Embed Size (px)
Citation preview

Temperature Compensation through Systems Biology
Peter Ruoff 1 *, Maxim Zakhartsev2# and Hans V. Westerhoff3
1Department of Mathematics and Natural Science, University of Stavanger, N-4036
Stavanger, Norway, 2Biochemical Engineering, International University of Bremen,
Campus Ring 1, D-28759 Bremen, Germany, EU, 3Manchester Centre for Integrative
Systems Biology, School for Chemical Engineering and Analytical Sciences, The
University of Manchester, Manchester M601QD, UK, EU and BioCentrumAmsterdam, BioCentre Amsterdam, FALW, Free University, NL-1081 HV Amsterdam,The Netherlands, EU.
Running title:Temperature Compensation of Fluxes
Subdivision:Systems biology
*Corresponding author:P. Ruoff, Department of Mathematics and Natural Science, Faculty of Science and
Technology, University of Stavanger, N-4036 Stavanger, Norway. Email:
[email protected]; fax: (+47) 51841750; url: http://www.ux.uis.no/~ruoff
Keywords:Temperature compensation, gene expression, metabolic regulation, control
coefficients, systems biology
#present address: Department of Marine Animal Physiology, Alfred Wegener Institute
for Marine and Polar Research (AWI), Building E, office 1555, Am Handelshaven 12,D-27570 Bremerhaven, Germany.

- 2 -
Summary
Temperature has a strong influence on most individual biochemical reactions.
Despite this, many organisms have the remarkable ability to keep certain physiological
fluxes approximately constant over an extended temperature range. In this paper it is
shown how temperature compensation can be considered as a pathway phenomenon
rather than the result of a single-enzyme property. By using metabolic control analysis
reaction networks that exhibit temperature compensation can be identified. Because
most activation enthalpies are positive, temperature compensation of a flux can occur
when certain control coefficients are negative. This can be achieved in networks with
branching reactions or if the first irreversible reaction is regulated by a feedback loop.
Hierarchical control analysis shows that networks that are dynamic through regulated
gene expression or signal transduction may offer additional possibilities to bring the
apparent activation enthalpies close to zero and leading to temperature compensation.
A calorimetric experiment with yeast provides evidence that such a dynamic
temperature adaptation can actually occur.
Introduction
Temperature is an environmental factor, which influences most of the
chemical processes occurring in living and nonliving systems. Van’t Hoff’s rule states
that reaction rates increase by a factor (the Q10) of two or more when the temperature
is increased by ten centigrades [1]. Despite this strong influence of temperature on
individual reactions, many organisms have the remarkable ability to keep some of
their metabolic fluxes at an approximately constant level over an extended
temperature range. Examples are the oxygen consumption rates of ectoterms living in
costal zones [2] and of fish [3], the period lengths of all circadian [4] and some

- 3 -
ultradian [5, 6] rhythms, photosynthesis in cold adapted plants [7, 8], homeostasis
during fever [9], or the regulation of heat shock proteins [10].
In 1957 Hastings and Sweeney suggested that in biological clocks such
temperature compensation may occur as the result of opposing reactions within the
metabolic network [11]. A later kinetic analysis of the problem [12] came essentially
to the same conclusion, and predictions of the theory were recently tested by
experiments using Neurospora’s circadian clock [13] and chemical oscillators [14,
15].
In this paper we use metabolic and hierarchical control analysis [16-22] to
show how certain steady state fluxes in static reaction networks can be temperature
compensated according to a similar principle and how dynamic networks have an
additional repertoire of mechanisms. The content of this paper is mostly theoretical,
but we use the temperature adaptation of yeast cells and the temperature adaptation of
photosynthesis as illustrations. These and other systems (for example adaptation of
gene expression) warrant experimental studies in their own right, which we wish to
cover in subsequent work.
Results and Discussion
Global Condition for Temperature Compensated Flux
In this section the condition for temperature compensation for a global
reaction kinetic network is derived. We refer to this condition as global, because the
network is assumed to contain all biochemical processes (at the genetic and metabolic
levels) that occur in the system. Consider a set of N elementary single step reactions
describing a global network. Each reaction i is assigned a rate constant ki [23] and a
steady state flux (reaction rate) Ji with associated activation enthalpy
€
Eaki . Here

- 4 -
reversible reactions may be considered as two separate reactions; an alternative is to
look upon ki as a parameter that proportionally affects both the forward and the
reverse reaction of the step [23]. Rate constants and absolute temperature are
connected through the Arrhenius equation
€
ki = Aie−Eaki
RT , where activation enthalpies
€
Eaki are considered to be independent of temperature. The Arrhenius factor Ai
subsumes any structural activation entropy. Flux Jj (i.e., the flux of elementary
reaction “j”) becomes temperature compensated within a temperature interval around
a reference temperature Tref (at which parameters and rate constants are defined) if the
following balancing equation is satisfied (for derivation, see Supplementary Material):
€
d lnJ j
d lnT=1RT
*CiJ j Ea
ki = 0i=1
N
∑ (1a)
€
*CiJ j is the global control coefficient [19, 21] of flux with respect to the rate constant
ki defined as
€
*CiJ j =
∂ lnJ j
∂ lnki.
€
*CiJ j measures the change in flux for a fractional increase
in ki, therein comprising the effects of changes in gene expression or signal
transduction that may affect the concentration and activity of the enzyme catalyzing
step. In general, the global control coefficients obey the summation theorem
€
*CiJ j =1
i=1
N
∑ [19, 21]. Because the activation enthalpies (
€
Eaki ) are positive, temperature
compensation is only possible if some of the global control coefficients are negative.
The condition for temperature compensation using metabolic control coefficients
Sometimes a biochemical system is described only at its metabolic level of
organization. In this case, one can use metabolic control coefficients denoted by
capital C without the asterisk [19, 21], which is a set addressing the control by all the

- 5 -
enzymes/steps at the metabolic level and do not include transcriptional, translational
processes or signal transduction events. The effects of these at the metabolic level
should be made explicit in terms of changes in amount or covalent modification state
of the enzymes. Accordingly, the ‘balancing equation’ is given by (see Supplementary
Material for derivation):
€
d lnJ j
d lnT= Cki
catJ j Ea
kicat
RT+ Ckm
catJ j RT
em
m∑ +
i∑ RKl
J j
l∑ Ea
Kl
RT= 0 (1b)
The first term on the right-hand side of Eq. 1b describes the contribution of
€
kicat
(turnover number), where
€
CkicatJ j =
∂ lnJ j
∂ lnkicat is the metabolic control coefficient and
€
Eakicat
is the corresponding activation enthalpy of the turnover number
€
kicat . The second term
is the contribution due to the variation of the concentration of active enzyme m (em)
by altered gene expression, translation, or signal transduction. It contains the
temperature response coefficient of the activity of that step
€
RTem ≡
d lnemd lnT
. If one is not
aware of the change in enzyme activity due to these hierarchical mechanisms, one
may measure an apparent activation enthalpy
€
Ea,apparentkicat
= Eakicat
+ RT ⋅ RTei . With this
Eq. 1b reduces to:
€
d lnJ j
d lnT= Cki
catJ j Ea,apparent
kicat
RT+
i∑ RKl
J j
l∑ Ea
Kl
RT= 0 (1c)
If an increase in temperature leads to a decreased expression level of the enzyme
catalyzing step m, the temperature response coefficient of the enzyme becomes
negative. The apparent activation enthalpy of that step in a metabolic network may
have zero or negative values.
The final term in Eq. 1b describes the contribution due to changes in the rapid
equilibria or steady states that the enzyme is engaged in with substrates, inhibitors,

- 6 -
and activators. For substrates Xl , Kl is the (apparent) Michaelis constant and
€
EaKl is
the formation enthalpy
€
ΔHl0 associated with Kl [1, 24].
€
EaKl tends to be positive in
sign, favoring dissociation at higher temperatures [1, 24].
€
RKl
J j =∂ lnJ j∂ lnKl
is the
response coefficient of the flux with respect to an increase in the Michaelis constant
[25].
An example of temperature compensation through and of an enzyme’s
expression level
In the following example we illustrate the usage of global and metabolic
control coefficients to get temperature compensation in a enzyme’s activity and steady
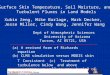
state level. Fig. 1 shows a model of an enzyme’s expression (translation and
transcription), where the enzyme catalyzes the reaction S → P. For the sake of
simplicity, we assume that transcription, translation and the degradation processes
have pseudo first-order kinetics with respect to their substrates, i.e. we neglect
saturation effects for the RNA polymerase catalyzing transcription, the RNase
catalyzing the breakdown of RNA, the ribosomes catalyzing the synthesis of E, and
the proteasomes/proteases catalyzing the degradation of E. The steady state flux (J5)
through step 5 for producing P is described by
€
J5 =k5catess[S]KM + [S]
=k1k3k5
cat
k2k4⋅
[S]KM + [S]
(2)
where ess is the steady state level of E, i.e. the level attained after all processes in the
system have relaxed, including those of transcription and translation. The global
control coefficients calculated from this equation are
€
*Ck5catJ5 =1 and
€
*CkiJ5 =1 for i=1, 3
and −1 for i=2, 4 while the (global) response coefficient with respect to the Michaelis

- 7 -
constant amounts to
€
*RKM
J5 =∂ lnJ5∂ lnKM
−KM
KM + [S]. Assuming an Arrhenius temperature
dependence of rate constants ki and
€
k5cat , J5 can be temperature compensated due to
the negative control coefficients of reactions 2 and 4. If the enthalpy of formation of
the enzyme substrate complex is negative (which is the more common case), such
compensation may also derive from the negative response coefficient.
Likewise, when describing the system at the metabolic level we get
€
Ck5catJ5 =1
and the (metabolic) response coefficient
€
RKM
J5 =∂ lnJ5∂ lnKM
−KM
KM + [S]. Because
€
d lnJ5dT
and the activation and formation enthalpies are unaffected whether the description
occurs globally or at a metabolic level, an expression for the temperature variation of
the enzyme’s steady state level can be found by comparing the global and metabolic
balancing equations (Eqs. 1a, 1b)
€
Ea,apparentk5cat
− Eak5cat
RT=d lnessd lnT
=1RT(Ea
k1 + Eak3 − Ea
k2 − Eak4 ) (3)
showing that ess can become temperature compensated when the sum of activation
enthalpies in Eq. 3 becomes zero.
Rules for temperature compensated and uncompensated flux in fixed networks
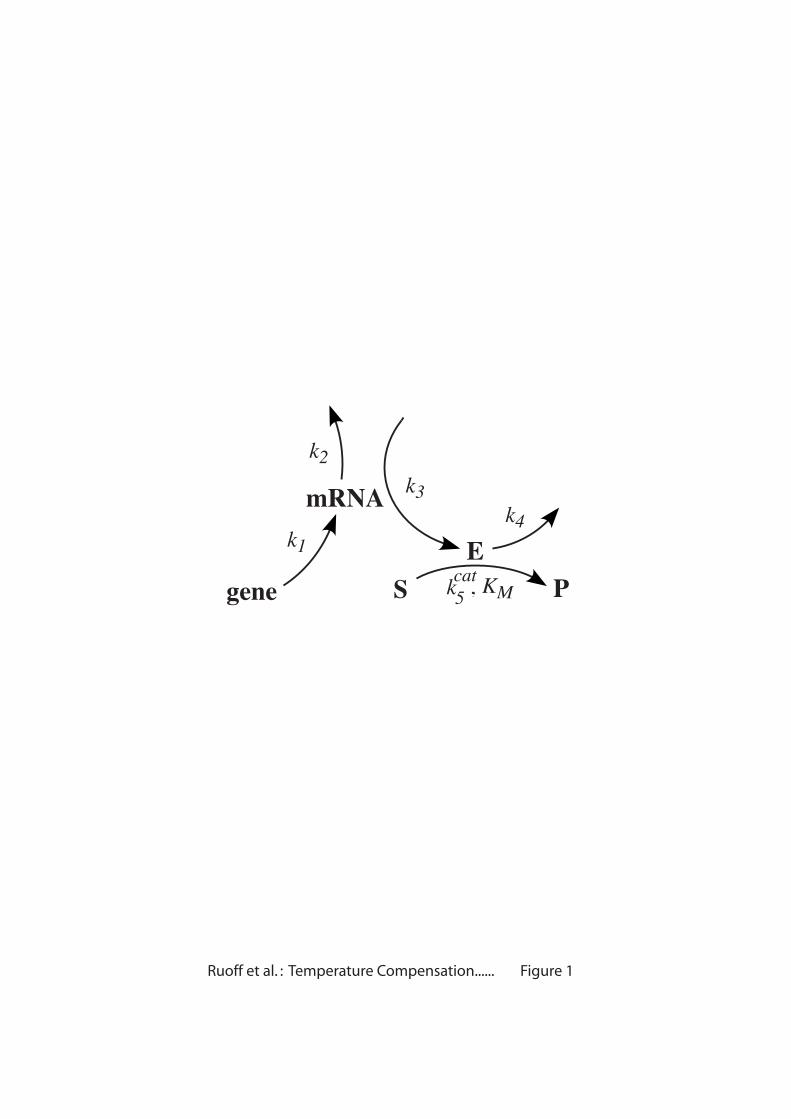
We now investigate the conditions for temperature compensation in simple
reaction networks. The fluxes (reaction rates) can be characterized as input, internal,
and output fluxes (Fig. 2a). Under what conditions can a certain (output) flux (say J’)
become temperature compensated? In order to keep such an analysis tractable the
number of reaction intermediates are limited to four. In addition, input fluxes were
limited to one with one or several output fluxes. An overview of the networks is
shown in Figs. 2b and 2c. It may be noted that these networks do not represent a

- 8 -
complete set of all possible networks containing four intermediates, but represent
examples for which temperature compensation of flux J’ becomes possible or not.
However, based on these networks it is possible to derive some general rules (see
below). For the sake of simplicity we assume that the considered networks consist of
first-order reactions (except when including feedback loops). In a metabolic context,
this would reflect the view that under physiological conditions the enzymes which
catalyze each reaction step are not saturated by their substrates [26]. Positive
feedforward or feedback loops from an intermediate I to process i are described by
substituting the original rate constant ki with kik[I]n, where k is an activation constant
and n is the cooperativity (Hill coefficient). Negative feedback or feedforward loops
from intermediate I to process i are described by substituting ki with ki/(KI+[I]m),
where KI is an inhibitor constant and m is the cooperativity. For each network the
steady state output flux J’ (indicated by the dashed box in each scheme) is examined
in terms of whether temperature compensation is possible. The tested networks (see
Supplementary Material) were then divided into those where J’ is unable to exhibit
temperature compensation (Fig. 2b) and those where J’ can be compensated (Fig. 2c).
The following rules can be stated. Temperature compensation of an output
flux is not possible for (i) any (reversible or irreversible) chain or loop of (first-order)
reactions or a branched network which has only one output flux and a product
insensitive first step (Fig. 2b, schemes (1)-(3)); (ii) networks with only one output flux
having in addition positive and/or negative feedback loops which are assigned to
internal fluxes or to an output flux, but not to the first step (Fig. 2b schemes (4)-(7)).
In all schemes of Fig. 2b we have that C1=1, while all the other control coefficients
are zero. Temperature compensation of an output flux becomes possible when (i) the
network has more than one output flux (Fig. 2c, schemes (8)-(10)) or (ii) the networks

- 9 -
have positive and/or negative feedback loops which are assigned to at least one input
flux (Fig. 2c, schemes (11)-(12)).
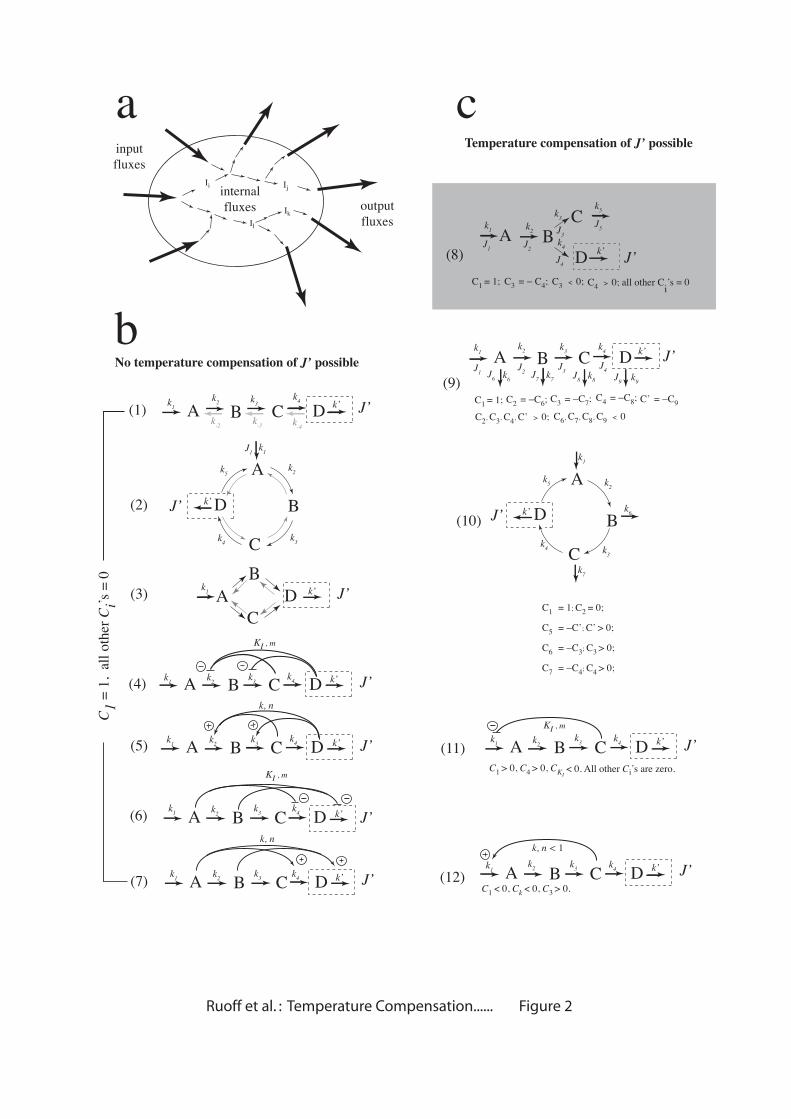
From static to dynamic temperature compensation
In the derivation of Eqs. 1a and 1b we assumed that activation enthalpies are
constants and independent of temperature. While this assumption is realistic for
single-step elementary reactions, at the metabolic level of description activation
enthalpies of enzyme catalyzing steps may depend on temperature as enzymes may be
affected by temperature dependent processes such as phosphorylation,
dephosphorylation, conformational changes, etc. Due to these different levels of
description we distinguish between static and dynamic temperature compensation. By
static temperature compensation we mean that all activation enthalpies are assumed to
be temperature independent and constant, and together fulfill the balancing equation
for a certain reference temperature. To illustrate static and dynamic temperature
compensation as well as uncompensated behavior we use scheme 8 (Fig. 2c) as an
example. This scheme is one of the simplest models that can show temperature
compensation of output flux J’. The control coefficients can be easily calculated (see
Supplementary Material). We have taken a set of arbitrary chosen rate constant
values, and it may be noted that the behavior shown below are not specific for the
chosen rate constant values. Similar behavior can be obtained with any set of rate
constants. Independent of the chosen rate constant values uncompensated behavior is
obtained when all activation enthalpies are chosen to be equal, for example
€
Ea' . In
this case, using the summation theorem
€
*CiJ ' =1
i=1
N
∑ , Eq. 1a can be expressed as
€
d lnJ 'd lnT
=Ea'
RT, showing that flux J’ is highly dependent on temperature. Such

- 10 -
uncompensated behavior is shown in Fig. 3a (open squares) when all activation
enthalpies in scheme (8) (Fig. 2c) are set to 67 kJ/mol. In this case J’ shows an
exponential increase with temperature. In Fig. 3b the increase in J’ is seen when a
15°C → 35°C temperature step is applied to the uncompensated system. In static
temperature compensation the activation enthalpies have been chosen such that Eq. 1a
is approximately fulfilled at Tref (25°C) at which the rate constants have been defined
and the control coefficients have been evaluated (open diamonds, Fig. 3a). The
condition for static temperature compensation of scheme (8) reads:
€
Eak1 + C4
J ' (Eak4 − Ea
k3 ) ≅ 0 with
€
C4J ' =
k3k3 + k4
(see Supplementary Material). Because
the control coefficients depend on the rate constants and therefore also on
temperature, the static compensated flux J’ will gradually change over an extended
temperature range as shown in Fig. 3a.
In dynamic compensation, one (or several) of the apparent (see above) or real
activation enthalpies is allowed to change as a function of temperature. Processes that
may lead to this include posttranslational processing of proteins such as
phosphorylation, dephosphorylation, or conformational changes, splice variation, etc.
[27, 28]. For example, when
€
Eak1 increases with temperature as shown in the inset of
Fig. 3a, J’ becomes practically independent of temperature (solid circles, Fig. 3a).
Incidentally, temperature compensation means that a steady state flux (or the
period length of an oscillatory flux, as for example in circadian rhythms) is (virtually)
the same at different but constant temperatures. However, when a sudden change in
temperature is applied, either as a step or as a pulse, even in temperature compensated
systems transient kinetics are observed as illustrated in Fig. 3c. By applying a
temperature step, J’ undergoes an excursion and relaxes back to its steady state. The
time scale of relaxation will be dependent on the rate constants, i.e., metabolic

- 11 -
relaxation typically occurs in the subminute range. When gene expression adaptation
is involved, relaxation may be much slower.
Fig. 3d shows how in the static compensated case of Fig. 3a the various fluxes
Ji depend on temperature. Although input flux J1 increases exponentially with
temperature (
€
J1 = k1 = A1e−Eak1
RT ), flux J4=J’ becomes compensated because J3 (which
also increases exponentially with temperature) removes just enough flux from J1 (i.e.,
J4=J1-J3) and thus ”opposes” or ”balances” J1’s contribution to J4. In a static
regulated network internal branching of the flux leading to two output fluxes may
enable temperature compensation. There is experimental evidence for such a
mechanism. For example in fish, the administration of [14C]glucose in the presence of
citrate showed a dramatic increase in the [14C]lipid/14CO2 ratio as a function of
temperature, whereas the carbon flow through the citric acid cycle was characterized
by a Q10 of less than 1 between 22°C and 38°C. This was attributed to an increased
sensitivity of acetyl-CoA carboxylase to citrate activation at higher temperatures,
resulting in elevated levels of fatty acid biosynthesis and a much lower than otherwise
expected increase in carbon flow through the citric acid cycle [2, 29].
With an increasing number of output fluxes more output fluxes can be
temperature compensated simultaneously. In scheme (9, Fig. 2c) it is easy to see that
J6 can be compensated by J2, J7 by J3, J8 by J4 and J’ by J9. In the Supplementary
Material a description is given how activation enthalpies can be found in order to
(statically) temperature compensate these output fluxes simultaneously.
Because at steady states internal fluxes are related to input and output fluxes,
the above principles of how to temperature compensate one or several output fluxes
can also be applied to internal fluxes. In networks with branch points (for example

- 12 -
scheme (8, Fig. 2c)), at least one of the downstream internal fluxes after the branch
point can be temperature compensated, while none of the upstream fluxes can show
temperature compensation unless there are more branch points upstream. The same
applies also to cyclic networks. Testing for example the irreversible clockwise scheme
(2, Fig. 2b), fluxes J2, J3, J4 and J5 can be temperature compensated while J1 and J’
cannot show temperature compensation.
Dynamic temperature compensation/adaptation in yeast
There is experimental evidence that dynamic temperature compensation
occurs. As an example we show our experimental results obtained for yeast, but
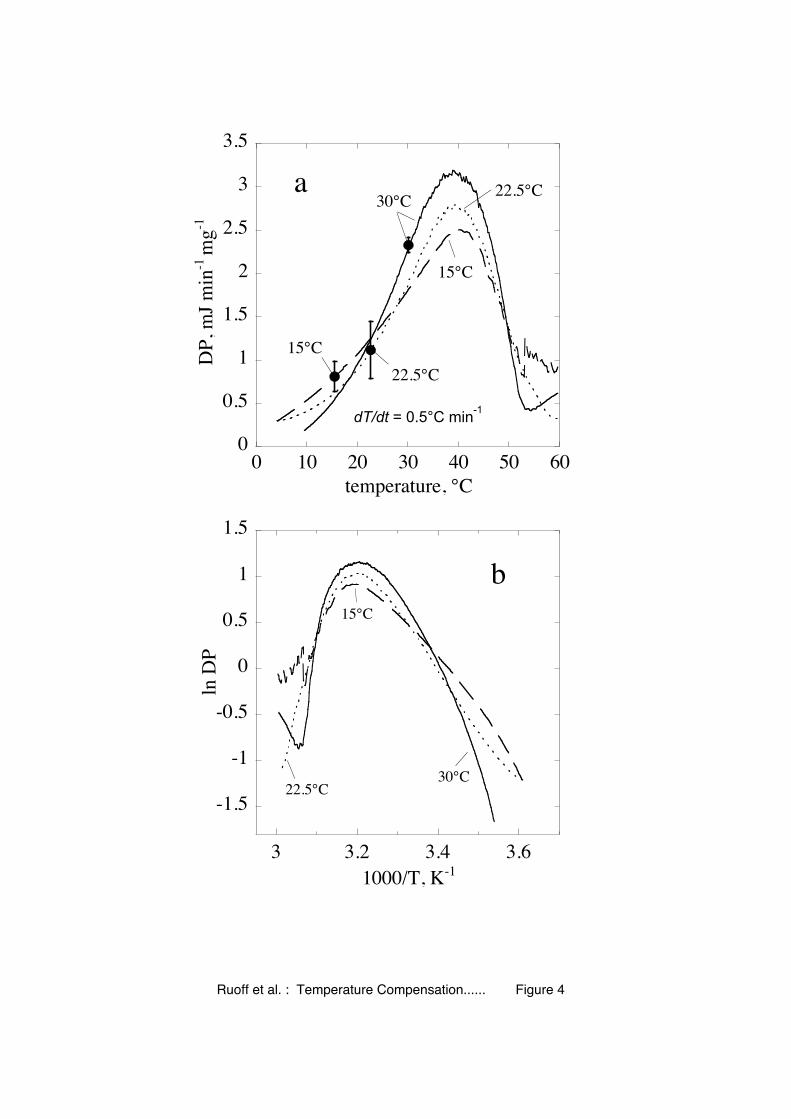
similar results have been reported for other organisms [30]. In Fig. 4 yeast cells were
acclimated at three different temperatures (15°C, 22.5°C and 30°C) and the overall
metabolic rate (measured as the heat released from the cells) was determined as a
function of temperature.
When cells adapted to 30°C were cooled down to 15°C, a 4.6-fold decrease in
metabolic rate was observed, suggesting the virtual absence of static temperature
compensation. However, when the cells are allowed to acclimate at 15°C, the
decrease in flux is only 2.2-fold, indicating a substantial temperature compensation.
As indicated above this decrease in overall activation energy may be related to a
variety of processes, such as altered gene expression or post-transcriptional
modification of proteins/enzymes, but the mechanisms behind such adaptation are still
not well understood.

- 13 -
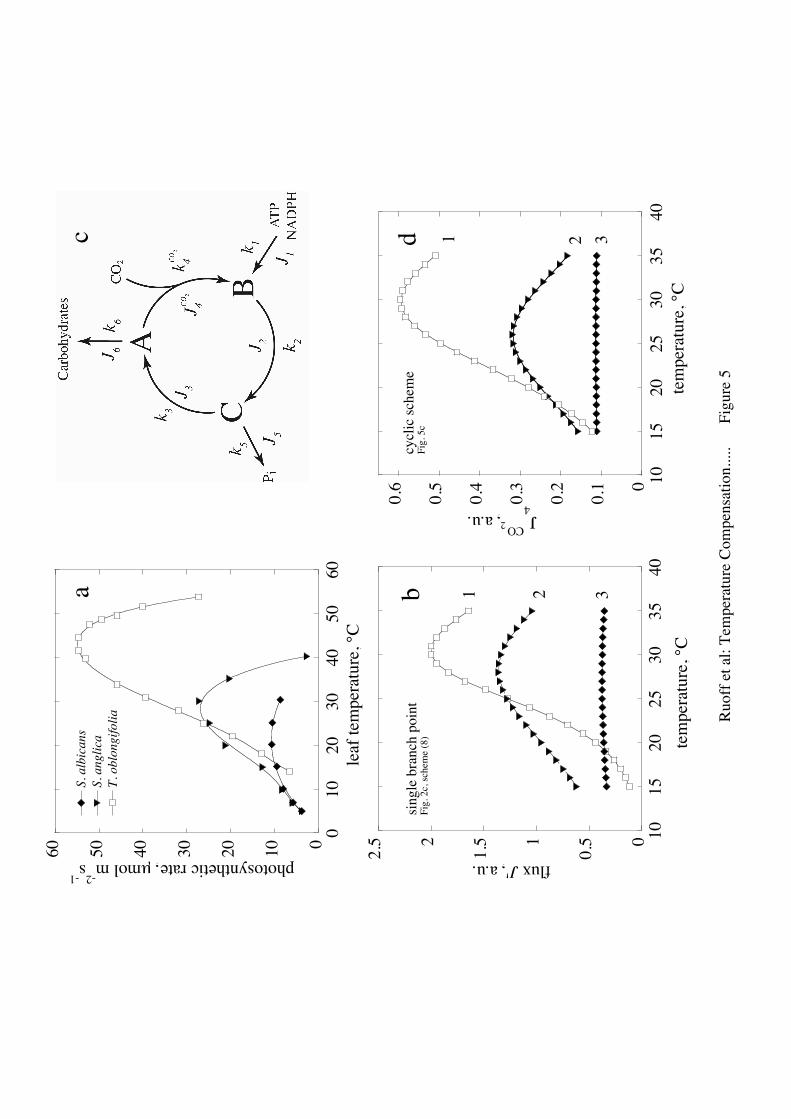
Temperature compensation in photosynthesis
Photosynthesis, i.e. the assimilation of carbon dioxide by plants, is a process
that adapts to a plant’s environment. Plants growing at low temperatures tend to have
a relatively low but often temperature compensated photosynthetic activity, while in
plants living at high temperatures photosynthesis is uncompensated with a typical
bell-shaped form. Fig. 5a shows the photosynthetic rate (in terms of CO2 uptake) for
three plant species adapted to hot (T. oblongifolia), temperate (S. anglica) and cold (S.
albicans) thermal environments [7]. Typically, the hot-adapted plant shows a relative
large variation in its photosynthetic response with a maximum at a relative high
temperature, while the cold-adapted plant shows only a small variation in its
photosynthetic response (temperature compensation). We have analyzed these
temperature responses in terms of a single branch point (because this is the simplest
system that can show temperature compensation) and in terms of a “minimal Calvin
Benson cycle”.
Considering first the single branch-point, an analysis of scheme (8, Fig. 2c)
showed that the extent by which J4 and J’ becomes temperature compensated, mainly
depends on the activation enthalpy for the influx J1 and the activation enthalpy of the
outflux J3 which ”competes with” the compensated flux J4 = J’ for intermediate B. To
get an uncompensated bell-shaped response (Fig. 5b, curve 1), activation enthalpies E3
and E4 need to be large. Reducing these activation enthalpies eventually leads to
temperature compensation (Fig. 5b, curves 2, 3).
A more realistic model is shown in Fig. 5c, when the steady states of a simple
representation of the Calvin Benson cycle are considered. This includes the balance
between the input fluxes of ATP, NADPH and CO2, and the output fluxes of ADP,
NADP+, Pi and carbohydrates. A steady states analysis of this model is given in the

- 14 -
Supplementary Material section showing that the assimilation of CO2 (
€
J4CO2 ) can be
temperature compensated, because of the balance of fluxes J1, J3, J4 (positive
contributions) with fluxes J5 and J6 (negative contributions). When the activation
enthalpies of J1 and J3 dominate
€
J4CO2 shows the bell-shaped response for hot-adapted
species (curve 1, Fig. 5d). Temperature compensation can be achieved when the
positive contributions balance the negative, i.e., when the activation enthalpies of J1
and J3 are reduced (curve 3, Fig. 5d).
Conclusion
In this paper we have derived a general relationship how temperature
compensation of biochemical steady state flux can occur by means of the balancing
equations (1a-c). Our focus here was primarily how dynamic temperature
compensation can occur through systems biology mechanisms [31]. The analysis
shows that certain network topologies need to be met in order to get negative control
coefficients. These negative control coefficients oppose the overall positive
contributions of the control coefficients as indicated by the summation theorem
€
*CiJ j
i=1
N
∑ =1 (or
€
CkicatJ j
i∑ =1 at the metabolic level). This can be achieved by various
means: positive and negative feedforward and/or feedback loops, signal transduction
events (for example by phosphorylation, dephosphorylation) and by adaptation
through gene expression. As a special case of the derived principle temperature
compensation can occur for a single enzyme (”instantaneous temperature
compensation” [2]) when balancing occurs for example between the enzyme’s
Michaelis constant (KM, KD) and its turnover number [32]. In this case, mechanisms
which include enzyme-substrate interactions, enzyme modulator interactions,

- 15 -
metabolic branch points, or conformational changes [2, 27, 28] may be involved.
While quantum mechanical tunneling is principally temperature independent, studies
with methylamine dehydrogenase showed a strong temperature dependence of the
enzyme catalyzed process where thermal activation or “breathing” of the protein
molecule is required to facilitate the tunneling reaction [33].
A challenge in applying realistic models will be the description of how
apparent activation enthalpies change with temperature and of the actual mechanisms
involved in these processes.
Materials and Methods
Determination of Yeast Metabolic Activities. The wild type yeast strain
Saccharomyces cereviciae SPY509 (from the European Saccharomyces cerevisiae
Archive of Functional Analysis (EUROSCARF, http://web.uni-
frankfurt.de/fb15/mikro/euroscarf/) with genotype MAT or a, his3_1, leu2_0, lys2_0,
ura3_0 were grown in 250 ml flasks in 100 ml of complex YPD media (10 g yeast
extract, 20 g peptone, and 20 g glucose in 1000 ml of the media) under constant
nitrogen bubbling through the media and agitated with 250 rpm at various acclimation
temperatures [34, 35]. Yeast cultures were always kept at early exponential growth
phase (OD660 < 2) by diluting the culture with fresh media at each particular
temperature. The acclimation time was for at least 10-14 days. A differential scanning
calorimeter (VP-DSC, MicroCal, USA) was used for measurements of heat
production by living yeast. The yeast cells were washed in a 100 mM glucose solution
(pH 5.5) at the relevant acclimation temperature under nitrogen bubbling so as to
remove the YPD media, which has a high specific heat capacity, resuspended in 100
mM glucose solution to 10 g wet cell biomass per liter, and incubated at the

- 16 -
acclimation temperature under a nitrogen atmosphere for 1 hour before the
measurements.
Before the measurements all solutions were degassed, including the
suspension of living cells. 100 mM glucose solution was used as the reference for the
differential scanning calorimeter (DSC) measurements. The heat production was
determined between 4°C and 60° using a scanning rate of 0.5°C min-1. Two
independent sets of cells were scanned starting from the acclimation temperature
either down to 4°C or up to 60°. The heat production has been expressed in units of
differential power (DP) per mg of wet cell biomass (mJ min-1 mg-1). After each
measurement the yeast suspension was replaced with a freshly prepared one. About
80% of the metabolic activity of the yeast cells was estimated to correspond to
anaerobic glycolysis.
Model Calculations. Numerical calculations were performed using the FORTRAN
subroutine LSODE (Livermore Solver of Ordinary Differential Equations) [36]. Some
analytical solutions of steady state fluxes were obtained with the help of MATLAB
(www.mathworks.com).
Abbreviations and Symbols
Ci abbreviation used in Supplementary Material for
€
∂ lnJ '∂ lnki
=kiJ'
∂J'∂ki
.
€
*CiJ j global control coefficient defined as
€
∂ lnJ j
∂ lnki=kiJ j
∂J j
∂ki
.
€
CkicatJ j metabolic control coefficient defined as
€
∂ lnJ j
∂ lnkicat =
kicat
J j
∂J j
∂kicat
.
ei concentration of enzyme catalyzing process i.

- 17 -
ess steady state concentration of enzyme E in Fig. 1; see also Eq. 3.
Ei abbreviation used in Supplementary Material for
€
Eaki .
€
Eaki activation enthalpy of elementary component process with rate constant ki.
€
Eaki and ki are related by the Arrhenius equation
€
ki = Aie−Eaki
RT .
€
Eakicat
activation enthalpy of turnover number
€
kicat of enzyme catalyzed process i.
€
EaKi is the formation enthalpy
€
ΔHi0 of the rapid equilibrium between enzyme and
substrate in enzyme catalyzed process i. The temperature dependence of Ki(or other equilibrium constants such as KM, KI) is analogous to the Arrhenius
equation, i.e.,
€
Ki = eΔSi
0
R e−ΔHi
0
RT .
Jj, flux (reaction rate) of elementary component process j, or flux of enzymecatalyzed process j.
k activation constant used in positive feedforward/feedback loop.
ki rate constant of elementary component process i.
€
kicat turnover number of enzyme catalyzed process i.
Ki rapid equilibrium (dissociation) constant between enzyme and substrate inenzyme catalyzed process i.
KI inhibition constant used in negative feedforward/feedback loop. For itstemperature dependence see also above description of
€
EaKi and description of
scheme (11) in Supplementary Material.
KM Michaelis constant (Fig. 1).
m Used as an index for enzyme-catalyzed processes or used to describe thecooperativity (Hill coefficient) in negative feedforward/feedback loops actingfrom intermediate I on reaction i by replacing ki with ki/(KI+[I]m).
n Cooperativity (Hill coefficient) in positive feedforward/feedback loops actingfrom intermediate I on reaction i by replacing ki with kik[I]n.
R gas constant.
€
RTem metabolic response coefficient defined as
€
d lnemd lnT
.

- 18 -
€
RKi
J j metabolic response coefficient defined as
€
d lnJ j
d lnKi
.
€
*RKi
J j global response coefficient defined as
€
d lnJ j
d lnKi
.
T temperature.
Tref reference temperature at which rate constants and parameters are defined.

- 19 -
References1. Laidler, K. J. & Meiser, J. H. (1995) Physical Chemistry. Second Edition,
Houghton Mifflin Company, Geneva (Illinois).2. Hazel, J. R. & Prosser, C. L. (1974) Molecular Mechanisms of Temperature
Compensation in Poikilotherms, Physiol Rev. 54, 620-677.3. Zakhartsev, M. V., De Wachter, B., Sartoris, F. J., Portner, H. O. & Blust, R.
(2003) Thermal physiology of the common eelpout (Zoarces viviparus), J Comp
Physiol [B]. 173, 365-78.4. Bünning, E. (1963) The Physiological Clock, Springer-Verlag, Berlin.
5. Lloyd, D. & Murray, D. B. (2005) Ultradian metronome: timekeeper for
orchestration of cellular coherence, Trends Biochem Sci. 30, 373-7.6. Iwasaki, K., Liu, D. W. & Thomas, J. H. (1995) Genes that control a temperature-
compensated ultradian clock in Caenorhabditis elegans, Proc Natl Acad Sci U S A. 92,10317-21.
7. Baker, N. R., Long, S. P. & Ort, D. R. (1988) Photosynthesis and temperature,
with particular reference to effects on quantum yield, Symp Soc Exp Biol. 42, 347-75.8. Cabrera, H. M., Rada, F. & Cavieres, L. (1998) Effects of temperature on
photosynthesis of two morphologically contrasting plant species along an altitudinalgradient in the tropical high Andes, Oecologia. 114, 145-152.
9. Pollheimer, J., Zellner, M., Eliasen, M. M., Roth, E. & Oehler, R. (2005) Increased
susceptibility of glutamine-depleted monocytes to fever-range hyperthermia: the roleof 70-kDa heat shock protein, Ann Surg. 241, 349-55.
10. Peper, A., Grimbergen, C. A., Spaan, J. A., Souren, J. E. & van Wijk, R. (1998)A mathematical model of the hsp70 regulation in the cell, Int J Hyperthermia. 14, 97-
124.
11. Hastings, J. W. & Sweeney, B. M. (1957) On the mechanism of temperatureindependence in a biological clock, Proc Natl Acad Sci U S A. 43, 804-811.
12. Ruoff, P. (1992) Introducing temperature-compensation in any reaction kineticoscillator model, J Interdiscipl Cycle Res. 23, 92-99.
13. Ruoff, P., Loros, J. J. & Dunlap, J. C. (2005) The relationship between FRQ-
protein stability and temperature compensation in the Neurospora circadian clock,Proc Natl Acad Sci U S A. 102, 17681-6.
14. Kovács, K., Hussami, L. L. & Rábai, G. (2005) Temperature Compensation in the
Oscillatory Bray Reaction, J Phys Chem A. 109, 10302-10306.

- 20 -
15. Kóvacs, K. M., Rábai, G. (2002) Temperature-compensation in pH-oscillators,
Phys Chem Chem Phys. 4, 5265-5269.16. Fell, D. (1997) Understanding the Control of Metabolism, Portland Press,
London and Miami.17. Heinrich, R. & Schuster, S. (1996) The Regulation of Cellular Systems, Chapman
and Hall, New York.
18. Kacser, H. & Burns, J. A. (1973) The control of flux, Symp Soc Exp Biol. 27, 65-104.
19. Kahn, D. & Westerhoff, H. V. (1991) Control theory of regulatory cascades, JTheor Biol. 153, 255-85.
20. Kell, D. & Westerhoff, H. (1986) Metabolic Control Theory: its role in
microbiology and biotechnology, FEMS Microbiol Rev. 39, 305-320.21. Westerhoff, H. V., Koster, J. G., Van Workum, M. & Rudd, K. E. (1990) On the
Control of Gene Expression in Control of Metabolic Processes (Cornish-Bowden, A.,
ed) pp. 399-412, Plenum, New York.22. Cornish-Bowden, A. (2004) Fundamentals of Enzyme Kinetics. Third Edition,
Portland Press, London.23. Brown, G. C., Westerhoff, H. V. & Kholodenko, B. N. (1996) Molecular control
analysis: control within proteins and molecular processes, J Theor Biol. 182, 389-96.
24. Ruoff, P., Christensen, M. K., Wolf, J. & Heinrich, R. (2003) Temperaturedependency and temperature-compensation in a model of yeast glycolytic oscillations,
Biophys Chem. 106, 179-192.25. Chen, Y. & Westerhoff, H. V. (1986) How do Inhibitors and Modifiers of
Individual Enzymes affect steady-state Fluxes and Concentrations in Metabolic
Systems?, Math Modell. 7, 1173-1180.26. Dixon, M., Webb, E. C., Thorne, C. J. R. & Tipton, K. F. (1979) Enzymes,
Longman, London.27. Hochachka, P. W. & Somero, G. N. (2002) Biochemical Adaptation. Mechanism
and Process in Physiological Evolution, Oxford University Press, Oxford.
28. Somero, G. N. (1995) Proteins and temperature, Annu Rev Physiol. 57, 43-68.29. Hochachka, P. W. (1968) Action of temperature on branch points in glucose and
acetate metabolism, Comp Biochem Physiol. 25, 107-18.

- 21 -
30. Hikosaka, K., Ishikawa, K., Borjigidai, A., Muller, O. & Onoda, Y. (2006)
Temperature acclimation of photosynthesis: mechanisms involved in the changes intemperature dependence of photosynthetic rate, J Exp Botany. 57, 291-302.
31. Alberghina, L. & Westerhoff, H. V. (2006) Systems Biology. Definitions and
Perspectives, Springer-Verlag, Berlin.
32. Andjus, R. K., Dzakula, Z., Marjanovic, M. & Zivadinovic, D. (2002) Kinetic
properties of the enzyme-substrate system: a basis for immediate temperaturecompensation, J Theor Biol. 217, 33-46.
33. Sutcliffe, M. J. & Scrutton, N. S. (2000) Enzyme catalysis: over-the-barrier orthrough-the-barrier?, Trends Biochem Sci. 25, 405-8.
34. Burke, D., Dawson, D. & Stearns, T. (2000) Methods in Yeast Genetics. A Cold
Spring Harbor Laboratory Course Manual., Cold Spring Harbor Laboratory Press,Cold Spring Harbor, New York.
35. Sambrook, J., Fritsch, E. F. & Maniatis, T. (1989) Molecular Cloning: A
Laboratory Manual, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NewYork.
36. Radhakrishnan, K. & Hindmarsh, A. C. (1993) Description and Use of LSODE,
the Livermore Solver for Ordinary Differential Equations. NASA Reference
Publication 1327, Lawrence Livermore National Laboratory Report UCRL-ID-
113855., National Aeronautics and Space Administration, Lewis Research Center,Cleveland, OH 44135-3191.
37. Horton, H. R., Moran, L. A., Scrimgeour, K. G., Perry, M. D. & D., R. J. (2006)Principles of Biochemistry, Pearson Prentice Hall, Upper Saddle River, New Jersey.

- 22 -
Supplementary Material
Derivation of Eq. 1a. Assume Jj (the flux of elementary reaction “j”) is a function of
the rate constants of N component reactions:
Jj= Jj (k1, k2,...., ki,...., kN) (4)
The derivative with respect to temperature T is calculated by using the chain rule,
where index i runs over all elementary (single-step) component reactions:
€
dJ j
dT=
∂J j
∂ki
i∑ ∂ki
∂T
(5)
The last term in Eq. 5 can be explicitly calculated from the Arrhenius equation, i.e.,
€
ki = Aie−Eaki
RT ⇒
€
∂ki∂T
=Ea
ki
RT 2(Aie
−Eaki
RT ) =Ea
ki
RT 2ki (6)
which gives
€
dJ j
dT=
∂J j
∂ki
i∑ Ea
ki
RT 2ki =
∂J j
∂ lnki
i∑ Ea
ki
RT 2 (7)
by using ∂ki/ki = ∂lnki. Multiplying Eq. 7 by T/Jj and observing that dJj/Jj = dlnJj, and
T/dT=1/dlnT, Eq. 7 can be written as:
€
TJ j
∂J j
∂T=d lnJ j
d lnT=1RT
∂ lnJ j
∂ lnki
i∑ Ea
ki =1RT
*CiJ j
i∑ Ea
ki (8)
This shows that the activation enthalpy of flux Jj is equal to the weighted average of
the activation enthalpies of the steps, where the enthalpies are normalized by RT and
the weighting factors are the flux control coefficients.

- 23 -
Derivation of Eq. 1b. Each reaction step i is now considered to be catalyzed by an
enzyme and for the sake of simplicity we assume that each step can be described byMichaelis-Menten kinetics with non-saturating enzymes following first-order kinetics
with respect to substrates, i.e.
€
J j = J j (k1cate1K1
, k2cate2K2
,...., kicateiKi
,....., kNcateNKN
) (9)
where
€
kicat is the turnover number, ei the concentration of enzyme i, and Ki is a
dissociation constant of a rapid equilibrium (or a dynamic equilibrium constant)between enzyme and substrates. Using chain rules for the variation in turnover
numbers, enzyme concentrations, and rapid equilibrium constants (between enzymes
and substrates) one obtains
€
d lnJ j
dT=1J j
∂J j
∂kicat
∂ki
cat
∂T
i∑ +
1J j
∂J j
∂em
∂em∂T
m∑ +
1J j
∂J j
∂Kl
∂Kl
∂T
l∑ (10)
Because
€
kicat and Kl have an Arrhenius-type temperature dependence [1, 24], the first
and the last terms in Eq. 10 can be written as (see Eqs. (6)-(8))
€
d lnJ j
dT=
Eakicat
RT 2∂ lnJi∂ lnki
cat
i∑ +
1J j
emem
∂J j
∂em
∂em∂T
m∑ +
EaKi
RT 2∂ lnJ j
∂ lnKl
l∑ (11)
Because
€
emJ j
∂J j
∂em=kmcat
J j
∂J j
∂kmcat =
∂ lnJ j
∂ lnkmcat = Ckm
catJ j we can finally write
€
d lnJ j
dT=
Eakicat
RT 2Cki
catJ j
i∑ +
1J j
CkmJ j d lnem
dT
m∑ +
EaKi
RT 2∂ lnJ j
∂ lnKl
l∑ (12)
Multiplication with T and using the definition of the response coefficients
€
RTem =
d lnemd lnT
,
€
RKl
J j =d lnJ j
d lnKl
yields Eq. 1b.

- 24 -
Reaction schemes where J’ cannot be temperature compensated (Fig. 2b)
For the sake of simplicity we use global control coefficients with the following
notation
€
Ci =kiJ'
∂J'∂ki
,
€
C'= k 'J '
∂J'∂k'
, and
€
CiJ j =
kiJ j
∂J j
∂ki
. Ei are activation enthalpies
for reaction step i with rate constant ki.
Scheme (1). We consider the irreversible case, but the analysis can easily be applied
to the reversible situation. The rate equations at steady state conditions are:
€
d[A]dt
= k1 − k2[A] = 0
d[B]dt
= k2[A]− k3[B] = 0
d[C]dt
= k3[B]− k4[C] = 0
d[D]dt
= k4[C]− J '= 0
(13)
Summing up the four equations leads to J’=k1 showing that C1=1, while the othercontrol coefficients are zero.
Scheme (2). We are looking at the irreversible clockwise loop. The rate equations at
steady state conditions are:
€
d[A]dt
= k1 + k5[D]− k2[A] = 0
d[B]dt
= k2[A]− k3[B] = 0
d[C]dt
= k3[B]− k4[C] = 0
d[D]dt
= k4[C]− J '−k5[D] = 0
(14)
Summing up the four equations leads to J’=k1 showing that C1=1, while the other
control coefficients are zero.
Scheme (3). Although this scheme contains a branch point, flux J’ = k1 showing that
C1=1, while the other control coefficients are zero.

- 25 -
Scheme (4). Reaction species D inhibits process 2 by replacing k2 by
€
k2KI + [D]m
.
€
d[A]dt
= k1 −k2[A]
KI + [D]m= 0
d[B]dt
=k2[A]
KI + [D]m− k3[B] = 0
d[C]dt
= k3[B]− k4[C] = 0
d[D]dt
= k4[C]− J '= 0
(15)
The sum of the four equations leads to J’=k1 showing that C1=1, while the othercontrol coefficients are zero.
Scheme (5). Like in scheme (4), the contributions of the (positive) feedback loop
cancel and flux J’=k1 leading to C1=1 and the other Ci’s = 0.
Scheme (6). We consider a negative feedforward to the output flux J’. The rate
equations read:
€
d[A]dt
= k1 − k2[A] = 0
d[B]dt
= k2[A]− k3[B] = 0
d[C]dt
= k3[B]− k4[C] = 0
d[D]dt
= k4[C]−k '
KI + [B]m[D] = 0
(16)
The steady state output flux J’ is given by:
€
J'= k'KI + [B]SS
m [D]SS = k1 (17)
showing that C1=1, while the other control coefficients are zero.

- 26 -
Scheme (7). Reaction species A activates reaction 4 (positive feedforward).
€
d[A]dt
= k1 − k2[A] = 0
d[B]dt
= k2[A]− k3[B] = 0
d[C]dt
= k3[B]− k4k[A]n[C] = 0
d[D]dt
= k4k[A]n[C]− J'= 0
(18)
The sum of the four equations leads to J’=k1 showing that C1=1, while the other
control coefficients are zero. The same result we get for the activation of reaction 4 by
B or the activation of reaction 3 by A.
The rate equations of a positive feedforward from B to an output flux read:
€
d[A]dt
= k1 − k2[A] = 0
d[B]dt
= k2[A]− k3[B] = 0
d[C]dt
= k3[B]− k4[C] = 0
d[D]dt
= k4[C]− k 'k[B]n[D] = 0
(19)
The steady state output flux J’ is given by:
€
J'= k 'k[B]SSn [D]SS = k1 (20)
leading to
€
C1 =1, while the other control coefficients are zero.

- 27 -
Reaction schemes where J’ can be temperature compensated (Fig. 2c)
Scheme (8). Calculating the steady state values:
€
d[A]dt
= k1 − k2[A] = 0 ⇒ [A]SS =k1k2
d[B]dt
= k2[A]− (k3 + k4 )[B] = 0 ⇒ [B]SS =k1
k3 + k4d[C]dt
= k3[C]− k5[C] = 0 ⇒ [C]SS =k3k5[B]SS
d[D]dt
= k4[B]− J '= 0 ⇒ [D]SS =k4k'[B]SS =
k1k4k'(k3 + k4 )
(21)
Because J’=k’[D]SS, we get
€
J'= k1k4k3 + k4
. We can write
€
C3 =∂ lnJ '∂ lnk3
=k3J'⋅∂J'∂k3
= −k3
k3 + k4
and
€
C4 =∂ lnJ '∂ lnk4
=k4J'⋅∂J'∂k4
= +k3
k3 + k4 showing that C3=−C4. Likewise it can be shown
that C1=1.
Scheme (9).
€
d[A]dt
= k1 − (k2 + k6)[A] = 0
d[B]dt
= k2[A]− (k3 + k7)[B] = 0
d[C]dt
= k3[B]− (k4 + k8)[C] = 0
d[D]dt
= k4[C]− (k'+k9)[D] = 0
(22)
By successive elimination of the steady state values for A, B, C, and D in the equations,
we get:
€
J'= k '[D]SS = k' k1k2k3k4(k2 + k6)(k3 + k7)(k4 + k8)(k '+k9)
(23)
Calculating the Ci’s, one get that C1=1 and
€
C2 = −C6 =k6
k2 + k6;
€
C3 = −C7 =k7
k3 + k7;
€
C4 = −C8 =k8
k4 + k8;
€
C'= −C9 =k9
k'+k9. In order
to temperature compensate fluxes J6, J7, J8 and J’ simultaneously, we observe that when

- 28 -
compensating first J6, this flux depends only on k1, k2 and k6 with
€
C1J6 =1 and
€
C2J6 = −C6
J6 = −k2
k2 + k6 while all the other control coefficients are zero. This leads to the
balance equation
€
E1 + C2J6 (E2 − E6) = 0 (24)
which determines the values of E1, E2 and E6. The condition to temperature compensateJ7 is:
€
E1 + C2J7 (E2 − E6) + C3
J7 (E3 − E7) = 0 (25)
Because
€
C2J6 is generally different from
€
C2J7 , the sum
€
E1 + C2J7 (E2 − E6) in Eq. 20 need
not to be zero, but E3 and E7 can be chosen such that Eq. 20 is satisfied. In this way J6,
J7 and J8 and J’ can be simultaneously temperature compensated when solving thebalance equations 19-22.
€
E1 + C2J8 (E2 − E6) + C3
J8 (E3 − E7) + C4J8 (E4 − E8) = 0 (26)
€
E1 + C2J ' (E2 − E6) + C3
J ' (E3 − E7) + C4J ' (E4 − E8) + CJ ' (E '−E9) = 0 (27)
Scheme 10. The rate equations of this cyclic scheme with steady state conditions read:
€
d[A]dt
= k1 − k2[A]+ k5[D] = 0
d[B]dt
= k2[A]− (k3 + k6)[B] = 0
d[C]dt
= k3[B]− (k4 + k7)[C] = 0
d[D]dt
= k4[C]− (k'+k5)[D] = 0
(28)
By successively eliminating the steady state values [A]SS, [B]SS and [C]SS, an expression
for [D]SS and J’ can be obtained:
€
J'= k ' k1k3k4N
(29)
where N=k’k6k7 + k’k4k6 + k’k3k7 + k’k3k4 + k5k6k7 + k4k5k6 + k3k5k7. Calculating the
control coefficients as
€
Ci =kiJ'
∂J'∂ki
, we get:

- 29 -
C1 = 1, C2 = 0,
€
C3 =k6(k 'k7 + k'k4 + k5k7 + k5k4 )
N,
€
C4 =k7(k 'k6 + k'k3 + k5k6 + k5k3)
N,
€
C5 = −k5(k6k7 + k6k4 + k3k7)
N, C6 = −C3, C7 = −C4, C’ = −C5 with
€
Ci + C'=1i=1
7
∑ .
Scheme (11). Inhibition of J1 by any intermediate. We take as an example the inhibitionof J1 by C with Hill coefficient m = 1. The rate equations with steady state conditions
are:
€
d[A]dt
=k1
KI + [C]1− k2[A] = 0
d[B]dt
= k2[A]− k3[B] = 0
d[C]dt
= k3[B]− k4[C] = 0
d[D]dt
= k4[C]− k '[D] = 0
(30)
leading to
€
J'= k '[D]SS =k1
KI + [C]SS. Eliminating the variables A and B and determining
the positive solution of the steady state value of C:
€
[C]SS =−k4KI + (k4KI )
2 + 4k1k42k4
(31)
J’ can be expressed as:
€
J'= 2k1k4k4KI + k4M
; M = (k4KI2 + 4k1) (32)
Calculating the control coefficients as
€
Ci =kiJ'
∂J'∂ki
, we get:
€
C1 =k4 (KI k4M + k4KI
2 + 2k1)(k4KI + k4M ) ⋅ k4M
,
€
C4 =2k1k4
(k4KI + k4M ) ⋅ k4M,
€
CKI= −
k4KI
(k4KI + k4M );
C2 = C3 = C’ = 0. Note, temperature compensation is only possible because the inhibitor
constant KI (which can be interpreted as a dissociation constant in rapid equilibriumbetween the enzyme that catalyzes step 1 and the inhibitor C) is assumed to be
temperature dependent. The temperature dependence of KI can be described analogously
to the Arrhenius equation by substituting the activation enthalpy with the enthalpy of
formation
€
ΔHI0. In this case the pre-exponential factor AI can still be treated as
temperature independent with AI = exp(−
€
ΔSI0/R) [1]. To describe the temperature

- 30 -
dependence of KI, KM, and Ki in an uniform manner, the enthalpy of formation, for
example for
€
ΔHi0 has been substituted by
€
EaK I in the main text (see also description of
€
EaK I in the Abbreviations and Symbols section). It should also be noted that when
treating KI as a temperature dependent parameter, then the sum of the control coefficients
is generally not one. In this case we get:
€
Cii=1
4
∑ + CKI+ C'= 4k1k4
(k4KI + k4M ) k4M (33)
Scheme (12). Activation of J1 by any intermediate. In order to avoid an exponential
increase of concentrations, n needs to be lower than 1. As an example, we look at theactivation of reaction 1 by intermediate C. The rate equations with steady state conditions
are:
€
d[A]dt
= k1k[C]n − k2[A] = 0, 0 < n <1
d[B]dt
= k2[A]− k3[B] = 0
d[C]dt
= k3[B]− k4[C] = 0
d[D]dt
= k4[C]− k '[D] = 0
(34)
Summing up all steady state equations leads to
€
J'= k '[D]SS = k1k[C]SSn (35)
When adding only the first three equations, we get
€
k1k[C]SSn − k4[C]SS = 0 . Solving for
[C]SS from the last equation and multiplying [C]SS with k4, we get:
€
J'= k3k1kk3
−1n−1
(36)
Calculating the control coefficients as
€
Ci =kiJ'
∂J'∂ki
, we get:
€
C1 = Ck = −1n −1
,
€
C3 =nn −1
, while all other Ci are zero. The sum
€
Ci + C'+Cki=1
4
∑ =n − 2n −1
shows that in
case of a feedback activation of an input flux using n < 1 the summation theorem is notobeyed.

- 31 -
A Simple Representation of the Calvin Benson CycleFig. 5c shows a simple representation of the Calvin Benson cycle with its 3 stages: (i)the reduction phase (flux J2) due to influx of ATP and NADPH produced by the light
reaction together with the output flux of Pi, (ii) the regeneration phase (flux J3) with theoutput flux J6 forming carbohydrates, and (iii) the ATP/NADPH-driven carboxylation
phase (flux
€
J4CO2 ) assimilating CO2 [37]. The rate constant
€
k4CO2 is assumed to be
dependent on the partial but constant CO2 pressure.
The rate equations and steady state conditions are given by the following equations:
€
d[A]dt
= k3[C]− (k4CO2 + k6)[A] = 0
d[B]dt
= k4CO2 [A]+ k1 − k2[B] = 0
d[C]dt
= k2[B]− (k3 + k5)[C] = 0
(37)
Eliminating variables C and B and calculating
€
J4CO2=
€
k4CO2 [A]SS leads to
€
J4CO2=
€
k4CO2k1k3L
, L = k5k4CO2 + k5k6 + k3k6 (38)
The control coefficients
€
Ci =kiJ4CO2
∂J4CO2
∂ki
are C1 = 1, C2 = 0,
€
C3 = −C5 =k5(k4 + k6)
L,
and
€
C4 = −C6 =k6(k5 + k3)
L. The assimilation of CO2 can be temperature compensated
because of the ”balance” between the input and output fluxes.

- 32 -
Figure legends
Fig. 1. Simple model of transcription (mRNA synthesis with rate constant k1) and
translation (protein synthesis with rate constant k3) of an enzyme E which catalyzes
the conversion of S → P. All reactions are considered to be first-order, except reaction
rate
€
J5 =d[P]dt
=k5catess[S]KM + [S]
. The other constants are: k2, rate constant for mRNA
degradation; k4, rate constant of enzyme degradation. KM and
€
k5cat are the Michaelis
constant and the turnover number, respectively.
Fig. 2. Network models. (a) General scheme depicting input, internal and output
fluxes. (b) Reaction schemes where the steady state flux J’ cannot be temperaturecompensated. (c) Reaction schemes where J’ can be temperature compensated (see
Supplementary Material). For the sake of simplicity global control coefficients
(without an asterisk) are used and defined as
€
Ci =kiJ'
∂J'∂ki
,
€
C'= k 'J '
∂J'∂k'
, and
€
CiJ j =
kiJ j
∂J j
∂ki
, where ki is the rate constant of reaction step i. Positive/negative signs
indicate positive/negative feedforward or feedback loops leading to activation or
inhibtion of a particular process. For a description about the kinetics using activationconstant k and inhibition constant KI in positive or negative feedforward/feedback
loops, see main text. Control coefficients with respect to k and KI are defined as
€
Ck =kJ'
∂J'∂k
and
€
CKI=KI
J'∂J'∂KI
.
Fig. 3. Kinetics of compensated and uncompensated network (8) (Fig. 2c). At 25°C
the rate constants have the following (arbitrary chosen) values k1=1.7 (time units)−1,
k2=0.1 (time units)−1, k3=0.5 (time units)−1, k4=1.5 (time units)−1, k5=1.35 (time units)−1,
k6=0.7 (time units)−1. Initial concentrations of A, B, C, and D (at t=0) are zero. The

- 33 -
control coefficients (as defined in the legend of Fig. 2) at 25°C are
€
C1J '=1.0,
€
C3J '=−0.250,
€
C4J '=0.250. (a) Open squares show the exponential increase of J’ as a
function of temperature for the uncompensated network (all activation enthalpies
being taken 67 kJ/mol; note:
€
Eak2 ,
€
Eak5 and
€
Eak6 do not matter, because the associated
control coefficients are zero). Open diamonds show the effect of static temperature
compensation of J’ when using
€
Eak1 =26 kJ/mol,
€
Eak3 =120 kJ/mol and
€
Eak4 =22 kJ/mol.
Solid circles show the effect of dynamic temperature compensation by keeping
€
Eak3
and
€
Eak4 constant but increasing
€
Eak1 (described as
€
E1dynamic) with increasing
temperatures as shown in the inset. For each temperature T
€
E1dynamic was estimated
according to the equation
€
E1dynamic (T) = Ea
k1 − 0.5 CiJ ' (T)
i∑ Ea
ki , where the Ci(T)’s were
calculated at temperature T. (b) Transient kinetics of the uncompensated network (Fig.
3a) when applying a 15°C→35°C temperature step at t=300 time units. The inset
shows the details of the response kinetics. The difference in the J’ steady state levels
at 15°C and 35°C is clearly seen. (c) Transient kinetics of the static temperature
compensated network (Fig. 3a) when applying a 15°C→35°C temperature step at
t=300. The inset shows transient kinetics when a 35°C→15°C temperature step is
applied. (d) Fluxes J1 and J3 as a function of temperature in the static temperature
compensation of J’ (Fig. 3a).
Fig. 4. Experimental evidence for dynamic temperature compensation. (a)
Temperature dependent metabolic activity of S. cerevisiae. The cells were acclimated
at anaerobic conditions to 15°C, 22.5°C and 30°C. The anaerobic metabolic activity
of the cells was measured as the overall generated differential power DP in mJ min-1
per mg of wet cell biomass using Differential Scanning Microcalorimetry

- 34 -
(0.5°C/min). The curves are the average of n=4 at 15°C, n=16 at 22.5°C, and n=5 at
30°C. The large dots indicate metabolic activities at the corresponding acclimation
temperatures with standard deviations. For temperatures higher than 40°C the
produced heat was lowered by cell death. (b) Arrhenius plots for the three acclimation
temperatures. Activation enthalpies were estimated by linear regression between 4°C
(0.0036 K-1) and 40°C (0.0032 K-1) for acclimation at 15°C and 22.5°C, and by
linear regression between 10°C (0.0035 K-1) and 40°C (0.0032 K-1) for acclimation
at 30°C. Estimated activation enthalpies are: 44.8 kJ/mol (acclimation at 15°C), 52.6
kJ/mol (acclimation at 22.5°C), and 75.6 kJ/mol (acclimation at 30°C).
Fig. 5. Mimicking temperature compensation and temperature adaptation of
photosynthesis in higher plants. (a) Photosynthetic flux of plant species living in hot
(S. albicans), temperate (S. anglica) and cold environments (T. oblongifolia).
Redrawn from Ref. [7]. (b) Temperature response of a single branch point (flux J4 of
scheme (8)) with different activation enthalpy combinations. For the sake of
simplicity Ei are activation enthalpies for reaction step i with rate constant ki. For
details see Supplementary Material. (1) E1=190 kJ/mol, E3=290 kJ/mol, E4=20 kJ/mol;
(2) E1=70 kJ/mol, E3=190 kJ/mol, E4=20 kJ/mol; (3) E1=20 kJ/mol, E3=93 kJ/mol,
E4=23 kJ/mol. In addition, the value of k1 at 25°C has been reduced from 1.7 (time
units)−1 to 0.5 (time units)−1. All other rate constants and Tref were as described in Fig.
3. (c) A minimal model of the Calvin Benson Cycle with reduction phase (fluxes J1,
J2, J5), regeneration phase (fluxes J3, J6) and carbon dioxide assimilation (flux
€
J4CO2 ).
(d)
€
J4CO2 as a function of temperature for 3 parameter set combinations (curves 1-3).
Joint rate constant values for all 3 curves (defined at Tref=25°C): k2=0.1 (time units)−1,

- 35 -
k3=0.5 (time units)−1,
€
k4CO2=1.5 (time units)−1
(concentration units)−1 , k5=1.35 (time
units)−1 , k6=0.7 (time units)−1. Ci values (at Tref=25°C): C1 = 1, C2 = 0, C3 = −C5 =
0.895, C4 = −C6 = 0.390. k1 values and activation enthalpy combinations: (1) k1=2.2
(concentration units) (time units)−1 E1 = 92 kJ/mol, E3 = 90 kJ/mol, E4 = 40 kJ/mol, E5
= 50 kJ/mol, E6 = 220 kJ/mol; (2) k1=1.4 (concentration units) (time units)−1 E1 = 62
kJ/mol, E3 = 70 kJ/mol, E4 = 40 kJ/mol, E5 = 50 kJ/mol, E6 = 220 kJ/mol; (3) k1=0.5
(concentration units) (time units)−1 E1 = 30.5 kJ/mol, E3 = 39 kJ/mol, E4 = 40 kJ/mol,
E5 = 60 kJ/mol, E6 = 70 kJ/mol with
€
Cii=1
6
∑ Ei = 0.012 kJ/mol. Note, because C2=0, k2
and activation enthalpy E2 do not influence the steady state value of
€
J4CO2 and its
temperature profile. Also note, because C1 = 1,
€
J4CO2 values can be changed by
changing k1, but without changing the form of the temperature profile for
€
J4CO2 .
mRNA
geneE
S P
k1
k2k3
k4
k5 , KM
Ruoff et al. : Temperature Compensation...... Figure 1
cat
A B C Dk1 kʼ (1)
No temperature compensation of Jʼ possible
IkIl
IjIi
inputfluxes
outputfluxes
a
b
cTemperature compensation of Jʼ possible
(8)A B
C
D
k1 k2 k3
k4 kʼ
k5
C1 = 1; C3 = − C4; C3 < 0; C4 > 0; all other Ciʼs
= 0
J1 J2
J5 J3
J4 Jʼ
internalfluxes
C1
= 1,
all
othe
r Ciʼs
= 0
A B C Dk1 kʼ
(9)
k2 k3 k4
k6 k7 k8 k9
C2 = −C6; C3 = −C7; C4 = −C8; Cʼ = −C9
C2, C3, C4, Cʼ > 0; C6, C7, C8, C9 < 0C1 = 1;
J1 J6 J2 J7
J3 J4 J8 J9
Jʼ
Jʼ
B
k2
k3
A
C
D
k1
k4
kʼ
k5
k6
k7
(10)
C1 = 1; C2 = 0;
C5 = −Cʼ; Cʼ > 0;
C6 = −C3; C3 > 0;
C7 = −C4; C4 > 0;
Jʼ(2)
A
B
C
D
k1
kʼ
J1
k2
k3 k4
k5
Jʼ
(3) AB
CDk1 kʼ Jʼ
C1 > 0, C4 > 0, CKI
A B C Dk1 k2 k3 k4 kʼ (11)KI , m
< 0. All other Ciʼs are zero.
Jʼ
A B C Dk1 k2 k3 k4 kʼ (12)
k, n < 1
C1 < 0, Ck < 0, C3 > 0. Jʼ
(4) A B C Dk1 k2 k3 k4 kʼ
KI , m
Jʼ
(5) A B C Dk1 k2 k3 k4 kʼ
k, n
Jʼ
Ruoff et al. : Temperature Compensation...... Figure 2
A B C Dk1 k2 k3 k4 kʼ (6)
KI , m
Jʼ
(7) A B C Dk1 k2 k3 k4 kʼ
k, n
Jʼ
k2 k3 k4
k-2 k-3 k-4
0
2
4
6
8
0 100 200 300 400 500 600
flux
J', a
.u.
35°C
c
15°C
time, a.u.
(static)compensation
0
0.5
1
1.5
2
2.5
3
3.5
15 20 25 30 35temperature, °C
a
flux
J', a
.u.
uncompensated
'static'compensation
'dynamic'compensation
15
20
25
30
35
40
15 20 25 30 35
E 1dyna
mic, k
J/mol
temperature, °C
0
0.5
1
1.5
2
2.5
3
3.5
4
0 100 200 300 400 500 600flu
x J',
a.u
.time, a.u.
b
uncompensated
35°C15°C
00.51
1.52
2.53
3.5
290 300 310 320
flux
J', a
.u.
time, a.u.
15°C 35°C
00.20.40.60.81
1.2
200 250 300 350 400
flux
J', a
.u.
time, a.u.
35°C 15°C
0
0.5
1
1.5
2
2.5
15 20 25 30 35
flu
x, a
.u.
temperature, °C
J1 = J2 = J3 + J
4
J' = J1 − J
3 = J4
J3 = J
5
d
Ruoff et al. : Temperature Compensation...... Figure 3
0
0.5
1
1.5
2
2.5
3
3.5
0 10 20 30 40 50 60
DP,
mJ m
in-1
mg-1
temperature, °C
15°C
30°C
dT/dt = 0.5°C min-1
a 22.5°C
15°C22.5°C
-1.5
-1
-0.5
0
0.5
1
1.5
3 3.2 3.4 3.6
ln D
P
1000/T, K-1
b15°C
22.5°C30°C
Ruoff et al. : Temperature Compensation...... Figure 4
c
0102030405060
010
2030
4050
60
S. a
lbic
ans
S. a
nglic
aT.
obl
ongi
folia
photosynthetic rate, µmol m-2
s-1
leaf
tem
pera
ture
, °C
a
0
0.51
1.52
2.5 10
1520
2530
3540
tem
pera
ture
, °C
1 2 3b
flux J', a.u.
singl
e br
anch
poi
ntFi
g. 2
c, sc
hem
e (8
)
0
0.1
0.2
0.3
0.4
0.5
0.6 10
1520
2530
3540
J4
CO , a.u. 2
tem
pera
ture
, °C
1 2dcy
clic
sche
me
Fig.
5c
3
Ruof
f et a
l: Te
mpe
ratu
re C
ompe
nsat
ion.
....
F
igur
e 5







![Transport driven by eddy momentum fluxes in the Gulf ...oceanrep.geomar.de/10017/1/2010GL045473.pdf · model to steady state. 3. Results [6] Figure 1 shows the transport streamfunction](https://img.pdfslide.us/doc/110x75/5e8f6c3bed5951508a698d5f/transport-driven-by-eddy-momentum-fluxes-in-the-gulf-model-to-steady-state.jpg)