Embed Size (px)
Citation preview

Experimental Hematology 2011;39:457–472
Targeting of active mTOR inhibits primary leukemia T cells and synergizeswith cytotoxic drugs and signaling inhibitors
Ana Batistaa,c, Jo~ao T. Baratad, Elke Raderschalla, Stephen E. Sallanb, Nadia Carlessoe,Lee M. Nadlera, and Angelo A. Cardosoa,c
aDepartment of Medical Oncology, Dana-Farber Cancer Institute, Harvard Medical School, Boston, Mass., USA; bPediatric Oncology, Dana-Farber
Cancer Institute, Harvard Medical School, Boston, Mass., USA; cDivision of Hematology/Oncology and Indiana University Simon Cancer Center,
Indiana University School of Medicine, Indianapolis, Ind., USA; dInstitute of Molecular Medicine, Lisbon, Portugal; eHerman B Wells Center for
Pediatric Research, Indiana University School of Medicine, Indianapolis, Ind., USA
(Received 13 December 2010; revised 22 December 2010; accepted 5 January 2011)
Offprint requests t
Hematology/Oncology
ana University Scho
R3-C321J, Indianapol
Supplementary data
online version at doi
0301-472X/$ - see fro
doi: 10.1016/j.exph
Objective. Rationally designed therapies aim at the specific disruption of critical signalingpathways activated by malignant transformation or signals from the tumor microenviron-ment. Because mammalian target of rapamycin (mTOR) is an important signal integratorand a key translational regulator, we evaluated its potential involvement in T-cell acutelymphoblastic leukemia (T-ALL) and whether mTOR blockade synergizes with chemothera-peutic agents or other signaling antagonists to inhibit primary leukemia T cells.
Materials and Methods. mTOR signaling status was assessed using biochemical, immunostain-ing, and molecular regulation studies and functional assays performed to assess the impact ofmTOR blockade on T-ALL proliferation, survival, and cell cycle.
Results. We observed that mTOR signaling is highly activated in all T-ALL patients tested,with phosphorylation of its downstream substrates eIF4G and S6 ribosomal protein. mTORactivation was detected in vivo and was further increased in vitro by stimulation withinterleukin-7, a potentially leukemogenic cytokine normally produced by the bone marrowmicroenvironment. In T-ALL cells, mTOR blockade was associated with accumulation ofthe cyclin-dependent kinase inhibitor p27kip1, which preferentially adopted a nuclear localiza-tion. Functional studies using rapamycin or CCI-779 showed a dominant inhibitory effect ofmTOR blockade on interleukin-7Linduced proliferation, survival, and cell-cycle progressionof T-ALL cells. Furthermore, mTOR blockade markedly potentiated the antileukemia effectsof dexamethasone and doxorubicin, and showed highly synergistic interactions in combinationwith specific inhibitors of phosphatidylinositol 3-kinase/Akt and Janus kinase 3 signaling.
Conclusions. This study shows activation of mTOR signaling in primary T-ALL cells evolvingin the leukemic bone marrow, and supports the inclusion of mTOR antagonists in currenttherapeutic regimens for this cancer. � 2011 ISEH - Society for Hematology and StemCells. Published by Elsevier Inc.
Increasing evidence supports the therapeutic potential ofstrategies targeting molecules that regulate critical cellfunctions or tumorigenic processes. Because deregulationof signal transduction pathways is a common feature of
o: Angelo A. Cardoso, M.D., Ph.D., Division of
and Indiana University Simon Cancer Center, Indi-
ol of Medicine, 980 W. Walnut Street, Room
is, IN 46202; E-mail: [email protected]
associated with this article can be found in the
: 10.1016/j.exphem.2011.01.005.
nt matter. Copyright � 2011 ISEH - Society for Hematolo
em.2011.01.005
human cancer [1,2], it is essential to identify and charac-terize the signaling machinery critical for the survival,proliferation, and drug resistance of tumor cells. Recentstudies stress the importance of factors from the tumor-supportive microenvironment in engaging critical signalingevents in malignant cells. Tumor cells become dependenton signal transduction deregulation or on these extrinsicsignals to survive and, therefore, become more sensitiveto its disruption [3,4]. Since these pathways frequentlysignal within cross-talk networks, molecules that integratedistinct signaling cascades may constitute effective thera-peutic targets.
gy and Stem Cells. Published by Elsevier Inc.

458 A. Batista et al./ Experimental Hematology 2011;39:457–472
The mammalian target of rapamycin (mTOR) is a serine/threonine kinase that serves as a critical integrator ofmultiple signal transduction pathways. It senses mitogenicstimuli and nutritional and energy status, coupling thesesignals with cell growth and proliferation by controllingthe translation of key regulatory proteins [5,6]. Throughphosphorylation, mTOR mediates the inactivation of trans-lation initiation factor binding protein 4E-BP1 and activa-tion of the ribosomal kinase S6K1. Inactivation of 4E-BP1enables the formation of the multi-subunit eIF4F complex,which promotes translation initiation; whereas activationof S6K1 mediates cell growth [7–9]. Experimental evidencedemonstrated that mTOR is modulated by upstream events,including positive regulation by phosphatidylinositol 3-kinase (PI3K)/Akt pathway [10,11] and negative regulationby the tuberous sclerosis tumor suppressor complex (TSC1/TSC2) [12]. Both PI3K/Akt and TSC1/TSC2, as well as themTOR pathway, are often deregulated in cancer [13]. mTORis specifically inhibited by rapamycin and its analogues,binding to FKBP12 to form a complex that blocks mTORkinase activity [14]. Blockade of mTOR has shown anti-tumor activity both as a single intervention and in combina-tion with chemotherapeutic agents [15,16], and seems toovercome resistance to cytotoxic drugs [17–19]. Its potentialas a significant molecular target in hematological malignan-cies has been suggested [20,21].
We have been investigating the role of the tumormicroen-vironment in the biology of T-cell acute lymphoblasticleukemia (T-ALL). In particular, we dissected molecularand functional responses of leukemia T cells to interleukin-7 (IL-7), a cytokine present in the bone marrow (BM) andthymic microenvironments [22,23], and that has been impli-cated in T-cell development, leukemogenesis, and T-ALLbiology [24]. We have shown that IL-7 stimulation of pedi-atric T-ALL modulates the survival, cell-cycle progression,growth, and glucose uptake of primary leukemia T cellsthrough a mechanism requiring PI3K/Akt [25]. IL-7 alsoengages mitogen-activated protein kinase/extracellularsignal-regulated kinase and Janus kinase (Jak)/signal trans-ducers and activators of transcription signals in these cells,although their functional relevance needs further clarifica-tion. Other reports have implicated other signaling cascadesin T-cell leukemia biology, namely the constitutive activationof Notch [26]. Recently, it has been suggested that Notchsignals regulate mTOR activation in leukemia T-cell linesand in mouse models of T-ALL [27,28]. However, little isknown about how these distinct signals are integrated withinprimary leukemia cells, particularly in response to stimulifrom the leukemic microenvironment.
Here, we show that mTOR pathway is activated in allT-ALL patients tested, and that IL-7 stimulation ofleukemia T cells further engages this signaling cascade.Blockade of mTOR by rapamycin or CCI-779 inhibits theproliferation, survival, and cell-cycle progression ofT-ALL cells. Importantly, blockade of mTOR synergizes
with other signaling inhibitors and conventional cytotoxicagents to effectively inhibit leukemia T cells. This studysupports mTOR pathway as a valid target in primaryT-ALL cells and provides the rational for the use of mTORinhibitors in therapeutic regimens for this malignancy.
Materials and methods
Primary leukemia and TAIL7 cellsPrimary T-ALL cells were obtained from diagnostic BM or periph-eral blood specimens of pediatric patients with high leukemiainvolvement (O90%) after appropriate informed consent and Insti-tutional Review Board approval. After gradient centrifugation, cellswere washed in RPMI-1640 plus 10% fetal bovine serum (RPMI-10). Normal blood T cells were purified from healthy donors bypositive selection using CD3 microbeads (Miltenyi, Auburn, CA,USA). TAIL7, a human IL-7�dependent leukemia T-cell line thatmimics the functional and molecular properties of primary T-ALLcells, was established in our laboratory [29]. TAIL7 cellsweremain-tained in RPMI-10 plus IL-7 (10 ng/mL; R&D Systems, Minneap-olis, MN, USA), and viable cells were separated on Ficoll andrecultured in freshmedia every 7 days.When necessary, TAIL7 cellswere starved in serum-, IL-7�free RPMI for 72 hours before use.
ImmunohistochemistryPrimary T-ALL or TAIL7 cells (5 � 106) were injected intrave-nously in irradiated nonobese diabetic/severe combined immunode-ficient mice (NOD.CB17-Prkdcscid/J; Jackson Laboratory, BarHarbor,ME,USA),with animals sacrificedwhenmoribund, accord-ing to protocols approved by the Dana Farber Cancer Institute’sAnimal Care and Use Committee. Femurs were collected and BManalyzed for tumor engraftment by flow cytometry (CytomicsFC500; Beckman-Coulter) after staining with antibodies for humanCD45, CD7, or CD2, and mouse CD45 (BD-Biosciences, San Jose,CA,USA). For histology/immunohistochemistry, femurswere fixedin 10% formalin, dehydrated (ethanol/xylene), and embedded inparaffin; some slides were stained with hematoxylin/eosin. Activa-tion status of mTOR signaling was assessed by immunohistochem-istry using a phospho-S6 ribosomal protein (S6RP)�specificantibody (1:200; Cell Signaling), followed by staining with horse-radish peroxidase�conjugated goat-anti-rabbit antibody. Speci-mens from an epithelial cancer with constitutive mTOR activationwere used as control. Images were acquired on a Nikon EclipseE800 microscope (Nikon Instruments, Melville, NY, USA) usinga Nikon camera.
ImmunofluorescenceTAIL7 cells were cultured in RPMI-10, IL-7 (10 ng/mL), or IL-7plus rapamycin (10 nM; Calbiochem, San Diego, CA, USA) for96 hours. Cells (5 � 105) were harvested, washed, and resus-pended in phosphate-buffered saline (PBS), dropped onto glassslides, and air dried. After centrifugation, the preparations werefixed in absolute methanol (30 minutes, �20�C), and briefly rinsedin ice-cold acetone. Slides were washed with PBS, blocked witha 10% fetal bovine serum solution (1 hour) and incubated withphospho-p53 (1:50; Cell Signaling) or p27kip1 antibody (1:50;Transduction Laboratories, BD Biosciences) overnight at 4�C.Phospho-p53(Ser46) and p27kip1-labeled slides were incubated(1 hour, 37�C) with phycoerythrin-conjugated rabbit or fluorescein

459A. Batista et al./ Experimental Hematology 2011;39:457–472
isothiocyanate�conjugated mouse antibodies (1:200; Abcam),respectively. The preparations were counterstained with 40-6-diamidino-2-phenylindole (1 mg/mL, 2 � saline-sodium citrate,1 minute), and the slides were mounted in glycerol (90%); Tris-HCl (0.1 M [pH 8.0]); and 1,4-diazobicyclo-2,2,2-octane (2.3%).Images were acquired using a Nikon Eclipse E800.
ImmunoblottingLysates from primary T-ALL, TAIL7 cells, or normal T lympho-cytes were prepared in lysis buffer containing Tris-HCl (50 mM[pH 7.4]); NaCl (150 mM); NP-40 (1%); sodium dodecyl sulfate(0.1%); Na-deoxycholate (0.5%); EDTA (1 mM); plus 1% phos-phatase inhibitor cocktails I and II (Sigma-Aldrich, St Louis,MO, USA), and 1% protease inhibitor cocktail (Roche, Penzberg,Germany). For assaying mTOR and cell-cycle status, cells wereresuspended in RPMI-10 and incubated with rapamycin (10 nM)or CCI-779 (10 nM; Wyeth, Madison, NJ, USA; generous giftfrom Dr. John Ryan) for the indicated time points. All TAIL7experiments were performed using IL-7 (10 ng/mL). For assayingPI3K/Akt-mTOR signaling, quiescent TAIL7 cells were pretreated(2 hours) with rapamycin (10 nM) or LY294002 (PI3K/Akt inhib-itor, 10 mM; Calbiochem), and then stimulated with IL-7 (10 ng/mL, 15 minutes; 37�C). Reactions were stopped with cold PBSand cell lysates prepared as described. Equal amounts of protein(50 mg/sample) were resolved by sodium dodecyl sulfate poly-acrylamide gel electrophoresis, transferred onto nitrocellulosemembranes, and immunoblotted with phosphospecific antibodiesfor eIF4G(Ser1108); S6RP(Ser235/236); PDK1(Ser241); Akt(Ser473); GSK3b(Ser9); FKHR(Ser256); Cdk2(Thr160); p53(Ser46) (Cell Signaling); PRAS40(Thr246) (BioSource); or anti-bodies for Cyclin D3; Cdk6; p27kip1 (Cell Signaling); or for totalS6RP, total Akt (Cell Signaling), or actin (Santa Cruz Biotech-nology) as loading controls. Immunodetection was performed byincubation with horseradish peroxidase�conjugated antibodiesfor mouse, rabbit, or goat IgG (Promega, Madison, WI, USA), fol-lowed by chemiluminescence developing (Western Lightning; Per-kin Elmer, Boston, MA, USA). Determination of relative proteinintensity was performed by densitometry analysis (Kodak Molec-ular Imaging, Rochester, NY, USA), using total S6RP, total Akt, oractin expression as control.
Cell-cycle, apoptosis, and cell-size analysisFor cell-cycle analysis, quiescent TAIL7 cells were cultured inRPMI-10medium alone, with IL-7 (10 ng/mL) or with IL-7 plus ra-pamycin or CCI-779 (10 nM) for 72 and 96 hours. Cell-cycle profilewas assessed by measuring DNA content by propidium iodide (PI;BD-Biosciences) staining. Cells (1 � 106) were resuspended inPBS, fixed in ice-cold 80% ethanol (1 hour, 4�C), pretreated withRNase (50 mg/mL, 30 minutes), and incubated with PI (2.5 mg/mL,30 minutes). Analysis was carried immediately after staining ona Cytomics-FC500 flow cytometer, and data analyzed on MCycle32 software (Phoenix Flow, San Diego, CA, USA).
For apoptosis studies, cells (5 � 105) were resuspended inbinding buffer, stained with fluorescein isothiocyanate�conjugatedAnnexin V (1 mL/mL; BD-Biosciences) and PI (5 mg/mL,15 minutes), and analyzed by flow cytometry. Alterations in cellsize were determined by analysis of live cells (gated as AnnexinV/PI-negative) in forward scatter vs. side scatter plots; percentageof large-sized cells was calculated by defining a threshold gate
that excluded most of the bulk, small-sized cells in the control(RPMI-10) condition.
Proliferation and viability assaysCell proliferation was determined by DNA synthesis quantificationusing 3H-thymidine incorporation. Primary T-ALL or quiescentTAIL7 cells (2 � 106 cells/mL) were cultured in RPMI-10, IL-7(10 ng/mL), or IL-7 plus IL-9 (10 ng/mL; R&D Systems). For inhi-bition experiments, indicated concentrations of rapamycin, CCI-779, LY294002, WHI-P131 (Jak3 inhibitor; Calbiochem), or thechemotherapy drugs dexamethasone (American PharmaceuticalPartners, Schaumburg, IL, USA) or doxorubicin (Bedford Labora-tories, Bedford, OH, USA) were used as single agents or two-agentcombinations (mTOR inhibitor plus second agent). All cultureswere performed in triplicate (37�C, 96 hours) and 3H-thymidine(1 mCi/well) was added for the last 18 hours of culture. Cellswere harvested into glass-fiber filters using an automated harvesterand analyzed in a liquid scintillation counter (Microbeta TriLux,Wallac; Perkin-Elmer).
Proliferation was also assessed by staining using the stable fluo-rochrome 5,6-carboxylfluorescein-diacetate-succinimidyl ester(CFSE; Molecular Probes, Eugene, OR, USA), the fluorescenceintensity of which halves with each cell division, allowing todiscriminate successive generations of proliferating cells. TAIL7cells (1 � 106 cells/mL) were starved, washed in PBS, and stainedwith CFSE (0.2 mM, 5 minutes, 37�C), with the reaction stoppedby the addition of excess RPMI-10. A sample of CSFE-labeled cellswas collected and fixed in 1% formaldehyde, as day-0 stainingcontrol. The remaining cells were plated in RPMI-10 plus IL-7(10 ng/mL) in the presence of rapamycin or CCI-779 (10 nM), at37�C, for analysis on day 7. Cells were harvested, acquired byflow cytometry (Cytomics FC500) and data analyzed using FlowJo(Tree Star, Inc., Ashland, OR, USA).
Cell viability and, indirectly, cell proliferation, were assessedby quantifying metabolic active cells through measurements ofcellular contents of adenosine triphosphate using the CellTiter-Glo luminescence assay (Promega, Madison, WI, USA).
Drug interaction and statistical analysesThe interaction between two drugs was evaluated by applying theChou-Talalay method [30] to dose-response cell proliferationcurves, using the CalcuSyn software (Biosoft, Cambridge, UK).The method, based on the use of the median-effect andcombination-index equations, generates a drug Combination Index(CI), where CI! 1,5 1, orO1 values indicate synergism, additiveeffect, or antagonism, respectively.
Differences between experimental study conditions were eval-uated using the statistical tests indicated in the respective figurelegends. Differences of means were considered statistically signif-icant for p ! 0.05.
Results
mTOR pathway is activated in primary T-ALL cellsmTOR pathway is often activated in cancer and moleculesupstream and downstream of mTOR seem to contributedirectly to the initiation and progression of several tumortypes. To determine the potential involvement of mTOR

460 A. Batista et al./ Experimental Hematology 2011;39:457–472
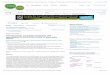
in T-cell ALL, 12 pediatric primary leukemia were analyzedfor the status of activation of mTOR signaling. Similar anal-ysis was performed in the representative T-ALL patient-derived cell line TAIL7, which is IL-7�dependent andmimics the functional and molecular properties of primaryT-ALL cells [29]. In all cases analyzed, leukemia cellsshowed a marked phosphorylation of the mTOR down-stream effectors eIF4G and S6RP, with significant higherintensity than normal T cells (Fig. 1A, B; SupplementaryFigure E1A; online only, available at www.exphem.org).To investigate whether this activation is present in malignantcells residing in the leukemic BM microenvironment, weevaluated the phosphorylation status of S6RP in BM biop-sies of nonobese diabetic/severe combined immunodeficientmice engrafted with human leukemia T cells. As shown byimmunohistochemistry analyses, most leukemia cell blastsexpressed phosphorylated S6RP (Fig. 1C), indicating theengagement of the mTOR pathway in tumor cells evolvingin the leukemic BM in vivo. Overall, these results provideevidence for a persistent activation of mTOR signaling inT-ALL.
Engagement of mTOR signaling in T-ALL is associatedwith activation of PI3K/AktBiochemical and genetic experimental data support theconcept that mTOR signaling is engaged in response tomito-genic stimuli through activation of the PI3K/Akt/TSC/Rhebaxis [31]. Also, tumors with aberrantly high PI3K/Aktsignaling seem to be particularly sensitive to mTORblockade. Therefore, we evaluated whether the mTOR acti-vation observed in leukemia T cells may be attributed toPI3K/Akt signaling. First, we determined the activationstatus of several downstream PI3K targets by analyzing theirphosphorylation state. All patients’ specimens showed robustphosphorylation of PDK1, a direct positive regulator of Akt(Fig. 2A). Fittingly, phosphorylation of Akt was observedin most patients (five of six patients), although some hetero-geneity was seen (Fig. 2A). Most cases exhibited engage-ment of the Akt downstream substrates GSK3, FKHRtranscription factors, and PRAS40, as well (Fig. 2A). Simul-taneous phosphorylation of all Akt substrates was not consis-tently observed in the patients analyzed, likely reflecting thesignal wiring heterogeneity seen on other cancers [32].Second, we analyzed the effects of PI3K/Akt blockade onmTOR signaling in T-ALL cells by specifically inhibitingPI3K using LY294002 (10 mM), in comparison to directmTOR blockade with rapamycin (10 nM). PI3K inhibitionwith LY294002 and the direct mTOR blockadewith rapamy-cin exerted a comparable effect on the S6RP phosphorylation(Fig. 2B), indicating that signals from PI3K/Akt pathwayinput on mTOR function.
IL-7 is a cytokine critical for T-cell development and it isimplicated in T-cell leukemogenesis [24]. In the IL-7�dependent TAIL7 cells, IL-7 stimulation significantlyincreased phosphorylation of S6RP (Fig. 2B; lane 2 vs.
lane 1; and Supplementary Figure E2A, E2B; online only,available at www.exphem.org). Importantly, inhibition ofS6RP phosphorylation by rapamycin was dominant even inthe presence of IL-7 (lane 6 vs. lane 5; and SupplementaryFigure E2A, E2B; online only, available at www.exphem.org), demonstrating that these mTOR antagonists are ableto overcome stimulatory signals provided by the leukemicmicroenvironment. Interestingly, treatment with rapamycinresulted in partial reduction of Akt phosphorylation(Fig. 2B; lane 6 vs. lane 2; and Supplementary Figure E2C;online only, available at www.exphem.org), suggesting theexistence of divergent regulatory loops influencing Akt andmTOR signaling, as supported by studies implicating thecomplex mTOR/Rictor as PDK2 [33].
Blockade of mTOR by rapamycin or CCI-779 inhibitsT-ALL proliferationTo determine the functional effects of mTOR blockade onleukemia T cells, experiments were conducted using thespecific mTOR inhibitors rapamycin and CCI-779. Westarted by performing biochemical analyses to confirm thatthese agents effectively inhibit mTOR signaling inmalignantT cells. TAIL7 cells were treated for 15 to 120 minutes with10 nM of each drug, a concentration correspondent toa known pharmacologically relevant dose of rapamycin. Asshown in Figure 2C (and Supplementary Figure E3A; onlineonly, available at www.exphem.org), both agents downregu-lated the phosphorylation levels of S6RP in a time-dependentmanner, with comparable kinetics. Importantly, both rapa-mycin and CCI-779 (10 nM) markedly downregulatedS6RP phosphorylation in primary leukemia T cells, althoughsome degree of inter-patient variation was observed(Fig. 2D). These results show that rapamycin and CCI-779have comparable efficacy in blocking mTOR signaling inT-ALL cells.
Next, we assessed the impact of mTOR blockade onleukemia T-cell proliferation in response to IL-7. Cells weretreated with increasing doses of rapamycin or CCI-779(0.01 to 103 nM) in the presence of IL-7 (10 ng/mL), andproliferation was determined by measuring 3H-thymidineuptake. Both rapamycin and CCI-779 inhibited proliferationof TAIL7 in a dose-dependent manner, with an IC50 ofapproximately 10 nM for both agents (Fig. 3A). Over therange of effective doses, the drug solvent (dimethyl sulf-oxide) showed no significant toxicity on these cells(Supplementary Figure E3B; online only, available atwww.exphem.org). In the absence of IL-7, TAIL7 cells arequiescent and the effect of mTOR blockade on cell viabilitywas minimal; conversely, mTOR inhibitors significantlyantagonized the stimulatory effects of IL-7 on T-ALL cells,inducing efficient arrest of cells fully engaged in IL-7�mediated proliferation (Fig. 3B). The antiproliferative effectsof mTOR blockade on leukemia T cells were also evaluatedusing aCFSE-based assay. As shown in Figure 3C, IL-7 stim-ulation resulted in a higher than twofold increase in the
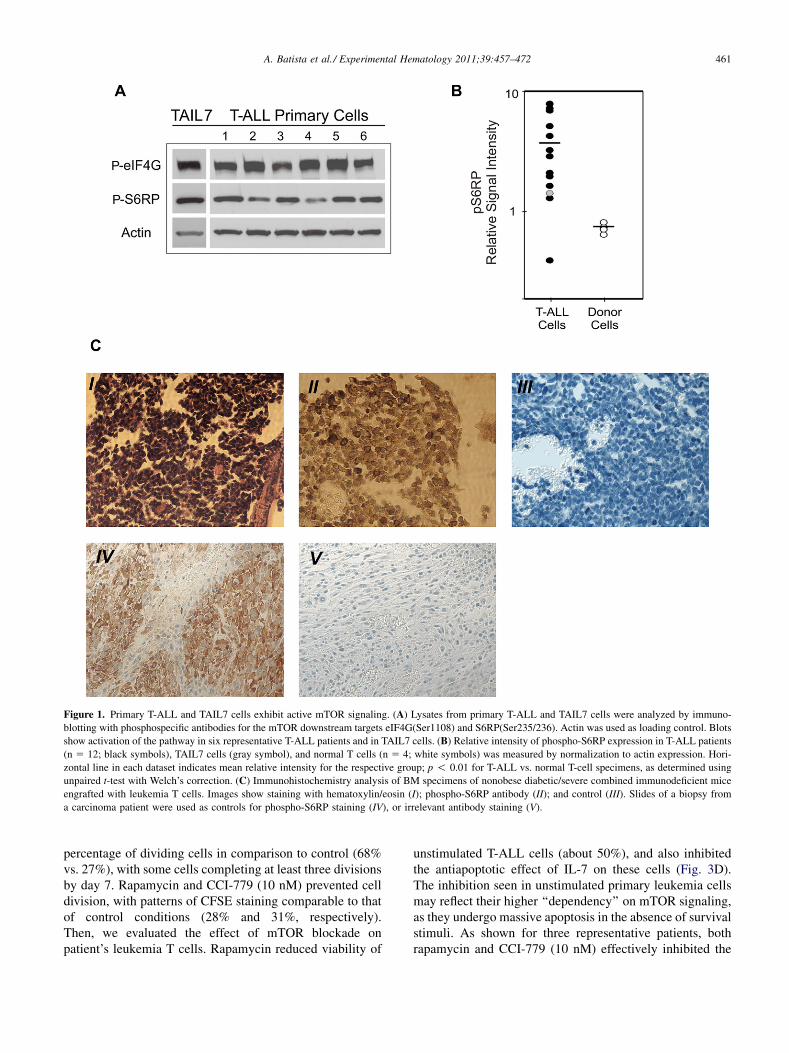
Figure 1. Primary T-ALL and TAIL7 cells exhibit active mTOR signaling. (A) Lysates from primary T-ALL and TAIL7 cells were analyzed by immuno-
blotting with phosphospecific antibodies for the mTOR downstream targets eIF4G(Ser1108) and S6RP(Ser235/236). Actin was used as loading control. Blots
show activation of the pathway in six representative T-ALL patients and in TAIL7 cells. (B) Relative intensity of phospho-S6RP expression in T-ALL patients
(n 5 12; black symbols), TAIL7 cells (gray symbol), and normal T cells (n 5 4; white symbols) was measured by normalization to actin expression. Hori-
zontal line in each dataset indicates mean relative intensity for the respective group; p ! 0.01 for T-ALL vs. normal T-cell specimens, as determined using
unpaired t-test with Welch’s correction. (C) Immunohistochemistry analysis of BM specimens of nonobese diabetic/severe combined immunodeficient mice
engrafted with leukemia T cells. Images show staining with hematoxylin/eosin (I); phospho-S6RP antibody (II); and control (III). Slides of a biopsy from
a carcinoma patient were used as controls for phospho-S6RP staining (IV), or irrelevant antibody staining (V).
461A. Batista et al./ Experimental Hematology 2011;39:457–472
percentage of dividing cells in comparison to control (68%vs. 27%), with some cells completing at least three divisionsby day 7. Rapamycin and CCI-779 (10 nM) prevented celldivision, with patterns of CFSE staining comparable to thatof control conditions (28% and 31%, respectively).Then, we evaluated the effect of mTOR blockade onpatient’s leukemia T cells. Rapamycin reduced viability of
unstimulated T-ALL cells (about 50%), and also inhibitedthe antiapoptotic effect of IL-7 on these cells (Fig. 3D).The inhibition seen in unstimulated primary leukemia cellsmay reflect their higher ‘‘dependency’’ on mTOR signaling,as they undergo massive apoptosis in the absence of survivalstimuli. As shown for three representative patients, bothrapamycin and CCI-779 (10 nM) effectively inhibited the
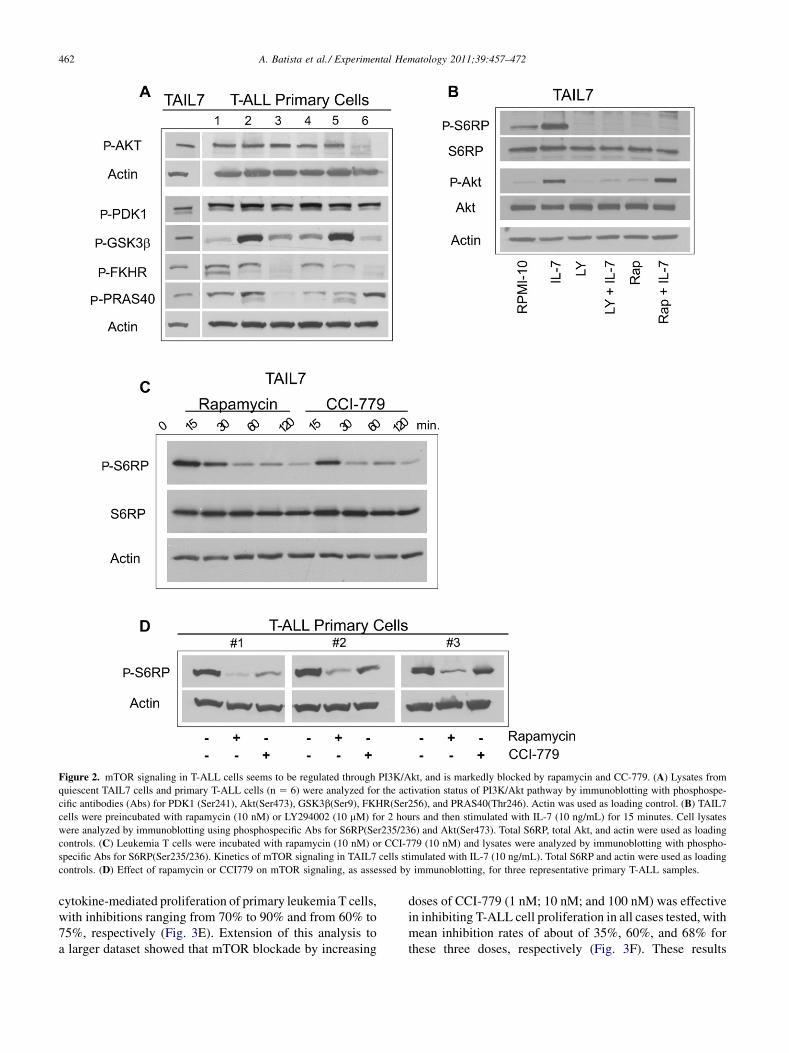
Figure 2. mTOR signaling in T-ALL cells seems to be regulated through PI3K/Akt, and is markedly blocked by rapamycin and CC-779. (A) Lysates from
quiescent TAIL7 cells and primary T-ALL cells (n 5 6) were analyzed for the activation status of PI3K/Akt pathway by immunoblotting with phosphospe-
cific antibodies (Abs) for PDK1 (Ser241), Akt(Ser473), GSK3b(Ser9), FKHR(Ser256), and PRAS40(Thr246). Actin was used as loading control. (B) TAIL7
cells were preincubated with rapamycin (10 nM) or LY294002 (10 mM) for 2 hours and then stimulated with IL-7 (10 ng/mL) for 15 minutes. Cell lysates
were analyzed by immunoblotting using phosphospecific Abs for S6RP(Ser235/236) and Akt(Ser473). Total S6RP, total Akt, and actin were used as loading
controls. (C) Leukemia T cells were incubated with rapamycin (10 nM) or CCI-779 (10 nM) and lysates were analyzed by immunoblotting with phospho-
specific Abs for S6RP(Ser235/236). Kinetics of mTOR signaling in TAIL7 cells stimulated with IL-7 (10 ng/mL). Total S6RP and actin were used as loading
controls. (D) Effect of rapamycin or CCI779 on mTOR signaling, as assessed by immunoblotting, for three representative primary T-ALL samples.
462 A. Batista et al./ Experimental Hematology 2011;39:457–472
cytokine-mediated proliferation of primary leukemia T cells,with inhibitions ranging from 70% to 90% and from 60% to75%, respectively (Fig. 3E). Extension of this analysis toa larger dataset showed that mTOR blockade by increasing
doses of CCI-779 (1 nM; 10 nM; and 100 nM) was effectivein inhibiting T-ALL cell proliferation in all cases tested, withmean inhibition rates of about of 35%, 60%, and 68% forthese three doses, respectively (Fig. 3F). These results
Figure 3. mTORblockade inhibits proliferation of primary T-ALL and TAIL7 cells. (A) Quies-
cent TAIL7 cells were cultured in RPMI-10 plus IL-7 (10 ng/mL) in the presence of increasing
doses of rapamycin orCCI-779 (range, 0.01�103 nM) for 96hours. Proliferationwas assessed by3H-thymidine incorporation anddata are expressed as a percentage of theRPMI-10þ IL-7 condi-
tion. (B) Quiescent TAIL7 were cultured in RPMI-10 with/without IL-7 (10 ng/mL) and mTOR
inhibitors (rapamycin, 10 nM; CCI-779, 10 nM) for 96 hours. The number of viable cells was
determined by measurement of cellular adenosine triphosphate levels, and data normalized to
the RPMI-10 control condition. (C) TAIL7 cells were cultured with rapamycin (10 nM) or
CCI-779 (10 nM) and cell division was assessed by CSFE staining after 7 days; RPMI-10 was
used as control condition. (D) Primary T-ALL cells were cultured with rapamycin (10 nM) in
the presence/absence of IL-7 (10 ng/mL). Viability was determined by measuring adenosine
triphosphate levels and datawere normalized to theRPMI-10 condition. (E) PrimaryT-ALLcells
were cultured with rapamycin (10 nM) or CCI-779 (10 nM) for 96 hours in the presence of cyto-
kine stimuli (IL-7, 10 ng/mL; IL-9, 10 ng/mL); proliferation was measured by 3H-thymidine
uptake and data expressed as percentage of the control RPMI-10þ IL-7/IL-9 condition. Results
are shown for three leukemia patients. (F) Proliferation rates for 10 primary T-ALL samples
treated for 96 hours with increasing doses of CCI-779 (1 nM; 10 nM; 100 nM); data expressed
as a percentage of proliferation observed in the control in RPMI-10 þ IL-7/IL-9 condition.
463A. Batista et al./ Experimental Hematology 2011;39:457–472
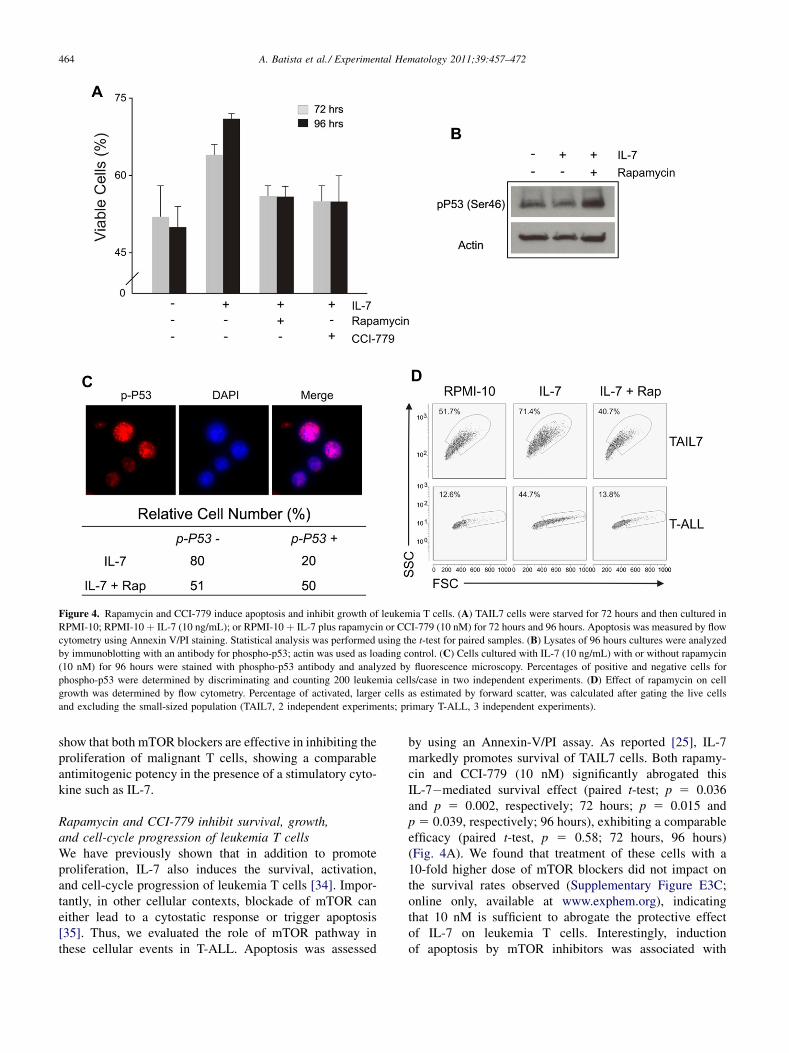
Figure 4. Rapamycin and CCI-779 induce apoptosis and inhibit growth of leukemia T cells. (A) TAIL7 cells were starved for 72 hours and then cultured in
RPMI-10; RPMI-10þ IL-7 (10 ng/mL); or RPMI-10þ IL-7 plus rapamycin or CCI-779 (10 nM) for 72 hours and 96 hours. Apoptosis was measured by flow
cytometry using Annexin V/PI staining. Statistical analysis was performed using the t-test for paired samples. (B) Lysates of 96 hours cultures were analyzed
by immunoblotting with an antibody for phospho-p53; actin was used as loading control. (C) Cells cultured with IL-7 (10 ng/mL) with or without rapamycin
(10 nM) for 96 hours were stained with phospho-p53 antibody and analyzed by fluorescence microscopy. Percentages of positive and negative cells for
phospho-p53 were determined by discriminating and counting 200 leukemia cells/case in two independent experiments. (D) Effect of rapamycin on cell
growth was determined by flow cytometry. Percentage of activated, larger cells as estimated by forward scatter, was calculated after gating the live cells
and excluding the small-sized population (TAIL7, 2 independent experiments; primary T-ALL, 3 independent experiments).
464 A. Batista et al./ Experimental Hematology 2011;39:457–472
show that both mTOR blockers are effective in inhibiting theproliferation of malignant T cells, showing a comparableantimitogenic potency in the presence of a stimulatory cyto-kine such as IL-7.
Rapamycin and CCI-779 inhibit survival, growth,and cell-cycle progression of leukemia T cellsWe have previously shown that in addition to promoteproliferation, IL-7 also induces the survival, activation,and cell-cycle progression of leukemia T cells [34]. Impor-tantly, in other cellular contexts, blockade of mTOR caneither lead to a cytostatic response or trigger apoptosis[35]. Thus, we evaluated the role of mTOR pathway inthese cellular events in T-ALL. Apoptosis was assessed
by using an Annexin-V/PI assay. As reported [25], IL-7markedly promotes survival of TAIL7 cells. Both rapamy-cin and CCI-779 (10 nM) significantly abrogated thisIL-7�mediated survival effect (paired t-test; p 5 0.036and p 5 0.002, respectively; 72 hours; p 5 0.015 andp 5 0.039, respectively; 96 hours), exhibiting a comparableefficacy (paired t-test, p 5 0.58; 72 hours, 96 hours)(Fig. 4A). We found that treatment of these cells with a10-fold higher dose of mTOR blockers did not impact onthe survival rates observed (Supplementary Figure E3C;online only, available at www.exphem.org), indicatingthat 10 nM is sufficient to abrogate the protective effectof IL-7 on leukemia T cells. Interestingly, inductionof apoptosis by mTOR inhibitors was associated with
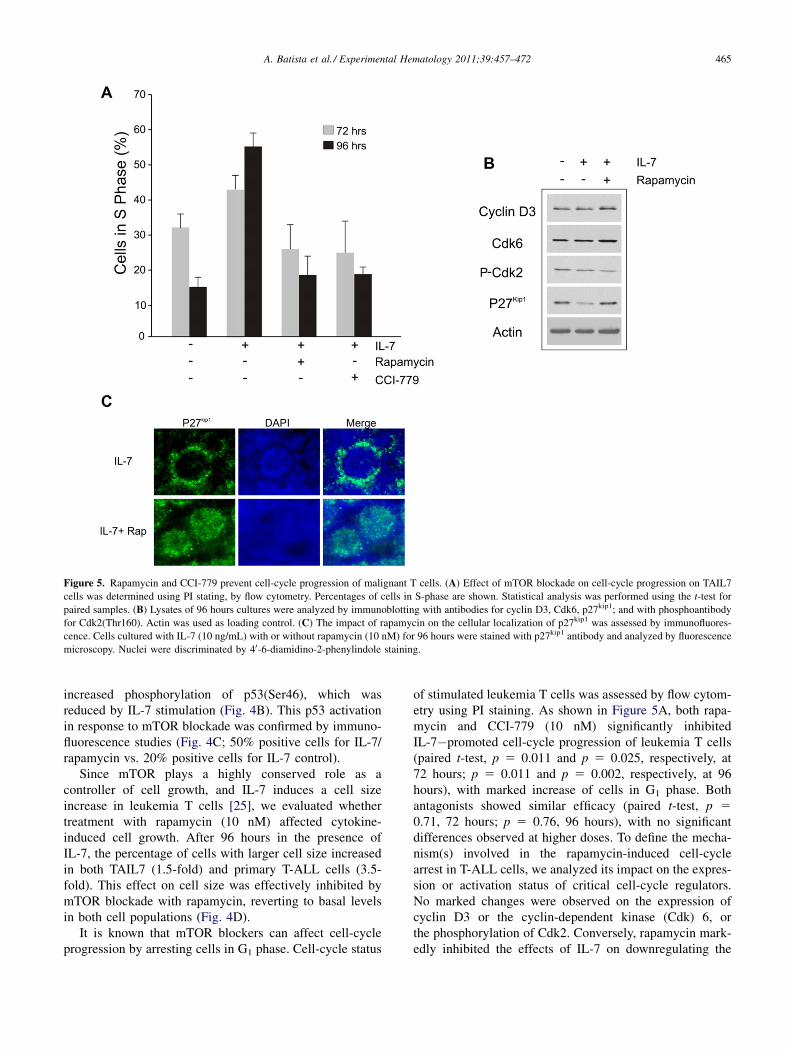
Figure 5. Rapamycin and CCI-779 prevent cell-cycle progression of malignant T cells. (A) Effect of mTOR blockade on cell-cycle progression on TAIL7
cells was determined using PI stating, by flow cytometry. Percentages of cells in S-phase are shown. Statistical analysis was performed using the t-test for
paired samples. (B) Lysates of 96 hours cultures were analyzed by immunoblotting with antibodies for cyclin D3, Cdk6, p27kip1; and with phosphoantibody
for Cdk2(Thr160). Actin was used as loading control. (C) The impact of rapamycin on the cellular localization of p27kip1 was assessed by immunofluores-
cence. Cells cultured with IL-7 (10 ng/mL) with or without rapamycin (10 nM) for 96 hours were stained with p27kip1 antibody and analyzed by fluorescence
microscopy. Nuclei were discriminated by 40-6-diamidino-2-phenylindole staining.
465A. Batista et al./ Experimental Hematology 2011;39:457–472
increased phosphorylation of p53(Ser46), which wasreduced by IL-7 stimulation (Fig. 4B). This p53 activationin response to mTOR blockade was confirmed by immuno-fluorescence studies (Fig. 4C; 50% positive cells for IL-7/rapamycin vs. 20% positive cells for IL-7 control).
Since mTOR plays a highly conserved role as acontroller of cell growth, and IL-7 induces a cell sizeincrease in leukemia T cells [25], we evaluated whethertreatment with rapamycin (10 nM) affected cytokine-induced cell growth. After 96 hours in the presence ofIL-7, the percentage of cells with larger cell size increasedin both TAIL7 (1.5-fold) and primary T-ALL cells (3.5-fold). This effect on cell size was effectively inhibited bymTOR blockade with rapamycin, reverting to basal levelsin both cell populations (Fig. 4D).
It is known that mTOR blockers can affect cell-cycleprogression by arresting cells in G1 phase. Cell-cycle status
of stimulated leukemia T cells was assessed by flow cytom-etry using PI staining. As shown in Figure 5A, both rapa-mycin and CCI-779 (10 nM) significantly inhibitedIL-7�promoted cell-cycle progression of leukemia T cells(paired t-test, p 5 0.011 and p 5 0.025, respectively, at72 hours; p 5 0.011 and p 5 0.002, respectively, at 96hours), with marked increase of cells in G1 phase. Bothantagonists showed similar efficacy (paired t-test, p 50.71, 72 hours; p 5 0.76, 96 hours), with no significantdifferences observed at higher doses. To define the mecha-nism(s) involved in the rapamycin-induced cell-cyclearrest in T-ALL cells, we analyzed its impact on the expres-sion or activation status of critical cell-cycle regulators.No marked changes were observed on the expression ofcyclin D3 or the cyclin-dependent kinase (Cdk) 6, orthe phosphorylation of Cdk2. Conversely, rapamycin mark-edly inhibited the effects of IL-7 on downregulating the

466 A. Batista et al./ Experimental Hematology 2011;39:457–472
cyclin-dependent kinase inhibitor p27kip1 (Fig. 5B). Interest-ingly, we observed that, in addition to blocking its down-regulation, rapamycin also displaces p27kip1 from acytoplasmic into a nuclear localization (Fig. 5C). Theseresults may explain the effect of mTOR blockade on cellcycle and are in agreement with our previous work showingthat p27kip1 downregulation in T-ALL cells by IL-7 promotessurvival and cell-cycle progression [34]. Overall, theseobservations indicate that the mTOR pathway plays animportant role in the response of malignant T cells tosurvival, growth, and cell-cycle progression signals fromtheir microenvironment, and that the use of mTORblockers may inhibit or abrogate these functional advantagesof malignant cells.
mTOR blockade potentiates the antileukemia effectsof chemotherapy drugsToaddress the potential benefit of includingmTOR inhibitorsin treatment regimens for T-ALL, we evaluated whethermTOR blockade impacts on the antileukemia activity ofdexamethasone and doxorubicin, two drugs commonlyused in this malignancy. Single drug-inhibition curves weregenerated by treating TAIL7 cells with increasing clinicallyachievable concentrations of dexamethasone and doxoru-bicin (0.01�103 nM). Fifty-percent proliferation inhibitiondoses were determined as 10 nM for dexamethasone and100 nM for doxorubicin (Fig. 6A, C, respectively; graphinserts; single dose curves, open ovals). Rapamycin and theindividual chemotherapeutic drugs were tested in combina-tion at doses ranging from 6fourfold of their respectiveIC50 ratios, and the Chou-Talalay method was used to deter-mine potential drug synergisms. The combination of rapamy-cin with dexamethasone or with doxorubicin generatedCI values ranging from 0.002 to 0.021 and 0.011 to 0.199,respectively, thus indicating highly synergistic drug inter-action. Over the range of concentrations tested, TAIL7 pro-liferation was inhibited between 80% and 100% withrapamycinþdexamethasone, and between 70% and 100%with rapamycinþdoxorubicin (Figs. 6A, C, respectively;combined drugs curves). Then, we evaluated the effect ofthe combination of rapamycin and these chemotherapeuticdrugs on primary T-ALL cells, using combined concentra-tions two and five times lower than their respective IC50
values. As seen in Figures 6B and D, blockade of mTORclearly increased the antimitogenic effects of both drugs,with marked decrease on leukemia cell proliferation. Similarresults were obtained with CCI-779 (data not shown).
mTOR blockade synergizes with other signalingantagonists to effectively inhibit T-ALL cellsIncreasing evidence suggests that the simultaneous targetingof distinct signaling pathways, or of different effectorswithinthe same pathway, may represent a more effective strategyto treat cancer. We evaluated whether mTOR blockadecan sensitize leukemia T cells to signaling inhibitors
targeting pathways active in these cells [25]. First, wedetermined the individual effects of blockade of PI3K(LY294002), and of Jak-3 (WHI-P131) on IL-7�promotedproliferation of TAIL7 cells, using increasing doses of eachantagonist (0.01�103 mM). These antagonists inhibited cellproliferation in a dose-dependent manner, with an IC50 ofapproximately 10 mM (Figs. 7A, C; graph inserts; singledrug curves, open ovals). Second, rapamycin was combinedwith each inhibitor at their equipotent ratios, using theapproach described for conventional drugs. The combinationof rapamycin with LY294002, or WHI-P131 generated CIvalues ranging from 0.063 to 0.162 and 0.176 to 0.213,respectively, indicating a very highly synergistic interactionbetween rapamycin and these signaling antagonists. Over theIC50 6 fourfold concentration range tested, TAIL7 prolifer-ation was inhibited between 90% and 100% with rapa þLY294002 and 70% and 100% with rapa þ WHI-P131(Fig. 7A and C, respectively; combined drugs curves). Inall cases, maximum inhibition was achieved with concentra-tions of rapamycin and inhibitors that individually showa non-cytotoxic submaximal effect. Finally, these drugcombinations were tested in primary leukemia T cells, usingcombined concentrations two- and fivefold lower than theIC50 value. As shown in Figure 7B and D for two representa-tive patients, marked drug interactions were observed, withmTOR blockade increasing the antileukemia effects of theindividual inhibitors and effectively preventing T-ALLproliferation. Comparable results were observed usingCCI-779 (data not shown). Taken together, these observa-tions indicate that, in combinationwith other agents (conven-tional or specific signaling antagonists), blockade of mTORsignaling may be a valid approach for the targeted therapy ofT-cell leukemia.
DiscussionmTOR is an attractive molecule for rational targeted therapybecause it integrates a multiplicity of upstream growth-promoting signals conveyed bydistinct pathways, is often de-regulated in cancer, and can exist in a hyperactivated state incancer cells. Here, we provided in situ and molecularevidence that primary leukemia T cells from all patientstested exhibit active mTOR signaling and are sensitive tomTOR blockade. Importantly, we show that mTOR antago-nists can also inhibit leukemia cell survival, cell-cycleprogression, and growth in the presence of the protectiveeffect of IL-7, which is present in the leukemic BM.
The causes leading tomTOR activation or hyperactivationin T-cell leukemia are largely unknown. Aberrant PI3K/Aktactivation and loss of regulation by the tumor suppressorPTEN are well-known oncogenic events that input onmTOR [10,11,36]. Defective PTEN function, stemmingfrom deletions, silencing, or mutations, is associated witha wide spectrum of tumors [37,38], and seems to rendertumors more sensitive to mTOR blockade [39]. We have
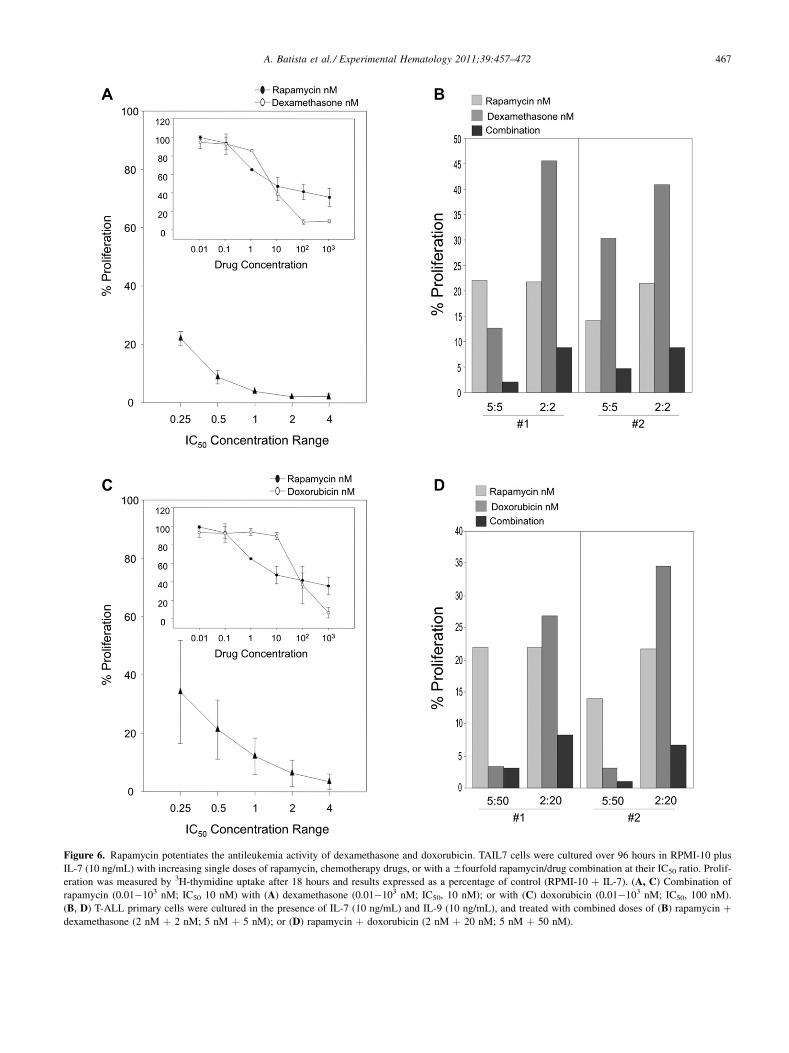
Figure 6. Rapamycin potentiates the antileukemia activity of dexamethasone and doxorubicin. TAIL7 cells were cultured over 96 hours in RPMI-10 plus
IL-7 (10 ng/mL) with increasing single doses of rapamycin, chemotherapy drugs, or with a 6fourfold rapamycin/drug combination at their IC50 ratio. Prolif-
eration was measured by 3H-thymidine uptake after 18 hours and results expressed as a percentage of control (RPMI-10 þ IL-7). (A, C) Combination of
rapamycin (0.01�103 nM; IC50 10 nM) with (A) dexamethasone (0.01�103 nM; IC50, 10 nM); or with (C) doxorubicin (0.01�103 nM; IC50, 100 nM).
(B, D) T-ALL primary cells were cultured in the presence of IL-7 (10 ng/mL) and IL-9 (10 ng/mL), and treated with combined doses of (B) rapamycin þdexamethasone (2 nM þ 2 nM; 5 nM þ 5 nM); or (D) rapamycin þ doxorubicin (2 nM þ 20 nM; 5 nM þ 50 nM).
467A. Batista et al./ Experimental Hematology 2011;39:457–472
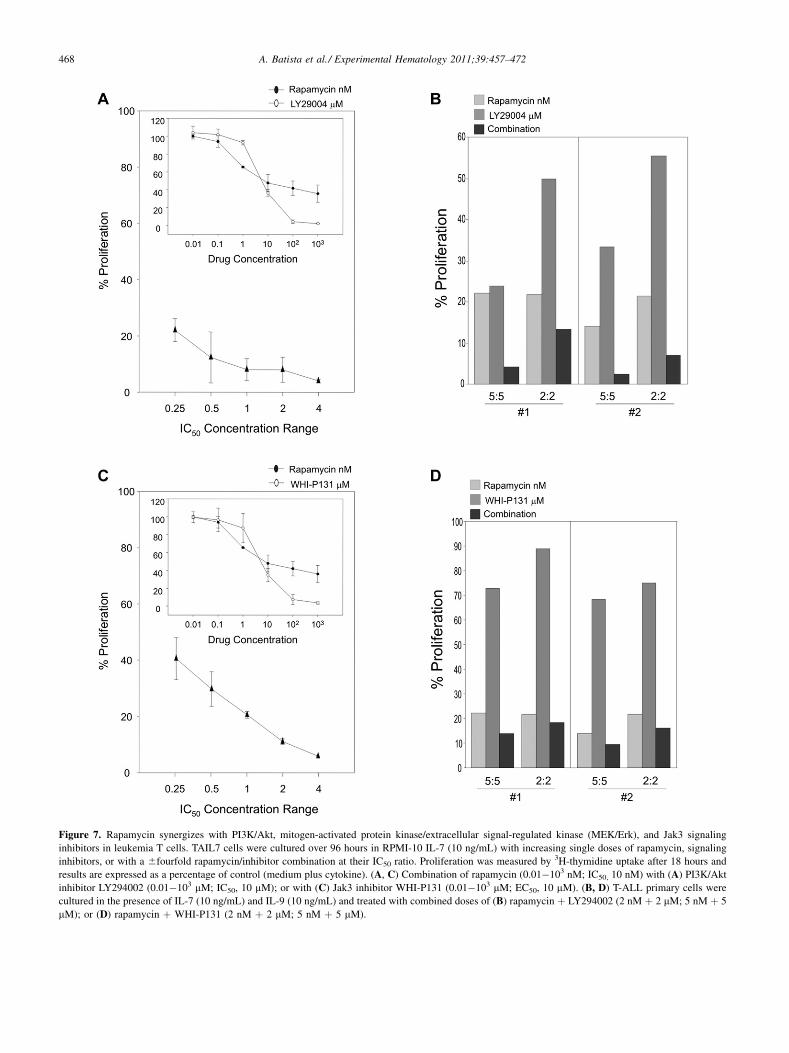
Figure 7. Rapamycin synergizes with PI3K/Akt, mitogen-activated protein kinase/extracellular signal-regulated kinase (MEK/Erk), and Jak3 signaling
inhibitors in leukemia T cells. TAIL7 cells were cultured over 96 hours in RPMI-10 IL-7 (10 ng/mL) with increasing single doses of rapamycin, signaling
inhibitors, or with a 6fourfold rapamycin/inhibitor combination at their IC50 ratio. Proliferation was measured by 3H-thymidine uptake after 18 hours and
results are expressed as a percentage of control (medium plus cytokine). (A, C) Combination of rapamycin (0.01�103 nM; IC50, 10 nM) with (A) PI3K/Akt
inhibitor LY294002 (0.01�103 mM; IC50, 10 mM); or with (C) Jak3 inhibitor WHI-P131 (0.01�103 mM; EC50, 10 mM). (B, D) T-ALL primary cells were
cultured in the presence of IL-7 (10 ng/mL) and IL-9 (10 ng/mL) and treated with combined doses of (B) rapamycin þ LY294002 (2 nM þ 2 mM; 5 nM þ 5
mM); or (D) rapamycin þ WHI-P131 (2 nM þ 2 mM; 5 nM þ 5 mM).
468 A. Batista et al./ Experimental Hematology 2011;39:457–472

469A. Batista et al./ Experimental Hematology 2011;39:457–472
shownPI3K/Akt hyperactivation inT-ALL [25] and observedthat it is associated with PTEN loss of function through non-mutational, post-transcriptional mechanisms [40].
The studies presented here indicate that PI3K/Aktengagement is critical for mTOR activation in T-ALL cellsand that, reciprocally, mTOR partially regulates Akt in thesecells (Fig. 2B; and Supplementary Figure E2C; online only,available at www.exphem.org). The heterogeneity observedin the engagement of the Akt substrates in primary leukemiaspecimens may result from patient-specific wiring of intra-cellular signaling networks, leading to modulation of onlysome of the components of a signaling cascade in responseto defined stimuli, such as oncogenic events or cytokine-triggered signals [32]. Importantly, the fact that simulta-neous blockade of mTOR and PI3K synergize to inhibitleukemia T cells indicates that functional uncoupling ofthese pathways exists in T-ALL. A significant percentageof T-ALL tumors, from all major genotypes, harbor acti-vating mutations of Notch1 [26]. A recent study showedthat in a subgroup of immortalized leukemia T-cell lines,Notch positively regulates mTOR activation througha PI3K/Akt-independent, c-myc�dependent mechanism[27]. Because these observations were not verified onpatient’s specimens, which, in contrast to immortalizedcell lines, are dependent on extrinsic signals, further studiesare necessary to define the role of Notch/mTOR cross-signaling in T-ALL biology. In independent studies, weobserved that Notch blockade by g-secretase inhibitorsdoes not affect mTOR signaling in primary leukemia T cellsand that constitutive Notch activation does not confer factor-independence to these cells (Batista et al., manuscriptsubmitted).
An aspect frequently dismissed in T-ALLand othermalig-nancies is that malignant cells reside in specialized tissueniches and are responsive to, and in many instances depen-dent on, extrinsic signals provided by their microenviron-ment, which can contribute to the activation of variousmolecular cascades, including PI3K/Akt signaling. Wereported that active engagement of PI3K/Akt signaling isessential for IL-7�mediated stimulation of growth, survival,cell-cycle progression, glucose metabolism, and mainte-nance of mitochondrial integrity in T-ALL cells [25]. PI3K/Akt and mTOR signaling are also involved in theleukemia-promoted activation of BM endothelial cells,which reciprocally support leukemia cells through solublefactors (such as IL-7) and cell-to-cell contact mechanisms[41,42]. Other microenvironmental cues known to act onleukemia cells need to be assessed as potential activatorsof PI3K/Akt/mTOR signaling in T-ALL. For example,CXCR4 cross-linking by stromal cell-derived factor�1/CXCL12 triggers PI3K/Akt activation, and this axis hasbeen implicated in the homing and maintenance of leukemiacells in the BM, namely on their association with the endo-thelial niche [43]. Also, the thymic chemokine TECK/CCL25, which is expressed by BM endothelium (Cardoso,
unpublished observations) has been implicated in the biologyof T-ALL [44] and has been shown to activate the PI3K/Aktcascade [45].
Thus, the mechanisms of mTOR persistent activation inT-ALL and how it couples with PI3K/Akt and othersignaling pathways need further clarification, particularlyin primary leukemia cells. Another aspect requiring furtheranalysis is the regulatory feedback role of mTOR on PI3K/AKT signaling in T-ALL, namely the effects of mTORblockade on the modulation of Akt status. Recent work indi-cates that mTOR inhibition can trigger dual effects on Aktactivation, possibly in a cell-specific manner. In some cases,rapamycin restrains a negative feedback loop from activatedS6K1 to Akt, thus increasing Akt activation [46]. In othercontexts, prolonged rapamycin administration downregu-lates Akt activation by interfering with the equilibrium ofthe formation of the mTOR/Rictor complex, the putativekinase PDK2 that phosphorylates Akt at Ser473 [33].Understanding the prevalence of these mechanisms iscrucial to the clinical translation of mTOR blockers inT-cell ALL, specifically for their use as single or as partof combinatorial targeted strategies.
Despite clear functional responses to mTOR blockade,a consistent difference in the modulation of mTOR down-stream pathways was seen in our studies; while the S6K1activation was rapidly abolished, we only observed partialinhibition of 4E-BP1 (Supplementary Figure E1B; onlineonly, available at www.exphem.org). This difference insensitivity has been reported earlier [47], and may eitherreflect substrate selectivity by mTOR inhibitors, as consis-tent with previous studies [48]; a different threshold for thephosphorylation of S6K1 and 4E-BP1; or the presence inT-ALL of a rapamycin-insensitive pathway that also signalsthrough 4E-BP1. At the functional level, mTOR disruptionby rapamycin or CCI-779 consistently resulted in compa-rable antiproliferative effects on leukemia T cells. Impor-tantly, the range of estimated EC50 values indicatesa high sensitivity of T-ALL cells to mTOR blockade.
In this study, we show that mTOR blockade bypasses theprotective effect of IL-7, with inhibition of proliferationbeing accompanied by induction of apoptosis and cell-cycle arrest. Although rapamycin has been shown to inhibitleukemia cells and to affect T-cell homeostasis [34,49–51],the ability of IL-7 to directly trigger mTOR signaling hasnot been reported previously. Our observation that inhibi-tion of mTOR overcomes the survival advantage conferredby IL-7 is in contrast with findings in B-cell precursor ALL[50], in which IL-7 could reverse mTOR inhibition, asS6K1 is suggested as a downstream target of both cytokineand rapamycin. It is possible that in T-ALL cells, IL-7signaling may be restricted to events upstream of mTOR.Alternatively, the strength of the signal may be insufficientto prevail over the response of cells to mTOR blockade.
Mechanistically, the inhibitory effects of mTORblockade in T-ALL are associated with upregulation of

470 A. Batista et al./ Experimental Hematology 2011;39:457–472
p27kip1 and phosphorylation of p53(Ser46). We reportedpreviously that IL-7 sustains survival and cell-cycle progres-sion of leukemia T cells through a p27kip1-dependent mech-anism, and that enforced expression of p27kip1 inhibitedIL-7�stimulated cdk2 activation and reversed Rb hyper-phosphorylation [34]. It is known that rapamycin blockscell-cycle progression at G1 phase by upregulating p27kip1
to a threshold triggering the assembly of cyclin E/Cdk2-p27kip1 complex [52]. The present work provides evidencethat rapamycin increases p27kip1 to levels above thoserequired to prevent cell division, thus overcoming IL-7 stim-ulation. We also show that IL-7 stimulation of T-ALL cellsresults not only in p27kip1 downregulation but also in itsdisplacement to the cytoplasm; whereas mTOR blockaderestores nuclear p27kip1 and its inhibitory activity. Oncogenicactivation of Ras, PI3K/Akt, or mitogen-activated proteinkinase has been implicated in engaging p27kip1 downregula-tion mechanisms. In particular, activated Akt can directlyphosphorylate p27kip1 NLS motif, impairing nuclear importand causing cytoplasmatic accumulation [53]. It is reason-able that a similar regulation exists in T-ALL cells.
Therapies using rationally designed inhibitors can haveremarkable efficacy with limited secondary effects, asproven with BRC/ABL targeted inhibition in chronicmyeloid leukemia [54]. Preclinical studies in hematologicalmalignancies showed that mTOR blockers can exert anti-proliferative, apoptotic, and antiangiogenic activity, andcan slow the growth and reduce the size of tumors[49,50,55–57], supporting their therapeutic use. A substan-tial limitation to the success of targeted monotherapies isthat prolonged drug use frequently generates mechanismsof resistance, with tumor cells acquiring beneficial muta-tions or taking advantage of signaling redundancies orcross-signaling. Combinatory strategies have been sug-gested as an approach to contour this hindrance. In vitrostudies and animal models in myeloma, acute myeloidleukemia, and BCR-ABL transformed cells showed thatmTOR blockade synergized with tyrosine kinase inhibitorsand Revlimid and sensitized cells to dexamethasone or eto-poside [47,58,59]. A recent study in T-ALL showed that thedual PI3K/Akt inhibitor PI-103 inhibits leukemia T cells,inducing caspase-3 activation [60]. Here, we demonstratethat mTOR inhibition cooperated with different signalinginhibitors and chemotherapy drugs with high degrees ofsynergism observed for the combined blockade of mTORwith PI3K/Akt and Jak/signal transducers and activatorsof transcription pathways, as well as with the chemothera-peutic agents dexamethasone or doxorubicin. Importantly,these synergisms allowed effective inhibition of primaryT-ALL proliferation at doses up to five times lower thantheir estimated IC50, suggesting that it may be translatedinto clinical benefit.
In conclusion, this report provides evidence for persis-tent activation of mTOR signaling in malignant T cellswithin the leukemic microenvironment and demonstrates
that mTOR blockade significantly inhibits the survivaland proliferation of T cells, overcoming the stimulatoryand protective effects of IL-7. Also, it suggests that theuse of mTOR inhibitors can be maximized in rationalcombinatory therapies. Finally, it supports the specific tar-geting of mTOR signaling as a valid strategy for the treat-ment of pediatric T-cell ALL.
AcknowledgmentsWe thank Dr. Sabina Signoretti and the Pathology Core for theirassistance on the immunohistochemistry studies. Supported bygrants from the National Institutes of Health (P01-CA68484)and from the Fundac~ao para Ciencia e Tecnologia, Portugal(FCT; SAU/13240 and CBO/34914). A.B. was supported bya scholarship FCT-Portugal.
Conflict of interest disclosureNo financial interest/relationships with financial interest relatingto the topic of this article have been declared.
References1. Hahn WC, Weinberg RA. Rules for making human tumor cells.
N Engl J Med. 2002;347:1593–1603.
2. Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:
57–70.
3. Sherr CJ. The Pezcoller lecture: cancer cell cycles revisited. Cancer
Res. 2000;60:3689–3695.
4. Weinstein IB. Cancer. Addiction to oncogenesdthe Achilles heal of
cancer. Science. 2002;297:63–64.
5. Fingar DC, Blenis J. Target of rapamycin (TOR): an integrator of
nutrient and growth factor signals and coordinator of cell growth
and cell cycle progression. Oncogene. 2004;23:3151–3171.
6. Schmelzle T, Hall MN. TOR, a central controller of cell growth. Cell.
2000;103:253–262.
7. Burnett PE, Barrow RK, Cohen NA, Snyder SH, Sabatini DM. RAFT1
phosphorylation of the translational regulators p70 S6 kinase and 4E-
BP1. Proc Natl Acad Sci U S A. 1998;95:1432–1437.
8. Gingras AC, Raught B, Sonenberg N. eIF4 initiation factors: effectors
of mRNA recruitment to ribosomes and regulators of translation. Annu
Rev Biochem. 1999;68:913–963.
9. Ruvinsky I, Sharon N, Lerer T, et al. Ribosomal protein S6 phosphor-
ylation is a determinant of cell size and glucose homeostasis. Genes
Dev. 2005;19:2199–2211.
10. Sekulic A, Hudson CC, Homme JL, et al. A direct linkage between the
phosphoinositide 3-kinase-AKT signaling pathway and the mamma-
lian target of rapamycin in mitogen-stimulated and transformed cells.
Cancer Res. 2000;60:3504–3513.
11. Vogt PK. PI 3-kinase, mTOR, protein synthesis and cancer. Trends
Mol Med. 2001;7:482–484.
12. Inoki K, Li Y, Zhu T, Wu J, Guan KL. TSC2 is phosphorylated and
inhibited by Akt and suppresses mTOR signalling. Nat Cell Biol.
2002;4:648–657.
13. Altomare DA, Testa JR. Perturbations of the AKT signaling pathway
in human cancer. Oncogene. 2005;24:7455–7464.
14. Abraham RT, Wiederrecht GJ. Immunopharmacology of rapamycin.
Annu Rev Immunol. 1996;14:483–510.
15. Geoerger B, Kerr K, Tang CB, et al. Antitumor activity of the rapa-
mycin analog CCI-779 in human primitive neuroectodermal

471A. Batista et al./ Experimental Hematology 2011;39:457–472
tumor/medulloblastoma models as single agent and in combination
chemotherapy. Cancer Res. 2001;61:1527–1532.
16. Mondesire WH, Jian W, Zhang H, et al. Targeting mammalian
target of rapamycin synergistically enhances chemotherapy-induced
cytotoxicity in breast cancer cells. Clin Cancer Res. 2004;10:7031–
7042.
17. Yan H, Frost P, Shi Y, et al. Mechanism by which mammalian target
of rapamycin inhibitors sensitize multiple myeloma cells to
dexamethasone-induced apoptosis. Cancer Res. 2006;66:2305–2313.
18. Wei G, Twomey D, Lamb J, et al. Gene expression-based chemical
genomics identifies rapamycin as a modulator of MCL1 and glucocor-
ticoid resistance. Cancer Cell. 2006;10:331–342.
19. Grunwald V, DeGraffenried L, Russel D, Friedrichs WE, Ray RB,
Hidalgo M. Inhibitors of mTOR reverse doxorubicin resistance
conferred by PTEN status in prostate cancer cells. Cancer Res. 2002;
62:6141–6145.
20. Hagner PR, Schneider A, Gartenhaus RB. Targeting the translational
machinery as a novel treatment strategy for hematologic malignancies.
Blood. 2010;115:2127–2135.
21. Chapuis N, Tamburini J, Green AS, et al. Perspectives on inhibiting
mTOR as a future treatment strategy for hematological malignancies.
Leukemia. 2010;24:1686–1699.
22. Funk PE, Stephan RP, Witte PL. Vascular cell adhesion molecule
1-positive reticular cells express interleukin-7 and stem cell factor in
the bone marrow. Blood. 1995;86:2661–2671.
23. Sakata T, Iwagami S, Tsuruta Y, et al. Constitutive expression of
interleukin-7 mRNA and production of IL-7 by a cloned murine
thymic stromal cell line. J Leukoc Biol. 1990;48:205–212.
24. Barata JT, Cardoso AA, Boussiotis VA. Interleukin-7 in T-cell acute
lymphoblastic leukemia: an extrinsic factor supporting leukemogen-
esis? Leuk Lymphoma. 2005;46:483–495.
25. Barata JT, Silva A, Brandao JG, Nadler LM, Cardoso AA,
Boussiotis VA. Activation of PI3K is indispensable for interleukin
7-mediated viability, proliferation, glucose use, and growth of T cell
acute lymphoblastic leukemia cells. J Exp Med. 2004;200:659–669.
26. Weng AP, Ferrando AA, Lee W, et al. Activating mutations of
NOTCH1 in human T cell acute lymphoblastic leukemia. Science.
2004;306:269–271.
27. Chan SM, Weng AP, Tibshirani R, Aster JC, Utz PJ. Notch signals
positively regulate activity of the mTOR pathway in T-cell acute
lymphoblastic leukemia. Blood. 2007;110:278–286.
28. Cullion K, Draheim KM, Hermance N, et al. Targeting the Notch1 and
mTORpathways in amouse T-ALLmodel. Blood. 2009;113:6172–6181.
29. Barata JT, Boussiotis VA, Yunes JA, et al. IL-7-dependent human
leukemia T-cell line as a valuable tool for drug discovery in T-ALL.
Blood. 2004;103:1891–1900.
30. Chou TC, Talalay P. Quantitative analysis of dose-effect relationships:
the combined effects of multiple drugs or enzyme inhibitors. Adv
Enzyme Regul. 1984;22:27–55.
31. Hay N, Sonenberg N. Upstream and downstream of mTOR. Genes
Dev. 2004;18:1926–1945.
32. Irish JM, Hovland R, Krutzik PO, et al. Single cell profiling of poten-
tiated phospho-protein networks in cancer cells. Cell. 2004;118:
217–228.
33. Sarbassov DD, Ali SM, Sengupta S, et al. Prolonged rapamycin treat-
ment inhibits mTORC2 assembly and Akt/PKB. Mol Cell. 2006;22:
159–168.
34. Barata JT, Cardoso AA, Nadler LM, Boussiotis VA. Interleukin-7
promotes survival and cell cycle progression of T-cell acute lympho-
blastic leukemia cells by down-regulating the cyclin-dependent kinase
inhibitor p27(kip1). Blood. 2001;98:1524–1531.
35. Easton JB, Houghton PJ. mTOR and cancer therapy. Oncogene. 2006;
25:6436–6446.
36. Vivanco I, Sawyers CL. The phosphatidylinositol 3-kinase AKT
pathway in human cancer. Nat Rev Cancer. 2002;2:489–501.
37. Stiles B, Groszer M, Wang S, Jiao J, Wu H. PTENless means more.
Dev Biol. 2004;273:175–184.
38. Sansal I, Sellers WR. The biology and clinical relevance of the PTEN
tumor suppressor pathway. J Clin Oncol. 2004;22:2954–2963.
39. Neshat MS, Mellinghoff IK, Tran C, et al. Enhanced sensitivity of
PTEN-deficient tumors to inhibition of FRAP/mTOR. Proc Natl
Acad Sci U S A. 2001;98:10314–10319.
40. Silva A, Yunes JA, Cardoso BA, et al. PTEN posttranslational inacti-
vation and hyperactivation of the PI3K/Akt pathway sustain primary
T cell leukemia viability. J Clin Invest. 2008;118:3762–3774.
41. Costa LF, Balcells M, Edelman ER, Nadler LM, Cardoso AA. Proan-
giogenic stimulation of bone marrow endothelium engages mTOR and
is inhibited by simultaneous blockade of mTOR and NF-kappaB.
Blood. 2006;107:285–292.
42. Veiga JP, Costa LF, Sallan SE, Nadler LM, Cardoso AA. Leukemia-
stimulated bone marrow endothelium promotes leukemia cell survival.
Exp Hematol. 2006;34:610–621.
43. Sipkins DA, Wei X, Wu JW, et al. In vivo imaging of specialized bone
marrow endothelial microdomains for tumour engraftment. Nature.
2005;435:969–973.
44. Qiuping Z, Jei X, Youxin J, et al. CC chemokine ligand 25 enhances
resistance to apoptosis in CD4þT cells frompatients with T-cell lineage
acute and chronic lymphocytic leukemia by means of livin activation.
Cancer Res. 2004;64:7579–7587.
45. Youn BS, Kim YJ, Mantel C, Yu KY, Broxmeyer HE. Blocking of
c-FLIP(L)dindependent cycloheximide-induced apoptosis or Fas-
mediated apoptosis by the CC chemokine receptor 9/TECK interaction.
Blood. 2001;98:925–933.
46. O’Reilly KE, Rojo F, She QB, et al. mTOR inhibition induces
upstream receptor tyrosine kinase signaling and activates Akt. Cancer
Res. 2006;66:1500–1508.
47. Mohi MG, Boulton C, Gu TL, et al. Combination of rapamycin and
protein tyrosine kinase (PTK) inhibitors for the treatment of leukemias
caused by oncogenic PTKs. Proc Natl Acad Sci U S A. 2004;101:
3130–3135.
48. McMahon LP, Choi KM, Lin TA, Abraham RT, Lawrence JC Jr.
The rapamycin-binding domain governs substrate selectivity by
the mammalian target of rapamycin. Mol Cell Biol. 2002;22:7428–
7438.
49. Avellino R, Romano S, Parasole R, et al. Rapamycin stimulates
apoptosis of childhood acute lymphoblastic leukemia cells. Blood.
2005;106:1400–1406.
50. Brown VI, Fang J, Alcorn K, et al. Rapamycin is active against B-
precursor leukemia in vitro and in vivo, an effect that is modulated
by IL-7-mediated signaling. Proc Natl Acad Sci U S A. 2003;100:
15113–15118.
51. Sharma D, Kumar SS, Raghu R, Khanam S, Sainis KB. Differential
modulation of mitogen driven proliferation and homeostasis driven
proliferation of T cells by rapamycin, Ly294002 and chlorophyllin.
Mol Immunol. 2007;44:2831–2840.
52. Kawamata S, Sakaida H, Hori T, Maeda M, Uchiyama T. The upregu-
lation of p27Kip1 by rapamycin results in G1 arrest in exponentially
growing T-cell lines. Blood. 1998;91:561–569.
53. Liang J, Zubovitz J, Petrocelli T, et al. PKB/Akt phosphorylates p27,
impairs nuclear import of p27 and opposes p27-mediated G1 arrest.
Nat Med. 2002;8:1153–1160.
54. Giles FJ, Cortes JE, Kantarjian HM. Targeting the kinase activity of
the BCR-ABL fusion protein in patients with chronic myeloid
leukemia. Curr Mol Med. 2005;5:615–623.
55. Frost P, Moatamed F, Hoang B, et al. In vivo antitumor effects of the
mTOR inhibitor CCI-779 against human multiple myeloma cells in
a xenograft model. Blood. 2004;104:4181–4187.
56. Recher C, Beyne-Rauzy O, Demur C, et al. Antileukemic activity
of rapamycin in acute myeloid leukemia. Blood. 2005;105:2527–
2534.

472 A. Batista et al./ Experimental Hematology 2011;39:457–472
57. Teachey DT, Obzut DA, Cooperman J, et al. The mTOR inhibitor CCI-
779 induces apoptosis and inhibits growth in preclinical models of
primary adult human ALL. Blood. 2006;107:1149–1155.
58. Raje N, Kumar S, Hideshima T, et al. Combination of the mTOR
inhibitor rapamycin and CC-5013 has synergistic activity in multiple
myeloma. Blood. 2004;104:4188–4193.
59. Xu Q, Thompson JE, Carroll M. mTOR regulates cell survival after eto-
poside treatment in primary AML cells. Blood. 2005;106:4261–4268.
60. Chiarini F, Fala F, Tazzari PL, et al. Dual inhibition of class IA phos-
phatidylinositol 3-kinase and mammalian target of rapamycin as a new
therapeutic option for T-cell acute lymphoblastic leukemia. Cancer
Res. 2009;69:3520–3528.

Supplementary Figure E1. mTOR signaling is upregulated in malignant T cells as compared to normal T-lymphocytes. (A) Lysates from purified, resting
normal T cells (T1-4) and TAIL7 cells were analyzed by immunoblotting using a phosphospecific antibody (Ab) for S6RP(Ser235/236); actin was used as
loading control and for normalization. Although normal blood T cells are not the best counterparts for T-cell ALL, it has been shown that in comparison to
normal thymocytes, T-ALL cells exhibit markedly increased activation of PI3K/Akt signaling [40], which is upstream of mTOR signaling. (B) Blot was
probed with a phospho-Ab for IeF4G(Ser1108), and actin was used as loading control.
472.e1A. Batista et al./ Experimental Hematology 2011;39:457–472
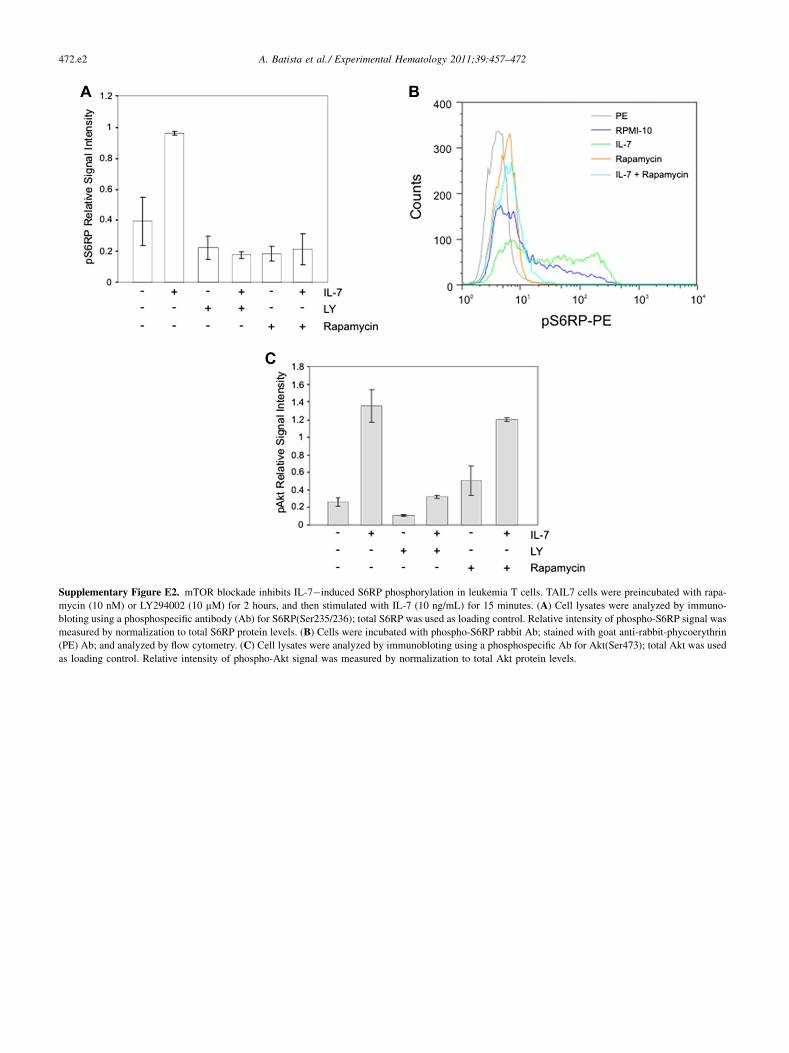
Supplementary Figure E2. mTOR blockade inhibits IL-7�induced S6RP phosphorylation in leukemia T cells. TAIL7 cells were preincubated with rapa-
mycin (10 nM) or LY294002 (10 mM) for 2 hours, and then stimulated with IL-7 (10 ng/mL) for 15 minutes. (A) Cell lysates were analyzed by immuno-
bloting using a phosphospecific antibody (Ab) for S6RP(Ser235/236); total S6RP was used as loading control. Relative intensity of phospho-S6RP signal was
measured by normalization to total S6RP protein levels. (B) Cells were incubated with phospho-S6RP rabbit Ab; stained with goat anti-rabbit-phycoerythrin
(PE) Ab; and analyzed by flow cytometry. (C) Cell lysates were analyzed by immunobloting using a phosphospecific Ab for Akt(Ser473); total Akt was used
as loading control. Relative intensity of phospho-Akt signal was measured by normalization to total Akt protein levels.
472.e2 A. Batista et al./ Experimental Hematology 2011;39:457–472
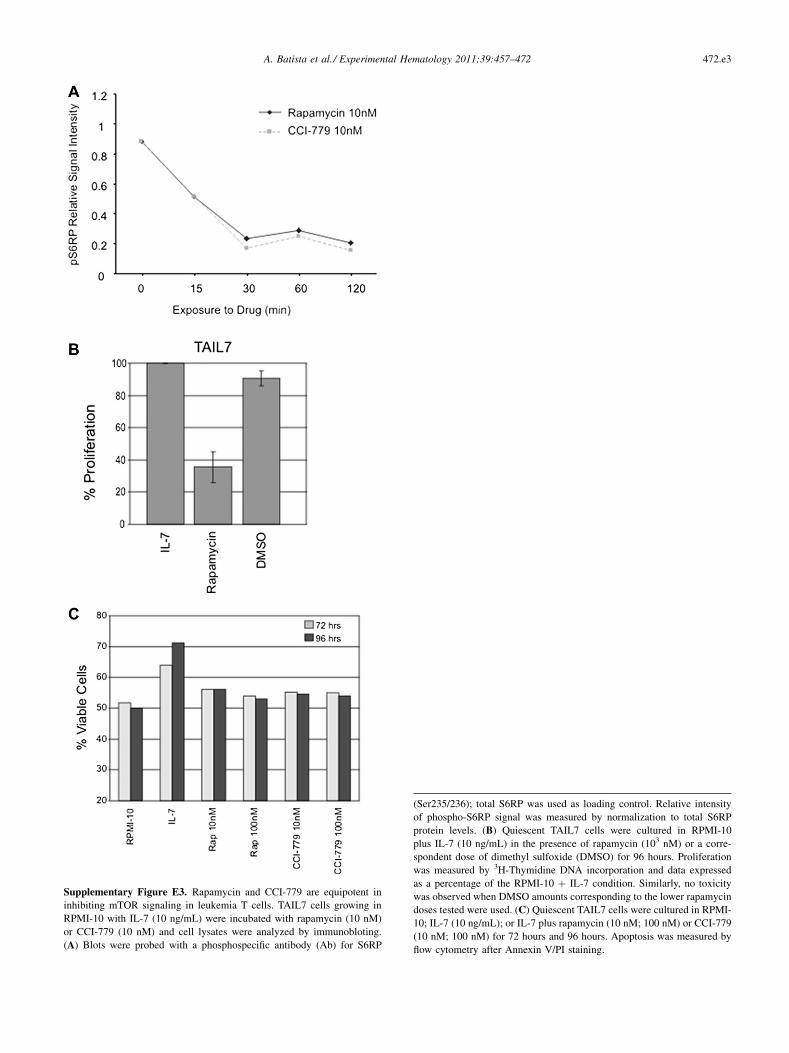
Supplementary Figure E3. Rapamycin and CCI-779 are equipotent in
inhibiting mTOR signaling in leukemia T cells. TAIL7 cells growing in
RPMI-10 with IL-7 (10 ng/mL) were incubated with rapamycin (10 nM)
or CCI-779 (10 nM) and cell lysates were analyzed by immunobloting.
(A) Blots were probed with a phosphospecific antibody (Ab) for S6RP
(Ser235/236); total S6RP was used as loading control. Relative intensity
of phospho-S6RP signal was measured by normalization to total S6RP
protein levels. (B) Quiescent TAIL7 cells were cultured in RPMI-10
plus IL-7 (10 ng/mL) in the presence of rapamycin (103 nM) or a corre-
spondent dose of dimethyl sulfoxide (DMSO) for 96 hours. Proliferation
was measured by 3H-Thymidine DNA incorporation and data expressed
as a percentage of the RPMI-10 þ IL-7 condition. Similarly, no toxicity
was observed when DMSO amounts corresponding to the lower rapamycin
doses tested were used. (C) Quiescent TAIL7 cells were cultured in RPMI-
10; IL-7 (10 ng/mL); or IL-7 plus rapamycin (10 nM; 100 nM) or CCI-779
(10 nM; 100 nM) for 72 hours and 96 hours. Apoptosis was measured by
flow cytometry after Annexin V/PI staining.
472.e3A. Batista et al./ Experimental Hematology 2011;39:457–472
![The natural compound forskolin synergizes with ... · PDF fileThe natural compound forskolin synergizes with dexamethasone to ... (50mM Tris [pH7.5], 150mM NaCl ... The natural compound](https://img.pdfslide.us/doc/110x75/5abc4b217f8b9ab1118e03fe/the-natural-compound-forskolin-synergizes-with-natural-compound-forskolin-synergizes.jpg)