Embed Size (px)
Citation preview

Tt
XWa
b
c
d
a
ARRAA
KASDCB
1
wcmr[mffi5su[
z
h0
International Journal of Biological Macromolecules 70 (2014) 284–291
Contents lists available at ScienceDirect
International Journal of Biological Macromolecules
j ourna l ho me pa g e: www.elsev ier .com/ locate / i jb iomac
he atmospheric and room-temperature plasma (ARTP) method onhe dextranase activity and structure
iaobei Wanga,c, Mingsheng Lub,c, Shujun Wangb,c,∗∗, Yaowei Fangb,c, Delong Wanga,ei Rend,c, Gengmao Zhaoa,∗
Key Laboratory of Marine Biology, Nanjing Agricultural University, Nanjing, Jiangsu 210095, ChinaJiangsu Marine Resources Development Research Institute, Lianyungang, Jiangsu 222005, ChinaSchool of Marine Science and Technology, Huaihai Institute of Technology, Lianyungang, Jiangsu 222005, ChinaCollege of Food Science and Engineering, Dalian Ocean University, Dalian, Liaoning 116023, China
r t i c l e i n f o
rticle history:eceived 6 May 2014eceived in revised form 5 July 2014ccepted 7 July 2014vailable online 11 July 2014
a b s t r a c t
A novel atmospheric and room-temperature plasma (ARTP) method was used to breed high-yieldingmutations of Arthrobacter KQ11. Mutagenesis produced two mutations, 4-1 and 4-13, which increasedenzyme activity by 19 and 30%, respectively. Dents on the cell envelope were observed under scanningelectron microscopy (SEM). The optimal temperature and pH of the wild strain were 45 ◦C and 5.5 andthose of the mutant strains were 45 ◦C, pH 6.0 (4-1) and 50 ◦C, pH 6.0 (4-13). Under optimal enzyme
eywords:RTPEMextranaserystal structureiofilm
production conditions of the wild and mutant strains, the dextranase activity of 4-13 was 50% higherthan that of the wild strain. Through amino acid alignment, several nucleotides of the mutant strainswere found to have changed. Experiments performed in vitro suggested that this endo-dextranase mayinhibit biofilm formation by Streptococcus mutans.
© 2014 Elsevier B.V. All rights reserved.
. Introduction
Dextranase can hydrolyze the �-1,6-glycosidic bond of glucanithin or at the end of the chain. This gives dextranase significant
ommercial value. It is widely used in the sugar industry, in theodification of alternan, in the synthesis of isomaltooligosaccha-
ide (IMO), and in the prevention and treatment of dental plaques1,2]. Artificially produced dextranases have already been used in
any fields, but most dextranases are from fungi [3]. Dextranaserom Arthrobacter KQ11 has many advantages over dextranasesrom fungi, such as a shorter fermentation period and fewer safetyssues. The optimal pH of dextranase from Arthrobacter KQ11 is.5–6.5, its optimal temperature is 50 ◦C, and its stability is verytrong [4]. Dextranase from Arthrobacter KQ11 is more suitable for
se in the prevention of dental plaques than other dextranases are5].∗ Corresponding author. Tel.: +86 02584396678; fax: +86 02584396678.∗∗ Corresponding author. Tel.: +86 051885895421; fax: +86 051885895429.
E-mail addresses: [email protected] (S. Wang), [email protected],[email protected] (G. Zhao).
ttp://dx.doi.org/10.1016/j.ijbiomac.2014.07.006141-8130/© 2014 Elsevier B.V. All rights reserved.
However, the dextranase activity of Arthrobacter KQ11 producedon an industrial-scale still fails to satisfy demand. Obtaining a high-yield strain capable of enzyme activity would be very useful. Thisneed developed side by side with mutation. Although genetic engi-neering can be used to increase enzyme activity, there are somepossible safety hazards. In this present paper, ARTP was chosen asa mutagenic method.
In 1928, Stadler [6] reported that X-rays could be used in themutagenesis of barley and maize. Many other kinds of physical andchemical mutagenesis processes have since become widely used[7,8]. However, these traditional methods have many disadvan-tages, such as low mutagenic efficiency and safety issues. Plasmamutagenesis is not subject to these problems, and it has drawnresearchers’ attention [9,10]. It is more effective, less toxic, andless costly than other methods [11]. It has also been successfullyused on several microorganisms, such as Bacillus subtilis, whoseantibiotic production increased by 23% [12], Clostridium beijerinckii,whose butanol production increased by 37% [13], and Streptomycesavermitilis, whose avermectin production increased by 21% [10].
However, there have been few reports of ARTP in improvement ofenzyme activity. In this project, strain Arthrobacter oxydans KQ11,which was isolated from sea mud, was subjected ARTP in order toincrease the activity of dextranase.
iologi
2
2
lymb(p
2
mtwonip(1cotht1Oef1ct
2
tsfTc
2
tuw3daod4[I
2
it
X. Wang et al. / International Journal of B
. Materials and methods
.1. Microbes and medium
A. oxydans KQ11 was isolated from sea mud and stored in theaboratory. The seed medium (g l−1) was as follows: peptone 5,east powder 1, sodium chloride 4, pH 7.0. Early screening cultureedium (g l−1) contained the following: peptone 5, yeast extract 1,
lue dextran 2, NaCl 4, agar 20, pH 7.0. Enzyme production mediumg l−1) was as follows: peptone 5, yeast extract 1, dextran 7, NaCl 4,H 7.0.
.2. Experimental protocol
The strain KQ11 was cultivated at 30 ◦C and 150 × g to logarith-ic phase (16 h). Then 1 ml of bacteria was moved on to sterile
ubes and the concentration of bacteria was adjusted to OD600 = 0.1ith sterile saline. Then 10 �l of bacteria were removed and spread
n a 1-cm plate. This plate was later exposed to the ARTP system’sozzle exit (Siqingyuan, Wuxi, China) [14]. In this study, the work-
ng parameters such as Pin, Pout, and the distance (D) between thelasma torch nozzle exit and the sample plate were set to 120 WPin), 20 W (Pout), and 2 mm (D). The flow rate of helium gas was5 slm. After mutation, the plate was placed in a new sterile tubeontaining 300 �l of sterile water, and 100 �l of bacteria was spreadn the early screening culture medium, which contained blue dex-ran. After 48 h of cultivation at 30 ◦C, colonies that exhibited largealos were selected and inoculated into 250 ml shake flasks con-aining 50 ml of medium. Cultivation was performed at 30 ◦C and50 × g for 16 h. The concentration of bacteria was adjusted toD600 = 1 with sterile saline. Then the bacteria were inoculated intonzyme production medium and cultivated at 30 ◦C and 150 × gor 24 h. After fermentation, the culture broth was centrifuged at2,000 × g for 10 min to remove the bacterial cells. The cell-freeulture supernatant was used to calculate the enzyme activities ofhe mutant strains.
.3. Subcultures of mutation strains
The genetic stability of the mutations was evaluated by subcul-uring, as described below [13]. The mutations were incubated onlant medium and cultivated for 12 h at 30 ◦C. Bacteria were takenrom one slant and inoculated on the next for seven generations.he enzyme activity of every generation was measured in a 250-mlonical flask containing 50 ml of enzyme production medium.
.4. Morphological changes in the cells
Then 10 �l of strain KQ11, 4-1 and 4-13, which were cultivatedo logarithmic phase, were moved to cover glasses, then roastedntil as dry as velum on a super clean bench. The cover glassesere immersed in liquid containing 2.5% (v/v) glutaraldehyde for
–4 h. Then the cover glasses were immersed in 1% (w/v) osmic acidissolved in cacodylated buffer for 1–1.5 h. After this step, gradu-ted dehydration was performed with ethanol. The concentrationf ethanol ranged from 50 to 100% with a gradient of 10%. Every gra-ient took 10 to 15 min. All these procedures were carried out at◦C. The cover glasses were air-dried and sputter-coated with gold
15]. The sample was examined by SEM (Hitachi S-4000; Hitachinstruments Inc., San Jose, CA, USA).
.5. Protein molecular weight
The wild strain KQ11 and mutant strains 4-1 and 4-13 werenoculated into seed medium and cultivated at 30 ◦C and 150 × go logarithmic phase. The concentration of bacteria was adjusted
cal Macromolecules 70 (2014) 284–291 285
to OD600 = 1 with sterile saline. Then the bacteria were inoculatedinto enzyme production medium and cultivated at 30 ◦C and 150 × gfor 28 h. Then 10 ml of bacteria were centrifuged at 12,000 × g for10 min to remove thallus, and the cell-free culture supernatant wasconcentrated to 1 ml using Millipore 30000. The molecular massof dextranase was estimated using 10% sodium dodecyl sulfatepolyacrylamide gel (SDS-PAGE) containing 0.5% blue dextran. Afterelectrophoresis, staining was performed using 0.1% Coomassie bril-liant blue R-250 (Bio-Rad, USA), and protein bands were detected.For dextranase activity staining, the blue gel was washed for 30 minat room temperature in 20 mM Tris–HCl buffer (pH 7.5) containing2.5% (v/v) Triton X-100 before being incubated in 20 mM Tris–HClat 37 ◦C until the white bands were visible [16].
2.6. Kinetic studies and enzyme production conditions of wildand mutant strains
Optimal temperature: The optimal temperatures for dextranaseactivity were determined by incubating the enzymes with sub-strate (3% dextran 20000) at different temperatures (30, 35, 40, 45,50, and 55 ◦C) for 15 min to study the influence of temperature ondextranases from wild and mutant strains.
Optimal pH: To study the influence of pH on dextranases fromwild and mutant strains, the substrate (3% dextran 20000) was dis-solved in three different buffers, 50 mM acetic acid sodium acetatebuffer (pH 5.5, 6.0), 50 mM phosphate buffer (pH 6.5, 7.0), and50 mM Tris–HCl buffer (pH 7.5, 8.0). Then the dextranases fromthe wild and mutant strains were incubated with the substrate for15 min.
In order to study the enzyme production conditions of the wildand mutant strains, different carbon sources, such as dextran, cornstarch, sweet potato starch, and glucose and different nitrogensources such as yeast extract, soybean meal, peptone, and NaNO3were used to study the enzyme production conditions. Then theoptimal cultivate temperature and pH of wild and mutant strainswere characterized.
Optimal cultivation temperature: The wild strain KQ11 andmutant strains 4-1 and 4-13 were inoculated into the seed mediumand cultivated at 30 ◦C, 150 × g to logarithmic phase. The concentra-tion of bacteria was adjusted to OD600 = 1 with sterile saline. Thenthe bacteria were inoculated into enzyme production medium andcultivated at 20, 25, 30, and 35 ◦C at 150 × g for 28 h and the cell-freeculture supernatant was used to calculate the enzyme activity.
Optimal cultivation pH: The same method was used to prepareseed liquid. The bacteria were inoculated to enzyme productionmedium with different pH, 50 mM acetic acid sodium acetate buffer(pH 5.5, 6.0), 50 mM phosphate buffer (pH 6.5, 7.0), and 50 mMTris–HCl buffer (pH 7.5, 8.0), and cultivated at 30 ◦C and 150 × g for28 h. Then the enzyme activity was calculated.
2.7. Mutant sites on the protein
Genomic DNA was obtained from mutant strains 4-1 and 4-13.The dextranase gene was amplified using polymerase chain reac-tion. Sense primer 5′-CGCGGATCCCAGGAGCCCCGCTGCGACAGA-3′. Antisense primer 5′-CCCAAGCTTCCACGCGTTCCA TTATCCA-3′.The reaction procedure was set as follows: (94 ◦C, 3 min) ×1 cycle,(98 ◦C, 10 s; 58 ◦C, 30 s; 68 ◦C, 2 min) ×35 cycles, and (68 ◦C, 5 min)×1 cycle. The products were separated using 1% agarose gels and theproducts, which were approximately 2000 bp in size, were excised,
purified, and joined to T-tailed vector PMD-19. The recombinantplasmids were transformed into DH 5�-competent cells. Then cellswhich contained recombinant plasmid were used for sequencing.All steps were carried out according to the manufacturer’s protocol.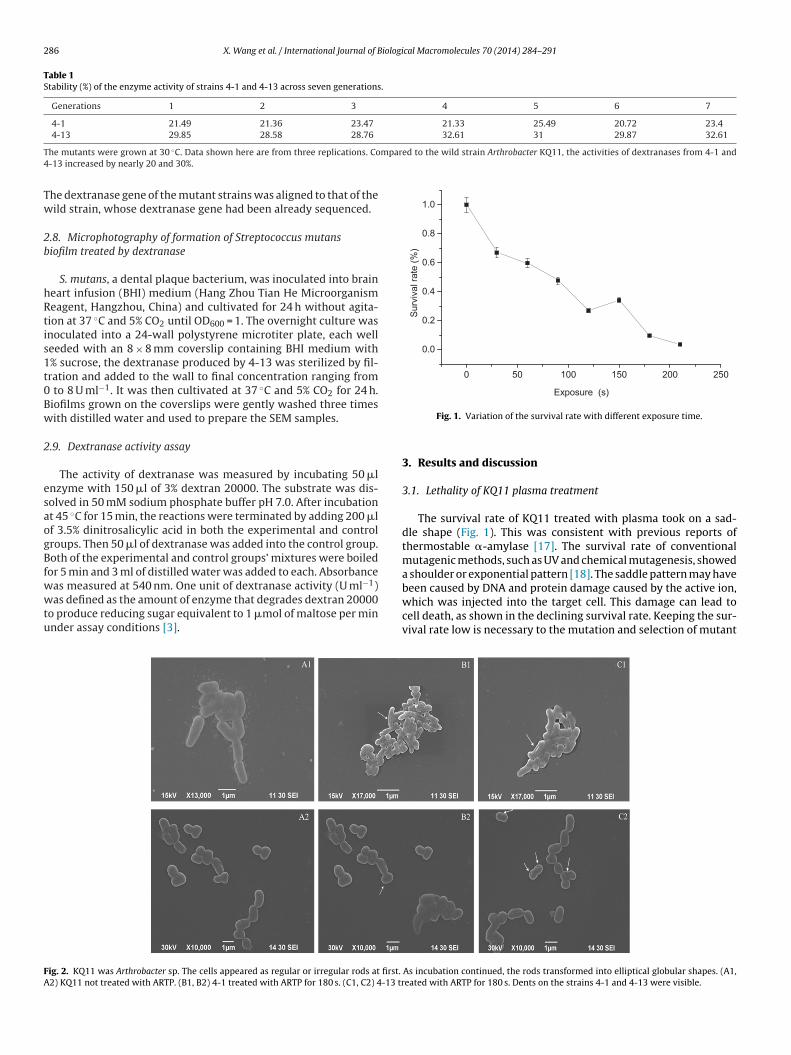
286 X. Wang et al. / International Journal of Biological Macromolecules 70 (2014) 284–291
Table 1Stability (%) of the enzyme activity of strains 4-1 and 4-13 across seven generations.
Generations 1 2 3 4 5 6 7
4-1 21.49 21.36 23.47 21.33 25.49 20.72 23.44-13 29.85 28.58 28.76 32.61 31 29.87 32.61
T mpared to the wild strain Arthrobacter KQ11, the activities of dextranases from 4-1 and4
Tw
2b
hRtis1t0Bw
2
esaogBfwwtu
250200150100500
0.0
0.2
0.4
0.6
0.8
1.0
Surv
ival
rate
(%)
Expo sure (s )
FA
he mutants were grown at 30 ◦C. Data shown here are from three replications. Co-13 increased by nearly 20 and 30%.
he dextranase gene of the mutant strains was aligned to that of theild strain, whose dextranase gene had been already sequenced.
.8. Microphotography of formation of Streptococcus mutansiofilm treated by dextranase
S. mutans, a dental plaque bacterium, was inoculated into braineart infusion (BHI) medium (Hang Zhou Tian He Microorganismeagent, Hangzhou, China) and cultivated for 24 h without agita-ion at 37 ◦C and 5% CO2 until OD600 = 1. The overnight culture wasnoculated into a 24-wall polystyrene microtiter plate, each welleeded with an 8 × 8 mm coverslip containing BHI medium with% sucrose, the dextranase produced by 4-13 was sterilized by fil-ration and added to the wall to final concentration ranging from
to 8 U ml−1. It was then cultivated at 37 ◦C and 5% CO2 for 24 h.iofilms grown on the coverslips were gently washed three timesith distilled water and used to prepare the SEM samples.
.9. Dextranase activity assay
The activity of dextranase was measured by incubating 50 �lnzyme with 150 �l of 3% dextran 20000. The substrate was dis-olved in 50 mM sodium phosphate buffer pH 7.0. After incubationt 45 ◦C for 15 min, the reactions were terminated by adding 200 �lf 3.5% dinitrosalicylic acid in both the experimental and controlroups. Then 50 �l of dextranase was added into the control group.oth of the experimental and control groups’ mixtures were boiled
or 5 min and 3 ml of distilled water was added to each. Absorbance
as measured at 540 nm. One unit of dextranase activity (U ml−1)as defined as the amount of enzyme that degrades dextran 20000o produce reducing sugar equivalent to 1 �mol of maltose per minnder assay conditions [3].
ig. 2. KQ11 was Arthrobacter sp. The cells appeared as regular or irregular rods at first.
2) KQ11 not treated with ARTP. (B1, B2) 4-1 treated with ARTP for 180 s. (C1, C2) 4-13 tr
Fig. 1. Variation of the survival rate with different exposure time.
3. Results and discussion
3.1. Lethality of KQ11 plasma treatment
The survival rate of KQ11 treated with plasma took on a sad-dle shape (Fig. 1). This was consistent with previous reports ofthermostable �-amylase [17]. The survival rate of conventionalmutagenic methods, such as UV and chemical mutagenesis, showeda shoulder or exponential pattern [18]. The saddle pattern may havebeen caused by DNA and protein damage caused by the active ion,
which was injected into the target cell. This damage can lead tocell death, as shown in the declining survival rate. Keeping the sur-vival rate low is necessary to the mutation and selection of mutantAs incubation continued, the rods transformed into elliptical globular shapes. (A1,eated with ARTP for 180 s. Dents on the strains 4-1 and 4-13 were visible.
iological Macromolecules 70 (2014) 284–291 287
st
3
cs4o
wcalmis
mrcDmaTi
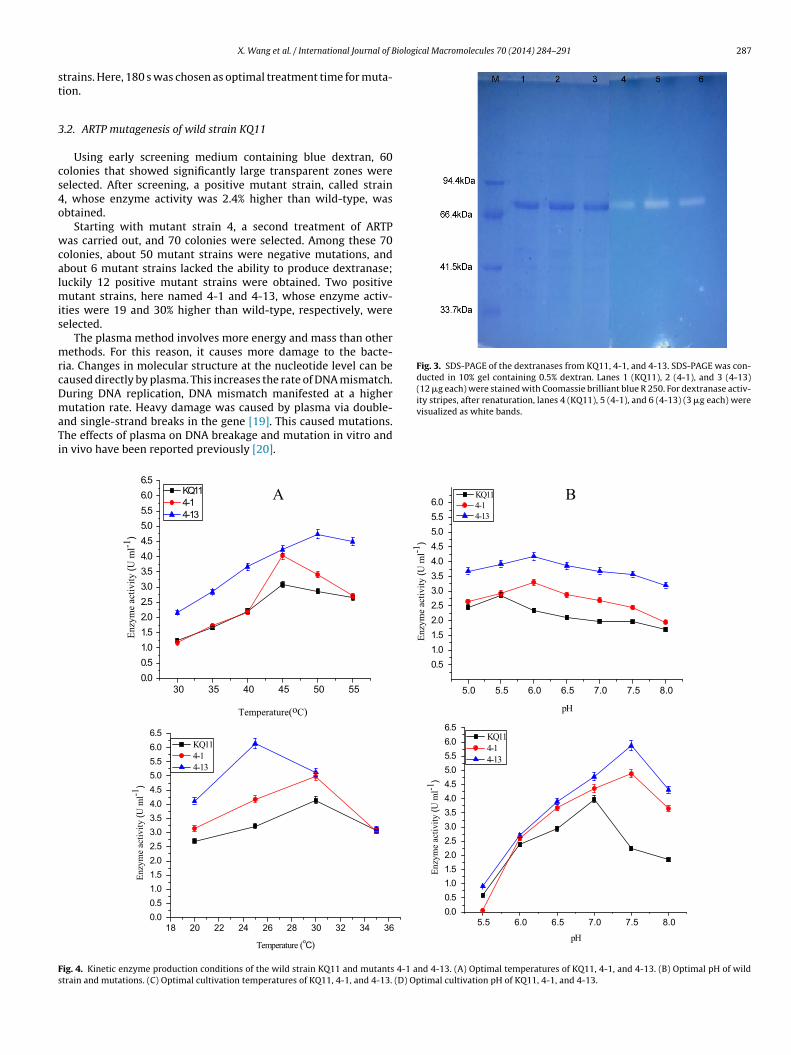
Fig. 3. SDS-PAGE of the dextranases from KQ11, 4-1, and 4-13. SDS-PAGE was con-ducted in 10% gel containing 0.5% dextran. Lanes 1 (KQ11), 2 (4-1), and 3 (4-13)(12 �g each) were stained with Coomassie brilliant blue R 250. For dextranase activ-ity stripes, after renaturation, lanes 4 (KQ11), 5 (4-1), and 6 (4-13) (3 �g each) were
Fs
X. Wang et al. / International Journal of B
trains. Here, 180 s was chosen as optimal treatment time for muta-ion.
.2. ARTP mutagenesis of wild strain KQ11
Using early screening medium containing blue dextran, 60olonies that showed significantly large transparent zones wereelected. After screening, a positive mutant strain, called strain, whose enzyme activity was 2.4% higher than wild-type, wasbtained.
Starting with mutant strain 4, a second treatment of ARTPas carried out, and 70 colonies were selected. Among these 70
olonies, about 50 mutant strains were negative mutations, andbout 6 mutant strains lacked the ability to produce dextranase;uckily 12 positive mutant strains were obtained. Two positive
utant strains, here named 4-1 and 4-13, whose enzyme activ-ties were 19 and 30% higher than wild-type, respectively, wereelected.
The plasma method involves more energy and mass than otherethods. For this reason, it causes more damage to the bacte-
ia. Changes in molecular structure at the nucleotide level can beaused directly by plasma. This increases the rate of DNA mismatch.uring DNA replication, DNA mismatch manifested at a higher
utation rate. Heavy damage was caused by plasma via double-nd single-strand breaks in the gene [19]. This caused mutations.he effects of plasma on DNA breakage and mutation in vitro andn vivo have been reported previously [20].
visualized as white bands.
5550454035300.00.51.01.52.02.53.03.54.04.55.05.56.06.5
Enzy
me
activ
ity (U
ml-1
)
Temperature(oC)
KQ11 4-1 4-13
8.07.57.06.56.05.55.0
0.51.01.52.02.53.03.54.04.55.05.56.0
Enzy
me
activ
ity (U
ml-1
)
pH
KQ11 4-1 4-13
363432302826242220180.00.51.01.52.02.53.03.54.04.55.05.56.06.5
Enzy
me
activ
ity (U
ml-1
)
Temperature (oC)
KQ 11 4-1 4-13
8.07.57.06.56.05.50.00.51.01.52.02.53.03.54.04.55.05.56.06.5
Enzy
me
activ
ity (U
ml-1
)
pH
KQ11 4-1 4-13
A B
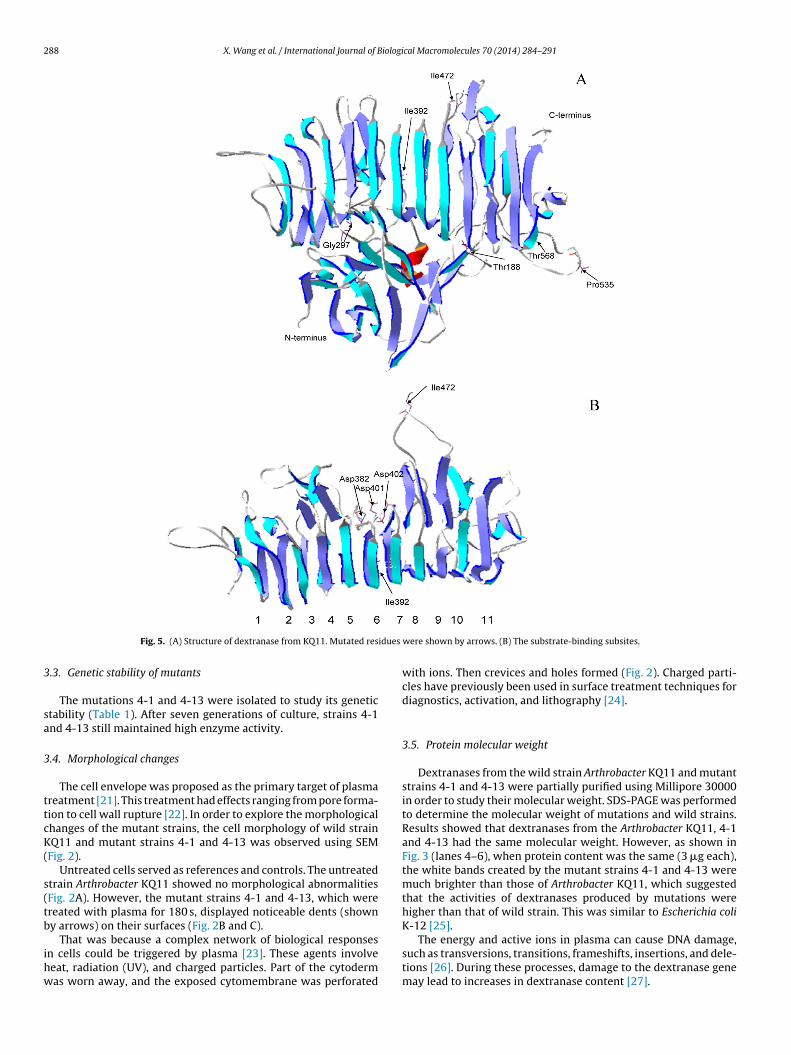
ig. 4. Kinetic enzyme production conditions of the wild strain KQ11 and mutants 4-1 and 4-13. (A) Optimal temperatures of KQ11, 4-1, and 4-13. (B) Optimal pH of wildtrain and mutations. (C) Optimal cultivation temperatures of KQ11, 4-1, and 4-13. (D) Optimal cultivation pH of KQ11, 4-1, and 4-13.
288 X. Wang et al. / International Journal of Biological Macromolecules 70 (2014) 284–291
idues
3
sa
3
ttcK(
s(tb
ihw
K-12 [25].
Fig. 5. (A) Structure of dextranase from KQ11. Mutated res
.3. Genetic stability of mutants
The mutations 4-1 and 4-13 were isolated to study its genetictability (Table 1). After seven generations of culture, strains 4-1nd 4-13 still maintained high enzyme activity.
.4. Morphological changes
The cell envelope was proposed as the primary target of plasmareatment [21]. This treatment had effects ranging from pore forma-ion to cell wall rupture [22]. In order to explore the morphologicalhanges of the mutant strains, the cell morphology of wild strainQ11 and mutant strains 4-1 and 4-13 was observed using SEM
Fig. 2).Untreated cells served as references and controls. The untreated
train Arthrobacter KQ11 showed no morphological abnormalitiesFig. 2A). However, the mutant strains 4-1 and 4-13, which werereated with plasma for 180 s, displayed noticeable dents (showny arrows) on their surfaces (Fig. 2B and C).
That was because a complex network of biological responsesn cells could be triggered by plasma [23]. These agents involveeat, radiation (UV), and charged particles. Part of the cytodermas worn away, and the exposed cytomembrane was perforated
were shown by arrows. (B) The substrate-binding subsites.
with ions. Then crevices and holes formed (Fig. 2). Charged parti-cles have previously been used in surface treatment techniques fordiagnostics, activation, and lithography [24].
3.5. Protein molecular weight
Dextranases from the wild strain Arthrobacter KQ11 and mutantstrains 4-1 and 4-13 were partially purified using Millipore 30000in order to study their molecular weight. SDS-PAGE was performedto determine the molecular weight of mutations and wild strains.Results showed that dextranases from the Arthrobacter KQ11, 4-1and 4-13 had the same molecular weight. However, as shown inFig. 3 (lanes 4–6), when protein content was the same (3 �g each),the white bands created by the mutant strains 4-1 and 4-13 weremuch brighter than those of Arthrobacter KQ11, which suggestedthat the activities of dextranases produced by mutations werehigher than that of wild strain. This was similar to Escherichia coli
The energy and active ions in plasma can cause DNA damage,such as transversions, transitions, frameshifts, insertions, and dele-tions [26]. During these processes, damage to the dextranase genemay lead to increases in dextranase content [27].

X. Wang et al. / International Journal of Biological Macromolecules 70 (2014) 284–291 289
Fig. 6. Dextranase on biofilm formation by S. mutans.

2 iological Macromolecules 70 (2014) 284–291
3K
atdttadthpwf6
otf[pp
tttge5ewsweyn
bptt4tatebciTp
3
sAtotAt
d
Table 2Sites of mutation of mutant strains induced by ARTP relative to wild strain KQ11,the signal peptide which contain 22 amino acid has already been removed.
Strains Nucleotide Number Amino acid Number
4-1 A → G 347G → A 449G → A 521G → A 1175 M → I 392T → C 1397
4-13 A → G 347G → A 521T → C 562 I → T 188A → G 889 E → G 297T → C 1397G → A 1415 M → I 472
90 X. Wang et al. / International Journal of B
.6. Kinetic and enzyme production conditions of ArthrobacterQ11, 4-1, and 4-13
Dextranase activities from the wild strain Arthrobacter KQ11nd mutant strains 4-1 and 4-13 were measured across a broademperature range. The optimal temperatures of wild and mutantextranase were found to be different (Fig. 4A). Dextranase fromhe wild strain showed maximum activity at 45 ◦C and those fromhe mutant strains 4-1 and 4-13 showed maximum activity at 45nd 50 ◦C, respectively. After exceeding optimum temperature, aecrease in the enzyme activity was observed. This was caused byhe instability of the dextranase. The disturbances were caused byigher temperatures through intra-molecular attractions betweenolar and non-polar groups within the protein structure, whichere broken and generated by these disturbances. The dextranases
rom fungi generally showed maximum activity between 55 and0 ◦C [28].
Three different buffers were used to determine the optimum pHf dextranases from both wild and mutant strains. Results showedhat the optimal pH for both wild and mutant strains was also dif-erent (Fig. 4B). This is also true of dextran-synthesizing enzyme29]. Dextranase from the wild strain showed maximum activity atH 5.5, and the dextranases from the mutant strains 4-1 and 4-13eaked at pH 6.0.
Different sources of carbon and nitrogen were used to studyhe enzyme production of both wild and mutant strains to iden-ify ideal enzyme production conditions. Results showed that whenhe carbon sources were dextran and corn starch and the nitro-en sources were soybean meal, NaNO3, and yeast extract, thenzyme activity levels of the wild and mutant strains were 4.13,.08, and 6.27 U ml−1. Under the same fermentation conditions,nzyme production by mutant strain 4-13 was 1.5-fold that of theild strain. No enzyme activity was observed when only corn starch
erved as a carbon source. This indicated that enzyme productionas inducible, as reported previously [30]. Moreover, when yeast
xtract was added to the fermentation medium, the dextranaseield increased immediately, suggesting that a single source ofitrogen was insufficient for higher enzyme production [31].
Experiments were performed with the same concentrations ofacteria and the same inoculum size but different cultivation tem-eratures and pH levels. Results showed the optimal cultivationemperatures of KQ11, 4-1, and 4-13 to be 30, 30, and 25 ◦C, respec-ively (Fig. 4C). The optimal cultivation pH levels of KQ11, 4-1, and-13 were 7.0, 7.5, and 7.5, respectively (Fig. 4D). After reachinghe optimal temperature, the enzyme activity started to decrease,nd the decline in dextranase production may have been caused byhe low multiplication rate of the bacteria, which led to decreasednzyme yield. It has already been reported that when fungi are incu-ated at suboptimal temperatures, the decrease in fungal biomassould lead to decreases in enzyme production [32]. As the pHncreased or decreased, the enzyme production declined sharply.hese results are consistent with those of studies on dextranaseroduction by fungal species [33].
.7. Mutant sites on the protein
Dextranase genes from the wild and mutant strains wereequenced. Through amino acid alignment, dextranase fromrthrobacter KQ11 (GenBank accession no. KJ571608) was foundo belong to GH 49. After nucleotide alignment, several nucleotidesf the dextranase gene of mutant strains 4-1 and 4-13 were foundo differ from those of the wild strain KQ11 (Table 2). Overall, G to
transversion was dominant among base substitutions to both ofhe mutant strains.
The structure of dextranase of Arthrobacter KQ11 was pre-icted by SWISS-MODEL and analyzed by the SPDBV software
T → C 1603 L → P 535G → A 1701 A → T 568
(http://swissmodel.expasy.org/). Dextranase from ArthrobacterKQ11 consists of two domains, domain N and domain C (Fig. 5A).Domain N consists of nearly 200 residues forming 13 �-strands.Domain C has a right-handed parallel �-helix folded into a large,right-handed cylinder. At the N-terminus of the �-helix, there areseveral residues that connect the two domains. The substrate-binding subsites are located at the center of domain C around coils6 and 7, forming a narrow catalytic groove (Fig. 5B). The model indi-cates that Asp 382, Asp 401, and Asp 402 are the catalytic residues(Fig. 5B). Asp residues of enzymes are essential to enzyme activityinvolving hydrolysis of glucans [34,35]. The Asp 353, Asp 372, andAsp 373 residues have been shown to be essential for the catalyticactivity of Aspergillus niger Isopollulanase, a member of GH 49 [36].Asp 395 is the catalytic amino acid of dextranase from Penicilliumminioluteum [37]. Asp residues were found to hydrogen bond towater molecules if the nucleophilic attack on the anomeric carbonwas positioned properly [37].
The model indicates (Fig. 5B) that mutated residues (Table 2) Ile392 (mutant strain 4-1) and Ile 472 (mutant strain 4-13) are close tothe catalytic site, and moreover, Ile 472 located in the loop close tothe catalytic groove may have direct contact to bound sugar. Maybethis is the main reason of enzyme activity was increased to someextent. However, the other mutated residues (mutant strain 4-13),such as Thr188, Gly 297, Pro 535, and Thr 568, are located far fromthe catalytic site (Fig. 5A) and they could hardly affect the catalyticactivity.
3.8. Microphotography of formation of S. mutans biofilm treatedby dextranase
The formation of S. mutans was inhibited by dextranase in vitro,as shown in Fig. 6. Different concentrations of dextranase producedby 4-13 were incubated with S. mutans for 24 h, and the inhi-bition increased gradually with the concentrations of dextranaseenhanced. Because endo-dextranase can hydrolyze the �-1,6 gly-cosidic bond of glucan within or at the end of the chain, the matrixbecame less adhesive. When the concentration of dextranase was5 U ml−1, the inhibition of the biofilm formation was approximately90%. The inhibition of the biofilm was more obvious than the dex-tranase production by A. oxydans KQ11 [38]. The results suggestedthat the dextranase produced by 4-13 was effective in inhibitingthe biofilm formation.
4. Conclusion
Marine microorganisms are a new protein resource that humanscan develop. These newly discovered proteins have considerableapplication value. In this study, a strain called KQ11, which wasisolated from sea mud, was studied. After mutagenesis, two positive

iologi
m1mIf1a
A
DtNauofPJP
R
[
[
[
[
[
[
[
[[
[
[
[
[
[
[[
[[
[[
[[
[
[
[
[[
210–220.
X. Wang et al. / International Journal of B
utations, 4-1 and 4-13, were obtained. Their enzyme activity was9 and 30% higher than wild-type, respectively. In terms of the bothutations, visible dents were found on the surfaces of the strains.
n addition, the kinetic parameters of the mutants were differentrom those of the wild strain KQ11. The optimal temperature of 4-3 was 50 ◦C, while that of KQ11 was 45 ◦C. The optimal pH of 4-1nd 4-13 was 6.0, but that of KQ11 was 5.5.
cknowledgements
We are grateful to the National High Technology Research andevelopment Program of China (863 Program) (2011AA09070302);
he National Natural Science Foundation of China (31271929); theational Natural Science Foundation of China (Study on structurend function of dextranase from marine Arthrobacter and molec-lar modification); the National Key Technology R&D Programf China (2012BAC07B03); the National marine research specialunds for public welfare projects of China (201205020-8); Jiangsurovince and Technology Support Program (BE2013662); Theiangsu Government Scholarship Funding; the Priority Academicrogram Development of Jiangsu Higher Education Institutions.
eferences
[1] Y.-M. Kim, M.-Y. Seo, H.-K. Kang, K. Atsuo, D. Kim, Enzyme and Microbial Tech-nology 44 (2009) 159–164.
[2] T.D. Leathers, M.S. Nunnally, G.L. Côté, Carbohydrate Polymers 81 (2010)732–736.
[3] G. Eggleston, A. Monge, Process Biochemistry 40 (2005) 1881–1894.[4] D. Wang, M. Lu, X. Wang, Y. Jiao, Y. Fang, Z. Liu, S. Wang, Carbohydrate Polymers
103 (2014) 294–299.[5] S. Duarte, S. Gregoire, A.P. Singh, N. Vorsa, K. Schaich, W.H. Bowen, H. Koo, FEMS
Microbiology Letters 257 (2006) 50–56.[6] L.J. Stadler, Proceedings of the National Academy of Sciences of the United
States of America 14 (1928) 69–75.[7] M. Kurowska, A. Labocha-Pawlowska, D. Gnizda, M. Maluszynski, I. Szarejko,
Mutation Research-Fundamental and Molecular Mechanisms of Mutagenesis738 (2012) 52–70.
[8] Z. Liu, K. Zhang, J.-F. Lin, L.-Q. Guo, Scientia Horticulturae 130 (2011) 18–24.[9] T. Nakajima, H. Yasuda, H. Kurita, K. Takashima, A. Mizuno, IJ PEST 5 (2011)
42–49.10] L.Y. Wang, Z.L. Huang, G. Li, H.X. Zhao, X.H. Xing, W.T. Sun, H.P. Li, Z.X. Gou, C.Y.
Bao, Journal of Applied Microbiology 108 (2010) 851–858.11] S. Preis, D. Klauson, A. Gregor, Journal of Environmental Management 114
(2013) 125–138.
[
[
cal Macromolecules 70 (2014) 284–291 291
12] H. Chen, Z. Chen, M. Wu, S. Deng, Journal of Applied Microbiology 108 (2010)96–103.
13] T. Guo, Y. Tang, Y.-L. Xi, A.-Y. He, B.-J. Sun, H. Wu, D.-F. Liang, M. Jiang, P.-K.Ouyang, Biotechnology Letters 33 (2011) 2379–2383.
14] Y. Lu, L. Wang, K. Ma, G. Li, C. Zhang, H. Zhao, Q. Lai, H.-P. Li, X.-H. Xing, Bio-chemical Engineering Journal 55 (2011) 17–22.
15] V. Chagas-Moutinho, V. Sant’Anna, A. Oliveira-Menezes, W. De Souza, ActaTropica 130 (2013) 162–166.
16] E. Khalikova, P. Susi, N. Usanov, T. Korpela, Journal of Chromatography B 796(2003) 315–326.
17] X. Li, J. Zhang, S. Zhu, Genetics and Molecular Research 10 (2011) 2181–2189.18] L.-Z. Kang, F. Han, J.-F. Lin, L.-Q. Guo, W.-F. Bai, Scientia Horticulturae 151 (2013)
97–102.19] T. Akpa, K. Weber, E. Schneider, J. Kiefer, M. Frankenberg-Schwager, R. Har-
bich, D. Frankenberg, International Journal of Radiation Biology 62 (1992)279–287.
20] B.E. Sandström, M. Granström, S.L. Marklund, Free Radical Biology and Medicine16 (1994) 177–185.
21] D. Dobrynin, G. Fridman, G. Friedman, A. Fridman, New Journal of Physics 11(2009) 1–26.
22] T. Winter, J. Winter, M. Polak, K. Kusch, U. Maeder, R. Sietmann, J. Ehlbeck,S. van Hijum, K.-D. Weltmann, M. Hecker, H. Kusch, Proteomics 11 (2011)3518–3530.
23] J. Ehlbeck, U. Schnabel, M. Polak, J. Winter, T. von Woedtke, R. Brandenburg,T. von dem Hagen, K.D. Weltmann, Journal of Physics D: Applied Physics 44(2011) 13002–13019.
24] H.I. Smith, IEE Proceedings 62 (1974) 1361–1387.25] S. Lin, R. Liang, X. Meng, H. OuYang, H. Yan, Y. Wang, G.S. Jones, International
Journal of Biological Macromolecules 68 (2014) 173–177.26] F. Yatagai, Uchu Seibutsu Kagaku 18 (2004) 224–234.27] A. Tanaka, N. Shikazono, Y. Hase, Journal of Radiation Research 51 (2010)
223–233.28] E.R. Jiménez, Sugar Technology 11 (2009) 124–134.29] N.N. Siddiqui, A. Aman, S.A. Ul Qader, Carbohydrate Polymers 91 (2013)
209–216.30] J. Fukumoto, H. Tsuji, D. Tsuru, Journal of Biochemistry 69 (1971) 1113–1121.31] S.A.U. Qader, S. Bano, S. Iqbal, N. Syed, A. Azhar, Türk Biyokimya Dergisi 31
(2006) 135–140.32] T. Subasioglu, E. Cansunar, Hacettepe Journal of Biology and Chemistry 38
(2010) 159–164.33] D.K. Das, S.K. Dutta, The International Journal of Biochemistry and Cell Biology
28 (1996) 107–113.34] K. Funane, M. Shiraiwa, K. Hashimoto, E. Ichishima, M. Kobayashi, Biochemistry
32 (1993) 13696–13702.35] R. Russell, J. Ferretti, Journal of General Microbiology 136 (1990) 803–810.36] M. Mizuno, A. Koide, A. Yamamura, H. Akeboshi, H. Yoshida, S. Kamitori, Y.
Sakano, A. Nishikawa, T. Tonozuka, Journal of Molecular Biology 376 (2008)
37] A.M. Larsson, R. Andersson, J. Stahlberg, L. Kenne, T.A. Jones, Structure 11 (2003)1111–1121.
38] Y.-L. Jiao, S.-J. Wang, M.-S. Lv, B.-H. Jiao, W.-J. Li, Y.-W. Fang, S. Liu, Journal ofIndustrial Microbiology and Biotechnology 41 (2014) 17–26.


















![Ao Atmospheric Corrosion - [DePa] Departamento de ...depa.fquim.unam.mx/labcorr/publicaciones/AtmosphericCorrosion.pdf · Ao Atmospheric Corrosion M. TULLMIN ... Atmospheric corrosion](https://img.pdfslide.us/doc/110x75/5b78a8f87f8b9a331e8c0cd9/ao-atmospheric-corrosion-depa-departamento-de-depafquimunammxlabcorrpublicaciones.jpg)










