Embed Size (px)
Citation preview

Taking a Numeric Path
Idan Szpektor

The Input
A partial description of a molecule: The atoms The bonds The bonds lengths and angles
Spatial constraints on some of the atoms of a molecule.

The Questions
Is there a conformation of the molecule that answers the spatial constraints?
Provide explicit description of such conformations.
How many “really” different conformations are there? Which one is the best?

Hard life…
Torsion angles are (usually) unconstrained
a large number of degrees of freedom
The answers to the questions are not easy to find analytically. They are also resource consuming (running time).

A Numeric Path
Two algorithms take different approaches to limit the space of possible conformations to search in: Numeric randomized algorithm Semi numeric algorithm on a simpler problem

A Randomized Kinematics-Based Approach to Pharmacophore-Constrained Conformational Search and Database Screening
S. M. Lavalle, P. W. Finn, L. E. Kavraki, J. C. Latombe (2000)

The Pharmacophore Problem
The problem: to find a molecule conformation that satisfies a set of constraints out of a database of flexible molecules (e.g. for ligand docking).
Constraints: Different atoms and their features in the molecule
(types, charges etc.) The bond length and angles between bonds Static bond torsion angles Relative locations of some atoms from an anchor
atom

The Molecule Model – no rings

The Molecule Model (Cont…)
An atom ai carries standard information
A bond bi carries the following information: li – the bond length αi – the angle from the previous bond The set of possible torsion angles θi [0, 2π)
All info besides the torsion angles is fixed. Θ is the m dimensional vector of variables that
defines the conformation

The Pharmacophore Model
A finite set of features corresponding to a subset of atoms
Constraints on the relative positions between features (the atoms), when one of the features is designated as aanch, the origin of a global xyz coordinate

The Kinematic Model
The bond length, angles and the torsion angles Θ can be used to give the positions of all of the atoms, relative to aanch
We look at each atom center as a local coordinate frame. We would like to use the transformation from one coordinate frame to the another.

The Kinematic Model (Cont…)

The Kinematic Model (Cont…)
The homogeneous transformation is:
1 1 1 1
1 1 1 1
cos sin 0 0
sin cos cos cos sin sin
sin sin cos sin cos cos
0 0 0 1
i i
i i i i i i ii
i i i i i i i
lT
l

The Kinematic Model (Cont…)
The xyz position of atom ai is given by:
1 2
0
0
0
1 1
i
x
yTT T
z

The Kinematic Model (Cont…)
The coordinate frame of the molecule could be rotated with respect to the global coordinate frame of the pharmacophore feature positions
Another global coordinate frame transformation is needed

The Kinematic Model (Cont…)
Global rotation transformation based on Euler angles γ,φ,ψ:
( , , )
c s c c c s c s 0
s c c c s s c s c c s s 0
s c s s c 0
0 0 0 1
RT

The Kinematic Model (Cont…)
The complete xyz position of atom ai is given by:
1 2
0
0( , , )
0
1 1
R i
x
yT TT T
z

The Kinematic Error Function
Given Θ, γ,φ and ψ, the total amount of error between the requested feature positions G and the actual feature positions g can be measured as:
1
21
( , , , ) ( , , , )N
i i
i
d G g

The Energy Function
In a sense, the energy function measures the likelihood that the molecule will achieve a conformation in nature.
An example for an energy function:
2 21 12 2
12 6
,
( ) (1 cos( ))
( ) ( )
4
dtorsions
b abonds angles
ij ij i jij
i j ij ij ij
e K n
K R R K
q q
r r r

Two Questions
For a given molecule from a database and a pharmacophore:
Can the molecule achieve a low-energy conformation that satisfies the given pharmacophore?
What are the “distinct” low-energy conformations that satisfy the pharmacophore?

Randomized Conformation Search with Constraints – The Motivation
For the problem, the system of equations is generally under constrained, which leads to a complicated multidimensional solution set.
This consideration, and the need for efficiency, led to the choice of a numerical randomized technique.
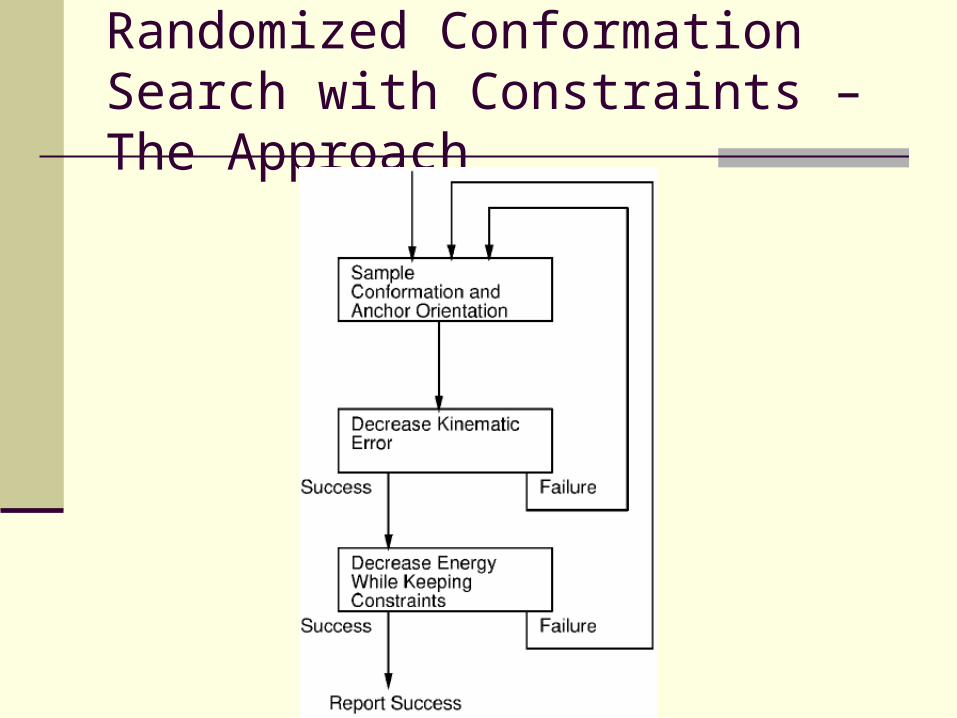
Randomized Conformation Search with Constraints – The Approach

The Search – Gradient Descent
Randomly sample the neighborhood of Pi
Search for a point Pi+1 such that f(Pi+1) < f(Pi)
If such Pi+1 is found, move to Pi+1 and repeat the search

Distance Minimization

Energy Minimization

Integrating into a Database Search
A small set (~several hundreds) of candidate molecules (configurations) are chosen from a database (using 2D information)
Kinematics-based conformational search is performed to further reduce the set of candidates

Search in a Set of Candidates
Each time a sample conformation fails to match, the likelihood that the molecule will ever succeed decrease
On the other hand, after any number of ‘fail’ iterations, it is impossible to conclude that the molecule will never succeed

Search in a Set of Candidates (cont…)
Perform one attempt (random sample + match search) per molecule
Go through all the molecules in the set and repeat
Stop when a requested number of matches was found or a maximum number of iterations was reached

Conformation Clustering
In general, having alternative low-energy conformations is useful because in many cases it is not the lowest-energy conformation that results in docking.

Conformation Clustering (cont...)
Use a metric m(θ1, θ2), such as RMS of the displacements of the atoms between two conformations
A threshold mmax is the maximum distance that still regards two conformations as identical (in the same cluster)

Clustering Algorithm – Idea
Always keep one representative for each cluster
The representative is the conformation with the lowest energy

Clustering Algorithm – Detailed
for a new conformation θi:
if exists another conformation θk such that m(θk, θi) ≤ mmax and e(θk) ≤ e(θi), discard θi
otherwise: add θi as a new cluster
remove all θk such that m(θk, θi) ≤ mmax

Experiments
2 different pharmacophores, for ACE and Thermolysin inhibitors 6 different molecules for each pharmacophore, kept in the
database in randomly picked conformations The docked conformations are known

Experiments (Cont…)
Cluster distance mmax=1.5 Ǻ
20 iterations for the different candidates

Experiments (Cont…)
In general, conformations’ energies are within 2–7 kcal/mol of the energy of the known docked conformation.
The RMS distance of the conformations from the known docked conformations: Thermolysin inhibitors – 0.50, 2.96, 0.59, 0.81,
2.40, and 2.56 Å. ACE inhibitors – 1.26, 1.79, 0.94, 2.03, 1.87,
and 1.98 Å.

Experiments (Cont…)
A sufficient clustering record for a single molecule required about 5–20 min.
A claim: “Our previous work with randomized techniques has shown that if we continue iterating our algorithm we increase our chances of covering the conformational space of the molecule, and hence, our chances of providing exhaustive information about the constrained conformations of the molecule.”

A Cluster record

Cyclic coordinate descent: A robotics algorithm for protein loop closure
A. A. Canutescu and R. L. Dunbrack Jr. (2003)

The Loop Closure Problem
The problem: matching a given loop to a given backbone (e.g. for Homology modeling).
Constraint: connecting the two protein segments on either end of the loop, termed N and C-terminal anchors.

Loop Closure Problem (Cont…)

Previous Solutions
Number of available conformation is enormous.
Analytical: for 6 degrees of freedom.
Numerical: changing all torsion angles at once to the next “best” position.
Numerical methods are computationally expensive and sometimes unstable.

Cyclic Coordinate Descent (CCD)
Originally developed for robotics.
An iterative relaxation algorithm.
Adjust only one degree of freedom at a time.

CCD Algorithm
Proceed in an iterative fashion along the chain of degrees of freedom.
Modify each torsion angle so that the end of the loop gets as close as possible to the desired position.

CCD Simplicity
One equation in one unknown for each degree of freedom.
The equation provides: Optimum setting for the variable First and second derivatives

The Main Equation

The Main Equation (Cont...)
2 2 2
1 1 2 2 3 3(1) S FM F M F M
ˆ ˆ(2) cos sini i i i i i i iFM r r r s f d
22 2 ˆ ˆ(3) 2 cos ( ) 2 sin ( )i i i i i i i i id r f r f r r f s
(4) cos sinS a b c

The Main Equation (Cont...)
Multiplying the last two terms by:
Defining:
We get:
2 2
2 2
b c
b c
2 2 2 2cos ,sin
b c
b c b c
2 2(5) cos( )S a b c
(4) cos sinS a b c

The Main Equation (Cont...)
2
ˆ ˆ(6) 2 sin ( ) 2 cos ( )i
i i i i i i
d dr f r r f s
d
ˆ( )(7) tan
ˆ( )
i i ii
i i ii
f s r
f r r

CCD Benefits
Computationally Fast.
Analytically simple - no singularities.
Constraints can be placed on any degree of freedom.
Using derivatives, small increments in change can be done in preference to of large changes.

Test 1 – Success Percentage
2752 different loops.
100 randomly different starting conformations for each loop.
A match (closed loop) is when distance from the terminals is less than 0.08 Ǻ.
Maximum 5000 iterative cycles (through all torsion angles) per search.

Test 1 (cont…)
Two test frameworks: No constraints Using a Ramachandran Map
When using the map: for a new proposed angle θnew, find (ψnew,θnew)
in the map and compare to (ψold,θold)
Change to θnew with probability:map( , )
map( , )new new
old old

Results for Test 1

Analysis of Test 1
Loops that fail to converge arrive to a local minimum (usage of Monte Carlo method to escape local minimum).
CCD favors large changes in the first residues.
Loops that failed to converge are usually extended loops.

Test 2 – Minimum RMS to Original
CCD is meant to be a component of a matching algorithm that should also include loop generation and energy function.
How well CCD alone converge to the original loop conformation.
30 original loops. For each loop, 5000 random conformations were generated and closed.
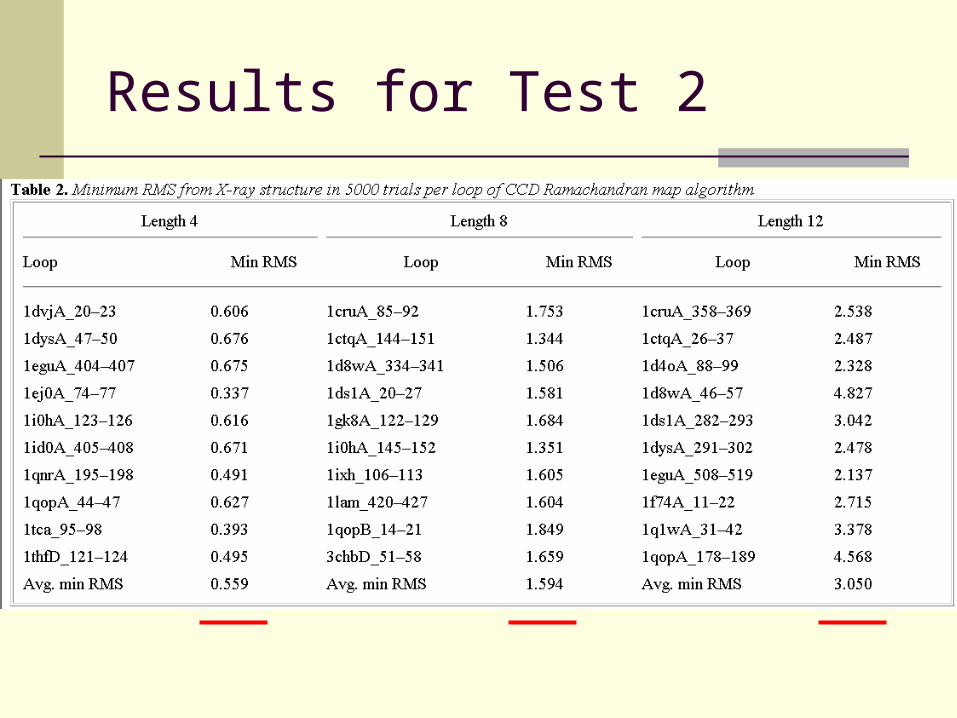
Results for Test 2

Results for Test 2 (Cont...)

Test 3 – Structure Convergence
Using Ramachandran map and starting with the same loop conformation.
Each time using different random generated numbers.
500 tests. do all final conformations are the same?
Compared to 500 closures of the same loop with different starting conformations.

Test 3 – Results

Comparison to Random TWEAK
5000 trails (10 loops, 500 initial configurations each). Closure when RMS < 0.08 Ǻ.
CCD – closed 5000. ~7 min running time
TWEAK – closed 4841. ~40 min running time





























