Embed Size (px)
Citation preview

Molecular Genetics Of Wilms' Tumor:
The Analysis Of The WTI And Hl9 Tumor Suppressor Genes.
Gang Hu
Department of Biochemistry
McGill University, Montreal
August 1996
A Thesis submitted to the Faculty of Graduate
Studies and Research in partial fulfilment of the
requirements of the degree of Masters of Science.
Gang Hu, 1996

IY&lUUllal LIUIW Y ~IUIIUII I W ~ U W r icriiui lai= 1-1 of Canada du Canada
Acquisitions and Acquisitions et Bibliographie Services services bibliographiques
395 Wellington Street 395, nie Wellington Ottawa ON K1A ON4 Ottawa ON K I A ON4 Canada Canada
The author has granted a non- L'auteur a accordé une licence non exclusive licence allowing the exclusive permettant à la National Library of Canada to Bibliothèque nationale du Canada de reproduce, loan, distribute or sell reproduire, prêter, distribuer ou copies of this thesis in microform, vendre des copies de cette thèse sous paper or electronic formats. la fome de microfiche/film, de
reproduction sur papier ou sur format électronique.
The author retains ownership of the L'auteur conserve la propriété du copyright in this thesis. Neither the droit d'auteur qui protège cette thèse. thesis nor substantial ' extracts fiom it Ni la thèse ni des extraits substantie1s may be printed or otherwise de celle-ci ne doivent être imprimés reproduced without the author's ou autrement reproduits sans son permission. autorisation.

The pl6/MTS 1 (multiple tumor suppressor l), p 15lMTS2
and Hl9 genes have been recently proposed as candidate
tumor suppressor genes. In this study, we determined the
mutation status of these three genes in 82 Wilms' tumors b y
the polymerase chain reaction and single-strand conformation
polymorphism(PCR-SSCP) analysis. The results suggested tha t
pl6lMTSl and p15IMTS2 genes are not involved in Wilms'
tumor, and that the H l 9 gene may be altered at a low fequency
in Wilms ' tumorigenesis.
We also analyzed the involvement of the WT1-
3'untranslated region(UTR) in the translational regulation of
the WTl gene expression. The data revealed that the WT1-
3'UTR does not affect WTl gene expression at the translational
Ievel in vitro.
These studies contribute to the molecular genetic
understanding of Wilms' tumorigenesis.

Les gènes p l6/MTS 1 (multiple tumor suppressor 1 ), pWMTS2 et Hl9 ont été récemment proposés comme gbnes suppresseur de tumeurs. Dans cette étude, nous avons examiné si ces trois gènes sont mutés dans des tumeurs de Wilms' e t ceci à l'aide des expériences de polymérisation en chaîne (PCR) et de conformation polymorphique de l'ADN simple brin (SSCP). Les résultats de nos travaux portant sur l'étude de 82 cas d e tumeurs de Wilms' suggerant que les gbnes p16/MTS 1 e t pWMTS2 ne sont pas impliqués dans l'étiologie de la t umeur de Wilms', par contre, le gène H l 9 pourrait être altéré, mais a faible fréquence, dans des tumeurs de Wilms'.
Nous avons egalement examiné le rôle de la région 3' non- traduite (3'-UTR) du gène de tumeur de Wilms' (WT1) dans s a propre rkgulation traductionnelle. Nos données ont indiqué q u e la région 3'-UTR du gène WTI n'est pas impliqué dans l e processus de traduction du gène, du moins in vitro.
Les résultats de nos études ont amené une compréhension relativement significative sur la génétique moléculaire de la tumeur de Wilms'.

ACKNOWLEOGEMENTS
First of all, 1 wish to express my appreciation to Dr. Jerry
Pelletier for supporting, directing my studies, and for his
guidance and helpful discussions. Many thanks to Nabeel
Bardeesy, 1 am very grateful to him for his helpful suggestions
and discussions concerning this manuscript. And I also wish to
thank other members in Our lab for their help and friendship.
Finally, with al1 of my heart, 1 thank my husband and m y
family for their encouragement and support during m y
graduate studies.

TABLE OF CONTENTS
ImODUCTION
Wilms' Tumor And Associated Syndromes
WAGR Syndrome
Beckwith-Wiedemann Syndrome(BWS)
Denys-Drash Syndrome(DDS)
Genetics In Wilms' Tumor-Syndromes
WT1-Wilms' Tumor Suppressor Gene
Association Of Other Tumor Suppressor Genes
In Wilms' Tumor
p53 Mutations In Wilms' Tumor
Association Of The Hl9 Gene In Wilms'
Tumor
pl6/MTS1 And p15/MTS2 Genes
Projects
Evaluating The Mutation Status Of MTS
And Hl9 Genes In Wilms' Tumor
Analysis Of The Putative Role Of The
WT1-3'UTR(Untranslated Region)
Translational Regulation
MflTER IClLS flND METHODS
Genomic DNA Of Wilrns' Tumor Source
PCR-SSCP Analysis
Plasmids And Construction
In Vitro Transcription And Translation
RESULTS

Searching Mutation Of MTS And Hl9 Genes
In Wilms' Tumor
Analysis Of The Function Of The WTld'UTR
DISCUSSION
Low Rate Of Mutations Of MTS And Hl9 Genes
In W T
Absence Of The MTS Gene Mutations In
Wilms' Tumor
Low Rate Of Polymorphism Of The Hl9
Gene In Wilms' Tumor
The WT1-3'UTR 1s Not Involved In WT1
Translational Regulation
SUMMfiRY
REFERENCES

LIST OF FIGURES
FI G. 1 Knudson-Strong Two Hit Hypothesis.
Fig. 2 Schematic diagram of genomic organization of the H l 9
gene and oligonucleotide primer paires used to amplify
the exonic regions.
Fig. 3 Effect of human WT1-3'UTR deletion on translation in
vitro.
Fig. 4 Effect of mouse WT1-3'UTR deletion on translation in
vitro.
Fig. 5 PCR-SSCP analysis of H l 9 exon 1 and exon 5 in DNA
sample No.Sth and No.60.

Table 1 Summary of the polymorphism of the H l 9 gene in WT
cases.

LIST OF ABBREUIATIONS
A- adenine
ATP- adenosine triphosphate
AUIRE- AU rich element
AWTA- association of Wilms' tumor-aniridia
bp- base pair(s)
BWS- Beckwith-Wiedemann Syndrome
CDK- cyclin-dependent kinases
CpG- cytosine-guanine dinucleotide
CTP- cytidine triphosphate
DDS- Denys-Drash Syndrome
DNA- deoxyribonucleic acid
EGR gene(s)- Early Growth Response genecs)
G1- the gap 1 period of interphase in a ce11 cycle
GTP- guanosine triphosphate
GU- genitourinary
kbp- kilobase pairs
kDa- kilodaltons
LOH- loss of heterozygosity
mRNA- messenger ribonucleic acid
MTS gene(s)- multiple tumor suppressor genes
PAX gene- a paired box- and homeobox-containing gene
PCR- polymerase chain reaction
S- the synthetic phase of the interphase in a ceIl cycle
SSCP- single strand conformational polymorphism

T- thymine
TNF- a- cachectin-tumor necrosis factor a
U- uracil
UTP- uridine triphosphate
IJTR- untranslated region
WT- Wilms' Tumor

Molecular Genetics Of Wilms' Tumor: The Analysis Of The WT1 And H l 9 Tumor
Suppressor Genes.
Wilms' Tumor And Associated Svndromes
Wilms' tumor or Nephroblastoma û a fairly amimon renal tumor in
childhood. It typically affects about 1 in 1 0,000 children under the age of 1 5
occurring primarily between 3 and 6 years of age (1-4). In most cases,
Wilms' tumors corne to medical attention after abdominal
swelling or an abdominal mass is detected by parents. Some
children may experience fever, abdominal pain or hematuria.
Wilms' tumor is a solid multilobulated tumor mas s,
cornposed of persistent blastema, dysplastic tubules a n d
supporting stroma (5, 6). It develops either from one pole o r
from the central body of the immature kidney, replacing t h e
renal parenchyma and producing a gross appearance of
encapsulation due to compression and scarring of t h e
surrounding tissue.
Most Wilms' tumor cases are sporadic and unilateral,
however 5-10% are bilateral and 1% of the cases are familial.
The age of onset of patients with bilateral tumors is much earl ier

than in unilateral cases. In hereditary cases of WT, t h e
proportion of bilateral cases is higher than in the sporadic form,
and the age of onset of hereditary unilateral tumors is muc h
lower than for sporadic cases (7). These data have been
interpreted by Knudson-Strong's "two step" mutation hypothesis
(8 and see fig.1). This mode1 suggests that two mutational s teps
are required for the development of Wilms' tumor. The bilateral
and hereditary cases are presumed to arise and have an earlier
age of onset, because the first predisposing mutation is
postulated to be present already in the germ-line cell, and only
one additional somatic mutation is required for tumorigenesis. In
sporadic cases, two independent genetic mutations have to
accumulate postzygotically within the same target ce11 for t h e
development of a tumor.
Wilms' tumor occurs either as an isolated cancer or in
association with congenital anomalies comprising at least four
different syndromes: WAGR syndrome; Beckwith-Wiedemann
syndrome (BWS); Denys-Drash syndrome @DS) and Perlman
syndrome. When Wilms' tumor occurs in combination with other
congenital anomalies, the incidence of the bilateral neoplasm i s
greater than cases without congenital anomalies (9).
WAGR Svndrome
WAGR syndrome is a constellation of Wilms' Tumor, Aniridia,
Genitourinary (GU) Anomalies, and Mental Retardation. Aniridia

Hereditary Sporadic
germline
one mutation inherited (germline)
1 2nd somatic mutation
tumor formation
no mutation
germline
1 two somatic mutations
tumor formation
Fig. 1 Knudson-Strong Two Hit Hypothesis. The bilateral and hereditary cases are presumed to arise and have an earlier age of onset, since the first predisposing mutation is postulated to be already present in the germ-line cell, and only one additional somatic mutation is required for tumorigenesis. In sporadic cases, two independent genetic mutations have to accumulate postzygotically within the same target cell. The asterisk (*) denotes a mutation of the WT1 gene.

is defined as congenital absence of part or al1 of the iris.
Generally, this anomaly is bilateral, and the frequency of aniridia
in the general population is between 1150,000 and 111 00 ,000 .
(10, 1 1). The association of Wilms' tumor-aniridia (AWTA) w a s
first reported by Brusa and Torricelli (12). Miller et al. in 1964
(13), noted the frequency of aniridia arnong patients with WT
was 1 in 73, thus establishing that aniridia is much more
frequent in patients with Wilms' tumor than in the general
population. By the same token, Wilms' turnor arises much more
frequently in patients with aniridia than in the general
population, and also, the bilateral neoplasm arises in 20% of t h e
cases, when Wilms' tumor occurs in combination with aniridia
(14).
With more recent clinical investigation, the association of
Wilms' tumor-aniridia was enlarged to include ment a l
retardation and genitourinary (GU) anomalies. The mental
retardation is defined as well as delayed somatic growth a n d
cognitive and motor impairment (4). GU anomalies may invo lve
the kidney, collecting system and external genitalia. In clinical
observation, GU anomalies include renal h ypoplasia, uni1 ateral
renal agenesis, horseshoe kidney s, urethral atresia, bifid ur e ter s ,
hypospadias, cryptorchidism and ambiguous genitalia. The
incidence of GU anomalies in association with sporadic WT is
approximately threefold higher than in the general population.
GU anomalies are more often associated with bilateral WT t han
aniridia (1, 4, 13). The incidence of congenital abnormalities i n

children with bilateral WT is at least ten times higher than that
with unilateral neoplasm (3, 4).
Beckwith-Wiedemann Svndrome (B W SI
In the 196OYs, Beckwith and Wiedemann described a
syndrome consisting of the association of macroglossia,
ornphalocele, h yperplastic visceromegaly, neonatal
hypoglycemia, cytomegaly of the adrenal cortex, renal medull ary
dysplasia and fetal gigantism (15, 16). Since then, the syndrome
has been extensively described and reviewed. The frequency of
occurrence of this syndrome has been estimated about 7 per
100,000 births (17, 18). Individuals with this syndrome are a t
increased risk for the development of neoplasrns. Approximatel y
20% of patients with BWS eventually develop cancers such as
Wilms' tumor, adrenocortical carcinomas and hepatoblas toma
(17, 18, 19).
Most BWS cases are sporadic, but there is a subset of familial
cases. Genetic linkage analysis demonstrated that chromosome
1 lp15 is associated with the familial form of BWS (20, 21).
Denys-Drash Syndrome IDDS)
The observations of Wilms' tumor occurring i n
hermaphroditic children have been reported for many years. 1 n

1967, Denys et al. first described the association of male
pseudohermaphroditism, Wilms' tumor, and renal failure, in a
child with XX/XY mosaicism (22). Three years later, Drash et al.
also noted two unrelated children evaluated for abnormal sexual
differentiation. These two children had negative buccal sme ar,
but one had a 46, XY karyotype on peripheral leucocytes, both
were diagnosed with Wilms' tumor and eventually died from
progressive renal failure (23). Since these original observations,
new case descriptions of the association between male
pseudohermaphroditisrn, Wilms' tumor and nephropathy have
appeared (24, 25) and this syndrome was designated as Denys-
Drash syndrome by Garfunkel in 1985 (26).
The Wilms' tumors in patients with Denys-Drash syndrome
may be bilateral, unilateral or unilateral but multinodular (22,
23, 27-34).
Genetics In W W Tumor-Svndromes
A chromosomal abnormality was long suspected in some
patients with Wilms' tumor. This suspicion was based on the
association of Wilms' tumor with various congenital anomalies.
Consequently, the investigation of chromosome abnormalities i n
Wilms' tumor has been studied extensively.
The chromosome abnormalities such as deletions or O t her
mutational events sometimes involve loss of the homologue of a
chromosomal segment, and DNA polymorphisms di s tri bu ted

along the chromosome can be used to determine the extent of
the loss of heterozygosity (LOH) i n individual tumors. The
analysis of LOH is an indirect method to determine if a mu ta ted
tumor suppressor gene is in the region identified by LOH. Also,
LOH analysis can detect deletions of a small chromosomal region
that cannot be found by cytogenetic studies. Therefore, LûH
analysis has been used to identify chromosomal regions
frequently deleted in tumors, indicating areas which may harbor
tumor suppressor genes. The study of LOH in Wilms' tumor h a s
demonstrated that the most common chromosome structural
abnormalities occur on chromosomes 1 , 7, 1 l p and 16. So far, n o
specific regions of chromosome 1 and 7 have been identified fo r
having conclusive involvement in tumorigenesis. Chromosome
1 l p exhibits LOH in 30-40% of al1 Wilms' tumors (35).
Chromosome bands l l p 1 3 and l lp15 both are associated
with Wilms' tumor in distinct syndromes. Deletions within 1 l p 13
have led to the isolation of the WT suppressor gene, WT1 (36).
Chromosome 1 lp15 is involved in the predisposition to BWS an d
harbors a second Wilms' tumor gene (37). In addition, the long
arm of chromosome 16 is thought to harbor a gene involved i n
progression since 20% of Wilms' tumor specimens of LOH
restricted to this region (38).
In summary, LOH studies, cytogenetic analysis and clinical
investigation consistently indicate that Wilms' tumors involve
multiple genetic alterations to give rise to a fully malignant
phenotype, hence, the discovery of WT1 gene and genetic studies

of other loci is important for our understanding of the genetic
cornplexity for this , pediatric tumor.
WT1-Wilms' Tumor Suppressor Gene
Cytogenetic analysis of Wilms' tumors frequently revealed a
deletion within the short arm of chromosome 11 at band pl3 i n
the WAGR syndrome (36). This constitutional deletion provided
the first clue to the location of a gene involved in t h e
development of Wilms' tumor. It is now known that the WAGR
deletion encompasses a number of contiguous genes, including
the Wilms' tumor suppressor gene WT1 and the aniridia gene
PAX6. Loss of one allele of the PAX6 gene is responsible for
aniridia (39), whereas mutations of one WTI allele are not only
responsible for Wilms' tumor predisposition, but also produce
genitourinary defects (40, 4 1).
The WT1 gene has been isolated by a positional cloning
approach. It maps to chromosome l lp13, and spans
approximately 50 kbp of DNA, composed of 10 exons (42). WT 1
codes for an 3 kb mRNA of containing a coding region of 1.4 kb.
The gene encodes four alternatively spliced transcripts which
yield distinct polypeptides. The transcripts are characterized b y
the presence or absence of exon 5 which encodes for 17 amino
acids, and the selection of a downstream splice donor si te
inserting three codons between exons 9 and 10 (43). The
products are 49-54 kDa nuclear proteins (44, 45) and contain

two known functional domains with homology to transcription
factors. The amino terminus contains a proline and glutamine
rich domain and mediates transcription repression (46, 47). The
carboxy terminus has four cysteine-histidine zinc fingers a n d
mediates DNA binding (48). This region shows significant
homogy to two early growth regulated mammalian polypeptides,
EGRl and EGR2 (49, 50) and can bind to the EGRl DNA
recognition site (S'GCGGGGGCG3') (51). These features suggested
that product of WT1 gene acts as a transcription factor and may
be involved in the growth proliferation response of nephroblasts
during renal development. Recent studies have shown the W T l
encoded products repress expression of insulin like grow th
factor 2 and the A chain of platelet-derived growth factor at t h e
transcription level (52, 53). The ability to suppress t h e
expression of these growth-associated genes may contribute t O
the function of WTI as a tumor suppressor gene. These genes
are expressed at high levels in Wilms' tumor, and it has been
suggested that the overexpression of these genes due to
inactivating mutations in WTI could contribute to tumor
phenotypes.
Mutation studies have demonstrated that WT1 deletions or
mutations occur in 10-158 of Wilms' tumor cases (54-61).
Practically, the constitutional WTI mutations are often found i n
children with an inherited predisposition to Wilms' tumor. Hence
in total, 5-7% of al1 Wilms' tumors are caused by an inherited
mutation in the WTl gene (62). In patients with WAGR-
associated deletions, the second WT1 allele is always mutated

within the tumor (63-65), and mutations in the WT1 gene have
been found in almost 100% of DDS patients (66, 67).
The WTl gene is expressed in specific types of cells (68), and
highly expressed in the developing kidney. During em bry onic
development, WTI is expressed in condensing blastemic cells,
renal vesicles, and glomerular epithelium(69), al1 thought to b e
the sites of origin of Wilms' tumor. WT1 expression peaks
around the time of birth in the murine renal system (68). I n
contrast, the expression of WT1 is at high levels in the
differentiating testis and ovary, and continues at high levels
throughout adult life (70). These observations indicate that the
WT1 gene product is not only involved in development of t h e
renal systern, but also implicates WT1 in genitourinary
development.
WT1 transcripts are also found in the spleen, in specific
regions of the spinal cord, brain and body wall muscles (71), but
the role that the WT1 protein plays in these tissues is unclear. I n
addition, WT1 expression has been detected in hematopoietic ce11
lines such as HL-60 promyelocytes and K562 cells (69, 70, 71).
Recently studies have demonstrated that WT1 is mutated i n
about 15% of leukemias (72).
In most Wilms' tumors, the WTl gene is expressed by t h e
same cellular constituents as during normal nephrogenesis (73),
and primarily occurs in blastema, immature tubular structures
and glomeruloid bodies. The levels of WT1 expression is highly
variable in Wilms' tumor (74-76), which seems to be related to
the quantity of each ce11 type present in each specimen.

Association Of Other Tumor Su~messor Genes In W h s ' Tumor
p53 Mutations In Wilms' Tumor
The p53 gene is a tumor suppressor gene, located on t h e
short arm of chromosome 17. It encodes a 53 kDa nuclear
phosphoprotein which appears to act as a negative regulator of
ce11 proliferation (77, 78). The p53 protein probably acts as a
transcriptional activator that suppresses abnormal ce11
proliferation by acting as a G1 ce11 cycle checkpoint control fo r
DNA damage (79). p53 gene alterations are the most f requen t
genetic events in human cancers, and have been reported i n
almost every type of adult and pediatric sporadic neoplasm (80-
83). Mutation analysis showed that p53 mutations were p resen t
in 8 of 140 Wilms' tumors (84). The small proportion with p 5 3
mutations were found to be the subset of nephroblastomas
known as anaplastic Wilms' tumor, which have a very poor
prognosis (82). The majority of WTs do not harbor p53 gene
mutations. Therefore, alterations of p53 gene can be used as a
molecular marker for anaplastic Wilms' tumor. Recently,
Bardeesy et al. reported that p53 gene mutations are associated
with the clona1 progression of Wilms' tumor cells showing
favorable histology to anaplastic cells, and suggested that tu m o r
cells with p53 mutations show attenuated apoptosis, indicating

that such lesions may provide a selective advantage in vivo b y
decreasing ce11 death ( 85).
Association Of Hl9 Gene In Wilms' Tumor
As previously mentioned, genetic mapping of BWS patients
and LOH studies of Wilms' tumors have dernonstrated that a
second WT gene locus is present at chromosome 1 l p15 .
Interestingly, the LOH for chromosome 1 lp15 markers in WT
always involved loss of the materna1 alleles (86) , raising t h e
possibility that a genomically imprinted gene is involved in BWS
and Wilms' tumor predisposition.
Genomic imprinting is a phenomenon in which some genes
are silenced when transmitted through a particular parent a l
germ line. Genes subject to imprinting are therefore
monoallelically expressed. Several imprinted genes have b e e n
documented in humans, the Hl9 gene is one such irnprinted gene
and maps to chromosome 1 lp15 (87, 88).
The Hl9 gene was originally cloned in screenings aimed a t
isolating imprinted genes. It codes for a spliced a n d
polyadenylated RNA, which is highly expressed in m a n y
developing organs such as fetal kidney, and has reduced b u t
persistent expression in certain human post-natal organs,
including juvenile and adult kidney (89-9 1). Transcription of
H l 9 is highly regulated and remains detectable in differentiated
myoblasts in culture (92). Hl9 RNA is a major transcript i n

expressing tissues, however, the lack of a long or evolutionarily
conserved translational reading frame and the failure t O
identify peptide products have impeded attempts to predict a
biological function. The H l 9 gene shares some characteristic a s
other mRNA: it is transcribed by RNA polymerase II, a n d
processed by RNA splicing and polyadeny lated. T h e s e
observations suggested that the Hl9 gene product may be a n
RNA molecule, in other words, the biologically active product
might be the H l 9 RNA itself (93). In 1991, Brunkow et al.
reported that inappropriate expression of an Hl9 transgene i n
mice caused fetal mortality (94), and later, Hao et al. described
that the expression of an human Hl9 expression construct i n
certain human embryonic tumor ce11 lines such as RD@ cells
derived from an embryonal rhabdomyosarcoma) and G40 1 (G40 1
cells derived from a malignant rhabdoid tumor), had cau sed
growth retardation and morphological changes, and had an t i -
clonogenic and anti-tumourigenic effects (95). Based on t he se
observations, the H l 9 gene was proposed to be a candidate
tumor suppressor gene. According to this hypothesis, Moulton
and CO-workers analyzed H l9 expression, CpG-methylation, a n d
1 l p 15.5 allelic status in 25 primary WTs. They found that H 1 9
RNA expression was reduced at least 20-fold from fetal k idney
levels in 18 of Wilms' tumor cases. Ten of the expression-
negative tumors retained 1 1 p 15.5 heterozygosity. In nine of
these, H l 9 DNA was biallelically hypermethylated and in t w o
cases hypermethylation locally restricted to H l 9 sequences w a s
also present in the non-neoplastic kidney parenchyma (96).

These observations revealed genetic and epigenetic inactivation
of the H l 9 gene in Wilms' tumorigenesis, suggesting that H 19
plays a tumor suppressor role in the pathogenesis of WT. These
finding are suggestive of a possible biological role of the H l 9
gene in tumorigenesis.
pl6/MTSl And ~15 /MTs2 Genes
Recently, two tumor suppressor genes MTS(multip1e t um or
suppressor)l and MTS2 genes have been demonstrated to b e
involved in many tumor types (97). These two genes a r e
tandemly located within 25 kb of each other at chromosome
9p21, where the region showed the high frequency of deletions
in multiple tumor types. The sequence of the two genes was
determined, and they encode proteins that are cy clin-dependent
kinases inhibitors p l6fMTS 1 and pl 5iMTS2. These proteins ac t
as negative ce11 cycle regulators to inactivate specific cyclin-
protein kinase complexes that are required for progression
through the ce11 cycle (98, 99). The p l6 protein inhibits ras -
induced proliferation and cellular transformation (100). The
MTSl and 2 genes have a high degree of structural a n d
functional homology of each other. They are both encoded b y
two exons, and, 44% and 97% of the MTS2 exons, respectively,
are identical to the first and second exon of MTSl (97).
Functionally, pl6 or p15, each can bind to CDK4 and CDK6, a n d
inhibit their kinase activation, causing the blockade of the ce11

cycle between G1 and S, and resulting in suppression of cellular
proliferation (98, 99). The MTSl and 2 genes were analyzed for
deletions in ce11 lines derived frorn 12' different turnor types. The
analysis demonstrated that MTSl is homozygously deleted a t
high frequency in ce11 lines derived from tumors of lung, breast,
brain, bone, skin, bladder, kidney, ovary and lymphocyte, and a t
least 75% melanoma ce11 lines contain mutant MTSl or have lost
the gene from both homologs. In addition, nonsense and splice-
junction mutations in MTSl have been observed in primary
melanomas and bladder tumors (97). According to these
findings, it has been inferred that MTS mutations are involved in
tumor formation in wide range of tissues and the MTSl and 2
genes are designated as candidate tumor suppressor genes.

Evaluatine The Mutation Status Of MT'S And Hl9 Genes In Wilms'
Turnor
Cancer has been commonly linked to aberrant proliferation
and a failure of the transformed cells to differentiate. Cancer-
associated genes generally can be divided into two types: the
activated proto-oncogenes and the inactivated tumor suppre s sor
genes. The inactivation of a tumor suppressor gene could result
from deletions of the gene or from nonsense mutations that
result in truncated, nonfunctional protein. In these cases bot h
alleles are usually altered In some cases, tumor suppressor
genes are mutated on only one allele producing a protein which
acts in a dominant negative manner inhibiting the function of
the other normal allele. Therefore, the frequency of mutation
and LOH provides persuasive evidence that inactivation of
candidate tumor suppressor genes is necessary for a ce11 to
become cancerous.
As mentioned earlier, genetic analysis has revealed the
MTSl and 2 genes as candidate tumor suppressor genes, and t h e
deletions of these two genes occurs frequently in many cancer
ce11 lines and in certain malignant neoplasms (97), although,
Wilms' tumors have not been previously analyzed. Hence, in
order to assess the possible involvement of the MTS gene

alterations in Wilms' tumor, we searched for deletions or somatic
mutations in the genomic DNAs of Wilrns' tumor.
The H l 9 gene has been suggested as a candidate t u m o r
suppressor gene involved in Wilms' tumor, and the locus of t h e
H l 9 gene where is assumed the second WT suppressor gene, i s
associated in the predisposition of BWS and WT. Based on t he se
considerations, we explored the mutational status of the H l 9
gene in DNA samples from Wilms' tumor and BWS patients.
Analvsis Of The Putative Role Of The WT1-3'UTR(Untranslated Re
Translational Replat ion
The regulation of gene expression may occur at many levels,
including transcriptional initiation, elongation, mRNA splicing,
transport and stability, and translational efficiency.
Transcriptional efficiency is regulated by enhancers, inducible
factors and other transcription factors. A number of t h e
components regulating WT1 transcription have been identified
(101). Post-transcriptional regulation of WTI however is not a
well studied mechanism. Translational regulation often involves
the participation of sequences in the untranslated regions of
mRNA (102). The 5'-untranslated region (5'-UTR) strongly
influences the efficiency of translation. Recently, the 3'-UTR h a s
also been reported to function in translational regulation (102,
103), as well as play a role in affecting mRNA stability (103).
Examples of inhibition of translation by interactions with in t h e

3'-UTR include p-interferon in Xenopus oocytes (104) a n d
creatine kinase in U937 cells (105). The 3'-UTR may also
function to increase protein translation. Studies with ornithine
decarboxylase, which is under translational control b y
polyamines (106), have demonstrated coordinated regulation of
translation by both the S'-and 3'-UTRs. Whereas ornithine
decarboxylase translation initiation is inhibited by sequences in
the 5'-UTR of the mRNA, the 3'-UTR functions to augment, and
partially negate, this inhibition of translation (107).
The importance of the 3'-UTR in regulation of gene
expression inspired us to analyze the translational regulation of
the WT1 gene, and determine what function the 3'-UTR of WTI
has in the expression. Work from Our laboratory had previously
demonstrated elements of the WTl-S'UTR which had a strong
effect on translational efficiency (108). Accordingly, we dec ided
to investigate the function of the WT13'UTR in the translational
efficiency of the WTl gene using an in vitro translation assay.

MATERIRLS AND METHODS
Genomic DNA Of Wilmç' Tumor Source
Al1 the genomic DNA of Wilms' Tumors in Our analysis were
supplied from Dr. Benhard Zabel, Department of Pediatrics,
University of Mainz, Germany. All of DNA samples were isolated
from sporadic tumors, there is no any anaplastic tumor case i n
these specimens.
PCR-SSCP Andysis
To detect possible deletions, insertions or point mutations i n
the Hl9 gene and MTS gene in Wilms' tumor, we used
polymerase chain reaction-single strand conformation
polymorphism(PCR-SSCP) assays.
For amplification of the H l 9 gene, 12 oligonucleotide pairs
were designed to enable coverage of the entire Hl9 coding
region. The primers are summarized in the Fig. 2.
In the analysis of exon 1 & 2 of MTSUpl6, the prirners were
designed as follows: For exon 1: 2F. S'GAAGAAAGAGGAG-
GGGCïG3', 1 1 08R. SGCGCTACCTGATTCCAATïC3'; Exon2: 42F.
S'GGAAAnGGAAACTGGAAGC3'' 55 1 R. S'TCTGAGCTlTGGAAGC-
TCT3'. For exon2 of MTS2jp15 gene, the oligonucleotide pair is:
89F. S'TGAGTTTAAACCTGAAGGTGG3', 50R. SGGGTGGGAAATT'G-
GGTAAG3'.

Primer Primer Pair
1
2
3
4
5
6
7
8
9
1 O
11
12
Sequence Annealing Exon Temperature OC Scanned
PCR-product
Generated, bp

Fig. 2 Schematic diagram of genomic organization of the H l 9
gene and oligonucleotide primer pairs used to amplify the
exonic regions. The black boxes represent the 5 exons of the
H l 9 gene and each exon is numbered. Oligonucleotide primer
pairs used to amplify specific regions of H l 9 are represented
by bidirectional arrows under the exon segments. Primer
names, their sequence, annealing temperature, and size of the
PCR product generated are tabulated below the H l 9 gene.

19
Exonic sequences of the Hl9 gene and the MTSl and 2 genes
were amplified from genomic DNA of Wilms' tumor samples b y
the polymerase chah reaction(PCR).
Each PCR was performed in 20pl volumes containing 50ng of
genomic DNA as template, 0.5 pmoles of each oligonucleotide,
0 final concentration dimethyl sulfoxide, 1 OOpM
deoxynucleoside triphosphates, 0.2unit of Taq I polymerase an d
l x PCR buffer [lOmM Tris-HCL(PH8.4), 50mM KCi, 1 SmM MgCl*,
0.01% Gelatin, 0.01% NP-40 and 0.01% Tween-201. One of the
primer pair was radiolabelled with Y - ~ ~ P - A T P and T4
polynucleotide kinase, 50pC of 3 2 ~ was used to label 20 pmoles
of oligo. The amplification reactions were performed for 3 0 - 3 5 ,
cycles.
The PCR products were directly analyzed by the SSCP(sing1e
strand cornformation polymorphism) method. 2p1 of the KR
products were mixed with 8pl of the formamide dye mix (95%
formamide, 20mM EDTA, 0.05 $6 bromophenol blue, 0.05 % x y le n e
cyan01 FF), boiled for 5 minutes, and then placed on ice for 5
minutes. 2pl samples were loaded onto 8% polyacrylamide (50:l
acrylamidelbisacrylamide) gels and the gels were
electrophoresed for 3-5 hours at 30W in the cold room (4 OC).
Before loading the samples, the polyacrylamide gels were pre-
electrophoresed for 30 minutes in 1X TBE (90mM Tris, 90mM
boric acid, 2.5 mM EDTA) at 30W in the cold room. The gels w e re
dried for 30 minutes at 80 OC with filter paper and exposed to
Kodak X-OMAT film at -70 OC for 12-24 hours with or without an
intensifying screen.

Plasmids And Construction
For human WTl cDNA clones: Plasmid WT33 contains a
truncated WT1 fragment (1024bp-3335bp), pBSNiWT1 contains
the WTl coding region but lacks a 3'-UTR (568bp-2080bp).
pSP65/hWT(+3') was generated as follows: WT33 was digested
with NcoI (in WTl) and XbaI (in pKSII vector), pBSN/WT1 w as
digested with NcoI (in WT1) and EcoRI(in pKSII vector), the two
WT1 fragments were ligated at the NcoI site, to obtain the ful l
length WTI from 390bp-3335 bp (EcoRI-Xbai), and this in sert
was introduced into the transcription vector pSP65-38A.( see fig . 3). The 2.9kb fragment of WTl which lacks a 3'-UTR was
obtained by digestion of pBSNlWTl with EcoRI (two EcoRI are in
the pKSII vector), then ligation of this WT1 fragment wit h
pSP65-38A, to generate pSP651hWTl(-3') (see fig. 3).
For mouse WTl cDNA clones: Plasmid pKSIIWT(-I-) contai ns
the entire coding region of the WTl murine cDNA (3200bp)
(Pelletier J. 199 1). To generate truncations of the WT 1 -3'UTR,
pKS8IWT(-I-) was digested with EcoRI in the 5' end of WT1, and
with EcoRI, HincII, BglII, XbaI or Sau3AI at the 3' end of WTI.
These inserts were introduced into the EcoRI-EcoRI, EcoRI-
HincII, EcoRI-BglII, EcoRI-XbaI and EcoRI-Sau3AI sits of pSP65-
38A. The resulting plasmids were referred to as pS P65/WTe,
pSP65/WTh, pSP65/WTb, pSP65IWTx and pSP65/WTs(see fig.
4) -
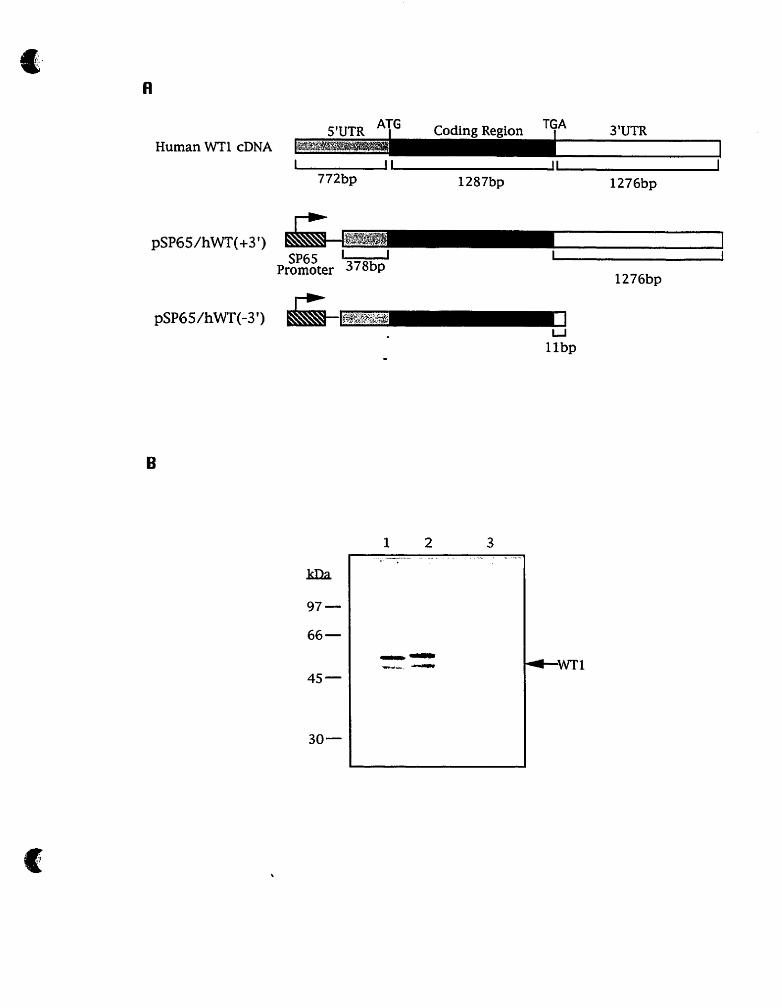
5'UTR A?G Codina Region 3'UTR Hurnan WT1 cDNA
U l lbp

Fig. 3 Effect of human WTI-3'UTR deletion on translation in
vitro.
A. diagram of deletions generated within the human WT1-
3'UTR. The hatched boxes represent the SP6 RNA polymerase
promoters, the stippled boxes represent the S'UTR, the black
boxes indicate the coding region, and the white boxes represent
the 3'UTR.
B. In vitro translation of human WT1 mRNA constructs.
Lane 1. pSP65/hWT(+3'); Lane 2. pSP6ShWT(-3'); Lane 3. no
input RNA. After in vitro translation reaction, 5p1 translation
products were loaded onto a 10% SDSlpolyacrylamide gel,
following electrophoresis, the gel was treated with ance, ce, dried, and exposed to X-Omat (Kodak) film at -70 OC with an
intensifying screen overnight.
pSP65/hWT(+3') does not. contain the WTI alternative
splice, pSP65/hWT(-3') contains the alternative splice. So, the
bands of the translation products in lane 1 and 2 do not appear
to line up.

SPG U Promoter 180bp
Xba I
1 n
u I l t b p

Fig. 4 Effect of mouse WT1-3'UTR deletion on translation in
vitro.
A. diagram of deletions generated within the WT1-
3'UTR.The hatched boxes represent the SP6 RNA polymerase
promoters, the stippled boxes represent the S'UTR, the black
boxes indicate the coding region, and the white boxes represent
3'UTR.
B. In vitro translation of WT1 mRNA constructs. Lane 1.
pSP6S/mWTe; Lane 2. pSP65/mWTb; Lane 3. pSP65lmWTh;
Lane 4. pSP6S/rnWTx; Lane 5. pSP65fmWTs; Lane 6. no input
RNA. After in vitro translation reaction, 5ul translation
products were loaded onto a 109 SDSlpolyacrylamide gel,
following electrophoresis, the gel was treated with pro an ce, dried, and exposed to X-Omat (Kodak) film at -70 OC with an
intensifying screen overnight.

The plasmid pKS8/WT(-1-) and WT33 were constructed b y
Dr.Jerry Pelletier, pBSNlWT1 was a gift of Dr. Benhard Zabel
(Germany). The transcription vector pSP65-38A was modified a s
described ( Pelletier et al 1991).
The restriction enzymes were purchased frorn New England
Biolabs and were used according to the manufacturer ' s
directions.
Plasmid selection were performed by using the DHl OP
bacteria strain (GibcoBRL). DHlOP was grown in LB media. The
protocol of competent DH10B preparation, transformation w i t h
plasmid, and plasmid isolation, were as described (109).
Zn Vitro Transcri~tion And Translation
For in vitro transcription, the plasmids pSP65/WTe,
pSP65/WTh, pSP65IWTb, pSP65tWTx, pSP6S/WTs,
pSP65/hWT(+3') and pSP65/hWT(-3') were linearized w i t h
HindZII. After digestion, linearized plasmids were extracted b y
phenol-chloroform( 1 : 1 ; v/v), purified by passing over the G-50
spun-dialysis column, and precipitated by ethanol. In 1 0 0 ~ 1
transcription reactions, 500pM m7GpppG and 100prn GTP w a s
added in the reaction mix, in order to synthesize capped mRNA
transcripts. 1mM ATP, CTP and UTP were included in the mix,
10pC 5-'H-CTP was added in the reaction mix as a trace label to
quantify mRNA yields. SP6 polymerase was used in the reaction
for the mRNA synthesis. The incubation tirne was 2hr at 4 2 ' ~ .

After incubating, the samples were extracted b y
phenollchloroform 1 : l v : ) passed over G-50 spun-dialysis
column and ethanol precipitated. 2p1 of the products of t h e
transcription reactions were measured by scintillation counting
to quantify mRNA synthesis yields. 4p1 of the products were
loaded ont0 1% formaldehyde agarose gels for analysis of t h e
integrity of the RNA products.
For translation reactions, 1 pg RNA templates as translated i n
50p1 rabbit reticulocyte lysate(Promega Biotech) reaction mix.
1mM amino acid mixture (minus Methionine) and 16pglml of
[ 3 5 ~ ] Methionine (final concentration) were added in the mix. The
translation control was set by a mix lacking RNA templates. The
translation reactions were performed at 30°c for lhr. After
incubation, 5p1 of translation products were loaded ont0 a 10%
SDS-polyacryamide gel for analysis of protein produc ts.
Following electrophoresis, the gel was treated with ance, ce, dried, and exposed to Fuji x-ray film at -70 OC for 12 hr.

RESULTS
Searching Mu ta tion Of MTS Arad Hl9 Genes In Wilmç' Tumor
To detect possible deletions, insertions or point mutations of
the H l 9 gene and MTS genes in Wilms' tumor, we used the PCR-
SSCP assay. The SSCP assay is based on the property of single
stranded nucleic acid to assume sequence-specific conformations
due to intramolecular hydrogen bonding when analyzed u n der
non-denaturing conditions. A difference of even a single base
pair can cause the single stranded DNA to adopt different
conformations (conformers). The resulting conformers have a
characteristic electrophoretic mobility and the differe n t
conformer displays a mobility shift in the presence of sequence
polymorphisms. In the SSCP assay, radiolabelled DNA fragments
of the gene of interest are generated by PCR. Subsequently the
DNA is denatured, the single strands are elec trophoresed
through a non-denaturing polyacrylamide gel. The mobility of
conformers is detected by autoradiography. The differences i n
migration of polyrnorphic sequences are observed as a "shift" on
the autoradiogram.
The SSCP method is efficient, rapid, and has a high mutation
detection rate (1 10- 1 12), which allows simple and rapid analysis
of a large number of samples. Additionally, the mu tational

detection ability of this method is not interfered with b y
contamination with the normal product (frorn normal DNA) i n
the PCR, hence heterozygous mutations are detectable.
During Our mutational detection for the Hl9 gene and MTS
genes in Wilms' tumor, the fragments of approximately 200bp of
the Hl9 gene and MTS genes were amplified by polymerase
chah reaction (PCR) and the products are analyzed by SSCP. The
PCR-SSCP analysis for each product was performed tw ice
alternating the identity of the radiolabelled oligonucleotides
primer. This is because the sequence context in which a
mutation is located can have striking influences on the shift
detected by SSCP analysis, thus it is optimal to analyze both
strands of the gene of interest when searching for mutations.
For analysis of mutations in the MTS genes, we used
ofigonucleotide pairs designed to enable coverage of the
sequences of exon 1 and 2 of the pl6/MTS1 gene and exon 2 of
the p151MTS2 gene. Exon 1 and 2 of the pl6lMTS1 gene
displayed a high rate of mutation frequency in melanoma ce11
lines (97), eighteen mutations distributed in 14 of 34 melanoma
lines. In our WT specimens, we decided to scan the mutation
frequency of these three exon regions of MTS genes in Wilms
tumors. We analyzed 28 genomic DNA samples isolated f rom
sporadic Wilms' tumors. N65 is a genomic DNA from a normal
individual which was used as a standard for the intact normal
MTS genes in the PCR-SSCP analysis. In these 28 cases, no
fragment showing mobility shifts for the MTS genes was found,
and the data was consistent with the results of Southern blot

analysis for these genes, that did not display any deletions of
these genes in Wilms' tumor (Dr.Jerry Pelletier, unpublished).
For analysis of the Hl9 gene, we designed oligonucleotide
primer pairs which allowed us to amplify the entire Hl9 exonic
region. In total, we scanned genomic DNAs of 54 Wilms' tumors
for the H l 9 gene mutations, of which 47 genomic DNAs w e r e
derived from sporadic Wilms' tumors. Considering whether t h e
Hl9 gene is involved in the predisposition to BWS and Wilms'
tumor, we examined 7 cases of familial BWS DNA samples
(Table.1). We also tested 22 other DNA samples which w e r e
isolated from normal blood cells for cornparison. The PCR-SSCP
analysis showed that two tumor DNA samples had mobility shifts
(see Fig. 5). N0.60 DNA was derived from a sporadic Wilms'
tumor and showed mobility shifts with primer pairs 4A/4B a n d
1 I A/ 1 1 B. No.Sth DNA was derived from a BWS patient, a n d
showed a mobility shift with the primer pairs 4A14B. No o the r
DNA samples showed evidence of H l 9 gene alterations. We d id
not sequence these two SSCP shifts. Since, genetic studies h a v e
revealed that 50% of WTs show LOH restricted to chromosome
1 l p l 5 and thus the Wilms' tumor locus located at 1 l p l 5 i s
expected to be altered at high frequency. Therefore, we expected
the alteration of the H l 9 gene in WT should be at a very high
frequency. During the course of these studies, deletion analysis
of WTs using markers from 1 l p 15 identified specimens w i t h
small deletions 1Mbp centromeric to Hl9 (J, Pelletier,
unpublished data). In Our data, only two DNA samples showed a
mobility shift out of 54 WT specimens. The low rate of t h e

unaffec ted
N0.W
unaffected
' unaffected
No.GO
unaffected
unaffec ted
No.0
unaffected
N0.60
unaffected *
'JoSth

Fig. 5 PCR-SSCP analysis of H l 9 exon 1 and exon 5 in DNA
sample No.Sth and No.60. PCR product from an unaffected
individual were included as an interna1 standard for the
normal allele. Mobility shifts are indicated by arrowheads.
(A). Mutational analysis of exon 1 of the H l 9 gene in turnor
DNA samples No.Sth and No.60. The segments of H l 9 exon 1
were amplified with oligonucleotide primer pair 4A/4B, and .
the PCR products were amplified from these two DNA samples
showing the mobility shift (arrowhead). (a) Oligonucleotide 4A
was radiolabeled and (b) oligonucleotide 4B was radiolabeled
when PCR products were amplified.
(B). Mutational analysis of exon 5 of the H l 9 .gene in tumor
DNA sample No.60. Exon 5 of the Hl9 gene was amplified with
oligonucleotide primer pair 1 1 A/11 B, and the PCR product
display an mobility shift (arrowhead). (a) Oligonucleotide 11A
was radiolabeled, and (b) oligonucleotide 11B was radiolabeled -
for amplifications.

mobility shift was not as significant as we expected, which
prompted us to reconsider the significance of the mutation status
of the Hl9 gene in WT.
Analvsis Of The Function Of The WT13'UTR
To understand what function the 3'-UTR of WT1 has in the
control of translation, we developed a deletion strategy to
investigate the involvment of different elements of WT1-3'UTR
for the efficiency of in vitro translation.
The full length cDNA of human and mouse WTl gene is
3335bp and 3138bp. The length of human and mouse WT1-
3'UTR is 1277bp and 1263bp respectively, and parts of these
regions were deleted in a series of in vitro
transcriptiodtranslation constructs ( see Fig. 3& 4 ) The
constructs contain part of the 5'-UTR, the full length coding
region and different deletions of the WTl-3'UTR.
pSP65/hWT(+3') contains the intact human WT1 coding region
with complete 3'-UTR (the length is 2.9kb), pSP65/hWT(-3')
contains human WTl coding region without the 3'-UTR (the
length is 1630bp) (see Fig.3). pSP65lWTe contains the mouse
WT1 coding region with the entire WT1-3'UTR (total size:
2803bp). pSP651WTh' pSP65/WTb, pSP65TWTx and pSP6WWTs
contains the truncated mouse WTl gene, deleted respectively a t
HincII(2655bp), BglII(2243bp), XbaI(1765bp) and
Sau3AI(1640bp) sites in WTl-3'UTR (see Fig.4). After

construction of these plasmids, they were linearized and used a s
templates in in vitro transcription experiments. mRNA w a s
successfully synthesized and the size of the RNA products w e r e
consistent with the inserts of these plasmids. Equal quantities of
RNA products were translated in the rabbit reticulocyte ly sate
system. The results showed that al1 RNA products were
translated with equal efficiencies (see Fig.3 & 4 ) and thus the
3'-UTR of WT1 does not affect translation in vitro.

DISCUSSION
Low Rate Of Mutations Of MTS And Hl9 Genes h WT
Absence Of The MTS Gene Mutations In Wilms' Tumor.
Since the MTS genes were discovered and were found to b e
mutated in a wide range of ceIl lines, a number of investigators
have evaluated the frequency of the MTS genes alterations in
prirnary human tumor material. The results of examinations of
primary tumors have conflicted in their support for the role of
the MTS genes as tumor suppressors. Studies of the MTS genes,
in particular p l6lMTS 1 have shown high frequencies of
homozygous deletions or point mutations in some tumor types,
suggesting that involvement of the p16fp15 genes is a common
event in certain human primary tumors (97, 1 13-1 18). Studies
of other cancers have demonstrated low mutation rates ( 1 19-
123). Indeed, the frequency of p l6 gene mutation in primary
tumors ranges from 0% in breast cancer (120) to 52% in
esophageal squamous ce11 carcinomas (1 13). During Our studies,
no point mutations or deletions of the MTS genes were observed
in 28 Wilms' tumor cases, either by PCR-SSCP analysis o r
Southern blot analysis. These observations suggest that t h e
mutation or deletion on the MTS genes does not contribute to

Wilms' tumor onset or progression. In addition, reviewing t h e
recent reports of LOH studies in Wilms' tumor, common
chromosome structural abnormalities are found occurring O n
chromosomes 1,7, 11 and 16, excluding chromosome 9 where t h e
MTS genes are located. The MTS genes are thus not commonly
involved in Wilms' tumor carcinogenesis.
Low Rate Of Pol~morphisim Of The H l 9 Gene In Wilms' Tumor
As mentioned earlier, previous functional, imprinting a n d
mapping data have implicated H l 9 as a candidate WT
suppressor gene. Proof of this hypothesis requires evidence f rom
the molecular analysis of primary tumors. We investigated t h e
mutational status of the H l 9 gene in Wilms' tumor cases. In 5 4
Wilms' tumor genomic DNAs, only 2 cases were observed to have
mobility shifts ( table 1). This data suggested the low frequency
of the H l 9 gene alterations in WT. Possible explanations for t h e
low rate of alteration of the Hl9 gene are (i) That a gene o the r
than H l 9 is the Wilms' tumor suppressor gene located a t
chromosome 1 lp15. (ii) Either, the alteration of Hl9 gene m a y
not play a main role in Wilms' tumorigenesis, or (iii) Since H l 9 i s
irnprinted, and therefore can be inactivated by a single genetic
- hit at one locus, the evidence of tumor suppression activity m a y
differ from that obtained for other tumor suppressor genes. 1 n
particular, if genetic inactivation is through mitotic
recombination or to alterations in an "imprinting center'' which is

Table.1 Summary of the polymorphism of the H l 9 gene in WT cases.
Wilms' tumor type DNA sarnple No. Polymorphism Affected Exon Oligo Pair
sporadic WT 1-36 - - -
- 1 and 5
-
W T with BWS Sth 1 1 4M4B
San - - - 1 Sch - - -
SI0 - Bca - ScAL
Newc

adjacent to the gene then the criterion of intragenic mutat ion
which has been useful for identifying classical tumor suppressor
genes may not apply to imprinted genes.
The WTlô'UTR 1s Not Involved In WT1 Translational
Remdation.
WT 1 is a developmentally regulated transcription factor th a t
functions as a tumor suppressor gene, and has a highly t issue-
specific pattern of gene expression, but the regulatory
mechanism of WTl expression is still as a rnystery. In this s tudy,
we demonstrated that WT1-3'UTR does not play a functional role
in the regulation of WT1 expression and translation in vitro (see
fig.3 & 4). As protein product levels in the absence and presence
of the WTl-3'UTR were not significantly different in the rabbi t
reticulocyte translation system. It remains formally possible t ha t
the in vitro translation system does not contain factors necessory
for mimicking the in vitro regulation of WT1 translation. The
factors may thernselves be developmentally regulated and t hu s
absent in reticulocyte which do not normally express WTl.
Motifs found in 3'UTRs are known to influence translational
efficiency in particular AU-rich (usually AUUUA) elements. AU
rich elements (3'AURE) affect the efficiency of translation
include that of the cachectin-tumor necrosis factor a (TNF-a)
chimeric constructs which are inhibited in mouse macrophage-
derived cells grown in the absence of endotoxin but show

increased expression in cells grown in the presence of inducer,
and a differential effect on translation has been attributed to the
3'AURE of the TNF-a transcript (1 24). Both cDNA of human a n d
mouse WT1 contain AU-rich elements. However, according to Our
data, effects of the 3'AURE for WT 1 translation regulation w e r e
not observed in vitro. But it does not eliminate the function of
the 3'AURE of WT1 in vivo. Perhaps, the 3'AURE affects t h e
translational efficiency and or mRNA stability of WTl b y
interacting with certain cellular proteins of the WT1 transcrip ts
i n cellular environment.

In summary, we evaluated the mutationai status of t h e
p16IMTS 1 and p15/MTS2 genes in 28 sporadic Wilms' tumor
cases, no point mutations or deletions were observed, and these
results were consistent with the result of Southern blot analysis
of these genes in Wilms' tumor. Taken together, p l6/MTS1 a n d
p15IMTS2 are not frequently associated in the Wilms'
tumorigenesis. Likewise, only two polymorphisims were found in
54 Wilms' tumor genomic DNAs during the mutation analysis of
the H l 9 gene, suggesting the low alteration frequency of the H l 9
gene in Wilms' tumor cases. We demonstrated the WT 1 -3'UTR
does not function in the translational expression of the WTI gene
in a simple in vitro system. Al1 of these study results contribute
to the molecular genetic understanding of Wilms' tumorigenesis.

REFERENCES
1. Pendergrass, T.M. 1976. Congenital anomalies in children
with Wilms' tumor, a new survey. Cancer 37: 403.
2. Breshow NE, Beckwith SB, Ci01 M, et al. 1988. Age
distribution of Wilms' tumor: Report From the National
Wilms' Tumor Study. Cancer Res 48: 1653.
3. Bond J.V.1975. Bilateral Wilms' tumor. Age at diagnosis,
associated congenital anomalies, and possible pattern of
inheritance. Lancet II: 482
4. Breslow N.E, Beckwith J.B, 1982. Epidemiological features of
Wilms' tumor: Results of the National Wilms' Tumor Study. J.
Natl. Cancer Inst. 68: 429
5. Bove K.E, Koffler H, McAdams A.J. 1969. Nodular renal
blastema. Definition and possible significance. Cancer 24:323.
6. Bennington J.L, BeckwithJB, 1975. Tumors of the kidney,
renal pelvis and ureter. In Atlas of tumor pathology, series
2, fascile 12. Armed Forces Institute of Pathology,
Washington,D.C.

7. Matsunaga E. 1981. Genetics of Wilms' tumor. Hum. Genet.
57: 231.
8. Knudson A G , Jr. and L.C. Strong. 1972. Mutation and cancer:
A mode1 for Wilms' tumor of the kidney. J. Natl. Cancer Inst.
48: 313.
9. Nakagome Y, Ise T, Sakurai M, Nakajo T, Okamoto E, Takano
T, Saito S, et a1.1984. High-resolution studies in patients
with aniridia-Wilms' tumor association, Wilms' tumor or
related congenital abnormalities. Hum. Genet .67: 245.
10. Shaw M.W, Falls H.F, Neel J.V. 1960. Congenital aniridia.
Am. J. Hum. Genet 12: 389.
I l . Francois J, Coucke D, Coppieters R. 1977. Aniridia-Wilms'
tumor syndrome. Ophthalmology 174: 35.
12.Brusa P. Torricelli C. 1953. Nefroblastoma di Wilms' ed
affezioni renale congenite nella casistica dell' IPPAI di
Milano. Minerva Pediatr. vol. 5: 457.
13. Miller,R.W., J.F. Fraumeni, and M.D. Manning. 1964.
Association of Wilms' tumor with aniridia,
hemihypertrophy and other congenital malformations. N.
Eng. J. Med. 270: 922.

14. Fraumeni IF, Glass AG. 1968. Wilms' tumro and congenital
aniridia. J. Am. Med. Assoc. 206: 825,
15. Beckwith J.B. 1963. Extreme cytomegaly of the fetal adrenal
cortex, omphalocoele, hyperplasia of kidneys and pancreas,
and Leydig-ce11 hyperplasia. Another syndrome? Presented
a t the Annual Meeting of Western Society for Pediatric
Research, Los Angeles, California, Nov. I l .
16. Wiedemann H.R. 1964. Complexe malformatif familial avec
hernie ombilicale et macroglossie-Un syndrome nouveau? J.
Genet. Hum. 13: 223.
17. Higurashi M, Ijima K, Sugimoto Y, Ishikawa N, Yoneyarna K.
1980. The birth prevalence of malformatin syndromes in
Tokyo infants. Am. J. Med.Genet. 6:189.
18. Pettenati M.J, Haines J.L, Higgins R.R, Wappner R.S, Palmer
C.G. Weaver D.D. 1986. Wiedemann-Beckwith syndrome:
Presentation of clinical and cytogenetic data on 22 new
cases and review of the literature. Hum. Genet. 74: 143.
19. Sotelo-Avila C, Gonzalez-Crussi F, Fowler J.W. 1980.
Complete and incomplete forrns of Beckwith-Wiedemann
syndrome: Their oncogenic potential. J. Pediatr. 96: 47.
20. Koufos A., Grundy P., Morgan K. 1989. Familial Wiedemann-

Beckwith syndrome and a second Wilms' tumor locus both
map to 1 lp15.5. Am. J. Hum. Genet 44: 71 1.
21. Ping AJ, Reeve AE, Law DJ. 1989. Genetic linkage of
Beckwith-Wiedemann syndrome to 1 lp15. Am. J. Hum.
Genet 44: 720.
22. Denys P, Malvaux P. van den Berghe H, Tanghe W,
Proesmans W. 1967. Association d'un syndrome anatomo-
pathologique de pseudohermaphrodisme masculin, d'une
tumeur de Wilms', d'une nephropathie parenchymateuse et
d'un mosaicisme X X I X Y . Arch. Fr. Pediatr. 24: 729.
23. Drash A, Sherman F, Hartmann WH, Blizzard RM. 1970. A
syndrome of pseudohermaphroditism, Wilms' tumor,
hypertension, and degenerative renal disease. J. Pediatr. 76:
5 85.
24. Habib R, Loiret C, Gubler M.C, Niaudet P, Bensman A, Levy
M, Broyer M. 1985. The nephropathy associated with male
pseudo hermaphroditism and Wilms' tumor (Drash
syndrome): A distinct glomerular lesion-Report of 10 cases.
Clin. Nephrol.24: 269.
25. Jadresic L, Leake J, Gordon I, Dillon M.J, Grant D.B. Pritchard
J, Risdon R.A, Barrat T.M. 1990. Clinicopathologic review of
twelve children with nephropathy, Wilms' tumor. and

genital abnormalities (Drash syndrome). J. Pediatr. 1 17:
26. Garfunkel J.M. 1985. Editors comments on Edidin, D.V.
Pseudohermaphroditism, glomerulopathy, and Wilms'
tumor (Drash syndrome). J. Pediatr. 107: 988.
27. Spear G.S, Hyde T.P, Gruppo R.A, Slusser R. 1971.
Pseudohermaphroditism, glomerulonephritis with nephrotic
syndrome, and Wilms' tumor in infancy. J. pediatr. 79: 677.
28. Barakat A.Y, Papadopoulou Z.L, Chandra R.S, Hollerman C.E,
Calcagno P.L. 1974. Pseudohermaphroditisrn, nephron
disorder and Wilms' tumor: A unifying concept. Pediatrics
54: 366.
29. McCoy F.E, Jr., Franklin W.A, Aronson A.J, and Spargo B.H.
1983. Glomerulonephritis associated with male
pseudohermaphroditism and nephroblastoma. Am. J. Surg.
Pathol. 7: 387.
30. Habib R, Loriat C, Gubler MC, Niaudet P, Bensman A, Levy
M, and Broyer M. 1985. The nephropathy associated with
male pseudohermaphroditism and Wilms' tumor (Drash
syndrome): A distinct glomerular lesion-Report of 10 cases.
Clin. Nephrol. 24: 269.

31. Gallo G, Chemes H.E. 1987. The association of Wilms' tumor,
male pseudohermaphroditism and diffuse glomerular
disease (Drash syndrome). Report of eight cases with
clinical and morphologic findings and review of the
literature. Pediatr. Pathol. 7: 175.
32. Jensen J.C, Ehrlich R.M, Hanna M.K, Fine RN, Grunberger
1.1989. A report of 4 patients with the Drash syndrome
and a review of the literature. J. UroI. 141: 1174.
33. Jadresic L, Leake J, Gordon 1, Dillon M.J, Grant D.B, Pritchard
J, Barrat T.M. 1990. Clinicopathologic review of twelve
chidren with nephropathy, Wilrns' tumor, and genital
abnormalities (Drash syndrome). J. Pediatr. 1 17: 7 17.
34. Tank E.S. and Melvin T. 1990. The association of Wilms'
tumor with nephrologic disease. J. Pediatr. Surg. 25: 724.
35. Pelletier J, Munroe D., and Housman D.1991. Molecular
Genetics of Wilms' tumor. Genome Analysis Volume 3:
Genes and Phenotypes. 135.
36. Francke U, Holmes L.B, Atkins L, and Riccardi V.M.1979.
Aniridia-Wilms' tumor association: Evidence for specific
deletion of 1 lp13. Cytogenet.Cel1 Genet. 24: 185.

37. Henry 1, Jeapierre M, Couillin P, Barichard F, Serre JL, et al.
1989. Molecular definition of the 1 1 pl 5.5 region involved
in Beckwith-Wiedemann syndrome and probably in
predisposition to adrenocortical carcinpma. Hum. Genet. 8 1 :
273.
38. Marion A. Maw, Paul E. Grundy, Lynn J. Millow, Michael R.
Eccles, et al 1992. A third Wilrns' tumor locus on
chromosome l6ql. Cancer Res 52: 3094.
39. Ton CCT, Hirvonen H, Miwa H, et al. 1991. Positional cloning
and characterization of a paired box- and homeobox-
containing gene from the aniridia region. Ce11 67: 1059.
40. Pelletier J, Bruening W, Li FP, Haber DA, Glaser T, Housman
DE. 1991. WTl mutations contribute to abnormal genital
system development and hereditary Wilms' tumor. Nature.
353: 431.
41. Pelletier J, Bruening W, Kashtan CE, et al. 1991. Germline
mutations in the Wilms' tumor suppressor gene are
associated with abnormal urogenital development in Denys-
Drash syndrome. Ce11 67: 437.
42. Haber DA., Sohn RL., Buckler AJ. 1991. Alternative splicing
and genomic structure of the Wilms' tumor gene WTI. Proc.
Natl. Acad. Sci Usa 88: 9618.

43. Brenner B, Wildhardt G, Schneider S.1992. RNA
polymerasechain reaction detects different levels of four
alternativel~ spliced WT 1 transcripts in Wilms' tumor.
Oncogene 7: 143 1.
44. M o ~ ~ s JI, Madden SL, Tournay OE. 1991. Characterization of
the zinc finger protein encoded by the WT1 Wilms' tumor
locus. Oncogene 6: 2339.
45. Telerman A, Dodernont H, Degraef C. 1992. Identification of
the cellular protein encoded by the human Wilms' tumor
(WTl) gene. Oncogene 7: 2545.
46. Mitchell PJ, Tijan T.1989. Transcriptional regulation in
mammaliam cells by seqence specific DNA binding proteins.
Science 24: 371.
47. Madden SC, Cook DM, Moms JF. 1991. Transcriptional
repression mediated by the WT1 Wilms' tumor gene
product. Science 253: 1550.
48. Evans B., Hollenferg S. 1988. Zinc fingers: Gilt by association.
Ce11 52: 1.
49. Joseph LJ, LeBean MM, Jamieson GA.1988. Molecular
cloning seqencing and mapping of EGR-2, a human early

growth response gene encodeing a protein with zinc-
binding finger structure. Proc. Natl. Acad Sci USA 85: 7164.
50. Sukhatime VP., Cao X., Chang LC. 1988. A zinc finger
encoding gene coregulated with c-fox during growth and
differntiation and after cellular depolarization. Ce11 53: 37.
51. Rauscher FJ, Morris JF, Tournay 0E.1990. Binding of the
Wilms' tumor locus zinc finger protein to the EGR-1
consensus seqence. Science 250: 1259.
52. Gashler AL, Bonthron DT, Madden SL.1992. Human
plateltderived growth factor A chain is transcriptionally
repressed by the Wilms' tumor suppressor WT1. Proc. Natl.
Acad. Sci. USA 89: 6010.
53. Drummond IA., Madden SL-, Rohwer-Nutter P. 1992.
Repression of the insulin-like growth factor II gene by the
Wilms' tumor supressor WTl. Science 257: 674.
54. Brown KW, Wilmore HP, Watson JE, Mott MG, Berry J, et
al. 1993. Low frequency of mutations in the WTl coding
region in Wilms' tumor. Genes Chromosomes Cancer 8: 74.
55. Cowell JK, Wadey RB, Haber DA, Cal1 KM, Housman DE,
Pritchard J. 1991. Structural rearrangements of the WTl
gene in Wilms' tumor cells. Oncogene 6: 595.

56. Gessler M, Konig A, Arden K, Grundy P, Orkin S, et a1.1994.
Infrequent mutation of the WTl gene in 77 Wilms' tumors.
Hum. Mutat. 3: 212,
57. Glaser T, Lane J, Housman D.1990. A mouse mode1 of the
aniridia-Wilms' tumor deletion syndrome. Science 250:
823.
58. Little MH, Prosser J, Condie A, Smith PJ, Van Heyningen V,
Hastie ND. 1992. Zinc finger point mutations within the WT1
gene in Wilms' tumor patients. Proc. Natl. Acad. Sci. U.S.A.
89: 4791.
59. Royer-Pokora B, Ragg S, Heckl-Ostricher B, Held M, Loos U,
et al. 1991. Direct pulsed field gel electrophoresis of Wilms'
tumors shows that DNA deletions in l lp13 are rare. Genes
Chromosomes Cancer 3: 89.
60. Tadokoro K, Fujii H, Ohshima A, Kakizawa Y, Shimizu K, et al.
1992. Intragenic homozygous deletion of the WT1 gene in
Wilms' turnor. Oncogene 7: 1215.
61. Varanasi R, Bardeesy N, Ghahremani M, Petruzzi MJ, Nowak
N, et al. 1994. Fine structure analysis of the WT.1 gene in
sporadic Wilms' tumors. Proc. Natl. Acad. Sci. USA. 91:

62. Hastie N.D.1994. The Genetics of Wilms' tumor-A case of
disrupted development. Annu. Rev. Genet. 28: 523.
63. Baird PN, Groves N, Haber DA, Housman DE, Cowell JK.1992.
Identification of mutations in the WTl gene in turnors from
patients with the WAGR syndrome. Oncogene 7: 2141.
64. Brown KW, Watson JE, Poirier V, Mott MG, Berry PJ,
Maitland NJ. 1992. Inactivation of the remaining allele of
the WT1 gene in a Wilms' tumor from a WAGR patient.
Oncogene 7: 763.
65. Gessler M, Konig A, Moore J, Qualman S, Arden K, et al.
1993. Homozygous inactivation of WTl in a Wilms' tumor
associated with the WAGR syndrome. Genes Chtomosornes
Cancer 7: 131.
66. Baird P.N., Santos A., Groves N., Jadresic L., Cowell J.K. 1992.
Constitutional mutations in the WT1 gene in patients with
Denys-Drash syndrome. Hum. Molec. Genet. l(5): 301.
67. Pelletier J, Bruening W, Kashtan C.E, Mauer S.M, Manivel J.C,
Housman D, et a1.1991. Germline Mutations in the Wilms'
tumor suppressor gene are associated with abnormal
urogenital development in Denys-Drash syndrome. Ce11 67:
43 7.

68. Armstrong JF., Pritchard-Jones K., Bickmore WA., Hastie ND.,
Bard JBL. 1992. The expression of the Wilms tumor gene
WTl in the developing mammalian embryo. Mech. Dev. 40:
85.
69. Pritchard-Jones K, Fleming S, Davidson D, Bickrnore W,
Porteous D, Pelletier J, Housman D, et a1.1990. The candidate
Wilms' tumor gene is involved in genitourinary development.
Nature 346: 194.
70. Pelletier J, Schalling M, Buckler AJ, Rogers A, Haber DA,
Housman DE. 1991. Expression of the Wilrns' tumor gene
WTI in the murine urogenital system. Genes & Dev 5:1345.
71. Sharrna PM, Yang X, Bowrnan M, Roberts V, Sukumar S.
1992. Molecular cloning of rat Wilms' tumor complementary
DNA and a study of messenger RNA expression in the
urogenital system and the brain. Cancer Res. 52: 6407.
72. King-Underwood L., Renshaw J. and Pritchard-Jone K. 1996.
Mutations in the Wilms' tumor gene WT1 in leukemias.
Blood. Vo1.87. No.6: 2171.
73. Pritchard-Jones K, Fleming S. 199 1. Ce11 types expressing the
Wilms' tumor gene (WT I ) in Wilms' tumor: Implications for
tumor histo-genesis. Oncogene 6: 22 1 1.

74. Gerald WL, Gramling S, Sens DA, et a1.1992. Expression of
the 1 lp13 Wilms' tumor gene, WT1, correlates with
histologie category of Wilms' tumor. AmJ Pathol 140: 1031.
75. Miwa H, Tomlinsom GE, Timmons CF, et a1.1992. RNA
expression of the WTl gene in Wilms' tumors in relation to
histology. J Natl Cancer Inst 84: 18 1.
76. Yeger H, Cullinane C, Flenniken A, et a1.1992. Coordinate
expression of Wilms' tumor genes correlates with Wilms'
tumor phenotypes. Ce11 Growth Different. 3: 855.
77. Leine A.L, Momand J, Finlay C.A.1991. The p53 tumor
suppressor gene. Nature (Lond.). 351: 453.
78. Vogelstein B, Kinzler K.W.1992. p53 function and
dysfunction. Cell. 70: 523.
79. Kartan M.B, Onyekwere O, Sidransky D, Vogelstein B, Craig
R.W. 1991. Participation of p53 protien in the cellular
response to DNA damage. Cancer Res. 51: 6304.
80. Nigro JM, Baker SJ, Preisinger AC, Vogelstein B, et a1.1989.
Mutations in the p53 gene occure in diverse hurnan tumor
types. Nature (lond.). 342: 705.

81. Hollstein M, Sidranky D, Vogestein B,, Harris CC. 1991. p53
mutations in human cancers. Science (Washington DC). 253:
49.
82. Imantura J, Bartram CR, Koeffler HP.1993. Mutation of the
p53 gene in neuroblastoma and its rekationship with N-myc
amplification. Cancer Res. 53: 4053.
83. Vogan K, Bernstein M, Leclerc J .M, Pelletier J, Gros P. 1993.
Absence of p53 gene mutations in primary neuroblastomas.
Cancer Res. 53: 5269.
84. Bardeesy N, Falkoff D, Petruzzi MJ, Nowak N, Zabel B,
Pelletier J. 1994. Anaplastic Wilms' tumor, a subtype
displaying poor prognosis, harbours p53 gene mutations.
Nature Genet. 7: 91.
85. Bardeesy N, Beckwith J.B, Pelletier J. 1995. Clona1 expansion
and attenuated apoptosis in Wilms' tumors are associated
with p53 gene mutation. Cancer Research 55: 215.
86. Schroeder WT, Chao LY, Dao DD, Strong LC, Pathak S, et al.
1987. Non-random loss of materna1 chromosome I l allele in
Wilms' tumors. Am. J. Hum. Genet. 40: 413.

87. Bartolomei M, Zemel S, Ti;ghman SM.1991. Parental
imprinting of the mouse Hl9 gene. Nature 351: 153.
88. Rachrnilewitz J, Goshen R, Ariel 1, Schneider T, et a1.1992.
Parental imprinting of the human H l 9 gene. FEBS Lett. 309:
25.
89. Pachnis V, Brannan CI, Tilghman SM, 1988. The structure
and expression of a novel gene activated in early mouse
embryogenesis. EMBO J. 7: 673.
90. Poirier F, Chan C-T3, Timmons PM, Roberson El, Evans MJ,
Rigby PWJ. 1991. The murine H l 9 gene is activated during
embryonic stem ce11 differentiation in vitro and at the time
of implantation in the developing ernbryo. Development.
113: 1105.
91. Rachmilewitz J, Gileadi O, Eldar-Geva T, Schneider T, et al.
l99Za. Transcription of the H l 9 gene in differentiating
cytotrophoblasts from human placenta. Mol Reprod Dev. 32:
196.
92. Davis RL, Weintraub H, Lassar AB. 1987. Expression of a
single transfected cDNA converts fibroblasts to myoblasts.
Cell. 51: 987.

93. Brannan CI, Dees EC, Ingrarn RS. 1990. The product of the
Hl9 gene may function as an RNA. Molecular and Cellular
Biology. Jan: 28.
94. Brunkow ME, Tilghman SM. 1991. Ectopic expression of the
Hl9 gene in mice causes prenatal lethality. Genes &
Development 5: 1092.
95. Hao Y, Crenshaw T, Tycko B, et al. 1993. Tumour-suppressor
activity of Hl9 RNA. Nature 365: 764.
96. Moulton T, Crenshaw T, Moosikasuwan J, Tycko B, et al.
1994. Epigenetic lesions at the H l 9 locus in Wilms' tumour
patients. Nature Genetics 7: 440.
97. Kmab A, Gruis A.N, Weaver-Feldhaus J, Liu Q, Harshman K,
et al. 1994. A ce11 cycle regulator potentially involved in
genesis of many tumor types. Science. 264: 436.
98. Hannon GJ, Beach D. 1994. pl5 INK4B is a potential effector
of TGF-B-induced ce11 cycle arrest. Nature 371: 257.
99. Serrano M, Hannon GJ, Beach D. 1993. A new regulatory
motif in cell-cycle control causing specific inhibition of
cyclin D/CDK4. Nature 366: 704.

100. Serrano M, Gomez-Lahoz E, Barsagi D, et a1.1995.
Inhibition of Ras-induced prolifefation and cellular
transformation by p l6INK4. Science 267: 249.
101. Wu Ying-Jin, Fraizer Gai1 C., Saunders Grady F. 1995. Gata-
1 trans activates the WTl hematopoietic specific enhancer.
J. Bio. Chem. 270: 5944.
102. Kozak M.1992. Annu. Rev. Ce11 Bio1.8:197.
103. Jackson R.J, Standart N. 1990. D o the poly(A) tail and
3' untranslated region control mRNA translation? Ce11 62:
15.
104. Kruys V, Wathelket M, Poupartt P, Contreras R, Flers W,
Huez G. 1987. The 3' untranslated region of the human
interferon-B mRNA has an inhibitory effect on translation.
Proc. Natl. Acad. Sci. U.S.A. 84. 6030.
105. Ch'ng JL, Shoemaker DL. Schimmel P. Holmes EW. 1990.
Reversal of creatine kinase translational repression by 3'
untranslated sequences. Science 245: 1003.
106. Kahana C, Nathans D. 1985. Translational regulation of
mammalin ornithine decarboxylase by polyamine. J. Bio.
Chem. 260: 5390.

107. Grens A, Scheffler IE, 1990. The 5 ' - and 3'- untranslated
regions of ornithine decarboxylase mRNA affect the
translational efficiency. J. Bio. Chem. 265: 1 18 10.
108. Wendy Bruening and Jerry Pelletier. 1996. A non-AUG
translational initiation event generates novel WT 1
isoform. J. Bio. Chem. 271: 8646.
109. Sarnbrook J., Fritsch E.F., Maniatis T. 1989. Molecular
Cloning. A Laboratory Manual. Cold Spring Harbor
Laboratory Press, Cold Spring Harbor, New York.
110. Sheffield V.C., Beck J.S., Kwitek A.E., Sandstrom D.W., Stone
E.M. 1993. The sensitivity of single-strand conformation
polymorphism analysis for the detection of single base
substitutions. Genomics 16: 325.
11 1. Grompe M. 1993. The rapid detection of unknown
mutations in nucleic acids. Nature Genet. 5: 11 1.
112. Orita M., Iwahana H., Kanazawa H., Hayashi K., Sekiya T.
1989. Detection of polymorphisms of human DNA by gel
electrophoresis as single-strand conformation
polymorphisms. Proc. Natl. Acad. Sci. USA 86: 2766
113. Mori T, Miura K, Aoki T, Nishihira T, Mori S, Nakamura Y.
1994. Frequent somatic mutation of the MTS 1KDK4I

(multiple . tumor suppressor/cyclin-dependent kinase 4
inhibitor) gene in esophageal squamous ce11 carcinoma.
Cancer Res. 54: 3396.
114. Zhou X, Tarmin L, Yin J, et al. 1994. The MTSl gene is
frequently mutated in primary human esophageal tumors.
Oncogene 9: 3737.
115. Hayashi N, Yoshihisa S, Tsuchiya E, Ogawa M, Nakamura Y.
1994. Somatic mutations of the MTSl (multiple tumor
suppressor l)ICDK41 (cyclin-dependent kinase-4
inhibitor) gene in human primary non-srna11 ceIl lung
carcinomas. Biochem Biophys Res Commun 202: 1426.
116. Giani C, Finocchario G. 1994. Mutation rate of the CDKN2
gene in malignant gliomas. Cancer Res 54: 6338.
117. Walker DG, Duan W, Popovic EA, Kaye AH, Tomlinson FH,
Lavin M.1995. Homozygous deletions of the multiple
tumor suppressor gene 1 in the progrssion of human
astrocytomas. Cabcer Res. 55: 20.
118. Hebert J, Cayuela JM, Berkeley J, Sigaux F. 1994. Candidate
tumor suppressor genes MTS 1 (p16) and MTS2 (p15)
display frequent homozygous deletions in primary cells
from T- but not from B ce11 lineage acute lymphoblastic
leukemias. Blood 84: 4038,

OntaM, Nagai H, Shimizu M, et al. 1994. Rarity of somatic
and germline mutations of the cyclin-dependent kinase 4
inhibitor gene, CDK41, in melanoma. Cancer Res 54: 5269.
Xu L, Sgroi D, Sterner CJ, et al. 1994. Mutation analysis of
CDKN2 (MTSlIP16) in human breast carcinomas. Cancer
Res 54: 5262.
Zhang S, Kiein-Szanto AJP, Sauter ER, et al. 1994. Higher
frquency of alterations in the p16fCDKN2 gene in
squamous ce11 carcinoma ce11 lines than in primary tumors
of the head and neck. Cancer Res 54: 5050.
Peiffer SL, Bartsch D, Whelan AJ, Mutch DG, Herzog TJ,
Goodfellow PJ. 1995. Low frequency of CDKN2 mutation in
endometrial carcinomas. Mol Carcinog 13: 2 10.
123. Ueki K, Rubio M. Ramesh V, et al. MTSllCDKN2 gene
mutations are rare in primary human astrocytomas with
allelic loss of chromosome 9p. Hum Mol Genet 3: 1841.
124. Han J, Brown T, Beutler B. 1990. Endotoxin-Responsive
Sequences Control Cachectin/Tumor Necrosis Factor
Biosynthesis At The Translational Level. J.Exp. Med. 171.
465.

APPLIED t IMAGE, lnc 1653 East Main Street - i Rochester, NY 14609 USA
-LI - - = Phone: 7161482-0300 -- -- - - Fax: 73 61288-5989
@ 1993. Applied Imge, Inc.. All Rights Resewed





























