogeneous, unimolecular gas reaction, the rate constant being 4 x 1012 exp (60,000/RT). Assuming that the recombination of NH2 radicals does not involve any energy of activation, it is found that D(NH2-NH,) = 60 + 3 kcal./mole. This value in conjunction with the relevant thermochemical data leads to the heat of formation of NH2 radical as 41 kcal./mole and to D(NH2-H) = 104 ? 2 kcal./mole. The latter is good evidence in support of Gaydon's value of 225 kcal./mole for the heat of dis- sociation of N,.
The autoxidation of tetraltn 267
Bamford, C. H. & Dewar, M. J. S. I949 Nature, 163, 256.
Bolland, J. L. 1946 Proc. Roy. Soc. A, 186, 218.
Bolland, J. L. 1948 Trans. Faraday Soc. 44, 669.
Bolland, J. L. & Gee, G. I946 Trans. Faraday Soc. 42, 236, 244. Bolland, J. L. & ten Have, P. 1947a Trans. Faraday Soc. 43, 201. Bolland, J. L. & ten Have, P. I947b Disc. Faraday Soc. 2, 252. Briers, F., Chapman, D. L. & Walters, E. 1926 J. Chem. Soc. p. 562. Burnett, G. M. & Melville, H. W. 1947a Proc. Roy. Soc. A, 189, 456. Burnett, G. M. & Melville, H. W. I947b Proc. Roy. Soc. A, 189, 474. Fischer, F. & Pfleiderer, G. 1922 Z. anorg. Chem. 124, 61.
George, P. 1946a Proc. Roy. Soc. A, 185, 337.
George, P. I946b Trans. Faraday Soc. 42, 210.
George, P., Rideal, E. K. & Robertson, A. 1946 Proc. Roy. Soc. A, 185, 288. George, P. & Robertson, A. x946a Proc. Roy. Soc. A, 185, 309.
George, P. & Robertson, A. 9I46b Trans. Faraday Soc. 42, 217.
Gregg, R. A. & Mayo, F. R. 1947 Disc. Faraday Soc. 2, 328. Lewis, F. M., Walling, C., Cummings, W., Briggs, E. R. & Mayo, F. R. I948 J. Amer. Chem.
Soc. 70, 1519.
Noyes, W. A. & Leighton, P. A. 194I Photochemistry of gases. New York: Reinhold. Robertson, A. & Waters, W. A. 1946 Trans. Faraday Soc. 42, 201. Robertson, A. & Waters, W. A. 1948a J. Chem. Soc. p. 1574. Robertson, A. & Waters, W. A. x948b J. Chem. Soc. p. 1578. Robertson, A. & Waters, W. A. I948c J. Chem. Soc. p. 1585. Swain, C. G. & Bartlett, P. D. I946 J. Amer. Chem. Soc. 68, 2381. Weber, G. 1948 Trans. Faraday Soc. 44, 185.
The dissociation energy of the N-N bond in hydrazine BY M. SZWARC, Chemistry Department, University of Manchester
(Communicated by M. G. Evans, F.R.S.-Received 19 January 1949)
The thermal decomposition of hydrazine was investigated over a temperature range of 630 to 780? C and at pressures of a few mm. Hg. The experiments were carried out in a flow system, toluene being used as a carrier gas. This technique makes it possible to discriminate between the heterogeneous decomposition of hydrazine
3N2H4- N2 + 4NH,, (1)
and 2N2H4- H2 + N2 + 2NH (2)
and the homogeneous decomposition to
N2H4-> 2NH2.. (3)
The NH2 radicals produced by the latter process were removed, in the presence of excess toluene, by the rapid reaction (4)
C6H5. CH, + NH2,- C6H5. CH2. + NH3. (4) Thus the rate of formation of dibenzyl measures the rate of reaction (3). The study of the stoichiometry of the overall process, of the kinetics of various steps and of the effect of packing the reaction vessel led to the conclusion that (3) is a homogeneous, unimolecular gas reaction, the rate constant being 4 x 1012 exp (60,000/RT). Assuming that the recombination of NH2 radicals does not involve any energy of activation, it is found that D(NH2-NH,) = 60 + 3 kcal./mole. This value in conjunction with the relevant thermochemical data leads to the heat of formation of NH2 radical as 41 kcal./mole and to D(NH2-H) = 104 ? 2 kcal./mole. The latter is good evidence in support of Gaydon's value of 225 kcal./mole for the heat of dis- sociation of N,.
This content downloaded from 169.229.32.137 on Thu, 8 May 2014 03:34:34 AMAll use subject to JSTOR Terms and Conditions





























