TABLETTablet is solid unit dosage form in which one usual dose
is accurately placed. It is basically oral route of administration
other route of tablets include vaginal tablets, rectal and
implantation tablets.TYPES OF TABLETSORAL TABLETS FOR INGESTION
These tablets are meant to be swallowed intact along with a
sufficient quantity of potable water. Exception is chewable tablet.
Over 90% of the tablets manufactured today are ingested orally.
This shows that this class of formulation is the most popular world
wide and the major attention of the researcher is towards this
direction.Standard compressed tabletsMultiple compressed
tabletsCompression coated tabletLayered tabletInlay tabletModified
Release tabletDelayed action tabletTargeted tabletFloating
tabletColon targeting tabletChewable tabletDispersible tablet
2- TABLETS USED IN THE ORAL CAVITY The tablets under this group
are aimed release API in oral cavity or to provide local action in
this region. The tablets under this category avoids first-pass
metabolism, decomposition in gastric environment, nauseatic
sensations and gives rapid onset of action. The tablets formulated
for this region are designed to fit in proper region of oral
cavity.Lozenges and trochesSublingual tabletBuccal tabletDental
conesMouth dissolved tablet3- TABLETS ADMINISTERED BY OTHER ROUTES
These tablets are administered by other route except for the oral
cavity and so the drugs are avoided from passing through gastro
intestinal tract. These tablets may be inserted into other body
cavities or directly placed below the skin to be absorbed into
systemic circulation from the site of application.Vaginal
tabletImplants
4- TABLETS USED TO PREPARE SOLUTION The tablets under this
category are required to be dissolved first in water or other
solvents before administration or application. This solution may be
for ingestion or parenteral application or for topical use
depending upon type of medicament used.Effervescent tabletSoluble
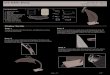
tabletOral tablets for ingestion:1- Standard compressed
tabletsThese are the standard uncoated tablets made by either
direct compression or wet granulation or dry granulation or double
compaction.
FIGURE.1. STANDARD COMPRESSED TABLETThey may be used for local
action in gastro-intestinal tract or systemic action. When the
tablet exert local action, they are formulated as more water
insoluble by means of selecting slow dissolving excipients and thus
provide local action for long time period. e.g., antacids and
adsorbents. The drugs that produce systemic action have some
aqueous solubility and designed to disintegrate and dissolve
quickly so that the drug can be quickly absorbed and produce
systemic action.
2- Multilayered tabletsWhen two or more active pharmaceutical
ingredients are needed to be administered simultaneously and they
are incompatible, the best option for the formulation pharmacist
would be to formulate multilayered tablet. It consists of several
different granulations that are compressed to form a single tablet
composed of two or more layers and usually each layer is of
different colour to produce a distinctive looking tablet. Each
layer is fed from separate feed frame with individual weight
control. Dust extraction is essential during compression to avoid
contamination. Therefore, each layer undergoes light compression as
each component is laid down. This avoids granules intermixing if
the machine vibrates.For example, admixture containing
Phenylephedrin HCL and Ascorbic Acid with ParacetamolCompression
coated tabletsThis type of tablet has two parts, internal core and
surrounding coat. The core is small porous tablet and prepared on
one turret. For preparing final tablet, a bigger die cavity in
another turret is used in which first the coat material is filled
to half and then core tablet is mechanically transferred, again the
remaining space is filled with coat material and finally
compression force is applied. This tablet readily lend itself in to
a repeat action tablet as the outer layer provides the initial dose
while the inner core release the drug later on. But, when the core
quickly releases the drug, entirely different blood level is
achieved with the risk of over dose toxicity. To avoid immediate
release of both the layers, the core tablet is coated with enteric
polymer so that it will not release the drug in stomach while, the
first dose is added in outer sugar coating. Even so, coating
operation requires interpretation while manufacturing and dawdling
the manufacturing process. Sometimes, inner core may be of liquid
formulation to provide immediate release of core after the coat
gets dissolved.
FIGURE.3. COMPRESSION COATED TABLETLayered tabletsA layered
tablet for the controlled release of active substances in a liquid
medium comprising at least one active substance-containing, layered
matrix with contact surfaces to the liquid medium which are at
least partially provided with a cover layer delaying or preventing
the active substance release, is characterized by the fact that the
cover layer is at least one additional layer lying with thickness
gradients on contact surfaces of the layered, prefabricated matrix,
or that the matrix is at least one additional layer lying with
thickness gradients on contact surfaces of the layered,
prefabricated cover layer, which additional layer is applied by
pressing powdery or granular material on the layered, prefabricated
matrix or on the layered, prefabricated cover layer.
Inlay tabletsA type of layered tablet in which instead the core
tablet being completely surrounded by coating, top surface is
completely exposed. While preparation, only the bottom of the die
cavity is filled with coating material and core is placed upon it.
When compression force is applied, some coating material is
displaced to form the sides and compress the whole tablet. It has
some advantages over compression coated tablets:i) Less coating
material is required.ii) Core is visible, so coreless tablets can
be easily detected.iii) Reduction in coating forms a thinner tablet
and thus freedom from capping of top coating.
FIGURE.4. INLAY TABLETS3- Modified release tabletThe main aim
behind formulation of this dosage form is to release the medicament
slowly for long time duration after administration of a single
tablet.More over, these type of formulations are generally used to
target the site specific releases.
FIGURE.5. GRAPHICAL COMPARISON OF BLOOD CONCENTRATION V/S TIMEA
widespread use of this type of tablet is seen in present scenario,
as well as many researchers have concentrated their attention in
this direction. 4- Delayed action tabletEnteric coated tablet is
such an example of delayed action tablet. This formulation is
preferred when,i) Drugs that irritates gastric mucosa e.g., aspirin
or strong electrolytesii)Drugs that produce nausea and
vomiting.iii)Drugs is sensitive to low pH e.g., erythromyciniv)When
its necessary to release the drug undiluted. e.g., intestinal
antibacterial, antiseptic agents, intestinal vermifuge, etc.The
commonly used coating agents are: Cellulose acetate phthalate,
Hydroxy methyl propyl phthalate, polyvinyl acetate phthalate,
Eudragit, etc. This dosage form is intended to hydrate and begin to
dissolve in duodenum (pH 4 to 6) or in small intestine where pH
increases to 7 to 8. The presence of esterases or bile salts like
surface active agents plays a role in drug release.5- Targeted
tabletWhen we need to release the drug at a specific site in the
elementary tract, targeted drug delivery is a preferred option.
Depending upon the composition and release mechanism of a tablet,
the drug is delivered to a particular region. Under this category,
we have two types of tablet:I.Gastro retentive Tablet (floating
tablets)This type of dosage form is to be opted when API release is
desired in stomach (Antacids, APIs used against H.pylori infection)
or site of absorption is either stomach or upper part of small
intestine.
FIGURE.10. FLOATING TABLETII. Colonic tabletsWhen the aim is to
deliver the drug into colon without dilution in other regions of
gastrointestinal tract or the drug has poor absorption in stomach
or small intestine, colonic drug delivery is an answer of choice.
The pH in this region varies from 6.4 - 7 and presence of microbial
flora plays as important role in drug release especially in this
region. Various mechanisms are adopted for drug release in this
area are coating with pH sensitive polymer e.g., EudragitS100,
Eudragit L100, biodegradable polymer like polymers which are
sensitive to colonic bacteria, bioadhesive polymers which
selectively sticks to colonic mucosa e.g., polycarbophils or
polyethans, redox sensitive polymers that respond to redox
potential in colon which expresses the total metabolic and
bacterial action.6- Chewable tabletThe patients who have difficulty
in swallowing tablets whole or for children who have not yet learnt
to swallow a tablet, chewable tablet serves as an attractive
alternative. The added advantage of this medication is that it can
be taken at any time or when water is not available. Mannitol is
normally used as a base due to low hygroscopy and more importantly,
it gives pleasant, cooling sensation. Antacid tablets are
invariably prepared as chewable to obtain quick ingestion relief as
well as the antacid dose is too large to swallow and the activity
is related to particle size. Another example is multivitamin tablet
which a patient can take as a daily dose.
7- Dispersible tabletThese tablets disintegrate either rapidly
in water, to disperse instantaneously in the mouth to be swallowed
without the aid of water. So, its preferred for pediatric patients
who cannot swallow a solid dosage form. Also helpful for patients
having prolonged illness who are prone to nauseatic sensations if
they have to swallow a tablet. The added advantage of this
formulation is faster onset of action as compared to standard
compressed tablet. The properties of the water dispersible tablet,
such as porosity, hardness, disintegration time and increase in
viscosity after dispersion are necessary to investigate during
manufacturing which decides the product performance. The common
examples of API formulated in this dosage form are analgesics e.g.,
aspirin, ibuprofen, etc.
Tablets used in the oral cavity:1- Lozenges and trochesThe
tablet is a flat faced at least about 18mm in diameter and meant to
suck and dissolves in the mouth. The compressed tablet is called
troches and the tablets produced by fusion or candy molding process
are called lozenges. Flavours and sweeteners are added to make
tablets palatable. The tablet generally contains sucrose or lactose
and gelatin solution to impart smooth taste. Lozenges for local
action in mouth/ throat are: antiseptics, antibiotics, demulcents,
antitussive agents or astringents. To produce systemic action:
multivitamin tablet.2- Sublingual tabletThey are to be placed under
the tongue and produce immediate systemic effect by enabling the
drug absorbed directly through mucosal lining of the mouth beneath
the tongue.
FIGURE.11. SUBLINGUAL TABLETSThe drug absorbed from stomach goes
to mesenteric circulation which connects to stomach via portal
vein. Thus, absorption through oral cavity avoids first-pass
metabolism. The tablets are usually small and flat, compressed
lightly to keep them soft. The tablet must dissolve quickly
allowing the API to be absorbed quickly. Its designed to dissolve
in small quantity of saliva. After the tablet is placed in the
mouth below the tongue, the patient should avoid eating, drinking,
smoking and possibly talking in order to keep the tablet in place.
Swallowing of saliva should also be avoided since the saliva may
contain dissolved drug. Bland excipients are used to avoid salivary
stimulation. Due to inconvenience in administration, this dosage
form is prepared only for those API(s) for which the only
satisfactory nonparenteral method is this route. For example,
Glyceryl trinitrate (vasodilator) and Isoprinosine sulphate
(bronchodilator).3- Buccal tabletCompleteness of drug absorption is
desired but fast drug absorption is not intended. The tablets are
designed not to disintegrate. They are flat elliptical or capsule
shaped tablets as it can be easily held between gum and cheek. Its
placed near the opening of parotid duct to provide the medium to
dissolve the tablet.
FIGURE.12. BUCCAL TABLETSSince this tablet is to be kept for
30-60 minutes in oral cavity, care should be taken to see that all
the ingredients are finely divided to avoid gritty or irritating
sensation. This tablet is most often used when replacement hormonal
therapy is to be administered. Antifungal drugs are preferred to be
administered by this route. e.g., Miconazole under preclinical
trial still not in market.4- Dental conesThese tablets are designed
to be loosely packed in the empty socket remaining following a
tooth extraction.
FIGURE.13. DENTAL CONES
Main purpose behind the use of this tablet is either to prevent
multiplication of bacteria in the socket by employing a slow
releasing antibacterial compound or to reduce bleeding by an
astringent or coagulant containing tablet. Its formulated to
dissolve or erode slowly in presence of a small volume of serum or
fluid over 20-40 minutes period.5- Mouth Dissolved tablets/ Rapidly
Dissolving tablets:Known as orally disintegrating tablets, they are
also called mouth-dissolving, fast-dissolving, rapid-melt, porous,
orodispersible, quick dissolving. These kinds of tablets are
preferred when fast action or relief is desired. Most commonly used
drugs under this formulation are the agents active against
migraine. The tablets are designed to disintegrate as well as
dissolve within one minute or some within 10 seconds of oral
administration in limited quantity of saliva. They liquefy on
tongue and patient swallows the liquid, without the need of water.
A number of techniques are used to prepare these tablets, including
lyophilization, soft direct compression. Tablets administered by
other routes:1- Vaginal tabletThis tablet undergoes slow
dissolution and drug release in vaginal cavity of women. The shape
is kept ovoid or pear shaped to facilitate retention in vagina. The
tablet should be made compatible with plastic tube inserters which
are designed to place the tablet in the upper region of vaginal
tract. These tablets generally release antibacterial, antiseptics
or astringents to treat vaginal infections or release steroids for
systemic absorption.2- ImplantsThese tablets are inserted into
subcutaneous tissue by surgical procedures where they are very
slowly absorbed over a period of a month or a year. A special
injector with a hollow needle and plunger is used to administer the
rod shaped tablet for other shapes, surgery is required. The
tablets may be pellet, cylindrical or rosette shaped with diameter
not more than 8mm. They are sterile formulation without excipients
and made hard with large particle size to achieve gradual drug
release. The tablets are produced by a sterile single punch hand
operated machine in which the die cavity is filled with hand since
the material does not normally flow well. Mainly, these tablets are
prepared to deliver growth hormones to food producing animals and
ear is the preferred site for administration of the drug.Tablets
used to prepare solution:Effervescent tabletThe oral dosage forms
are the most popular way of taking medication despite having some
disadvantages like slow absorption and thus onset of action is
prolong. This can be overcome by administrating the drug in liquid
from but, many drugs have limited level of stability in liquid
form. So, effervescent tablets acts as an alternative dosage form.
The tablet is added into a glass of water just before
administration and the drug solution or dispersion is to be drunk
immediately. The tablet is quickly broken apart by internal
liberation of CO2 in water due to interaction between tartaric acid
and citric acid with alkali metal carbonates or bicarbonates in
presence of water.
FIGURE.14. EFFERVESCENT TABLETSDue to liberation in CO2 gas, the
dissolution of API in water as well as taste masking effect is
enhanced. The advantages of effervescent tablets compared with
other oral dosage forms includes an opportunity for formulator to
improve taste, a more gentle action onSoluble tabletTablets are
solids of uniform shape and dimensions, usually circular, with
either flat or convex faces, the distance between faces being less
than the diameter. Water soluble tablets are intended for
application after dissolution in water and contain an active
ingredient should be totally soluble in water at used
concentrations. All the excipients used to formulate these tablets
are required to be completely soluble in water including the
glidants, binders, etc.
Advantages and disadvantages of tablet as a dosage form. The
advantages are listed below:I.Large scaleManufacturing is feasible
in comparison to other dosage forms. Therefore,economy can be
achieved.II.Accuracy of doseis maintained since tablet is a solid
unit dosage form.III. Tailor made release profile can be
achieved.IV. Longer expiry period and minimum microbial
spillageowing to lower moisture content.V. As tablet is not a
sterile dosage form, stringentenvironmental conditions are not
required in the tablet department.VI. Ease of packaging (blister or
strip) and easyhandling over liquid dosage form.VII. Easy
totransport in bulk. Emergency supply supplies can be carried by
patients.VIII.Organolepticproperties (taste, appearance and odour)
are best improved by coating oftablet.IX. Product identification is
easy and markings done withthe help of grooved punches and printing
with edible ink.X. Different types of tablets are available like
buccal,floating, colon targeting, effervescent, dispersible,
soluble, and chewable,etc.XI. In composition to parenterals dosage
form, a doctoror a nurse is not required for administration. I.e.
self administration ispossible.XII. In comparison tocapsules,
tablets are more tamperproof.The disadvantages are listed
below:I.It is difficultto convert a high dose poorly compressible
API into a tablet of suitable sizefor human use.II.Difficult
toformulate a drug with poor wettability, slow dissolution into a
tablet.III. Slow onset of action as compared to parenterals,liquid
orals and capsules.IV. The amount of liquid drug (e.g. Vitamin
E,Simethicone) that can be trapped into a tablet is very less.V.
Difficult to swallow for kids, terminally ill andgeriatric
patients.
Tablets Manufacturing IntroductionThe manufacture of oral solid
dosage forms such as tablets is a complex multi-stage process under
which the starting materials change their physical characteristics
a number of times before the final dosage form is produced.
Traditionally, tablets have been made by granulation, a process
that imparts two primary requisites to formulate: compactibility
and fluidity. Both wet granulation and dry granulation (slugging
and roll compaction) are used. Regardless of weather tablets are
made by direct compression or granulation, the first step, milling
and mixing, is the same; subsequent step differ. Numerous unit
processes are involved in making tablets, including particle size
reduction and sizing, blending, granulation, drying, compaction,
and (frequently) coating. Various factors associated with these
processes can seriously affect content uniformity, bioavailability,
or stability.
Manufacturing process
VARIOUS UNIT OPERATION SEQUENCES IN TABLET MANUFACTURING
Manufacturing process1- Dispensing(weighing and
measuring)Dispensing is the first step in any pharmaceutical
manufacturing process. Dispensing is one of the most critical steps
in pharmaceutical manufacturing; as during this step, the weight of
each ingredient in the mixture is determined according to dose.2-
SizingThe sizing (size reduction, milling, crushing, grinding,
pulverization) is an impotent step (unit operation) involved in the
tablet manufacturing.In manufacturing of compressed tablet, the
mixing or blending of several solid ingredients of pharmaceuticals
is easier and more uniform if the ingredients are approximately of
same size. This provides a greater uniformity of dose. A fine
particle size is essential in case of lubricant mixing with
granules for its proper function.3- Powder blendingIn the tablet
pressing process, the main guideline is to ensure that the
appropriate amount of active ingredient is in each tablet. Hence,
all the ingredients should be well-mixed. If a sufficiently
homogenous mix of the components cannot be obtained with simple
blending processes. The successful mixing of powder is acknowledged
to be more difficult unit operation because, unlike the situation
with liquid, perfect homogeneity is practically unattainable.
BLENDIND MACHINEMethod:Doubling method: If we have to mix
different type of material (active ingradient or excipients ) with
different quantity. Firstly mix smallest quantity material with
same quantity of other material then mix that quantity which we
gained after mixing material with other same quantity of powder and
so on. By adopting this method uniform mixing can be achieved.
4- GranulationIn the pharmaceutical industry, granulation refers
to the act or process in which primary powder particles are made to
adhere to form larger, multiparticle entities called granules. It
is the process of collecting particles together by creating bonds
between them. Bonds are formed by compression or by using a binding
agent. Granulation is extensively used in the manufacturing of
tablets.The granulation process combines one or more powders and
forms a granule that will allow tableting process to be within
required limits. This way predictable and repeatable process is
possible and quality tablets.Why Granulation?Granulation is carried
out for various reasonsTO make the material flowable: Many powders,
because of their small size, irregular shape or surface
characteristics, are cohesive and do not flow well. Granules
produced from such a cohesive system will be larger and more
isodiametric, both factors contributing to improved flow
properties.To prevent segregation: Segregation is due to
differences in the size or density of the component of the mix.
Normally, the smaller and/or denser particles tend to concentrate
at the base of the container with the larger and/or less dense ones
on the top. An ideal granulation will contain all the constituents
of the mix in the correct proportion in each granule and
segregation of granules will not occur.To make it compressible:
Some powders are difficult to compact even if a readily compactable
adhesive is included in the mix, but granules of the same powders
are often more easily compacted. This is associated with the
distribution of the adhesive within the granule and is a function
of the method employed to produce the granule.For example, if one
were to make tablets from granulated sugar versus powdered sugar,
powdered sugar would be difficult to compress into a tablet and
granulated sugar would be easy to compress. Powdered sugars small
particles have poor flow and compression characteristics. These
small particles would have to be compressed very slowly for a long
period of time to make a worthwhile tablet. Unless the powdered
sugar is granulated, it could not efficiently be made into a tablet
that has good tablet characteristics such as uniform content or
consistent hardness.Ideal characteristics of granulesThe ideal
characteristics of granules include uniformity, good flow, and
compactibility. These are usually accomplished through creation of
increased density, spherical shape, narrow particle size
distribution with sufficient fines to fill void spaces between
granules, adequate moisture (between 1-2%), and incorporation of
binder, if necessary.The effectiveness of granulation depends on
the following propertiesi) Particle size of the drug and
excipientsiii) Volume of binder (less or more)iv) Wet massing time
( less or more)v) Amount of shear applied to distribute drug,
binder and moisture.vi) Drying rate ( Hydrate formation and
polymorphism)Wet granulation1 The most widely used process of
agglomerationing pharmaceutical industry is wet granulation. Wet
granulation process simplyinvolves wet massing of the powder blend
with a granulating liquid, wet sizingand drying Imortant steps
involved in the wet granulation. i) Mixing of the drug and
excipient.ii) Preparation of binder solutioniii) Mixing of binder
solution with powder mixture to formwet mass.iv) Coarse screening
of wet mass using a suitable sieve.v) Drying of moist granules.vi)
Screening of dry granules through a suitable sieve.vii) Mixing of
screened granules with disintegrant, glidant, and lubricant.
Limitation of wet granulationi) The greatest disadvantage of wet
granulation is its cost. It is an expensive process becauseof
labor, time, equipment, energy and space requirements.ii) Loss of
material during various stages of processing iii) Stability may be
major concern for moisture sensitive or thermo labile drugs iv)
Multiple processing steps add complexity and make validation and
control difficult.v) An inherent limitation of wet granulation is
that any incompatibility between formulation components is
aggravated.Dry granulationIn dry granulation process the powder
mixture is compressed without the use of heat and solvent. It is
the least desirable of all methods of granulation. The two basic
procedures are to form a compact of material by compression and
then to mill the compact to obtain a granules. Two methods are used
for dry granulation. The more widely used method is slugging, where
the powder is precompressed and the resulting tablet or slug are
milled to yield the granules. The other method is to precompress
the powder with pressure rolls using a machine such as
Chilosonator.AdvantagesThe main advantages of dry granulation or
slugging are that it uses less equipments and space. It eliminates
the need for binder solution, heavy mixing equipment and the costly
and time consuming drying step required for wet granulation.
Slugging can be used for advantages in the following situations:i)
For moisture sensitive material( Active ingradient) .ii) For heat
sensitive material.iii)For improved disintegration since powder
particles are not bonded together by a binder. Disadvantagesi) It
requires aspecialized heavy duty tablet press to form slugii) It
does not permit uniform colour distribution as can be. iii)
Achieved with wet granulation where the dye can be incorporated
into binder liquid.iv) The process tends to create more dust than
wet granulation, increasing the potential contamination. steps in
dry granulationi) Milling of drugs and excipientsii) Mixing of
milled powdersiii) Compression into large, hard tablets to make
slugiv) Screening of slugsv) Mixing with lubricant and
disintegrating agentvi) Tablet compressionTwo main dry granulation
processesSlugging processGranulation by slugging is the process of
compressing dry powder of tablet formulation with tablet press
having die cavity large enough in diameter to fill quickly. The
accuracy or condition of slug is not too important. Only sufficient
pressure to compact the powder into uniform slugs should be used.
Once slugs are produced they are reduced to appropriate granule
size for final compression by screening and milling.Factors which
determine how well a material may slugi)Compressibilityor
cohesiveness of the materii) Compression ratio of powderiii)
Density of the powderiv) Machine typev) Punch and die sizevi) Slug
thicknessvii) Speed of compressionviii) Pressure used to produce
slug
Roller compactionThe compaction of powder by means of pressure
roll can also be accomplished by a machine called chilsonator.
Unlike tablet machine, the chilsonator turns out a compacted mass
in a steady continuous flow. The powder is fed down between the
rollers from the hopper which contains a spiral auger to feed the
powder into the compaction zone. Like slugs, the aggregates are
screened or milled for production into granules.Formulation for dry
granulationThe excipients used for dry granulation are basically
same as that of wet granulation or that of direct compression. With
dry granulation it is often possible to compact the active
ingredient with a minor addition of lubricant and disintegrating
agent. Fillers that are used in dry granulation include the
following examples: Lactose, dextrose, sucrose, MCC, calcium
sulphate, Sta-Rx etc.Granule lubricationAfter granulation, a final
lubrication step is used to ensure that the tableting blend does
not stick to the equipment during the tableting process. This
usually involves low shear blending of the granules with a powdered
lubricant, such as magnesium stearate or stearic acid.
5- DryingDrying is a most important step in the formulation and
development of pharmaceutical product. It is important to keep the
residual moisture low enough to prevent product deterioration and
ensure free flowing properties. The commonly used dryer includes
Fluidized bed dryer, Vacuum tray dryer, Microwave dryer, Spray
dryer, Freeze dryer, Turbo - tray dryer, Pan dryer, etc.6- Tablet
compressionAfter the preparation of granules (in case of wet
granulation) or sized slugs (in case of dry granulation) or mixing
of ingredients (in case of direct compression), they are compressed
to get final product. The compression is done either by single
punch machine (stamping press) or by multi station machine (rotary
press).
Single punch compression machine
Rotary Press
7- Tablet Coating Coated tablets are defined as tablets covered
with one or more layers of mixture of various substances such as
natural or synthetic resins ,gums ,inactive and insoluble filler,
sugar, plasticizer, polyhydric alcohol ,waxes ,authorized colouring
material and some times flavoring material .Coating may also
contain active ingredient. Substances used for coating are usually
applied as solution or suspension under conditions where vehicle
evaporates.Type of tablet coating process1- Sugar coating :
Compressed tablets may be coated with coloured or uncoloured sugar
layer 30-50% size also multi stage process2- Film coating is
deposition of a thin film of polymer surrounding the tablet core
(spry). Film coating is more favored over sugar coating 2-3 % over
all size, sigle stage process.
TABLE.26. COMPARISON BETWEEN FILM COATING AND SUGAR COATING
FEATURESFILM COATINGSUGAR COATING
Tablet:AppearanceWeight increase because of coating materialLogo
or break linesRetain contour of original core. Usually not as shiny
as sugar coat type2-3%PossibleRounded with high degree of
polish30-50%Not possible
ProcessOperator training requiredAdaptability to GMPProcess
stagesFunctional coatingsProcess tends itself to automation and
easy training of operatorHighUsually single stageEasily adaptable
for controlled releaseConsiderableDifficulty may ariseMultistage
processNot usually possible apart from enteric coating
Aspects of tablet coatingi) Avoid irritation of oesophagus and
stomachii) Avoid bad tasteiii) Avoid inactivation of drug in the
stomachiv) Improve drug effectivenessv) Prolong dosing intervalvi)
Improve dosing intervalvii) Improve patient complianceII.
Technologyi) Reduce influence of moistureii) Avoid dust
formationiii) Reduce influence of atmosphereiv) Improve drug
stability
8- PackagingPharmaceutical manufacturers have to pack their
medicines before they can be sent out for distribution. The type of
packaging will depend on the formulation of the medicine.What is
packaging ?Packaging is defined as the collection different
components which surround the pharmaceutical product from the time
of production until its use.Importance of packaging Protect against
all adverse external influencesthat can alter the properties of the
product. Protect against biological contamination. Protect against
physical damage. Carry the correct information and identificationof
the product.
Functions of packagingContainmentNot to leak, nor allow
diffusion and permeationStrong enough to hold the contents during
handlingProtectionLightMoistureOxygenBiological
contaminationMechanical damageCounterfeiting
Material characteristicsAdditional qualities required It must
preserve the physical properties of all dosage forms and protect
them against damage or breakage. It must not alter the identity of
the product. It must preserve the characteristics properties of the
product to comply specifications. It must protect product against
undesirable chemical, biological or physical entities.
Choosing appropriate primary packProduct
characteristics/sensitivity Hygroscopicity Physical degradation
Chemical degradation Drug release properties Mechanical properties
Photosensitivity Gas liberation tendency Dimensional aspects
Selection of packaging material Moisture barrier requirements
Light barrier requirements Gas barrier requirements Chemical
properties Type of packingStrip packing of colored sugar coated
tabletsSelection of special vials for special productsProper design
of blisters
Evaluation of tablets Introduction The quantitative evaluation
and assessment of a tablets chemical, physical and bioavailability
properties are important in the design of tablets and to monitor
product quality. These properties are important since chemical
breakdown or interactions between tablet components may alter the
physical tablet properties, and greatly affect the bioavailability
of the tablet system.There are various standards that have been set
in the various pharmacopoeias regarding the quality of
pharmaceutical tablets. These include the diameter, size, shape,
thickness, content, content uniformity, weight, hardness,
disintegration and dissolution characters. The diameters and shape
depends on the die and punches selected for the compression of
tablets. The remaining specifications assure that tablets do not
vary from one production lot to another. The following standards or
quality control tests should be carried out on compressed tablets .
1. General appearance 2. Size and shape 3. Organoleptic properties
4. Content5. Content uniformity 6. Weight variation 7. Mechanical
strength(hardness, friability) 8. Disintegration 9. Dissolution
General Appearance The general appearance of tablets, its visual
identity and overall elegance is essential for consumer acceptance,
control of lot-to-lot uniformity and general tablet-to-tablet
uniformity and for monitoring the production process. The control
of general appearance involves measurement of attributes such as a
tablets size, shape, color, presence or absence of odour, taste,
surface texture and consistency . According to Drugs And Cosmetics
Act, 1940 : General requirements shall includes compliance with
colour, consistency, clarity, stability, freedom from foreign
matter or fungal growth defects like chipping and capping of
tablets, motteled apperance tablets, cracking of coating and other
characterstics defects that can be precieved by visual
inspection.
Tablets Size and shape The shape and dimensions of compressed
tablets are determined by the type of tooling during the
compression process. At a constant compressive load, tablets
thickness varies with particle size distribution and packing of the
powder mix being compressed and with tablet weight. Tablet
thickness is consistent from batch to batch or within a batch only
if the tablet granulation or powder blend is adequately consistent
in particle size and particle size distribution, if the punch
tooling is of consistent length, and if the tablet press is clean
and in good working condition. The thickness of individual tablets
may be measured with a micrometer, which permits accurate
measurements and provides information of the variation between
tablets. Tablet thickness should be controlled within a 5%
variation of a standard value. Any variation in thickness within a
particular lot of tablets or between manufacturers lots should not
be apparent to the unaided eye for consumer acceptance of the
product. In addition, thickness must be controlled to facilitate
packaging. The physical dimensions of the tablet along with the
density of the material in the tablet formulation and their
proportions, determine the weight of the tablet. The size and shape
of the tablet can also influence the choice of tablet machine to
use, the best particle size for granulation, production lot size
that can be made, the best type of tableting processing that can be
used, packaging operations, and the cost of production.
Organoleptic properties Color is a vital means of identification
for many pharmaceutical tablets and is also usually important for
consumer acceptance. The color of the product must be uniform
within a single tablet, from tablet to tablet and from lot to lot.
Non uniformity of coloring not only lack esthetic appeal but could
be associated by the consumer with non uniformity of content and
general poor product quality. Non uniformity of coloring is usually
referred to as mottling. The eye cannot differentiate small
differences in color nor can it precisely define color and efforts
have been made to quantitate color evaluations. Reflectance
spectrophotometry, colorimetric measurements and micro reflectance
photometer have been used to measure color uniformity and gloss on
a tablet surface . Odor may also be important for consumer
acceptance of tablets and can provide an indication of the quality
of tablets.As the presence of an odor in a batch of tablets could
indicate a stability problem, such as the characteristic odor of
acetic acid in degrading aspirin tablets. However, the presence of
an odor may be characteristic of the drug (e.g. vitamins), added
ingredients (e.g. flavoring agent) or the dosage form (e.g.
film-coated lets). Taste is also important for consumer acceptance
of certain tablets (e.g. chewable tablets) and many companies
utilize taste panels to judge the preference of different flavors
and flavor levels in the development of a product. Taste preference
is however subjective and the control of taste in the production of
chewable tablets is usually based on the presence or absence of a
specified taste.
Content of active ingradientsAs mentioned earlier a physical
sound tablet may not produce the desired effect. To evaluate
tablets potential for efficacy, the amount of drug per tablet need
to be moniterd from tablet to tablet and batch to batch. Determine
the amount of active ingradient by the method like %age purity and
calculate the amount of active ingradient per tablet. According to
Drugs And Cosmetics Act, 1940 : The content of active ingradients,
other than vitamins, enzymes and antibiotics, in patent or
proprietary medicines shall not less than 90 per cent and not more
than 110 per cent of labeled content ; however, for enzymes and
vitamins, only for lower limit of 90 per cent shall apply. In all
dry formulations containing antibiotics, the limit shall be 90 to
130 percent of labeled contents and in case of liquid antibiotics
formulations, the limit shall be 90 to 140 per cent of labellad
contents.Method: According to I.P :Determine the amount of active
ingradient by the method described in assay and calculate the
amount of active ingradient per tablet. The result lies within the
range of active ingradient stated in the monograph of specific
tablet. This range is based on the requirement that 20 tablets, are
used in assay. Where 20 tablets can not be obtained, a smaller
number, which must not be less than 5, may be used, but to allow
for sampling errors the tolerances are windened in accordance with
Table 1 apply when the stated limits are between 90 and 110
percent. For limits other than 90 to 110 per cent, proportionately
smaller or larger allowances should be made. Table 1Weigth of
active ingradient in each tablet Subtract from lower limit for
samples of Add to the upper limit for samples of
0.12 g or less
More than o.12 g But less than 0.3g
0.3 g or more
1510 5 15 10 5
0.2
0.2
0.1 0.7
0.5
0.2 1.6
1.2
0.8 0.3 0.3
0.2
0.8 0.6
0.4
1.8
1.5
1.0
Content Uniformity Test For TabletsThe content uniformity test
is used to ensure that every tablet contains the amount of drug
substance intended with little variation among tablets within a
batch. Due to increased awareness of physiological availability,
the content uniformity test has been included in the monographs of
all coated and uncoated tablets and all capsules intended for oral
administration.Factor effecting uniformity of contens:
(1)Nonuniform distribution of drug substance throuhtout the powder
mixture or granulation.(2)segregation of the powder mixture or
granulation during the various manufacturing processes.(3)Tablet
weight variation.
It is official test which is prescribed by I.P. Test is
applicable on what type of tablets??This test is applicable to
tablets that contain very potent drugs like digoxin, digitoxin etc.
Mostly this test is applied when these type of tablets cantain 10
mg or less than 10 mg or less than 10 per cent w/w of active
ingradient. For tablets containing more than one active ingradient
carry out the test for each active ingardient.The test for
uniformity of content is not applicable to tablets containing
multivitamins and trace elements.Method:Determine the content of
active ingradient in each of 10 dosage units taken at random using
the method given in the monograph or by any suitable analytical
method.Aceptance limits:The preparation complies with the test if
each individual content is 85 to 115 per cent of average content.
The preparation fails to comply with the test if more than one
individual content is outside these limits or if one individual
content is outside the limits of 75 to 125 per cent of the average
content. If one individual content is outside the limits of 85 to
115 per cent of average content but within the limits of 75 to 125
percent, Repeat the determination using another 20 dosage units.The
preparation complies with the test if not more than one of the
individual content of the total sample of 30 dosage units is
outside 85 to 115 per cent of the average content and none is
outside the limits of 75 to 125 per cent of the average
content.
Weight variation Tablet degisned to contain a specific amount of
drug in a specific tablet formula, the weight of tablet being made
is routinely measured to help ensure that tablet contain proper
drug. It is method to check the uniformity of weight of
tablet.Importance:Weight variation is quality control method to
check the uniformity of tablets in lot to lot and batch to batch.
It is official method prescribed by I.P. Method:For weight
variation weigh individually 20 tablets. average weight is
calculated by adding weight of all individual twenty tablets
divided by no. of tablets here no. of tablets are twenty so divided
by 20. The individual tablet weights are then compared to the
average weight.The IP has provided limits for the average weight of
uncoated compressed tablets. Twenty tablets are weighed
individually and the average weight is calculated. The individual
tablet weights are then compared to the average weight. Not more
than two of the tablets must differ from the average weight by not
more than the percentages stated in Table 1. No tablet must differ
by more than double the percentage. Average weight (Uncoated and
film coated tablets) Percent difference
80mg or less than 80mg10
More than 80 mg but less than 250 mg7.5
More than 250 mg 5
Table 1: Weight variation requirements Example:Weight variation
of Tablet Arthgesic.Weigh individually 20 tablets. Calculate its
average weight and then apply the limits according to I.P.
According to I.P. more than 250 mg tablet average weight percentage
difference can be 5%. So apply 5% percentage
difference.Sr.No.Weight of tablet
123 780mg 820mg
4 780mg
5 830mg
6 810mg
7891011121314 15 800mg 780mg 770mg 810mg 830mg 830mg 830mg 770mg
790mg
16 790mg
17 780mg
18 790mg
19 780mg
20 830mg
Average weight = total weight/no.of tablets =16110/20 = 805.5 so
average weight of tablet is 805.5 apply I.P. standard. According to
I.P. more than 250 mg tablet average weight percentage difference
can be 5%. So apply 5% percentage difference. 805.5-40.275=765.2
805.5=+40.275=845.77 Range is b/w 765.2 845.77 Now compare each
tablet within this range. During comparing no tablet is out of
computing range. So sample is passed.
Mechanical strength of tablets The mechanical strength of a
tablet provides a measure of the bonding potential of the material
concerned and this information is useful in the selection of
excipients. An excessively strong bond may prevent rapid
disintegration and subsequent dissolution of a drug. If bonding is
very weak it may break very easily during handling packaging,
transporatation.The mechanical properties of pharmaceutical tablets
are quantifiable by the friability, hardness or crushing strength ,
crushing strength-friability values, tensile strength and brittle
fracture Index. Hardness or Crushing strength Friability
The strength of a tablet plays a very important role in its
marketing and dissolution. The mechanical strength of tablet or
granules can be determined by its hardness and through friability
test.
Hardness of TabletsHardness may be defined as the resistance of
tablets to capping, abrasion or breakage under conditions of
storage, transportation and handling.Tablet hardness have been
associated with other tablet properties such as density and
porosity. Hardness generally increase with normal storage of
tablets and depends on the shape, chemical properties, binding
agent and pressure applied during compression . It is non-official
quality control method. It is not prescribed by I.P.Hardness
test:The small and portable hardness tester was manufactured and
introduced by Monsanto in the Mid 1930s. It is now designated as
either the Monsanto or Stokes hardness tester. The instrument
measures the force required to break the tablet when the force
generated by a coil spring is applied diametrally to the
tablet.
MONSANTO HARDNESS TESTERThe Strong-Cobb Pfizer and Schleuniger
apparatus which were later introduced measures the diametrically
applied force required to break the tablet.
PFIZER TESTERHardness which is now more appropriately called
crushing strength determinations are made during tablet production
and are used to determine the need for pressure adjustment on
tablet machine.The strength of a tablet can be determined by
breaking it between second and third fingers with the thumb acting
as a fulcrum. If there is a click sound, the tablet is deemed to
have acceptance strength.If the tablet is too hard, it may not
disintegrate in the required period of time to meet the dissolution
specifications; if it is too soft, it may not be able to withstand
the handling during subsequent processing such as coating or
packaging and shipping operations. The force required to break the
tablet is measured in kilograms and a crushing strength of 4Kg is
usually considered to be the minimum for satisfactory tablets. Oral
tablets normally have a hardness of 4 to 10kg ; however, hypodermic
and chewable tablets are usually much softer ( 3 kg ) and some
sustained release tablets are much harder (10 -20 kg ).
Friability Test for TabletsFriabiltyIt is may be defined as the
excessive breakness of tablets during mechanical shocks of handling
in manufacture, packaging, and shipping.Friction and shock are the
forces that most often cause tablets to chip, break.Why we test
friability? Tablet hardness is not absolute indicator of strength
since some formulations, when compressed into very hard tablets,
tend to cap on attrition, losing there crown portions. Therefore
another measure of tablets strength, its friability is often
measured.They not only lack elegance and consumer acceptance but
also spoil the areas of manufacturing such as coating and
packaging.In friability test the tablets are prone to abrasion
hence enabling us to check for the tablet strength under
application of force in different manner.Friability is affected by
various external and internal factors like:1) Punches that are in
poor condition or worn at their surface edges, resulting in
whiskering at the tablet edge and show higher than normal
friability values.2) Friability test is influenced by internal
factors like the moisture content of tablet granules and finished
tablets. Moisture at low and acceptable level acts as a
binderFriability is official method because it is prescribed by
I.P.
Tablet Friability Tester
Method :The friability test is closely related to tablet
hardness and is designed to evaluate the ability of the tablet to
withstand abrasion in packaging, handling and shipping. It is
usually measured by the use of the Roche friabilator. Ten tablets
are weighed and placed in the apparatus where they are exposed to
rolling and repeated shocks as they fall 6 inches in each turn
within the apparatus. After four minutes of this treatment or 100
revolutions, the tablets are weighed and the weight compared with
the initial weight. The loss due to abrasion is a measure of the
tablet friability. The value is expressed as a percentage. A
maximum weight loss of not more than 1% of the weight of the
tablets being tested during the friability test is considered
generally acceptable and any broken or smashed tablets are not
picked up . Normally, when capping occurs, friability values are
not calculated. A thick tablet may have less tendency to cap
whereas thin tablets of large diameter often show extensive
capping, thus indicating that tablets with greater thickness have
reduced internal stress . FRIABILITY TEST FOR TABLETS:The
friability test is carried out in an instrument called a
friabilator. A friability testing apparatus should stimulate the
conditions that the product will be exposed to during the process
of production. This test is a method to determine physical strength
of uncoated tablets upon exposure to mechanical shock.The commonly
used friabilator in laboratories is the Roche friabilator .
ROCHE FRIABILATORThis instrument consists of a plastic chamber
for placing the tablets which is attached to a horizontal axis. The
drum has an inside diameter of 287mm and is about 38mm in depth,
made of a transparent synthetic polymer with polished internal
surface. A set of pre weighed tablets [if one tablet weigh 650mg or
less then approx 6.5g of total weight should be taken and for more
than 650mg/tablet weight, 10 tablets should be taken] (3) are
placed in the plastic chamber revolving at 24-25rpm for 4 min. The
tablets are subjected to combined effects of abrasion and shock.
The tablets are dropped at a distance of six inches on each
revolution. If the tablet size or shape becomes irregular adjust
the drum so that base forms an angle of about 10 degrees with bench
top and the tablets fall freely when drum is rotated.The instrument
is operated for 100 revolutions after which the tablets are dusted
and reweighed.Conventional compressed tablets that lose less than
0.5% to 1% of weight are considered acceptable.Most effervescent
tablets and some chewable tablets undergo high friability weight
loss which is an indication for the special stack packing that is
required for these types of tablets.In case of hygroscopic tablets
a humidity-controlled environment (relative humidity less than 40%)
is required for testing.Tablets prone to capping during the test
are considered unfit for commercial use.
Tablet Disintegration For a drug to be absorbed from a solid
dosage form after oral administration, it must first be in
solution, and the first important step toward this condition is
usually the break-up of the tablet; a process known as
disintegration. In the past, the only release index required for a
tablet was its disintegration time which does not necessarily
measure the physiological availability of the drug in a patient.
Studies have shown that the agitation of the gastric contents
during normal contractions is quite mild in contrast to the
turbulent agitation produced in the disintegration test apparaus .
The low order magnitude of agitation in the stomach produces
substantially higher disintegration in vivo than those obtained
using the USP apparatus. Furthermore, the particles of the
disintegrated tablets are not dispersed throughout the stomach but
remains as an aggregate. Thus, the tablet disintegration test is
limited to manufacturing control of lot-to-lot variations in
individual products and is not a measure of bioavailability.
Nevertheless, it is used to provide a simple and useful means for
monitoring and controlling the quality of tablets. Theories of
disintegration Disintegration Test Disintegration Tests of Dosage
forms Disintegration Tests : Pharmacopoeial comparison
Disintegration ApparatusTheories of disintegration of Tablets
:Several mechanisms of tablet disintegration have been proposed.
Some of these are given below. Even though these concepts are
listed separately, inter-relationships probably occur in almost all
tablet formulations. (i) Evolution of gas If a gas is evolved by a
chemical reaction when the tablet comes into contact with water,
then the tablet will disintegrate. This is the basis for the
manufacture of effervescent tablets. An example of such a reaction
is of sodium bicarbonate with citric and tartaric acids, which
yields carbon dioxide. Peroxides incorporated in certain
formulations decompose in the presence of oxygen and this also
causes disintegration. (ii) Heat of wetting The heat produced when
a tablet is immersed in water causes the entrapped air in the
tablet to expand and exert sufficient pressure to disintegrate the
tablet . (iii) Effect of water absorption The water absorbed by the
tablet initiate disintegration, but this depends on the solubility
of the drug and other ingredients present . If tablet have high
solubility its disintegration increase due to initiation of
breakage of binding between the granules or molecules of
tablet.(iv) Swelling The grains of the disintegrant, particularly
of starches, swell in the presence of water and exert pressure on
the granules to force them apart. Shangraw et al reported that
tablets of water insoluble drugs disintegrated faster with starches
than those of water soluble drugs due to the diminished water
absorption capacity of the starches in the latter case. (v)
Porosity of tablets It has been shown that penetration of water
into a tablet is proportional to its mean pore diameter or
porosity. The porosity and permeability of tablets decrease as the
tabletting pressure is increased , and as the porosity decreases,
the disintegration time increases. Though no quantitative
relationships have been reported between disintegration and
penetration times, generally short disintegration times are
associated with rapid fluid penetration. Disintegration
Test:Disintegration test is widely used in the pharmaceutical
industry for evalution of disintigration capability of formulations
(ex:tablets) and quality control of different dosage forms.
Applications of Disintegration test : 1.Disintegration test is a
simple test which helps in the preformulation stage to the
formulator.2. It helps in the optimisation of manufacturing
variables, such as compressional force time3.This test is also a
simple in-process control tool to ensure uniformity from batch to
batch and among different tablets.4.It is also an important test in
the quality control of tablets.Advantages of Disintegration
tests:This test is simple in concept and in practice.It is very
useful in preformulation, optimisation and in quality
control.Disadvantages:Disintegration test cannot be relied upon for
the assurance of bioavailability.
Disintegration Tests : Pharmacopoeial comparison Disintegration
tests are performed as per the pharmacopoeial standards.
Disintegration is a measure of the quality of the oral dosage form
like tablets and capsules. Each of the pharmacopoeia like the USP,
BP, IP etc each have their own set of standards and specify
disintegration tests of their own. USP, European pharmacopoeia and
Japanese pharmacopoeia have been harmonised by the International
conference on Harmonisation and are interchangeable. The
disintegration test is performed to find out the time it takes for
a solid oral dosage form like a tablet or capsule to completely
disintegrate. The time of disintegration is a measure of the
quality. This is because, for example, if the disintegration time
is too high; it means that the tablet is too highly compressed or
the capsule shell gelatin is not of pharmacopoeial quality or it
may imply several other reasons. And also if the disintegration
time is not uniform in a set of samples being analysed, it
indicates batch inconsistency and lack of batch uniformity.
DISINTEGRATION METHOD:The disintegration test is a measure of the
time required under a given set of conditions for a group of
tablets to disintegrate into particles which will pass through a 10
mesh screen. Generally, the test is useful as a quality assurance
tool for conventional dosage forms. The disintegration test is
carried out using the disintegration tester which consists of a
basket rack holding 6 plastic tubes, open at the top and bottom,
the bottom of the tube is covered by a 10-mesh screen. The basket
is immersed in a bath of suitable liquid held at 37oC, preferably
in a 1L beaker. For compressed uncoated tablets, the testing fluid
is usually water at 37o C but some monographs direct that simulated
gastric fluid be used. If one or two tablets fail to disintegrate,
the test is repeated using 12 tablets. For most uncoated tablets,
the BP requires that the tablets disintegrate in 15minutes
(although it varies for some uncoated tablets) while for coated
tablets, up to 2hours may be required. The individual drug
monographs specify the time disintegration must occur to meet the
Pharmacopoeial standards. The disintegration test for each dosage
form is given in the pharmacopoeia. There are some general tests
for typical types of dosage forms. However, the disintegration test
prescribed in the individual monograph of a product is to be
followed. If the monograph does not specify any specific test, the
general test for the specific dosage form may be employed. Some of
the types of dosage forms and their disintegration tests
are:1.Uncoated tablets-Tested using distilled water as medium at
37+/-2 C at 29-32 cycles per minute; test is completed after 15
minutes. It is acceptable when there is no palpable core at the end
of the cycle (for at least 5 tablets or capsules) and if the mass
does not stick to the immersion disc.2.Coated tablets-The same test
procedure is adapted but the time of operation is 30
minutes.3.Enteric coated/ Gastric resistant tablets-The test is
carried out first in distilled water (at room temperature for 5
min.; USP and no distilled water per BP and IP), then it is tested
in 0.1 M HCL(upto 2 hours; BP) or Stimulated gastric fluid (1 hour;
USP) followed by Phosphate buffer, pH 6.8 (1 hour; BP) or
Stimulated intestinal fluid without enzymes (1 hour; USP).
Disintegration Test Apparatus(as per I.P.):The apparatus
consists of a basket rack assembly, a 1-litre beaker, a
thermostatic arrangement for heating the fluid and a mechanical
device for raising and lowering the basket in the immersion fluid
at a constant frequency rate.Basket-rack assembly: The basket-rack
assembly is rigid and supports six cylindrical glass tubes, 77.52.5
mm long, 21.5 mm in internal diameter and with a wall thickness of
about 2mm. The tubes are held vertically by two superimposed
transparent plastic plates, 902mm in diameter and 6.751.75 mm thick
perforated by six holes having the same diameter as the tubes. The
holes are equidistant from the centre of the plate and equally
spaced from one another. Attached to the under side of lower plate
is woven stainless steel wire cloth with a plain square weave with
2.00.2 mm mesh apertures and with a wire diameter of 0.650.045mm.
The upper plate is covered with a stainless steel disc perforated
by six holes, each about 24+or-2 mm in diameter, which fits over
the tubes and holds them between the plastic plate and the upper
open ends of the glass tubes. A suitable means is provided to
suspend the basket rack the entire basket-rack assembly is movable
by reciprocating motor which is fixed to the apex of the
basket-rack assembly from the raising and lowering device using a
point on its axis. This motor device for raising or lowering it
smoothly at a constant frequency of between 28 and 32 cycles per
minute through a distance of 50 to 60 mm. The time required by
upward stroke is equal to time required for downward stroke
direction should be smooth not abrupt. Discs: a cylindrical disc
for each tube, each 20.70.15mm thick in diameter and 9.5 0.15mm
thick, made of transparent plastic with a relative density of 1.18
to 1.20, and pirced with five holes, each 2 mm in diameter. One in
the center and the other four spaced equally on a circle of radius
6mm from the center of the disc. Four equally-spaced grooves are
cut in the lateral surface of the disc in such a way that at the
upper surface of the disc they aye 9.5 mm wide and 2.55 mm deep and
the lower surface 1.6 mm square.
DISINTEGRTION APPARATUSMedium: The assembly in suspended in the
liquid medium in a suitable vessel, preferably a 1 litre beaker.
The volume of the liquid is such that the wire mesh at its highest
point is at least 25mm below the surface of the liquid, and at its
lower point is at least 25 mm above the bottom of the beaker. At no
time should the top of the basket-rack assembly become submerged.
There is a thermostatic arrangement for heatingthe liquid and
maintaining the temperature at 37 oC 2 degree
celsius.Disintegration Test Method as per I.P. Introduce one tablet
or capsule into each tube and, if directed in the appropriate
general monograph, add a disc to each tube. Suspend the assembly in
the beaker containing the tube specified liquid and operate the
apparatus for the specified liquid and operate the apparatus for
the specified time given in individual monograph. Remove the
assembly from the liquid. The tablet or capsules pass the test if
all of them have disintegrated.If the 1 or 2 tablet fail to
disintegrate, repeat the test on 12 additional tablets; Not less
than 16 of the total of 18 tablets tested disintegrate.If the
tablet adhere to the disc and the preparation under examination is
fail to comply, repeat the test omitting the disc. The preparation
complies with the test if all the tablets in the repeat test is
disintegrate. For uncoated tablets :Use water as the liquid. Add a
disc to each tube. Operate the apparatus for 15 minutes. Unless
otherwise stated in individual monograph. Examine the state of the
tablets fail to comply because of adherence to discs, repeat the
test on a further 6 tablets omitting the discs. The tablets comply
with the test if all 6 tablets have disintegrated.For coated
tablets :Use water as the liquid. Add a disc to each tube. Operate
the apparatus for minutes. Unless otherwise stated in individual
monograph. Examine the state of the tablets if any of tablets has
not disintegrated, repeat the test with0.1 M hydrochloric acid. The
tablets comply the with the test if all 6 tablets have
disintegrated in the acidic medium.For coated tablets :Carry out
the test described above but operate the apparatus for 30 minutes.
Unless otherwise stated in individual monograph.If coated tablets
fail to comply because of adherence to discs, repeat the test on a
further 6 tablets omitting the discs. The tablets comply with the
test if all 6 tablets have disintegrated.For enteric coated tablets
:Apparatus: Use the apparatus for the tablets described
above.Method: Put one tablet into each tube, suspend the assembly
in the beaker containing 0.1M hydrochloric acid and operate without
the discs for 2 hours, unless otherwise stated in the individual
monograph. Remove the assembly from the liquid. No tablet shows
signs of cracks that would allow the escape of the content of
disintegration, apart from fragments of coating.Replace the liquid
in the beaker with mixed phosphate buffer pH 6.8, add a disc to
each tube and operate the apparatus for a further 60 minutes.
Remove the assembly from the liquid. If the tablet fails to comply
because of adherence to discs, repeat the test on a further 6
tablets without the discs. The tablets pass the test if all six
have disintegrated.
Dissolution testDissolution: Dissolution is the process by which
a solid solute enters a solution. Dissolution is pharmaceutically
defined as the rate of mass transfer from a drug substance into the
dissolution medium or solvent under standardized conditions of
liquid/solid interface, temperature and solvent composition. It is
a dynamic property that changes with time and explains the process
by which a homogenous mixture of a solid or a liquid can be
obtained in a solvent. It happens to chemically occur by the
crystal break down into individual ions, atoms or molecules and
their transport into the solvent.
Dissolution is considered one of the most important quality
control tests performed on pharmaceutical dosage forms and is now
developing into a tool for predicting bioavailability, and in some
cases, replacing clinical studies to determine bioequivalence.
Dissolution behaviour of drugs has a significant effect on their
pharmacological activity. In fact, a direct relationship between in
vitro dissolution rate of many drugs and their bioavailability has
been demonstrated.Solid dosage forms may or may not disintegrate
when they interact with gastrointestinal fluid following oral
administration depending on their design (Figure ). For
disintegrating solid oral dosage forms, disintegration usually
plays a vital role in the dissolution process since it determines
to a large extent the area of contact between the solid and liquid.
However it is well known that considerable dissolution of the drug
can take place before complete disintegration of the dosage form, a
phenomenon which depends largely on the mechanism of disintegration
and certain physicochemical properties of the drug, such as its
solubility. This could be important when considering the motility
of the drug or dosage form, and the release of the drug at specific
sites, in the gastrointestinal tract. Thus, correlations have been
established between disintegration times and dissolution rates for
various pharmaceutical tablets. It should be noted, however, that
there is not always an automatic correlation between disintegration
and dissolution, especially for drugs with very low dissolution
rates. For many drugs, particularly those that are poorly soluble
in the gastric fluid, the rate-limiting step in the absorption
process is the dissolution rate and a dissolution rate
determination can therefore be a useful guide to comparative
bioavailability. Since drug absorption and physiological
availability depend on the availability of the drug substance in
the dissolved state, suitable dissolution characteristics are
important property for a satisfactory tablet. The dissolution test
measures the amount of time required for certain percentage of the
drug substance in a tablet to go into solution under a specified
set of conditions. It describes a step towards physiological
availability of the drug substance, but it is not designed to
measure the safety or efficacy of the tablet being tested. It
provides in vitro control procedure to eliminate variation among
production batches. The dissolution medium must be aqueous and the
pH of the medium should be controlled and should simulate in vivo
conditions. The dissolution medium should be 0.1M HCl and pH 6.8
buffer to simulate the biological extremes.. Studies have shown
that low agitation must be used (i.e. in the order of 50rpm) and
that the tablet must not be subjected to abrasion in keeping with
the mild agitation in the gastrointestinal tract 45 .
Figure : Schematic diagram of the dissolution process
Dissolution testing is widely used in the pharmaceutical industry
for optimization of formulation and quality control of different
dosage forms. Dissolution test is mainly used for poor soluble
drugs(tablets or capsules).Dissolution test as per I.P. Dissolution
apparatus: There are two types of aaparatus. Dissolution apparatus
1: An assembly consisting of the foolowing.
DISSOLUTION APPARATUS FIGUREa. A cylindrical vessel, A, made of
borosilicate glass or any other suitable transparent material, with
a hemispherical bottom and with a nominal capacity of 1000ml and an
inside diameter of 98-106mm.the vessel has a flanged upper rim and
is fitted with a lid that has a number of opening one of which is
central.b. A motor with a speed of rotation of the paddle within
the 4% of that specified in the individual monograph. The motor is
fitted with a stirring element which consists of a drive shaft and
blade forming a paddle, BThe blade passes through the diameter of
the shaft so that the bottom of the blade is flush with the bottom
of the shaft. The shaft is flush with the bottom of the shaft. The
shaft is positioned so that its axis is within 2mm of the axis of
the vessel and lower edge of the blede is 23 to 27mm from the
inside bottom of the vessel. The apparatus operates insuch a way
that the paddle rotates smoothy.c. A water bath set to maintain the
dissolution medium at 36.5o to 37.5 o. the bath liquid is kept in
constant and smooth motion during the test. The vessel is securely
clamped in the water bath in such a way that the displacement
vibration from other equipment, including the water circulation
device, is minimused.Apparatus 2:
figure 2The assembly is same as in apparatus1 except that in the
stirring element the paddle is replaced by basket, D (figure-2).
The metallic shaft rotates smoothly. The basket consists of two
components. The top part, with a vent, is attached to the shaft C,
it it fitted with three spring clips, or other suitable means, that
allow removal of the lower part of introduction of the prepration
under examination and that firmly hold the lower part for
introduction of the prepration under examination and that firmly
hold the lower part of the basket concentric with the axis of the
vessel during rotation. The lower detachable part of the basket is
made of welded-steam cloth, with a wire thickness of 0.254mm
diameter with narrow rim of sheet metal around the top and the
bottom. The basket may be plated with a 2.5mm layer of gold for use
with acidic media. The distance between the inside bottom of the
vessel and the basket is maintained at 23 to 27 mm during the
test.Time. Where a single time specification is given in the
monograph, the test may be concluded in a shorter period if the
requirement for the minimum amount dissolved is met. If two or more
times are specified, specimen are to be withdrawn only at the
stated times, within a tolerance of 2 per cent.MethodConventional
and proloned-release solid dosage formsPlace the stated volume of
the dissolution medium. Free from dissolved air, in to the vessel
of the apparatus. Assemble the apparatus and warm the dissolution
medium to 36.5o to 37.5 o . unless otherwise stated, place one
dosage unit in the apparatus, taking care to exclude air bubbles
from the surface of the dosage unit. When apparatus 1 is used,
allow the tablet to sink to the bottom of the vessel prior to the
rotation of the paddle. A suitable device such as a wire of glass
helix may be used to keep horizontal at the bottom of the vessel
tablets that would other wise float.When Apparatus 2 is used, place
the tablet in a dry basket at the beginning of each test. Lower the
basket into position before rotation. Operate the apparatus
immediately at the speed of rotation specified in the individual
monograph within the time interval specified in the individual
monograph. Withdraw a specimen from a zone midway between the
surface of the dissolution medium and the top of the roating blade
or basket, not less than 10 mm from the wall of the vessel. Except
in the case of single sampling, add a volume of the samples
withdrawn. Where two or more tablets are directed to be placed
together in the apparatus, carry out six replicate tests.For each
of the tablet tested, calculate the amount of dissolved active
ingradient in solution as a percentage of the stated amount. Where
two or more tablets are placed together, determine for each test
the amount of active ingradient in solution per tablet and
calculate as a percentage of the stated amount.Acceptance
criteriaConventional-release dosage formsUnless otherwise
specified, the requirements are met if the quantities of active
substance dissolved from the dosage units conform to table 1. If
the results do not conform to the requirement at stage S1 given in
table, continue testing with additional dosage units through stages
S2 and S3 unless the results conform at stage s2. Table
1StageNumber TestedAcceptance Criteria
S16Each unit is not less than D*+ 5%.
S26Average of 12 units (S1 + S2) is equal to or greater than D,
and no unit is less than D* 15%.
S312Average of 24 units (S1 + S2 + S3) is equal to or greater
than D, not more than 2 units are less than D15%, and no unit is
less than D25%.
*D is the amount of dissolved active ingradient specified in the
individual monograph, expressed as a percentage of the labeled
content.**Percentages of the labeled content.Table 2 StageNumber
TestedAcceptance Criteria
L16No individual value lies outside each of the stated ranges
and no individual value is less than the stated amount at the final
test time.
L26The average value of the 12 units( L1 +L2) lies within each
of the stated ranges and is not less than stated amount at the
final test time, none is more than 10 per cent of the labeled
content outside each of the stated ranges and none is more than 10
percent of labeled amount below the stated amount of final test
time.
L312The average value of the 24 units (L1 + L2 + L3) Lies within
each of the stated ranges, and is not less than the stated amount
at the final test time; not more than 2 of 24 units are more than
10 per cent of labeled content outside each of the stated ranges;
not more than 2 of the 24 units are more than 10 percent of labeled
content below the stated amount at the final test time; and none of
the units is the more than 20 per cent of labeled content outside
each of the stated ranges or more than 20 per cent of labelled
content below the stated amount at the final test time.
Prolonged-release dosage formsUnless otherwise specified, the
requirements are met if the quantities of acive substance dissolved
from the dosage units conform to table 2. If the result do not
conform to the reuirements at stage L1given in the table, continue
testing with additional dosage units through stages L2 and L3
unless unless the test conform at stage L2. The limits embrace each
value of D, the amount dissolved at each specified dosing
interval.Modified-release dosage forms. Use method A or Method
B.Method AAcid stage. Place 750 ml of 0.1 M hydrochloric acid in
the vessel, and assemble the apparatus. Warm the dissolution medium
to 36.5o to 37.5 o. Place one dosage unit in the apparatus, cover
the vessel and operate the apparatus at the specified rate. After 2
hours of operation in the acidic medium, withdraw an aliquot of the
liquid and proceed immediately as directed under buffer stage.
Perform the analysis of aliquot using suitable assay method.Buffer
stage. Complete the operations of adding the buffer and adjusting
the pH within 5 minutes. With the apparatus operating at the rate
specified, add to the medium in vessel 250 ml of a 0.2 m solution
of trisodium phosphate dodecahydrate that has been warmed to 36.5o
to 37.5 o. adjust if necessary with 2M hydrochloric acid or 2M
sodium hydroxide to a Ph of 6.8 0.05.Method B Acid stage. Place
1000 ml of 0.1 M hydrochloric acid in the vessel, and assemble the
apparatus. Warm the dissolution medium to 36.5o to 37.5 o. Place
one dosage unit in the apparatus, cover the vessel and operate the
apparatus at the specified rate. After 2 hours of operation in the
acidic medium, withdraw an aliquot of the liquid and proceed
immediately as directed under buffer stage. Perform the analysis of
aliquot using suitable assay method.Buffer stage. Use buffer that
has previously been warmed to36.5o to 37.5 o. Drain the acid from
the vessel and add 1000 ml of pH 6.8 buffer, prepared by mixing 3
volumes of 0.1M hydrochloric acid with 1 volume of 0.2 M solution
of trisodium phosphate dodecahydrate and adjusting, if ne necessary
with 2M hydrochloric acid or 2M sodium hydroxide to a Ph of 6.8
0.05. Continue to operate the apparatus for 45 minutes Or for
specified time. At the end of the period, withdraw an aliquot of
the liquid and perform the analysis aliquot using suitable assay
method.
Acceptation criteriaAcid stage. Unless otherwise specified, the
requirements of this part of the test are met if the quantities,
based on the percentage of labelled content of active substance
dissolved from the units tested conform to table 3. Continue the
testing through the 3 levels unless the results of both acid and
bufeer stages conform at an earier level. Table 3StageNumber
TestedAcceptance Criteria
A16No individual value exceeds 10 per cent dissolved.
A26Average of 12 units (A1 + A2) is not more than 10 per cent
dissolved, and no individual unit is greater than 25 per cent
dissolved.
A312Average of 24 units (A1 + A2+ A3) is not more than 10 per
cent dissolved, and no individual unit is greater than 25 per cent
dissolved.
Buffer stage. Unless otherwise specified, the requirements of
this part of the test are met if the quantities, based on the
percentage of the labelled content of active substance dissolved
from the units tested conform to table 4. Continue the testing
through 3 level unless the test of both acid and buffer stages
conform at an earlier level. The value of D in table 4is 75 per
cent dissolved unless otherwise specified. The quantity D is
specified total amount of active substance dissolved in both the
acid and buffer stages, expressed as a percentage of the labelled
content.
Table 4StageNumber TestedAcceptance Criteria
S16Each unit is not less than D*+ 5%.
S26Average of 12 units (S1 + S2) is equal to or greater than D,
and no unit is less than D* 15%.
S312Average of 24 units (S1 + S2 + S3) is equal to or greater
than D, not more than 2 units are less than D15%, and no unit is
less than D25%.
RefrencesText book of Industrial pharmacy Theory and Pratice by
Leon and lachman.Dispensing pharmacy by Copper and Gunn.Indian
pharmacopeia 2005.Usp
pharmacopeia.www.wikipedia.comwww.google.comwww.ncbi.com
1