Embed Size (px)
Citation preview

1
“Isoimidas monosustituidas como inhibidores de la Acetilcolinesterasa”
T E S I S QUE PARA OBTENER EL GRADO DE:
MAESTRÍA EN CIENCIAS CON ESPECIALIDAD EN
FARMACOLOGÍA
P R E S E N T A:
Q.F.B. Juan Alberto Guevara Salazar
Instituto Politécnico Nacional
Escuela Superior de Medicina
México, D.F. DICIEMBRE DEL 2006

2

3

4
ÍNDICE
Índice 4
Abreviaturas 7
Summary 11
I. RESUMEN 12
II. INTRODUCCIÓN 12
II.1 Relación Cuantitativa Estructura-Actividad (QSAR) 12
II.1.1 Simplificación del modelo 13
II.1.2 Asociación de dos o más moléculas 13
II.1.3 Replicación moduladora 13
a) Apertura o cierre de anillos 14
b) Introducción de enlaces múltiples 14
c) Homología 15
d) Introducción de grupos voluminosos 15
e) Bioisosterismo 16
1) Descriptor electrónico 18
2) Descriptor estérico 19
3) Descriptor hidrófobo 21
4) Métodos para establecer relaciones cuantitativas estructura-actividad
biológica
22
II.2 Química Computacional 23
II.2.1 Métodos computacionales 23
II.2.2 Docking 26
II.2.2a Estructura tridimensional de la molécula blanco 28
II.2.2b Sitio de unión con el blanco 28
II.2.2c Selección de los ligandos 29
II.2.2d Simulación de la interacción ligando-receptor y formación del
complejo
30
II.3 Química de amidas e imidas 32
II.3.1 Amidas 32
II.3.2 Imidas 33
II.4 Sistema Nervioso 34
II.4.1 Funciones generales del Sistema Nervioso Autónomo 37

5
II.4.2 Transmisión simpática 39
II.4.3 Transmisión parasimpática 42
II.4.4 Acetilcolinesterasa 46
II.5 Enfermedad de Alzheimer 50
II.5.1 Generalidades 50
II.5.2 Fisiología de la memoria y el aprendizaje 50
II.5.3 Manifestaciones clínicas 56
II.5.4 Causas de la enfermedad de Alzheimer 57
II.5.5 Fisiopatología 58
II.5.6 Tratamiento de la enfermedad de Alzheimer 59
II.6 Cinética enzimática 61
II.6.1 Generalidades 61
II.6.2 Catálisis enzimática. Modelo de Michaelis-Menten 63
II.6.3 Inhibición enzimática 67
III. ANTECEDENTES 72
IV. JUSTIFICACIÓN 78
V. HIPÓTESIS 78
VI. OBJETIVOS 79
VI.1 Objetivo general 79
VI.2 Objetivos particulares 79
VII. PARTE EXPERIMENTAL 79
VII.1 Instrumentación 79
VII.2 Reactivos 80
VII.3 Procedimiento general de síntesis de los ácidos fenilmaleámicos y
fenilisomaleimidas para monosustituidas
81
VII.3.1 Síntesis del ácido fenilmaleámico 81
VII.3.2 Síntesis del ácido p-metoxifenilmaleámico 82
VII.3.3 Síntesis del ácido p-nitrofenilmaleámico 83
• Procedimiento de purificación del tetrahidrofurano (THF) 83
VII.3.4 Síntesis de la fenilisomaleimida 84
VII.3.5 Síntesis de la p-metoxifenilisomaleimida 85
VII.3.6 Síntesis de la p-nitrofenilisomaleimida 86
• Procedimiento de purificación del diclorometano (CH2Cl2) 88

6
VII.4 Determinación de la actividad enzimática de la Acetilcolinesterasa 89
VII.4.1 Procedimiento general de medición 89
VII.4.2 Normalización del método 92
VII.4.2a Determinación de la concentración óptima de enzima [E] 93
VII.4.2b Determinación del tiempo óptimo de actividad 93
VII.4.3 Determinación de la constante de MIchaelis-Menten (Km) y la
rapidez máxima (rmáx)
93
VII.4.4 Ensayos de inhibición sobre AChE de Electrophorus electricus 94
VII.4.4a Neostigmina 94
VII.4.4b Fenilisomaleimida, p-metoxifenilisomaleimida y p-
nitrofenilisomaleimida 96
VII.5 Docking 96
VIII. RESULTADOS Y DISCUSIÓN 98
VIII.1 Síntesis de los ácidos p-X-fenilmaleámicos 98
VIII.2 Síntesis de las p-X-fenilisomaleimidas 102
VIII.3 Determinación de la actividad enzimática de la Acetilcolinesterasa 108
VIII.3.1 Normalización del método 108
VIII.3.1a Determinación de la concentración óptima de enzima [E] 108
VIII.3.1b Determinación del tiempo óptimo de actividad 109
VIII.3.2 Determinación de la constante de MIchaelis-Menten (Km) y la
rapidez máxima (rmáx)
111
VIII.3.3 Neostigmina 113
VIII.3.4 Fenilisomaleimida 117
VIII.3.5 p-metoxifenilisomaleimidas 123
VIII.3.6 p-nitrofenilisomaleimidas 131
VIII.4 Docking 137
IX. CONCLUSIONES 141
X. REFERENCIAS 142
XI. ANEXOS 147

7
ABREVIATURAS
5-HT: 5-hidroxitriptamina
(Serotonina)
a, a0: Interceptos u ordenadas al
origen
A: Absorbancia
Å: Amstrongs
AA: Ácido araquidónico
AACS: Sitio activo selectivo a unión
a aromáticos
ABS: Sitio de unión a acilo
AC: Adenilato ciclasa
AcOEt: Acetato de etilo
Ach: Acetilcolina
AChE: Acetilcolinesterasa
Ala: Alanina
AM: Anhídrido maleico
AMPA: L-α-amino-3-hidroxi-5-metil-
4-isoxazolpropionato
AMPc: Adenosín-monofosfato
cíclico
anh.: Anhidro
apoE: Apolipoproteína E
APP: Proteína β amiloide
AS: Sitio aniónico
Asp: Aspartato
ATP: Adenosín-trifosfato
AP: Potencial de Acción
BuChE: Butirilcolinesterasa 13C: Carbono 13
°C: Grados Celsius
C: Concentración
CA: Complejo activado
CAM/KII: Protein-cinasa II
dependiente de Ca2+-Calmodulina
c.c.f.: Cromatografía en capa fina
ChAT: Acetilcolintransferasa
CICATA: Centro de Investigación
en Ciencia Aplicada y Tecnología
Avanzada
CINVESTAV: Centro de
Investigación Avanzada
COMT: Catocol-O-metil-transferasa
COX-2: Ciclooxigenasa inducible
C.V.: Capital Variable
D1-5: Receptor de dopamina subtipo
1, 2, 3, 4 y 5
DA: Dopamina
DAG: Diacilglicerol
DCC: Diciclohexilcarbodiimida
DCU: Diciclohexilurea
DE50: Dosis efectiva 50
D.F.: Distrito Federal
DL50: Dosis letal 50
DMSO: Dimetilsulfóxido
DNA: Ácido desoxirribonucleico
DRX: Difracción de Rayos X
e: Número de Euler (neperiano) [E]: Concentración de enzima
E: Adrenalina o Epinefrina
EA: Enfermedad de Alzheimer
E.C.: Enzyme Classification
Edit.: Editorial
EE.UU.: Estados Unidos de América

8
EM: Espectrometría de Masas
EPSP: Potencial postsináptico
excitatorio
Es: Descriptor estérico de Taft
ES: Sitio esterático
ESM: Error Estándar de la Media
E.S.M.: Escuela Superior de
Medicina f: Función
FDA: Food and Drugs
Administration
FES: Facultad de Estudios
Superiores
FIMI: Fenilisomaleimida
FMA: Ácido fenilmaleámico
FMI: Fenilmaleimida
GABA: Ácido γ-amino-butírico
Gi: Proteína Gi (inhibition)
Glu: Glutamato
Gly: Glicina
Gq: Proteína Gq
Gs: Proteína Gs (stimulation)
Grb2: Factor de crecimiento unido a
receptor 2
h: Constante de Planck 1H: Hidrógeno 1
H1: Receptor de histamina subtipo 1
Hex: Hexano
HOMO: Highest Occupied Molecular
Orbital
His: Histidina
I.C.: Intervalo de Confianza
IP3: Trifosfato de inositol
I.P.N.: Instituto Politécnico Nacional
IPSP: Potencial postsináptico
inhibitorio
IR: Infrarrojo
J: Constante de acoplamiento
k+1: Constante experimental de
rapidez intrínseca de formación del
complejo k-1: Constante experimental de
rapidez intrínseca de disociación del
complejo
k2: Constante experimental de
rapidez intrínseca 2
K3: Constante experimental de
rapidez intrínseca 3
kB: Constante de Boltzmann
K, Keq: Constante de equilibrio
Kd: Constante de disociación
kDa: Kilo-Daltons
Ki: Constante de inhibición
Km: Constante de Michaelis-Menten Km
app: Constante de Michaelis-
Menten aparente Ks: Constante de afinidad l: Longitud de la celda
L-aArDC: L-aminoácido aromático
descarboxilasa
LUMO: Lowest Unoccupied
Molecular Orbital
M1-5: receptor muscarínico subtipos
1, 2, 3, 4 y 5
mn, mi, m0: Pendientes
MAO: Monoaminooxidasa

9
MAPK: Protein-cinasa activada por
mitógeno
MAP’s: Proteínas asociadas a
microtúbulos
MB: Mega-bytes
MEK: Cinasa de reconocimiento de
tirosín/treonín cinasas
MFIMI: p-metoxifenilisomaleimida
MFMA: Ácido p-metoxifenilmaleámico
MFMI: p-metoxifenilmaleimida
mRNA: Ácido ribonucleico
mensajero
n: Número de experimentos
nH: Coeficiente de Hill
NM: Receptor nicotínico tipo M NN: Receptor nicotínico tipo N ND: No Determinado
NE: Noradrenalina o Norepinefrina
NFIMI: p-nitrofenilisomaleimida
NFMA: Ácido p-nitrofenilmaleámico
NFMI: p-nitrofenilmaleimida
NMDA: N-metil-D-aspartato NMN: Normetanefrina
OH: Sitio oxianiónico org: Organization
p: Probabilidad
P: Producto
PAS: Sitio aniónico periférico
PDB: Protein Data Bank
p.f.: Punto de fusión
Phe: Fenilalanina
PIP2: Fosfatidil inositol difosfato PKA: Protein-cinasa A
PKC: Protein-cinasa C
PLA2: Fosfolipasa A2
PLC: Fosfolipasa C
PLD2: Fosfolipasa D2 p.p.: Páginas
QSAR: Quantitative Structure-
Activity Relationship
r, r: Coeficiente de correlación lineal
r2: Coeficiente de determinación
r, r0: Rapidez o velocidad
rmáx: Rapidez máxima
R: Constante de los gases ideales
Raf1: Cinasa Raf1
Ras: Familia de proto-oncogenes
RE: Retículo endoplásmico
RMN: Resonancia Magnética
Nuclear
RNA: Ácido ribonucleico
ROS: Especies reactivas de
oxígeno
RTK: Receptor de tirosín-cinasa
S: Sustrato
S.A.: Sociedad Anónima
Ser: Serina
Shc: Proteín-cinasa activada por
estrés
SNA: Sistema Nervioso Autónomo
SNC: Sistema Nervioso Central
SNP: Sistema Nervioso Periférico
SNS: Sistema Nervioso Somático
Sos: Son of sevenless
t: Tiempo
T: Temperatura

10
T3M: Tirosina-3-monooxigenasa
THF: Tetrahidrofurano
Trad.: Traducción
Trp: Triptofano
TrpH: Triptofano hidroxilasa
Tyr: Tirosina
TyrH: Tirosina hidroxilasa
U.N.A.M.: Universidad Nacional
Autónoma de México
UV: Ultravioleta
V: Volumen
Vis: Visible
Vol. Volumen (literatura) VTA: Área central del tegmento www: World Wide Web
α1-2: Receptor simpático alfa
subtipos 1 y 2
β1-3: Receptor simpático beta
subtipos 1, 2 y 3
δ: Desplazamiento químico
ΔG: Energía Libre de Gibbs
ε: Coeficiente de absortividad molar
λmáx: Longitud de onda máxima
ν: Número de onda
π: Descriptor hidrófobo o tipo de
interacción intermolecular
σ: Sigma de Hammett
τ: Proteína tau

11
SUMMARY Alzheimer’s disease causes an irreversible degeneration of mental capacities
which include a gradual loss of memory as well as personality and behavioural
changes. Worldwide the number of people with this disorder increases year by
year, above all in developed countries in Western Europe and North America
(including Mexico). There are various behavioural and pharmacological
strategies to mitigate the effects of this disease on the patients and their
families, the latter being based on antagonist of the NMDA receptors or
inhibitors of acetylcholinesterase (AChE). The inhibitors compensate for the
deficit of the neurotransmitter acetylcholine in the central nervous system. This
study is part of the look for new, more selective inhibitors through the rational
design of compounds. Three new phenylisomaleimides compounds were
designed and synthesized with three different substituents in the para position:
1) a strong electro withdrawing substituent (-NO2), 2) an electron donating
substituent (-OCH3) and 3) a lead compound with a neutral subtituent (-H).
Docking studies suggest that the compound with the –OCH3 substituent would
have greater affinity for the enzyme, while those with the –NO2 and –H
substituents would be practically the same in this respect. However, in the in
vitro assays the compound with the –NO2 substituent had a grater affinity for the
enzyme than both –H and –OCH3.

12
I. RESUMEN La enfermedad de Alzheimer es una enfermedad cerebral irreversible y
progresiva que implica la pérdida gradual de la memoria, cambios de
personalidad y el comportamiento, así como alteración severa de las
capacidades intelectuales. El número de pacientes con este padecimiento se
incrementa año con año a nivel mundial, sobre todo, en países desarrollados
de Europa occidental y Norteamérica (incluido México); así como en algunos
países en vías de desarrollo; no obstante, existen diversas estrategias para
paliar la enfermedad y retrasar los severos efectos que tiene sobre los
pacientes y la convivencia con sus familiares. Las estrategias seguidas
incluyen a la terapia conductual y la farmacológica, esta última, en general,
envuelve a fármacos antagonistas a los receptores NMDA de glutamato y a los
inhibidores de la acetilcolinesterasa (AChE), los cuales actúan de manera
parasimpaticomimética para compensar el déficit del neurotransmisor
acetilcolina en el Sistema Nervioso Central. Hoy por hoy, se han intentado
buscar nuevos fármacos más selectivos a blancos biológicos, y la AChE no es
la excepción, debido a la gran incidencia de la enfermedad de Alzheimer; por
tal motivo, este documento trata sobre la síntesis de nuevas moléculas capaces
de unirse e inhibir a la acetilcolinesterasa mediante un diseño racional de
interacción sobre el blanco. Se propuso obtener 3 compuestos tipo
fenilisomaleimidas cuyos grupos sustituyentes en posición para incluyen a un
grupo fuertemente electroatractor (-NO2), uno electrodonador (-OCH3) y uno de
referencia (-H). Los resultados obtenidos incluyen una transformación
química de las moléculas sintetizadas en el ensayo in vitro con AChE, cuyos
resultados por simulación de la interacción ligando-enzima sugieren que la
molécula con sustituyente –OCH3 es más afín a la enzima, mientras que con –
NO2 y –H son prácticamente iguales; sin embargo, experimentalmente la
molécula con el sustituyente –NO2 es más afín que –H, y a su vez que –OCH3.
II. INTRODUCCIÓN II.1 Relación Cuantitativa Estructura-Actividad (QSAR)4 Los medicamentos están considerados como el descubrimiento más importante
del siglo XX; actualmente la inmensa cantidad de fármacos que dispone la

13
humanidad han sido desarrollados a partir de un número pequeño de prototipos
denominados “cabezas de serie”4.
El descubrimiento y desarrollo de un nuevo fármaco considera una serie de
etapas las cuales son:
a) Búsqueda de un modelo
b) Manipulación del modelo
c) Determinación de formas farmacéutica de dosificación
La búsqueda del modelo o “cabeza de serie” supone encontrar una actividad
biológica nueva en un compuesto químico. Se trata de encontrar nuevas
estructuras químicas que sirvan de referencia para su modificación molecular.
El descubrimiento de una nueva cabeza de serie puede realizarse de diferentes
maneras, así pues, los modelos realizados más interesantes se encuentran en
los metabolitos secundarios de los organismos vivos y planteamientos
bioquímicos específicos con el fin de sintetizar sus análogos estructurales.
Desde el punto de vista general, pueden considerarse tres tipos de estrategias
para manipular una cabeza de serie, las cuales son:
II.1.1 Simplificación del modelo
Es un método conocido como variación estructural disyuntiva la cual consiste
en simplificar la estructura química de productos naturales de estructura
compleja4.
II.1.2 Asociación de dos o más moléculas
Es la unión de dos moléculas con determinada actividad, con el propósito de
potenciar aquella. Entre las uniones más conocidas, se encuentran la “adición
molecular” definida como la asociación de moléculas distintas por interacciones
débiles; “la replicación molecular” que consiste en asociar dos o más moléculas
iguales por unión covalente, y la “hibridación molecular” que es la unión de 2 ó
más moléculas distintas por unión covalente4.
II.1.3 Replicación moduladora
Es la sustitución de determinados grupos de la estructura modelo. Es la
estrategia más frecuente de manipulación de la cabeza de serie4.
La aplicación de los procedimientos que anteceden no suelen llevarse de una
forma aleatoria, sino a través de una serie de criterios que tienen ciertas
posibilidades de conducir a mejoras terapéuticas:

14
a) Apertura o cierre de anillos
Se trata de una modificación que ha conducido con frecuencia a compuestos
que retienen o rebasan la actividad del modelo. En algunos casos, las
variaciones en los anillos conducen a estructuras con mecanismos de acción
diferentes (Figura 1).
CH3
NH2
NH2
ANFETAMINA TRANILCIPROMINA Figura 1. Formación de un anillo. Anfetamina: Simpaticomimético, es un falso
neurotransmisor de catecolaminas e inhibidor de la recaptura de 5-HT. Tranilcipromina: Antidepresivo inhibidor de la MAO.
b) Introducción de enlaces múltiples
La introducción de un enlace doble o triple puede alterar la forma de la
molécula en el sentido del incremento de su rigidez, y por tanto, sus
propiedades fisicoquímicas. En relación a este tipo de modificación, cabe
mencionar un criterio de uso frecuente, denominado “Principio de vinología”, en
el cual dos sustituyentes X y Y unidos por una cadena vinílica o polivinílica
(incluido el anillo de benceno), se comportan desde el punto de vista de
distribución electrónica, como si estuvieran unidos directamente (Figura 2)4.
X Y XY
n
X Y X
Y
H2N
O
O
N
H2N
O
O
N
H2N
O
O
N
PROCAÍNA
VINÍLOGOSDEPROCAÍNA
Figura 2. Principio de vinología. Procaína: anestésico local bloqueador de canales de Na+
voltaje-dependientes. Sus vinílogos son activos.
La aplicación del principio de vinología al diseño de estructuras activas fracasa
si los factores electrónicos no son decisivos en la actividad e intervienen otros,
como factores estéricos, distancias entre grupos determinados, etc4.

15
c) Homología
Un homólogo de un determinado compuesto es el análogo que resulta de la
adición de un carbono a una cadena o anillo. Este cambio suele ir acompañado
por un incremento en la lipofilicidad. Los comportamientos más comunes a la
homología son:
• La actividad crece al aumentar el número de carbonos hasta alcanzar un
máximo, a partir del cual vuelve a disminuir4.
• La actividad crece hasta alcanzar una meseta, manteniéndose más o
menos constante para varios términos y descendiendo después; por
ejemplo los bloqueadores ganglionares (Figura 3)4.
N N
CH3
H3C
H3C
CH3
CH3
CH3n
n = 4-6
Figura 3. Bloqueadores ganglionares.
• La acción farmacológica cambia si la homologación afecta a una parte
de la molécula que participa en su unión con el receptor (Figura 4).
N
S
N
CH3
CH3
CH3N
S
N
CH3
CH3
N
CO2C2H5C6H5
CH3
N
C6H5
CO2C2H5
CH3
PROMETAZINA CLORPROMAZINA
MEPERIDINA ETOHEPTAZINA Figura 4. Cambio en la acción farmacológica por homologación. Prometazina: antihistamínico H1; Clorpromazina: neuroléptico. Meperidina: hipnoanalgésico;
Etoheptazina: hipnoanalgésico
d) Introducción de grupos voluminosos
En algunas ocasiones la introducción de grupos voluminosos a una estructura
capaz de provocar una respuesta por unión a un determinado receptor puede
conducir a un antagonista de dicho receptor4. Este fenómeno puede atribuirse a
la alta lipofilicidad de dichos grupos funcionales (Figura 5).

16
NOH3C
H3C
H3C
CH3
O
ACETILCOLINA(Agonista colinérgico)
H3CN
CH3
H3C
H3C
CH3
O
O
O
PROPANTELINIO(Antagonista colinérgico)
Figura 5. Introducción de grupos voluminosos.
e) Bioisosterismo
El isosterismo es un concepto químico que se ha aplicado al desarrollo de
nuevas moléculas con actividad farmacológica. En la definición original de
Langmuir, este término se utilizaba para describir la semejanza en propiedades
físicas y químicas que presentan una serie de iones y moléculas que contienen
el mismo número de átomos y electrones de valencia. La llamada ley de
desplazamiento de hidruro enunciada por Grimm que dice que un átomo es
isóstero con la especie que resulta al añadir un hidrógeno y dos electrones al
átomo que le precede en el sistema periódico.
El fenómeno por el que dos compuestos de estructuras relacionadas presentan
propiedades biológicas semejantes fue descrito por Friedman como
bioisosterismo; por tanto, pueden definirse como bioisóteros a las moléculas o
grupos que debido a poseer propiedades físicas o químicas análogas,
producen una respuesta farmacológica semejante u opuesta (Figura 6).
N N
N
CH3H3C
CH3
CH3
O
N N
CH3H3C
CH3
CH3
O
AMINOPIRINA(Analgésico-antipirético)
AMINOFENAZONA(Analgésico-antipirético)
Figura 6. Bioisosterismo clásico.
Como hemos visto, las estrategias de modificación molecular son necesarias
para hallar moléculas con probable acción farmacológica; por tanto, su
manipulación constituye el establecimiento de relaciones estructura-actividad,
que es uno de los objetivos más importantes de la química farmacéutica.

17
Las relaciones estructura-actividad tienen como primer propósito establecer el
descubrimiento del grupo farmacóforo, es decir, la estructura mínima
responsable de la acción farmacológica a través de la interacción con
receptores o enzimas; como segundo objetivo es el agregar sustituyentes al
grupo farmacóforo de una forma estratégica generando así compuestos con
propiedades terapéuticas óptimas.
Una relación estructura actividad se establece cuando un conjunto de
propiedades de una serie de compuestos explica su actividad o respuesta
biológica:
Actividad Biológica = f (Propiedades) Los datos más adecuados para establecer una relación estructura-actividad
son los que determinan la dosis para una respuesta biológica, como son la
DE50 y DL50, que indican la dosis que se requiere para alcanzar el 50% de la
respuesta máxima en términos de eficacia o mortalidad; así como las
constantes de disociación y e rapidez (k2. kcat, Kd, Ki, etc.).
Análogamente a los descriptores de las propiedades físicas de las moléculas,
los descriptores de las propiedades biológicas se expresan como el logaritmo
negativo de la dosis o concentración (C) para producir una respuesta
determinada en los ensayos in vivo, in vitro o in silico. En las medidas in vitro,
se relacionan con el logaritmo negativo de la constante de disociación ligando-
receptor, así como los experimentos in silico.
Puede aceptarse que la actividad biológica de un compuesto es dependiente
de tres factores principales: fisicoquímicos, estructurales y teóricos:
Actividad Biológica = f (Fisicoquímicos) + f (Estructurales) + f (Teóricos) El análisis cuantitativo de las relaciones estructura-actividad (QSAR) surge de
la interpretación sistemática de esa representación, y fue introducida por
primera vez por Corwin Hansch en 19634. Establecer este tipo de relación tipo
QSAR consta de seis etapas fundamentales:
• Planteamiento de los objetivos
• Determinación de la actividad biológica
• Descripción de los compuestos
• Análisis estadístico
• Interpretación de la relación establecida

18
• Predicción de actividades
Los descriptores electrónicos, estéricos e hidrófobos constituyen el factor
fisicoquímico que relaciona a la molécula con su actividad biológica.
1) Descriptor Electrónico. El análisis QSAR se basa en la demostración hecha por Hammett en 1937 de
la influencia constante de un sustituyente particular en las propiedades
fisicoquímicas de moléculas diferentes. Hammett aportó la primera definición
de constante de sustituyente, encontró que en el caso de los derivados
bencénicos meta y para sustituidos, se observa una relación lineal entre
logaritmos de las constantes de disociación del ácido benzoico (K´) en abscisas
y fenilacéticos (K) en ordenadas, diversamente sustituidos (pKa). Los puntos
siguen aproximadamente la relación que establece la ecuación [1], donde A es
la pendiente y C la ordenada al origen:
[1] log K = A logK’ + C La pendiente A fue llamada por Hammett �, y depende de la reacción
considerada (en este caso, la disociación), por lo que para un sustituyente X, la
ecuación [1] puede expresarse como la ecuación [2], y cuando X = H, como la
[3]:
[2] log KX = ρ log K’X + C
[3] log KH = ρ log K’H + C
Restando las ecuaciones anteriores se tiene [4]:
[4] log (KX/KH) = ρ log (K’X/K’H)
Hammett escogió como reacción estándar, con un valor de r igual a 1, la
ionización de los ácidos benzoicos diversamente sustituidos en agua a 25 °C, y
definió como constante de Hammett de un sustituyente al logaritmo del
conciente entre la constante de ionización del ácido benzoico sustituido y del
ácido benzoico, ecuación [5], estableciendo el valor de σ = 0 para X = H:
[5] σ = log (K’X/K’H)
Esta ecuación significa que los sustituyentes electroatractores tendrán valores
de σ > 0, mientras que los electrodonadores tendrán valores σ < 0. A partir de
la ecuaciones [4] y [5] se deduce la ecuación [6]:
[6] log (K’X/K’H) = ρσ

19
La constante de sustituyente mide el efecto electrónico o polar del mismo, que
incluye la electronegatividad (efecto de campo o inductivo) y la resonancia. De
la representación de cualquier σ de Hammett como una combinación lineal de
ambos, surgió en 1968 la escala de Swain y Lupton, con los parámetros e y Y,
que corresponden a una separación cuantitativa de los efectos de resonancia e
inductivo para cada sustituyente [8]:
[8] r e + f Y
Para comprender esta ecuación, es necesario ilustrarlo con un ejemplo;
supongamos que tenemos al –Cl y al -NO2; sus valores de Y son positivos para
ambos ya que su efecto es inductivo atrayente de electrones (I de Ingold),
mientras que el valor de e es positivo para -NO2 y negativo para –Cl, porque
sus efectos de resonancia (M de Ingold) en el anillo bencénico son opuestos.
Por tal motivo, los sustituyentes electroatractores y electrodonadores pueden
tener valores positivos o negativos inductivos y de resonancia dependiendo del
mismo sustituyente, así como de su posición en el anillo.
En QSAR, se usan otro tipo de descriptores electrónicos entre los que se
encuentran el pKa, desplazamientos químicos de RMN para 1H, 13C, 19F, 15N,
etc.; números de onda en infrarrojo, potenciales redox, momentos dipolares,
etc. También se pueden utilizar parámetros derivados de la química cuántica
obtenidos básicamente de programas computacionales que determinan la
función de onda de Schröedinger, tales como, cargas efectivas de los átomos,
cargas parciales, energías de ionización, energías de optimización de
geometría, energías de HOMO y LUMO, y potenciales electrostáticos
moleculares.
2) Descriptor Estérico.
Taft definió σ* para los sustituyentes en series alifáticas. Esta constante mide el
efecto inductivo de los sustituyentes X en la hidrólisis básica o ácida de los
ésteres sustituidos en la posición orto (Figura 7):
XCH2-COOR + H2O XCH2-COOH + ROH Figura 7. Hidrólisis básica y/o ácida de ésteres alifáticos utilizados por Taft.
Los efectos de los sustituyentes y, por tanto, la energía libre de activación del
proceso, se pueden tratar como la suma de las contribuciones independientes

20
de los efectos polares (ρσ), de resonancia (R) y estéricos (S), tanto en medio
básico (B) como en medio ácido (A) (ecuaciones 1 y 2):
1) log [k/k0]B = ρBσ*X + SB + RB
2) log [k/k0]A = ρAσ*X + SA + RA
Las constantes experimentales de rapidez k y k0 se refieren a las reacciones de
XCH2-COOR y CH3-COOR, respectivamente. La escala de σ* para
sustituyentes alifáticos está referida al grupo –CH3 y no al –H.
Por otra parte, los efectos polares de los sustituyentes son mucho mayores en
medio básico que en medio ácido. Esta hipótesis está basada en los valores de
ρ obtenidos por Hammett para la hidrólisis de benzoatos meta y para
sustituidos. En las hidrólisis básicas, ρB oscila entre +2.2 y +2.8, mientras que
en las ácidas, dicho valor varía de -0.2 a 0.0, lo que indica que ρB >> ρA. Se
puede asumir que ρB – ρA es un valor constante, que Taft escogió como 2.48
para colocar en una misma escala las constantes de reacción ρ en series
alifáticas y aromáticas, así como los parámetros σ* de Taft y s de Hammett (4):
4) log [k/k0]B - log [k/k0]A = 2.48 σ*X
Los valores de σ* de Taft, son positivos para los sustituyentes electroatractores
y negativos para los electrodonadores y se pueden referir también al hidrógeno
en lugar del metilo (5):
5) σ*(X) = 2.8 σ*(CH2X)
Una vez conocidos los efectos polares de los sustituyentes sobre las
constantes experimentales de rapidez de hidrólisis de los ésteres alifáticos
(ecuación 4), Taft utilizó esa información para determinar los efectos estéricos.
Si la ecuación (2) se sustituye en ρA por cero y además se designa a la suma
de S + R como ES, tendremos (ecuación 6):
6) log [k/k0]A = ES Esto hace suponer que la hidrólisis ácida de los ésteres alifáticos y la reacción
de esterificación inversa son insensibles a los efectos polares. Generalizando
este concepto se deduce la ecuación 7:
7) log (k/k0) = δ ES
Donde ES es un descriptor químico del efecto estérico primario, es decir, de la
interacción directa entre el grupo voluminoso y la función estudiada, y δ
describe la sensibilidad de la reacción a los efectos estéricos de los

21
sustituyentes. Se toma δ igual a 1 para la reacción de hidrólisis ácida de
ésteres alifáticos, y ES igual a cero cuando X = H, es decir, cuando el grupo
unido a la función éster es un metilo.
3) Descriptor Hidrófobo. El análisis QSAR sólo ha sido realmente posible después de que Hansch y
Fujita (Figura 8) descubrieran que la contribución de un sustituyente
determinado al logaritmo del coeficiente de partición aceite-agua es un valor
constante.
Figura 8. Corwin Hansch y Toshio Fujita.
Estos investigadores definieron la constante de sustituyentes πX para efectos
hidrofóbicos como se indica en la ecuación 1:
1. πX = log PX – log PH
Donde PX y PH son los coeficientes de partición octanol-agua de la forma
neutra de las moléculas sustituidas y sin sustituir, respectivamente.
Cuanto más positivo es π, el sustituyente es más lipofílico y viceversa. La
importancia de los valores de π radica en los procesos de absorción,
distribución y eliminación de un fármaco, así como su correlación con los
estudios de interacciones ligando-receptor.
Hay que recordar que el coeficiente de partición es la razón de concentraciones
de una especie única entre dos fases en equilibrio C1/C2, donde la fase 1
corresponde al agua y la fase 2 al octanol u otro hidrocarburo no miscible con
el agua.
Es importante considerar la ionización de la molécula ya que este fenómeno
complica la determinación del logP. A un pH dado, la forma ionizada de la
molécula disminuirá el valor del coeficiente de partición si predomina sobre su
forma no ionizada; por tal motivo es importante hablar de coeficientes de

22
distribución (D), que considera todas las especies existentes: neutras e iónicas.
Ambos coeficientes están relacionados por las ecuaciones I y II para un ácido y
una base, cuando la especie iónica no es soluble en la fase orgánica:
I. log D = log P – log [1 + 10(pH-pKa)] II. log D = log P – log [1 + 10(pKa-pH)]
Cabe mencionar que en un análisis QSAR a veces se incluyen variables
indicadoras de la presencia o ausencia de grupos funcionales o estructurales
específicos, por ejemplo, que existan o no sustituyentes en posición orto de un
centro de reacción; que existen dos sustituyentes contiguos o no; etc. Otras
variables describen posibles centros para la interacción fármaco-receptor, que
no pueden ser expresados por ninguno de los parámetros descritos
previamente. Por ejemplo, la presencia de un grupo dador de puentes de
hidrógeno en la molécula puede aumentar o disminuir la actividad biológica.
4) Métodos para establecer relaciones cuantitativas estructura-actividad biológica. Las metodologías usadas para ver qué parámetros se relacionan mejor con la
actividad biológica son muy diversas. La más popular es el análisis de
regresión lineal múltiple, que consiste en encontrar por el método de mínimos
cuadrados los coeficientes a0, a1, …, an de una ecuación del tipo <1>, que mejor
represente la variable dependiente y (La respuesta biológica como función de
variables independientes x1, x2,…, xn).
<1> y = a0 + a1x1 + a2x2 + … + anxn
El análisis de Hansch consiste en utilizar este procedimiento con los distintos
tipos de constante de sustituyente citados previamente y el valor de la actividad
biológica dada como log (1/C), siendo C la concentración necesaria para
obtener una respuesta biológica determinada. La ecuación generalizada se
expresa como <2> siendo xs un parámetro estérico, xe uno electrónico y xh uno
hidrofóbico, y a, b, c, d y e los coeficientes de la regresión:
<2> log (1/C) = a0 + a1xs + a2xe + a3xh ; es decir,
log (1/C) = a + b logP + dσ + eES
log (1/C) = a + bπ + dσ + eES
Cuando se encuentran ecuaciones de este tipo con un término cuadrático en el
parámetro hidrofóbico, se trata de una relación parabólica <3>:

23
<3> log (1/C) = a + b (logP) – c (logP)2 + dσ + eES
log (1/C) = a + bπ – cπ2 + dσ + eES
Siempre que definamos una correlación hemos de valorarla estadísticamente
en función del número de compuestos usados (n), que debe superar un mínimo
para que la correlación sea válida, del coeficiente de determinación múltiple o
r2, que toma valores entre 0 y 1 (cuanto más próximo a 1 mejor es la
correlación), y de la desviación estándar (S) (cuanto menor sea, más ajustada
será la correlación).
El valor de los coeficientes indica la importancia o aportación de cada término o
propiedad tal de la actividad biológica, y puede ocurrir que no todos los
coeficientes sean significativos; es decir, la actividad biológica dependerá
según los casos de todos los efectos: electrónicos, estéricos e hidrófobos, o
solo alguno de ellos.
II.2 Química Computacional II.2.1 Métodos Computacionales La química computacional es una rama de la química teórica que considera el
modelaje de los aspectos químicos por computadora; así como la simulación y
predicción de los procesos químicos.
La aplicación de esta rama, cubre otras áreas de las ciencias naturales, como
son: la química orgánica, inorgánica, analítica (UV, IR, RMN, EM, etc.),
bioquímica, física, farmacología, ciencias de los materiales, cinética química,
catálisis, etc.
La química computacional se divide en diferentes áreas para su estudio, las
cuales son: Modelado Molecular, Mecánica y Dinámica Molecular, Química
Cuántica, Base de datos, Desarrollo de M (Tabla 1):

24
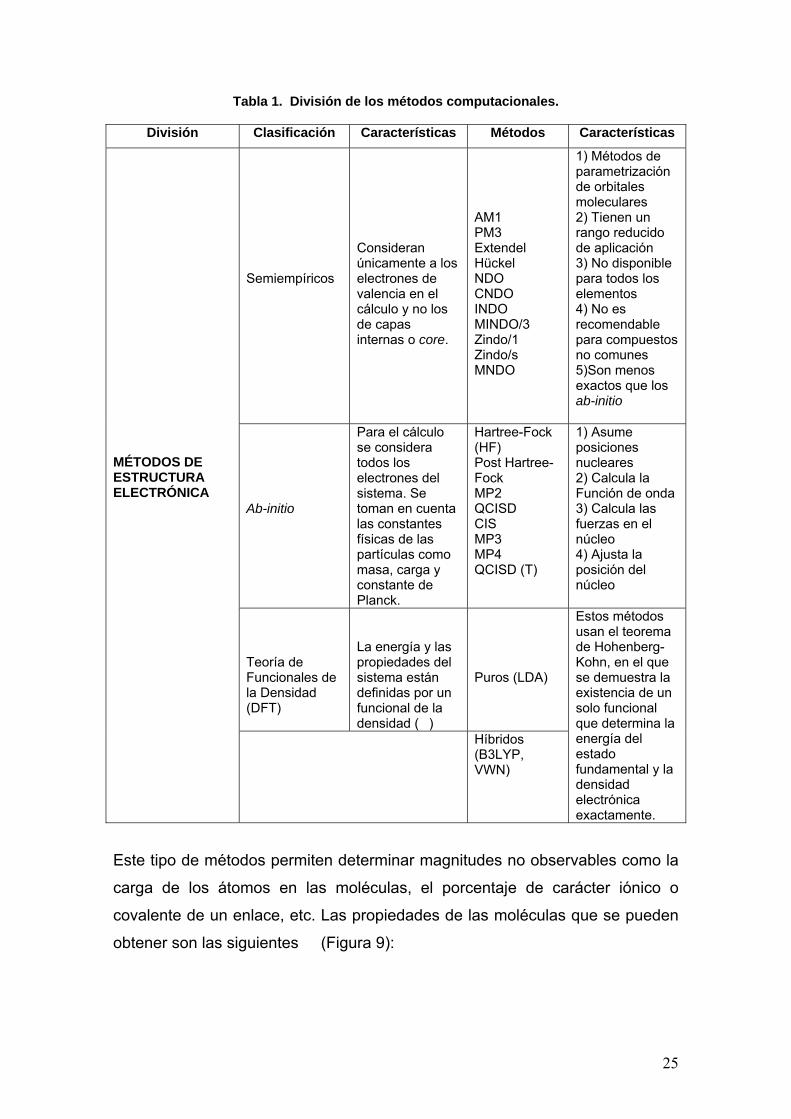
Tabla 1. División de los métodos computacionales
División Características
Modelado Molecular
Estudio de la relación entre la estructura molecular y las propiedades químicas de la materia.
Mecánica Molecular
Considera a la molécula como una colección de partículas, en donde la unión entre ellas se simula mediante la aproximación de resortes.
Dinámica Molecular
Permite simular partículas, involucrando explícitamente a la presión y a la temperatura como variables.
25
Tabla 1. División de los métodos computacionales.
División Clasificación Características Métodos Características
Semiempíricos
Consideran únicamente a los electrones de valencia en el cálculo y no los de capas internas o core.
AM1 PM3 Extendel Hückel NDO CNDO INDO MINDO/3 Zindo/1 Zindo/s MNDO
1) Métodos de parametrización de orbitales moleculares 2) Tienen un rango reducido de aplicación 3) No disponible para todos los elementos 4) No es recomendable para compuestos no comunes 5)Son menos exactos que los ab-initio
Ab-initio
Para el cálculo se considera todos los electrones del sistema. Se toman en cuenta las constantes físicas de las partículas como masa, carga y constante de Planck.
Hartree-Fock (HF) Post Hartree-Fock MP2 QCISD CIS MP3 MP4 QCISD (T)
1) Asume posiciones nucleares 2) Calcula la Función de onda 3) Calcula las fuerzas en el núcleo 4) Ajusta la posición del núcleo
Teoría de Funcionales de la Densidad (DFT)
La energía y las propiedades del sistema están definidas por un funcional de la densidad (�)
Puros (LDA)
MÉTODOS DE ESTRUCTURA ELECTRÓNICA
Híbridos (B3LYP, VWN)
Estos métodos usan el teorema de Hohenberg-Kohn, en el que se demuestra la existencia de un solo funcional que determina la energía del estado fundamental y la densidad electrónica exactamente.
Este tipo de métodos permiten determinar magnitudes no observables como la
carga de los átomos en las moléculas, el porcentaje de carácter iónico o
covalente de un enlace, etc. Las propiedades de las moléculas que se pueden
obtener son las siguientes (Figura 9):

26
Figura 9. Propiedades que pueden ser obtenidas de la química computacional.
Existen programas computacionales que desarrollan este tipo de métodos,
entre ellos se encuentran el programa Hyperchem, Gaussian, Spartan Pro, etc.
Los 3 programas tienen métodos de cálculo como mecánica molecular y
mecánica cuántica.5 II.2.2 Docking La mayoría de los fármacos conocidos han sido aislados de los productos
naturales, por síntesis o serendipia, así como sus análogos y derivados
químicos; sin embargo, hoy en día se dispone de tecnología más avanzada y
sofisticada para encontrar nuevas moléculas para uso terapéutico a través de
búsquedas sistemáticas6.
PROPIEDADES
Parámetros Geométricos
Propiedades Electrónicas
Propiedades Termodinámicas
Propiedades Espectroscópicas
Longitud de enlace Ángulo de enlace Ángulos diedros
Orbitales moleculares Distribución de carga Momento bipolar Afinidad electrónica Afinidad electrónica Potencial de ionización
Energía molecularPoblación Calor de formación Superficie de energía potencial ΔH, ΔG Energía de activación Caminos de reacción
Frecuencias vibracionales Intensidades de infrarrojo RMN (1H, 13C)

27
Para iniciar con la búsqueda es necesario encontrar un blanco biológico y
molecular en el cual será el punto de enfoque; entre los blancos más comunes
destacan las proteínas (receptores, enzimas, canales iónicos y receptores
nucleares), las hormonas y factores, y finalmente el DNA6.
Cabe mencionar que cuando se trabaja con proteínas y en particular con
enzimas es importante considerar tres aspectos fundamentales: especificidad y
selectividad por la enzima, afinidad con la cual se une el sustrato a la proteína y
la geometría del sitio activo.
Una vez escogido el blanco o receptor de estudio es necesario escoger una
serie de compuestos para identificar de ellos una molécula líder o cabeza de
serie4, a los cuales se les hace pasar por pruebas a microescala o screening
para seleccionar aquellas moléculas que tengan la mejor actividad biológica
sobre el blanco escogido; para eventualmente sintetizar nuevos análogos y
probarlos biológicamente con el fin de establecer una relación cuantitativa
estructura-actividad4, 6 (QSAR). Esta es la forma tradicional de hallar nuevas
moléculas activas; sin embrago, este trabajo requiere de mucho tiempo y de
recursos, por ello; se ha ideado metodologías más rápidas, eficientes y menos
costosas, como la simulación de la interacción ligando-receptor asistido por
computadora (Docking); a este tipo de ensayos se les denomina estudios in
silico.
La metodología Docking consta de una serie de etapas; la primera de ellas es
disponer de la estructura tridimensional de la molécula blanco; la segunda
consiste en tener una numerosa serie de ligando potenciales con estructura
tridimensional conocida; y la tercera etapa consiste en un algoritmo de
computadora que toma cada uno de los ligandos y los hace interaccionar con la
molécula blanco en el sitio de unión previamente escogido por el usuario
(Figura 10).

28
Figura 10. Etapas fundamentales para llevar a cabo un estudio de Docking6.
II.2.2a Estructura Tridimensional de la molécula blanco. Para hacer un estudio de docking, es necesario tener la estructura en tercera
dimensión determinada por difracción de Rayos X, por Resonancia Magnética
Nuclear (RMN) o Modelado molecular. La fuente principal de estructuras
tridimensionales de proteínas es el Protein Data Bank (PDB), el cual es de
acceso gratuito a través de la página web http:www.pdb.org. La mayoría de las
estructuras de proteínas que se descargan de esta base de datos no pueden
utilizarse directamente en el estudio de interacción con algún ligando; antes de
ello es necesario realizar un tratamiento de previo que consiste en la adición
de átomos de hidrógeno que constituyen casi la mitad de los átomos de la
proteína (ya que su posición no es detectada por la técnica de difracción de
rayos X); remover las moléculas de agua y las moléculas ajenas a la proteína,
exceptuando a los grupos prostéticos o coenzimas que están ocupando el sitio
de unión del receptor, y por último, la asignación de cargas eléctricas formales
y parciales a los átomos de los aminoácidos que constituyen a la proteína de
acuerdo al pH en la simulación.
II.2.2b Sitio de unión con el blanco. Una proteína es una molécula muy grande con respecto a los ligandos de
prueba; por tal motivo, el método de docking sugiere escoger una región de la
macromolécula para su exploración por los ligandos; ya que si no se escogiese
esta zona el cálculo se vuelve más complicado y su requerimiento en tiempo
aumenta considerablemente. Por lo regular; la región seleccionada es el sitio

29
activo de la enzima o receptor; sin embargo, las moléculas de prueba no
necesariamente interaccionan con el sitio activo, sino con uno alostérico; en
este caso la búsqueda de la mejor interacción se hace más complicada, no
obstante, existen dos alternativas para delimitar la selección de la región; una
de ellas consiste en revisar la superficie molecular con ciertas características
de tamaño, hidrofobicidad y grupos funcionales. La otra alternativa es
meramente experimental y sugiere determinar los sitios en donde las moléculas
de disolvente orgánico se albergan; y su determinación se hace constar con la
proteína cristalizada y visualizada en difracción de rayos X6. La ubicación del
disolvente sobre la proteína se consideran como potenciales sitios de unión a
ligando.
En general, a este tipo de búsqueda de sitios de unión a ligando se le conoce
como mapeo por rejillas (celdas) o “Grid Maps”, el cual consiste en una celosía
tridimensional de puntos centrados y alrededor de la región de interés. Las
distancias entre cada punto varía desde 0.2Å hasta 1.0Å7.
Cada punto en la celda debe tener coordenadas tridimensionales nx, ny y nz, las
cuales representan una energía potencial para cada átomo o grupo funcional
estudiado en la proteína (Figura 11).
Figura 11. Selección del sitio de unión a ligando7.
II.2.2c Selección de los ligandos. Los ligandos son una serie de moléculas que tienen ciertas características
químicas que le confieren interaccionar sobre una molécula blanco; estos
ligandos incluyen generalmente la cabeza de serie o líder y sus derivados.
Cada una de éstas moléculas deben pasar por un tratamiento computacional
previo a la interacción con el blanco; y esto consiste en minimizar u optimizar la

30
estructura de cada molécula, para obtener su confórmero más estable y de
menor energía5; así como añadir átomos de hidrógeno, estimación de cargas
formales y parciales, asignación del número de enlaces de rotación libre, masa
molar, hidrofobicidad, etc.
II.2.2d Simulación de la interacción ligando-receptor y formación del complejo. El objetivo de este paso, es hacer interaccionar el ligando con la región del
blanco previamente seleccionada; el programa computacional toma al ligando
haciéndolo explorar dentro del sitio de unión, mediante esquemas numéricos
de puntaje en función de la orientación adoptada por el ligando en una
interacción. Durante una interacción el ligando adopta una orientación con un
cierto número de contactos con la macromolécula, si estos contactos son
permisibles y positivos para la interacción tendrá un alto puntaje; de lo
contrario esa orientación e interacción son desechadas, es decir, el programa
va seleccionando aquellas interacciones con el más alto número de contactos
permisibles (en función de las distancias y cargas) hasta encontrar la mejor; a
este tipo de esquema numérico se le conoce como metodología Simplex6.
Una de las formas para obtener puntajes confiables es tener programas de
regularización de geometría empleando ecuaciones de la física clásica que
determinen la energía no covalente de la unión ligando-receptor como función
de las distancias atómicas entre moléculas; por ejemplo se puede utilizar la
ecuación de Coulomb, la cual calcula la energía electrostática entre dos átomos
cargados:
E = k [(q1.q2)/(r1-2)2]
Donde k, es una constante (1/(4πε0)), q1 es la carga de un átomo del blanco y
q2 la carga de un átomo del ligando, y r es la distancia entre ambos átomos6.
Cuando se trata de una interacción con distancias cercanas entre los átomos,
la ecuación de la energía potencial de Van der Waals es necesaria:
E = (Ae-br/r) – (C6/r6) Donde (Ae-br/r) representa el intercambio de energía repulsiva y – (C6/r6)
representa la dispersión de energía atractiva entre los átomos. En general el
término (Ae-br/r) es aproximadamente (C12/r12), quedando la ecuación:
E = (C12/r12) – (C6/r6) E = (Cm/rm) – (Cn/rn)

31
Donde C12 y C6, son constantes que dependen de la intensidad de energía y la
separación de equilibrio entre los núcleos de los átomos. 6 y 12, son los
parámetros de Lennard-Jones (m = 12, n = 6)7, que se refieren al modelo de las
fuerzas de van der Waals. (Figura 12).
Figura 12. Diagrama de Energía-radio para el modelo de fuerzas de Van der Waals.
reqm = Equilibrio de separación internuclear7. En un análisis de docking, existen algoritmos con los cuales es posible correr la
interacción entre el ligando y el receptor, entre los más comunes se encuentran
el método Monte Carlo que simula reconocimiento, los algoritmos genéticos y el
algoritmo genético Lamarckiano. En la actualidad los algoritmos genéticos son
los más socorridos para un análisis de docking y se basan en el lenguaje de
evolución natural genética y biológica. En el caso de docking los arreglos
espaciales de los ligandos y la proteína pueden ser definidos por el
establecimiento de valores descritos para la traslación, orientación y
conformación del ligando con respecto a la proteína; es decir, los ligandos
poseen estados variables que corresponden a un gen dentro del algoritmo
genético. El estado del ligando corresponde al genotipo, y sus coordenadas
atómicas corresponden a su fenotipo8.
El algoritmo toma un par de individuos (genes) que son cruzados por un
proceso de crossover para generar nuevos individuos con genes heredados del
padre y la madre; sin embargo, durante el crossover la progenie puede sufrir
mutaciones productos de la gran cantidad de aleatorizaciones ocurridas. Por tal
motivo, es necesario que el algoritmo haga una selección de la progenie
basada en el anclado de los individuos8.

32
El docking molecular o fitness es la energía de interacción total del ligando con
la proteína, y es evaluado a través de funciones de energía.
II.3 Química de Amidas e Imidas II.3.1 Amidas Las amidas son especies químicas que poseen un grupo funcional amida el
cual está constituida principalmente de un grupo carbonilo, y junto a éste un
nitrógeno, N, N-disustituido, N-sustituido o sin sustituir (Figura 13). O
R NH2
O
R NH
O
R NR' R'
R''
Amida nosustituida Amida N-sustituida Amida N, N-disustituida
Figura 13. Tipos de amidas.
Las amidas suelen obtenerse por la reacción de un cloruro de ácido con una
amina: amoníaco, aminas monosustituidas y aminas disustituidas9.
Industrialmente, se suelen preparar por calentamiento de las sales de amonio
de ácidos carboxílicos10.
Las amidas son mucho menos reactivas que los cloruros de ácido, anhídridos
de ácido o ésteres; por tal motivo, el enlace amida sirve como la unidad básica
a partir de la cual se forman las proteínas.
Estas moléculas pueden sufrir hidrólisis para formar ácidos carboxílicos mas
aminas cuando se calientan con ácidos o bases en disolución acuosa9, 10.
Las amidas no son básicas cuando se tratan con ácidos acuosos, sus
disoluciones son neutras, y su poder nucleofílico es débil; la razón principal de
esta característica es que la amida está estabilizada por la deslocalización del
par electrónico no compartido del nitrógeno al superponerse con los orbitales
del grupo carbonilo (Figura 14).
N
O
H
H
N
O
H
H Figura 14. Deslocalización del par de electrones libres del nitrógeno. Fenómeno de
resonancia.

33
El producto protonado de la amida se pierde por estabilización por resonancia,
por tanto la protonación está desfavorecida energéticamente (ΔG° positivo);
además esta amida protonada tiene mayor energía que una amina protonada
debido a que el grupo carbonilo atrayente de electrones desestabiliza de
manera inductiva la carga positiva vecina (Figura 15)9. O
R NH3δ
δ
Figura 15. Amida protonada.
Las reacciones más comunes para preparar aminas son las siguientes (Figura
16):
R OH
O
NH3
R Cl
O
2 NH3
R O
O
R
O
2 NH3
R OR'
O
NH3
R N H2OH3O
R NH2
O
R NH2
O
R NH2
O
R NH2
O
R NH2
O
H2O
NH4Cl
R'OH
R O
O
NH4
Ácidocarboxílico
AmoníacoAmida
Amoníaco AmidaClorurode ácido
AmoníacoAmidaAnhídrido
de ácido
Éster AmoníacoAmida
AmidaAguaNitrilo
200 °C
o NaOH, H2
Figura 16. Rutas de síntesis más frecuentes para obtener amidas9.
II.3.2 Imidas. Las imidas pueden prepararse a partir del ataque de amidas o sus sales sobre
anhídridos de acil halidas, de ácidos carboxílicos y ésteres. El mejor método de
síntesis para la preparación de imidas acíclicas es la reacción entre una amida
y un anhídrido a 100 °C catalizado con H2SO411.
Al igual que otros anhídridos, los cíclicos reaccionan con amoníaco para
obtener amidas. En este caso, el producto contiene los grupos –CONH2 y
–COOH. Si se calienta esta amida-ácido, se pierde una molécula de agua,
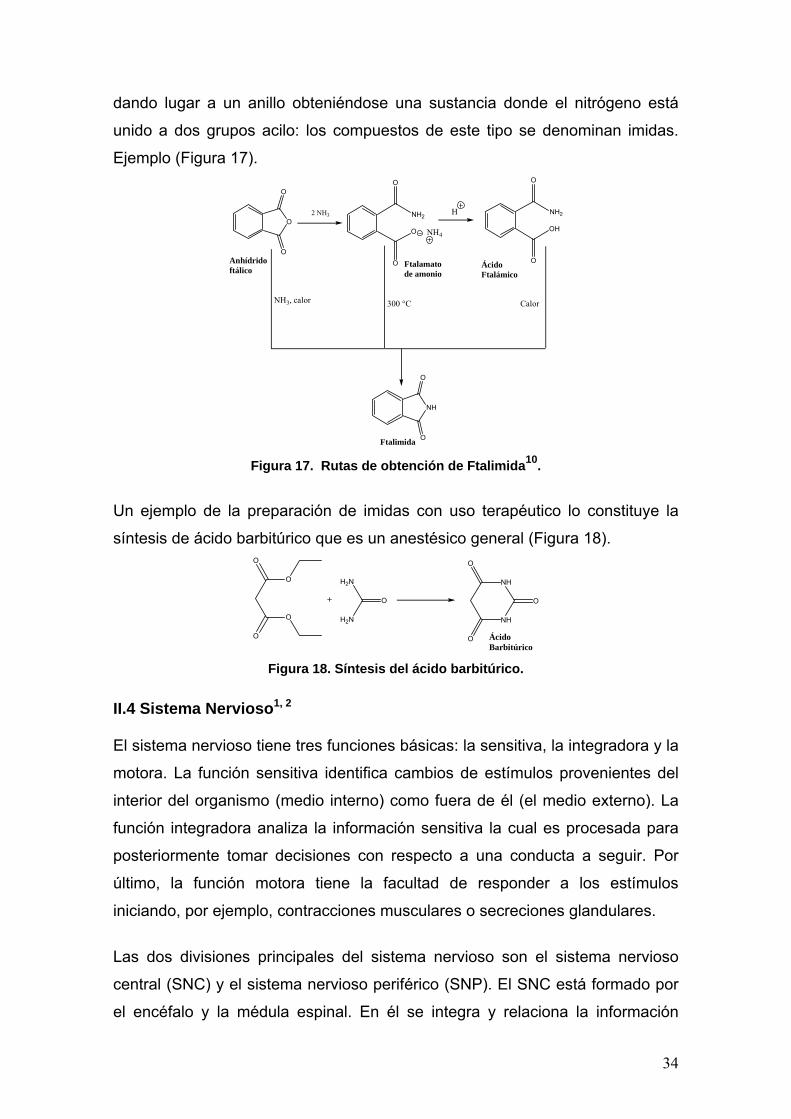
34
dando lugar a un anillo obteniéndose una sustancia donde el nitrógeno está
unido a dos grupos acilo: los compuestos de este tipo se denominan imidas.
Ejemplo (Figura 17).
O
O
O
2 NH3
O
O
NH2
O
NH
O
O
NH4
H NH2
O
O
OH
Anhídridoftálico
Ftalamatode amonio
ÁcidoFtalámico
Ftalimida
NH3, calor 300 °C Calor
Figura 17. Rutas de obtención de Ftalimida10.
Un ejemplo de la preparación de imidas con uso terapéutico lo constituye la
síntesis de ácido barbitúrico que es un anestésico general (Figura 18).
O
O
O
O
H2N
H2N
O
NH
NH
O
O
O
ÁcidoBarbitúrico
Figura 18. Síntesis del ácido barbitúrico.
II.4 Sistema Nervioso1, 2
El sistema nervioso tiene tres funciones básicas: la sensitiva, la integradora y la
motora. La función sensitiva identifica cambios de estímulos provenientes del
interior del organismo (medio interno) como fuera de él (el medio externo). La
función integradora analiza la información sensitiva la cual es procesada para
posteriormente tomar decisiones con respecto a una conducta a seguir. Por
último, la función motora tiene la facultad de responder a los estímulos
iniciando, por ejemplo, contracciones musculares o secreciones glandulares.
Las dos divisiones principales del sistema nervioso son el sistema nervioso
central (SNC) y el sistema nervioso periférico (SNP). El SNC está formado por
el encéfalo y la médula espinal. En él se integra y relaciona la información

35
sensitiva aferente, se generan los pensamientos y emociones y se forma y
almacena la memoria. La mayoría de los impulsos nerviosos que estimulan la
contracción muscular y las secreciones glandulares se originan en el SNC. El
SNC está conectado con los receptores sensitivos, los músculos y las
glándulas de las zonas periféricas del organismo a través del SNP. Este último
está formado por los pares craneales, que nacen en el encéfalo y los nervios
raquídeos, que nacen en la médula espinal. Una parte de estos nervios llevan
impulsos nerviosos hasta el SNC, mientras que otros salen del SNC.
El componente aferente del SNP consiste en células nerviosas llamadas
neuronas sensitivas o aferentes (ad = hacia; ferre = llevar). Conducen los
impulsos nerviosos desde los receptores sensitivos de varias partes del
organismo hasta el SNC. El componente eferente consiste en células nerviosas
llamadas neuronas motoras o eferentes ( ex = fuera de; ferre = llevar). Éstas se
originan en el interior del SNC y conducen los impulsos nerviosos desde éste a
los músculos y las glándulas.
El SNP puede subdividirse en sistema nervioso somático (SNS) (soma =
cuerpo) y sistema nervioso autónomo (SNA) (auto = propio; nomos = ley). El
SNS está formado por neuronas sensitivas que llevan información desde los
receptores cutáneos y los sentidos especiales, fundamentalmente de la
cabeza, la superficie corporal y las extremidades, hasta el SNC que conducen
impulsos sólo al sistema muscular esquelético. Como los impulsos motores
pueden ser controlados conscientemente, esta porción del SNS es voluntario.
El SNA está formado por neuronas sensitivas que llevan información desde
receptores situados fundamentalmente en las vísceras hasta el SNC, conducen
los impulsos hasta el músculo liso, el músculo cardíaco y las glándulas. Con
estas respuestas motoras no se encuentran normalmente bajo control
consciente, el SNA es involuntario (Figura 19).

36
Figura 19. Clasificación del Sistema Nervioso.
El SNA se presenta, para su división con bases anatómicas, en 2 porciones
principales: la división simpática (toraco-lumbar) y la parasimpático
(craneosacra). Ambas se originan en núcleos dentro del S.N.C. y emiten fibras
preganglionares eferentes que salen del tallo encefálico o la médula espinal y
terminan en los ganglios motores. Las fibras simpáticas preganglionares
abandonan el S.N.C. a través de los nervios raquídeos torácicos y lumbares.
Las fibras preganglionares parasimpáticas salen del S.N.C. a través de los
nervios craneales III, VII, IX, X; así como de la III y IV raíces sacras. (Figura
20).
Figura 20. Localización de las fibras eferentes del S.N.A.44
SISTEMA NERVIOSO
Sistema Nervioso Central (S.N.C.) Sistema Nervioso Periférico (S.N.P).
Sistema Nervioso Somático (S.N.S.)
Sistema Nervioso Autónomo (S.N.A.)
Sistema Nervioso Simpático (Toraco-lumbar)
Sistema Nervioso Parasimpático (Cráneo-sacro)
Sistema Nervioso Entérico

37
II.4.1 Funciones generales del Sistema Nervioso Autónomo3
La acción integradora del S.N.A. es de gran relevancia para el organismo ya
que regula aquellas estructuras que no se encuentran bajo control voluntario;
por ejemplo, la respiración, circulación, digestión, temperatura corporal,
metabolismo, sudación y secreciones de algunas glándulas endocrinas.
El sistema simpático y la médula suprarrenal no son esenciales para la vida; sin
embargo, en situaciones de estrés, se pone de manifiesto, ya que no se puede
regular la temperatura corporal cuando varía la temperatura ambiental; la
concentración de glucosa en sangre no se incrementa en reacción a
necesidades urgentes; no hay reacciones vasculares compensadoras a
hemorragias, privación de oxígeno, disminución de la resistencia a la fatiga;
etc.
En general el sistema simpático está en actividad continua y su grado de
dinamismo es variable de un momento a otro en ciertos órganos; por ejemplo:
durante la ira y el miedo, se acelera la frecuencia cardíaca, se incrementa la
presión arterial, los eritrocitos se vierten en la circulación desde el bazo, cambia
el flujo sanguíneo desde la piel y la región esplácnica hacia el músculo
esquelético, incremento de la concentración de glucosa en sangre, así como la
dilatación de bronquiolos y pupilas. Estos, y muchos efectos más son
resultados de la estimulación de adrenalina secretada por la médula
suprarrenal.
Por otro lado, el sistema parasimpático está organizado para la actividad
definida y localizada; se ocupa de manera primordial de la conservación de la
energía y la función orgánica durante los períodos de actividad mínima, su
eliminación es incompatible con la vida; por ejemplo, el corte del nervio vago da
lugar a infección pulmonar debido a la incapacidad de los cilios para eliminar
sustancia irritantes desde la vías respiratorias. El sistema parasimpático
lentifica frecuencia cardíaca, disminuye la presión arterial, estimula los
movimientos y las secreciones gastrointestinales, ayuda a la absorción de
nutrimentos, protege a la retina contra la luz excesiva y vacía la vejiga y el
recto.

38
Los impulsos nerviosos desencadenan reacciones en músculo liso, cardíaco y
esquelético, glándulas exocrinas y neuronas postsinápticas, mediante la
liberación de neurotransmisores químicos específicos.
El proceso de neurotransmisión es de gran importancia farmacológica ya que
los fármacos regulan una serie de fenómenos que acontecen de manera
natural, por ende, la neurotransmisión se divide en varias etapas (Figura 21).
A continuación se describirá brevemente las etapas de la neurotransmisión3: “1. El potencial de acción nervioso (AP) consiste en una inversión autopropagada
transitoria de la carga sobre la membrana axoniana (el potencial interno, Ei,
pasa desde un valor negativo, a través del potencial cero, hasta un valor
ligeramente positivo, sobre todo por incremento de la permeabilidad al Na+, y
posteriormente vuelve a valores de reposo mediante permeabilidad al K+).
Cuando el AP llega a la terminación presináptica, inicia la descarga del
transmisor excitador o inhibidor. La despolarización al nivel de la terminación
nerviosa y la entrada de Ca2+, inician el acoplamiento y, a continuación, la
fusión de la vesícula sináptica con la membrana de la terminación nerviosa. 2. La combinación del transmisor excitador con los receptores postsinápticos
produce despolarización local, el potencial postsináptico excitador (EPSP),
mediante un incremento de la permeabilidad de cationes, de manera notable el
Na+. El trasmisor inhibidor genera un incremento selectivo de la permeabilidad
a K+ o Cl-, lo cual da por resultado hiperpolarización local, el potencial
postsináptico inhibidor (IPSP). 3. El EPSP inicia un AP conducido en la neurona
NEUROTRANSMISIÓN
Conducción
Axoniana
Transmisión por
las uniones
Almacenamiento y liberación del
transmisor
Unión del transmisor con receptores
postsinápticos y producción del
potencial de acción
Inicio de la actividad
postsináptica
Destrucción o disipación del
transmisor
Figura 21. Etapas de la neurotransmisión

39
postsináptica; sin embargo, esto puede prevenirse mediante hiperpolarización
inducida por el IPSP concurrente. El transmisor disipa por destrucción
enzimática, recaptura en la terminación presináptica o las células gliales
adyacentes, o por difusión (Figura 22)”.
Figura 22. Fenómeno de sinapsis química3.
II.4.2 Transmisión Simpática. Las células que dan origen a las fibras preganglionares se encuentran en las
columnas intermediolaterales de la médula espinal, y se extienden desde el
primer segmento torácico hasta el segundo o el tercer segmentos lumbares.
Los axones provenientes de estas células corren por las raíces nerviosas
anteriores (ventrales), y hacen sinapsis con neuronas que se encuentran en los
ganglios simpáticos fuera del eje cefalorraquídeo. Los ganglios simpáticos tiene
tres localizaciones: paravertebral, prevertebral y terminal.
Los ganglios simpáticos paravertebrales comprenden 22 pares, cada uno
colocado a cada lado de la columna vertebral para formar cadenas laterales.
Los ganglios se conectan entre sí por troncos nerviosos y con los nervios
raquídeos, por medio de ramos comunicantes. Los ramos blancos se restringen
a los segmentos de la vía de salida toracolumbares; transportan a las fibras
mielínicas preganglionares que salen de la médula espinal por las raíces
raquídeas anteriores. Los ramos grises se originan en los ganglios y llevan
fibras posganglionares de vuelta hacia los nervios raquídeos para la
distribución hacia las glándulas sudoríparas y los músculos pilomotores, y hacia

40
los vasos sanguíneos del músculo estriado y la piel. Los ganglios
paravertebrales se encuentran en abdomen y pelvis, cerca de la superficie
ventral de la columna vertebral, y consisten en ganglios celiacos, mesentéricos
superiores, aortorrenales y mesentéricos inferiores. Los ganglios terminales se
localizan cerca de los órganos que inervan ganglios conectados con la vejiga
urinaria y el recto, así como, ganglios de la región cervical.
La transmisión adrenérgica incluye a la noradrenalina, neurotransmisor de la
mayor parte de las fibras simpáticas posganglionares y de ciertas vías del
S.N.C., y dopamina, transmisor predominante del sistema extrapiramidal del
mamífero y de diversas vías neuronales mesocorticales y mesolímbicas, lo
mismo que adrenalina, hormona principal de la médula suprarrenal. Sus
receptores están expresados en varios órganos efectores los cuales tienen
diferentes funciones (Tabla 2).
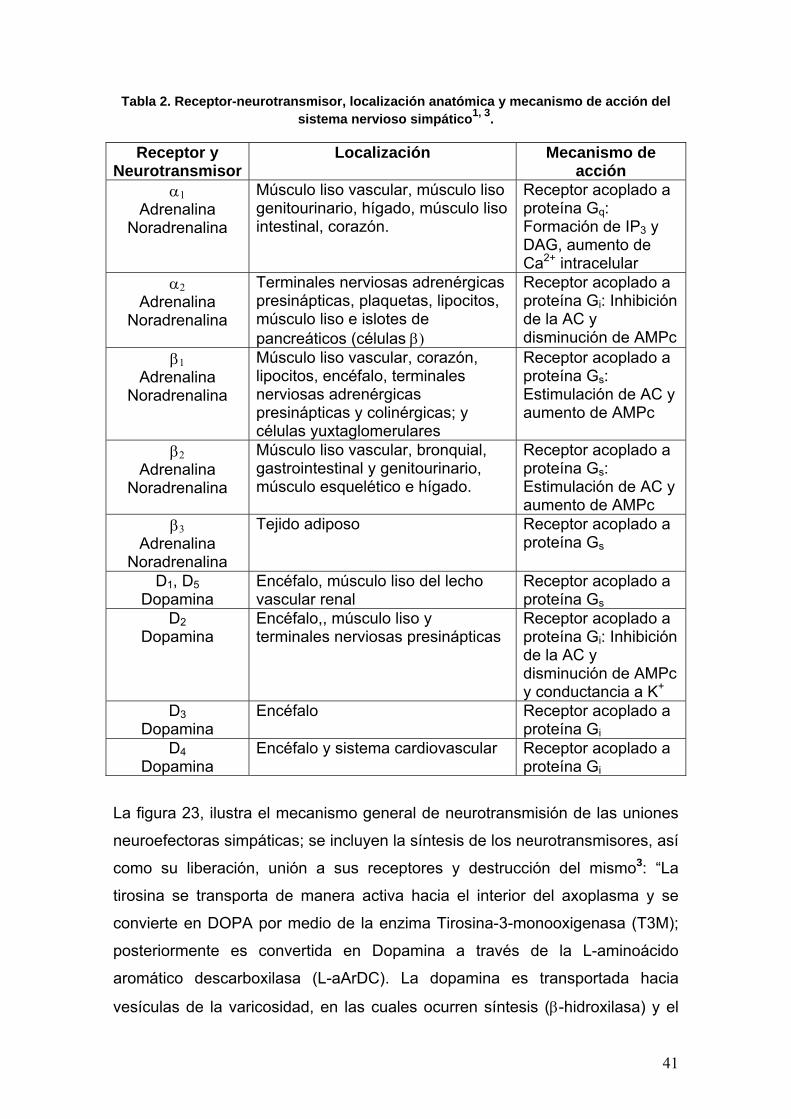
41
Tabla 2. Receptor-neurotransmisor, localización anatómica y mecanismo de acción del sistema nervioso simpático1, 3.
Receptor y
NeurotransmisorLocalización Mecanismo de
acción α1
Adrenalina Noradrenalina
Músculo liso vascular, músculo liso genitourinario, hígado, músculo liso intestinal, corazón.
Receptor acoplado a proteína Gq: Formación de IP3 y DAG, aumento de Ca2+ intracelular
α2 Adrenalina
Noradrenalina
Terminales nerviosas adrenérgicas presinápticas, plaquetas, lipocitos, músculo liso e islotes de pancreáticos (células β)
Receptor acoplado a proteína Gi: Inhibición de la AC y disminución de AMPc
β1 Adrenalina
Noradrenalina
Músculo liso vascular, corazón, lipocitos, encéfalo, terminales nerviosas adrenérgicas presinápticas y colinérgicas; y células yuxtaglomerulares
Receptor acoplado a proteína Gs: Estimulación de AC y aumento de AMPc
β2 Adrenalina
Noradrenalina
Músculo liso vascular, bronquial, gastrointestinal y genitourinario, músculo esquelético e hígado.
Receptor acoplado a proteína Gs: Estimulación de AC y aumento de AMPc
β3 Adrenalina
Noradrenalina
Tejido adiposo Receptor acoplado a proteína Gs
D1, D5 Dopamina
Encéfalo, músculo liso del lecho vascular renal
Receptor acoplado a proteína Gs
D2 Dopamina
Encéfalo,, músculo liso y terminales nerviosas presinápticas
Receptor acoplado a proteína Gi: Inhibición de la AC y disminución de AMPc y conductancia a K+
D3 Dopamina
Encéfalo Receptor acoplado a proteína Gi
D4 Dopamina
Encéfalo y sistema cardiovascular Receptor acoplado a proteína Gi
La figura 23, ilustra el mecanismo general de neurotransmisión de las uniones
neuroefectoras simpáticas; se incluyen la síntesis de los neurotransmisores, así
como su liberación, unión a sus receptores y destrucción del mismo3: “La
tirosina se transporta de manera activa hacia el interior del axoplasma y se
convierte en DOPA por medio de la enzima Tirosina-3-monooxigenasa (T3M);
posteriormente es convertida en Dopamina a través de la L-aminoácido
aromático descarboxilasa (L-aArDC). La dopamina es transportada hacia
vesículas de la varicosidad, en las cuales ocurren síntesis (β-hidroxilasa) y el

42
almacenamiento de noradrenalina (NE). Un potencial de acción produce
entrada de Ca2+ en la terminación nerviosa a través de canales de Ca2+ tipo L
dependientes de voltaje, lo que ocasiona la fusión de la vesícula y la membrana
plasmática y por ende, la liberación de la NE. El transmisor activa a los
receptores α y β adrenérgicos en la membrana de la célula postsináptica. La
NE que penetra en estas células se inactiva por acción de la enzima Catecol-O-
metiltransferasa (COMT), hasta normetanefrina (NMN). El mecanismo más
importante de la interrupción de la transmisión es la recaptura activa hacia el
interior del nervio y las vesículas de almacenamiento. Nota: La adrenalina se
sintetiza a partir de la NE por catálisis de la enzima Feniletanolamina-N-metil
transferasa.”
Tyr T3M DOPA
Tyr DβH
COMT
Ca
DA
NMN
Ca
Dopamina
L-aARDC
NE
2+
2+
NE NE
ββ
αα2
NE
NE
NE
Figura 23. Mecanismo general de transmisión simpática. + (Activación); - (Inhibición)
II.4.3 Transmisión Parasimpática3
Se constituye por fibras preganglionares que emergen de tres áreas del S.N.C.
y sus conexiones posganglionares. Las regiones de origen central son
mesencéfalo, bulbo raquídeo y parte sacra de la médula espinal. Las fibras
mesencefálicas, son aquellas que tienen origen del núcleo de Edinger-
Westphal del III par, y que pasan hacia el ganglio ciliar de la órbita (ver figura
20). Las fibras bulbares están constituidas por componentes parasimpáticos de
los pares craneales VII, IX y X. Las fibras del par VII, constituyen la cuerda del
tímpano, que inerva los ganglios que se encuentran sobre glándulas
submaxilares y sublinguales. Los componentes autónomos del par IX, inervan
al ganglio auditivo; mientras que las fibras parasimpáticos posganglionares

43
inervan esfínter del iris, músculo ciliar, glándulas salivales y lagrimales, así
como las glándulas mucosas de nariz, boca y faringe. Estas fibras también
incluyen a sus nervios vasodilatadores. El par X o vago, se origina en el bulbo
raquídeo y contiene fibras preganglionares, las cuales hacen sinapsis hasta
que llegan a ganglios pequeños que se encuentran sobre órganos del tórax y
abdomen. En la pared intestinal, las fibras vagales terminan alrededor de
células ganglionares en los plexos de Auerbach y Meissner. Las fibras
preganglionares son muy largas, mientras que las posganglionares son muy
cortas.
Las fibras sacras eferentes están constituidas por axones que se originan en
células que se encuentran en los segmentos, segundo, tercero y cuarto de la
médula sacra, y que viajan como fibras preganglionares para formar los nervios
pélvicos. Hacen sinapsis en los ganglios terminales que se encuentran cerca
de la vejiga, recto y órganos sexuales o en su interior. Las vías de salida
vagales y sacras brindan fibras motoras y secretoras a los órganos torácicos,
abdominales y pélvicos.
Las terminales de las neuronas colinérgicas contienen gran cantidad de
pequeñas vesículas con concentración alta de acetilcolina (ACh) cerca de la
porción sináptica. Las vesículas más alejadas de dicha porción, son más
grandes y su contenido de ACh es menor ya que en ellas se encuentran
también altas concentraciones de péptidos.
La ACh se sintetiza en el citoplasma a partir de la Acetil-CoA y colina por medio
de la catálisis de la enzima Acetilcolintransferasa (ChAT)1, 3. La acetil-CoA es
sintetizada en las mitocondrias a partir del piruvato y la colina es transportada
del exterior de la célula hacia el citosol a través de un transportador
dependiente de Na+. Una vez sintetizadas las moléculas de ACh, éstas son
almacenadas en las vesículas por medio de un antitransportador que elimina
H+; cada vesícula contiene entre 1000 y 50000 moléculas del neurotransmisor,
además que en cada terminación sináptica existen alrededor de 300000
vesículas.
La liberación del neurotransmisor es, como ya mencionamos, dependiente de
la entrada de Ca2+ extracelular a través de los canales de Ca2+ voltaje-
dependientes; ya que el catión desestabiliza a las vesículas y éstas se fusionan
con la membrana de la neurona por medio de proteínas de unión vesícula-
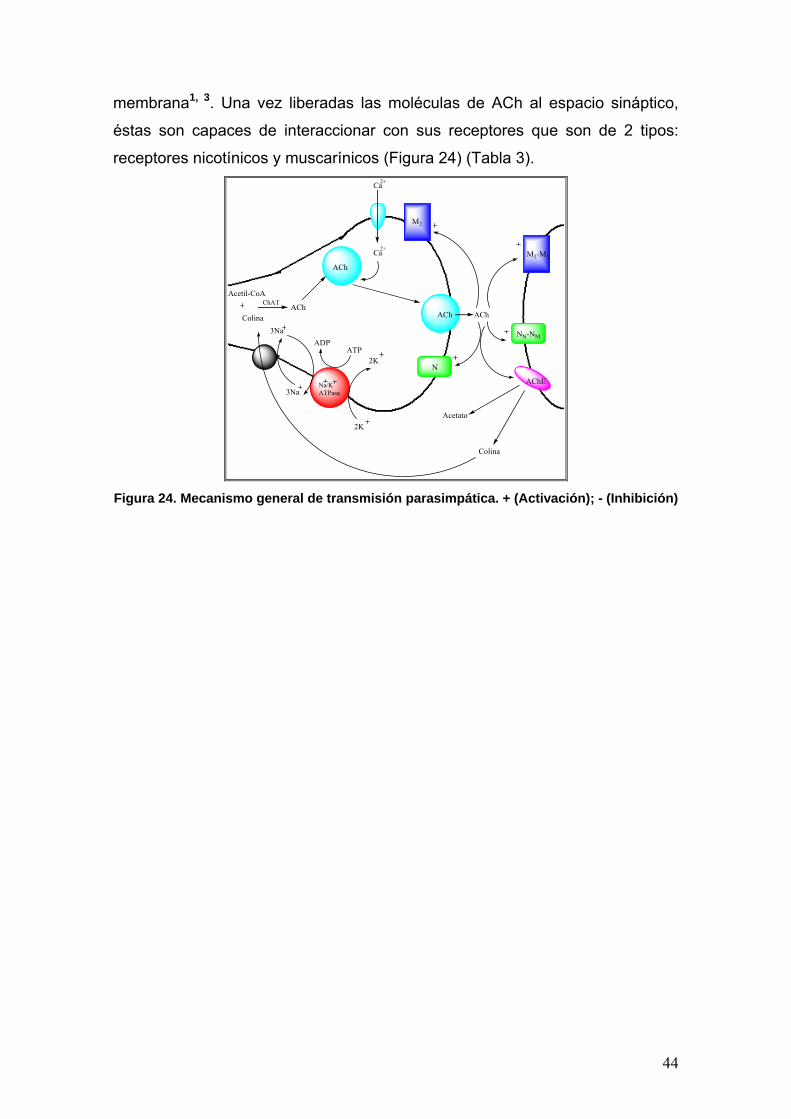
44
membrana1, 3. Una vez liberadas las moléculas de ACh al espacio sináptico,
éstas son capaces de interaccionar con sus receptores que son de 2 tipos:
receptores nicotínicos y muscarínicos (Figura 24) (Tabla 3). Ca
Ca
M2
3Na
3Na
2K
2KATP
ADP
N
M1-M5
NN-NM
Colina
Acetil-CoA
AChChAT
ACh
2+
2+
ACh ACh
Na/KATPasa
AChE
Colina
Acetato
Figura 24. Mecanismo general de transmisión parasimpática. + (Activación); - (Inhibición)
45
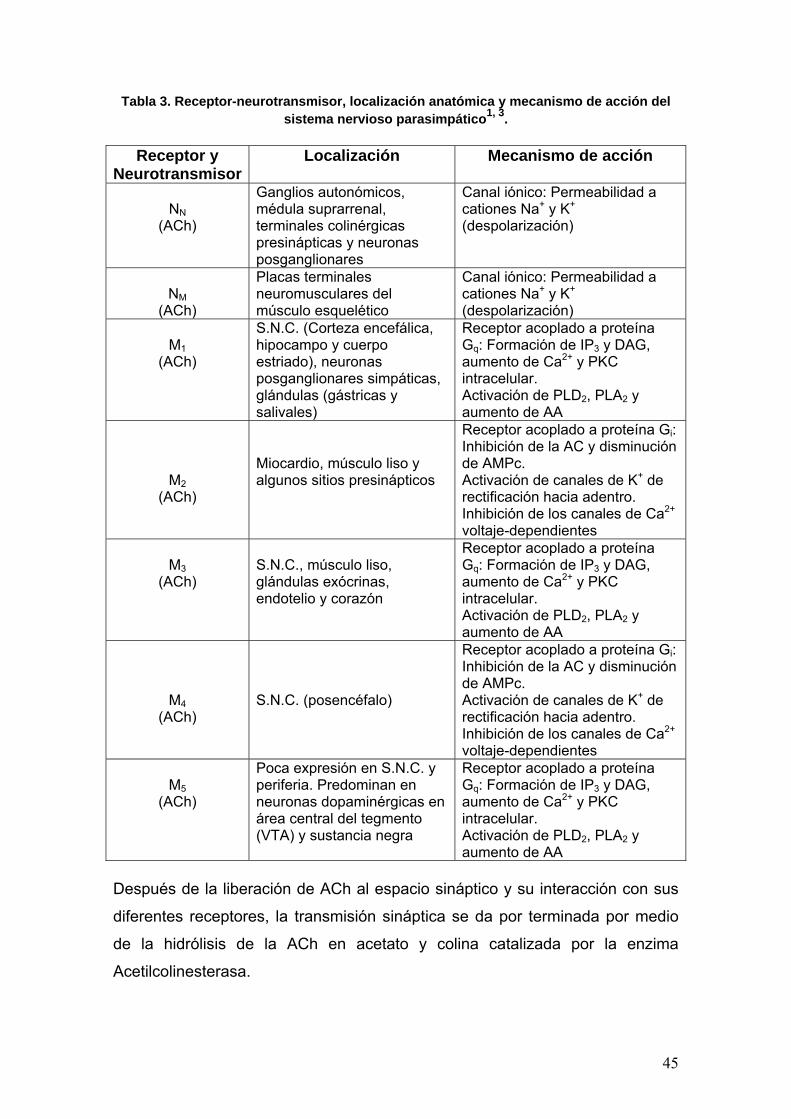
Tabla 3. Receptor-neurotransmisor, localización anatómica y mecanismo de acción del sistema nervioso parasimpático1, 3.
Receptor y
NeurotransmisorLocalización Mecanismo de acción
NN
(ACh)
Ganglios autonómicos, médula suprarrenal, terminales colinérgicas presinápticas y neuronas posganglionares
Canal iónico: Permeabilidad a cationes Na+ y K+
(despolarización)
NM
(ACh)
Placas terminales neuromusculares del músculo esquelético
Canal iónico: Permeabilidad a cationes Na+ y K+
(despolarización)
M1 (ACh)
S.N.C. (Corteza encefálica, hipocampo y cuerpo estriado), neuronas posganglionares simpáticas, glándulas (gástricas y salivales)
Receptor acoplado a proteína Gq: Formación de IP3 y DAG, aumento de Ca2+ y PKC intracelular. Activación de PLD2, PLA2 y aumento de AA
M2 (ACh)
Miocardio, músculo liso y algunos sitios presinápticos
Receptor acoplado a proteína Gi: Inhibición de la AC y disminución de AMPc. Activación de canales de K+ de rectificación hacia adentro. Inhibición de los canales de Ca2+ voltaje-dependientes
M3
(ACh)
S.N.C., músculo liso, glándulas exócrinas, endotelio y corazón
Receptor acoplado a proteína Gq: Formación de IP3 y DAG, aumento de Ca2+ y PKC intracelular. Activación de PLD2, PLA2 y aumento de AA
M4 (ACh)
S.N.C. (posencéfalo)
Receptor acoplado a proteína Gi: Inhibición de la AC y disminución de AMPc. Activación de canales de K+ de rectificación hacia adentro. Inhibición de los canales de Ca2+ voltaje-dependientes
M5
(ACh)
Poca expresión en S.N.C. y periferia. Predominan en neuronas dopaminérgicas en área central del tegmento (VTA) y sustancia negra
Receptor acoplado a proteína Gq: Formación de IP3 y DAG, aumento de Ca2+ y PKC intracelular. Activación de PLD2, PLA2 y aumento de AA
Después de la liberación de ACh al espacio sináptico y su interacción con sus
diferentes receptores, la transmisión sináptica se da por terminada por medio
de la hidrólisis de la ACh en acetato y colina catalizada por la enzima
Acetilcolinesterasa.

46
II.4.4 Acetilcolinesterasa12
Para que haya neurotransmisión en las sinapsis por parte de la acetilcolina, es
necesario que la molécula sea inactivada o eliminada para evitar su difusión e
interacción con receptores vecinos. Como antes mencionamos, la inactivación
de la ACh se lleva a cabo por hidrólisis catalizada por la enzima
Acetilcolinesterasa (Figura 25).
H3C O
O
NH3C
H3CCH3
AChE
H2OH3C O
O
+
OH
NH3C
H3CCH3
Acetilcolina
Acetato
Colina
Figura 25. Hidrólisis de ACh catalizada por la enzima AChE.
La distribución de la enzima es muy amplia, ya que se puede encontrar en las
neuronas colinérgicas (dendritas, pericarión y axones); así como en las uniones
neuromusculares; sin embargo, la acetilcolinesterasa no es la única
colinesterasa presente en el organismo, la butirilcolinesterasa (BuChE) es una
hidrolasa selectiva que cataliza el rompimiento del enlace éster de la
butirilcolina; y a diferencia de la AChE, la BuChE únicamente se encuentra a
bajas concentraciones en células gliales de S.N.C. y en suero ya que es
sintetizada en el hígado.12, 13 Por tal motivo, la BuChE no juega un papel
importante en las sinapsis neuronales, sino que su función principal es la
desintoxicación de ésteres de colina ingeridos en la dieta.13.
Por otra parte, la AChE, además de participar como factor para dar por
terminada la transmisión colinérgica, está implicada en otro tipo de funciones,
entre las que destacan la neuritogénesis, adherencia celular, sinaptogénesis,
ensamblado de fibras de amiloides, activación de receptores dopaminérgicos,
hematopoyesis y trombopoyesis12.
Actualmente la AChE ha sido estudiada ampliamente en mamíferos y en una
especie marina denominada Torpedo californica.
La estructura de la AChE en mamíferos y en T. californica, se dividen en 2
clases generales: oligómeros homoméricos simples de subunidades catalíticas

47
(monómeros, dímeros y tetrámeros) y vinculaciones heteroméricas de
subunidades catalíticas con subunidades estructurales.
Las formas homoméricas se encuentran en forma soluble en la células cuya
función radica en la exportación, o relacionadas con la forma exterior de la
célula por medio de aminoácidos hidrófobos intrínseca o de un
glucofosfolípido12, 13. Una forma heteróloga unida a la membrana externa de la
célula, se encuentra principalmente en las sinapsis neuronales; esta proteína
es un tetrámero de subunidades catalíticas enlazado por puente disulfuro a
una subunidad enlazada a lípidos cuya masa molar es de 20 kDa12, 13. Con
respecto a las formas heteroméricas, éstas están constituidas por tetrámeros
de subunidades catalíticas, enlazadas por puentes disulfuro a cada una de tres
bandas de subunidad estructural del tipo colágena; su masa molecular se
encuentra alrededor de 1000 kDa y se encuentra principalmente en la lámina
basal del músculo esquelético12, 13.
La clonación molecular indica que un solo gen codifica para AChE en los
vertebrados; sin embargo, existen varios productos génicos del proceso de
transducción del mRNA que difieren únicamente en sus terminaciones
carboxilo (exones 3A y 3H)13; mientras que en los exones 1 y 2, es conservada
y codifica solo para los primeros 535 aminoácidos13.
La estructura tridimensional de la AChE ha sido dilucidada en mamíferos y T.
californica por difracción de rayos X (DRX)14 en donde se detectó un centro
activo casi centrosimétrico en relación con cada subunidad, y que reside en la
base de una garganta de 20 Å aproximadamente. La garganta constituye el
sitio activo de la enzima y se ha demostrado que la estructura entre las dos
especies está altamente conservada en un 60%14 y sus aminoácidos entre una
y otra no difieren constitución sino en posición (Tabla 4).
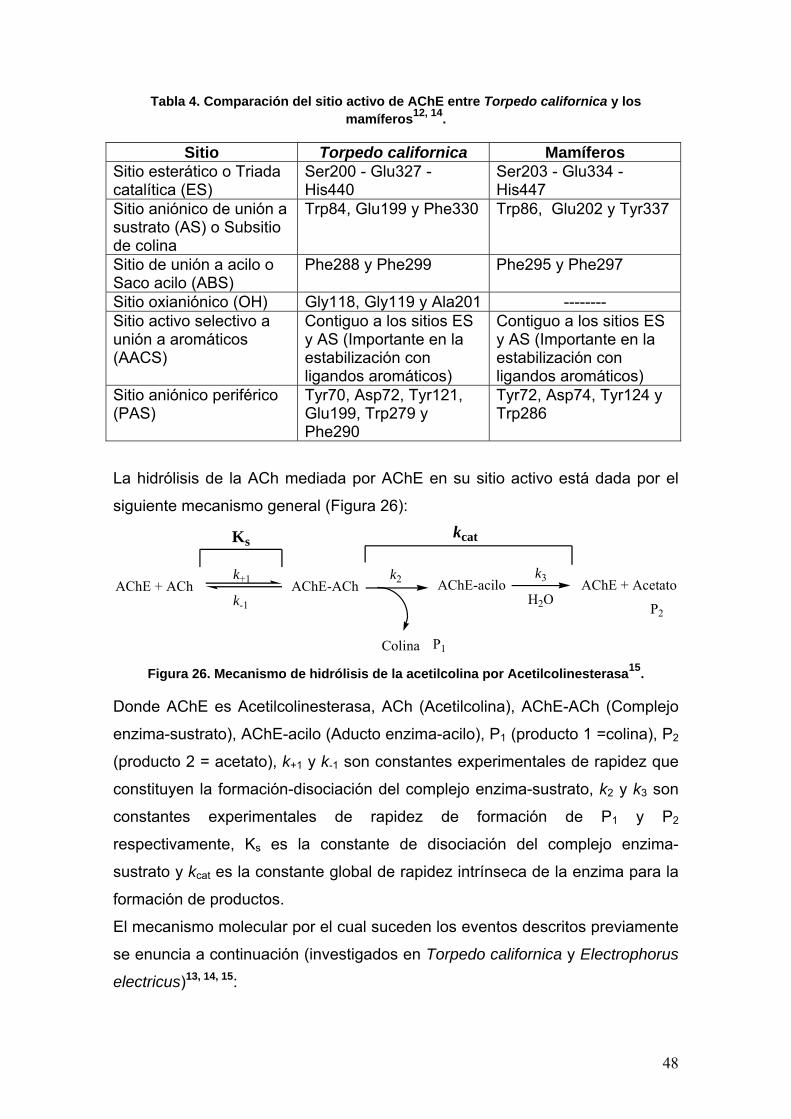
48
Tabla 4. Comparación del sitio activo de AChE entre Torpedo californica y los mamíferos12, 14.
Sitio Torpedo californica Mamíferos
Sitio esterático o Triada catalítica (ES)
Ser200 - Glu327 - His440
Ser203 - Glu334 - His447
Sitio aniónico de unión a sustrato (AS) o Subsitio de colina
Trp84, Glu199 y Phe330 Trp86, Glu202 y Tyr337
Sitio de unión a acilo o Saco acilo (ABS)
Phe288 y Phe299 Phe295 y Phe297
Sitio oxianiónico (OH) Gly118, Gly119 y Ala201 -------- Sitio activo selectivo a unión a aromáticos (AACS)
Contiguo a los sitios ES y AS (Importante en la estabilización con ligandos aromáticos)
Contiguo a los sitios ES y AS (Importante en la estabilización con ligandos aromáticos)
Sitio aniónico periférico (PAS)
Tyr70, Asp72, Tyr121, Glu199, Trp279 y Phe290
Tyr72, Asp74, Tyr124 y Trp286
La hidrólisis de la ACh mediada por AChE en su sitio activo está dada por el
siguiente mecanismo general (Figura 26):
P1
H2OP2
AChE + AChk+1
k-1
AChE-AChk2 AChE-acilo AChE + Acetato
k3
Colina
kcatKs
Figura 26. Mecanismo de hidrólisis de la acetilcolina por Acetilcolinesterasa15.
Donde AChE es Acetilcolinesterasa, ACh (Acetilcolina), AChE-ACh (Complejo
enzima-sustrato), AChE-acilo (Aducto enzima-acilo), P1 (producto 1 =colina), P2
(producto 2 = acetato), k+1 y k-1 son constantes experimentales de rapidez que
constituyen la formación-disociación del complejo enzima-sustrato, k2 y k3 son
constantes experimentales de rapidez de formación de P1 y P2
respectivamente, Ks es la constante de disociación del complejo enzima-
sustrato y kcat es la constante global de rapidez intrínseca de la enzima para la
formación de productos.
El mecanismo molecular por el cual suceden los eventos descritos previamente
se enuncia a continuación (investigados en Torpedo californica y Electrophorus
electricus)13, 14, 15:

49
La ACh entra a la garganta de AChE; cuyo arribo es con orientación acilo-
colina; una vez dentro de la garganta, el sitio ABS interacciona con el grupo
acilo, y el sitio AS con el amonio cuaternario de la ACh con el único fin de
estabilizar a la molécula. Otra interacción importante es la estabilización del
intermediario tetraédrico de la ACh por el sitio OH (Figura 27).
CH2 H2C
CH2
CO
OCH2
HN
CH3
O
O
NCH3
H3C
H3C
CH2
H2C
COO
H2C OH
H2C
N
HN
H3C
H H
CH2
H2CABS
PAS
AS
OH
Ala-201
Phe-288
Phe-299
Phe-290
Gly-118 Gly-119
Glu-327
Ser-200
His-440
Phe-
330
Glu
-199
Trp-84
Tyr-279
Asp-72
Trp-279
Tyr-121
ES Figura 27. Estabilización de la acetilcolina en el sitio activo de la Acetilcolinesterasa.
La estabilización de la ACh es muy importante, porque de ella depende el éxito
de la hidrólisis por la triada catalítica, ya que los sitios ABS y OH debilitan el
enlace éster, lo que favorece el ataque nucleofílico de la serina que a su vez
aumenta su poder nucleofílico gracias a los puentes de hidrógeno formados
entre el glutamato-histidina-serina (Figura 28)12, 13, 14, 15.

50
GluO
O
NN
His
HSer
OH
H3CO
O
N
CH3
CH3CH3
GluO
O
NN
His
HSer
O
CH3O
HON
CH3
CH3
CH3
P1 (Colina)
k2
GluO
O
NN
His
HSer
OH
H
O H
HO CH3
O
P2 (Acetato)
k3
Figura 28. Mecanismo molecular de la hidrólisis de ACh por la triada catalítica (ES).
La AChE es una de las enzimas más eficaces que se conocen ya que tiene la
capacidad de hidrolizar 600 000 moléculas de ACh en un minuto. Su Km está
alrededor de 50 a 100 μM12, 16 y su kcat a 1.4 x 104 s-1 16.
II.5 Enfermedad de Alzheimer II.5.1 Generalidades La Enfermedad de Alzheimer (EA) es la causa más común de demencia en
adultos mayores de 65 años y uno de los problemas más graves de salud
pública en países donde la población de adultos mayores va en incremento
(Europa Occidental, EE.UU. y países en vías de desarrollo). La incidencia
mundial de la Enfermedad está alrededor de 20 millones de personas
enfermas, mientras que en México la cifra rebasa los 350 000 casos17, 18.
II.5.2 Fisiología de la memoria y aprendizaje Actualmente no se conocen muy bien los mecanismos neurales de la memoria
y el aprendizaje; sin embargo, los esfuerzos de la comunidad científica
acumulan información acerca de estos aspectos.
La corteza cerebral y el hipocampo tienen diversas funciones entre las que
destacan las siguientes (Tabla 5, Figura 29):
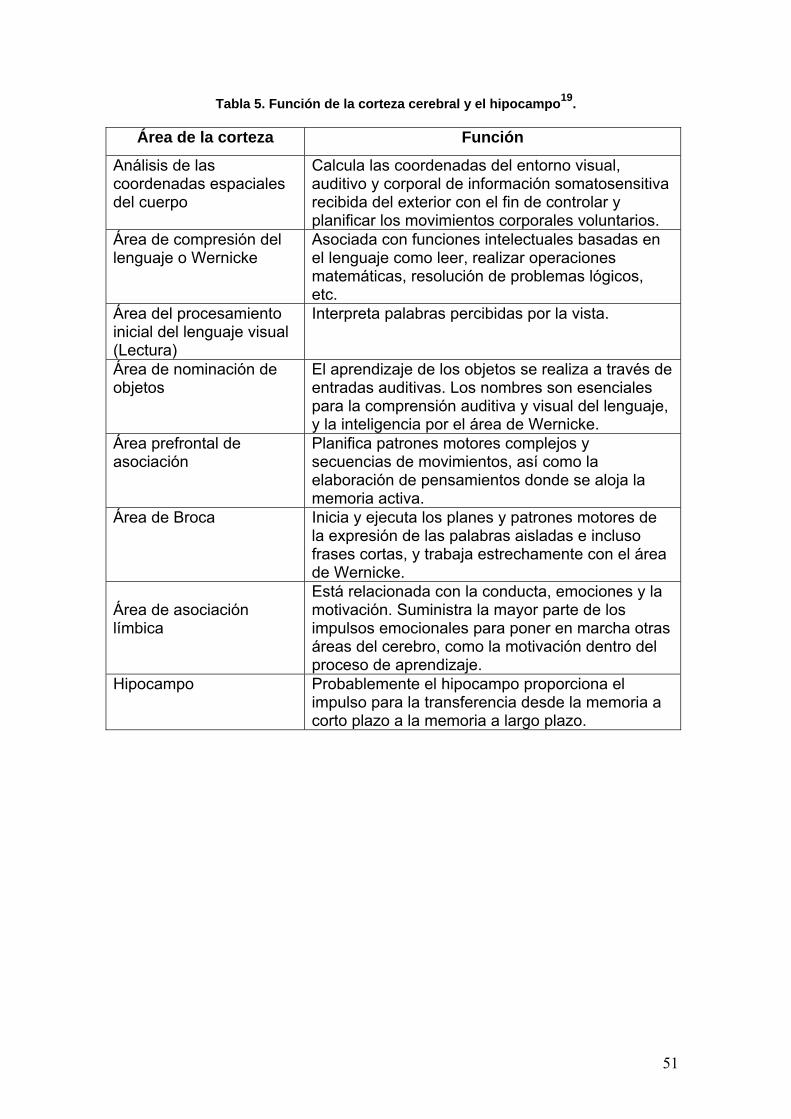
51
Tabla 5. Función de la corteza cerebral y el hipocampo19.
Área de la corteza Función
Análisis de las coordenadas espaciales del cuerpo
Calcula las coordenadas del entorno visual, auditivo y corporal de información somatosensitiva recibida del exterior con el fin de controlar y planificar los movimientos corporales voluntarios.
Área de compresión del lenguaje o Wernicke
Asociada con funciones intelectuales basadas en el lenguaje como leer, realizar operaciones matemáticas, resolución de problemas lógicos, etc.
Área del procesamiento inicial del lenguaje visual (Lectura)
Interpreta palabras percibidas por la vista.
Área de nominación de objetos
El aprendizaje de los objetos se realiza a través de entradas auditivas. Los nombres son esenciales para la comprensión auditiva y visual del lenguaje, y la inteligencia por el área de Wernicke.
Área prefrontal de asociación
Planifica patrones motores complejos y secuencias de movimientos, así como la elaboración de pensamientos donde se aloja la memoria activa.
Área de Broca Inicia y ejecuta los planes y patrones motores de la expresión de las palabras aisladas e incluso frases cortas, y trabaja estrechamente con el área de Wernicke.
Área de asociación límbica
Está relacionada con la conducta, emociones y la motivación. Suministra la mayor parte de los impulsos emocionales para poner en marcha otras áreas del cerebro, como la motivación dentro del proceso de aprendizaje.
Hipocampo Probablemente el hipocampo proporciona el impulso para la transferencia desde la memoria a corto plazo a la memoria a largo plazo.

52
Área de coordenadasespaciales corporalesy entorno
Área Motora
Planificación de movimientos complejosy elaboración depensamientos Área de
Broca
Área deasociaciónlímbica
Áreaauditiva
Área de Visión
ÁreaSomato-sensitiva
Área deWernicke
Figura 29. Mapa de las áreas funcionales de la corteza cerebral.19
La teoría holística de los pensamientos es el resultado de un patrón de
estimulación simultánea de muchas partes del sistema nervioso con una
secuencia definida, en la que intervienen la corteza cerebral, el tálamo, el
sistema límbico y la parte superior de la formación reticular del tronco
encefálico; las cuales determinan la naturaleza general del pensamiento,
atribuyéndole cualidades como placer, desagrado, dolor, comodidad,
modalidades toscas de sensación, etc. La estimulación de áreas concretas de
la corteza cerebral determinan las características del pensamiento, como la
localización precisa de las sensaciones en la superficie del cuerpo y de los
objetos situados en el campo visual, la sensación de textura, etc.
Para entender el proceso fisiológico de la memoria, es importante definir ciertos
conceptos, ya que facilitarán su estudio:
Los recuerdos se producen por variaciones de la sensibilidad de transmisión
sináptica de una neurona a la siguiente como resultado de la actividad neural
previa. Estas variaciones generan vías nuevas o facilitadas de transmisión de
las señales por los circuitos neuronales del cerebro; por definición, a este tipo
de de vías se les denominan Huellas de Memoria. Son importantes debido a
que, una vez establecidas, la mente puede activarlas para reproducir los
recuerdos.
La memoria puede ser de tipo positiva cuando el cerebro tiene la capacidad
automática y diferente de facilitar y almacenar las huellas de memoria de la
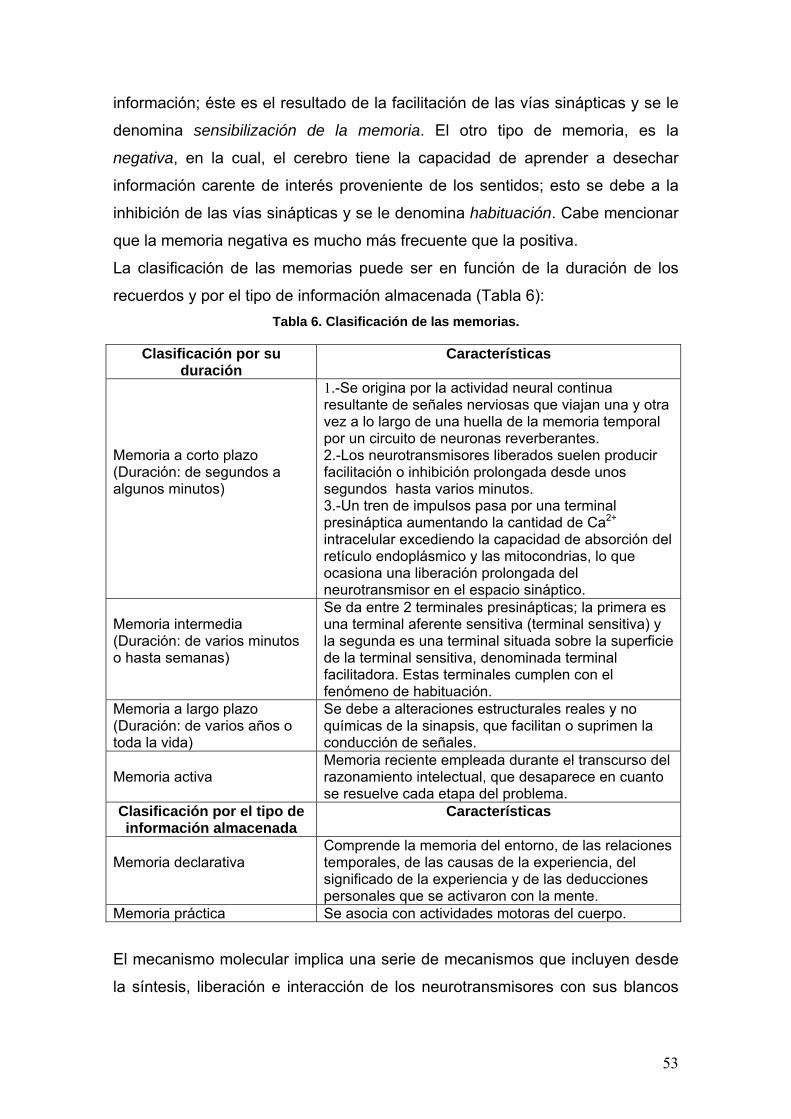
53
información; éste es el resultado de la facilitación de las vías sinápticas y se le
denomina sensibilización de la memoria. El otro tipo de memoria, es la
negativa, en la cual, el cerebro tiene la capacidad de aprender a desechar
información carente de interés proveniente de los sentidos; esto se debe a la
inhibición de las vías sinápticas y se le denomina habituación. Cabe mencionar
que la memoria negativa es mucho más frecuente que la positiva.
La clasificación de las memorias puede ser en función de la duración de los
recuerdos y por el tipo de información almacenada (Tabla 6): Tabla 6. Clasificación de las memorias.
Clasificación por su
duración Características
Memoria a corto plazo (Duración: de segundos a algunos minutos)
1.-Se origina por la actividad neural continua resultante de señales nerviosas que viajan una y otra vez a lo largo de una huella de la memoria temporal por un circuito de neuronas reverberantes. 2.-Los neurotransmisores liberados suelen producir facilitación o inhibición prolongada desde unos segundos hasta varios minutos. 3.-Un tren de impulsos pasa por una terminal presináptica aumentando la cantidad de Ca2+ intracelular excediendo la capacidad de absorción del retículo endoplásmico y las mitocondrias, lo que ocasiona una liberación prolongada del neurotransmisor en el espacio sináptico.
Memoria intermedia (Duración: de varios minutos o hasta semanas)
Se da entre 2 terminales presinápticas; la primera es una terminal aferente sensitiva (terminal sensitiva) y la segunda es una terminal situada sobre la superficie de la terminal sensitiva, denominada terminal facilitadora. Estas terminales cumplen con el fenómeno de habituación.
Memoria a largo plazo (Duración: de varios años o toda la vida)
Se debe a alteraciones estructurales reales y no químicas de la sinapsis, que facilitan o suprimen la conducción de señales.
Memoria activa
Memoria reciente empleada durante el transcurso del razonamiento intelectual, que desaparece en cuanto se resuelve cada etapa del problema.
Clasificación por el tipo de información almacenada
Características
Memoria declarativa
Comprende la memoria del entorno, de las relaciones temporales, de las causas de la experiencia, del significado de la experiencia y de las deducciones personales que se activaron con la mente.
Memoria práctica Se asocia con actividades motoras del cuerpo.
El mecanismo molecular implica una serie de mecanismos que incluyen desde
la síntesis, liberación e interacción de los neurotransmisores con sus blancos

54
moleculares, así como una serie de fosforilaciones de proteínas que en su
conjunto conforman el aprendizaje y la memoria.
El mecanismo general de la memoria a mediano plazo implica los siguientes
eventos:
La estimulación de la terminal facilitadora al mismo tiempo que la de la terminal
sensitiva libera serotonina (5-HT) en la sinapsis facilitadora de la superficie de
la terminal presináptica; este neurotransmisor interactúa con sus respectivos
receptores acoplados a proteína Gs, la cual activa a la enzima adenilato ciclasa
que incrementa la concentración de AMPc que a su vez activa a la protein-
cinasa A (PKA) que fosforila a canales de K+, lo que ocasiona bloqueo de la
conductancia del catión, por tanto, el potencial de acción se prolonga y esto
permite que el Ca2+ penetre a la neurona y así hay más liberación del
neurotransmisor.
En cuanto a la memoria a largo plazo, existen alteraciones estructurales en las
sinapsis; por ejemplo, el aumento del número de lugares de liberación de
vesículas para la secreción de la sustancia transmisora, aumento del número
de vesículas del transmisor, aumento del número de terminales presinápticas y
modificaciones de las estructuras de las espinas dendríticas. La figura 30,
muestra el mecanismo molecular general de la memoria.
Es importante mencionar otras funciones del hipocampo, ya que de ello
depende un entendimiento más claro de la enfermedad de Alzheimer, y esto
es; el daño al hipocampo ocasiona amnesia anterógrada que consiste en la
disminución de la memoria declarativa con incapacidad de almacenar la
información a mediano y a largo plazo. El daño al hipocampo puede producir
amnesia retrógrada que consta de una incapacidad para recuperar recuerdos
del pasado, es decir, de los compartimientos de la memoria a largo plazo. Si se
produce una amnesia retrógrada, quizá el grado de amnesia para los hechos
recientes sea mucho mayor que para los sucesos del pasado lejano; ya que
para este tipo de recuerdos ya se han establecido huellas de memoria y
cambio de estructura de las sinapsis19.
Cabe mencionar que la CAM/KII es una proteína que participa ampliamente en
el mecanismo de memoria, por ejemplo; regula la actividad de canales iónicos
de K+, NMDA y AMPA, así como receptores metabotrópicos de glutamato. Esta
proteína también está involucrada en la regulación de la síntesis y liberación de
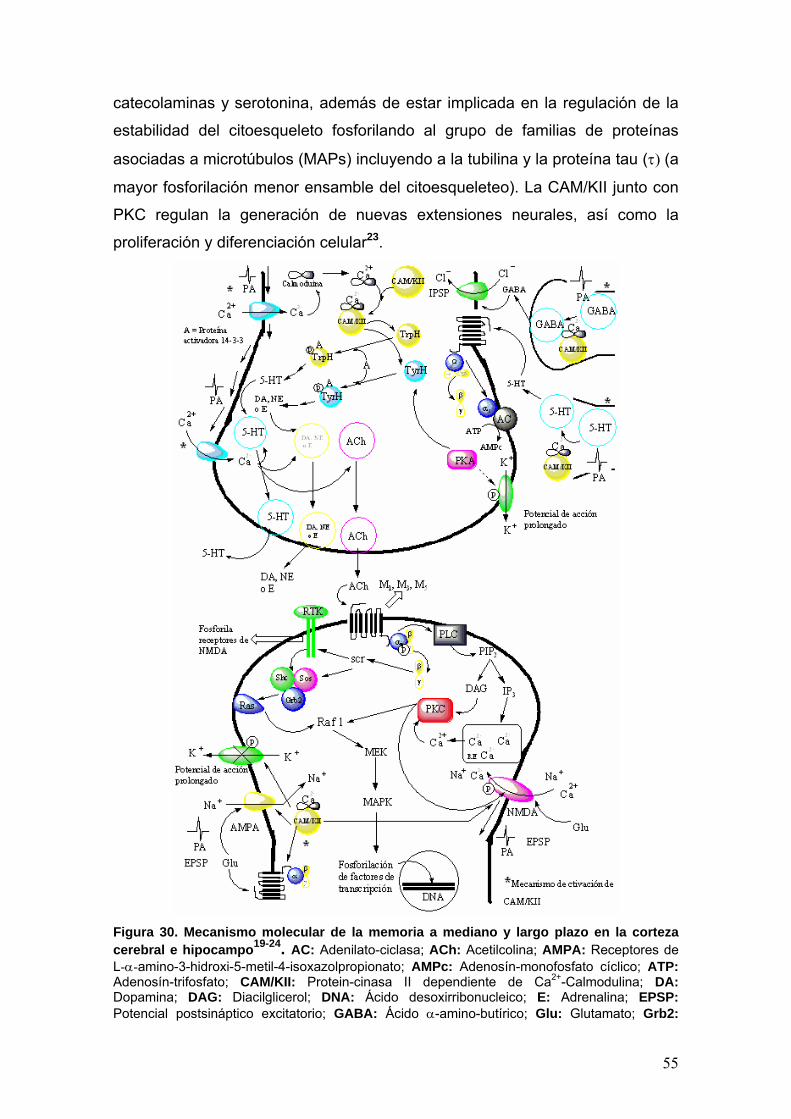
55
catecolaminas y serotonina, además de estar implicada en la regulación de la
estabilidad del citoesqueleto fosforilando al grupo de familias de proteínas
asociadas a microtúbulos (MAPs) incluyendo a la tubilina y la proteína tau (τ) (a
mayor fosforilación menor ensamble del citoesqueleteo). La CAM/KII junto con
PKC regulan la generación de nuevas extensiones neurales, así como la
proliferación y diferenciación celular23.
Figura 30. Mecanismo molecular de la memoria a mediano y largo plazo en la corteza cerebral e hipocampo19-24. AC: Adenilato-ciclasa; ACh: Acetilcolina; AMPA: Receptores de L-α-amino-3-hidroxi-5-metil-4-isoxazolpropionato; AMPc: Adenosín-monofosfato cíclico; ATP: Adenosín-trifosfato; CAM/KII: Protein-cinasa II dependiente de Ca2+-Calmodulina; DA: Dopamina; DAG: Diacilglicerol; DNA: Ácido desoxirribonucleico; E: Adrenalina; EPSP: Potencial postsináptico excitatorio; GABA: Ácido α-amino-butírico; Glu: Glutamato; Grb2:

56
Factor de crecimiento unido a receptor 2; 5-HT: Serotonina; IP3: Trifosfato de inositol; IPSP: Potencial presináptico inhibitorio; M1, M3, M5: Receptores muscarínicos; MAPK: Protein-cinasa activada por mitógeno; MEK: Cinasa de reconocimiento de tirosin/treonin cinasas; NE: Noradrenalina; NMDA: Receptores de N-metil-D-aspartato; PA: Potencial de acción; PIP2: Fosfatidil-inositol; PKA: Protein-cinasa A; PKC: Protein-cinasa C; PLC: Fosfolipasa C; Raf 1: Cinasa Raf 1; Ras: Familia de proto-oncogenes; RE: Retículo endoplásmico; RTK: Receptor de tirosin-cinasa; Shc: Protein-cinasa activada por estrés; Sos: Son of sevenless; TrpH: Triptofano hidroxilasa; TyrH: Tirosina hidroxilasa. II.5.3 Manifestaciones Clínicas En 1906 Alois Alzheimer (Figura 31) describió por primera vez a la enfermedad
en una paciente de 51 años de edad, y la describió como un trastorno de las
habilidades cognitivas con pérdida de la memoria de forma gradual y de
progreso irreversible. Actualmente la EA se sigue describiendo de esta forma, y
su primera manifestación se da por alteración de la memoria de
acontecimientos recientes, mientras que los recuerdos más antiguos se
preservan mejor. Conforme la enfermedad avanza el deterioro va siendo más
severo, al grado de que el paciente pierde habilidad para efectuar cálculos,
ejercicios, destrezas visuespaciales, manipulación de objetos comunes12, así
como el olvido de los nombres de sus familiares. El estado de alerta y el
sistema motriz del paciente no se altera hasta que la enfermedad está muy
avanzada. La muerte sobreviene por complicación de la inmovilidad, como
neumonía o embolia pulmonar. El paciente con EA suele sobrevivir en un lapso
de 6 a 12 años.
El diagnóstico de la enfermedad se basa en pruebas adecuadas de laboratorio
para excluir otros padecimientos que puedan dar la impresión de EA;
desgraciadamente aún no se encuentra una prueba clínica confiable para
confirmar la presencia de la enfermedad.
Figura 31. Alois Alzheimer.

57
II.5.4 Causas de la Enfermedad de Alzheimer En general las enfermedades neurodegenerativas (Enfermedades de
Huntington, Parkinson, Alzheimer y la Esclerosis lateral amiotrófica)12 tienen un
origen común (Figura 32, Tabla 7).
ToxinasAmbientales
MetabolismoNeuronal Envejecimiento
Formación de radicaleslibres, estrés oxidativoy excitotoxicosis
Vulnerabilidad selectivade las poblacionesneuronales
Daño al DNA Peroxidaciónde lípidos
Daño a proteínas
MUERTE CELULAR
Figura 32. Factores que causan enfermedades neurodegenerativas12.
Tabla 7. Descripción de los factores de causa de la Enfermedad de Alzheimer12.
Factores Características
Genéticos Mutaciones de los genes que codifican para la proteína precursora amiloide (APP) y las proteínas denominadas presenilinas* conducen a formas hereditarias de la EA. Individuos homocigotos de la isoforma** de la apolipoproteína E (apoE) para el alelo E4 tienen riesgo más alto que los homocigotos E2***.
Ambientales Se ha sugerido que lesiones cerebrales de origen traumático pueden desencadenar la EA.
Excitotoxicosis Lesión neural por exceso de glutamato mediado por receptores de NMDA. La entrada excesiva de Ca2+ puede activar proceso celulares destructivos.
Metabolismo y envejecimiento
La capacidad de las neuronas para el metabolismo oxidativo disminuye de manera progresiva al avanzar la edad debido a la acumulación de mutaciones en el genoma mitocondrial.
Estrés oxidativo
Producción de especies reactivas de oxígeno (ROS) como H2O2 y oxirradicales que pueden llegar a dañar al DNA; así como provocar peroxidación de lípidos en la membrana y por ende muerte neuronal.
Inflamación17 La inflamación juega un papel importante en el envejecimiento normal y en la EA. Las placas seniles tienen un componente inflamatorio que contribuye a la lesión neuronal.
Dieta17 Las concentraciones de homocisteína+ en sangre y concentraciones bajas de ácido fólico favorecen el desarrollo de la enfermedad.
*Probablemente participan en el procesamiento de APP. **Existen 4 isoformas de apoE, cuya función es transportar colesterol y lípidos en la sangre. ***Se desconoce el
mecanismo, pero se ha sugerido que E4 tiene una función secundaria de la proteína τ amiloide en el metabolismo de APP. + Abundante en carnes rojas.

58
II.5.5 Fisiopatología La EA se caracteriza por atrofia de la corteza cerebral y pérdida de las
neuronas corticales y subcorticales12, 17; es decir, las neuronas que actúan
sobre la memoria son las primeras afectadas, seguido de las neuronas
corticales del lenguaje y el razonamiento. Por otra parte, histológicamente se
ha observado en cortes de corteza cerebral de pacientes afectados, estructuras
anormales que presentan placas seniles y marañas neurofibrilares formadas
por proteínas de doble filamento y por la proteína � hiperfosforilada, debido a
una inadecuada regulación de la proteína CAM/KII23, lo que provoca colapso
del citoesqueleto y por ende dificultad en la comunicación celular y muerte
neuronal. Además de las marañas neurofibrilares y placas seniles, también se
encuentran formaciones esféricas del péptido β-amiloide (Figura 33). Este
péptido es altamente tóxico debido a que se adhiere a las membranas
neuronales, lo que constituye el inicio de formación de placas seniles. Su
formación radica en la división de la proteína precursora amiloide (APP).
La toxicidad de la β-amiloide se basa en la generación de radicales libres,
inflamación y decremento de irrigación sanguínea a las neuronas; además, se
ha observado que en dichas placas, la Acetilcolinesterasa (AChE) está
presente en altas concentraciones, lo que contribuye a la toxicidad por alterar la
comunicación colinérgica.
En la muerte neuronal por la formación de placas seniles y marañas
neurofibrilares disminuye dramáticamente las cantidades normales de
neurotransmisores, especialmente de ACh, ocasionando un déficit de la
molécula, lo que evita la interacción con sus receptores colinérgicos
Figura 33. A) Placa amiloide o senil en el hipocampo. Las estructuras triangulares son neuronas con marañas neurofibrilares. B) Marañas neurofibrilares17.

59
(principalmente M1); por lo tanto, rompe con la vía normal de señalización de la
memoria (Figura 30) en la corteza cerebral y el hipocampo. El exceso de AChE
contribuye a la muerte neuronal y a la baja interacción de ACh; estos
fenómenos constituyen la denominada “Hipótesis Colinérgica de la EA”.
Es notable señalar, que las neuronas más afectadas se encuentran en el
hipocampo y en la corteza cerebral, principalmente en el área de Broca, área
de Wernicke, área de coordenadas espaciales y del entorno, así como el área
de asociación límbica; lo que explica la pérdida de la memoria, la amnesia
anterógrada y retrógrada (cuando la enfermedad está muy avanzada) y la
habilidad para efectuar ejercicios sencillos y complejos; sin embargo, las áreas
auditiva y motoras no son prácticamente afectadas sino, hasta el final de la
enfermedad.
II.5.6 Tratamiento de la Enfermedad de Alzheimer Hoy en día el tratamiento de la EA, es de dos tipos: de rehabilitación a base de
terapias y farmacológico. El primero constituye el trato directo entre el paciente-
médico-familiares, para ayudar al primero (paciente) a recuperar algunas
funciones perdidas, así como los cuidados que deben tener sus familiares con
él. El segundo aspecto, lo conforman el tratamiento farmacológico que es a
base de fármacos para paliar la enfermedad y retrasar los efectos nocivos de la
enfermedad.
El primer cuadro de fármacos seleccionados son los inhibidores de AChE; que
evitan la hidrólisis de la ACh en el espacio sináptico y la toxicidad del exceso
de la enzima. Los fármacos comúnmente utilizados son: la Tacrina, Donepezilo,
Rivastigmina y la Galantamina (aprobados por la FDA); la Fisostigmina está en
desuso (Figura 34).

60
N
NH2
Tacrina
N
O
OCH3
OCH3
H
Donepezilo
ON
CH3
CH3
CH3
CH3
CH3
O
Rivastigmina
N
O
OH
CH3
OH3C
Galantamina
OHN
H3C
ON
(R)
(S)
N
H3C
H
CH3
CH3
Fisostigmina o Eserina Figura 34. Estructura química de los fármacos inhibidores de AChE para el tratamiento
de a EA.
• Tacrina12: Potente inhibidor de acción central que tiene efecto positivo moderado sobre el
rendimiento en la memoria cuando se administra conjuntamente con lecitina.
Los efectos adversos son dolor abdominal, anorexia, náusea, vómito y diarrea.
A pesar de estar aprobado por la FDA, el fármaco no es muy utilizado.
• Donepezilo12: Es un fármaco inhibidor selectivo de la AChE en el S.N.C.; produce mejoría
moderada de puntuaciones cognitivas con una vida media prolongada. Los
efectos adversos relacionados con el fármaco son: diarrea, náusea, vómito e
insomnio.
• Rivastigmina12: Tiene efectos positivos en recuperación de funciones cognitivas, ya que es un
inhibidor pseudoirreversible de la AChE y actúa principalmente en corteza
cerebral e hipocampo. Los efectos adversos son: diarrea, náusea, vómito e
insomnio.
• Galantamina12: Es un inhibidor reversible competitivo de la AChE con poca actividad sobre la
BuChE; tiene acción sobre receptores nicotínicos presinápticos, lo que facilita
la liberación de ACh en la terminal. Farmacodinámicamente tiene efectos
positivos en la recuperación de las funciones cognitivas de los pacientes. Los
efectos adversos son similares a los de la Rivastigmina.
• Fisostigmina12: Es un inhibidor pseudorreversible de la AChE de acción rápida; sus efectos
consisten en mejora en el proceso de aprendizaje y memoria. Actualmente el

61
fármaco está en desuso debido a que ocasiona síntomas de exceso
colinérgico, y su vida media es muy corta.
Otra estrategia para tratar la EA es el uso de la memantina (Figura 35) que es
un antagonista de los receptores de NMDA de glutamato; como se mencionó
anteriormente; el exceso de Ca2+ intracelular proveniente de los canales de
NMDA provoca excitotoxicosis.
Se utiliza principalmente para retrasar el deterioro neuronal y daño cognitivo
causado por Ca2+; sus efectos adversos incluyen cefalea y mareos.
CH3
NH2
H3CMemantina
Figura 35. Estructura de la memantina. Antagonista de los receptores de NMDA de glutamato.
En la actualidad se están investigando otro tipo de fármacos para tratar la EA; entre ellos están los inhibidores selectivos de COX-2, ya que la enfermedad tiene un componente inflamatorio12. Cabe mencionar que el consumo de ácido fólico, antioxidantes y vitaminas B6 y B12 en la dieta ayudan a prevenir la enfermedad, ya que son neuroprotectores17.
II.6 Cinética Enzimática II.6.1 Generalidades Hoy en día se sabe que las enzimas son catalizadores biológicos con gran
poder de acelerar reacciones químicas específicas; sin embargo, desde la
antigüedad se conocía su existencia empírica en civilizaciones como Babilonia,
Roma, Grecia, China e India. Estas civilizaciones utilizaban microorganismos
para ablandar la carne y en la producción de vinagre, quesos, bebidas
alcohólicas y pan (precisamente enzima deriva de la voz griega énzymos
(ένζυμη)48, cuyo significado se refiere al proceso de esponjamiento del pan, es
decir, en la levadura o dentro de la levadura)25.
Las enzimas fueron reconocidas hasta el siglo XIX con estudios sobre la
digestión de la carne por secreciones del estómago y la conversión del almidón
en azúcar por la saliva. En 1850 Louis Pasteur denominó fermentos a las
levaduras que convertían el azúcar en alcohol etílico, y para 1897 Eduard
Buchner realizó el mismo experimento pero con extractos de levadura y llegó a

62
la misma conclusión, con la diferencia que los fermentos de Pasteur ya habían
sido denominados como enzimas.
Actualmente se sabe que la mayoría de las enzimas son de naturaleza proteica
gracias a intensos estudios realizados en el siglo XX. Cabe mencionar que
existen enzimas cuya base molecular es RNA25, 26.
Las enzimas de tipo proteico tienen masas moleculares que van desde 12kDa
hasta 1000kDa. Por otra parte, su actividad depende de su estructura
tridimensional, ya que si se desnaturaliza pierde su capacidad catalítica; sin
embargo, en algunas de ellas no sólo depende de su estructura tridimensional,
sino de otros componentes llamados cofactores. Los cofactores pueden ser
uno o varios iones inorgánicos como Fe2+, Zn2+, Mg2+ o Mn2+; cuando el
cofactor es de naturaleza orgánica u organometálica, se le denomina
coenzima. Adicionalmente, algunas enzimas necesitan unión covalente con una
coenzima y uno ó más iones metálicos para optimizar su actividad; a este tipo
de cofactores se les conoce con el nombre de grupo prostético. Cuando la
enzima tiene a su o sus cofactores correspondientes y es completamente
activo se le denomina holoenzima, y a su vez a la parte proteica se le llama
apoenzima o apoporoteína.
En general, los cofactores funcionan como transportadores de partículas como
grupos funcionales, electrones, etc.
Las enzimas se pueden clasificar de acuerdo a la reacción que catalizan, y se
tienen identificadas 6 reacciones diferentes que pueden catalizar estas
proteínas (Tabla 8). Tabla 8. Clasificación internacional de las enzimas en función de la reacción de
catálisis26.
No. Clase Tipo de reacción catalizada. 1 Oxidorreductasas Transferencia de electrones, iones hidruro o átomos
de Hidrógeno. 2 Transferasas Reacciones de transferencia de grupos. 3 Hidrolasas Reacciones de hidrólisis (transferencia de grupos
funcionales al agua). 4 Liasas Adición de grupos o dobles enlaces, o formación de
dobles enlaces por eliminación de grupos. 5 Isomerasas Transferencia de grupos dentro de la molécula para
obtener formas isoméricas. 6 Ligasas Formación de enlaces C-C, C-S, C-O y C-N mediante
reacciones de condensación acopladas a la rotura de ATP.

63
La nomenclatura de las enzimas se basa en asignar números a la clase, subclase, nombre, grupo aceptor, sustrato como grupo aceptor. E.C. (Enzyme Classification): Clase. Subclase. Grupo aceptor. Sustrato aceptor del grupo (E.C. No. No. No. No.). Para facilitar la nomenclatura únicamente se utiliza el número de código y nombre según el tipo de reacción. Es importante señalar que una enzima se encuentra en condiciones óptimas,
cuando las condiciones del medio se lo permiten; es decir, cada enzima tiene
una actividad particular a ciertas condiciones ambientales, por ejemplo, a un
cierto pH, temperatura, concentración de sustrato, cantidad de enzima, etc.
II.6.2 Catálisis enzimática. Modelo de Michaelis-Menten Durante una reacción química de transformación de reactivos a productos,
existe un estado en el cual se llevan a cabo el rompimiento y formación de
enlaces, denominado estado de transición o complejo activado. La formación
del complejo activado es imprescindible para cualquier reacción,
independientemente del estado energético de los reactivos y los productos; es
decir, si la reacción es espontánea o requiere energía. Por lo tanto, el complejo
activado está asociado a una alta energía llamada energía de activación
(Figura 36). Cabe mencionar que una reacción energéticamente favorecida
hacia los productos (espontánea) no necesariamente es una reacción rápida.
En general, las enzimas son catalizadores biológicos cuya función es abatir la
energía de activación y por ende, acelerar la reacción del paso de reactivos a
productos; o de sustrato a producto.
Coordenadade reacción
Energía libre (R) Reactivo (P) Producto
CA*
R PΔG*
P RΔG*
ΔGº 'R
P
Figura 36. Diagrama de energía para una reacción espontánea de reactivos a productos, y formación del complejo en el estado de transición. CA: Complejo activado; ΔG*: Energía libre para alcanzar el estado de transición; ΔGº’: Energía libre estándar a pH = 7.026. Para una catálisis enzimática se tiene el siguiente sistema (Figura 37) de la
conversión de sustrato (S) a producto (P) por medio de una enzima (E).

64
E + S ES EP E + P
Coordenadade reacción
Energía libre
CA*
ΔG*
ΔG*S
P
no-cat
catES EP
CA*
Figura 37. Diagrama de energía para el sistema S P sin catalizar y con catálisis26.
Como se ha mencionado antes, la función de un catalizador es acelerar una
reacción química abatiendo la energía de activación, y en efecto, la teoría del
estado de transición relaciona la constante experimental de rapidez con la
energía del complejo activado a través de la constante de equilibrio del
sistema26 (Figura 38):
Keq' = [P]/ [S]
ΔGº ' = -RT ln Keq'
r = k[S]
k = [kBT/ h] e- (ΔG* '/ RT)
Figura 38. Relación entre la rapidez de la reacción y la energía libre del estado de transición. Keq’: Constante de equilibrio del sistema; ΔGº ‘: Energía libre en condiciones normales del sistema; ΔG*: Energía del estado de transición; kB: Constante de Boltzmann; h: Constante de Planck; k: Constante experimental de rapidez; R: Constante de los gases ideales; T: Temperatura. Esta relación de ecuaciones nos proporciona una gran información, a menor
energía de activación del complejo activado, la rapidez de la reacción
incrementa, ya que la ecuación indica una relación exponencial inversa de la
energía de transición con la constante experimental de rapidez.
Por otra parte, en general las reacciones enzimáticas se llevan a cabo bajo el
siguiente esquema:
S + E ES E + Pk+1
k-1
k2
A principios del siglo XX, Henri, Michaelis y Menten sentaron las bases de la
cinética enzimática actual, donde establecen que los reactivos (E + S) y el
complejo (ES) llegaban al equilibrio rápidamente durante una reacción

65
enzimática asumiendo que k2 << k-1. Posteriormente, Briggs y Haldane
reconocieron que el modelo de Henri-Michaelis-Menten podría ser descrito de
una forma más general que no requería que k2 << k-1. La forma general que
establecieron la denominaron “Teoría del Estado Estacionario”, que establece
que en un período de tiempo durante una reacción enzimática, la rapidez de
formación del complejo ES es exactamente igual al decaimiento de la enzima
libre y sus productos25.
A partir de este postulado, se llegó a la ecuación de Michaelis-Menten y la
gráfica de hipérbola rectangular (Figura 39 y 40).
A mayor concentración de sustrato, la enzima total se asocia al sustrato, por
tanto, la concentración de [ES] es constante debido a la saturación de la
enzima, y en ese momento se alcanza la rapidez máxima.
La determinación de los parámetros cinéticos de una enzima se pueden
determinar a partir de varios métodos, entre los que destacan: El método de la
doble recíproca de Lineweaver-Burk, Eadie-Hofstee, Hanes-Wolff y el Método
de Eisenthal-Cornish-Bowden.
El método de Lineweaver-Burk es el más utilizado para determinar Km y rmáx de
una enzima, y consiste en obtener una ecuación de línea recta a partir de la
transformación inversa de la ecuación de Michaelis-Menten:
r0 = rmáx [S]/ (Km + [S]), obteniendo la inversa: (1/r0) = (Km/rmáx)(1/[S]) + 1/rmáx
Donde la pendiente es el cociente entre Km y rmáx y la ordenada al origen es la
inversa de la rapidez máxima (Figura 41).

66
AL INICIO DE LA REACCIÓN
(d[ES]/ dt) = 0
[E]T = [E] + [ES] Cantidad de enzima total en el sistema
La rapidez de formación del complejo [ES] es constante en este período ya que cuando se forma un complejo se compensa con su descomposición a productos y enzima libre. [P] ~ 0; [S] ~ [S]inicial
r = k2[ES] La formación del producto se ajusta a una reacción de orden 1, ya que depende de [ES] bajo la rapidez intrínseca de la enzima k2
(d[ES]/ dt) = k+1[E][S] La formación de [ES] está gobernado por la k+1, y depende tanto de [E] y [S] libres; por tanto, es una reacción de orden 2.
-(d[ES]/ dt) = (k -1 + k2)[ES]La descomposición del complejo [ES] está gobernado por la disociación (k -1) y por la formación del producto (k2). La reacción es de orden 1 ya que depende únicamente de [ES]
k+1[E][S] = (k -1 + k2)[ES]A partir de esta igualdad se deduce la ecuación de Michaelis-Menten considerando que la Km se refiere al cociente de la rapidez de descomposición entre la de formación.
Km = (k -1 + k2)/ k+1 [ES] = [E]T[S]/ Km
r = k2[E]T[S]/ (Km + [S]) r = rmáx [S] / (Km + [S])
La rapidez máxima (rmáx) se refiere al producto de la constante experimental de rapidez intrínseca (k2) por la concentración enzima total [E]T. En este momento la curva de Michaelis-Menten se mantiene constante porque toda la enzima está ocupada con sustrato. Cinética de orden cero.
Figura 39. Deducción de la ecuación de Henri-Michaelis-Menten bajo la Teoría del Estado Estacionario25.
0
r0
[S]
rmáx
1/2 rmáx
Km
r0 = rmáx [S]/ (Km + [S])
Figura 40. Dependencia de la rapidez inicial con respecto a la concentración de sustrato.
Hipérbola rectangular que se ajusta al modelo Henri-Michaelis-Menten.
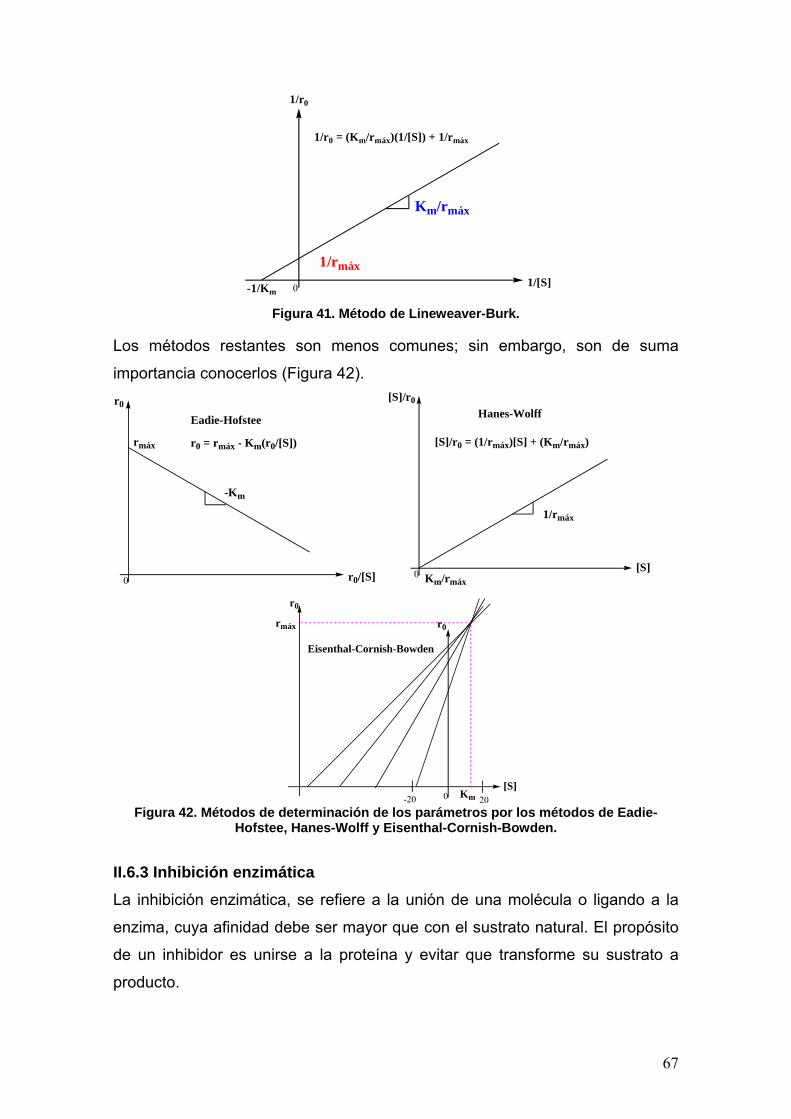
67
0
1/r0
1/[S]
Km/rmáx
1/rmáx
-1/Km
1/r0 = (Km/rmáx)(1/[S]) + 1/rmáx
Figura 41. Método de Lineweaver-Burk.
Los métodos restantes son menos comunes; sin embargo, son de suma
importancia conocerlos (Figura 42).
0 r0/[S]
r0
r0 = rmáx - Km(r0/[S])
-Km
rmáx
Eadie-Hofstee
0
[S]/r0
[S]
[S]/r0 = (1/rmáx)[S] + (Km/rmáx)
1/rmáx
Km/rmáx
Hanes-Wolff
0
r0
r0
20-20[S]
Km
rmáx
Eisenthal-Cornish-Bowden
Figura 42. Métodos de determinación de los parámetros por los métodos de Eadie-
Hofstee, Hanes-Wolff y Eisenthal-Cornish-Bowden.
II.6.3 Inhibición enzimática La inhibición enzimática, se refiere a la unión de una molécula o ligando a la
enzima, cuya afinidad debe ser mayor que con el sustrato natural. El propósito
de un inhibidor es unirse a la proteína y evitar que transforme su sustrato a
producto.

68
La unión del ligando con la enzima puede ser de diferente manera, de tal
suerte, que existen formas de diferenciar entre un tipo de inhibición y otro. La
figura 43 muestra la clasificación de los tipos de inhibición.
Figura 43. Clasificación de los tipos de inhibición25.
La reversibilidad se refiere a la capacidad de un sistema de pasar de un estado
inicial a uno o varios estados consecutivos, y al final de un ciclo, llega al estado
inicial sin perder materia y energía; por tanto, en cinética enzimática, esta regla
termodinámica no se viola ya que sucede lo antes mencionado, debido a que el
estado inicial de la enzima y el ligando tienen un estado basal de energía, y en
el momento de la unión adoptan un nuevo y varios estados energéticos
consecutivos durante la interacción, hasta llegar al estado inicial en el momento
de la disociación. Por lo tanto, la irreversibilidad se refiere del paso de un
estado energético inicial a uno final, sin regresar al estado inicial.
Con respecto a los fenómenos de competitividad, no competitividad y
acompetitividad, se aplican los mismos principios cinéticos y moleculares con la
diferencia de la reversibilidad del sistema.
En la figura siguiente (Figura 44) se presenta el diagrama general de inhibición
de una enzima, cuyo significado de las variables que se presentan es el
siguiente: Ks se refiere a la constante de disociación que existe entre la enzima
y el sustrato. Ki es la constante de disociación que establecen la enzima y el
inhibidor; kp es la constante experimental de rapidez con la que la enzima
convierte al sustrato en producto. El factor β es un coeficiente que modifica a Ks
y Ki en la formación del complejo ESI y refleja el efecto del inhibidor sobre las
TIPOS DE INHIBICIÓN
REVERSIBLE
IRREVERSIBLE
COMPETITIVA
NO COMPETITIVA
ACOMPETITIVA
COMPETITIVA
NO COMPETITIVA
ACOMPETITIVA

69
constantes. El factor β refleja el efecto del inhibidor sobre la kp que se refiere al
paso de producto a partir del complejo ESI25. Los inhibidores parciales tiene
valores de β que van de cero a uno (0 < β < 1); y los activadores enzimáticos
adoptan valores mayores de uno (β > 1)25.
E + SKs ES E + P
kp
+I
EI + S
Ki
ESI
+I
EI + P
αKi
βkpαKs Figura 44. Diagrama general de inhibición aplicable a inhibición competitiva, no
competitiva y acompetitiva.
En la tabla 9, se muestran las características cinéticas y moleculares de los
tipos de inhibición. Tabla 9. Características cinéticas y moleculares de los tipos de inhibición total.
Inhibición Ecuación Características α y β∗
Competitiva r0 =
rmáx [S]
[S] + Km 1 +[I]Ki
α = ∞; β = 0
No
competitiva r0 =
rmáx [S]
1 +[I]αKi
+ Km 1 +[I]Ki
[S]
α = # real +; β = 0
Acompetitiva r0 =
rmáx [S]
1 +[I]αKi
Km
1 +[I]αKi
+ [S]
α << 1; β = 0
*Las características cinéticas y moleculares se presentan en las figuras 45, 48 y 4925.
En una inhibición competitiva, el inhibidor compite junto con el sustrato por el
mismo sitio de la proteína (Sitio activo); por tal motivo, la gráfica de 1/r0 vs.
1/[S] presenta un aumento en las pendientes al incrementar la concentración
de inhibidor, porque este desplaza al sustrato en función de su afinidad, lo que
da por resultado un cambio en el valor de Km y la rapidez máxima no se ve
afectada (Figura 45).

70
1r0
1[S]0
[I] = 0
[I] > 0r0 =
rmáx [S]
[S] + Km 1 +[I]Ki
1r0
= 1rmáx
+ 1Kmrmáx
1 + [I]Ki[S]m0
m1 = Kmapp1
m2 = Kmapp2
E ESS
I
I
EI
S
Figura 45. Inhibición competitiva total reversible25.
Para obtener la Ki del sistema reversible, se puede utilizar la Ecuación de
Schild que relaciona el cociente de las pendientes con concentración de
inhibidor entre la pendiente sin inhibidor (mn/ m0) en función de la
concentración de inhibidor o el logaritmo del cociente (mn/ m0)-1 en función del
logaritmo de la concentración de inhibidor. A partir de la ecuación de Michaelis-
Menten y la ecuación de inhibición competitiva, se obtienen las 2 formas de la
ecuación de Schild igualando las dobles recíprocas de Lineweaver-Burk25
(Figura 46):
r0 =rmáx [S]
[S] + Km 1 + [I]Ki
1r0
= 1rmáx
+ 1Kmrmáx
1 + [I]Ki [S]
1r0
= 1rmáx
+ 1Kmrmáx [S]
r0 =rmáx [S]
[S] + Km
Doblerecíproca
Igualando:
1rmáx
+ 1[S] = 1
rmáx+ 1
1 + [I]Ki
[S]
α
mn
m0
m0
NOTA: mn = α m0
mn
Reordenandomnm0
α =
= α = 1 +[I]Ki
ECUACIÓNDE SCHILD
ó
log mnm0
-1 = nH log [I] - nH log Ki
Figura 46. Deducción de la ecuación de Schild.
Gráficamente, se obtiene una línea recta, cuya ordenada al origen es 1, y de
pendiente positiva que corresponde a Ki-1 (Figura 47). La otra forma de la
ecuación permite obtener el valor de nH (coeficiente de Hill) de la pendiente y
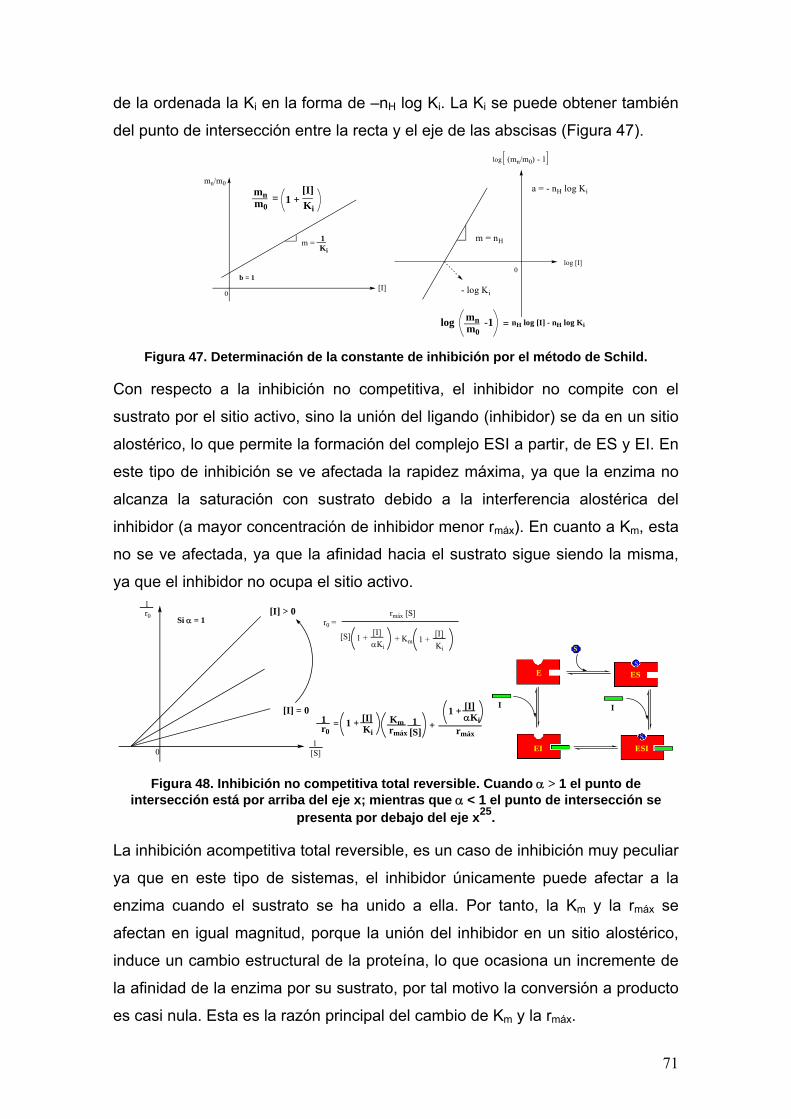
71
de la ordenada la Ki en la forma de –nH log Ki. La Ki se puede obtener también
del punto de intersección entre la recta y el eje de las abscisas (Figura 47).
0
mn/m0
[I]
m = 1Ki
b = 1
mnm0
1 +[I]Ki
=
log [I]
log (mn/m0) - 1
0
m = nH
a = - nH log Ki
- log Ki
log mnm0
-1 = nH log [I] - nH log Ki
Figura 47. Determinación de la constante de inhibición por el método de Schild.
Con respecto a la inhibición no competitiva, el inhibidor no compite con el
sustrato por el sitio activo, sino la unión del ligando (inhibidor) se da en un sitio
alostérico, lo que permite la formación del complejo ESI a partir, de ES y EI. En
este tipo de inhibición se ve afectada la rapidez máxima, ya que la enzima no
alcanza la saturación con sustrato debido a la interferencia alostérica del
inhibidor (a mayor concentración de inhibidor menor rmáx). En cuanto a Km, esta
no se ve afectada, ya que la afinidad hacia el sustrato sigue siendo la misma,
ya que el inhibidor no ocupa el sitio activo. 1r0
1[S]0
[I] = 0
[I] > 0Si α = 1 r0 =
rmáx [S]
1 +[I]αKi
+ Km 1 +[I]Ki
[S]
1r0
=rmáx
1Kmrmáx
1 + [I]Ki [S]
1 + [I]αKi+
I
SES
I
E
EIS
ESI
S
Figura 48. Inhibición no competitiva total reversible. Cuando α > 1 el punto de
intersección está por arriba del eje x; mientras que α < 1 el punto de intersección se presenta por debajo del eje x25.
La inhibición acompetitiva total reversible, es un caso de inhibición muy peculiar
ya que en este tipo de sistemas, el inhibidor únicamente puede afectar a la
enzima cuando el sustrato se ha unido a ella. Por tanto, la Km y la rmáx se
afectan en igual magnitud, porque la unión del inhibidor en un sitio alostérico,
induce un cambio estructural de la proteína, lo que ocasiona un incremente de
la afinidad de la enzima por su sustrato, por tal motivo la conversión a producto
es casi nula. Esta es la razón principal del cambio de Km y la rmáx.

72
1r0
1[S]0
[I] = 0
[I] > 0
r0 =
rmáx [S]
1 +[I]αKi
Km
1 +[I]αKi
+ [S]1r0
1r0
= rmáx1Km
rmáx [S]
1 + [I]αKi
+ 1
E
S
SES
I
S
ESI
Figura 49. Inhibición acompetitiva total reversible.
III. ANTECEDENTES A partir del descubrimiento empírico de la acetilcolinesterasa, se hicieron
estudios para caracterizar la función que tenía sobre el sistema nervioso
autónomo. Una de las primeras sustancias descubiertas que actúan sobre la
AChE fue la fisostigmina (Eserina), un alcaloide aislado de la semilla
Physostigma venenosum o nuez de ordalía que la utilizaban las tribus de África
Occidental para fines místicos (Rituales de brujería). Después de su
aislamiento en Inglaterra en 1840, fue introducida en la terapéutica en 1877 por
Laqueur para tratar el glaucoma3. En 1914 Sir Henry Dale sugirió que la enzima
que degradaba los ésteres de colina tenía un papel importante en la
neurotransmisión en el sistema nervioso autónomo y motor; a la cual se le
denominó Acetilcolinesterasa que era blanco biológico de la fisostigmina13.
En ese entonces, Alois Alzheimer describió por primera vez a la enfermedad
que lleva su nombre (1906), en una paciente de 51 años de nombre Auguste D.
en la XXXVII Conferencia de Psiquiatría del Sureste Alemán en Tübingen. Un
año después, Auguste D. perdió la vida a causa de la enfermedad y Alois
Alzheimer estudió las lesiones que presentaba su paciente y publicó sus
hallazgos en la Conferencia ya mencionada, con el título: “Sobre una
enfermedad específica de la corteza cerebral”27.
Mientras tanto, a partir de la investigación básica realizada por Stedman y cols.
sobre las bases químicas de la eserina, otros investigadores sintetizaron una
serie de ésteres aromáticos, de donde obtuvieron la neostigmina, la cual fue
incluida en la terapéutica por su gran actividad estimulante del tubo digestivo
en 1931.

73
Después de la introducción de la neostigmina, en el período comprendido entre
1932 y 1951, algunos científicos sintetizaron nuevos compuestos con fines para
combatir las plagas entre ellos se encuentran los compuestos organofosforados
(malatión y paratión) y los carbamatos aromáticos, que son inhibidores
irreversibles y reversibles de la AChE respectivamente. En tanto que en el
mismo período, también se desarrollaron compuestos muy tóxicos, cuyo
objetivo era usarlos en la “Guerra Química” durante la Segunda Guerra
Mundial debido a sus efectos de ahogamiento y visión borrosa3.
Por otra parte, Sjoerd L. Bonting y Robert M. Featherstone propusieron en 1955
una nueva técnica para medir la actividad de la AChE por un método
colorimétrico basado en la formación de un compuesto de coordinación
derivado de la formación previa de ácido hidroxámico (ACh remanente +
hidroxilamina) y cloruro férrico (FeCl3)29. No obstante, en 1960, George L.
Ellman y cols. propusieron otra técnica de medición basada también en un
método espectrofotométrico, en el cual se medía la actividad de la AChE
haciendo reaccionar la colina o tiocolina con ditiobisnitrobenzoato para formar
un compuesto color amarillo30 (Figura 50).
H3C O
O
NH3C
H3C CH3
H2O
OH
NH3C
H3C CH3
SS
O2N
O2N
OO
OO
OH
NH3C
H3C CH3
SO2N
O
O
O
NH3C
H3CCH3
H3C O
O
SH
O2NO
O
AChE +
++
AChColina
Colina
5-ditiobis-2-nitrobenzoato
5-tiol-2-nitro-benzoatoCOLOR AMARILLO
Figura 50. Método de Ellman.
A principios de los años 70’s se caracterizaron las diferentes estructuras de las
colinesterasas y se clasificaron como AChE y BuChE; así como el hallazgo de
los genes que las codifican y potenciales polimorfismos que presentan en
función de su localización13. En 1976, se descubre que en la EA existe un
déficit de acetilcolina (ACh) en la corteza cerebral, por tal motivo, se
introdujeron la fisostigmina y la tacrina (fármacos de primera generación) para

74
tratar la enfermedad. En base al descubrimiento del déficit colinérgico, se
emitió por primera vez la llamada “Hipótesis Colinérgica” para explicar la
patogenia del Alzheimer 1982; la cual postuló que la disminución cognitiva
estaba causada por la disminución de la síntesis de ACh cortical y que, en
consecuencia, si se lograba aumentar la concentración en el espacio sináptico,
se produciría una mejoría del estado de los enfermos28.
En 1983 se hicieron estudios sobre el mecanismo de hidrólisis ejercido por
AChE a la ACh basado en experimentos cinéticos15.
Entre 1990 y 1996 se reportó a la comunidad científica la estructura
tridimensional en rayos X de la AChE de Torpedo californica y se reconoció su
sitio activo. A partir de ese año, se introdujeron la segunda generación de
inhibidores de AChE, ya que la fisostigmina y la tacrina fueron retirados por la
FDA por su gran toxicidad que causaban en los pacientes. Los fármacos
introducidos y actualmente usados son el donepezilo (1996), rivastigmina
(1998) y galantamina (2000)12, 28.
En 1999 se estudiaron una serie de aril-carbamatos monosustituidos (grupos
electro donadores y electroatractores) como inhibidores de la AChE de
Electrophorus electricus, con el fin de establecer la relación estructura actividad
de los compuestos con la enzima. Los resultados obtenidos fueron una
inhibición irreversible de los compuestos en el sitio aniónico periférico (PAS),
debido a un cambio conformacional inducido por el ligando dentro del mismo
sitio, ya que se favorecen las interacciones intermoleculares entre anillos
aromáticos. Además hubo una correlación entre la constante de inhibición (Ki)
de los compuestos sustituidos en posiciones meta y para en función de la
constante de Hammett (σ); siendo los aril-carbamatos con sustituyentes
electrodonadores más activos que aquellos que tenían grupos
electroatractores14.
Hace 4 años se iniciaron estudios sobre nuevos compuestos derivados del
ácido m-aminobenzoico31 probados como inhibidores de AChE. Los resultados
obtenidos fueron los siguientes (Tabla 10):

75
Tabla 10. Resultados de los derivados del ácido m-aminobenzoico sobre AChE31.
Compuesto IC50 (nM) Tipo de inhibición OHO
NH
OHO O
92.5 + 5
Reversible
OHO
N
O
O
33.4 + 2
Reversible
OHO
NH
OHO O
357 + 10
Irreversible
OHO
N
O
O
256 + 13
Irreversible
Tacrina 50 + 4 Reversible Galantamina 360 + 10 Reversible
La relación existente entre el compuesto abierto (ácido o amida) y el
compuesto cerrado (imida) es claro, cuando están en forma imida, la actividad
se incrementa con respecto a la forma ácida. Con respectos a la forma de
inhibición, los compuestos que poseen el doble enlace (maleamida y
maleimida) tiene una inhibición irreversible ya que probablemente se lleva a
cabo una reacción tipo Michael en la garganta de la AChE con grupos
nucleófilos como tioles, alcoholes o imidazoles; mientras que cuando están
saturados (succinamida y succinimida) la inhibición es reversible31. En este
mismo documento, se realizó un estudio de Docking, donde se explica el tipo
de interacciones (π-π) que ocurren entre los ligandos y los residuos de
aminoácidos existentes en el sitio activo de la enzima.
Poco después, se sintetizaron derivados parecidos a los anteriores, cuyos
grupos sustituyentes eran en su mayoría –OH y –OCOCH3; y algunos otros con
–H, -CH2CH3 y –Cl sustituidos en posiciones orto, meta y para32.
En este trabajo se evaluaron 13 compuestos diferentes con las características
antes mencionadas utilizando como controles positivos a inhibidores conocidos
como edrofonio, neostigmina, tacrina y donepezilo (Tabla 11).

76
Tabla 11. Resultados del estudios de inhibición sobre AChE de eritrocito de bovino32.
Compuesto Ki (�M) Tipo de inhibición Ácido m-hidroxifenilsucciámico 46.0 + 0.3 Reversible Ácido p-hidroxifenilsucciámico 1.3 + 0.5 Reversible Ácido p-acetiloxifenilsucciámico 6.6 + 0.3 Reversible m-acetiloxifenilsuccinimida 40.0 + 0.3 Reversible p-acetiloxifenilsuccinimida 0.005 + 0.4 Reversible p-hidroxifenilsuccinimida 28.5 + 0.4 Reversible Ácido m-hidroxifenilmaleámico 2.3 + 1.2 Irreversible Ácido p-hidroxifenilmaleámico 1.2 + 1.3 Irreversible Ácido p-acetiloxifenilmaleámico 0.8 + 0.5 Irreversible m-acetiloxifenilmaleimida Inestable Inestable p-acetiloxifenilmaleimida 0.54 + 0.2 Irreversible Fenilmaleimida 2.7 + 1.5 Irreversible p-hidroxifenilmaleimida 1.8 + 1.0 Irreversible Edrofonio 18.5 Reversible Neostigmina 14.4 Reversible Tacrina 0.01 Reversible Donepezilo 0.003 Reversible
Los resultados muestran que los compuestos con el doble enlace (ácidos
maleámicos y maleimidas) tienen una inhibición irreversible, mientras aquellos
en el cual el doble enlace no está presente, la inhibición es de tipo reversible.
La relación estructura-actividad no está muy clara, sin embargo, la actividad de
la mayoría de los compuestos probados es mayor que la de los controles32.
Poco después, se realizó un estudio teórico de los derivados con grupos –OH y
–OCOCH3 sustituidos únicamente en posición para, en el cual se encontró que
las energía de HOMO y LUMO en la molécula están distribuidas en distintas
regiones. La energía de HOMO se encuentra principalmente en el anillo
aromático, mientras que la energía de LUMO en la porción ácida (amida) o
imida. Estos resultados evidenciaron una correlación lineal entre la actividad
inhibitoria y las energías HOMO y LUMO, es decir, que la inhibición depende
de las energías HOMO y LUMO, lo que hace pensar que el anillo aromático es
de gran importancia en el reconocimiento del sitio activo33.
Recientemente (2006) se publicó un estudio de inhibición enzimática de AChE
de eritrocito de bovino in vitro y una evaluación de ligando-receptor asistido por
computadores de AChE humana de derivados tipo isoimidas, cuyos grupos

77
sustituidos en posición para son: -OH y –OCOCH3. Los resultados obtenidos
se exponen en la Tabla 12:
Tabla 12. Estudio in vitro e in silico de derivados tipo isomaleimida34.
Compuesto Ki (μM) in vitro Kd (μM) Docking Tipo de inhibición
OH
N O
O
p-hidroxifenilisomaleimida
4.72 + 2.3
0.157
Irreversible
O
N O
O
CH3
O
p-acetiloxifenilisomaleimida
3.60 + 1.8
0.513
Irreversible
Como se puede apreciar en la tabla, no hay mucha diferencia entre las
constantes de inhibición para cada compuesto; sin embargo, si hay diferencia
entre la constante experimental y la teórica, probablemente se deba al tipo de
sistema empleado (condiciones distintas). Nuevamente se observa una
inhibición irreversible debido a la presencia del doble enlace en el anillo de 5
miembros. Las imágenes de docking proporcionan información del modo de
unión entre el ligando y el receptor34.
Cabe mencionar que los estudios de reversibilidad e irreversibilidad se
realizaron con el método propuesto por Kitz y Wilson35.

78
IV. JUSTIFICACIÓN Debido a la gran incidencia global y al incremento de la enfermedad de
Alzheimer en nuestro país y en el mundo, constituirá a mediano y largo plazo
un grave problema de salud pública debido al envejecimiento de la población,
cuya tendencia más evidente hoy en día, está representada por los países
más desarrollados (Europa Occidental y Estados Unidos de América)17, 18.
La terapéutica actual de la EA es de tipo paliativo, cuyo objetivo es retrasar los
efectos de la enfermedad. En general, los distintos tratamientos farmacológicos
han brindado mejorado la calidad de vida de los pacientes; sin embargo, siguen
teniendo ciertas desventajas, debido a la toxicidad que presentan, la cual es
secundaria a la carencia de selectividad hacia los blancos biológicos.
El grupo de fármacos más utilizados para paliar la enfermedad lo forman los
inhibidores de AChE, cuyas características antes descritas, no constituyen la
excepción de efectos adversos.
Esto justifica la búsqueda de mejores fármacos para inhibir a la enzima AChE
para el manejo terapéutico de la enfermedad, con un enfoque meramente
molecular, cuyos blancos farmacológicos estén bien definidos e identificados
para poder realizar su diseño de una manera sistemática y racional; con el fin
de obtener fármacos más selectivos y menos tóxicos.
V. HIPÓTESIS La AChE es una enzima fundamental para dar por terminada la transmisión de
tipo colinérgica en el sistema nervioso autónomo3, 12. Los estudios de rayos X
han ayudado a identificar en tercera dimensión la estructura de la proteína
(AChE), en la cual se ha identificado una oquedad (garganta) que constituye el
sitio activo de la enzima cuyos aminoácidos en su mayoría son de tipo
aromático, lo que le proporciona una alta densidad electrónica14; por tanto, se
propone sintetizar 3 compuestos de tipo fenilisomaleimidas monosustituidas en
posición para con 3 sustituyentes estratégicos: -OCH3 (fuerte grupo
electrodonador), -NO2 (grupo fuertemente electroatractor) y –H (grupo de
referencia) con el propósito de observar los efectos electrónicos que ejercen los
ligandos sobre la enzima. De este preámbulo, se presume que la
fenilisomaleimida sustituida con el grupo –NO2, tendrá mejor afinidad con la
proteína, debido a que este grupo deslocaliza una carga positiva en la

79
molécula, lo que favorecería interacciones de tipo π−π (π-de baja densidad
electrónica con π-de alta densidad electrónica).
VI. OBJETIVOS
VI.1 Objetivo General Sintetizar e identificar 3 Fenilisomaleimidas monosustituidas cuyos grupos
sustituyentes en posición para son: un compuesto de referencia (-H), un
compuesto con un grupo electroatractor (-NO2) y uno con un grupo
electrodonador (-OCH3), y eventualmente, probarlos como inhibidores de AChE
in vitro, y en simuladores de interacción ligando-receptor (docking).
VI.2 Objetivos Particulares
• Sintetizar los compuestos propuestos
• Identificar a los compuestos por técnicas simples como cromatografía en
capa fina (c.c.f.), punto de fusión (p.f.), solubilidad relativa y apariencia,
así como métodos espectroscópicos: espectrofotometría de Infrarrojo
(IR) y espectroscopía de Resonancia Magnética Nuclear (RMN) de 1H y 13C.
• Evaluar el efecto inhibitorio de los compuestos por cinética enzimática in
vitro, y caracterizar el tipo de inhibición que se lleva a cabo.
• Determinar la constante de disociación por simulación de interacciones
ligando-receptor asistido por computadora (Docking).
VII. PARTE EXPERIMENTAL VII.1 Instrumentación
• Las materias primas, productos y reactivos fueron pesados en la balanza
analítica Boeco Germany.
• Las reacciones fueron agitadas en una parrilla de calentamiento y
agitación magnética Corning.
• La concentración de los productos de reacción se realizó en un
rotaevaporador Yamato RE 50 con ayuda de una bomba de vacío Duo
Seal.

80
• Los puntos de fusión de las materias primas y los productos fueron
determinados en un aparato de punto de fusión Electrothermal 9300.
• Para la determinación cromatográfica de las materias primas y los
productos se emplearon cromatofolios de aluminio ALUGRAM SIL 20x20
cm recubiertos con gel de sílice 60F254 (Micherey-Nagel). El revelado se
realizó con lámpara de UV UVP Laboratory Products Epi Chem II
Darkroom.
• Los espectros de infrarrojo (IR) fueron tomados de un espectrofotómetro
Midac Corporation M Series.
• Los espectros de Resonancia Magnética Nuclear fueron determinados
en un espectroscopio JEOL Delta NMR a 270 MHz.
• Las absorbancias de los experimentos de cinética enzimática fueron
medidas en un espectrofotómetro UV-Vis Beackman Coulter DU 650.
• Los valores de pH de las disoluciones buffer de fosfatos fueron tomadas
de un potenciómetro Hanna Instruments pH 211 Microprocessor pH
Meter.
• Las muestras fueron agitadas con un vórtex Constant Speed Mixer,
marca Lab-Line.
• Las muestras de cinética enzimática fueron centrifugadas en una
centrífuga Universal 32R Hettich Zentrifugen.
• Los experimentos de cinética enzimática fueron incubados en un baño
de agua Termo-Baño Felisa.
VII.2 Reactivos Se utilizaron los siguientes reactivos para la síntesis de las isoimidas
monosustituidas, purificación, y la determinación espectral: Anilina (J. T.
Baker), p-metoxianilina (Sigma), p-nitroanilina (Aldrich), Anhídrido maleico
(Sigma), Tetrahidrofurano destilado (THF, ver procedimiento), Hexano
destilado, Diciclohexilcarbodiimida (DCC, Aldrich), Diclorometano destilado
(CH2Cl2, ver procedimiento), Sulfato de sodio anhidro (J. T. Baker), Sulfato de
calcio anhidro (CaSO4, Drierite), Bromuro de Potasio anhidro (KBr, Merck),
Agua deuterada (D2O, Aldrich), Cloroformo deuterado (CDCl3, Aldrich),
Dimetilsulfóxido deuterado (CD3SOCD3, Aldrich).

81
Para los experimentos de cinética enzimática se utilizaron los siguientes
reactivos: Fosfato dibásico de potasio (K2HPO4, Reactivos Químicos Monterrey
S.A. de C.V.), Fosfato monobásico de potasio (KH2PO4, J. T. Baker), Ioduro de
Acetilcolina (ACh+I-, Merck), Acetilcolinesterasa (AChE, EC 3.1.1.7 de
Electrophorus electricus, polvo liofilizado con 349 U/mg, Sigma-Aldrich),
Bromuro de Neostigmina (Sigma-Aldrich), Hidróxido de sodio (NaOH, Merck),
Cloruro férrico hexahidratado (FeCl3.6H2O, TSQ Tecquim), Clorhidrato de
hidroxilamina (NH2OH.HCl, J. T. Baker), Ácido Clorhídrico (HCl, Reactivos
Químicos Monterrey S.A. de C.V.), Acetona (Merck), Agua destilada Milli Q.
Todos los reactivos utilizados durante el proyecto tienen grado analítico de
pureza.
VII.3 Procedimiento general de síntesis de los ácidos fenilmaleámicos y fenilisomaleimidas para monosustituidas. La metodología de síntesis se basa en los experimentos reportados desde
1961 hasta nuestros días, la cual no ha sido modificada sustancialmente36-39. A
continuación se describe el método utilizado en este trabajo:
VII.3.1 Síntesis del ácido fenilmaleámico
NH2
+ O
O
O
NHOOH
O
THFanh. Agitación constantet = 24 hT = ambiente
Anilina AnhídridoMaleico
Ácido Fenilmaleámico Figura 51. Síntesis de ácido fenilmaleámico.
Se colocaron en un matraz bola (A) 5.0 mL (0.0548 mol) de Anilina disuelta en
(previamente destilada) 30 mL de THF anhidro, mientras que en otro matraz
(B), se pesaron 5.66 g + 10% de exceso (0.0548 mol + 10 %) de Anhídrido
Maleico (AM) que fueron disueltos en 30 mL de THF anhidro. Ambos matraces
fueron cubiertos con un tapón Septum y plástico Parafilm®. El matraz A se
colocó sobre una parrilla de agitación magnética, y por vía canulación el AM

82
(Matraz B) fue adicionado por goteo al matraz A. Una vez adicionado el AM, la
reacción se dejó en agitación constante a temperatura ambiente por 24 h.
Terminada la reacción, el producto fue filtrado al vacío ya que es insoluble en
THF. El ácido fenilmaleámico fue purificado por lavados sucesivos con hexano
frío y se dejó secar a temperatura ambiente por 24 h. Se obtuvo un polvo fino
color crema, cuyo punto de fusión fue de 204.2-205.7 ºC y un rendimiento de
98.21 %; además el producto fue identificado por métodos espectroscópicos de
Infrarrojo (IR) y Resonancia Magnética Nuclear (RMN) de 1H y 13C. (Ver Figura
54).
VII.3.2 Síntesis del ácido p-metoxifenilmaleámico
NH2
OCH3
O
O
O
NHOOH
O
OCH3
+THFanh. Agitación constantet = 24 hT = ambiente
p-metoxi-anilina
AnhídridoMaleico
Ácido p-metoxi-fenilmaleámico Figura 52. Síntesis del Ácido p-metoxifenilmaleámico.
Se pesaron 5.0 g (0.0406 mol) de p-metoxianilina disolviéndolos en 30 mL de
THF anhidro (Matraz A). En otro matraz (Matraz B), se pesaron 3.98 g + 10%
de exceso (0.04059 + 10% mol) de Anhídrido Maleico (AM) que fueron
disueltos en 30 mL de THF anhidro. Ambos matraces fueron cubiertos con un
tapón Septum y plástico Parafilm®. El matraz A se colocó sobre una parrilla de
agitación magnética, y por vía canulación el AM (Matraz B) fue adicionado por
goteo al matraz A. Una vez adicionado el AM, la reacción se dejó en agitación
constante a temperatura ambiente por 24 h.
Terminada la reacción, el producto fue filtrado al vacío, ya que este es insoluble
en THF. El ácido p-metoxifenilmaleámico fue purificado por lavados sucesivos
con hexano frío y se dejó secar a temperatura ambiente por 24 h. Se obtuvo un
polvo fino color amarillo intenso, cuyo punto de fusión y rendimientos fueron
189.0-190.0 ºC y 98.79 % respectivamente. El compuesto fue identificado por
métodos espectroscópicos de Infrarrojo (IR) y Resonancia Magnética Nuclear
(RMN) de 1H y 13C. (Ver Figura 54).

83
VII.3.3 Síntesis del ácido p-nitrofenilmaleámico
NH2
NO2
O
O
O
NHOOH
O
NO2
+THFanh. Agitación constantet = 48 hT = ambiente
p-nitro-anilina
AnhídridoMaleico
Ácido p-nitro-fenilmaleámico Figura 53. Síntesis del Ácido p-nitrofenilmaleámico.
Se pesaron 5.0 g (0.0362 mol) de p-nitroanilina disolviéndolos en 30 mL de
THF anhidro (Matraz A). En otro matraz (Matraz B), se pesaron 3.55 g + 10%
de exceso (0.0362 + 10 % mol) de Anhídrido Maleico (AM) que fueron disueltos
en 30 mL de THF anhidro. Ambos matraces fueron cubiertos con un tapón
Septum y plástico Parafilm®. El matraz A se colocó sobre una parrilla de
agitación magnética, y por vía canulación el AM (Matraz B) fue adicionado por
goteo al matraz A. Una vez adicionado el AM, la reacción se dejó en agitación
constante a temperatura ambiente por 48 h.
Terminada la reacción, el producto fue filtrado al vacío, ya que éste es insoluble
en THF. El ácido p-nitrofenilmaleámico fue purificado por lavados sucesivos
con hexano frío y se dejó secar a temperatura ambiente por 24 h. Se obtuvo un
polvo fino color amarillo claro, cuyo punto de fusión y rendimientos fueron
194.3-195.5 ºC y 95.52 % respectivamente. El compuesto fue identificado por
métodos espectroscópicos de Infrarrojo (IR) y Resonancia Magnética Nuclear
(RMN) de 1H y 13C. (Ver Figura 54).
• Procedimiento de purificación del tetrahidrofurano (THF) El disolvente (THF) utilizado en las reacciones previas, se dejó en agitación por
12 h en hidruro de litio (LiH) y aluminio bajo atmósfera de nitrógeno; una vez
transcurrido el tiempo se destiló. Después del proceso de destilación el THF fue
sometido a reflujo y agitación constante en presencia de Naº y benzofenona
hasta la aparición de un color violeta. Posteriormente a este proceso de
eliminación de oxígeno y humedad, el disolvente se destiló para su uso40.
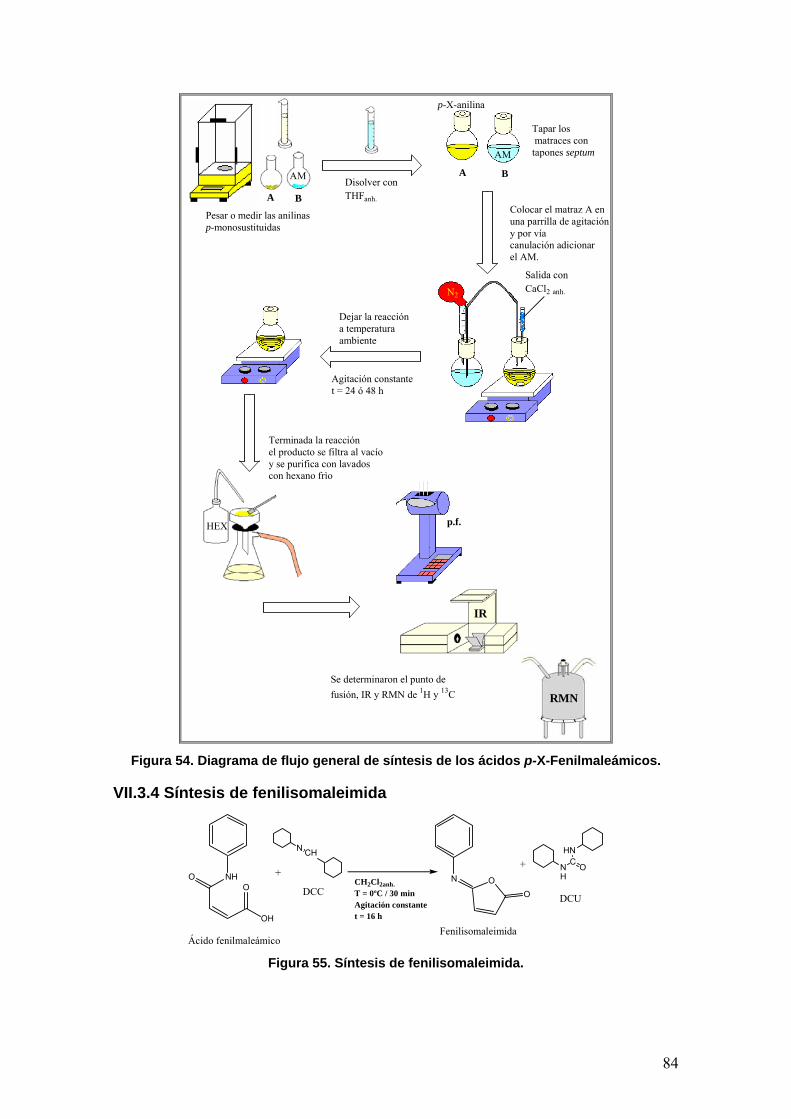
84
B
AM
A
N2
RMN
IR
AM
BA
Pesar o medir las anilinas p-monosustituidas
Disolver conTHFanh.
Tapar los matraces con tapones septum
p-X-anilina
Colocar el matraz A enuna parrilla de agitación y por víacanulación adicionarel AM.
Dejar la reaccióna temperatura ambiente
Agitación constantet = 24 ó 48 h
Salida con CaCl2 anh.
Terminada la reacciónel producto se filtra al vacíoy se purifica con lavadoscon hexano frìo
HEX
Se determinaron el punto defusión, IR y RMN de 1H y 13C
p.f.
Figura 54. Diagrama de flujo general de síntesis de los ácidos p-X-Fenilmaleámicos.
VII.3.4 Síntesis de fenilisomaleimida
NH
OH
OO
N CH
DCCN O
O
NH
CHN
O
DCU
++
Ácido fenilmaleámicoFenilisomaleimida
CH2Cl2anh.T = 0ºC / 30 minAgitación constantet = 16 h
Figura 55. Síntesis de fenilisomaleimida.

85
Se pesaron 3.0 g (0.0157 mol) de ácido fenilmaleámico suspendiéndolos en 50
mL de CH2Cl2 anhidro (Matraz A). En otro matraz (Matraz B), se pesaron 3.23
g + 10% de exceso (0.0157 mol) de Diciclohexilcarbodiimida (DCC) que fueron
disueltos en 50 mL de CH2Cl2 anhidro. Ambos matraces fueron cubiertos con
un tapón Septum y plástico Parafilm®. El matraz A se colocó en un baño de
hielo sobre una parrilla de agitación magnética, y por vía canulación la DCC
(Matraz B) fue adicionada por goteo al matraz A. Una vez adicionada la DCC,
la reacción se dejó en agitación constante a 0 ºC durante 30 min, y
posteriormente se dejó reaccionar a temperatura ambiente de 16 a 18 h.
Terminada la reacción, el producto fue filtrado al vacío para separar la
Diciclohexilurea (DCU, insoluble en CH2Cl2) formada durante la reacción de
deshidratación. La parte soluble fue filtrada a gravedad 2 veces lavando con
CH2Cl2 anhidro; posteriormente el producto fue concentrado en el
rotaevaporador. El producto en disolución con CH2Cl2 fue recristalizado con
hexano anhidro frío, obteniéndose un polvo color amarillo claro que fue filtrado
al vacío y lavado con hexano frío para eliminar el exceso de DCC. Finalmente,
se determinó el punto de fusión (60.0-62.0 ºC), rendimiento (97.69%) y el Rf.
(0.6029) en c.c.f. cuya fase móvil fue Hexano-AcOEt (1:1). El compuesto se
identificó mediante métodos espectroscópicos de Infrarrojo (IR) y Resonancia
Magnética Nuclear (RMN) de 1H y 13C. (Ver Figura 58).
VII.3.5 Síntesis de p-metoxifenilisomaleimida
NH
OH
OO
OCH3
N CH
DCCN O
O
OCH3
NH
CHN
O
DCU
++
Ácido p-metoxi-fenilmaleámico
p-Metoxi-Fenilisomaleimida
CH2Cl2anh.T = 0ºC / 30 minAgitación constantet = 16 h
Figura 56. Síntesis de p-metoxifenilisomaleimida.
Se pesaron 3.0 g (0.0135 mol) de ácido p-metoxifenilmaleámico
suspendiéndolos en 50 mL de CH2Cl2 anhidro (Matraz A). En otro matraz
(Matraz B), se pesaron 2.79 g + 10 % de exceso (0.0135 mol) de
Diciclohexilcarbodiimida (DCC) que fueron disueltos en 50 mL de CH2Cl2

86
anhidro. Ambos matraces fueron cubiertos con un tapón Septum y plástico
Parafilm®. El matraz A se colocó en un baño de hielo sobre una parrilla de
agitación magnética, y por vía canulación la DCC (Matraz B) fue adicionada por
goteo al matraz A. Una vez adicionada la DCC, la reacción se dejó en agitación
constante a 0 ºC durante 30 min, y posteriormente se dejó en proceso de
reacción entre 16 a 18 h a temperatura ambiente.
Terminada la reacción, el producto fue filtrado al vacío para separar la
Diciclohexilurea (DCU, insoluble en CH2Cl2) formada durante la reacción de
deshidratación. La parte soluble fue filtrada a gravedad 2 veces lavando con
CH2Cl2 anhidro; posteriormente el producto fue concentrado en el
rotaevaporador. El producto en disolución con CH2Cl2 fue recristalizado con
hexano anhidro frío, obteniéndose un polvo color amarillo intenso que fue
filtrado al vacío y lavado con hexano frío para eliminar el exceso de DCC.
Finalmente, se determinó el punto de fusión (72.0-73.0 ºC), rendimiento (99.59
%) y el Rf. (0.5441) en c.c.f. cuya fase móvil fue Hexano-AcOEt (1:1). El
compuesto se identificó mediante métodos espectroscópicos de Infrarrojo (IR) y
Resonancia Magnética Nuclear (RMN) de 1H y 13C. (Ver Figura 58).
VII.3.6 Síntesis de p-nitrofenilisomaleimida
NH
OH
OO
NO2
N CH
DCCN O
O
NO2
NH
CHN
O
DCU
++
Ácido p-nitro-fenilmaleámico
p-Nitro-Fenilisomaleimida
CH2Cl2anh.T = 0ºC / 30 minAgitación constantet = 16 h
Figura 57. Síntesis de p-nitrofenilisomaleimida.
Se pesaron 3.0 g (0.0127 mol) de ácido p-nitrofenilmaleámico suspendiéndolos
en 50 mL de CH2Cl2 anhidro (Matraz A). En otro matraz (Matraz B), se
pesaron 2.62 g + 10% de exceso (0.0127 mol) de Diciclohexilcarbodiimida
(DCC) que fueron disueltos en 50 mL de CH2Cl2 anhidro. Ambos matraces
fueron cubiertos con un tapón Septum y plástico Parafilm®. El matraz A se
colocó en un baño de hielo sobre una parrilla de agitación magnética, y por vía
canulación la DCC (Matraz B) fue adicionada por goteo al matraz A. Una vez

87
adicionada la DCC, la reacción se dejó en agitación constante a 0ºC durante 30
min, y posteriormente se dejó a temperatura ambiente de 16 a 18 h.
Terminada la reacción, el producto fue filtrado al vacío para separar la
Diciclohexilurea (DCU, insoluble en CH2Cl2) formada durante la reacción de
deshidratación. La parte soluble fue filtrada a gravedad 2 veces lavando con
CH2Cl2 anhidro; posteriormente el producto fue concentrado en el
rotaevaporador. El producto en disolución con CH2Cl2 fue recristalizado con
hexano anhidro frío, obteniéndose un polvo color amarillo intenso que fue
filtrado al vacío y lavado con hexano frío para eliminar el exceso de DCC.
Finalmente, se determinó el punto de fusión (107.5-108.6 ºC), rendimiento
(28.86 %) y el Rf. (0.4705) en c.c.f. cuya fase móvil fue Hexano-AcOEt (1:1). El
compuesto se identificó mediante métodos espectroscópicos de Infrarrojo (IR) y
Resonancia Magnética Nuclear (RMN) de 1H y 13C. (Ver Figura 58).
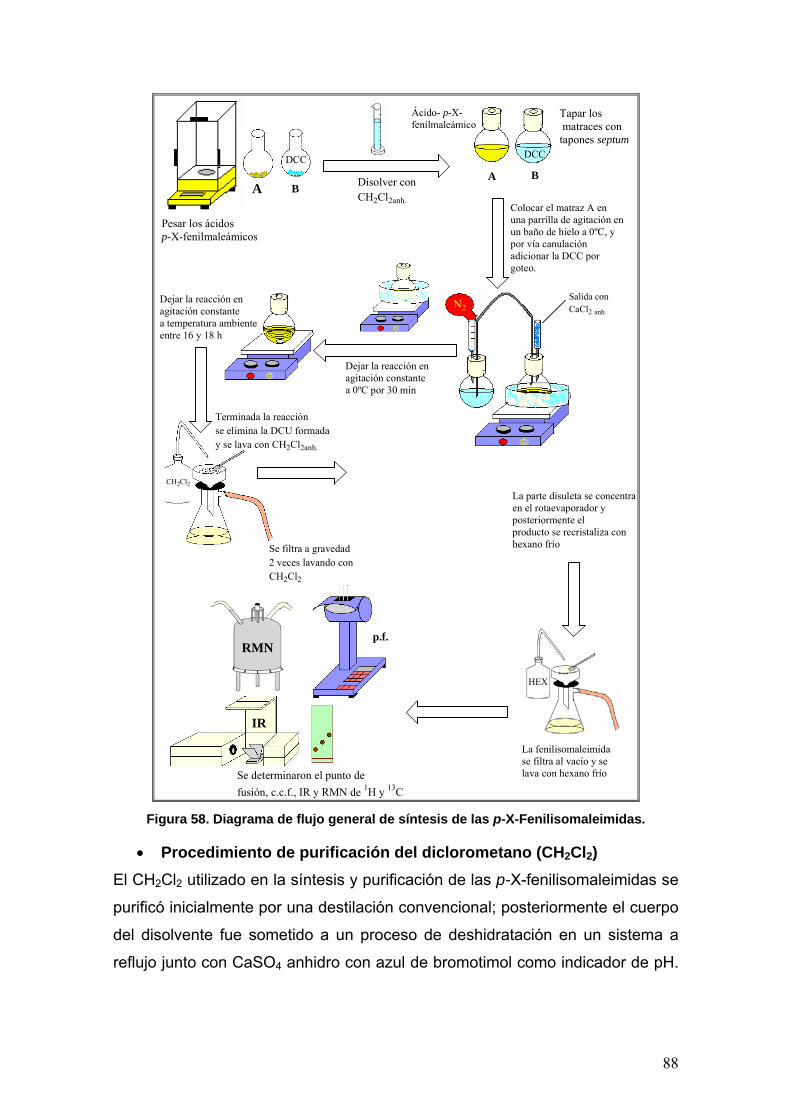
88
B
DCC
A
N2
DCC
B
IR
RMN
A
CH2Cl2
Tapar los matraces con tapones septum
Ácido- p-X-fenilmaleámico
Colocar el matraz A enuna parrilla de agitación en un baño de hielo a 0ºC, y por vía canulación adicionar la DCC por goteo.
Dejar la reacción enagitación constantea 0ºC por 30 min
Salida con CaCl2 anh.
Terminada la reacciónse elimina la DCU formaday se lava con CH2Cl2anh.
Se determinaron el punto defusión, c.c.f., IR y RMN de 1H y 13C
Pesar los ácidos p-X-fenilmaleámicos
Disolver conCH2Cl2anh.
Dejar la reacción enagitación constantea temperatura ambienteentre 16 y 18 h
La parte disuleta se concentra en el rotaevaporador y posteriormente elproducto se recristaliza con hexano fríoSe filtra a gravedad
2 veces lavando conCH2Cl2
HEX
La fenilisomaleimidase filtra al vacío y selava con hexano frío
p.f.
Figura 58. Diagrama de flujo general de síntesis de las p-X-Fenilisomaleimidas.
• Procedimiento de purificación del diclorometano (CH2Cl2)
El CH2Cl2 utilizado en la síntesis y purificación de las p-X-fenilisomaleimidas se
purificó inicialmente por una destilación convencional; posteriormente el cuerpo
del disolvente fue sometido a un proceso de deshidratación en un sistema a
reflujo junto con CaSO4 anhidro con azul de bromotimol como indicador de pH.

89
El sistema desecado se dejó por 3 h, para eventualmente destilarlo y utilizarlo
en las reacciones antes mencionadas.
VII.4 Determinación de la actividad enzimática de la Acetilcolinesterasa. El método utilizado es el propuesto por Sjored Bonting y Robert Featherstone
en 195629, el cual está basado en la formación de un compuesto de
coordinación derivado de la reacción dada entre acetilcolina remanente con
hidroxilamina alcalina, la cual forma ácido hidroxámico que a su vez genera un
complejo color violáceo en presencia de FeCl3 en medio ácido29 (Figura 59).
NaOHNH2OH.HCl
NaCl
NH3C
H3CCH3
H3C O
ON
OH
NH2OH
3 HCl Fe
ONHO
CH3
ONHO
CH3
HNO
OH3C
HOHN CH3
O
HCl
FeCl3Hidroxilaminaalcalina
Acetilcolina
ColinaÁcidoHidroxámico
Tris-(hidroxamato)-hierro III
+
Figura 59. Fundamento del método de ultramicroensayo de Bonting y Featherstone29, 41,
42.
VII.4.1 Procedimiento general de medición
• Se escogió un intervalo de concentraciones finales de sustrato
(Acetilcolina, ACh) que van de 0.1 a 6.4 mM.
• Se prepararon 2 disoluciones stock de ACh de concentraciones 0.005 M
y 0.08 M en buffer de fosfatos C = 0.1 M a pH = 8.0. El volumen
depende del número de curvas a realizar.
• Se preparó una disolución stock de AChE de concentración 0.6649 U/mL
en disolución buffer de fosfatos C = 0.1 M a pH = 8.0. Se preparó por
cada ensayo, y se incubó 10 min a 37 ºC antes de utilizarse. La
concentración final de enzima fue de 0.1 U/mL (ver normalización del
método).
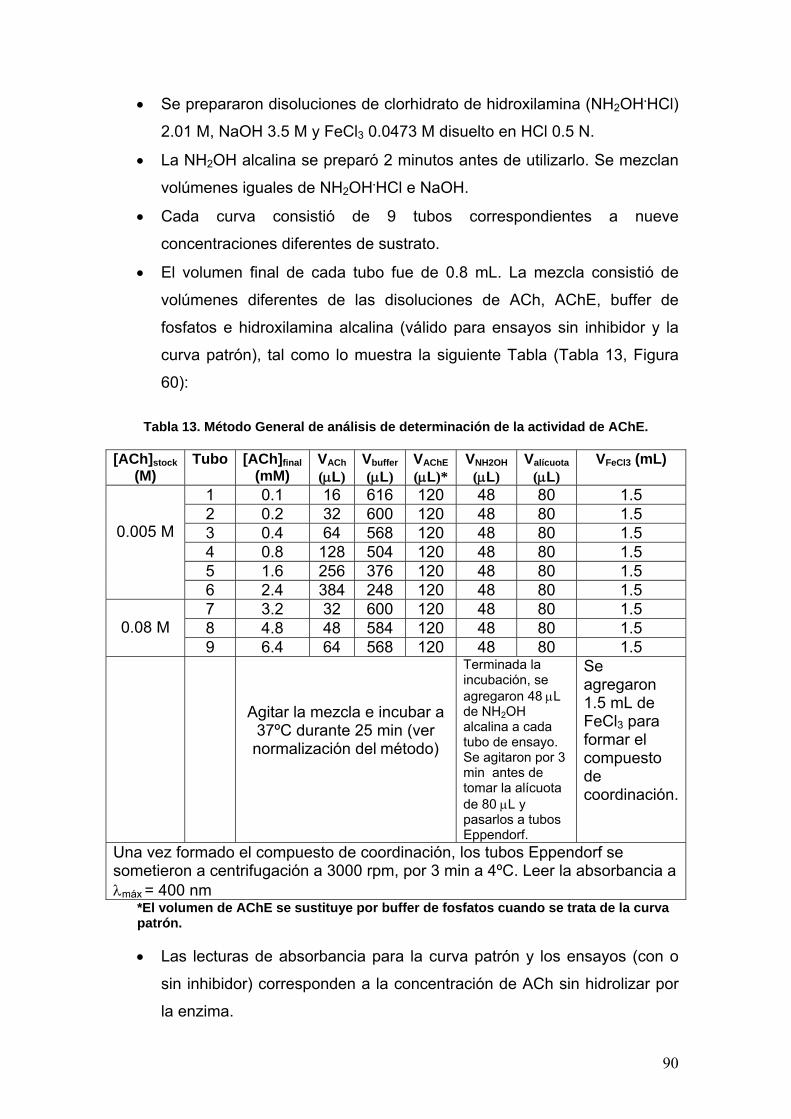
90
• Se prepararon disoluciones de clorhidrato de hidroxilamina (NH2OH.HCl)
2.01 M, NaOH 3.5 M y FeCl3 0.0473 M disuelto en HCl 0.5 N.
• La NH2OH alcalina se preparó 2 minutos antes de utilizarlo. Se mezclan
volúmenes iguales de NH2OH.HCl e NaOH.
• Cada curva consistió de 9 tubos correspondientes a nueve
concentraciones diferentes de sustrato.
• El volumen final de cada tubo fue de 0.8 mL. La mezcla consistió de
volúmenes diferentes de las disoluciones de ACh, AChE, buffer de
fosfatos e hidroxilamina alcalina (válido para ensayos sin inhibidor y la
curva patrón), tal como lo muestra la siguiente Tabla (Tabla 13, Figura
60):
Tabla 13. Método General de análisis de determinación de la actividad de AChE.
[ACh]stock (M)
Tubo [ACh]final (mM)
VACh (μL)
Vbuffer (μL)
VAChE (μL)*
VNH2OH (μL)
Valícuota (μL)
VFeCl3 (mL)
1 0.1 16 616 120 48 80 1.5 2 0.2 32 600 120 48 80 1.5 3 0.4 64 568 120 48 80 1.5 4 0.8 128 504 120 48 80 1.5 5 1.6 256 376 120 48 80 1.5
0.005 M
6 2.4 384 248 120 48 80 1.5 7 3.2 32 600 120 48 80 1.5 8 4.8 48 584 120 48 80 1.5
0.08 M
9 6.4 64 568 120 48 80 1.5
Agitar la mezcla e incubar a
37ºC durante 25 min (ver normalización del método)
Terminada la incubación, se agregaron 48 μL de NH2OH alcalina a cada tubo de ensayo. Se agitaron por 3 min antes de tomar la alícuota de 80 μL y pasarlos a tubos Eppendorf.
Se agregaron 1.5 mL de FeCl3 para formar el compuesto de coordinación.
Una vez formado el compuesto de coordinación, los tubos Eppendorf se sometieron a centrifugación a 3000 rpm, por 3 min a 4ºC. Leer la absorbancia a λmáx = 400 nm
*El volumen de AChE se sustituye por buffer de fosfatos cuando se trata de la curva patrón.
• Las lecturas de absorbancia para la curva patrón y los ensayos (con o
sin inhibidor) corresponden a la concentración de ACh sin hidrolizar por
la enzima.

91
• Las absorbancias de cada ensayo fueron restadas de las absorbancias
de la curva patrón correspondientes a cada concentración de sustrato.
La diferencia corresponde a la concentración de ACh hidrolizada por la
AChE.
ΔAACh-Hidr. = Acurva-Aensayo
• Cada ΔAACh-Hidr. se interpoló en la curva patrón descrita por la ley de
Lambert-Beer, con el fin de obtener la concentración de acetilcolina
hidrolizada.
A = εlC,
ε = Coeficiente de absortividad molar (pendiente)
l = Longitud de la celda (1cm) C = Concentración
CACh-Hidr. = ΔAACh-Hidr. - Intercepto
ε l • Una vez calculada la concentración de ACh hidrolizada, se determinó la
actividad mediante la siguiente ecuación:
Actividad=
CACh-Hidr. x Vfinal
1000 mLx
1000 μmol1 mmol
[E]final x Vfinal
1 mLx 25 min
• La actividad fue graficada en función de la concentración de sustrato, y
finalmente se realizó el procedimiento de de Lineweaver-Burk para
determinar la Km y la rmáx (sin inhibidor). Los resultados de la curva con
distintas concentraciones de inhibidor se determinaron de la misma
manera.
• Todos los experimentos fueron tratados mediante un análisis estadístico
de regresión lineal por mínimos cuadrados y t ‘Student’.
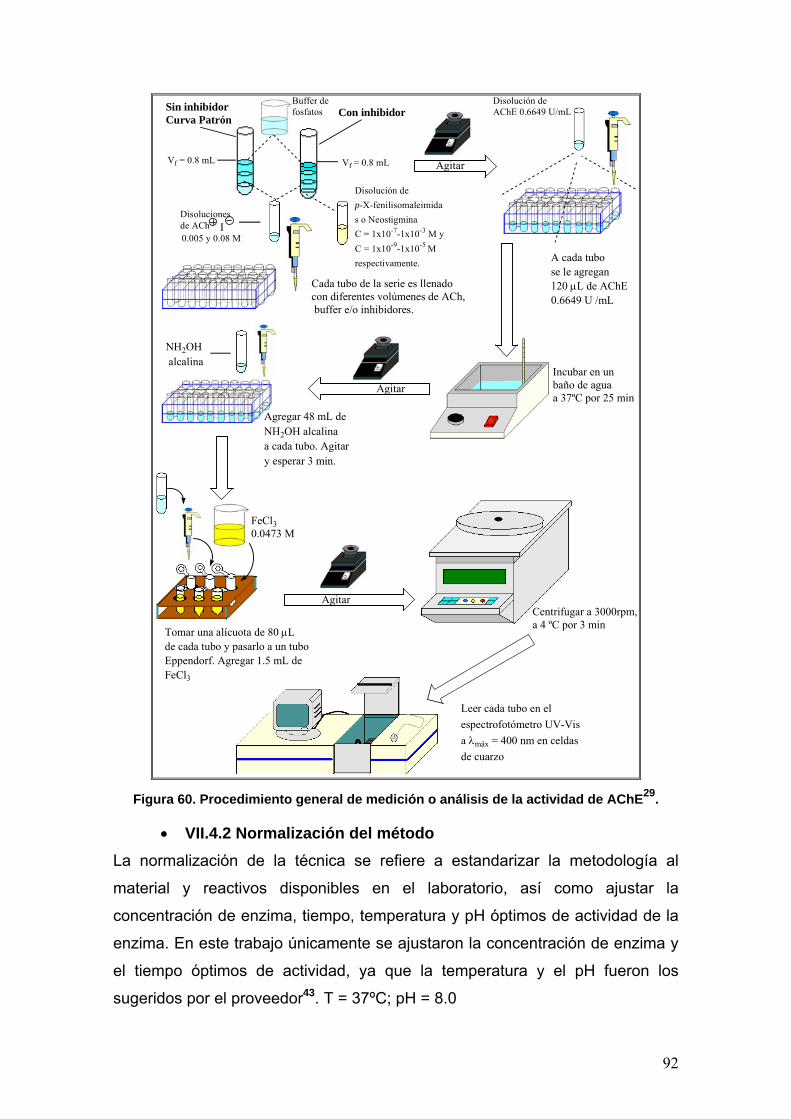
92
Vf = 0.8 mL Vf = 0.8 mL
Buffer defosfatos
Disolución dep-X-fenilisomaleimidas o NeostigminaC = 1x10-7-1x10-3 M y C = 1x10-9-1x10-5 M respectivamente.
Disoluciones de ACh I0.005 y 0.08 M
Disolución deAChE 0.6649 U/mL
Cada tubo de la serie es llenado con diferentes volúmenes de ACh, buffer e/o inhibidores.
Sin inhibidorCurva Patrón
Con inhibidor
A cada tubose le agregan120 μL de AChE 0.6649 U /mL
Incubar en unbaño de aguaa 37ºC por 25 min
Agregar 48 mL de NH2OH alcalinaa cada tubo. Agitary esperar 3 min.
NH2OH alcalina
Agitar
Agitar
FeCl30.0473 M
Tomar una alícuota de 80 μLde cada tubo y pasarlo a un tuboEppendorf. Agregar 1.5 mL deFeCl3
AgitarCentrifugar a 3000rpm,a 4 ºC por 3 min
Leer cada tubo en el espectrofotómetro UV-Visa λmáx = 400 nm en celdasde cuarzo
Figura 60. Procedimiento general de medición o análisis de la actividad de AChE29.
• VII.4.2 Normalización del método
La normalización de la técnica se refiere a estandarizar la metodología al
material y reactivos disponibles en el laboratorio, así como ajustar la
concentración de enzima, tiempo, temperatura y pH óptimos de actividad de la
enzima. En este trabajo únicamente se ajustaron la concentración de enzima y
el tiempo óptimos de actividad, ya que la temperatura y el pH fueron los
sugeridos por el proveedor43. T = 37ºC; pH = 8.0

93
VII.4.2a Determinación de la concentración óptima de enzima [E]
• Para determinar la concentración de enzima óptimo se utilizaron 3
concentraciones finales de prueba de enzima, las cuales fueron: 0.25 (n
= 2), 0.125 (n = 6) y 0.1 (n = 5) U/mL.
• El intervalo de concentraciones finales de sustrato escogidos fue: 0.1,
0.2, 0.4, 0.8, 1.6, 3.2, 6.4 mM para cada ensayo y la curva patrón.
• La metodología de medición para cada concentración de enzima y la
curva patrón fue la antes descrita en la sección VII.4.1.
VII.4.2b Determinación del tiempo óptimo de actividad
• Para determinar el tiempo óptimo de actividad se escogieron 3
concentraciones estratégicas de acetilcolina, las cuales fueron fijas
durante el ensayo. [ACh] = 0.8, 1.6, 3.2 mM
• La concentración de enzima final (AChE) fue de 0.1 U/mL (resultado de
la determinación de la concentración de enzima óptima).
• El intervalo de concentraciones finales de sustrato escogido para la
curva patrón fue: 0.1, 0.2, 0.4, 0.8, 1.6, 2.4, 3.2, 4.8 y 6.4 mM.
• Los experimentos fueron diseñados para observar la disminución de
sustrato (ACh) en función del tiempo, cuyos tiempos de toma de muestra
fueron: 5, 10, 15, 20, 25, 30, 40, 50 y 60 min.
• La medición de cada muestra se realizó mediante el método general de
análisis (VII.4.1).
VII.4.3 Determinación de la constante de Michaelis-Menten (Km) y la rapidez máxima (rmáx)
• La determinación de la actividad se realizó a través del procedimiento
general de medición o análisis descrito en la sección VII.4.1. Las
condiciones fueron: pH = 8.0, T = 37ºC, [E]final = 0.1 U/mL, t = 25 min de
incubación (resultado de la determinación del tiempo óptimo). El
experimento se repitió 37 veces ( n = 20)
• El intervalo de concentraciones finales de sustrato escogido para la
curva patrón y la determinación fue: 0.1, 0.2, 0.4, 0.8, 1.6, 2.4, 3.2, 4.8 y
6.4 mM.
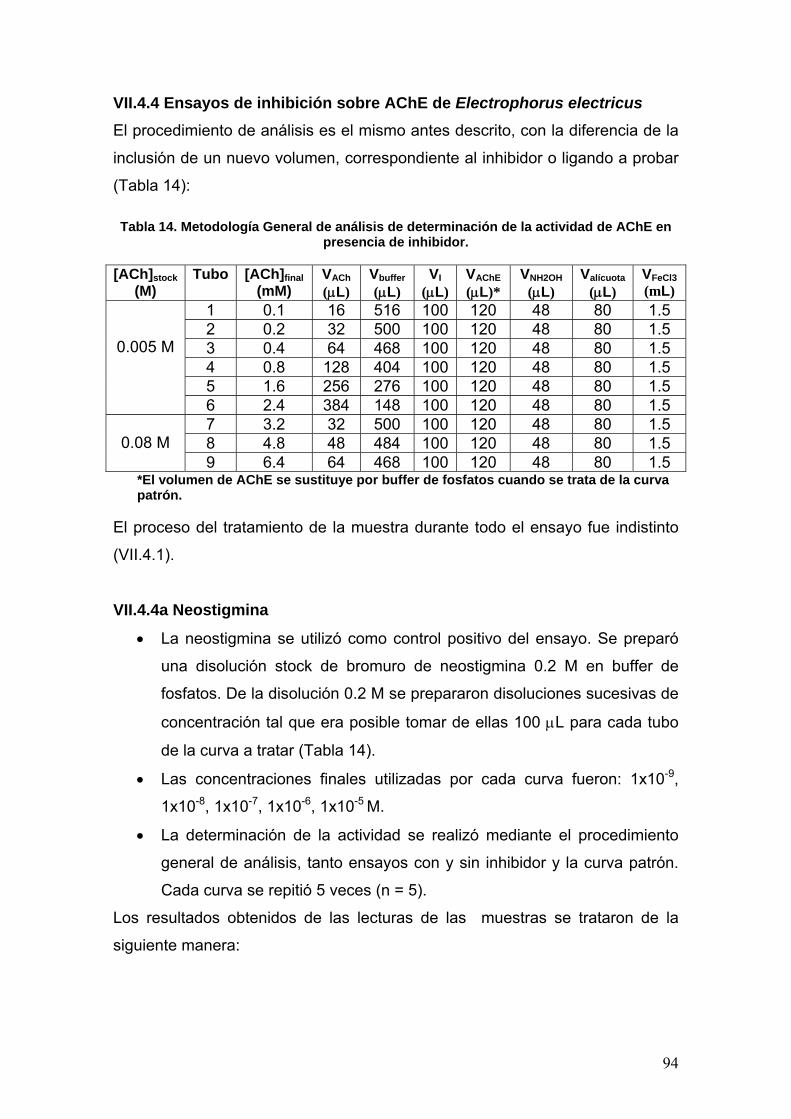
94
VII.4.4 Ensayos de inhibición sobre AChE de Electrophorus electricus
El procedimiento de análisis es el mismo antes descrito, con la diferencia de la
inclusión de un nuevo volumen, correspondiente al inhibidor o ligando a probar
(Tabla 14):
Tabla 14. Metodología General de análisis de determinación de la actividad de AChE en presencia de inhibidor.
[ACh]stock
(M) Tubo [ACh]final
(mM) VACh (μL)
Vbuffer (μL)
VI (μL)
VAChE (μL)*
VNH2OH (μL)
Valícuota (μL)
VFeCl3 (mL)
1 0.1 16 516 100 120 48 80 1.5 2 0.2 32 500 100 120 48 80 1.5 3 0.4 64 468 100 120 48 80 1.5 4 0.8 128 404 100 120 48 80 1.5 5 1.6 256 276 100 120 48 80 1.5
0.005 M
6 2.4 384 148 100 120 48 80 1.5 7 3.2 32 500 100 120 48 80 1.5 8 4.8 48 484 100 120 48 80 1.5
0.08 M
9 6.4 64 468 100 120 48 80 1.5 *El volumen de AChE se sustituye por buffer de fosfatos cuando se trata de la curva patrón.
El proceso del tratamiento de la muestra durante todo el ensayo fue indistinto
(VII.4.1).
VII.4.4a Neostigmina
• La neostigmina se utilizó como control positivo del ensayo. Se preparó
una disolución stock de bromuro de neostigmina 0.2 M en buffer de
fosfatos. De la disolución 0.2 M se prepararon disoluciones sucesivas de
concentración tal que era posible tomar de ellas 100 μL para cada tubo
de la curva a tratar (Tabla 14).
• Las concentraciones finales utilizadas por cada curva fueron: 1x10-9,
1x10-8, 1x10-7, 1x10-6, 1x10-5 M.
• La determinación de la actividad se realizó mediante el procedimiento
general de análisis, tanto ensayos con y sin inhibidor y la curva patrón.
Cada curva se repitió 5 veces (n = 5).
Los resultados obtenidos de las lecturas de las muestras se trataron de la
siguiente manera:

95
• Las lecturas de absorbancia para la curva patrón y los ensayos (con o
sin inhibidor) corresponden a la concentración de ACh sin hidrolizar por
la enzima.
• Las absorbancias de cada ensayo fueron restadas de las absorbancias
de la curva patrón correspondientes a cada concentración de sustrato.
La diferencia corresponde a la concentración de ACh hidrolizada por la
AChE.
ΔAACh-Hidr. = Acurva-Aensayo
• Cada ΔAACh-Hidr. se interpoló en la curva patrón descrita por la ley de
Lambert-Beer, con el fin de obtener la concentración de acetilcolina
hidrolizada.
A = εlC,
ε = Coeficiente de absortividad molar (pendiente)
l = Longitud de la celda (1cm) C = Concentración
CACh-Hidr. = ΔAACh-Hidr. - Intercepto
ε l • Una vez calculada la concentración de ACh hidrolizada, se determinó la
actividad mediante la siguiente ecuación:
Actividad=
CACh-Hidr. x Vfinal
1000 mLx
1000 μmol1 mmol
[E]final x Vfinal
1 mLx 25 min
• La actividad fue graficada en función de la concentración de sustrato, y
finalmente se realizó el procedimiento de Lineweaver-Burk para
determinar la Km y la rmáx (sin inhibidor). Los resultados de la curva con
distintas concentraciones de neostigmina se determinaron de la misma
manera (pendientes y ordenadas al origen modificadas por cada
concentración de inhibidor).
• Una vez obtenidas las pendientes de la rectas derivadas del tratamiento
de Lineweaver-Burk para el ensayo sin inhibidor y los casos con
inhibidor; se calculó la razón ([mi/m0], mi = pendiente para cada
concentración de inhibidor; m0 = pendiente sin inhibidor). Los cocientes

96
[mi/m0] fueron graficados en función de la concentración de inhibidor
para determinar la constante de inhibición (Ki) a través del método de
Schild.
• Todos los experimentos fueron tratados mediante un análisis estadístico
de regresión lineal por mínimos cuadrados y t ‘Student’. Los datos están
reportados como la media + ESM (Error estándar de la media).
VII.4.4b Fenilisomaleimida, p-metoxifenilisomaleimida y p-
nitrofenilisomaleimida
• Se preparó una disolución stock de p-X-fenilisomaleimida 0.1 M en
acetona grado analítico (G.A.). De esta disolución se prepararon
disoluciones sucesivas de concentraciones tales para poder tomar de
ellas 100 μL para cada tubo de la curva (Tabla 14). Las disoluciones
fueron aforadas con buffer de fosfatos.
• Las concentraciones finales utilizadas por cada curva fueron: 1x10-7,
1x10-6, 1x10-5, 1x10-4, 1x10-3 M. Los experimentos para los derivados
con sustituyentes –H y –NO2 se repitieron 3 veces (n = 3) y para el que
tiene el sustituyente –OCH3 4 veces (n = 4).
• La determinación de la actividad se realizó mediante el procedimiento
general de análisis, y el tratamiento de datos de igual forma que a la
neostigmina.
• Todos los experimentos que requirieron regresión lineal fueron tratados
mediante un análisis estadístico de regresión lineal por mínimos
cuadrados y t ‘Student’. Los datos están reportados como la media +
ESM (Error estándar de la media).
VII.5 Docking El estudio de simulación de la interacción ligando-receptor se efectuó mediante
el programa AutoDock 3.05 en plataforma Linux, en una PC Pentium IV a 1.6
GHz con 512 MB de memoria RAM.
La estructura cristalizada por DRX de la enzima AChE humana (1B41) fue
obtenida del Protein Data Bank (PDB); a la cual se le hizo un pre-tratamiento
con el programa RasMol, cuya función fue eliminar moléculas de agua y

97
ligandos ajenos a la proteína. Una vez eliminadas las moléculas sobrantes; se
le adicionaron átomos de hidrógeno (polares), cargas parciales y parámetros
de solvatación a la enzima, utilizando el programa AutoDockTools. Hechas las
modificaciones, los archivos fueron guardados como *pdbqs.
Por otra parte, los ligandos fueron dibujados y optimizados en los programas
Hyperchem 6.0 y Gaussian 98 respectivamente. El método de optimización
escogido fue un sistema ab initio con una base de datos Hartree Fock
restringida (RHF/6-31G*). Los archivos se cambiaron a formato PDB en el
programa Molekel 4.3.
Los ligandos también fueron sometidos a un proceso de optimización, el cual
consistió en adicionar cargas parciales a los átomos, así como la definición de
los átomos de hidrógeno polares mediante el programa AutoDockTools. Se
especificaron también los átomos de carbono aromáticos, las porciones rígidas
de la molécula y los enlaces que presentan mayor grado de libertad. Los
archivos se escribieron en formato *.pdbq para cada ligando.
El mapa de afinidad atómica se generó para el archivo *.pdbqs de la enzima
utilizando el programa AutoGrid 3.0. El procedimiento de asignación del mapa
de afinidad consistió en especificar cada uno de los distintos tipos de átomos
de los ligandos, así como la especificación del volumen a analizar de la enzima,
cuya función radicó en seleccionar el sitio activo de la AChE y calcular para
cada una de ellas la afinidad por cada átomo. Los archivos de este proceso
fueron guardados en formato *.gpf utilizando el mismo programa.
La ejecución del programa AutoDock requirió la estructura de la enzima en
formato *.pdbqs con sus respectivos mapas de afinidad; las estructuras de los
ligandos en formato *.pdbq y un archivo de parámetros *.dpf (éste se generó en
el programa AutoDockTools). Para iniciar el estudio de simulación de la
interacción ligando-receptor se utilizó el Algoritmo Genético Lamarckiano
implementado en AutoDock. Los parámetros utilizados fueron los siguientes:
100 corridas, 100 individuos en la población, 1x107 evaluaciones de energía y
27 000 generaciones.
El Autodock generó un archivo de salida *.dlg, el cual contenía las
conformaciones y energías libres de unión para cada una de las corridas
solicitadas. Esta información fue revisada usando el programa AutoDockTools,
en el cual también pudieron ser visualizadas las interacciones no covalentes y
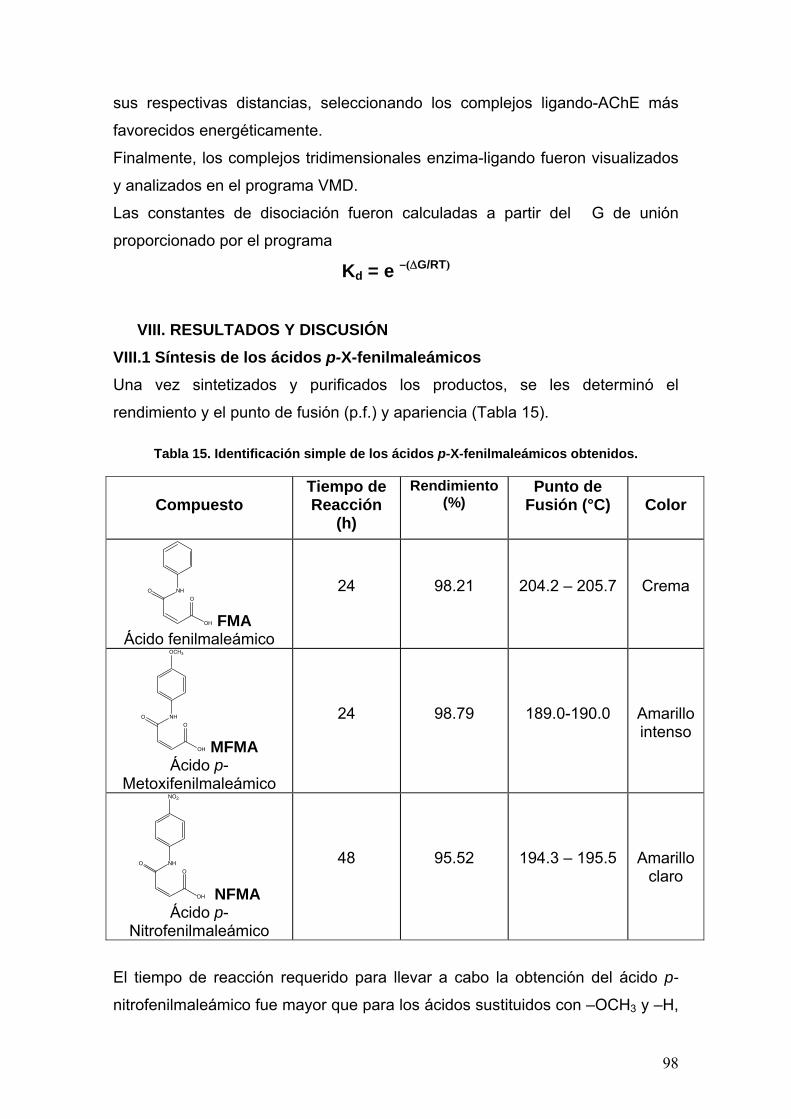
98
sus respectivas distancias, seleccionando los complejos ligando-AChE más
favorecidos energéticamente.
Finalmente, los complejos tridimensionales enzima-ligando fueron visualizados
y analizados en el programa VMD.
Las constantes de disociación fueron calculadas a partir del �G de unión
proporcionado por el programa
Kd = e –(ΔG/RT)
VIII. RESULTADOS Y DISCUSIÓN VIII.1 Síntesis de los ácidos p-X-fenilmaleámicos Una vez sintetizados y purificados los productos, se les determinó el
rendimiento y el punto de fusión (p.f.) y apariencia (Tabla 15).
Tabla 15. Identificación simple de los ácidos p-X-fenilmaleámicos obtenidos.
Compuesto
Tiempo de Reacción
(h)
Rendimiento (%)
Punto de Fusión (°C)
Color
NH
OH
OO
FMA Ácido fenilmaleámico
24
98.21
204.2 – 205.7
Crema
NH
OH
OO
OCH3
MFMA Ácido p-
Metoxifenilmaleámico
24
98.79
189.0-190.0
Amarillo intenso
NH
OH
OO
NO2
NFMA Ácido p-
Nitrofenilmaleámico
48
95.52
194.3 – 195.5
Amarillo claro
El tiempo de reacción requerido para llevar a cabo la obtención del ácido p-
nitrofenilmaleámico fue mayor que para los ácidos sustituidos con –OCH3 y –H,
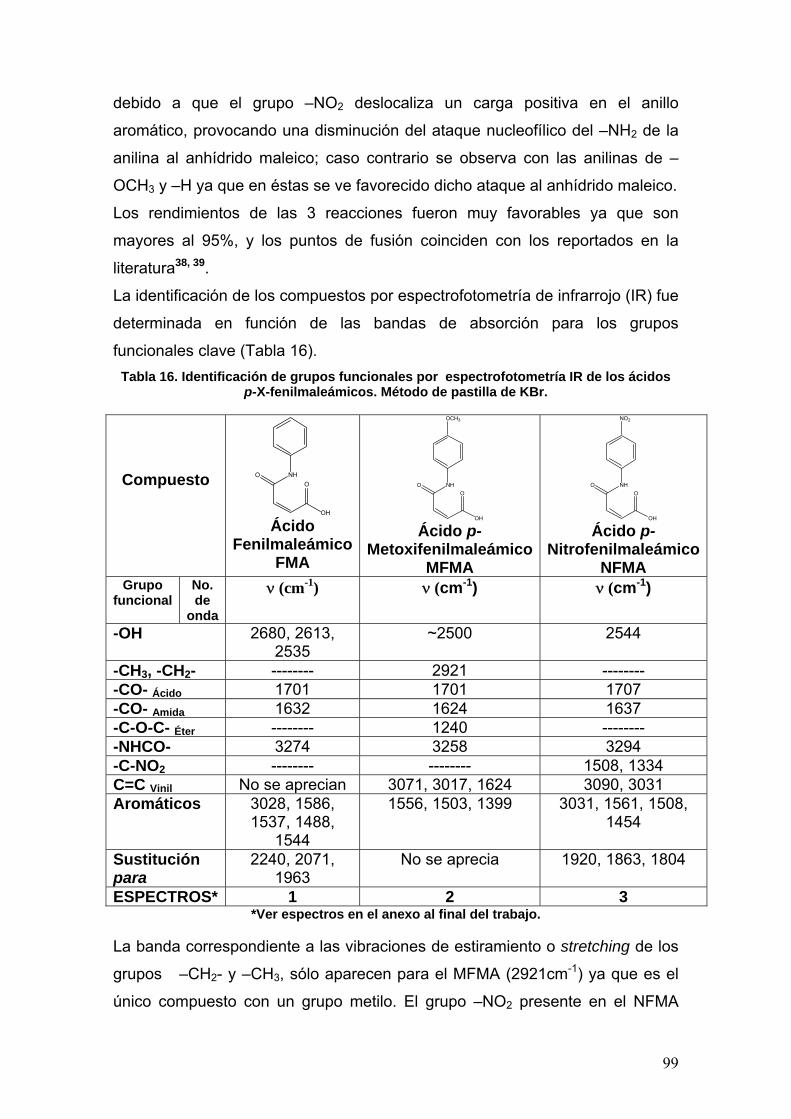
99
debido a que el grupo –NO2 deslocaliza un carga positiva en el anillo
aromático, provocando una disminución del ataque nucleofílico del –NH2 de la
anilina al anhídrido maleico; caso contrario se observa con las anilinas de –
OCH3 y –H ya que en éstas se ve favorecido dicho ataque al anhídrido maleico. Los rendimientos de las 3 reacciones fueron muy favorables ya que son
mayores al 95%, y los puntos de fusión coinciden con los reportados en la
literatura38, 39.
La identificación de los compuestos por espectrofotometría de infrarrojo (IR) fue
determinada en función de las bandas de absorción para los grupos
funcionales clave (Tabla 16). Tabla 16. Identificación de grupos funcionales por espectrofotometría IR de los ácidos
p-X-fenilmaleámicos. Método de pastilla de KBr.
Compuesto NH
OH
OO
Ácido
FenilmaleámicoFMA
NH
OH
OO
OCH3
Ácido p-
MetoxifenilmaleámicoMFMA
NH
OH
OO
NO2
Ácido p-
NitrofenilmaleámicoNFMA
Grupo funcional
No. de
onda
ν (cm-1)
ν (cm-1) ν (cm-1)
-OH 2680, 2613, 2535
~2500 2544
-CH3, -CH2- -------- 2921 -------- -CO- Ácido 1701 1701 1707 -CO- Amida 1632 1624 1637 -C-O-C- Éter -------- 1240 -------- -NHCO- 3274 3258 3294 -C-NO2 -------- -------- 1508, 1334 C=C Vinil No se aprecian 3071, 3017, 1624 3090, 3031 Aromáticos 3028, 1586,
1537, 1488, 1544
1556, 1503, 1399 3031, 1561, 1508, 1454
Sustitución para
2240, 2071, 1963
No se aprecia 1920, 1863, 1804
ESPECTROS* 1 2 3 *Ver espectros en el anexo al final del trabajo.
La banda correspondiente a las vibraciones de estiramiento o stretching de los
grupos –CH2- y –CH3, sólo aparecen para el MFMA (2921cm-1) ya que es el
único compuesto con un grupo metilo. El grupo –NO2 presente en el NFMA
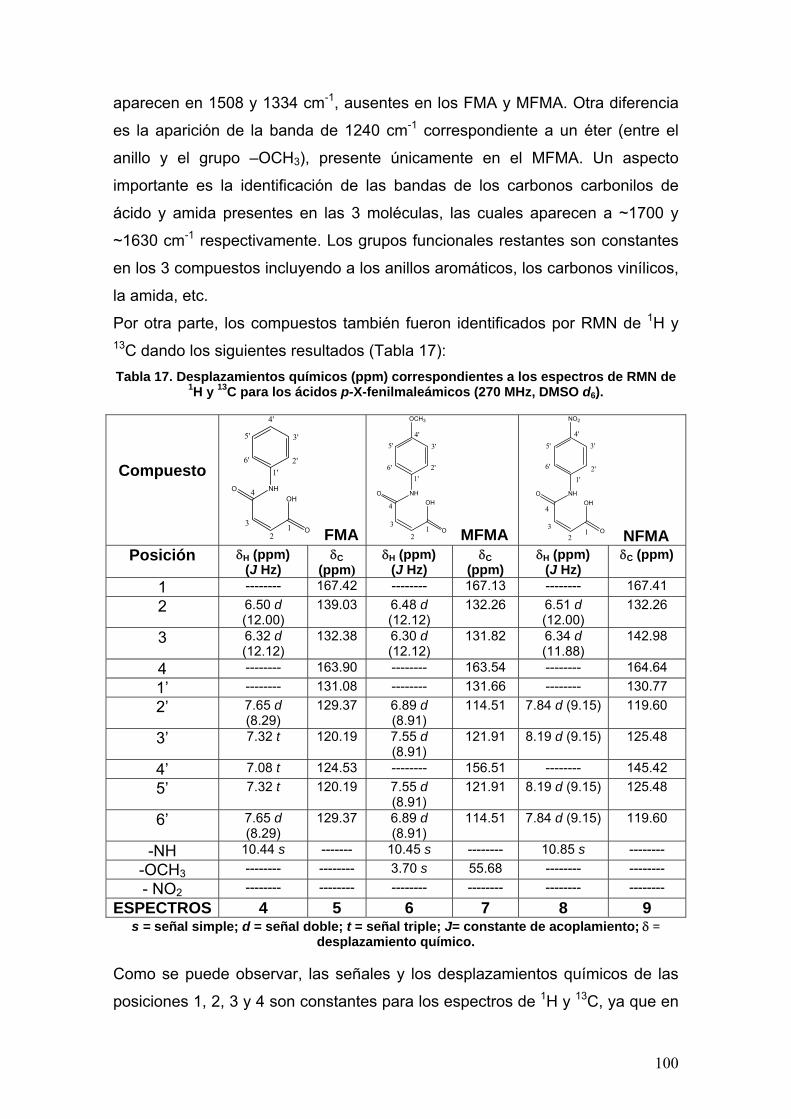
100
aparecen en 1508 y 1334 cm-1, ausentes en los FMA y MFMA. Otra diferencia
es la aparición de la banda de 1240 cm-1 correspondiente a un éter (entre el
anillo y el grupo –OCH3), presente únicamente en el MFMA. Un aspecto
importante es la identificación de las bandas de los carbonos carbonilos de
ácido y amida presentes en las 3 moléculas, las cuales aparecen a ~1700 y
~1630 cm-1 respectivamente. Los grupos funcionales restantes son constantes
en los 3 compuestos incluyendo a los anillos aromáticos, los carbonos vinílicos,
la amida, etc.
Por otra parte, los compuestos también fueron identificados por RMN de 1H y 13C dando los siguientes resultados (Tabla 17): Tabla 17. Desplazamientos químicos (ppm) correspondientes a los espectros de RMN de
1H y 13C para los ácidos p-X-fenilmaleámicos (270 MHz, DMSO d6).
Compuesto
NH
O
OOH
12
3
4
1'2'
3'
4'
5'
6'
FMA
NH
O
OOH
OCH3
12
3
4
1'2'
3'4'
5'
6'
MFMA
NH
O
OOH
NO2
12
3
4
1'2'
3'4'
5'
6'
NFMA Posición δH (ppm)
(J Hz) δC
(ppm) δH (ppm)
(J Hz) δC
(ppm) δH (ppm)
(J Hz) δC (ppm)
1 -------- 167.42 -------- 167.13 -------- 167.41 2 6.50 d
(12.00) 139.03 6.48 d
(12.12) 132.26 6.51 d
(12.00) 132.26
3 6.32 d (12.12)
132.38 6.30 d (12.12)
131.82 6.34 d (11.88)
142.98
4 -------- 163.90 -------- 163.54 -------- 164.64 1’ -------- 131.08 -------- 131.66 -------- 130.77 2’ 7.65 d
(8.29) 129.37 6.89 d
(8.91) 114.51 7.84 d (9.15) 119.60
3’ 7.32 t 120.19 7.55 d (8.91)
121.91 8.19 d (9.15) 125.48
4’ 7.08 t 124.53 -------- 156.51 -------- 145.42 5’ 7.32 t 120.19 7.55 d
(8.91) 121.91 8.19 d (9.15) 125.48
6’ 7.65 d (8.29)
129.37 6.89 d (8.91)
114.51 7.84 d (9.15) 119.60
-NH 10.44 s ------- 10.45 s -------- 10.85 s -------- -OCH3 -------- -------- 3.70 s 55.68 -------- -------- - NO2 -------- -------- -------- -------- -------- --------
ESPECTROS 4 5 6 7 8 9 s = señal simple; d = señal doble; t = señal triple; J= constante de acoplamiento; δ =
desplazamiento químico.
Como se puede observar, las señales y los desplazamientos químicos de las
posiciones 1, 2, 3 y 4 son constantes para los espectros de 1H y 13C, ya que en

101
esta porción de la molécula no se ve afectada por el sustituyente en el anillo
aromático. Es importante señalar que en esta porción las señales
correspondientes a los protones vinílicos dan una señal doble para cada protón
ya que se trata de un sistema acoplado, cuya constante es de J ≈ 12.00 Hz. El
carbono de la posición 1 (C-1) está ligeramente desplazado a campos más
bajos con respecto al carbono de la posición 4 (C-4), debido a que el C-1
corresponde al carbonilo del ácido que tiene átomo vecinos más
electronegativos que el C-4 correspondiente a la amida secundaria. El protón
correspondiente al –NH de la amida no difiere entre las moléculas y se
presenta como una señal simple entre 10 y 11 ppm.
Con respecto al anillo aromático, la diferencia más contundente entre las
moléculas se encuentra a partir de la posición 2’ hasta 6’. Las señales dobles (J
≈ 8-9 Hz) de las posiciones 2’ y 6’ no difieren significativamente en los 3
compuestos ya que no se ven muy afectadas por el sustituyente, por tal motivo
su señales son dobles con una J ≈ 8-9 Hz (acoplados con los protones de las
posiciones 3’ y 5’); sin embargo, la diferencia se encuentra en los
desplazamientos químicos, ya que el NFMA está más desplazado a campos
más bajos que los FMA y NFMA. El espectro de 1H de FMA presenta una señal
doble (2’-6’) correspondiente a dos protones aromáticos y 2 señales triples
correspondientes a los protones de las posiciones 3’-5’ (2 protones en el anillo)
y 4’ (protón asimétrico del anillo). Los espectros de MFMA y NFMA presentan
en posición 3’ y 5’ una señal doble acoplada con los protones de las posiciones
2’ y 6’ (J = 8-9 Hz). El MFMA presenta una señal simple que integra para 3
protones en 3.70 ppm, correspondiente a los hidrógenos del grupo metilo.
En relación a los espectros de 13C para la porción aromática, las diferencias
radican principalmente en las posiciones 3’, 4’ y 5’ ya que se ven afectadas por
el sustituyente. Evidentemente el FMA presenta en éstas posiciones
desplazamientos a campos altos con respecto a los MFMA y NMFA. Cabe
mencionar que MFMA presenta una señal del carbono del grupo metilo en el
espectro a 55.68 ppm.

102
VIII.2 Síntesis de las p-X-fenilisomaleimidas Los resultados de rendimiento, punto de fusión, tiempo de reacción, Rf y
apariencia de los compuestos p-X-fenilisomaleimidas se presentan en la Tabla
18.
Se puede apreciar en estos compuestos que el punto de fusión disminuyó
significativamente con respecto a los ácidos maleámicos correspondientes,
debido a que las interacciones intermoleculares entre las isomaleimidas son
menores a las que existen en los ácidos maleámicos. Esta variante estructural
reafirma la evidencia encontrada en los valores de Rf, ya que los 3 compuestos
son menos polares que los ácidos (por tal motivo existen menos interacciones).
Cabe mencionar que la polaridad de las isomaleimidas cambia en función del
grupo sustituyente (aumento de la polaridad): FIMI < MFIMI < NFIMI. La NFIMI
es la molécula más polar por que el grupo –NO2 tiene más cargas y se
encuentra en resonancia, por tal motivo su punto de fusión es más alto que la
FIMI y MFIMI debido a que la molécula tiene más oportunidades de
interaccionar con otras moléculas del mismo tipo.
En relación al tiempo de reacción, se utilizó como condición para las 3
reacciones un tiempo de 16 a 18 h para observar el efecto del sustituyente
sobre el rendimiento, y en efecto, la formación de NFIMI fue más lenta, ya que
se obtuvo un rendimiento de 28.68% debido a la deslocalización de una carga
positiva en el anillo inducida por el grupo –NO2 que debilita el ataque
nucleofílico del carbono carbonilo de la amida al carbonilo del ácido en la
formación del ciclo.
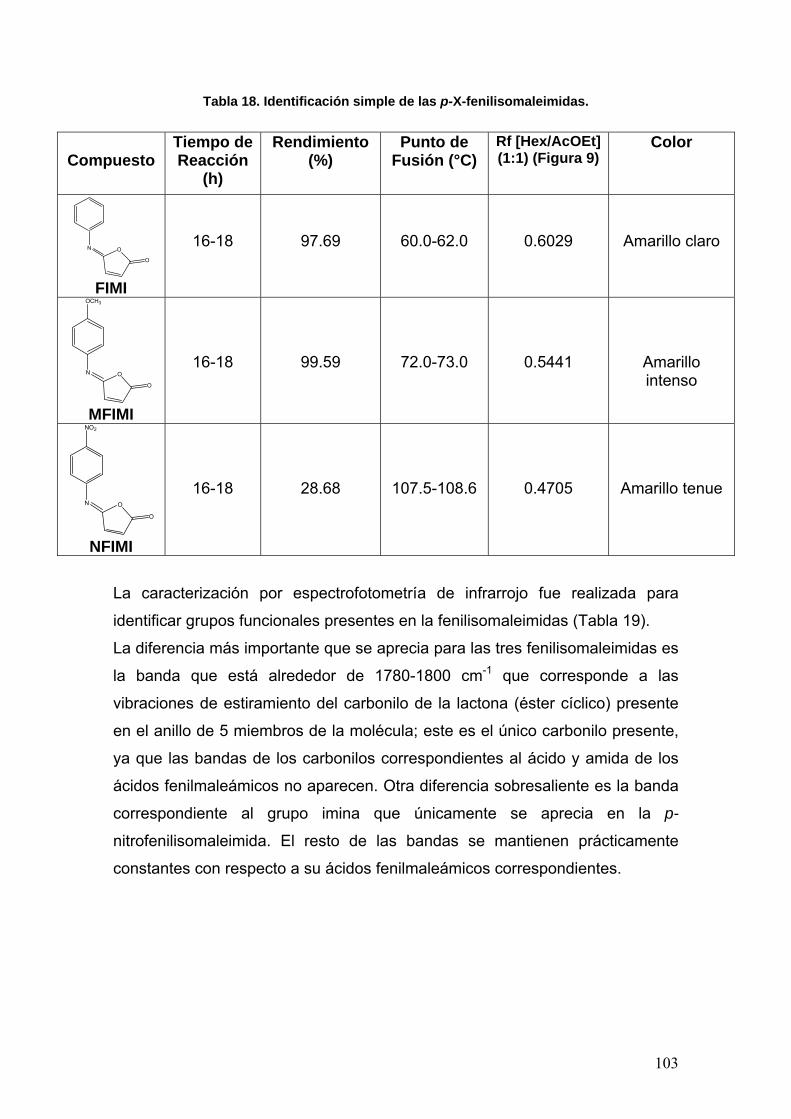
103
Tabla 18. Identificación simple de las p-X-fenilisomaleimidas.
La caracterización por espectrofotometría de infrarrojo fue realizada para
identificar grupos funcionales presentes en la fenilisomaleimidas (Tabla 19).
La diferencia más importante que se aprecia para las tres fenilisomaleimidas es
la banda que está alrededor de 1780-1800 cm-1 que corresponde a las
vibraciones de estiramiento del carbonilo de la lactona (éster cíclico) presente
en el anillo de 5 miembros de la molécula; este es el único carbonilo presente,
ya que las bandas de los carbonilos correspondientes al ácido y amida de los
ácidos fenilmaleámicos no aparecen. Otra diferencia sobresaliente es la banda
correspondiente al grupo imina que únicamente se aprecia en la p-
nitrofenilisomaleimida. El resto de las bandas se mantienen prácticamente
constantes con respecto a su ácidos fenilmaleámicos correspondientes.
Compuesto
Tiempo de Reacción
(h)
Rendimiento (%)
Punto de Fusión (°C)
Rf [Hex/AcOEt] (1:1) (Figura 9)
Color
N O
O
FIMI
16-18
97.69
60.0-62.0
0.6029
Amarillo claro
N O
O
OCH3
MFIMI
16-18
99.59
72.0-73.0
0.5441
Amarillo intenso
N O
O
NO2
NFIMI
16-18
28.68
107.5-108.6
0.4705
Amarillo tenue
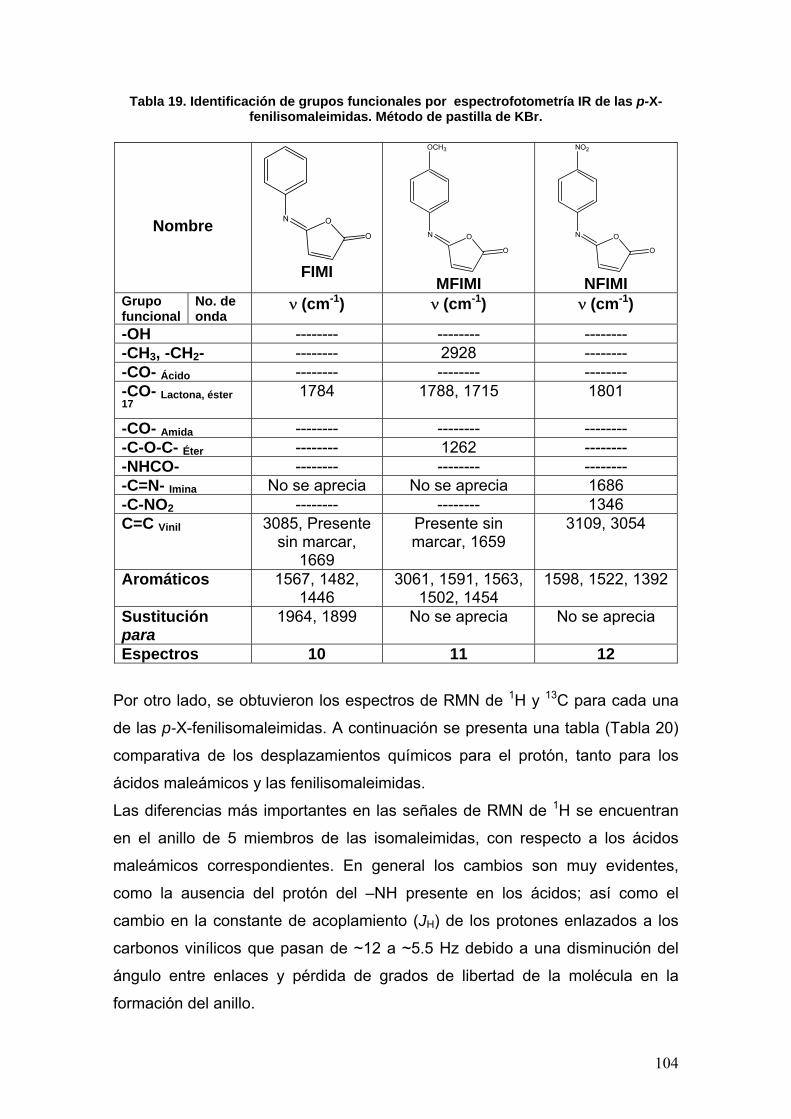
104
Tabla 19. Identificación de grupos funcionales por espectrofotometría IR de las p-X-fenilisomaleimidas. Método de pastilla de KBr.
Nombre N O
O
FIMI
N O
O
OCH3
MFIMI
N O
O
NO2
NFIMI
Grupo funcional
No. de onda
ν (cm-1) ν (cm-1) ν (cm-1)
-OH -------- -------- -------- -CH3, -CH2- -------- 2928 -------- -CO- Ácido -------- -------- -------- -CO- Lactona, éster 17
1784 1788, 1715 1801
-CO- Amida -------- -------- -------- -C-O-C- Éter -------- 1262 -------- -NHCO- -------- -------- -------- -C=N- Imina No se aprecia No se aprecia 1686 -C-NO2 -------- -------- 1346 C=C Vinil 3085, Presente
sin marcar, 1669
Presente sin marcar, 1659
3109, 3054
Aromáticos 1567, 1482, 1446
3061, 1591, 1563, 1502, 1454
1598, 1522, 1392
Sustitución para
1964, 1899 No se aprecia No se aprecia
Espectros 10 11 12
Por otro lado, se obtuvieron los espectros de RMN de 1H y 13C para cada una
de las p-X-fenilisomaleimidas. A continuación se presenta una tabla (Tabla 20)
comparativa de los desplazamientos químicos para el protón, tanto para los
ácidos maleámicos y las fenilisomaleimidas.
Las diferencias más importantes en las señales de RMN de 1H se encuentran
en el anillo de 5 miembros de las isomaleimidas, con respecto a los ácidos
maleámicos correspondientes. En general los cambios son muy evidentes,
como la ausencia del protón del –NH presente en los ácidos; así como el
cambio en la constante de acoplamiento (JH) de los protones enlazados a los
carbonos vinílicos que pasan de ~12 a ~5.5 Hz debido a una disminución del
ángulo entre enlaces y pérdida de grados de libertad de la molécula en la
formación del anillo.

105
En el caso particular de las señales de los protones del anillo aromático de
FIMI, sufren un cambio importante con respecto a FMA, el cual consta de la
desaparición de las señales triples con una consecuente formación de una
señal múltiple que se encuentra en 7.34-7.39 ppm. Cabe señalar que el protón
de la posición 3 se traslapa en la señal múltiple de los protones aromáticos de
las posiciones 2’, 3’, 5’ y 6’.
Las tres p-X-fenilisomaleimidas sufren un cambio importante en el
desplazamiento químico del protón de la posición 3 (anillo de 5 miembros) con
respecto a sus ácidos maleámicos correspondientes; pasando de valores de
~6.3 ppm a ~7.4 ppm debido a que este protón se ve más afectado por la
formación del anillo de 5 miembros y el grupo imina adyacente.
El resto de las señales permanecen prácticamente iguales para ambas
moléculas (ácidos maleámicos e isomaleimidas).
Con respecto a las señales de los espectros de 13C se tienen diferencias
importantes (Tabla 21) en las posiciones 2, 3, 4 y 1’ que son las más afectadas
en la formación del anillo de 5 miembros. Los carbonos de las posiciones C-2 y
C-4 en las tres moléculas (isomaleimidas) se desplazan a campos altos con
respecto a sus ácidos maleámicos, debido a la formación de la lactona a partir
del grupo carboxilo; sin embargo, sucede lo contrario en la posición 3, ya que la
señal del C-3 se desplaza a campos bajos debido al mismo fenómeno. El C-1’
se ve afectado de la misma manera (cambio de campos bajos a campos altos)
porque el nitrógeno perdió un protón y se formó la imina que conecta al anillo
de 5 miembros. Las señales correspondientes a los carbonos aromáticos y la
señal del carbono de –OCH3 no se ven afectadas.
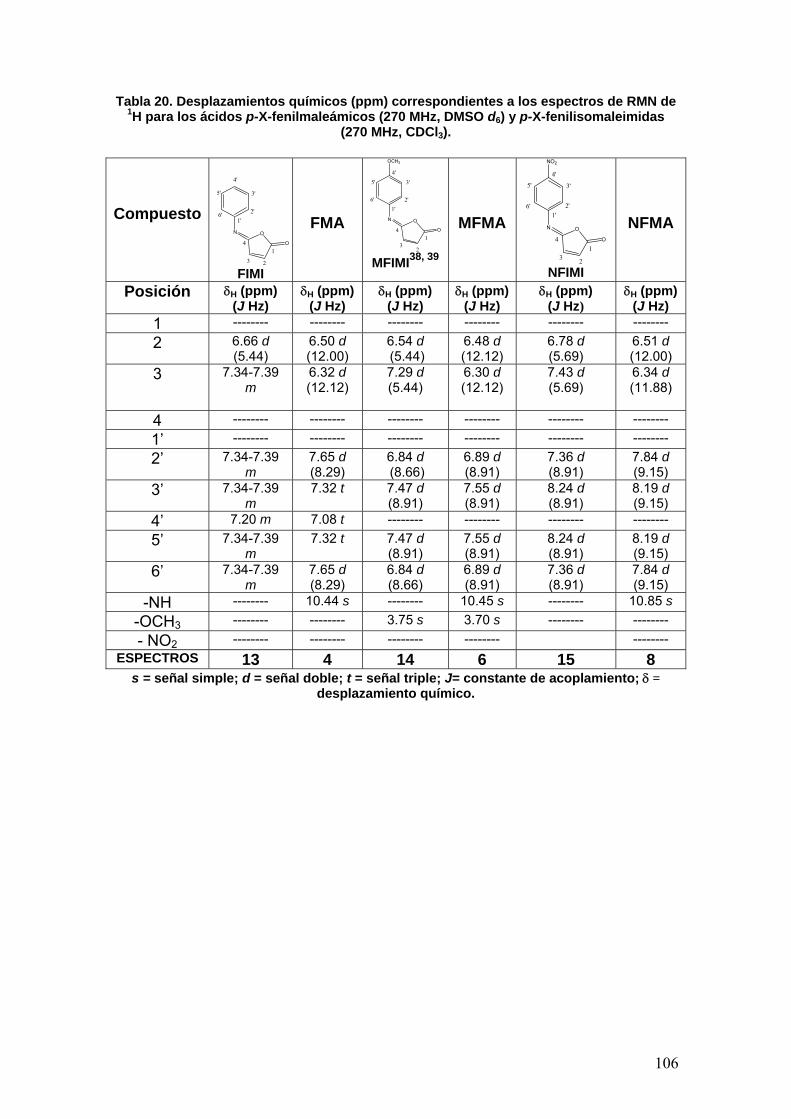
106
Tabla 20. Desplazamientos químicos (ppm) correspondientes a los espectros de RMN de 1H para los ácidos p-X-fenilmaleámicos (270 MHz, DMSO d6) y p-X-fenilisomaleimidas
(270 MHz, CDCl3).
Compuesto
N O
O1
23
4
1'2'
3'
4'
5'
6'
FIMI
FMA N O
O
OCH3
1
23
4
1'2'
4'
5' 3'
6'
MFIMI38, 39
MFMA N O
O
NO2
1
23
4
1'2'
3'
4'
5'
6'
NFIMI
NFMA
Posición δH (ppm) (J Hz)
δH (ppm) (J Hz)
δH (ppm) (J Hz)
δH (ppm) (J Hz)
δH (ppm) (J Hz)
δH (ppm) (J Hz)
1 -------- -------- -------- -------- -------- -------- 2 6.66 d
(5.44) 6.50 d (12.00)
6.54 d (5.44)
6.48 d (12.12)
6.78 d (5.69)
6.51 d (12.00)
3 7.34-7.39 m
6.32 d (12.12)
7.29 d (5.44)
6.30 d (12.12)
7.43 d (5.69)
6.34 d (11.88)
4 -------- -------- -------- -------- -------- -------- 1’ -------- -------- -------- -------- -------- -------- 2’ 7.34-7.39
m 7.65 d (8.29)
6.84 d (8.66)
6.89 d (8.91)
7.36 d (8.91)
7.84 d (9.15)
3’ 7.34-7.39 m
7.32 t 7.47 d (8.91)
7.55 d (8.91)
8.24 d (8.91)
8.19 d (9.15)
4’ 7.20 m 7.08 t -------- -------- -------- -------- 5’ 7.34-7.39
m 7.32 t 7.47 d
(8.91) 7.55 d (8.91)
8.24 d (8.91)
8.19 d (9.15)
6’ 7.34-7.39 m
7.65 d (8.29)
6.84 d (8.66)
6.89 d (8.91)
7.36 d (8.91)
7.84 d (9.15)
-NH -------- 10.44 s -------- 10.45 s -------- 10.85 s -OCH3 -------- -------- 3.75 s 3.70 s -------- -------- - NO2 -------- -------- -------- -------- --------
ESPECTROS 13 4 14 6 15 8 s = señal simple; d = señal doble; t = señal triple; J= constante de acoplamiento; δ =
desplazamiento químico.
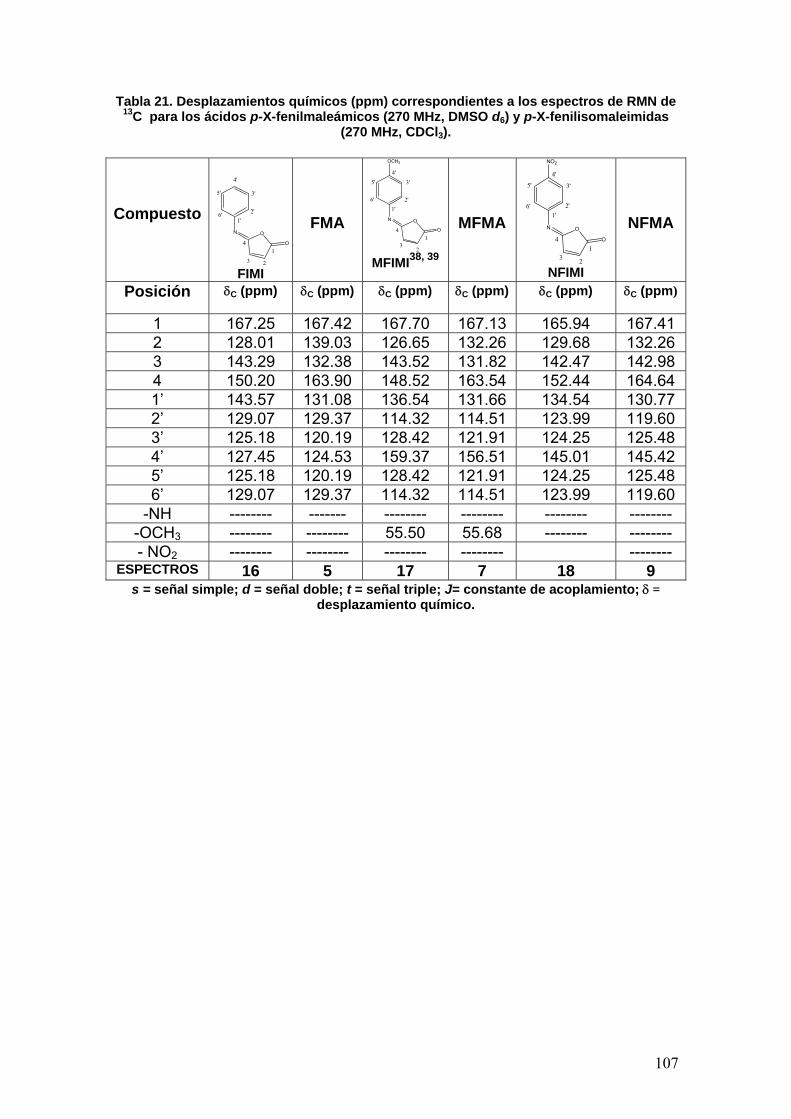
107
Tabla 21. Desplazamientos químicos (ppm) correspondientes a los espectros de RMN de 13C para los ácidos p-X-fenilmaleámicos (270 MHz, DMSO d6) y p-X-fenilisomaleimidas
(270 MHz, CDCl3).
Compuesto
N O
O1
23
4
1'2'
3'
4'
5'
6'
FIMI
FMA N O
O
OCH3
1
23
4
1'2'
4'
5' 3'
6'
MFIMI38, 39
MFMA N O
O
NO2
1
23
4
1'2'
3'
4'
5'
6'
NFIMI
NFMA
Posición δC (ppm) δC (ppm) δC (ppm) δC (ppm) δC (ppm)
δC (ppm)
1 167.25 167.42 167.70 167.13 165.94 167.41 2 128.01 139.03 126.65 132.26 129.68 132.26 3 143.29 132.38 143.52 131.82 142.47 142.98 4 150.20 163.90 148.52 163.54 152.44 164.64 1’ 143.57 131.08 136.54 131.66 134.54 130.77 2’ 129.07 129.37 114.32 114.51 123.99 119.60 3’ 125.18 120.19 128.42 121.91 124.25 125.48 4’ 127.45 124.53 159.37 156.51 145.01 145.42 5’ 125.18 120.19 128.42 121.91 124.25 125.48 6’ 129.07 129.37 114.32 114.51 123.99 119.60
-NH -------- ------- -------- -------- -------- -------- -OCH3 -------- -------- 55.50 55.68 -------- -------- - NO2 -------- -------- -------- -------- --------
ESPECTROS 16 5 17 7 18 9 s = señal simple; d = señal doble; t = señal triple; J= constante de acoplamiento; δ =
desplazamiento químico.
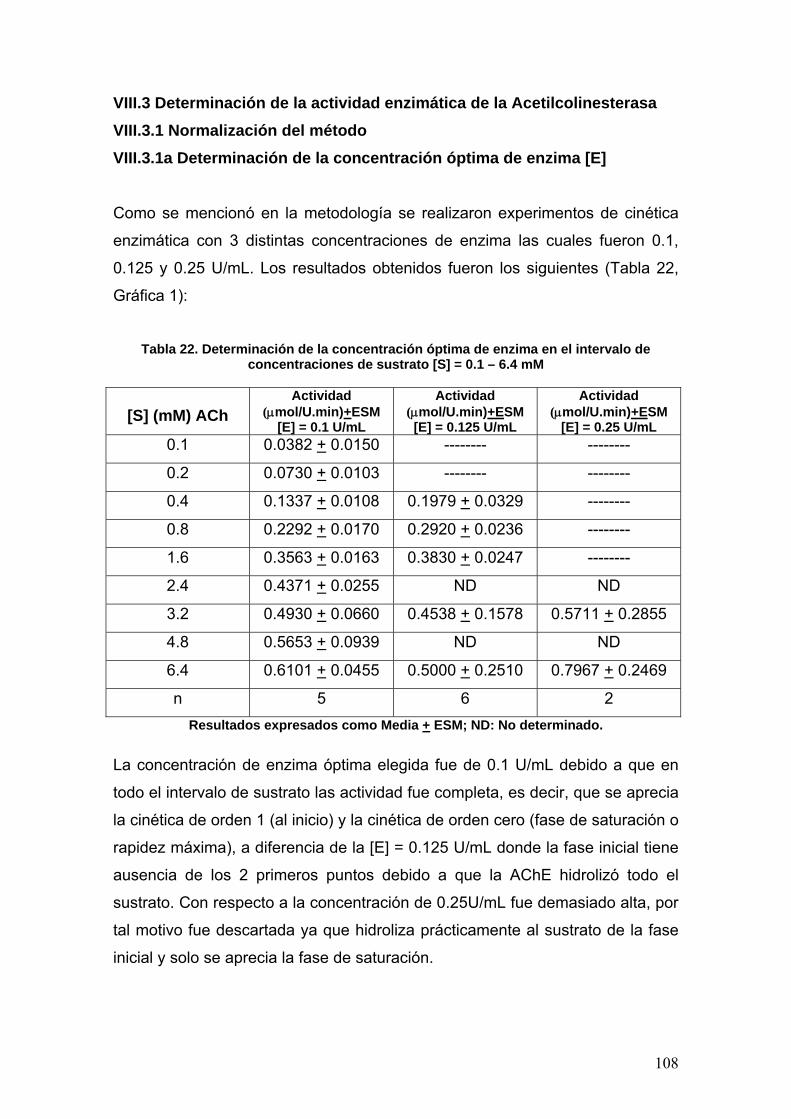
108
VIII.3 Determinación de la actividad enzimática de la Acetilcolinesterasa VIII.3.1 Normalización del método VIII.3.1a Determinación de la concentración óptima de enzima [E]
Como se mencionó en la metodología se realizaron experimentos de cinética
enzimática con 3 distintas concentraciones de enzima las cuales fueron 0.1,
0.125 y 0.25 U/mL. Los resultados obtenidos fueron los siguientes (Tabla 22,
Gráfica 1):
Tabla 22. Determinación de la concentración óptima de enzima en el intervalo de
concentraciones de sustrato [S] = 0.1 – 6.4 mM
[S] (mM) ACh
Actividad (μmol/U.min)+ESM
[E] = 0.1 U/mL
Actividad (μmol/U.min)+ESM [E] = 0.125 U/mL
Actividad (μmol/U.min)+ESM
[E] = 0.25 U/mL 0.1 0.0382 + 0.0150 -------- --------
0.2 0.0730 + 0.0103 -------- --------
0.4 0.1337 + 0.0108 0.1979 + 0.0329 --------
0.8 0.2292 + 0.0170 0.2920 + 0.0236 --------
1.6 0.3563 + 0.0163 0.3830 + 0.0247 --------
2.4 0.4371 + 0.0255 ND ND
3.2 0.4930 + 0.0660 0.4538 + 0.1578 0.5711 + 0.2855
4.8 0.5653 + 0.0939 ND ND
6.4 0.6101 + 0.0455 0.5000 + 0.2510 0.7967 + 0.2469
n 5 6 2 Resultados expresados como Media + ESM; ND: No determinado.
La concentración de enzima óptima elegida fue de 0.1 U/mL debido a que en
todo el intervalo de sustrato las actividad fue completa, es decir, que se aprecia
la cinética de orden 1 (al inicio) y la cinética de orden cero (fase de saturación o
rapidez máxima), a diferencia de la [E] = 0.125 U/mL donde la fase inicial tiene
ausencia de los 2 primeros puntos debido a que la AChE hidrolizó todo el
sustrato. Con respecto a la concentración de 0.25U/mL fue demasiado alta, por
tal motivo fue descartada ya que hidroliza prácticamente al sustrato de la fase
inicial y solo se aprecia la fase de saturación.
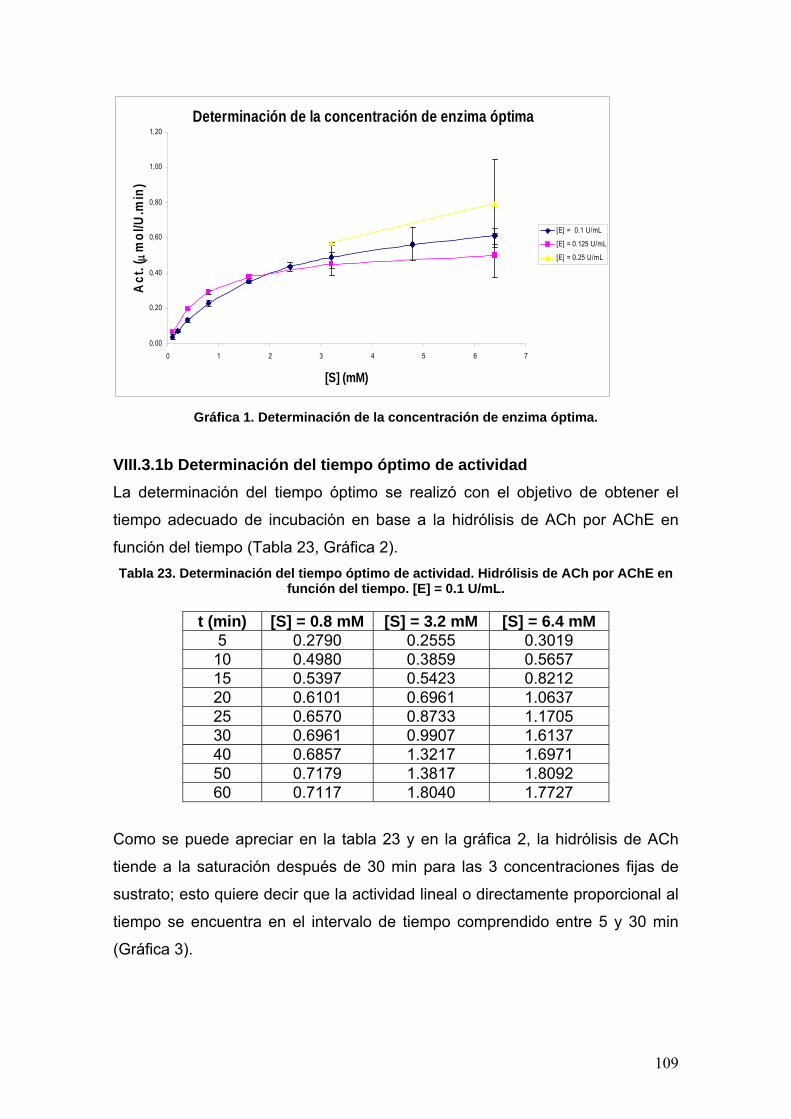
109
Determinación de la concentración de enzima óptima
0,00
0,20
0,40
0,60
0,80
1,00
1,20
0 1 2 3 4 5 6 7
[S] (mM)
Act
. (μ
mol
/U.m
in)
[E] = 0.1 U/mL
[E] = 0.125 U/mL
[E] = 0.25 U/mL
Gráfica 1. Determinación de la concentración de enzima óptima.
VIII.3.1b Determinación del tiempo óptimo de actividad La determinación del tiempo óptimo se realizó con el objetivo de obtener el
tiempo adecuado de incubación en base a la hidrólisis de ACh por AChE en
función del tiempo (Tabla 23, Gráfica 2). Tabla 23. Determinación del tiempo óptimo de actividad. Hidrólisis de ACh por AChE en
función del tiempo. [E] = 0.1 U/mL.
t (min) [S] = 0.8 mM [S] = 3.2 mM [S] = 6.4 mM 5 0.2790 0.2555 0.3019 10 0.4980 0.3859 0.5657 15 0.5397 0.5423 0.8212 20 0.6101 0.6961 1.0637 25 0.6570 0.8733 1.1705 30 0.6961 0.9907 1.6137 40 0.6857 1.3217 1.6971 50 0.7179 1.3817 1.8092 60 0.7117 1.8040 1.7727
Como se puede apreciar en la tabla 23 y en la gráfica 2, la hidrólisis de ACh
tiende a la saturación después de 30 min para las 3 concentraciones fijas de
sustrato; esto quiere decir que la actividad lineal o directamente proporcional al
tiempo se encuentra en el intervalo de tiempo comprendido entre 5 y 30 min
(Gráfica 3).
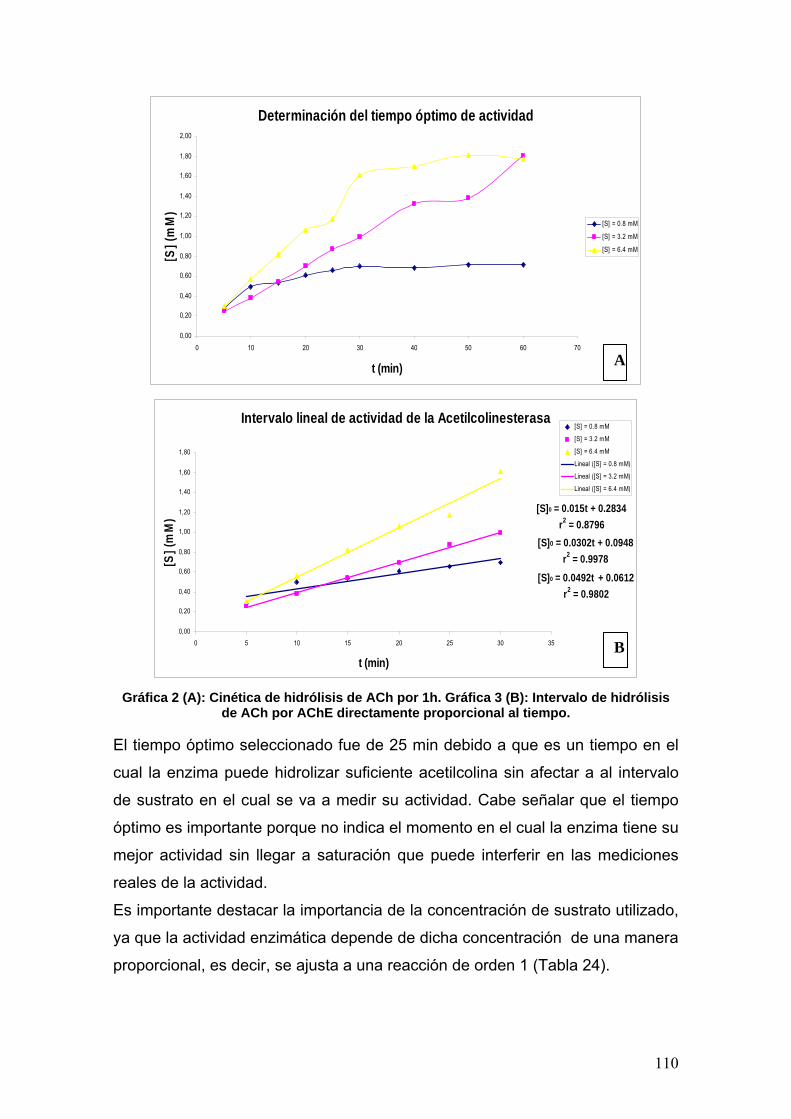
110
Determinación del tiempo óptimo de actividad
0,00
0,20
0,40
0,60
0,80
1,00
1,20
1,40
1,60
1,80
2,00
0 10 20 30 40 50 60 70
t (min)
[S] (
mM
)
[S] = 0.8 mM
[S] = 3.2 mM
[S] = 6.4 mM
Intervalo lineal de actividad de la Acetilcolinesterasa
[S]0 = 0.015t + 0.2834r2 = 0.8796
[S]0 = 0.0302t + 0.0948r2 = 0.9978
[S]0 = 0.0492t + 0.0612r2 = 0.9802
0,00
0,20
0,40
0,60
0,80
1,00
1,20
1,40
1,60
1,80
0 5 10 15 20 25 30 35
t (min)
[S] (
mM
)
[S] = 0.8 mM
[S] = 3.2 mM
[S] = 6.4 mM
Lineal ([S] = 0.8 mM)
Lineal ([S] = 3.2 mM)
Lineal ([S] = 6.4 mM)
Gráfica 2 (A): Cinética de hidrólisis de ACh por 1h. Gráfica 3 (B): Intervalo de hidrólisis
de ACh por AChE directamente proporcional al tiempo.
El tiempo óptimo seleccionado fue de 25 min debido a que es un tiempo en el
cual la enzima puede hidrolizar suficiente acetilcolina sin afectar a al intervalo
de sustrato en el cual se va a medir su actividad. Cabe señalar que el tiempo
óptimo es importante porque no indica el momento en el cual la enzima tiene su
mejor actividad sin llegar a saturación que puede interferir en las mediciones
reales de la actividad.
Es importante destacar la importancia de la concentración de sustrato utilizado,
ya que la actividad enzimática depende de dicha concentración de una manera
proporcional, es decir, se ajusta a una reacción de orden 1 (Tabla 24).
A
B
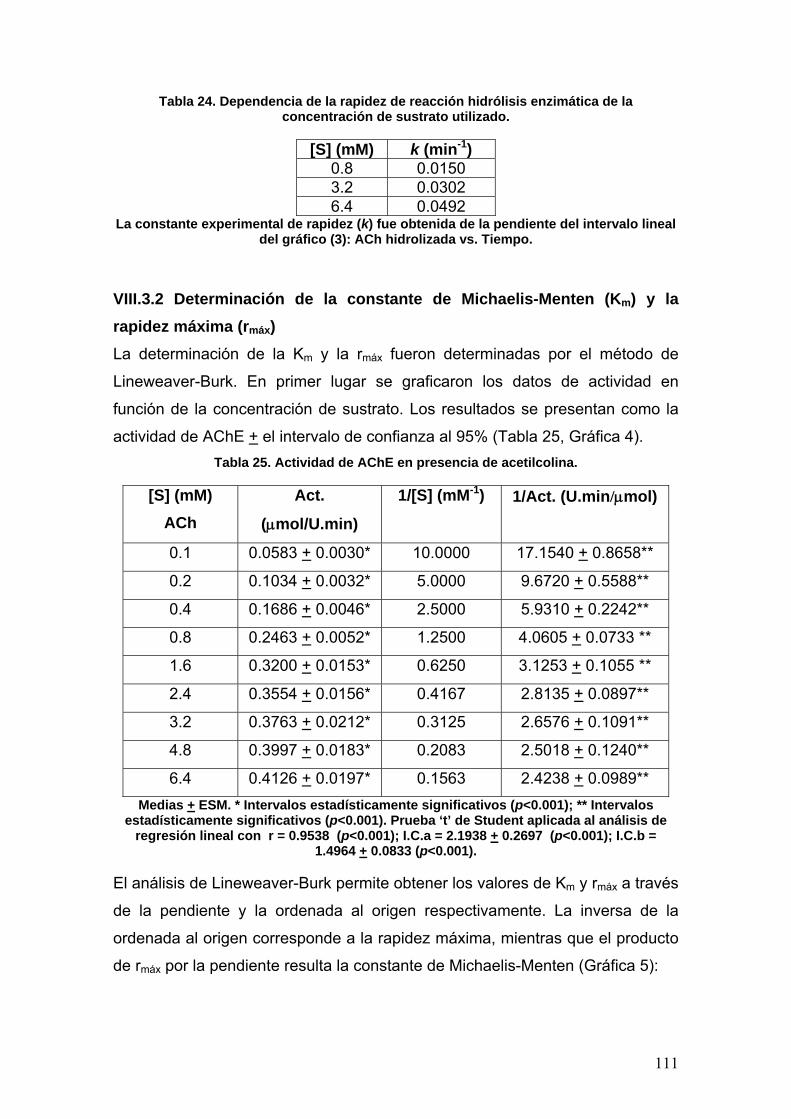
111
Tabla 24. Dependencia de la rapidez de reacción hidrólisis enzimática de la concentración de sustrato utilizado.
[S] (mM) k (min-1)
0.8 0.0150 3.2 0.0302 6.4 0.0492
La constante experimental de rapidez (k) fue obtenida de la pendiente del intervalo lineal del gráfico (3): ACh hidrolizada vs. Tiempo.
VIII.3.2 Determinación de la constante de Michaelis-Menten (Km) y la rapidez máxima (rmáx) La determinación de la Km y la rmáx fueron determinadas por el método de
Lineweaver-Burk. En primer lugar se graficaron los datos de actividad en
función de la concentración de sustrato. Los resultados se presentan como la
actividad de AChE + el intervalo de confianza al 95% (Tabla 25, Gráfica 4). Tabla 25. Actividad de AChE en presencia de acetilcolina.
[S] (mM)
ACh Act.
(μmol/U.min)
1/[S] (mM-1) 1/Act. (U.min/μmol)
0.1 0.0583 + 0.0030* 10.0000 17.1540 + 0.8658**
0.2 0.1034 + 0.0032* 5.0000 9.6720 + 0.5588**
0.4 0.1686 + 0.0046* 2.5000 5.9310 + 0.2242**
0.8 0.2463 + 0.0052* 1.2500 4.0605 + 0.0733 **
1.6 0.3200 + 0.0153* 0.6250 3.1253 + 0.1055 **
2.4 0.3554 + 0.0156* 0.4167 2.8135 + 0.0897**
3.2 0.3763 + 0.0212* 0.3125 2.6576 + 0.1091**
4.8 0.3997 + 0.0183* 0.2083 2.5018 + 0.1240**
6.4 0.4126 + 0.0197* 0.1563 2.4238 + 0.0989** Medias + ESM. * Intervalos estadísticamente significativos (p<0.001); ** Intervalos
estadísticamente significativos (p<0.001). Prueba ‘t’ de Student aplicada al análisis de regresión lineal con r = 0.9538 (p<0.001); I.C.a = 2.1938 + 0.2697 (p<0.001); I.C.b =
1.4964 + 0.0833 (p<0.001).
El análisis de Lineweaver-Burk permite obtener los valores de Km y rmáx a través
de la pendiente y la ordenada al origen respectivamente. La inversa de la
ordenada al origen corresponde a la rapidez máxima, mientras que el producto
de rmáx por la pendiente resulta la constante de Michaelis-Menten (Gráfica 5):
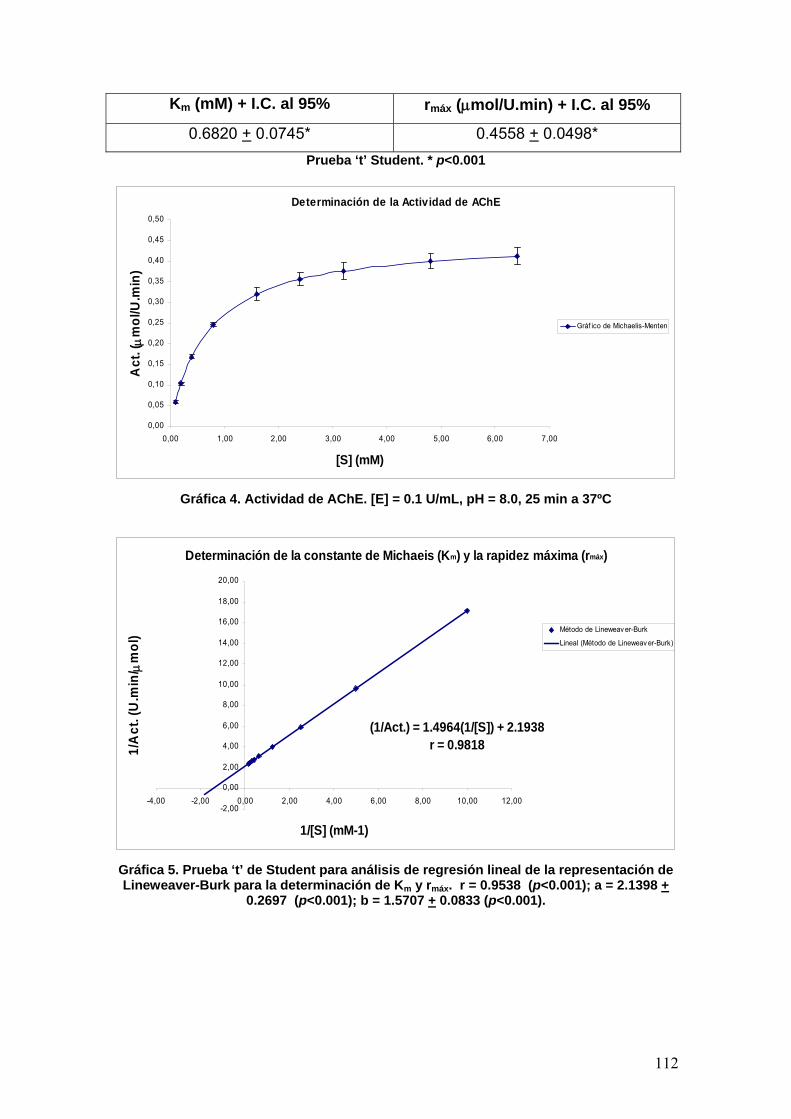
112
Km (mM) + I.C. al 95% rmáx (μmol/U.min) + I.C. al 95%
0.6820 + 0.0745* 0.4558 + 0.0498* Prueba ‘t’ Student. * p<0.001
Determinación de la Actividad de AChE
0,00
0,05
0,10
0,15
0,20
0,25
0,30
0,35
0,40
0,45
0,50
0,00 1,00 2,00 3,00 4,00 5,00 6,00 7,00
[S] (mM)
Act
. (μ
mol
/U.m
in)
Gráf ico de Michaelis-Menten
Gráfica 4. Actividad de AChE. [E] = 0.1 U/mL, pH = 8.0, 25 min a 37ºC
Determinación de la constante de Michaeis (Km) y la rapidez máxima (rmáx)
(1/Act.) = 1.4964(1/[S]) + 2.1938r = 0.9818
-2,00
0,00
2,00
4,00
6,00
8,00
10,00
12,00
14,00
16,00
18,00
20,00
-4,00 -2,00 0,00 2,00 4,00 6,00 8,00 10,00 12,00
1/[S] (mM-1)
1/A
ct. (
U.m
in/ μ
mol
) Método de Lineweav er-Burk
Lineal (Método de Lineweav er-Burk)
Gráfica 5. Prueba ‘t’ de Student para análisis de regresión lineal de la representación de Lineweaver-Burk para la determinación de Km y rmáx. r = 0.9538 (p<0.001); a = 2.1398 +
0.2697 (p<0.001); b = 1.5707 + 0.0833 (p<0.001).
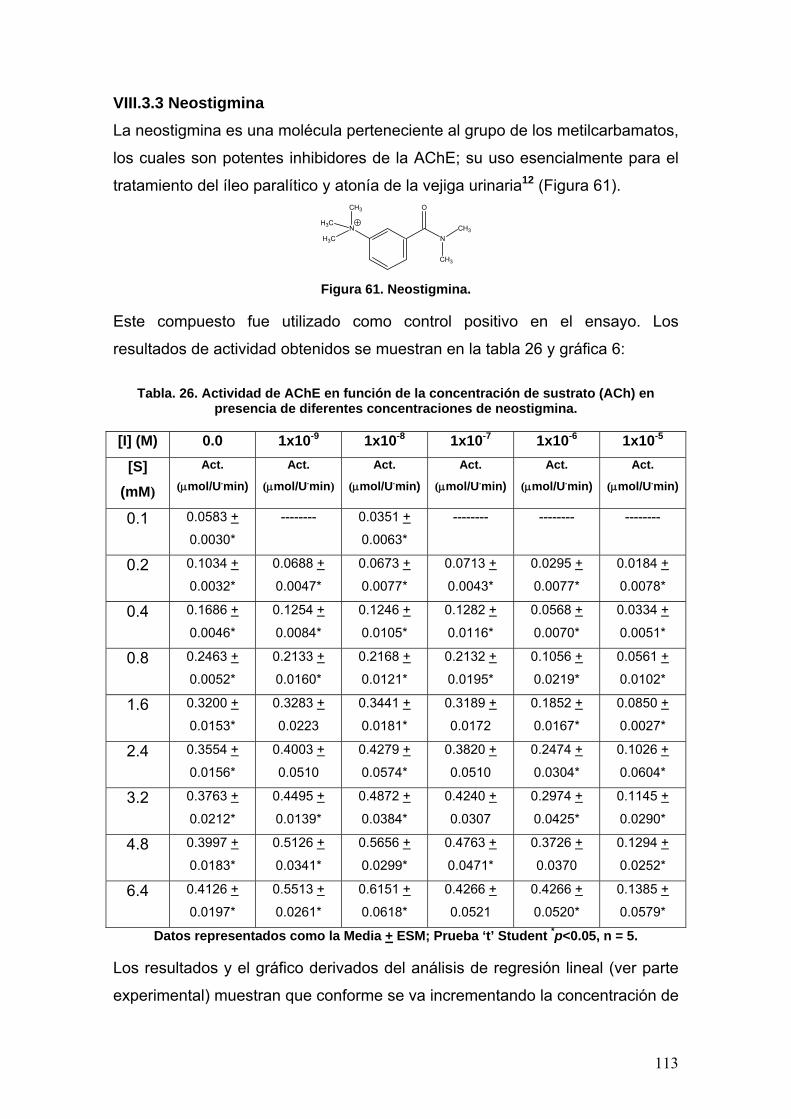
113
VIII.3.3 Neostigmina La neostigmina es una molécula perteneciente al grupo de los metilcarbamatos,
los cuales son potentes inhibidores de la AChE; su uso esencialmente para el
tratamiento del íleo paralítico y atonía de la vejiga urinaria12 (Figura 61).
N
O
CH3
CH3
NH3C
CH3
H3C
Figura 61. Neostigmina.
Este compuesto fue utilizado como control positivo en el ensayo. Los
resultados de actividad obtenidos se muestran en la tabla 26 y gráfica 6:
Tabla. 26. Actividad de AChE en función de la concentración de sustrato (ACh) en
presencia de diferentes concentraciones de neostigmina.
[I] (M) 0.0 1x10-9 1x10-8 1x10-7 1x10-6 1x10-5
[S] (mM)
Act.
(μmol/U.min)
Act.
(μmol/U.min)
Act.
(μmol/U.min)
Act.
(μmol/U.min)
Act.
(μmol/U.min)
Act.
(μmol/U.min)
0.1 0.0583 +
0.0030*
-------- 0.0351 +
0.0063*
-------- -------- --------
0.2 0.1034 +
0.0032*
0.0688 +
0.0047*
0.0673 +
0.0077*
0.0713 +
0.0043*
0.0295 +
0.0077*
0.0184 +
0.0078*
0.4 0.1686 +
0.0046*
0.1254 +
0.0084*
0.1246 +
0.0105*
0.1282 +
0.0116*
0.0568 +
0.0070*
0.0334 +
0.0051*
0.8 0.2463 +
0.0052*
0.2133 +
0.0160*
0.2168 +
0.0121*
0.2132 +
0.0195*
0.1056 +
0.0219*
0.0561 +
0.0102*
1.6 0.3200 +
0.0153*
0.3283 +
0.0223
0.3441 +
0.0181*
0.3189 +
0.0172
0.1852 +
0.0167*
0.0850 +
0.0027*
2.4 0.3554 +
0.0156*
0.4003 +
0.0510
0.4279 +
0.0574*
0.3820 +
0.0510
0.2474 +
0.0304*
0.1026 +
0.0604*
3.2 0.3763 +
0.0212*
0.4495 +
0.0139*
0.4872 +
0.0384*
0.4240 +
0.0307
0.2974 +
0.0425*
0.1145 +
0.0290*
4.8 0.3997 +
0.0183*
0.5126 +
0.0341*
0.5656 +
0.0299*
0.4763 +
0.0471*
0.3726 +
0.0370
0.1294 +
0.0252*
6.4 0.4126 +
0.0197*
0.5513 +
0.0261*
0.6151 +
0.0618*
0.4266 +
0.0521
0.4266 +
0.0520*
0.1385 +
0.0579*
Datos representados como la Media + ESM; Prueba ‘t’ Student *p<0.05, n = 5.
Los resultados y el gráfico derivados del análisis de regresión lineal (ver parte
experimental) muestran que conforme se va incrementando la concentración de
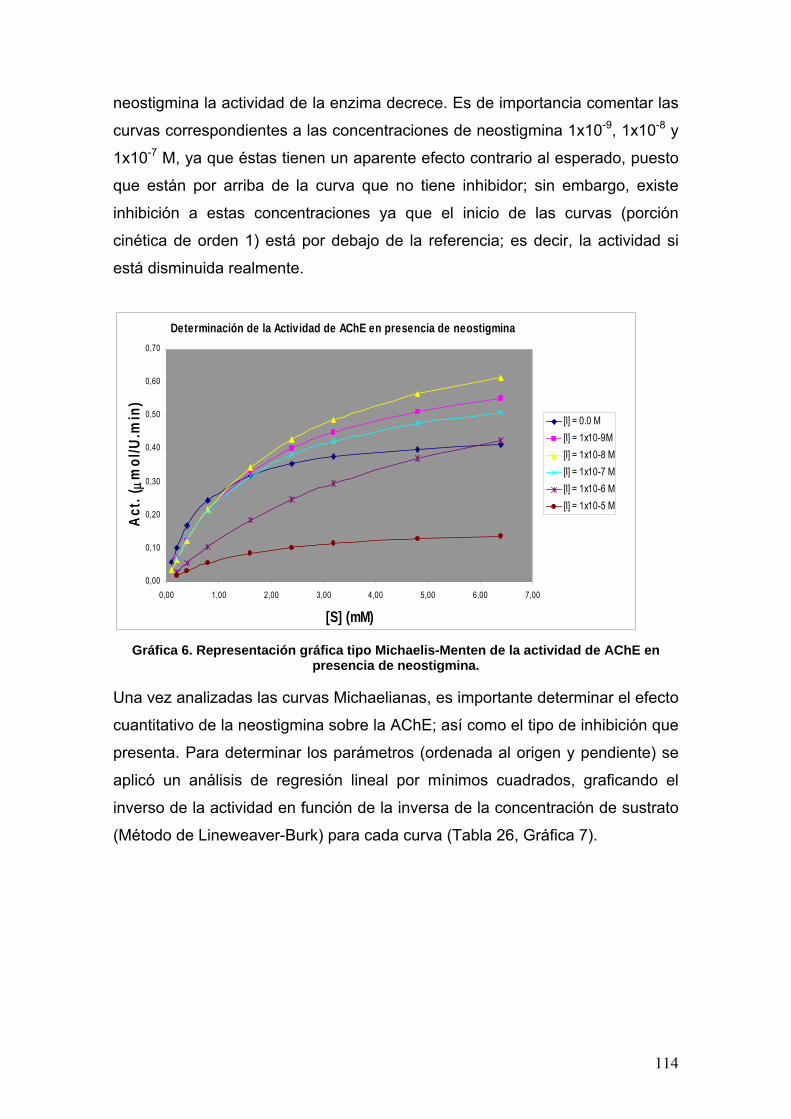
114
neostigmina la actividad de la enzima decrece. Es de importancia comentar las
curvas correspondientes a las concentraciones de neostigmina 1x10-9, 1x10-8 y
1x10-7 M, ya que éstas tienen un aparente efecto contrario al esperado, puesto
que están por arriba de la curva que no tiene inhibidor; sin embargo, existe
inhibición a estas concentraciones ya que el inicio de las curvas (porción
cinética de orden 1) está por debajo de la referencia; es decir, la actividad si
está disminuida realmente.
Determinación de la Actividad de AChE en presencia de neostigmina
0,00
0,10
0,20
0,30
0,40
0,50
0,60
0,70
0,00 1,00 2,00 3,00 4,00 5,00 6,00 7,00
[S] (mM)
Act
. (μm
ol/U
.min
)
[I] = 0.0 M[I] = 1x10-9M[I] = 1x10-8 M[I] = 1x10-7 M[I] = 1x10-6 M[I] = 1x10-5 M
Gráfica 6. Representación gráfica tipo Michaelis-Menten de la actividad de AChE en
presencia de neostigmina.
Una vez analizadas las curvas Michaelianas, es importante determinar el efecto
cuantitativo de la neostigmina sobre la AChE; así como el tipo de inhibición que
presenta. Para determinar los parámetros (ordenada al origen y pendiente) se
aplicó un análisis de regresión lineal por mínimos cuadrados, graficando el
inverso de la actividad en función de la inversa de la concentración de sustrato
(Método de Lineweaver-Burk) para cada curva (Tabla 26, Gráfica 7).
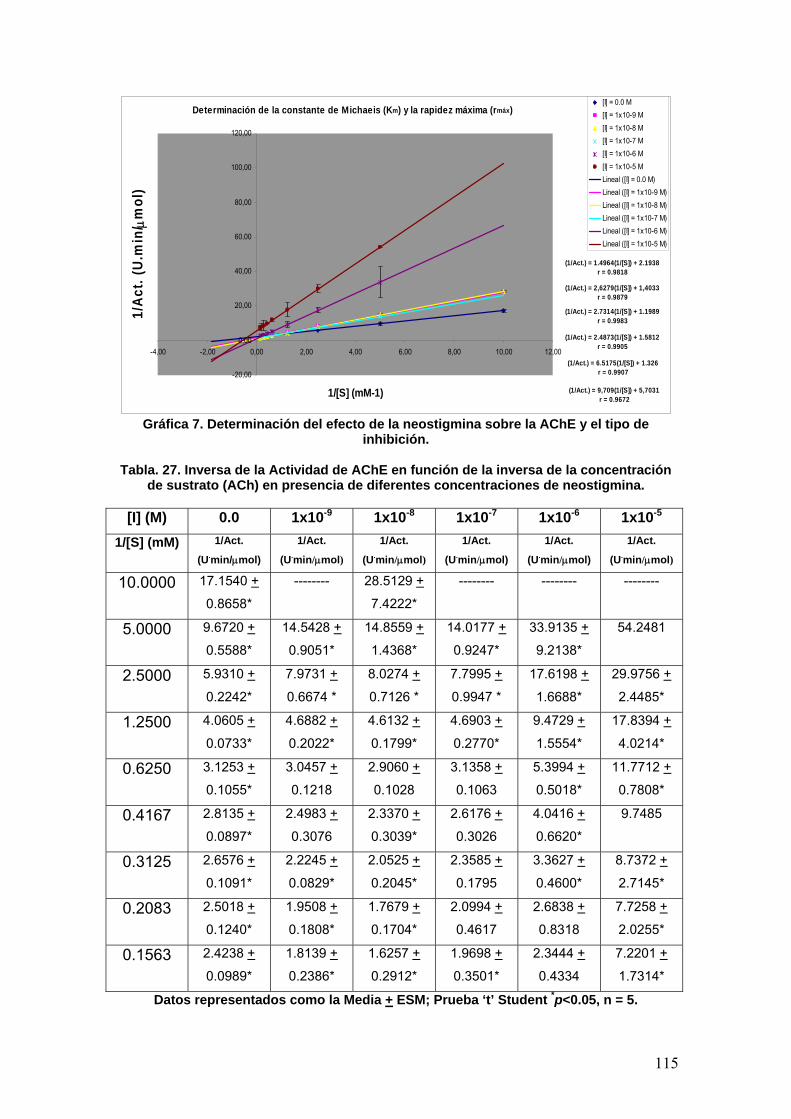
115
Determinación de la constante de Michaeis (Km) y la rapidez máxima (rmáx)
(1/Act.) = 1.4964(1/[S]) + 2.1938r = 0.9818
(1/Act.) = 2,6279(1/[S]) + 1,4033r = 0.9879
(1/Act.) = 2.7314(1/[S]) + 1.1989r = 0.9983
(1/Act.) = 2.4873(1/[S]) + 1.5812r = 0.9905
(1/Act.) = 6.5175(1/[S]) + 1.326r = 0.9907
(1/Act.) = 9,709(1/[S]) + 5,7031r = 0.9672
-20,00
0,00
20,00
40,00
60,00
80,00
100,00
120,00
-4,00 -2,00 0,00 2,00 4,00 6,00 8,00 10,00 12,00
1/[S] (mM-1)
1/A
ct. (
U.m
in/ μ
mol
)
[I] = 0.0 M[I] = 1x10-9 M[I] = 1x10-8 M[I] = 1x10-7 M[I] = 1x10-6 M[I] = 1x10-5 MLineal ([I] = 0.0 M)Lineal ([I] = 1x10-9 M)Lineal ([I] = 1x10-8 M)Lineal ([I] = 1x10-7 M)Lineal ([I] = 1x10-6 M)Lineal ([I] = 1x10-5 M)
Gráfica 7. Determinación del efecto de la neostigmina sobre la AChE y el tipo de
inhibición.
Tabla. 27. Inversa de la Actividad de AChE en función de la inversa de la concentración de sustrato (ACh) en presencia de diferentes concentraciones de neostigmina.
[I] (M) 0.0 1x10-9 1x10-8 1x10-7 1x10-6 1x10-5
1/[S] (mM) 1/Act.
(U.min/μmol)
1/Act.
(U.min/μmol)
1/Act.
(U.min/μmol)
1/Act.
(U.min/μmol)
1/Act.
(U.min/μmol)
1/Act.
(U.min/μmol)
10.0000 17.1540 +
0.8658*
-------- 28.5129 +
7.4222*
-------- -------- --------
5.0000 9.6720 +
0.5588*
14.5428 +
0.9051*
14.8559 +
1.4368*
14.0177 +
0.9247*
33.9135 +
9.2138*
54.2481
2.5000 5.9310 +
0.2242*
7.9731 +
0.6674 *
8.0274 +
0.7126 *
7.7995 +
0.9947 *
17.6198 +
1.6688*
29.9756 +
2.4485*
1.2500 4.0605 +
0.0733*
4.6882 +
0.2022*
4.6132 +
0.1799*
4.6903 +
0.2770*
9.4729 +
1.5554*
17.8394 +
4.0214*
0.6250 3.1253 +
0.1055*
3.0457 +
0.1218
2.9060 +
0.1028
3.1358 +
0.1063
5.3994 +
0.5018*
11.7712 +
0.7808*
0.4167 2.8135 +
0.0897*
2.4983 +
0.3076
2.3370 +
0.3039*
2.6176 +
0.3026
4.0416 +
0.6620*
9.7485
0.3125 2.6576 +
0.1091*
2.2245 +
0.0829*
2.0525 +
0.2045*
2.3585 +
0.1795
3.3627 +
0.4600*
8.7372 +
2.7145*
0.2083 2.5018 +
0.1240*
1.9508 +
0.1808*
1.7679 +
0.1704*
2.0994 +
0.4617
2.6838 +
0.8318
7.7258 +
2.0255*
0.1563 2.4238 +
0.0989*
1.8139 +
0.2386*
1.6257 +
0.2912*
1.9698 +
0.3501*
2.3444 +
0.4334
7.2201 +
1.7314*
Datos representados como la Media + ESM; Prueba ‘t’ Student *p<0.05, n = 5.
116
El gráfico de Lineweaver-Burk indica que el tipo de inhibición que posee la
neostigmina es de tipo competitivo ya que prácticamente todas las curvas
intersecan en el eje de las ordenadas, es decir, que la rapidez máxima no se ve
afectada por la presencia del inhibidor y únicamente la Km es afectada por el
factor � de una manera dosis dependiente.
Las regresiones lineales para cada curva ajustada fueron estadísticamente
significativas (p<0.001) con respecto a la curva que no tiene inhibidor
obteniéndose correlaciones lineales mayores a 0.96 (r>0.96). Las pendientes
fueron estadísticamente significativas (p<0.001) y las ordenadas al origen
difieren de cero (p<0.01) a excepción de la curva con concentración de
neostigmina 1x10-6 M. Cabe mencionar que las curvas correspondientes a las
concentraciones de neostigmina 1x10-9, 1x10-8 y 1x10-7 M no son significativas
entre ellas ya que los intervalos (media + ESM) se traslapan.
Una vez analizadas las curvas estadísticamente se determinó la constante de
inhibición de la neostigmina mediante el tratamiento matemático de Schild (ver
introducción). Se graficó el log [(mi/m0)-1] en función del log [I]; donde la
pendiente corresponde al coeficiente de Hill (nH) y la ordenada al origen a -
nHlogKi (Tabla 28, Gráfica 8).
mi = pendiente modificada por presencia de inhibidor o factor �
m0 = pendiente sin presencia de inhibidor
nH = coeficiente de Hill
Ki = constante de inhibición Tabla 28. Efecto de la presencia de neostigmina sobre la AChE. Procedimiento de Schild.
[I] (M) log [I] (M) mi mi/m0 log [m(mi/m0)-1]
0.0 -------- 1.4964 1.0000 --------
1x10-9 -9.00 2.6279 1.7561 -0.1214
1x10-8 -8.00 2.7314 1.8253 -0.0834
1x10-7* -7.00 2.4873 1.6622 -0.1790
1x10-6 -6.00 6.5175 4.355 0.5258
1x10-5 -5.00 9.7090 6.4882 0.7394 *La concentración de 1x10-7 M fue eliminado del tratamiento de Schild, ya que es un
punto que no afecta la regresión ya que no es estadísticamente significativo.

117
Determinación de la constante de inhibición de la neostigmina sobre AChE
log [(mi/m0) - 1]= 0,2331 log [I] + 1,8967r = 0,9817
-0,40
-0,20
0,00
0,20
0,40
0,60
0,80
-10,00 -9,00 -8,00 -7,00 -6,00 -5,00 -4,00 -3,00 -2,00 -1,00 0,00
log [I] (M)
log
[(mi /m
0 )-1]
Método de SchildLineal (Método de Schild)
Gráfica 8. Determinación de la constante de inhibición de la neostigmina mediante el
tratamiento de Schild.
Es importante destacar que esta línea recta fue analizada estadísticamente por
regresión lineal por mínimos cuadrados, la cual fue significativa (p<0.02) y r =
0.9817.
La constante de inhibición calculada fue de Ki = 7.28 x 10-9 M = 7.28 nM.
Ki = 7.28 + 4.76 nM La Ki reportada oscila entre 9.0 nM y 20 nM 45, 46.
La determinación de la constante de inhibición de la neostigmina es importante
desde el punto de vista metodológico, ya que el resultado obtenido coincide
con lo antes reportado.
VIII.3.4 Fenilisomaleimida Los resultados de actividad enzimática en presencia de FIMI se presentan en la
siguiente tabla (Tabla 29):
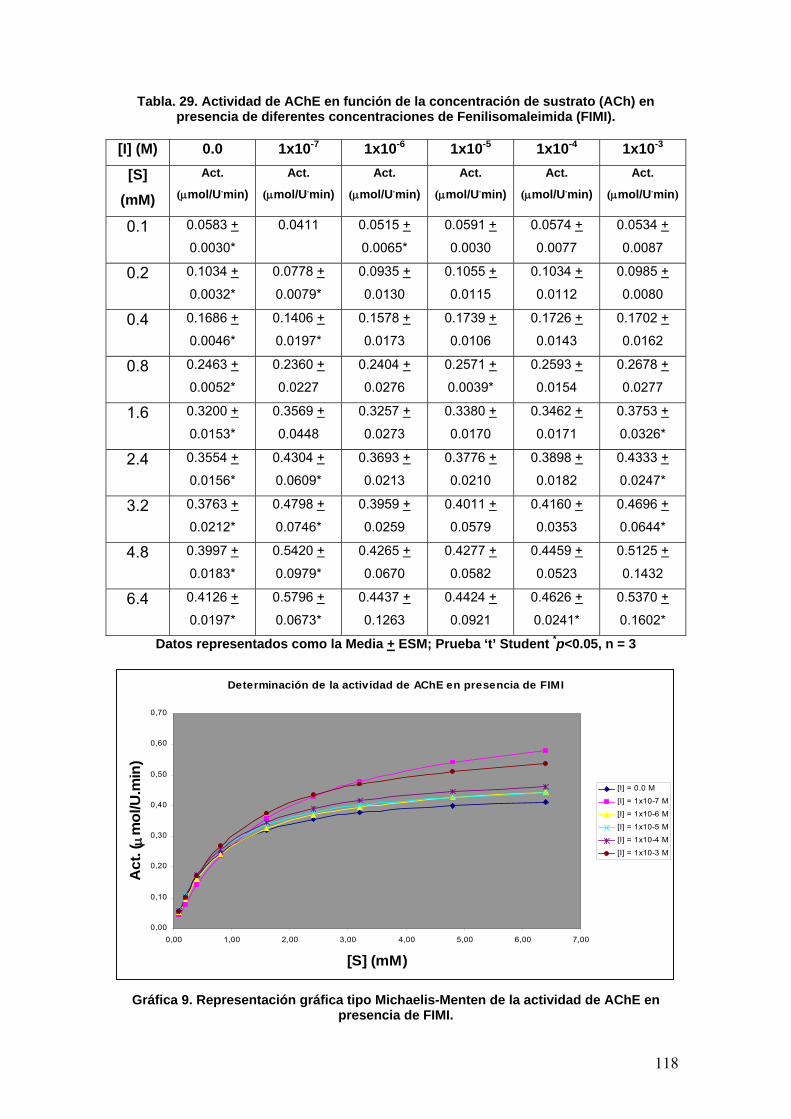
118
Tabla. 29. Actividad de AChE en función de la concentración de sustrato (ACh) en presencia de diferentes concentraciones de Fenilisomaleimida (FIMI).
[I] (M) 0.0 1x10-7 1x10-6 1x10-5 1x10-4 1x10-3
[S] (mM)
Act.
(μmol/U.min)
Act.
(μmol/U.min)
Act.
(μmol/U.min)
Act.
(μmol/U.min)
Act.
(μmol/U.min)
Act.
(μmol/U.min)
0.1 0.0583 +
0.0030*
0.0411 0.0515 +
0.0065*
0.0591 +
0.0030
0.0574 +
0.0077
0.0534 +
0.0087
0.2 0.1034 +
0.0032*
0.0778 +
0.0079*
0.0935 +
0.0130
0.1055 +
0.0115
0.1034 +
0.0112
0.0985 +
0.0080
0.4 0.1686 +
0.0046*
0.1406 +
0.0197*
0.1578 +
0.0173
0.1739 +
0.0106
0.1726 +
0.0143
0.1702 +
0.0162
0.8 0.2463 +
0.0052*
0.2360 +
0.0227
0.2404 +
0.0276
0.2571 +
0.0039*
0.2593 +
0.0154
0.2678 +
0.0277
1.6 0.3200 +
0.0153*
0.3569 +
0.0448
0.3257 +
0.0273
0.3380 +
0.0170
0.3462 +
0.0171
0.3753 +
0.0326*
2.4 0.3554 +
0.0156*
0.4304 +
0.0609*
0.3693 +
0.0213
0.3776 +
0.0210
0.3898 +
0.0182
0.4333 +
0.0247*
3.2 0.3763 +
0.0212*
0.4798 +
0.0746*
0.3959 +
0.0259
0.4011 +
0.0579
0.4160 +
0.0353
0.4696 +
0.0644*
4.8 0.3997 +
0.0183*
0.5420 +
0.0979*
0.4265 +
0.0670
0.4277 +
0.0582
0.4459 +
0.0523
0.5125 +
0.1432
6.4 0.4126 +
0.0197*
0.5796 +
0.0673*
0.4437 +
0.1263
0.4424 +
0.0921
0.4626 +
0.0241*
0.5370 +
0.1602*
Datos representados como la Media + ESM; Prueba ‘t’ Student *p<0.05, n = 3
Determinación de la actividad de AChE en presencia de FIMI
0,00
0,10
0,20
0,30
0,40
0,50
0,60
0,70
0,00 1,00 2,00 3,00 4,00 5,00 6,00 7,00
[S] (mM)
Act
. (μ
mol
/U.m
in)
[I] = 0.0 M
[I] = 1x10-7 M
[I] = 1x10-6 M
[I] = 1x10-5 M
[I] = 1x10-4 M
[I] = 1x10-3 M
Gráfica 9. Representación gráfica tipo Michaelis-Menten de la actividad de AChE en
presencia de FIMI.
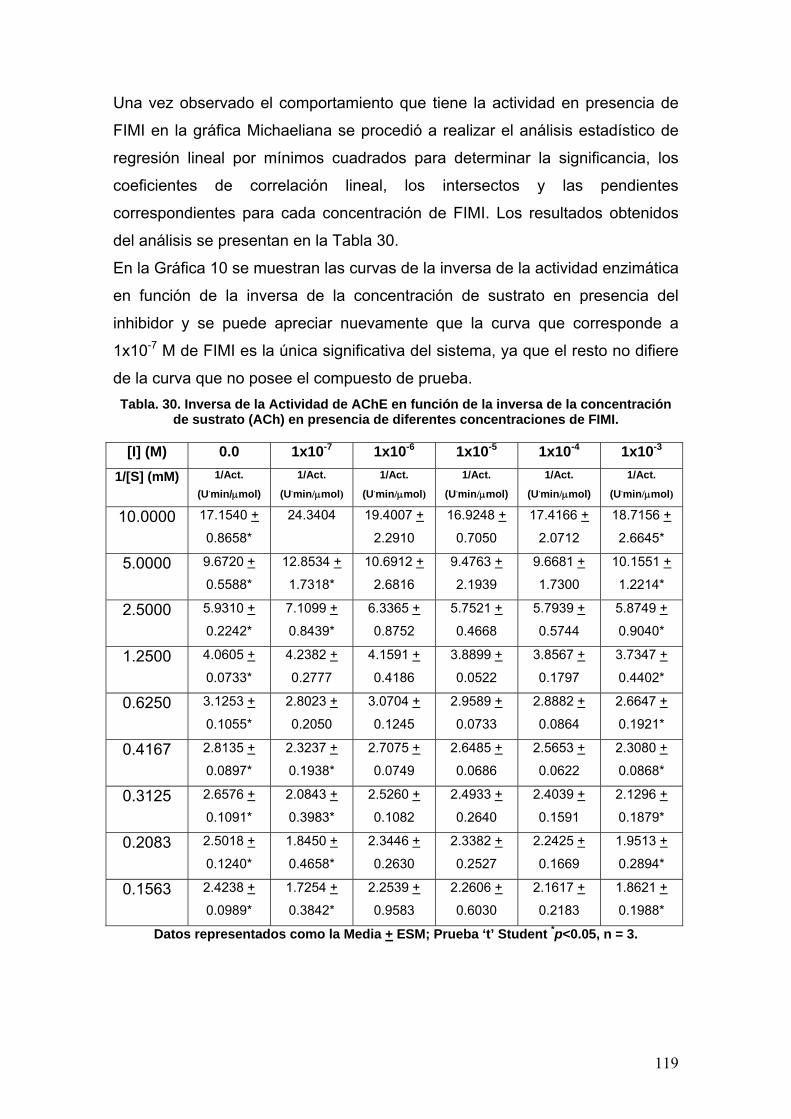
119
Una vez observado el comportamiento que tiene la actividad en presencia de
FIMI en la gráfica Michaeliana se procedió a realizar el análisis estadístico de
regresión lineal por mínimos cuadrados para determinar la significancia, los
coeficientes de correlación lineal, los intersectos y las pendientes
correspondientes para cada concentración de FIMI. Los resultados obtenidos
del análisis se presentan en la Tabla 30.
En la Gráfica 10 se muestran las curvas de la inversa de la actividad enzimática
en función de la inversa de la concentración de sustrato en presencia del
inhibidor y se puede apreciar nuevamente que la curva que corresponde a
1x10-7 M de FIMI es la única significativa del sistema, ya que el resto no difiere
de la curva que no posee el compuesto de prueba. Tabla. 30. Inversa de la Actividad de AChE en función de la inversa de la concentración
de sustrato (ACh) en presencia de diferentes concentraciones de FIMI.
[I] (M) 0.0 1x10-7 1x10-6 1x10-5 1x10-4 1x10-3 1/[S] (mM) 1/Act.
(U.min/μmol)
1/Act.
(U.min/μmol)
1/Act.
(U.min/μmol)
1/Act.
(U.min/μmol)
1/Act.
(U.min/μmol)
1/Act.
(U.min/μmol)
10.0000 17.1540 +
0.8658*
24.3404 19.4007 +
2.2910
16.9248 +
0.7050
17.4166 +
2.0712
18.7156 +
2.6645*
5.0000 9.6720 +
0.5588*
12.8534 +
1.7318*
10.6912 +
2.6816
9.4763 +
2.1939
9.6681 +
1.7300
10.1551 +
1.2214*
2.5000 5.9310 +
0.2242*
7.1099 +
0.8439*
6.3365 +
0.8752
5.7521 +
0.4668
5.7939 +
0.5744
5.8749 +
0.9040*
1.2500 4.0605 +
0.0733*
4.2382 +
0.2777
4.1591 +
0.4186
3.8899 +
0.0522
3.8567 +
0.1797
3.7347 +
0.4402*
0.6250 3.1253 +
0.1055*
2.8023 +
0.2050
3.0704 +
0.1245
2.9589 +
0.0733
2.8882 +
0.0864
2.6647 +
0.1921*
0.4167 2.8135 +
0.0897*
2.3237 +
0.1938*
2.7075 +
0.0749
2.6485 +
0.0686
2.5653 +
0.0622
2.3080 +
0.0868*
0.3125 2.6576 +
0.1091*
2.0843 +
0.3983*
2.5260 +
0.1082
2.4933 +
0.2640
2.4039 +
0.1591
2.1296 +
0.1879*
0.2083 2.5018 +
0.1240*
1.8450 +
0.4658*
2.3446 +
0.2630
2.3382 +
0.2527
2.2425 +
0.1669
1.9513 +
0.2894*
0.1563 2.4238 +
0.0989*
1.7254 +
0.3842*
2.2539 +
0.9583
2.2606 +
0.6030
2.1617 +
0.2183
1.8621 +
0.1988*
Datos representados como la Media + ESM; Prueba ‘t’ Student *p<0.05, n = 3.
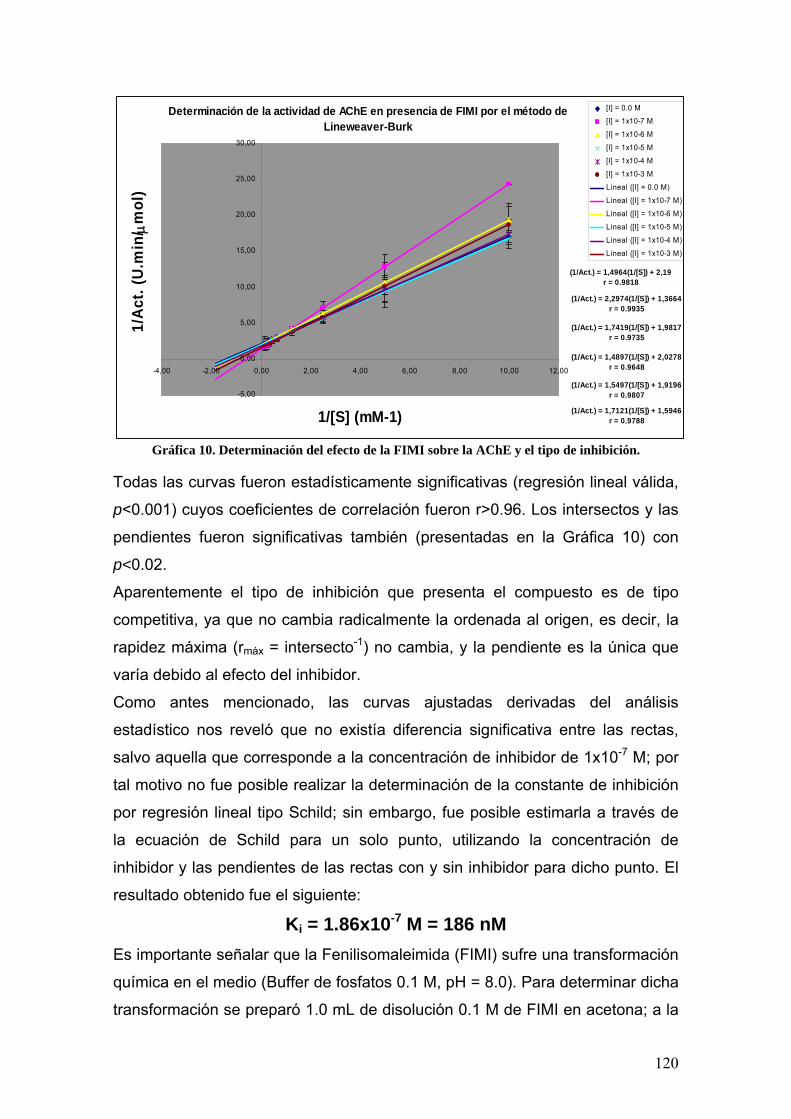
120
Determinación de la actividad de AChE en presencia de FIMI por el método de Lineweaver-Burk
(1/Act.) = 1,4964(1/[S]) + 2,19r = 0.9818
(1/Act.) = 2,2974(1/[S]) + 1,3664r = 0.9935
(1/Act.) = 1,7419(1/[S]) + 1,9817r = 0.9735
(1/Act.) = 1,4897(1/[S]) + 2,0278r = 0.9648
(1/Act.) = 1,5497(1/[S]) + 1,9196r = 0.9807
(1/Act.) = 1,7121(1/[S]) + 1,5946r = 0.9788
-5,00
0,00
5,00
10,00
15,00
20,00
25,00
30,00
-4,00 -2,00 0,00 2,00 4,00 6,00 8,00 10,00 12,00
1/[S] (mM-1)
1/A
ct. (
U.m
in/ μ
mol
)
[I] = 0.0 M
[I] = 1x10-7 M
[I] = 1x10-6 M
[I] = 1x10-5 M
[I] = 1x10-4 M
[I] = 1x10-3 M
Lineal ([I] = 0.0 M)
Lineal ([I] = 1x10-7 M)
Lineal ([I] = 1x10-6 M)
Lineal ([I] = 1x10-5 M)
Lineal ([I] = 1x10-4 M)
Lineal ([I] = 1x10-3 M)
Gráfica 10. Determinación del efecto de la FIMI sobre la AChE y el tipo de inhibición.
Todas las curvas fueron estadísticamente significativas (regresión lineal válida,
p<0.001) cuyos coeficientes de correlación fueron r>0.96. Los intersectos y las
pendientes fueron significativas también (presentadas en la Gráfica 10) con
p<0.02.
Aparentemente el tipo de inhibición que presenta el compuesto es de tipo
competitiva, ya que no cambia radicalmente la ordenada al origen, es decir, la
rapidez máxima (rmáx = intersecto-1) no cambia, y la pendiente es la única que
varía debido al efecto del inhibidor.
Como antes mencionado, las curvas ajustadas derivadas del análisis
estadístico nos reveló que no existía diferencia significativa entre las rectas,
salvo aquella que corresponde a la concentración de inhibidor de 1x10-7 M; por
tal motivo no fue posible realizar la determinación de la constante de inhibición
por regresión lineal tipo Schild; sin embargo, fue posible estimarla a través de
la ecuación de Schild para un solo punto, utilizando la concentración de
inhibidor y las pendientes de las rectas con y sin inhibidor para dicho punto. El
resultado obtenido fue el siguiente:
Ki = 1.86x10-7 M = 186 nM Es importante señalar que la Fenilisomaleimida (FIMI) sufre una transformación
química en el medio (Buffer de fosfatos 0.1 M, pH = 8.0). Para determinar dicha
transformación se preparó 1.0 mL de disolución 0.1 M de FIMI en acetona; a la

121
cual se le agregaron 1.4 mL de buffer de fosfatos. La mezcla se agitó
manualmente unos segundos hasta que la disolución se aclarara. Una vez
agitada, la disolución fue colocada en un matraz bola para evaporar la acetona
y el agua hasta sequedad a temperatura ambiente. La muestra totalmente seca
fue lleva a analizar a RMN de 1H y 13C para caracterizar lo que se había
formado, obteniéndose los resultados mostrados en la Tabla 31.
Los resultados obtenidos sugieren que la fenilisomaleimida (FIMI) se isomeriza
casi inmediatamente en el medio a la fenilmaleimida (FMI), puesto que el
espectro de RMN para FMI presenta en 6.84 ppm una señal simple que integra
para 2H, que corresponden a los protones vinílicos dada su alta simetría con
respecto a FIMI que presenta 2 señales dobles para el mismo sistema. Los
protones aromáticos no cambian notablemente en ambas moléculas.
En relación a las señales obtenidas en el espectro de 13C la FMI presenta
cambios contundentes en los carbonos C-2 y C-3. El C-2 de FMI no varía
mucho del C-2 de FIMI (~128.0 ppm) ya que prácticamente esa porción de la
molécula no sufre un cambio radical; sin embargo, el C-3 de FMI se desplaza a
campos más altos (129.23 ppm) con respecto a FIMI (143.29 ppm) ya que en la
FMI la molécula es simétrica y no tiene la influencia de la imina y la conjugación
electrónica que presenta la FIMI. Otro cambio importante se presenta en el C-1’
ya que el nitrógeno en la FIMI es exocíclico y el la FMI es intracíclico lo que
provoca un cambio en el desplazamiento químico de ese carbono. Los átomos
de carbono restantes no sufren un cambio significativo.
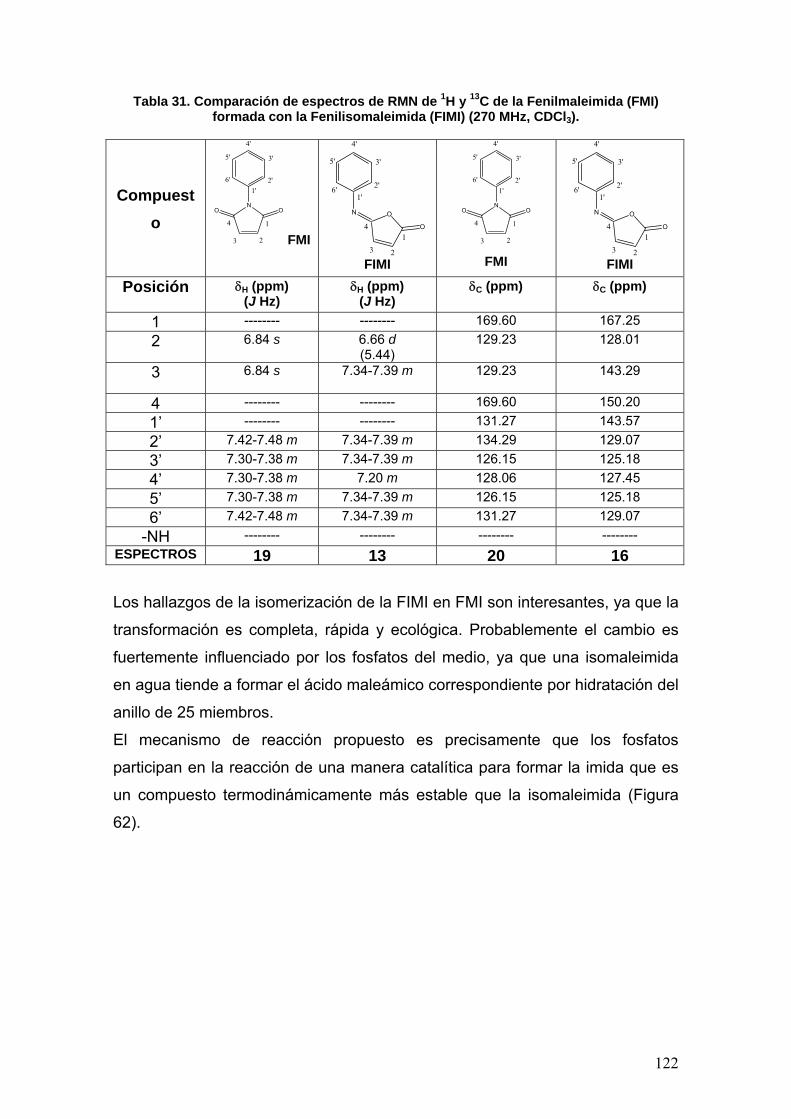
122
Tabla 31. Comparación de espectros de RMN de 1H y 13C de la Fenilmaleimida (FMI) formada con la Fenilisomaleimida (FIMI) (270 MHz, CDCl3).
Compuesto
NOO
1
23
4
1'2'
3'
4'
5'
6'
FMI
N O
O1
23
4
1'2'
3'
4'
5'
6'
FIMI
NOO
1
23
4
1'2'
3'
4'
5'
6'
FMI
N O
O1
23
4
1'2'
3'
4'
5'
6'
FIMI
Posición δH (ppm) (J Hz)
δH (ppm) (J Hz)
δC (ppm)
δC (ppm)
1 -------- -------- 169.60 167.25 2 6.84 s
6.66 d (5.44)
129.23 128.01
3 6.84 s
7.34-7.39 m
129.23 143.29
4 -------- -------- 169.60 150.20 1’ -------- -------- 131.27 143.57 2’ 7.42-7.48 m 7.34-7.39 m 134.29 129.07 3’ 7.30-7.38 m 7.34-7.39 m 126.15 125.18 4’ 7.30-7.38 m 7.20 m 128.06 127.45 5’ 7.30-7.38 m 7.34-7.39 m 126.15 125.18 6’ 7.42-7.48 m 7.34-7.39 m 131.27 129.07
-NH -------- -------- -------- -------- ESPECTROS 19 13 20 16
Los hallazgos de la isomerización de la FIMI en FMI son interesantes, ya que la
transformación es completa, rápida y ecológica. Probablemente el cambio es
fuertemente influenciado por los fosfatos del medio, ya que una isomaleimida
en agua tiende a formar el ácido maleámico correspondiente por hidratación del
anillo de 25 miembros.
El mecanismo de reacción propuesto es precisamente que los fosfatos
participan en la reacción de una manera catalítica para formar la imida que es
un compuesto termodinámicamente más estable que la isomaleimida (Figura
62).
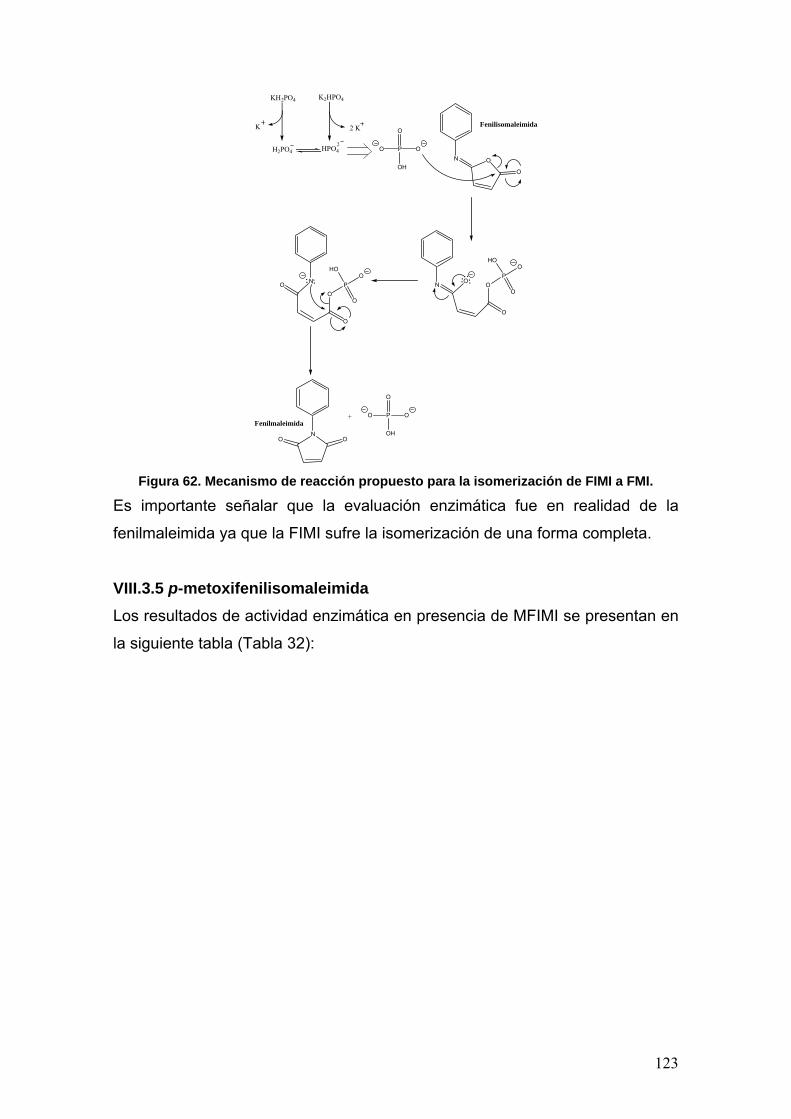
123
K2HPO4KH2PO4
K 2 K
HPO4N O
O
H2PO4 P
O
O O
OH
N OO
O
PO
O
HO
NOO
O
PO
O
HO
P
O
O O
OHNOO
2
+
Fenilisomaleimida
Fenilmaleimida
Figura 62. Mecanismo de reacción propuesto para la isomerización de FIMI a FMI.
Es importante señalar que la evaluación enzimática fue en realidad de la
fenilmaleimida ya que la FIMI sufre la isomerización de una forma completa.
VIII.3.5 p-metoxifenilisomaleimida Los resultados de actividad enzimática en presencia de MFIMI se presentan en
la siguiente tabla (Tabla 32):
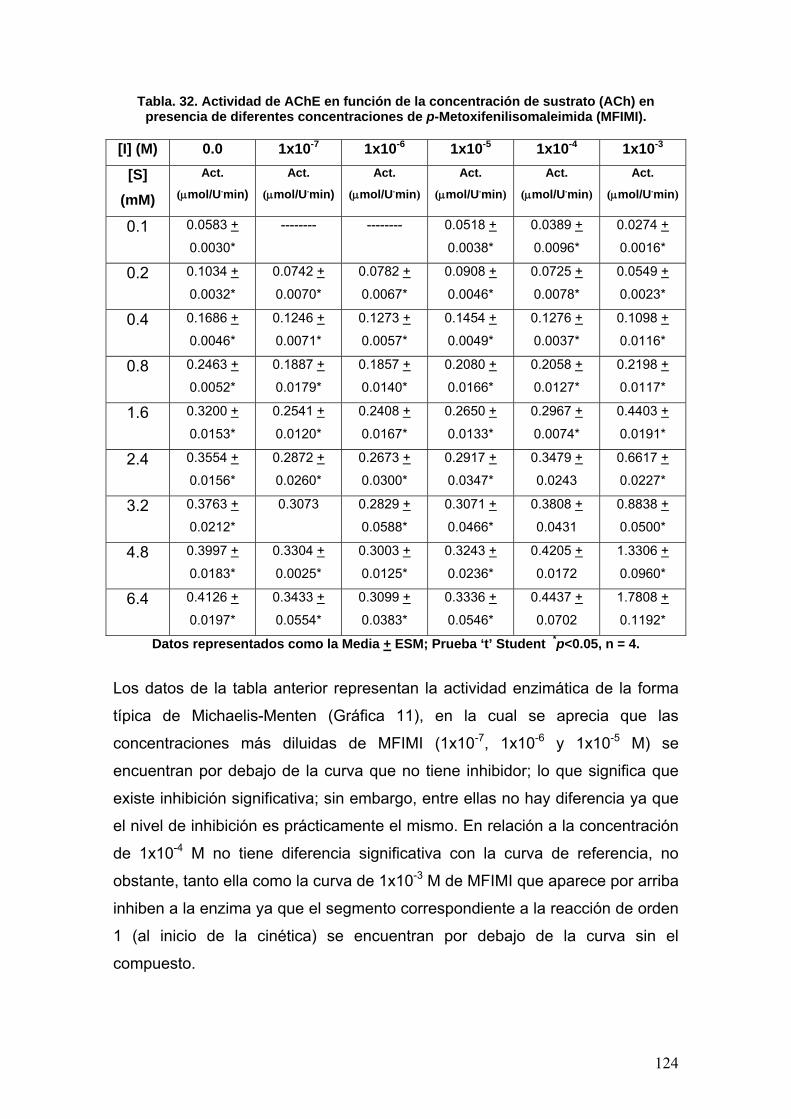
124
Tabla. 32. Actividad de AChE en función de la concentración de sustrato (ACh) en presencia de diferentes concentraciones de p-Metoxifenilisomaleimida (MFIMI).
[I] (M) 0.0 1x10-7 1x10-6 1x10-5 1x10-4 1x10-3
[S] (mM)
Act.
(μmol/U.min)
Act.
(μmol/U.min)
Act.
(μmol/U.min)
Act.
(μmol/U.min)
Act.
(μmol/U.min)
Act.
(μmol/U.min)
0.1 0.0583 +
0.0030*
-------- -------- 0.0518 +
0.0038*
0.0389 +
0.0096*
0.0274 +
0.0016*
0.2 0.1034 +
0.0032*
0.0742 +
0.0070*
0.0782 +
0.0067*
0.0908 +
0.0046*
0.0725 +
0.0078*
0.0549 +
0.0023*
0.4 0.1686 +
0.0046*
0.1246 +
0.0071*
0.1273 +
0.0057*
0.1454 +
0.0049*
0.1276 +
0.0037*
0.1098 +
0.0116*
0.8 0.2463 +
0.0052*
0.1887 +
0.0179*
0.1857 +
0.0140*
0.2080 +
0.0166*
0.2058 +
0.0127*
0.2198 +
0.0117*
1.6 0.3200 +
0.0153*
0.2541 +
0.0120*
0.2408 +
0.0167*
0.2650 +
0.0133*
0.2967 +
0.0074*
0.4403 +
0.0191*
2.4 0.3554 +
0.0156*
0.2872 +
0.0260*
0.2673 +
0.0300*
0.2917 +
0.0347*
0.3479 +
0.0243
0.6617 +
0.0227*
3.2 0.3763 +
0.0212*
0.3073 0.2829 +
0.0588*
0.3071 +
0.0466*
0.3808 +
0.0431
0.8838 +
0.0500*
4.8 0.3997 +
0.0183*
0.3304 +
0.0025*
0.3003 +
0.0125*
0.3243 +
0.0236*
0.4205 +
0.0172
1.3306 +
0.0960*
6.4 0.4126 +
0.0197*
0.3433 +
0.0554*
0.3099 +
0.0383*
0.3336 +
0.0546*
0.4437 +
0.0702
1.7808 +
0.1192*
Datos representados como la Media + ESM; Prueba ‘t’ Student *p<0.05, n = 4.
Los datos de la tabla anterior representan la actividad enzimática de la forma
típica de Michaelis-Menten (Gráfica 11), en la cual se aprecia que las
concentraciones más diluidas de MFIMI (1x10-7, 1x10-6 y 1x10-5 M) se
encuentran por debajo de la curva que no tiene inhibidor; lo que significa que
existe inhibición significativa; sin embargo, entre ellas no hay diferencia ya que
el nivel de inhibición es prácticamente el mismo. En relación a la concentración
de 1x10-4 M no tiene diferencia significativa con la curva de referencia, no
obstante, tanto ella como la curva de 1x10-3 M de MFIMI que aparece por arriba
inhiben a la enzima ya que el segmento correspondiente a la reacción de orden
1 (al inicio de la cinética) se encuentran por debajo de la curva sin el
compuesto.
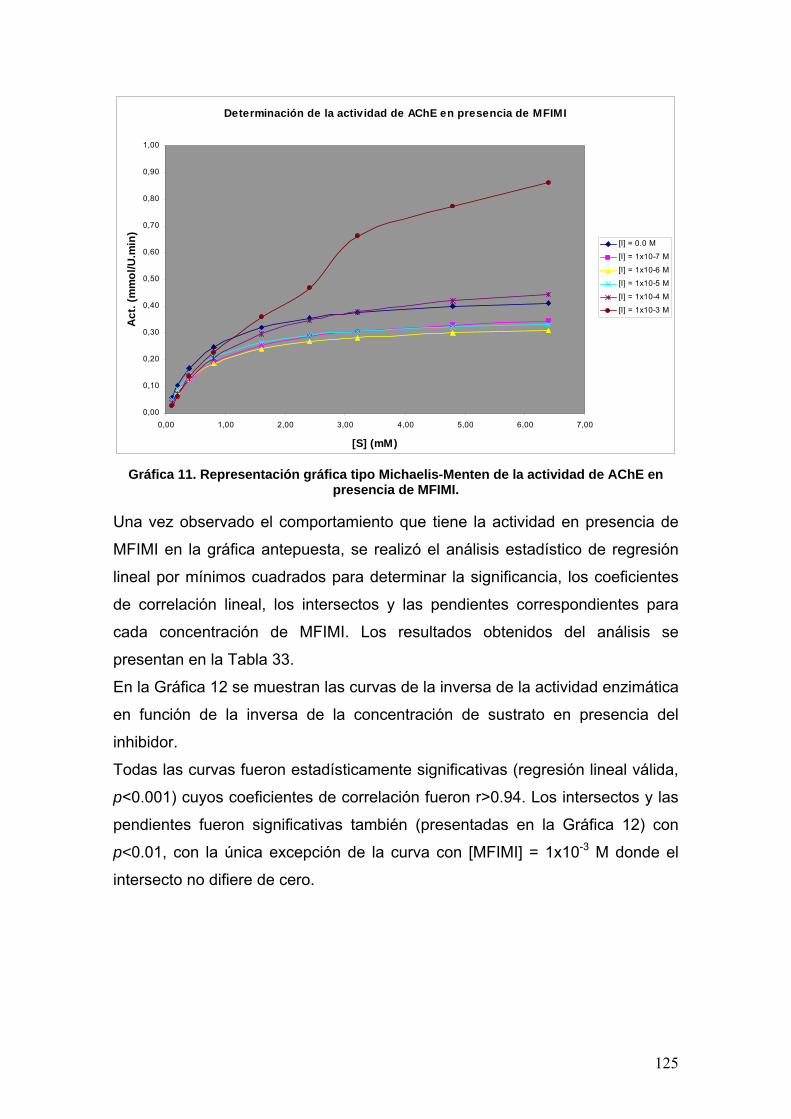
125
Determinación de la actividad de AChE en presencia de MFIMI
0,00
0,10
0,20
0,30
0,40
0,50
0,60
0,70
0,80
0,90
1,00
0,00 1,00 2,00 3,00 4,00 5,00 6,00 7,00
[S] (mM)
Act
. (m
mol
/U.m
in)
[I] = 0.0 M
[I] = 1x10-7 M
[I] = 1x10-6 M
[I] = 1x10-5 M
[I] = 1x10-4 M
[I] = 1x10-3 M
Gráfica 11. Representación gráfica tipo Michaelis-Menten de la actividad de AChE en
presencia de MFIMI.
Una vez observado el comportamiento que tiene la actividad en presencia de
MFIMI en la gráfica antepuesta, se realizó el análisis estadístico de regresión
lineal por mínimos cuadrados para determinar la significancia, los coeficientes
de correlación lineal, los intersectos y las pendientes correspondientes para
cada concentración de MFIMI. Los resultados obtenidos del análisis se
presentan en la Tabla 33.
En la Gráfica 12 se muestran las curvas de la inversa de la actividad enzimática
en función de la inversa de la concentración de sustrato en presencia del
inhibidor.
Todas las curvas fueron estadísticamente significativas (regresión lineal válida,
p<0.001) cuyos coeficientes de correlación fueron r>0.94. Los intersectos y las
pendientes fueron significativas también (presentadas en la Gráfica 12) con
p<0.01, con la única excepción de la curva con [MFIMI] = 1x10-3 M donde el
intersecto no difiere de cero.
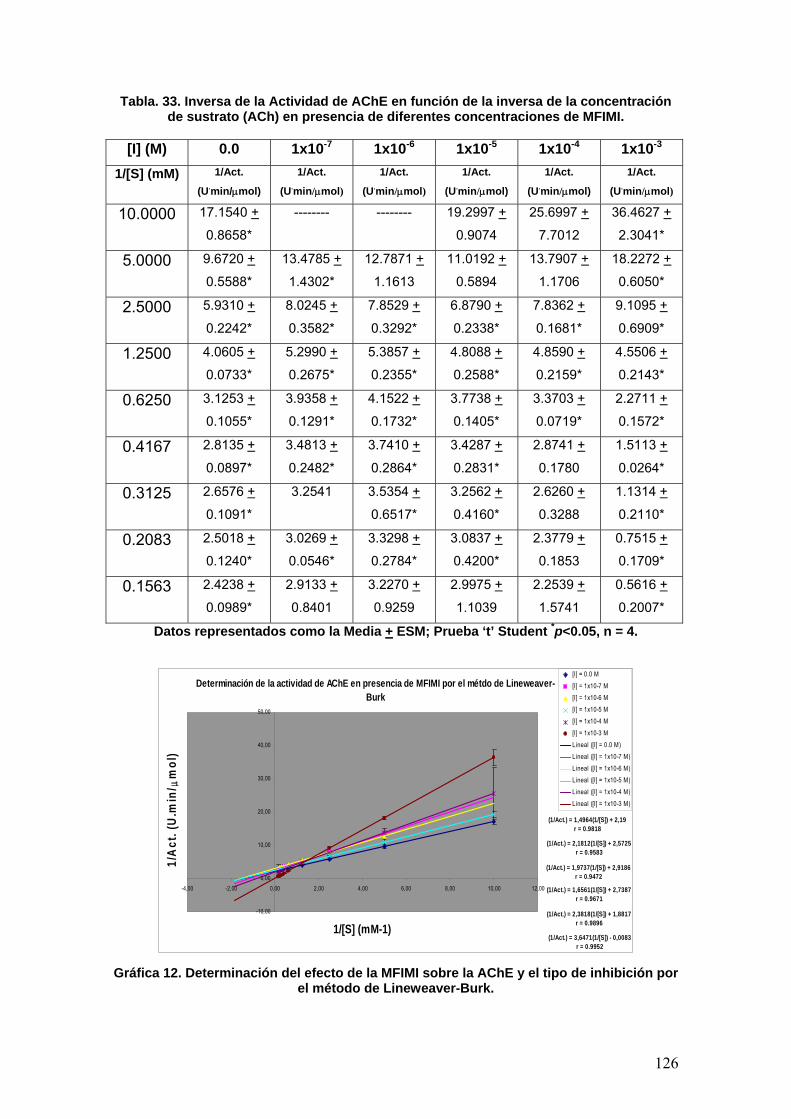
126
Tabla. 33. Inversa de la Actividad de AChE en función de la inversa de la concentración de sustrato (ACh) en presencia de diferentes concentraciones de MFIMI.
[I] (M) 0.0 1x10-7 1x10-6 1x10-5 1x10-4 1x10-3
1/[S] (mM) 1/Act.
(U.min/μmol)
1/Act.
(U.min/μmol)
1/Act.
(U.min/μmol)
1/Act.
(U.min/μmol)
1/Act.
(U.min/μmol)
1/Act.
(U.min/μmol)
10.0000 17.1540 +
0.8658*
-------- -------- 19.2997 +
0.9074
25.6997 +
7.7012
36.4627 +
2.3041*
5.0000 9.6720 +
0.5588*
13.4785 +
1.4302*
12.7871 +
1.1613
11.0192 +
0.5894
13.7907 +
1.1706
18.2272 +
0.6050*
2.5000 5.9310 +
0.2242*
8.0245 +
0.3582*
7.8529 +
0.3292*
6.8790 +
0.2338*
7.8362 +
0.1681*
9.1095 +
0.6909*
1.2500 4.0605 +
0.0733*
5.2990 +
0.2675*
5.3857 +
0.2355*
4.8088 +
0.2588*
4.8590 +
0.2159*
4.5506 +
0.2143*
0.6250 3.1253 +
0.1055*
3.9358 +
0.1291*
4.1522 +
0.1732*
3.7738 +
0.1405*
3.3703 +
0.0719*
2.2711 +
0.1572*
0.4167 2.8135 +
0.0897*
3.4813 +
0.2482*
3.7410 +
0.2864*
3.4287 +
0.2831*
2.8741 +
0.1780
1.5113 +
0.0264*
0.3125 2.6576 +
0.1091*
3.2541 3.5354 +
0.6517*
3.2562 +
0.4160*
2.6260 +
0.3288
1.1314 +
0.2110*
0.2083 2.5018 +
0.1240*
3.0269 +
0.0546*
3.3298 +
0.2784*
3.0837 +
0.4200*
2.3779 +
0.1853
0.7515 +
0.1709*
0.1563 2.4238 +
0.0989*
2.9133 +
0.8401
3.2270 +
0.9259
2.9975 +
1.1039
2.2539 +
1.5741
0.5616 +
0.2007*
Datos representados como la Media + ESM; Prueba ‘t’ Student *p<0.05, n = 4.
Determinación de la actividad de AChE en presencia de MFIMI por el métdo de Lineweaver-Burk
(1/Act.) = 1,4964(1/[S]) + 2,19r = 0.9818
(1/Act.) = 2,1812(1/[S]) + 2,5725r = 0.9583
(1/Act.) = 1,9737(1/[S]) + 2,9186r = 0.9472
(1/Act.) = 1,6561(1/[S]) + 2,7387r = 0.9671
(1/Act.) = 2,3818(1/[S]) + 1,8817r = 0.9896
(1/Act.) = 3,6471(1/[S]) - 0,0083r = 0.9952
-10,00
0,00
10,00
20,00
30,00
40,00
50,00
-4,00 -2,00 0,00 2,00 4,00 6,00 8,00 10,00 12,00
1/[S] (mM-1)
1/A
ct. (
U.m
in/ μ
mol
)
[I] = 0.0 M
[I] = 1x10-7 M
[I] = 1x10-6 M
[I] = 1x10-5 M
[I] = 1x10-4 M
[I] = 1x10-3 M
Lineal ([I] = 0.0 M)
Lineal ([I] = 1x10-7 M)
Lineal ([I] = 1x10-6 M)
Lineal ([I] = 1x10-5 M)
Lineal ([I] = 1x10-4 M)
Lineal ([I] = 1x10-3 M)
Gráfica 12. Determinación del efecto de la MFIMI sobre la AChE y el tipo de inhibición por
el método de Lineweaver-Burk.
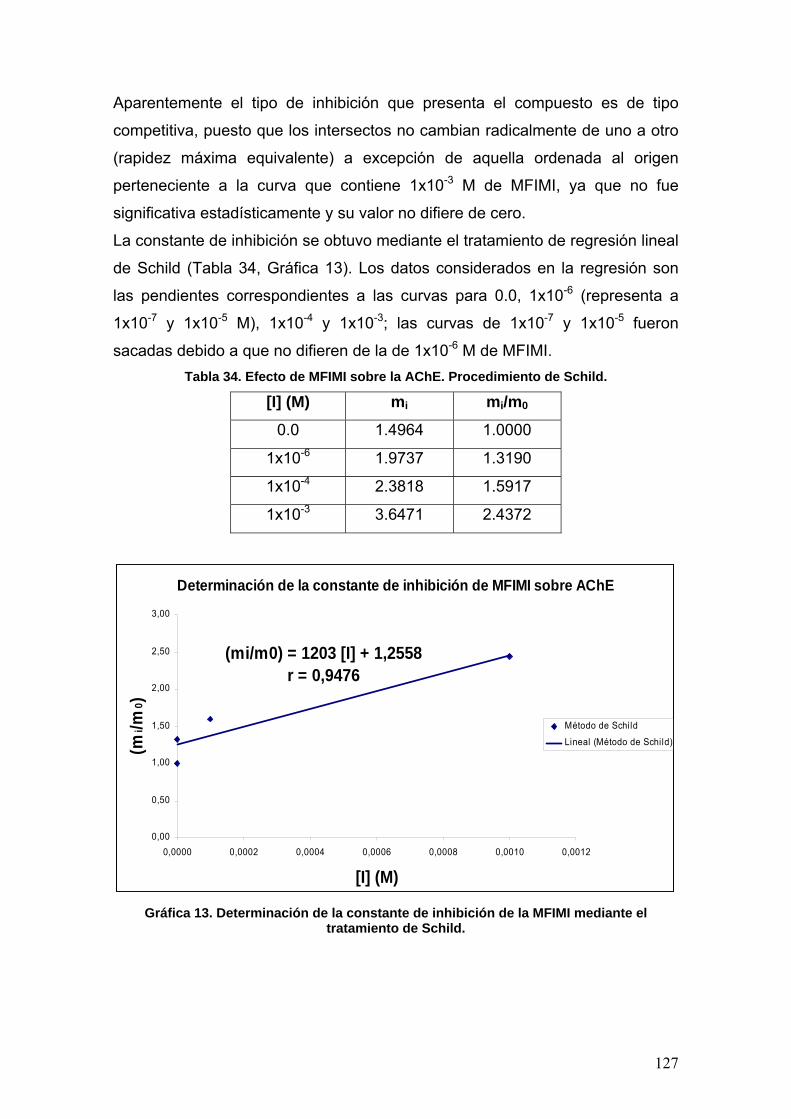
127
Aparentemente el tipo de inhibición que presenta el compuesto es de tipo
competitiva, puesto que los intersectos no cambian radicalmente de uno a otro
(rapidez máxima equivalente) a excepción de aquella ordenada al origen
perteneciente a la curva que contiene 1x10-3 M de MFIMI, ya que no fue
significativa estadísticamente y su valor no difiere de cero.
La constante de inhibición se obtuvo mediante el tratamiento de regresión lineal
de Schild (Tabla 34, Gráfica 13). Los datos considerados en la regresión son
las pendientes correspondientes a las curvas para 0.0, 1x10-6 (representa a
1x10-7 y 1x10-5 M), 1x10-4 y 1x10-3; las curvas de 1x10-7 y 1x10-5 fueron
sacadas debido a que no difieren de la de 1x10-6 M de MFIMI. Tabla 34. Efecto de MFIMI sobre la AChE. Procedimiento de Schild.
[I] (M) mi mi/m0
0.0 1.4964 1.0000
1x10-6 1.9737 1.3190
1x10-4 2.3818 1.5917
1x10-3 3.6471 2.4372
Determinación de la constante de inhibición de MFIMI sobre AChE
(mi/m0) = 1203 [I] + 1,2558r = 0,9476
0,00
0,50
1,00
1,50
2,00
2,50
3,00
0,0000 0,0002 0,0004 0,0006 0,0008 0,0010 0,0012
[I] (M)
(mi/m
0 )
Método de Schild
Lineal (Método de Schild)
Gráfica 13. Determinación de la constante de inhibición de la MFIMI mediante el
tratamiento de Schild.

128
Es importante destacar que la regresión lineal fue estadísticamente significativa
con r = 0.9476 (p<0.05).
La constante de inhibición calculada fue de Ki = 8.31x10-4 M = 831.24 μM.
Ki = 831.24 + 299.62 μM Es importante decir que en el medio de reacción enzimática (buffer de fosfatos
0.1 M, pH = 8.0) la p-Metoxifenilisomaleimida (MFIMI) sufre una transformación
química. Para determinarlo se preparó 1.0 mL de disolución 0.1 M de MFIMI en
acetona; a la cual se le agregaron 1.4 mL de buffer de fosfatos. La mezcla se
agitó manualmente obteniendo una parte sólida (derivada de una precipitación)
y otra soluble. El sólido fue filtrado a gravedad y se dejó secar a temperatura
ambiente, mientras que la parte en solución fue vertida en un matraz bola para
evaporar la acetona y el agua hasta sequedad a la misma temperatura. Las
muestras totalmente secas fueron analizadas por RMN de 1H para caracterizar
lo que se había formado, obteniéndose los resultados mostrados en la Tabla
35.
Los resultados obtenidos muestran restos de la MFIMI en la parte sólida y 2
compuestos más correspondientes al ácido p-metoxifenilmaleámico (MFMA,
parte soluble) y, la p-metoxifenilmaleimida (MFMI, también en la parte sólida).
Los espectros de RMN de 1H revelaron la formación del ácido p-
metoxifenilmaleámico (MFMA) en la parte soluble, mientras que en la parte
sólida se forma la p-metoxifenilmaleimida (MFMI). Es de importancia comentar
los cambios espectrales de la MFIMI y la MFMI formada ya que son
contundentes; los protones vinílicos H-2 y H-3 de la MFIMI presentan 2 señales
dobles con desplazamientos químicos 6.54 y 7.43 ppm respectivamente,
mientras que en el mismo sistema (hidrógenos vinílicos) la MFMI presenta solo
una señal simple que integra para 2 hidrógenos con una � de 7.14 ppm. El
sistema aromático no sufre cambios significativos entre las 2 moléculas. En
relación al MFMA formado a partir de MFIMI, los cambios son determinantes,
ya que presenta las señales típicas de la MFMA (ver Tabla 17).
Probablemente la formación de la mezcla (MFMA, MFMI y MFIMI) se deba
únicamente a un ligero aumento de la nucleofilicidad del átomo de nitrógeno
imídico (Figura 63) de la NFIMI por la influencia electrodonadora del grupo –
OCH3 que posee la molécula; por ello, los fosfatos llegan a reaccionar con la
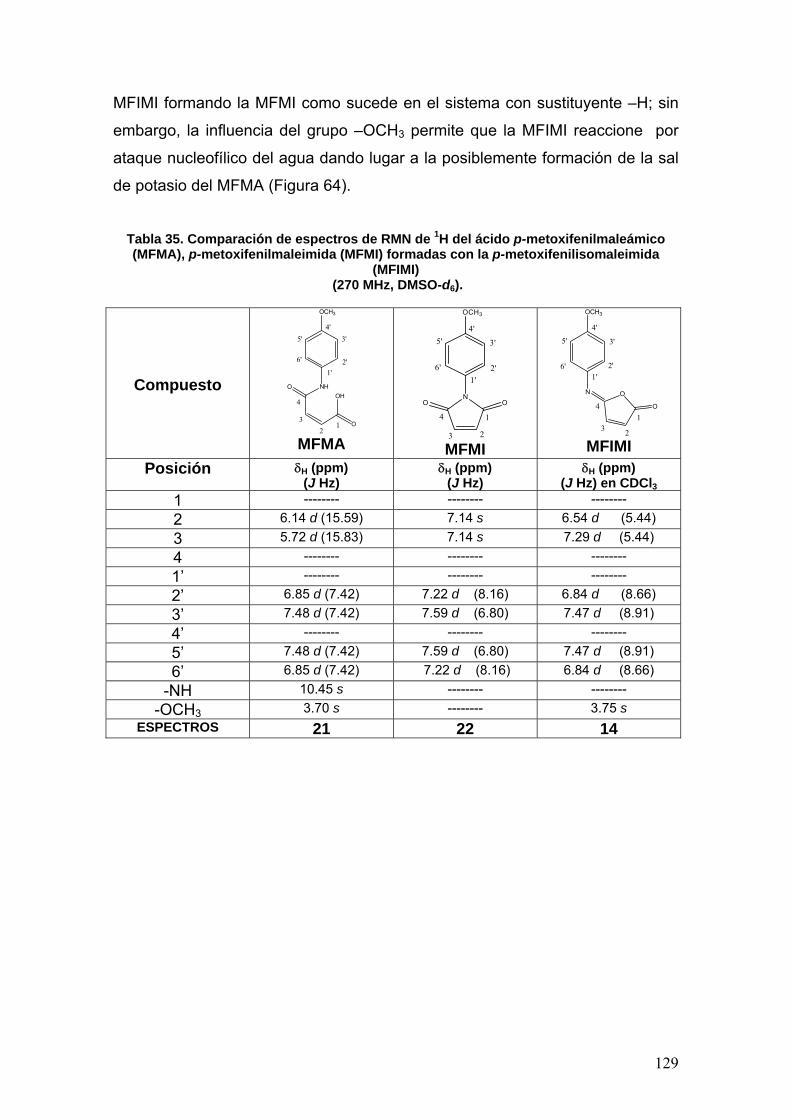
129
MFIMI formando la MFMI como sucede en el sistema con sustituyente –H; sin
embargo, la influencia del grupo –OCH3 permite que la MFIMI reaccione por
ataque nucleofílico del agua dando lugar a la posiblemente formación de la sal
de potasio del MFMA (Figura 64).
Tabla 35. Comparación de espectros de RMN de 1H del ácido p-metoxifenilmaleámico (MFMA), p-metoxifenilmaleimida (MFMI) formadas con la p-metoxifenilisomaleimida
(MFIMI) (270 MHz, DMSO-d6).
Compuesto NH
O
OOH
OCH3
12
3
4
1'2'
3'4'
5'
6'
MFMA
OCH3
NOO
1
23
4
1'2'
3'
4'5'
6'
MFMI
N O
O
OCH3
1
23
4
1'2'
3'
4'
5'
6'
MFIMI
Posición δH (ppm) (J Hz)
δH (ppm) (J Hz)
δH (ppm) (J Hz) en CDCl3
1 -------- -------- -------- 2 6.14 d (15.59) 7.14 s 6.54 d (5.44) 3 5.72 d (15.83) 7.14 s 7.29 d (5.44) 4 -------- -------- -------- 1’ -------- -------- -------- 2’ 6.85 d (7.42) 7.22 d (8.16) 6.84 d (8.66) 3’ 7.48 d (7.42) 7.59 d (6.80) 7.47 d (8.91) 4’ -------- -------- -------- 5’ 7.48 d (7.42) 7.59 d (6.80) 7.47 d (8.91) 6’ 6.85 d (7.42) 7.22 d (8.16) 6.84 d (8.66)
-NH 10.45 s -------- -------- -OCH3 3.70 s -------- 3.75 s
ESPECTROS 21 22 14
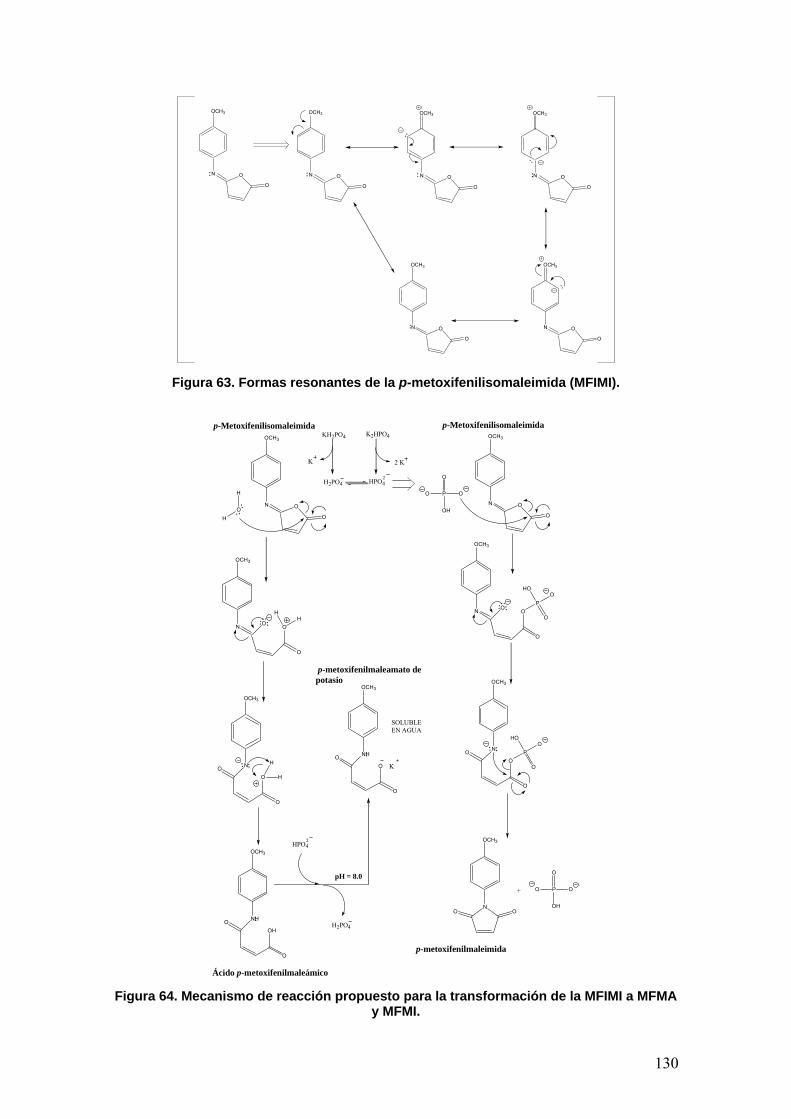
130
OCH3
N O
O
OCH3
N O
O
OCH3
N O
O
OCH3
N O
O
OCH3
N O
O
OCH3
N O
O
Figura 63. Formas resonantes de la p-metoxifenilisomaleimida (MFIMI).
H2PO4NHO
OH
O
OCH3
P
O
O O
OHNOO
OCH3HPO4
KNOO
O
OCH3
H
H
NHOO
O
OCH3
NOO
O
PO
O
HO
OCH3
N OO
O
OCH3
HH
N OO
O
PO
O
HO
OCH3
H
OH
P
O
O O
OHN O
O
OCH3
N O
O
OCH3
H2PO4 HPO4
2 KK
KH2PO4 K2HPO4
2
+
p-Metoxifenilisomaleimida
p-metoxifenilmaleimida
p-Metoxifenilisomaleimida
Ácido p-metoxifenilmaleámico
2
pH = 8.0
p-metoxifenilmaleamato depotasio
SOLUBLEEN AGUA
Figura 64. Mecanismo de reacción propuesto para la transformación de la MFIMI a MFMA
y MFMI.
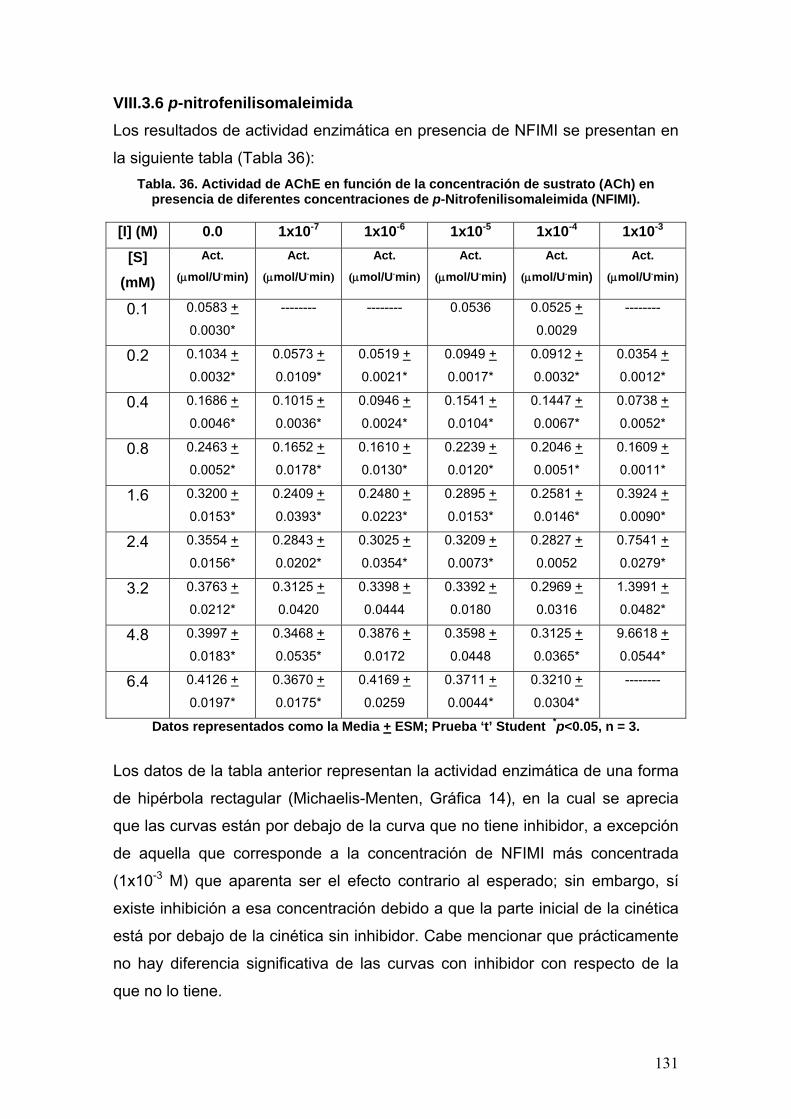
131
VIII.3.6 p-nitrofenilisomaleimida Los resultados de actividad enzimática en presencia de NFIMI se presentan en
la siguiente tabla (Tabla 36): Tabla. 36. Actividad de AChE en función de la concentración de sustrato (ACh) en
presencia de diferentes concentraciones de p-Nitrofenilisomaleimida (NFIMI).
[I] (M) 0.0 1x10-7 1x10-6 1x10-5 1x10-4 1x10-3
[S] (mM)
Act.
(μmol/U.min)
Act.
(μmol/U.min)
Act.
(μmol/U.min)
Act.
(μmol/U.min)
Act.
(μmol/U.min)
Act.
(μmol/U.min)
0.1 0.0583 +
0.0030*
-------- -------- 0.0536 0.0525 +
0.0029
--------
0.2 0.1034 +
0.0032*
0.0573 +
0.0109*
0.0519 +
0.0021*
0.0949 +
0.0017*
0.0912 +
0.0032*
0.0354 +
0.0012*
0.4 0.1686 +
0.0046*
0.1015 +
0.0036*
0.0946 +
0.0024*
0.1541 +
0.0104*
0.1447 +
0.0067*
0.0738 +
0.0052*
0.8 0.2463 +
0.0052*
0.1652 +
0.0178*
0.1610 +
0.0130*
0.2239 +
0.0120*
0.2046 +
0.0051*
0.1609 +
0.0011*
1.6 0.3200 +
0.0153*
0.2409 +
0.0393*
0.2480 +
0.0223*
0.2895 +
0.0153*
0.2581 +
0.0146*
0.3924 +
0.0090*
2.4 0.3554 +
0.0156*
0.2843 +
0.0202*
0.3025 +
0.0354*
0.3209 +
0.0073*
0.2827 +
0.0052
0.7541 +
0.0279*
3.2 0.3763 +
0.0212*
0.3125 +
0.0420
0.3398 +
0.0444
0.3392 +
0.0180
0.2969 +
0.0316
1.3991 +
0.0482*
4.8 0.3997 +
0.0183*
0.3468 +
0.0535*
0.3876 +
0.0172
0.3598 +
0.0448
0.3125 +
0.0365*
9.6618 +
0.0544*
6.4 0.4126 +
0.0197*
0.3670 +
0.0175*
0.4169 +
0.0259
0.3711 +
0.0044*
0.3210 +
0.0304*
--------
Datos representados como la Media + ESM; Prueba ‘t’ Student *p<0.05, n = 3.
Los datos de la tabla anterior representan la actividad enzimática de una forma
de hipérbola rectagular (Michaelis-Menten, Gráfica 14), en la cual se aprecia
que las curvas están por debajo de la curva que no tiene inhibidor, a excepción
de aquella que corresponde a la concentración de NFIMI más concentrada
(1x10-3 M) que aparenta ser el efecto contrario al esperado; sin embargo, sí
existe inhibición a esa concentración debido a que la parte inicial de la cinética
está por debajo de la cinética sin inhibidor. Cabe mencionar que prácticamente
no hay diferencia significativa de las curvas con inhibidor con respecto de la
que no lo tiene.
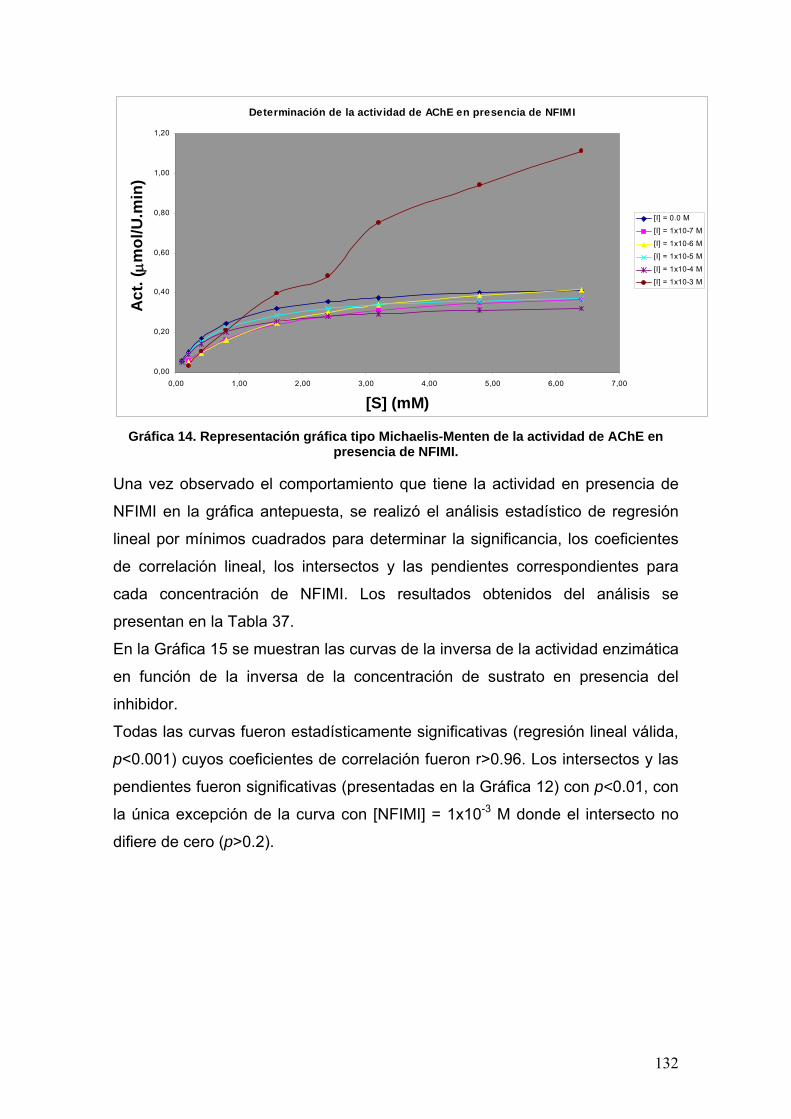
132
Determinación de la actividad de AChE en presencia de NFIMI
0,00
0,20
0,40
0,60
0,80
1,00
1,20
0,00 1,00 2,00 3,00 4,00 5,00 6,00 7,00
[S] (mM)
Act
. (μm
ol/U
.min
)
[I] = 0.0 M
[I] = 1x10-7 M
[I] = 1x10-6 M
[I] = 1x10-5 M
[I] = 1x10-4 M
[I] = 1x10-3 M
Gráfica 14. Representación gráfica tipo Michaelis-Menten de la actividad de AChE en
presencia de NFIMI.
Una vez observado el comportamiento que tiene la actividad en presencia de
NFIMI en la gráfica antepuesta, se realizó el análisis estadístico de regresión
lineal por mínimos cuadrados para determinar la significancia, los coeficientes
de correlación lineal, los intersectos y las pendientes correspondientes para
cada concentración de NFIMI. Los resultados obtenidos del análisis se
presentan en la Tabla 37.
En la Gráfica 15 se muestran las curvas de la inversa de la actividad enzimática
en función de la inversa de la concentración de sustrato en presencia del
inhibidor.
Todas las curvas fueron estadísticamente significativas (regresión lineal válida,
p<0.001) cuyos coeficientes de correlación fueron r>0.96. Los intersectos y las
pendientes fueron significativas (presentadas en la Gráfica 12) con p<0.01, con
la única excepción de la curva con [NFIMI] = 1x10-3 M donde el intersecto no
difiere de cero (p>0.2).
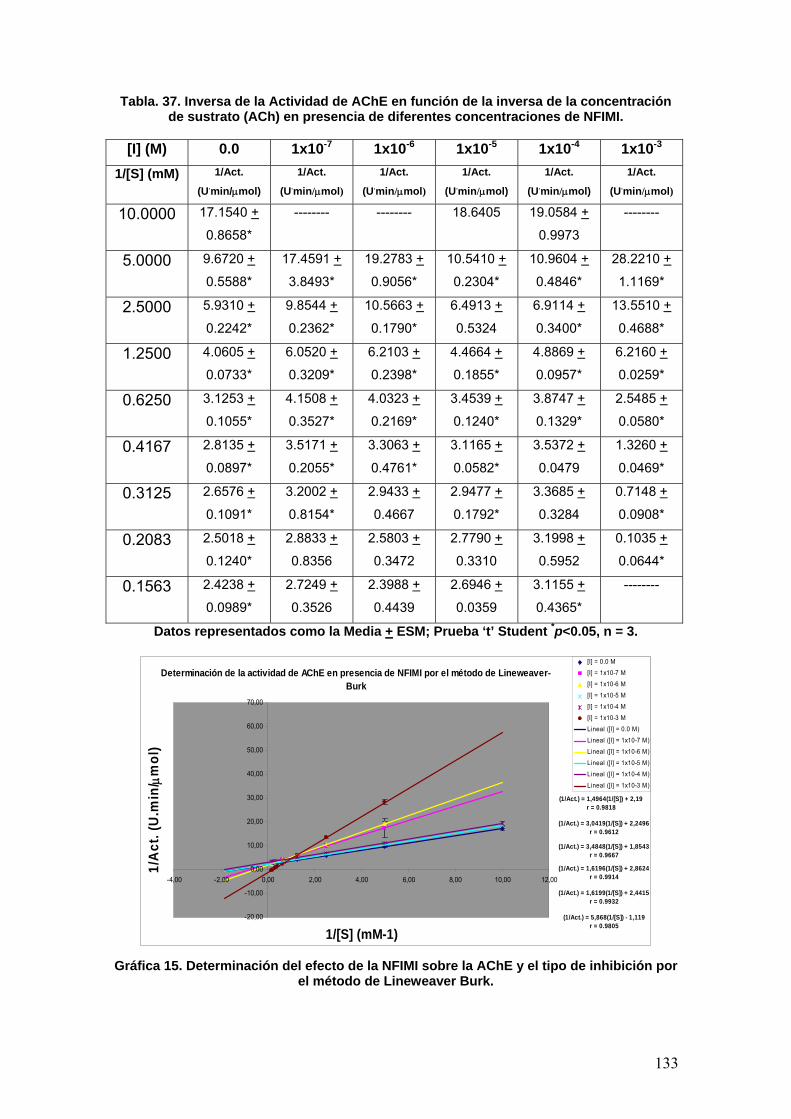
133
Tabla. 37. Inversa de la Actividad de AChE en función de la inversa de la concentración de sustrato (ACh) en presencia de diferentes concentraciones de NFIMI.
[I] (M) 0.0 1x10-7 1x10-6 1x10-5 1x10-4 1x10-3
1/[S] (mM) 1/Act.
(U.min/μmol)
1/Act.
(U.min/μmol)
1/Act.
(U.min/μmol)
1/Act.
(U.min/μmol)
1/Act.
(U.min/μmol)
1/Act.
(U.min/μmol)
10.0000 17.1540 +
0.8658*
-------- -------- 18.6405 19.0584 +
0.9973
--------
5.0000 9.6720 +
0.5588*
17.4591 +
3.8493*
19.2783 +
0.9056*
10.5410 +
0.2304*
10.9604 +
0.4846*
28.2210 +
1.1169*
2.5000 5.9310 +
0.2242*
9.8544 +
0.2362*
10.5663 +
0.1790*
6.4913 +
0.5324
6.9114 +
0.3400*
13.5510 +
0.4688*
1.2500 4.0605 +
0.0733*
6.0520 +
0.3209*
6.2103 +
0.2398*
4.4664 +
0.1855*
4.8869 +
0.0957*
6.2160 +
0.0259*
0.6250 3.1253 +
0.1055*
4.1508 +
0.3527*
4.0323 +
0.2169*
3.4539 +
0.1240*
3.8747 +
0.1329*
2.5485 +
0.0580*
0.4167 2.8135 +
0.0897*
3.5171 +
0.2055*
3.3063 +
0.4761*
3.1165 +
0.0582*
3.5372 +
0.0479
1.3260 +
0.0469*
0.3125 2.6576 +
0.1091*
3.2002 +
0.8154*
2.9433 +
0.4667
2.9477 +
0.1792*
3.3685 +
0.3284
0.7148 +
0.0908*
0.2083 2.5018 +
0.1240*
2.8833 +
0.8356
2.5803 +
0.3472
2.7790 +
0.3310
3.1998 +
0.5952
0.1035 +
0.0644*
0.1563 2.4238 +
0.0989*
2.7249 +
0.3526
2.3988 +
0.4439
2.6946 +
0.0359
3.1155 +
0.4365*
--------
Datos representados como la Media + ESM; Prueba ‘t’ Student *p<0.05, n = 3.
Determinación de la actividad de AChE en presencia de NFIMI por el método de Lineweaver-Burk
(1/Act.) = 1,4964(1/[S]) + 2,19r = 0.9818
(1/Act.) = 3,0419(1/[S]) + 2,2496r = 0.9612
(1/Act.) = 3,4848(1/[S]) + 1,8543r = 0.9667
(1/Act.) = 1,6199(1/[S]) + 2,4415r = 0.9932
(1/Act.) = 1,6196(1/[S]) + 2,8624r = 0.9914
(1/Act.) = 5,868(1/[S]) - 1,119r = 0.9805
-20,00
-10,00
0,00
10,00
20,00
30,00
40,00
50,00
60,00
70,00
-4,00 -2,00 0,00 2,00 4,00 6,00 8,00 10,00 12,00
1/[S] (mM-1)
1/A
ct. (
U.m
in/ μ
mol
)
[I] = 0.0 M
[I] = 1x10-7 M
[I] = 1x10-6 M
[I] = 1x10-5 M
[I] = 1x10-4 M
[I] = 1x10-3 M
Lineal ([I] = 0.0 M)
Lineal ([I] = 1x10-7 M)
Lineal ([I] = 1x10-6 M)
Lineal ([I] = 1x10-5 M)
Lineal ([I] = 1x10-4 M)
Lineal ([I] = 1x10-3 M)
Gráfica 15. Determinación del efecto de la NFIMI sobre la AChE y el tipo de inhibición por
el método de Lineweaver Burk.
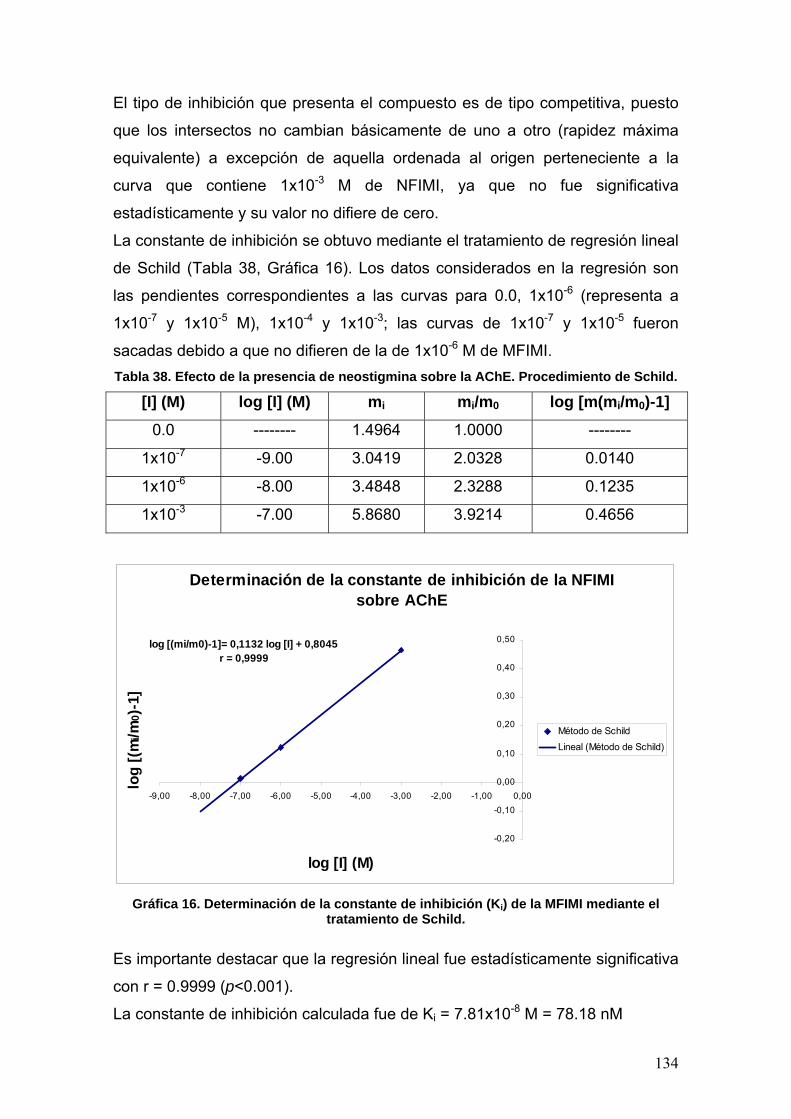
134
El tipo de inhibición que presenta el compuesto es de tipo competitiva, puesto
que los intersectos no cambian básicamente de uno a otro (rapidez máxima
equivalente) a excepción de aquella ordenada al origen perteneciente a la
curva que contiene 1x10-3 M de NFIMI, ya que no fue significativa
estadísticamente y su valor no difiere de cero.
La constante de inhibición se obtuvo mediante el tratamiento de regresión lineal
de Schild (Tabla 38, Gráfica 16). Los datos considerados en la regresión son
las pendientes correspondientes a las curvas para 0.0, 1x10-6 (representa a
1x10-7 y 1x10-5 M), 1x10-4 y 1x10-3; las curvas de 1x10-7 y 1x10-5 fueron
sacadas debido a que no difieren de la de 1x10-6 M de MFIMI. Tabla 38. Efecto de la presencia de neostigmina sobre la AChE. Procedimiento de Schild.
[I] (M) log [I] (M) mi mi/m0 log [m(mi/m0)-1]
0.0 -------- 1.4964 1.0000 --------
1x10-7 -9.00 3.0419 2.0328 0.0140
1x10-6 -8.00 3.4848 2.3288 0.1235
1x10-3 -7.00 5.8680 3.9214 0.4656
Determinación de la constante de inhibición de la NFIMI sobre AChE
log [(mi/m0)-1]= 0,1132 log [I] + 0,8045r = 0,9999
-0,20
-0,10
0,00
0,10
0,20
0,30
0,40
0,50
-9,00 -8,00 -7,00 -6,00 -5,00 -4,00 -3,00 -2,00 -1,00 0,00
log [I] (M)
log
[(m
i/m0)
-1]
Método de SchildLineal (Método de Schild)
Gráfica 16. Determinación de la constante de inhibición (Ki) de la MFIMI mediante el
tratamiento de Schild.
Es importante destacar que la regresión lineal fue estadísticamente significativa
con r = 0.9999 (p<0.001).
La constante de inhibición calculada fue de Ki = 7.81x10-8 M = 78.18 nM

135
Ki = 78.18 + 5.37 nM Cabe destacar que en el medio del análisis de cinética enzimática (buffer de
fosfatos 0.1 M, pH = 8.0) la p-Nitrofenilisomaleimida (NFIMI) sufre una
transformación química. Para determinar la estabilidad del compuesto en el
medio se preparó 1.0 mL de disolución 0.1 M de NFIMI en acetona; a la cual se
le agregaron 1.4 mL de buffer de fosfatos. La mezcla se agitó manualmente
obteniendo una parte sólida (derivada de una precipitación) y otra soluble. El
sólido fue filtrado a gravedad y se dejó secar a temperatura ambiente, mientras
que la parte en solución fue colocada en un matraz bola para evaporar la
acetona y el agua hasta sequedad a la misma temperatura. Las muestras
totalmente secas fueron analizadas por RMN de 1H para caracterizar lo que se
había formado, obteniéndose los resultados mostrados en la Tabla 39.
Los resultados obtenidos sugieren que la NFIMI se transforma en 2
compuestos al contacto con el buffer de fosfatos; uno de ellos corresponde al
ácido p-nitrofenilmaleámico (NFMA, parte soluble) y la p-nitrofenilmaleimida
(NFMI, parte sólida). Los espectros de RMN de 1H revelaron la formación del
ácido p-nitrofenilmaleámico (NFMA) en la parte soluble, mientras que en la
parte sólida se forma la p-nitrofenilmaleimida (NFMI). Es importante señalar los
cambios espectrales de la NFIMI y la NFMI formada ya que son contundentes;
los protones vinílicos H-2 y H-3 de la NFIMI presentan 2 señales dobles con
desplazamientos químicos 6.78 y 7.43 ppm respectivamente, mientras que en
el mismo sistema (hidrógenos vinílicos) la NFMI presenta solo una señal simple
que integra para 2 hidrógenos con una δ de 7.23 ppm39. El sistema aromático
no sufre cambios significativos entre las 2 moléculas. En relación al NFMA
formado a partir de NFIMI, los cambios son determinantes, ya que presenta las
señales típicas de la NFMA (ver Tabla 17).
Probablemente la formación de la mezcla (NFMA y NFMI) se deba a la
debilitación del átomo de nitrógeno imídico (Figura 65) de la NFIMI por la
influencia electroatractora del grupo –NO2 que posee la molécula; por ello, los
fosfatos llegan a reaccionar con la NFIMI formando la NFMI como sucede en el
sistema con sustituyente –H; no obstante, la influencia del grupo –NO2 permite
que la NFIMI reaccione por ataque nucleofílico del agua dando lugar a la
posiblemente formación de la sal de potasio del NFMA (Figura 66).
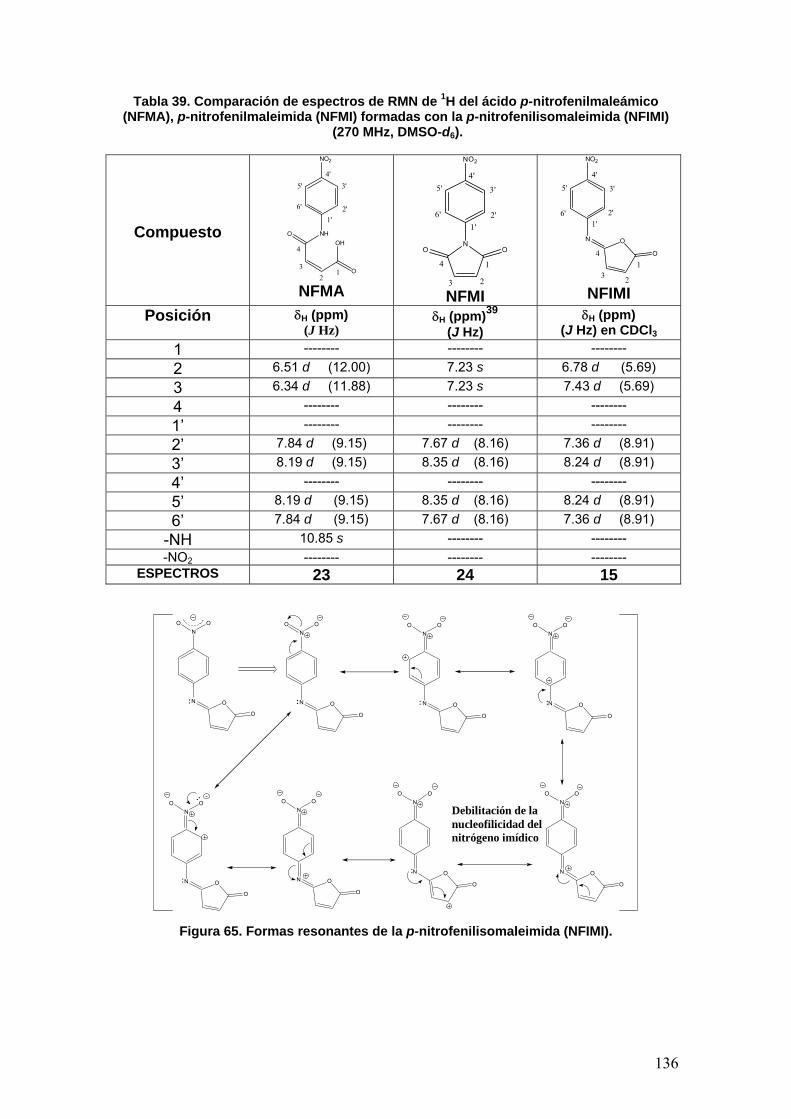
136
Tabla 39. Comparación de espectros de RMN de 1H del ácido p-nitrofenilmaleámico (NFMA), p-nitrofenilmaleimida (NFMI) formadas con la p-nitrofenilisomaleimida (NFIMI)
(270 MHz, DMSO-d6).
Compuesto NH
O
OOH
NO2
12
3
4
1'2'
3'4'
5'
6'
NFMA
NO2
NOO
1
23
4
1'2'
3'
4'5'
6'
NFMI
N O
O
NO2
1
23
4
1'2'
3'
4'
5'
6'
NFIMI
Posición δH (ppm) (J Hz)
δH (ppm)39 (J Hz)
δH (ppm) (J Hz) en CDCl3
1 -------- -------- -------- 2 6.51 d (12.00) 7.23 s 6.78 d (5.69) 3 6.34 d (11.88) 7.23 s 7.43 d (5.69) 4 -------- -------- -------- 1’ -------- -------- -------- 2’ 7.84 d (9.15) 7.67 d (8.16) 7.36 d (8.91) 3’ 8.19 d (9.15) 8.35 d (8.16) 8.24 d (8.91) 4’ -------- -------- -------- 5’ 8.19 d (9.15) 8.35 d (8.16) 8.24 d (8.91) 6’ 7.84 d (9.15) 7.67 d (8.16) 7.36 d (8.91)
-NH 10.85 s -------- -------- -NO2 -------- -------- --------
ESPECTROS 23 24 15
NOO
N O
O
NOO
N O
O
NOO
N O
O
NOO
N O
O
NOO
N O
O
Debilitación de lanucleofilicidad delnitrógeno imídico
NOO
N O
O
NOO
N O
O
NOO
N O
O
Figura 65. Formas resonantes de la p-nitrofenilisomaleimida (NFIMI).
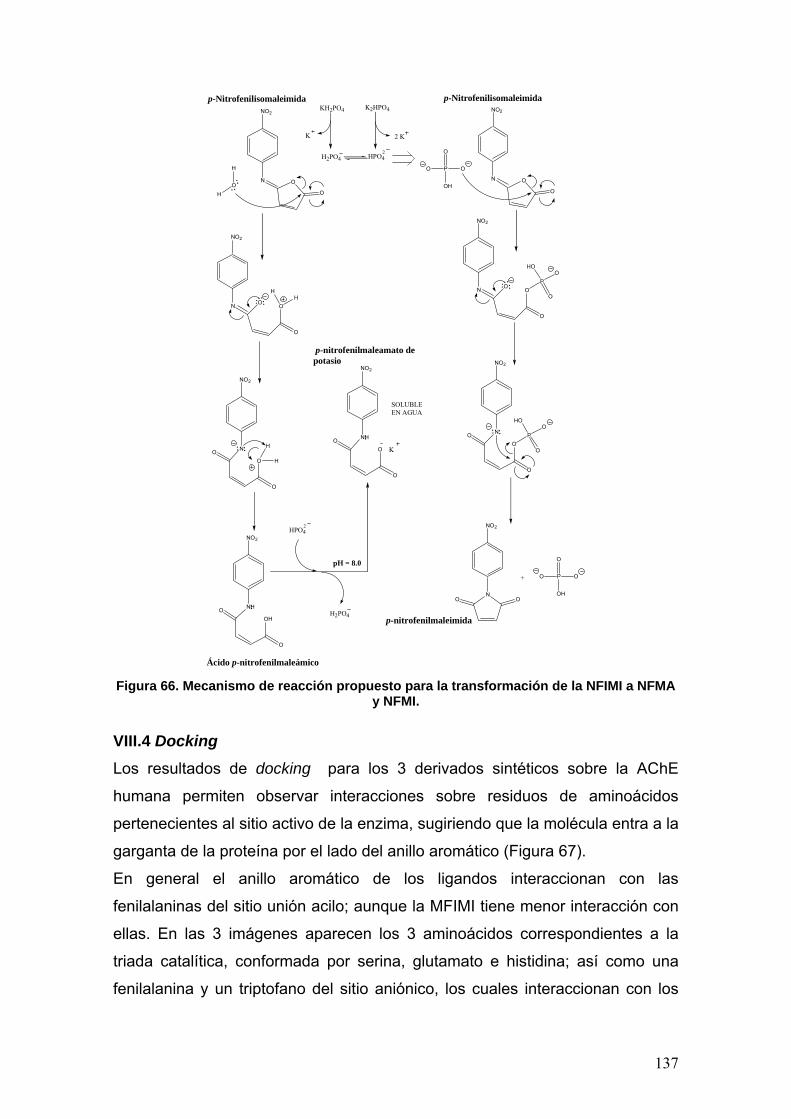
137
H2PO4NHO
OH
O
NO2
P
O
O O
OHNOO
NO2HPO4
KNOO
O
NO2
H
H
NHOO
O
NO2
NOO
O
PO
O
HO
NO2
N OO
O
NO2
HH
N OO
O
PO
O
HO
NO2
H
OH
P
O
O O
OHN O
O
NO2
N O
O
NO2
H2PO4 HPO4
2 KK
KH2PO4 K2HPO4
2
+
p-Nitrofenilisomaleimida
p-nitrofenilmaleimida
p-Nitrofenilisomaleimida
Ácido p-nitrofenilmaleámico
2
pH = 8.0
p-nitrofenilmaleamato depotasio
SOLUBLEEN AGUA
Figura 66. Mecanismo de reacción propuesto para la transformación de la NFIMI a NFMA
y NFMI.
VIII.4 Docking
Los resultados de docking para los 3 derivados sintéticos sobre la AChE
humana permiten observar interacciones sobre residuos de aminoácidos
pertenecientes al sitio activo de la enzima, sugiriendo que la molécula entra a la
garganta de la proteína por el lado del anillo aromático (Figura 67).
En general el anillo aromático de los ligandos interaccionan con las
fenilalaninas del sitio unión acilo; aunque la MFIMI tiene menor interacción con
ellas. En las 3 imágenes aparecen los 3 aminoácidos correspondientes a la
triada catalítica, conformada por serina, glutamato e histidina; así como una
fenilalanina y un triptofano del sitio aniónico, los cuales interaccionan con los
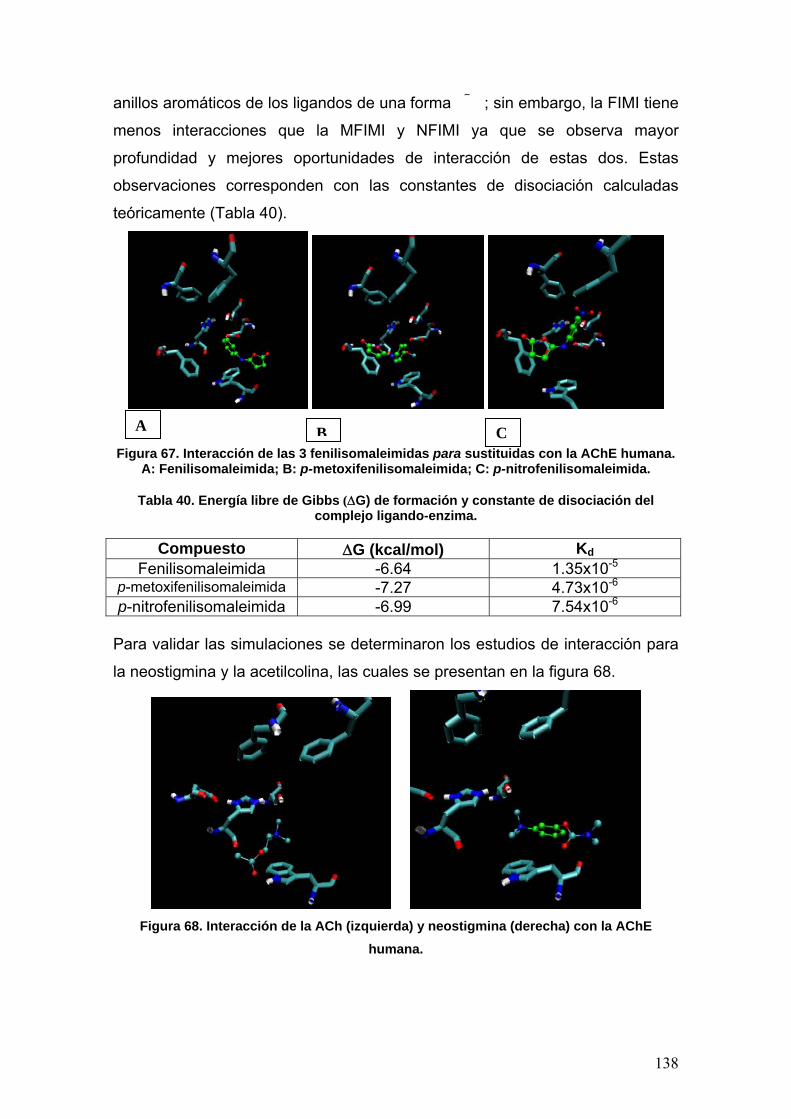
138
anillos aromáticos de los ligandos de una forma ��; sin embargo, la FIMI tiene
menos interacciones que la MFIMI y NFIMI ya que se observa mayor
profundidad y mejores oportunidades de interacción de estas dos. Estas
observaciones corresponden con las constantes de disociación calculadas
teóricamente (Tabla 40).
Figura 67. Interacción de las 3 fenilisomaleimidas para sustituidas con la AChE humana. A: Fenilisomaleimida; B: p-metoxifenilisomaleimida; C: p-nitrofenilisomaleimida.
Tabla 40. Energía libre de Gibbs (ΔG) de formación y constante de disociación del
complejo ligando-enzima.
Compuesto ΔG (kcal/mol) Kd Fenilisomaleimida -6.64 1.35x10-5
p-metoxifenilisomaleimida -7.27 4.73x10-6 p-nitrofenilisomaleimida -6.99 7.54x10-6
Para validar las simulaciones se determinaron los estudios de interacción para
la neostigmina y la acetilcolina, las cuales se presentan en la figura 68.
Figura 68. Interacción de la ACh (izquierda) y neostigmina (derecha) con la AChE
humana.
A B C

139
Como se puede observar la ACh interacciona con las fenilalaninas del seco
acilo, así como con la triada catalítica, mientras que la neostigmina se une a los
mismo aminoácidos pero favoreciendo las interacciones tipo � �.
Es importante comparar los datos experimentales como los teóricos, para
observar el comportamiento del sistema de una manera más analítica (Tabla
41).
Las fenilisomaleimidas para monosustituidas no se pudieron evaluar ya que
sufren una transformación química en el medio, sin embargo, fue posible
evaluar lo que se encontraba en la solución. Con respecto a FIMI se evaluó la
FMI cuya constante de inhibición es aproximadamente 160 veces más pequeña
que la teórica, debido a que los estudios de docking únicamente proporcionan
afinidad en el vacío y no toma en cuenta los factores experimentales que
pueden influir en la proteína, como en el ligando.
En relación al sistema MFIMI esta sufre una transformación a FMI que no se
puede evaluar debido a su insolubilidad en el medio, y a MFMA cuyas
constantes de disociación tanto experimental como teórica probablemente
coincidan, ya que la Ki determinada en cinética enzimática es menor, puesto
que en el ensayo sólo se evaluó la parte soluble, por tal motivo, las
concentraciones de MFMA son menores a las probadas. Lo mismo sucede con
el sistema NFIMI, donde la parte soluble fue evaluada (NFMA) y la parte
insoluble (NFMI) no pudo ser probada; sin embargo, las constantes de
disociación teórica y experimental no coinciden ya que la primera es por lo
menos ~450 veces más grande que la experimental; es decir, que la NFMA
tiene mayor afinidad sobre la AChE que la FIMI y MFIMI.
Con respecto a la ACh y neostigmina los datos experimentales y teóricos son
muy similares ya que la primera difiere ~20 veces una de otras, mientras que la
segunda aproximadamente 100 veces.

140
Tabla 41. Comparación de las constantes de disociación (Docking) e inhibición (experimental) de los compuestos de prueba.
Compuesto Kd (Docking) Ki (M)
N O
O
FIMI
1.35x10-5
ND
NOO
FMI
2.95x10-5 47
1.86x10-7
N O
O
OCH3
MFIMI
4.73x10-6
ND
NOO
OCH3
MFMI
1.41x10-5 47
ND*
NH
OH
OO
OCH3
MFMA
4.29x10-6 47
< 8.31x10-4
N O
O
NO2
NFIMI
7.54x10-6
ND
NOO
NO2
NFMI
1.18x10 47
ND*
NH
OH
OO
NO2
NFMA
3.54x10-5 47
< 7.81x10-8
H3C O
O
NH3C
H3CCH3 ACh
2.94x10-5
Km (M)
6.82x10-4
N
O
CH3
CH3
NH3C
CH3
H3C
Neostigmina
7.58x10-7
7.28x10-9
ND: No se puede determinar (inestables); ND*: No se puede determina por insolubilidad en el medio.

141
IX. CONCLUSIONES 1) Se sintetizaron e identificaron los tres ácidos p-X-fenilmaleámicos con
rendimientos mayores al 95%. La síntesis del ácido NFMA necesitó el doble de
tiempo de reacción que los ácidos FMA y MFMA debido al grupo electroatractor
que posee.
2) Se sintetizaron e identificaron las tres p-X-fenilisomaleimidas cuyos
rendimientos fueron mayores al 97% para FIMI y MFIMI, mientras que NFIMI
sólo alcanzó ~28% debido a la debilitación del ataque nucleofílico del nitrógeno
amídico del ácido NFMA influenciado por el grupo –NO2 que no permite
eficientemente la formación de NFIMI.
3) Se evaluaron todas y cada una de las fenilisomaleimidas en cinética
enzimática sobre AChE de Torpedo californica in vitro.
4) La FIMI no pudo ser evaluada ya que sufre una isomerización completa a
FMI en el medio (buffer de fosfatos 0.1 M, pH = 8.0). La FMI tuvo efectos
inhibitorios sobre la proteína de una manera competitiva.
5) La MFIMI no fue evaluada porque sufre reacciones químicas en el medio de
ensayo, obteniendo una parte insoluble (MFMI, no evaluada) y una parte
soluble correspondiente a la sal potásica de MFMA, la cual tuvo efecto
inhibitorio de tipo competitivo sobre la enzima.
6) La NFIMI sufre una transformación en el medio de reacción enzimática, lo
cual imposibilitó su evaluación; sin embargo, sólo pudo ser evaluada la sal
potásica de NFMA (parte soluble), la cual tiene efecto inhibitorio competitivo
sobre la AChE de T. californica. La parte insoluble en el medio corresponde la
NFMI.
7) Los compuestos fueron evaluados por simulación de la interacción ligando-
enzima asistido por computadora, la cual predijo que la FIMI era el ligando con
menor afinidad hacia la enzima, mientras que la MFIMI y NFIMI tenía
prácticamente la misma afinidad.
8) Experimentalmente, las constantes de inhibición de las partes evaluadas
(FMI y MFMA) se acercan en valor a lo reportado por docking; a excepción del
NFMA. Los datos de docking para las partes evaluadas (FMI, MFMA y NFMA)
predicen que el MFMA es el más afín de los tres compuestos (MFMA > FMI ≈
NFMA), pero experimentalmente el NFMA es el más afín del grupo, seguido por
FMI y MFMA (NFMA > FMI > MFMA).

142
9) Probablemente el compuesto con grupo –NO2 favorezca mejor las
interacciones π con alta densidad electrónica con π de baja densidad
electrónica; contrario al compuesto MFMA el cual posee un grupo
electrodonador (-OCH3), que probablemente desfavorece dichas interacciones
sobre la enzima.
X. REFERENCIAS
[1] Katzung Bertram G.; Farmacología básica y clínica; 9ª Edición; Trad. Monteón Batalla Ignacio de Jesús; Edit. El Manual Moderno, S.A. de C.V.; México, D.F.; (2005); p.p. 77-85
[2] http://www.herrera.unt.edu.ar/bioingenieria/Temas_inves/sist_nervioso/pagina1.htm /30/09/06/
[3] Hardman Joel G.; Limbird Lee E.; Las Bases Farmacológicas de la Terapéutica; 10ª Edición; Capítulo 6; “Neurotransmisión” Hoffman Brian B.; Taylor Palmer; p.p.125-129, 134-154, Capítulo 8; “Anticolinesterasas” Taylor Palmer; p.p. 183-190; Edit. McGraw-Hill; México, D.F.; (2004); Vol. I.
[4] Avendaño López María del Carmen; Introducción a la Química Farmacéutica; 1ª Edición; Edit. McGraw-Hill; España, Madrid; (1996); p.p. 31-35, 47-61, 73-92.
[5] Reyes Chumacera L. Antonio, Cruz Borbolla Julián, Nicolás Vázquez María Inés, Reyes Trejo Lino J.; Curso-Taller: Introducción a la Química Computacional; Manual; FES-Cuautitlán-U.N.A.M.; (2004); p.p. 1-13, 25 - 44.
[6] Padilla Zúñiga Jacqueline A., Rojo Domínguez Arturo; Simulación del Reconocimiento entre proteínas y moléculas orgánicas o Docking. Aplicación al diseño de fármacos; Mensaje Bioquímico, Vol. XXVI; Depto. De Bioquímica, Facultad de Medicina; U.N.A.M.; México, D.F. (2002); p.p. 129-145.
[7] http://www.csb.yale.edu/userguides/datamanip/autodock/html/Using_AutoDock_305.9.html#pgfId=34405 /14/10/06/
[8] Morris Garrett M., Goodsell David S., Halliday Robert S., Huey Ruth, Hart William E., Belew Richard K., Olson Arthur J.; “Automated Docking Using a Lamarckian Genetic Algorithm and Empirical Binding Free Energy Function”; Journal of Computational Chemistry; Vol. 19, 14; (1998); p.p. 1639-1662.
[9] McMurry John; Química Orgánica; 3a Edición; Trad. Pozas Horcasitas Rocío, García de la Mora Gustavo; Edit. Grupo Editorial Iberoamérica S.A. de C.V.; México, D.F.; (1994); p.p. 796-798, 808-811, 940-941.

143
[10] Thornton Morrison Robert, Neilson Boyd Robert; Química Orgánica; 3a Edición; Trad. Zugazagoitia Herranz Rosa, Fiedler Peter, Rock Cristina; Edit. Addison-Wesley- Iberoamérica; EE.UU. (1990); p.p. 855-857.
[11] Smith Michael B., March Jerry; March’s Advanced Organic Chemistry: Reactions, Mechanisms, and Structure; 5th Edition; Edit. A Wiley-Interscience Publication John Wiley & SONS., Inc.; U.S.A. MA; (2001); p.p. 514.
[12] Hardman Joel G.; Limbird Lee E.; Las Bases Farmacológicas de la Terapéutica; Capítulo 6; “Neurotransmisión” Westfall Thomas C., Westfall David P.; p.p. 152; Capítulo 8 “Anticolinesterasas” Taylor Palmer; p.p. 201-216; Capítulo 20 “Tratamiento de los trastornos degenerativos del S.N.C.” Standaert David G., Young Anne B.; p.p. 527-540; Trad. Benglio J. Rafael, Orizaga Samperio Jorge, Pérez-Tamayo Ruiz Ana María; Pinto 11ª Edición; Edit. McGraw-Hill; México, D.F.; (2006).
[13] Taylor Palmer; “The Cholinesterases”; The Journal of Biological Chemistry; Vol. 6; 5; (1991); p.p. 4025-4028.
[14] Lin Gialih, Lai Cheng-Yue and Liao Wei-Chung; “Molecular Recognition by Acetylcholinesterase at the Peripheral Anionic Site: Structure-Activity Relationships for Inhibitors by Aryl Carbamates”; Bioorganic & Medicinal Chemistry; 7; (1999); p.p. 2683-2689.
[15] Froede Harry C., Wilson Irwin B.; “Direct Determination of Acetyl-enzyme Intermediate in the Acetylcholinesterase-catalyzed Hydrolysis of Acetylcholine and Acetylthiocholine”; The Journal of Biological Chemistry; Vol. 257; 17; (1984); p.p. 11010-11012.
[16] Voet Donald, Voet Judith G.; Biochemistry; 2th Edition; Edit. John Wiley & Sons. Inc.; U.S.A.; (1995); p.p. 353.
[17] Rodríguez-Ithurralde Daniel, Olivera Silvia; “Enfermedad de Alzheimer: Preocupación de Todos” http: //iibce.edu.uy/neuromol/AlzheiPosdata.html /15/12/05/
[18] http://www.industria-farmaceutica.com/index.php?name=News&catid=&topic=20 /03/09/05/
[19] Guyton Arthur C., Hall John E.; Tratado de Fisiología Médica; 10a Edición; Trad. Agud Aparicio José Luis, Álvarez Baleriola Isabel, de Dios Perrino Consuelo, et. al.; Edit. McGraw-Hill Interamericana; México; D.F.; (2001); p.p. 799-829.
[20] Hamilton Susan E., Nathanson Neil M.; “The M1 receptors is required for muscarinic activation of Mitogen-activated protein (MAP) kinase in murine cerebral cortical neurons”; The Journal of Biological Chemistry; Vol. 276; 19; (2001); p.p. 15850-15853.

144
[21] Shinoe Toru, Matsui Minoru, Taketo Makoto M., Manabe Toshiya; “Modulation of synaptic plasticity by physiological activation of M1 muscarinic acetylcholine receptors in the mouse Hippocampus”; The Journal of Neurosciences; 25; 48; (2005); p.p. 11194-11200.
[22] Marino Michael J., Rouse Susan T., Levey Allan I., Potter Lincoln T., Conn Jeffrey; “Activation of the genetically defined m1 muscarinic receptor potentates N-methyl-D-aspartate (NMDA) receptor currents in hippocampal pyramidal cells”; Proc. Natl. Acad. Sci.; Vol. 95; p.p. 11465-11470.
[23] Yamauchi Takashi; “Neuronal Ca2+/Camodulin-Dependent Protein Kinase II – Discovery, progress in a quarter of a century, and perspective: Implication for learning and memory”; Biol. Pharm. Bull. 28; 8; (2005); 1342-1354.
[24] Alberts Bruce, Bray Dennis, Lewis Julian, Raff Martin, Roberts Keith, Watson James D.; Biología Molecular de la célula; 3a Edición; Trad. Durfort i Coll Mercè, Llobera i Sande Miquel; Edit. Ediciones Omega; España, Barcelona; (2002); p.p. 801-804.
[25] Copeland Robert A.; Enzymes: A practical introduction to structure, mechanism and data analysis; 2nd Edition; Edit. John Wiley & Sons. Inc.; U.S.A.; (2000); p.p. 1-5; 109-120; 128-135; 266-281.
[26] Lehninger Albert L., Nelson David L., Cox Michael M.; Principios de bioquímica; 2a Edición; Trad. Cuchillo Foix Claudi M., Vendrell i Roca Joseph; Edit. Ediciones Omega, S.A.; España, Barcelona; (1995); p.p.198- 236.
[27] http://html.rincondelvago.com/alzheimer.html /12/11/06/
[28] http://www.medicinainformacion.com/dmb_070904.htm /12/11/06/
[29] Bonting Sjored L., Featherstone Robert M.; “Ultramicro Assay of the Cholinesterases”; Archives of Biochemistry and Biophysics; 61; (1956); p.p. 89-98.
[30] Ellman George L.; Courtney K. Diane; Andres Valentino Jr., Featherstone Robert M.; “A new and rapid colorimetric determination of Acetylcholinesterase activity”; Biochemical Pharmacology; Vol. 7; (1961); p.p. 88-95.
[31] Trujillo Ferrara José, Montoya Cano Leticia, Espinoza Fonseca Michel; “Synthesis, Anticholinesterase activity of m-aminobenzoic acid derivatives”; Bioorganic & Medicinal Chemistry Letters; 13; (2003); p.p. 1825-1827.
[32] Trujillo Ferrara J., Vázquez Iván, Espinosa Judith, Santillán Rosa, Farfán Norberto, Höpfl Herbert; “Reversible and irreversible inhibitory of succinic and maleic acid derivatives on Acetylcholinesterase”; European Journal of Pharmaceutical Sciences; 18; (2003); p.p. 313-322.

145
[33] Flores Sandoval C. A., Zaragoza I. P., Marañón Ruiz V. F., Correa Basurto J., Trujillo Ferrara J.; “Theorical study succinic and maleic acid derivatives”; Journal of Molecular Structure: THEOCHEM; 713; (2005); p.p. 127-134.
[34] Correa Basurto José, Espinosa Raya Judith, González May Mario, Espinoza Fonseca Michel, Vázquez Alcántara Iván, Trujillo Ferrara José; “Inhibition of Acetylcholinesterase by two arylderivatives: 3a-Acetoxy-5H-pyrrolo(1,2-a) (3,1)benzoxazin-1, 5-(3aH)-diona and cis-N-p-Acetoxy-phenylisomaleimide”; Journal of Enzyme Inhibition and Medicinal Chemistry; 00 (0); (2006); p.p. 1-6.
[35] Kitz R, Wilson Irwin B.; “Esters of Methasulfonic acid as irreversible inhibitors of Acetylcholinesterase”; The Journal of Biological Chemistry; Vol. 237; 10; (1962); p.p. 3245-3249.
[36] Cotter Robert J., Sauers Carol K., Whelan John M.; “The synthesis of N-substituted isomaleimides”; The Journal Organic Chemistry; Vol. 26; 10; (1961); p.p. 10-15. [37] Oishi Tsutomu, Fujimoto Minoru, Yoshimoto Nobuko, Kimura Tadashi; “Anionic Polymerizations of N-substituted isomaleimide”; Polymer Journal; Vol. 21; 8; (1989); p.p. 655-659. [38] Barba Víctor, Hernández Cecilia, Rojas-Lima Susana, Farfán Norberto, Santillán Rosa; “Preparation of N-aryl-substituted spiro-β-lactams via Staudinger cycloaddition”; Can. Journal Chem.; 77; (1999); p.p. 2025-2032. [39] Fruk Ljiljana, Graham Duncan; “The electronic effects on the formation of N-aryl-maleimides and isomaleimides”; Heterocycles; Vol. 60; 10; (2003); p.p. 2305-2313. [40] Quintana Zavala Delia; Estudio preliminar para el desarrollo de una síntesis asimétrica de �-amino ácidos �-sustituidos. Preparación y alquilación altamente diastereoselectiva de N-N’-acetales quirales derivados del ácido �-aminopropiónico; Tesis para obtener el grado de maestro en ciencias; México, D.F. CINVESTAV-IPN; (1989); p.p. 41. [41] http://acpcommunity.acp.edu./Facultystaff/hass/oc2web/lab/exp/qa/class/alkfe3.pdf /23/11/06/ [42] Tsubouchi Akira, Langtao Shen, Yukihiro Hara, Masayasu Akiyama; “Iron (III) complexation behavior of benzene-centered tripodal mono-di- and tritopic ligands carrying a repeating –Ahe-(OH) Apr-sequence [Ahe=6-aminohexanoyl; (OH)Apr=3-(N-hydroxy)-aminopropanoyl]; New J. Chem.; 25; (2001); 275-282.
[43] http://www.sigmaaldrich.com/catalog/search/ProductDetail/SIGMA/C3389 /28/11/06/
[44] http://www.scartd.org/arxius/silva02.pdf /28/11/06/

146
[45] Endou Masayuki, Tanito Yasuto, Okumura Fukuichiro; “A comparison between chronotropic effects of neostigmine and edrophonium in isolated Guinea pig right atrium”; The Journal of Pharmacology and Experimental Therapeutics; Vol. 282; 3; (1997); p.p. 1480-1486. [46] Sagane Yoshimasa, Nakagawa Tomoyuki, Yamamoto Kosuke, Michikawa Soichi, Oguri Suguru, Momonoki Yoshie S.; “Molecular characterization of maize Acetylcholinesterase. A novel enzyme family in the plant Kingdom”; Plant Physiology; Vol. 138; (2005); p.p. 1359-1371. [47] Correa Basurto José, Flores Sandoval César, Marín Cruz Jesús, Rojo Domínguez Arturo, Espinoza Fonseca L. Michel, Trujillo Ferrara José G.; “Docking and quantum mechanic studies on cholinesterases and their inhibitors”; European Journal of Medicinal Chemistry; XX; (2006); p.p. 1-10. [48] Mateos Muñoz Agustín; Etimologías griegas del español; 22ª Edición; Edit. Editorial Esfinge, S.A. de C.V.; México; (1994); p.p. 85.





























