Embed Size (px)
Citation preview

Articleshttps://doi.org/10.1038/s41565-018-0227-7
1Department of Materials Science and Engineering, University of Virginia, Charlottesville, VA, USA. 2Center for Research on Advanced Fiber Technologies (CRAFT), Materials Research Institute, Pennsylvania State University, University Park , PA, USA. 3Department of Engineering Science and Mechanics, Pennsylvania State University, State College, PA, USA. 4NIST Center for Neutron Research, Gaithersburg, MD, USA. 5Department of Materials Science and Engineering, University of Maryland, College Park, MD, USA. 6Department of Biochemistry and Molecular Biology, Pennsylvania State University, University Park, PA, USA. 7Huck Institutes of the Life Sciences, Pennsylvania State University, University Park, PA, USA. 8Department of Mechanical and Aerospace Engineering, University of Virginia, Charlottesville, VA, USA. 9Department of Physics, University of Virginia, Charlottesville, VA, USA. 10These authors contributed equally: John A. Tomko, Abdon Pena-Francesch. *e-mail: [email protected]; [email protected]
The ability to actively manipulate the vibrational energy exchange and thermal conductivity of materials would enable novel directions of research in a wide range of nanoscale sci-
ence and technology fields. For example, a new class of thermally driven devices (such as thermal rectifiers, logic gates and transis-tors) could be realized with the ability to actively modulate the thermal conductivity1. Though the efficiency of these devices would not rival their electrical analogues, these devices could, in princi-ple, be powered ‘for free’ if the source of the ‘power’ is from wasted heat. Similarly, the ability to actively modulate the temperature of a device has been shown to create massive gains in thermoelectric efficiency, which would enable a new and resurgent direction to the decades-old thermoelectric problem2,3. Modulation of temperature gradients in materials could also lead to advances in all-solid-state electrocaloric or magnetic refrigeration1,4,5.
Numerous examples of variable, yet reversible, thermal con-ductivities in materials have been realized, albeit with only small modulations in their phonon or vibrational thermal conductivities. Prominent examples of such vibrational thermal switches include liquid crystal networks that can be oriented with an external mag-netic field (thus increasing their thermal conductivity from ∼ 0.20 to ∼ 0.35 W m−1 K−1; ref. 6), reversible delithiation of LiCoO2 cathode materials under electrochemical tuning (leading to reductions in the thermal conductivity from 5.4 to 3.7 W m−1 K−1; ref. 7), and an 11% increase in thermal conductivity in lead zirconate titanate thin films through manipulation of their nanoscale ferroelastic domains when subjected to external electric fields8. In these aforementioned stud-ies of thermal conductivity switches, the underlying mechanisms
driving the switching phenomena have been based on manipulating the thermal carrier populations or scattering rates. This approach of actively manipulating the lattice or vibrational thermal conductivity κ will be inherently limited to relatively small ∆ κ, because, under typical conditions (that is, not extreme pressures or temperatures), the scattering of only a relatively small window of the vibrational spectrum will be impacted. Here, we seek to not only enhance the ability to modulate thermal conductivity to enable on/off ratios much larger than the current state of the art, but also extend this dynamic control of thermal transport to biological, soft materials. We achieve this by moving beyond traditionally used approaches of actively impacting the vibrational scattering rates, and instead we utilize the unique nanocrystalline structures of networked proteins inspired from squid ring teeth (SRT), a unique structural protein, and actively manipulate the displacement amplitude of the vibra-tions in these crosslinked networks.
Indeed, precedent for actively tuning the thermal conductivity in soft materials is established in the orders of magnitude changes in thermal conductivity that have been predicted in polymeric systems based on strain, chain alignment and crystallinity. For example, record tunability, with a factor of 12 change in thermal conductiv-ity, has been calculated for polyethylene by combining the effects of strain and phase changes9. Experimentally, the recent discovery of high thermal conductivity in ordered or aligned polymers departs significantly from our conventional wisdom about relatively low thermal conductivities in polymeric systems10–13. This suggests the possibility for a large span in the variability of thermal conductivity when the system is altered from disordered to ordered, aligned or
Tunable thermal transport and reversible thermal conductivity switching in topologically networked bio-inspired materialsJohn A. Tomko1,10, Abdon Pena-Francesch2,3,10, Huihun Jung2,3, Madhusudan Tyagi4,5, Benjamin D. Allen6,7, Melik C. Demirel 2,3,7* and Patrick E. Hopkins1,8,9*
The dynamic control of thermal transport properties in solids must contend with the fact that phonons are inherently broad-band. Thus, efforts to create reversible thermal conductivity switches have resulted in only modest on/off ratios, since only a relatively narrow portion of the phononic spectrum is impacted. Here, we report on the ability to modulate the thermal conductivity of topologically networked materials by nearly a factor of four following hydration, through manipulation of the displacement amplitude of atomic vibrations. By varying the network topology, or crosslinked structure, of squid ring teeth-based bio-polymers through tandem-repetition of DNA sequences, we show that this thermal switching ratio can be directly programmed. This on/off ratio in thermal conductivity switching is over a factor of three larger than the cur-rent state-of-the-art thermal switch, offering the possibility of engineering thermally conductive biological materials with dynamic responsivity to heat.
NATurE NANoTECHNology | VOL 13 | OCTOBER 2018 | 959–964 | www.nature.com/naturenanotechnology 959
Articles NATure NANoTecHNology
crystalline phases. Thus, actively, rapidly and reversibly modulating degree of order in a soft material could in principle lead to these large changes in thermal conduction using these same concepts.
Squid ring teeth proteinsWe have identified a class of tandem-repeat (TR) protein that enables rapid and reversible changes in the dynamics of the protein chains via hydration. Our protein design is based on TR synthetic poly-peptides inspired by natural cephalopod proteins. Squid proteins have inspired the development of diverse technologies including camouflage coatings14, self-healing materials15, soft actuators16 and renewable bioplastics17. In particular, SRT proteins are high-strength biopolymers that are stabilized by a hydrogen-bonding network, and can be extracted from the suction cups of squids, or can be syntheti-cally produced via recombinant expression in bacteria18. We created a DNA assembly strategy that enables production of biomimetic SRT genes with variable length from a repetitive building block in a single cloning step, by using modified rolling-circle amplifica-tion, a known molecular biology technique19. We thus developed a library of TR sequences with a precisely controlled size distribution (TR-n4, TR-n7, TR-n11 and TR-n25, where n denotes the building block repeat number). Our SRT-inspired building block is based on the PAAASVSTVHHP crystal-forming polypeptide sequence and the YGYGGLYGGLYGGLGY amorphous polypeptide sequence, pro-ducing a self-assembled hydrogen-bonded network with crystalline physical crosslinks and amorphous disordered segments (Fig. 1b).
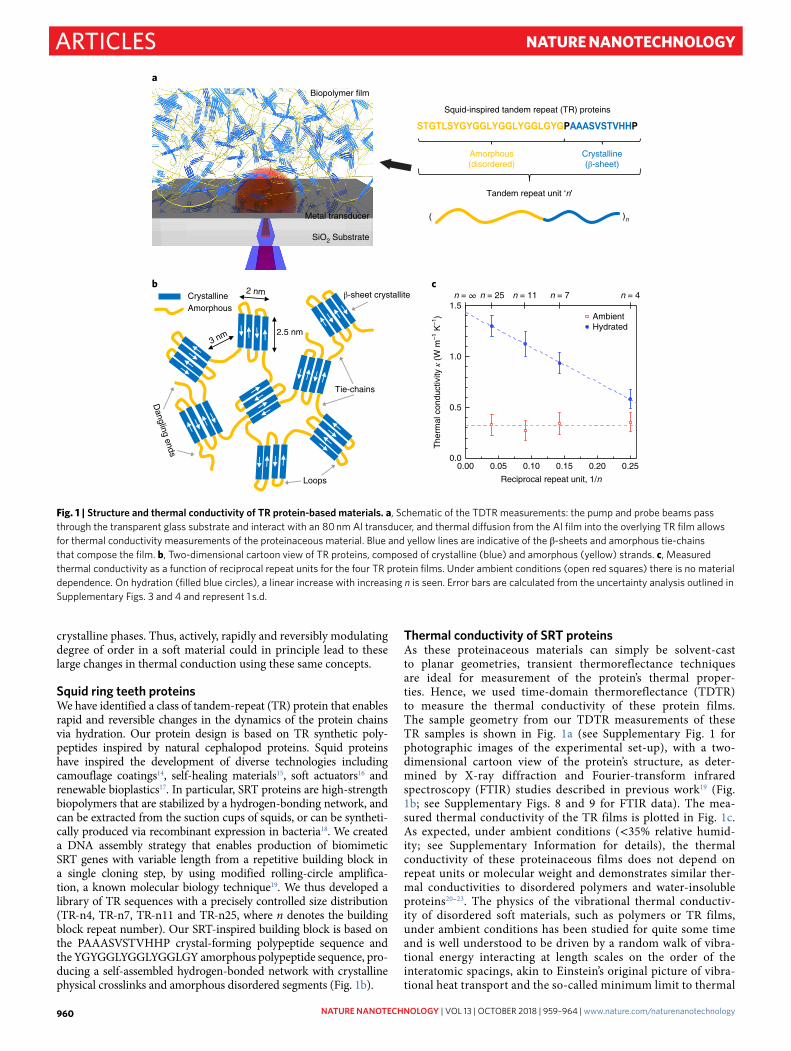
Thermal conductivity of SrT proteinsAs these proteinaceous materials can simply be solvent-cast to planar geometries, transient thermoreflectance techniques are ideal for measurement of the protein’s thermal proper-ties. Hence, we used time-domain thermoreflectance (TDTR) to measure the thermal conductivity of these protein films. The sample geometry from our TDTR measurements of these TR samples is shown in Fig. 1a (see Supplementary Fig. 1 for photographic images of the experimental set-up), with a two-dimensional cartoon view of the protein’s structure, as deter-mined by X-ray diffraction and Fourier-transform infrared spectroscopy (FTIR) studies described in previous work19 (Fig. 1b; see Supplementary Figs. 8 and 9 for FTIR data). The mea-sured thermal conductivity of the TR films is plotted in Fig. 1c. As expected, under ambient conditions (< 35% relative humid-ity; see Supplementary Information for details), the thermal conductivity of these proteinaceous films does not depend on repeat units or molecular weight and demonstrates similar ther-mal conductivities to disordered polymers and water-insoluble proteins20–23. The physics of the vibrational thermal conductiv-ity of disordered soft materials, such as polymers or TR films, under ambient conditions has been studied for quite some time and is well understood to be driven by a random walk of vibra-tional energy interacting at length scales on the order of the interatomic spacings, akin to Einstein’s original picture of vibra-tional heat transport and the so-called minimum limit to thermal
0.00 0.05 0.10 0.15 0.20 0.250.0
0.5
1.0
1.5n = ∞
cn = 4n = 7n = 11
AmbientHydrated
The
rmal
con
duct
ivity
κ (W
m–1
K–1
)
Reciprocal repeat unit, 1/n
n = 25
2.5 nm
CrystallineAmorphous
Loops
Tie-chains
β-sheet crystallite
Tandem repeat unit ‘n’
Amorphous(disordered)
Crystalline(β-sheet)
( )n
Squid-inspired tandem repeat (TR) proteins
aBiopolymer film
Metal transducer
SiO2 Substrate
b2 nm
3 nm
Dangling ends
Fig. 1 | Structure and thermal conductivity of Tr protein-based materials. a, Schematic of the TDTR measurements: the pump and probe beams pass through the transparent glass substrate and interact with an 80 nm Al transducer, and thermal diffusion from the Al film into the overlying TR film allows for thermal conductivity measurements of the proteinaceous material. Blue and yellow lines are indicative of the β -sheets and amorphous tie-chains that compose the film. b, Two-dimensional cartoon view of TR proteins, composed of crystalline (blue) and amorphous (yellow) strands. c, Measured thermal conductivity as a function of reciprocal repeat units for the four TR protein films. Under ambient conditions (open red squares) there is no material dependence. On hydration (filled blue circles), a linear increase with increasing n is seen. Error bars are calculated from the uncertainty analysis outlined in Supplementary Figs. 3 and 4 and represent 1 s.d.
NATurE NANoTECHNology | VOL 13 | OCTOBER 2018 | 959–964 | www.nature.com/naturenanotechnology960
ArticlesNATure NANoTecHNology
conductivity24,25. In terms of the TR films in this ambient state, this finding suggests that the thermal conductivity is limited by the amorphous regions, and any potential benefit of higher ther-mal conductivity in the nanocrystalline β -sheet ordered regions is lost from scattering in the amorphous domains. When these TR films are hydrated, the thermal conductivity not only increases compared to the ambient state, but a nearly linear dependency on 1/n also emerges, as shown in Fig. 1c and Supplementary Fig. 5. In the TR sample with the highest number of repeat units n (or low 1/n), we observe a nearly factor of 4 increase in thermal con-ductivity. The increase in thermal conductivity suggests that the amorphous chain conformation and overall network morphol-ogy, which are dependent on tandem repetition12,26, impact the overall thermal conductivity.
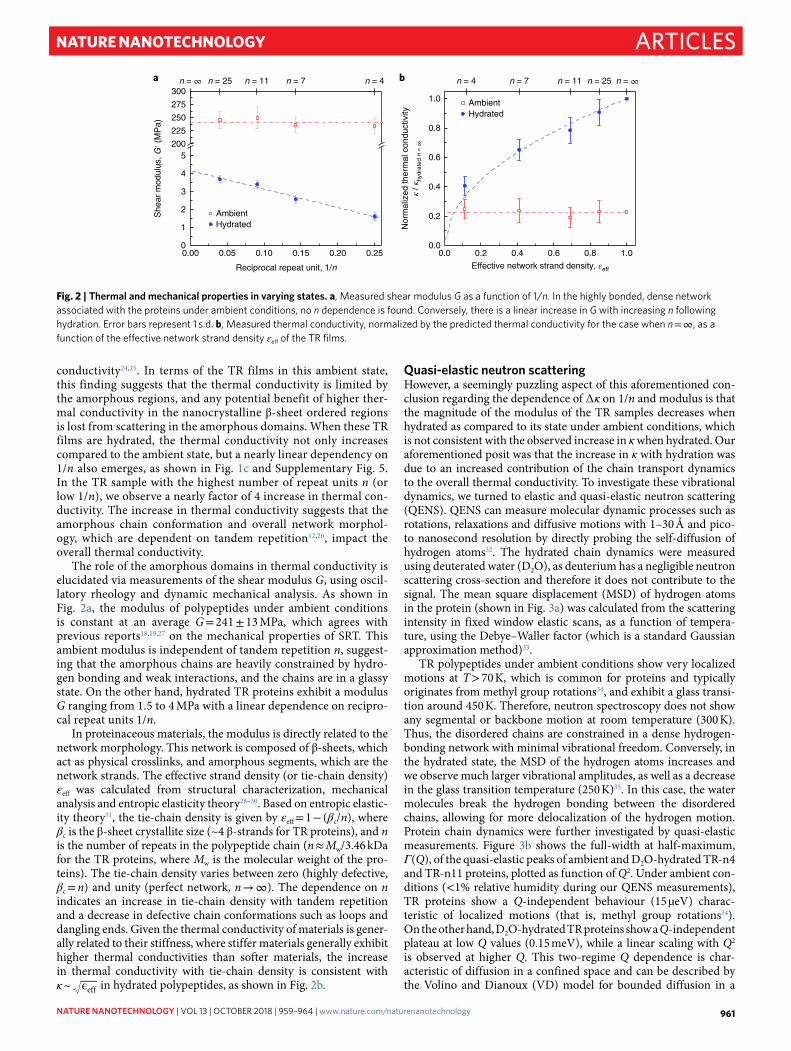
The role of the amorphous domains in thermal conductivity is elucidated via measurements of the shear modulus G, using oscil-latory rheology and dynamic mechanical analysis. As shown in Fig. 2a, the modulus of polypeptides under ambient conditions is constant at an average G = 241 ± 13 MPa, which agrees with previous reports18,19,27 on the mechanical properties of SRT. This ambient modulus is independent of tandem repetition n, suggest-ing that the amorphous chains are heavily constrained by hydro-gen bonding and weak interactions, and the chains are in a glassy state. On the other hand, hydrated TR proteins exhibit a modulus G ranging from 1.5 to 4 MPa with a linear dependence on recipro-cal repeat units 1/n.
In proteinaceous materials, the modulus is directly related to the network morphology. This network is composed of β -sheets, which act as physical crosslinks, and amorphous segments, which are the network strands. The effective strand density (or tie-chain density) εeff was calculated from structural characterization, mechanical analysis and entropic elasticity theory28–30. Based on entropic elastic-ity theory31, the tie-chain density is given by εeff = 1 − (βc/n), where βc is the β -sheet crystallite size (~4 β -strands for TR proteins), and n is the number of repeats in the polypeptide chain (n ≈ Mw/3.46 kDa for the TR proteins, where Mw is the molecular weight of the pro-teins). The tie-chain density varies between zero (highly defective, βc = n) and unity (perfect network, n → ∞ ). The dependence on n indicates an increase in tie-chain density with tandem repetition and a decrease in defective chain conformations such as loops and dangling ends. Given the thermal conductivity of materials is gener-ally related to their stiffness, where stiffer materials generally exhibit higher thermal conductivities than softer materials, the increase in thermal conductivity with tie-chain density is consistent with κ ~ ϵeff in hydrated polypeptides, as shown in Fig. 2b.
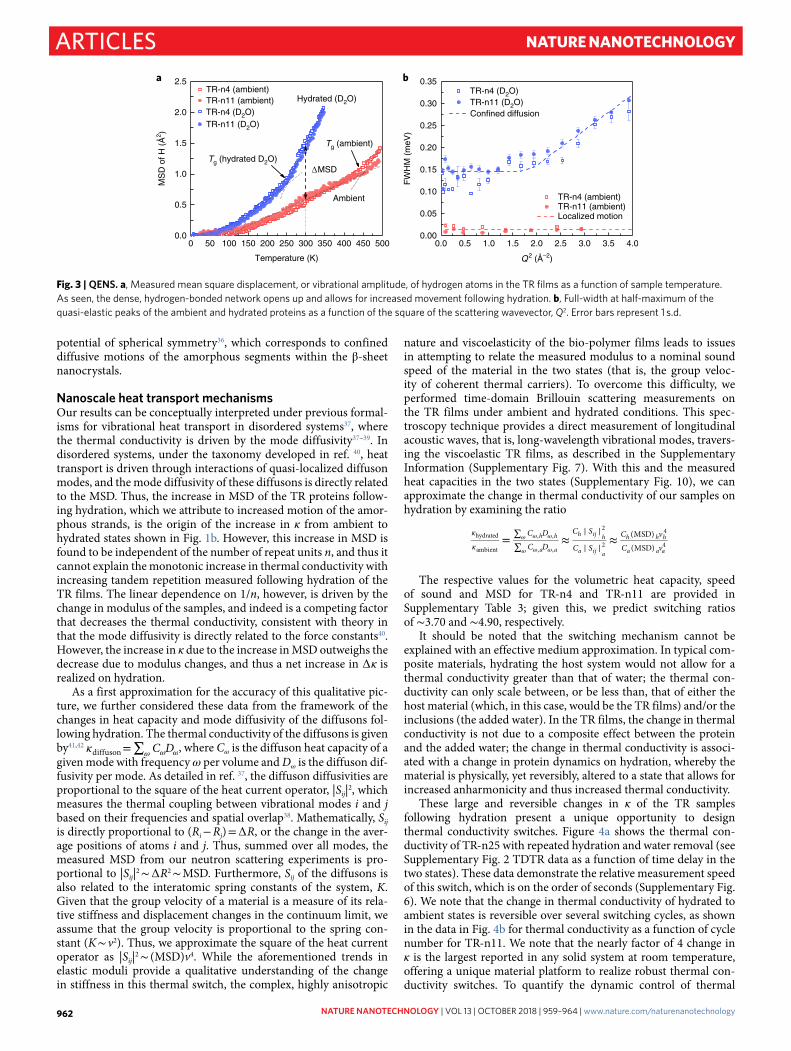
Quasi-elastic neutron scatteringHowever, a seemingly puzzling aspect of this aforementioned con-clusion regarding the dependence of ∆ κ on 1/n and modulus is that the magnitude of the modulus of the TR samples decreases when hydrated as compared to its state under ambient conditions, which is not consistent with the observed increase in κ when hydrated. Our aforementioned posit was that the increase in κ with hydration was due to an increased contribution of the chain transport dynamics to the overall thermal conductivity. To investigate these vibrational dynamics, we turned to elastic and quasi-elastic neutron scattering (QENS). QENS can measure molecular dynamic processes such as rotations, relaxations and diffusive motions with 1–30 Å and pico- to nanosecond resolution by directly probing the self-diffusion of hydrogen atoms32. The hydrated chain dynamics were measured using deuterated water (D2O), as deuterium has a negligible neutron scattering cross-section and therefore it does not contribute to the signal. The mean square displacement (MSD) of hydrogen atoms in the protein (shown in Fig. 3a) was calculated from the scattering intensity in fixed window elastic scans, as a function of tempera-ture, using the Debye–Waller factor (which is a standard Gaussian approximation method)33.
TR polypeptides under ambient conditions show very localized motions at T > 70 K, which is common for proteins and typically originates from methyl group rotations34, and exhibit a glass transi-tion around 450 K. Therefore, neutron spectroscopy does not show any segmental or backbone motion at room temperature (300 K). Thus, the disordered chains are constrained in a dense hydrogen-bonding network with minimal vibrational freedom. Conversely, in the hydrated state, the MSD of the hydrogen atoms increases and we observe much larger vibrational amplitudes, as well as a decrease in the glass transition temperature (250 K)35. In this case, the water molecules break the hydrogen bonding between the disordered chains, allowing for more delocalization of the hydrogen motion. Protein chain dynamics were further investigated by quasi-elastic measurements. Figure 3b shows the full-width at half-maximum, Γ(Q), of the quasi-elastic peaks of ambient and D2O-hydrated TR-n4 and TR-n11 proteins, plotted as function of Q2. Under ambient con-ditions (< 1% relative humidity during our QENS measurements), TR proteins show a Q-independent behaviour (15 µ eV) charac-teristic of localized motions (that is, methyl group rotations34). On the other hand, D2O-hydrated TR proteins show a Q-independent plateau at low Q values (0.15 meV), while a linear scaling with Q2 is observed at higher Q. This two-regime Q dependence is char-acteristic of diffusion in a confined space and can be described by the Volino and Dianoux (VD) model for bounded diffusion in a
0.00 0.00.0
0.2
0.4
0.6
0.8
1.0
0.2 0.4 0.6 0.8 1.00.05 0.10 0.15 0.20 0.250
1
2
3
4
5200
She
ar m
odul
us, G
′ (M
Pa)
Nor
mal
ized
ther
mal
con
duct
ivity
κ / κ
hydr
ated
n =
∞
225
250
275
300n = ∞a bn = 4n = 7n = 11
Reciprocal repeat unit, 1/n Effective network strand density, εeff
n = 25 n = 4 n = 7 n = 11 n = 25 n = ∞
AmbientHydrated
AmbientHydrated
Fig. 2 | Thermal and mechanical properties in varying states. a, Measured shear modulus G as a function of 1/n. In the highly bonded, dense network associated with the proteins under ambient conditions, no n dependence is found. Conversely, there is a linear increase in G with increasing n following hydration. Error bars represent 1 s.d. b, Measured thermal conductivity, normalized by the predicted thermal conductivity for the case when n = ∞ , as a function of the effective network strand density εeff of the TR films.
NATurE NANoTECHNology | VOL 13 | OCTOBER 2018 | 959–964 | www.nature.com/naturenanotechnology 961
Articles NATure NANoTecHNology
potential of spherical symmetry36, which corresponds to confined diffusive motions of the amorphous segments within the β -sheet nanocrystals.
Nanoscale heat transport mechanismsOur results can be conceptually interpreted under previous formal-isms for vibrational heat transport in disordered systems37, where the thermal conductivity is driven by the mode diffusivity37–39. In disordered systems, under the taxonomy developed in ref. 40, heat transport is driven through interactions of quasi-localized diffuson modes, and the mode diffusivity of these diffusons is directly related to the MSD. Thus, the increase in MSD of the TR proteins follow-ing hydration, which we attribute to increased motion of the amor-phous strands, is the origin of the increase in κ from ambient to hydrated states shown in Fig. 1b. However, this increase in MSD is found to be independent of the number of repeat units n, and thus it cannot explain the monotonic increase in thermal conductivity with increasing tandem repetition measured following hydration of the TR films. The linear dependence on 1/n, however, is driven by the change in modulus of the samples, and indeed is a competing factor that decreases the thermal conductivity, consistent with theory in that the mode diffusivity is directly related to the force constants40. However, the increase in κ due to the increase in MSD outweighs the decrease due to modulus changes, and thus a net increase in ∆ κ is realized on hydration.
As a first approximation for the accuracy of this qualitative pic-ture, we further considered these data from the framework of the changes in heat capacity and mode diffusivity of the diffusons fol-lowing hydration. The thermal conductivity of the diffusons is given by41,42 κ = ∑ω ω ωC Ddiffuson , where Cω is the diffuson heat capacity of a given mode with frequency ω per volume and Dω is the diffuson dif-fusivity per mode. As detailed in ref. 37, the diffuson diffusivities are proportional to the square of the heat current operator, |Sij|2, which measures the thermal coupling between vibrational modes i and j based on their frequencies and spatial overlap38. Mathematically, Sij is directly proportional to (Ri − Rj) = ∆ R, or the change in the aver-age positions of atoms i and j. Thus, summed over all modes, the measured MSD from our neutron scattering experiments is pro-portional to |Sij|2 ∼ ∆ R2 ∼ MSD. Furthermore, Sij of the diffusons is also related to the interatomic spring constants of the system, K. Given that the group velocity of a material is a measure of its rela-tive stiffness and displacement changes in the continuum limit, we assume that the group velocity is proportional to the spring con-stant (K ∼ v2). Thus, we approximate the square of the heat current operator as |Sij|2 ∼ (MSD)v4. While the aforementioned trends in elastic moduli provide a qualitative understanding of the change in stiffness in this thermal switch, the complex, highly anisotropic
nature and viscoelasticity of the bio-polymer films leads to issues in attempting to relate the measured modulus to a nominal sound speed of the material in the two states (that is, the group veloc-ity of coherent thermal carriers). To overcome this difficulty, we performed time-domain Brillouin scattering measurements on the TR films under ambient and hydrated conditions. This spec-troscopy technique provides a direct measurement of longitudinal acoustic waves, that is, long-wavelength vibrational modes, travers-ing the viscoelastic TR films, as described in the Supplementary Information (Supplementary Fig. 7). With this and the measured heat capacities in the two states (Supplementary Fig. 10), we can approximate the change in thermal conductivity of our samples on hydration by examining the ratio
= ≈ ≈κ
κ∑∑
∣ ∣
∣ ∣ω ω ω
ω ω ω
C D
C D
C S
C S
C v
C v
(MSD)
(MSD)h h
a a
h ij h
a ij a
h h h
a a a
hydrated
ambient
, ,
, ,
2
2
4
4
The respective values for the volumetric heat capacity, speed of sound and MSD for TR-n4 and TR-n11 are provided in Supplementary Table 3; given this, we predict switching ratios of ∼ 3.70 and ∼ 4.90, respectively.
It should be noted that the switching mechanism cannot be explained with an effective medium approximation. In typical com-posite materials, hydrating the host system would not allow for a thermal conductivity greater than that of water; the thermal con-ductivity can only scale between, or be less than, that of either the host material (which, in this case, would be the TR films) and/or the inclusions (the added water). In the TR films, the change in thermal conductivity is not due to a composite effect between the protein and the added water; the change in thermal conductivity is associ-ated with a change in protein dynamics on hydration, whereby the material is physically, yet reversibly, altered to a state that allows for increased anharmonicity and thus increased thermal conductivity.
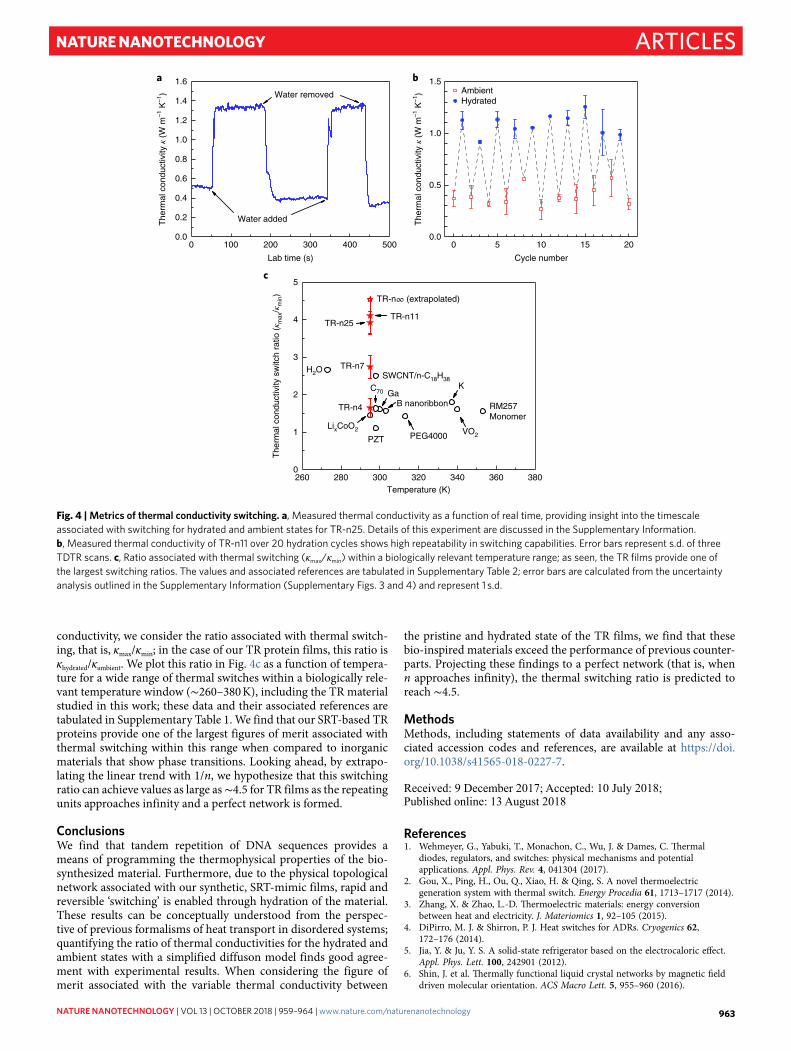
These large and reversible changes in κ of the TR samples following hydration present a unique opportunity to design thermal conductivity switches. Figure 4a shows the thermal con-ductivity of TR-n25 with repeated hydration and water removal (see Supplementary Fig. 2 TDTR data as a function of time delay in the two states). These data demonstrate the relative measurement speed of this switch, which is on the order of seconds (Supplementary Fig. 6). We note that the change in thermal conductivity of hydrated to ambient states is reversible over several switching cycles, as shown in the data in Fig. 4b for thermal conductivity as a function of cycle number for TR-n11. We note that the nearly factor of 4 change in κ is the largest reported in any solid system at room temperature, offering a unique material platform to realize robust thermal con-ductivity switches. To quantify the dynamic control of thermal
a b2.5 0.35
0.30
0.25
0.20
0.15
0.10
0.05
0.00
FW
HM
(m
eV)
2.0
1.5
1.0
0.5
MS
D o
f H (
Å2 )
0.00 50 100 150 200 250 300
Temperature (K) Q2 (Å–2)
Tg (hydrated D2O)
Ambient
Tg (ambient)
Hydrated (D2O)TR-n4 (ambient)TR-n11 (ambient)
TR-n4 (D2O)TR-n11 (D2O)Confined diffusion
TR-n4 (ambient)TR-n11 (ambient)Localized motion
TR-n4 (D2O)TR-n11 (D2O)
∆MSD
350 400 450 500 0.0 0.5 1.0 1.5 2.0 2.5 3.0 3.5 4.0
Fig. 3 | QENS. a, Measured mean square displacement, or vibrational amplitude, of hydrogen atoms in the TR films as a function of sample temperature. As seen, the dense, hydrogen-bonded network opens up and allows for increased movement following hydration. b, Full-width at half-maximum of the quasi-elastic peaks of the ambient and hydrated proteins as a function of the square of the scattering wavevector, Q2. Error bars represent 1 s.d.
NATurE NANoTECHNology | VOL 13 | OCTOBER 2018 | 959–964 | www.nature.com/naturenanotechnology962
ArticlesNATure NANoTecHNology
conductivity, we consider the ratio associated with thermal switch-ing, that is, κmax/κmin; in the case of our TR protein films, this ratio is κhydrated/κambient. We plot this ratio in Fig. 4c as a function of tempera-ture for a wide range of thermal switches within a biologically rele-vant temperature window (∼ 260–380 K), including the TR material studied in this work; these data and their associated references are tabulated in Supplementary Table 1. We find that our SRT-based TR proteins provide one of the largest figures of merit associated with thermal switching within this range when compared to inorganic materials that show phase transitions. Looking ahead, by extrapo-lating the linear trend with 1/n, we hypothesize that this switching ratio can achieve values as large as ∼ 4.5 for TR films as the repeating units approaches infinity and a perfect network is formed.
ConclusionsWe find that tandem repetition of DNA sequences provides a means of programming the thermophysical properties of the bio-synthesized material. Furthermore, due to the physical topological network associated with our synthetic, SRT-mimic films, rapid and reversible ‘switching’ is enabled through hydration of the material. These results can be conceptually understood from the perspec-tive of previous formalisms of heat transport in disordered systems; quantifying the ratio of thermal conductivities for the hydrated and ambient states with a simplified diffuson model finds good agree-ment with experimental results. When considering the figure of merit associated with the variable thermal conductivity between
the pristine and hydrated state of the TR films, we find that these bio-inspired materials exceed the performance of previous counter-parts. Projecting these findings to a perfect network (that is, when n approaches infinity), the thermal switching ratio is predicted to reach ∼ 4.5.
MethodsMethods, including statements of data availability and any asso-ciated accession codes and references, are available at https://doi.org/10.1038/s41565-018-0227-7.
Received: 9 December 2017; Accepted: 10 July 2018; Published online: 13 August 2018
references 1. Wehmeyer, G., Yabuki, T., Monachon, C., Wu, J. & Dames, C. Thermal
diodes, regulators, and switches: physical mechanisms and potential applications. Appl. Phys. Rev. 4, 041304 (2017).
2. Gou, X., Ping, H., Ou, Q., Xiao, H. & Qing, S. A novel thermoelectric generation system with thermal switch. Energy Procedia 61, 1713–1717 (2014).
3. Zhang, X. & Zhao, L.-D. Thermoelectric materials: energy conversion between heat and electricity. J. Materiomics 1, 92–105 (2015).
4. DiPirro, M. J. & Shirron, P. J. Heat switches for ADRs. Cryogenics 62, 172–176 (2014).
5. Jia, Y. & Ju, Y. S. A solid-state refrigerator based on the electrocaloric effect. Appl. Phys. Lett. 100, 242901 (2012).
6. Shin, J. et al. Thermally functional liquid crystal networks by magnetic field driven molecular orientation. ACS Macro Lett. 5, 955–960 (2016).
The
rmal
con
duct
ivity
κ (W
m–1
K–1
)
The
rmal
con
duct
ivity
κ (W
m–1
K–1
)
a b1.6
1.4
1.2
1.0
0.8
0.6
0.4
0.2
0.00 100 200
Lab time (s) Cycle number
00.0
1.0
0.5
AmbientHydrated
1.5
5 10 15 20
Water added
Water removed
300 400 500
The
rmal
con
duct
ivity
sw
itch
ratio
(κ m
ax/κ
min)
c5
TR-n∞ (extrapolated)
TR-n11TR-n25
TR-n7SWCNT/n-C18H38
TR-n4
LixCoO2
PZT PEG4000 VO2
B nanoribbonGa
C70K
RM257Monomer
H2O
4
3
2
1
0260 280 300 320 340 360 380
Temperature (K)
Fig. 4 | Metrics of thermal conductivity switching. a, Measured thermal conductivity as a function of real time, providing insight into the timescale associated with switching for hydrated and ambient states for TR-n25. Details of this experiment are discussed in the Supplementary Information. b, Measured thermal conductivity of TR-n11 over 20 hydration cycles shows high repeatability in switching capabilities. Error bars represent s.d. of three TDTR scans. c, Ratio associated with thermal switching (κmax/κmin) within a biologically relevant temperature range; as seen, the TR films provide one of the largest switching ratios. The values and associated references are tabulated in Supplementary Table 2; error bars are calculated from the uncertainty analysis outlined in the Supplementary Information (Supplementary Figs. 3 and 4) and represent 1 s.d.
NATurE NANoTECHNology | VOL 13 | OCTOBER 2018 | 959–964 | www.nature.com/naturenanotechnology 963

Articles NATure NANoTecHNology
7. Cho, J. et al. Electrochemically tunable thermal conductivity of lithium cobalt oxide. Nat. Commun. 5, 4035 (2014).
8. Ihlefeld, J. F. et al. Room-temperature voltage tunable phonon thermal conductivity via reconfigurable interfaces in ferroelectric thin films. Nano Lett. 15, 1791–1795 (2015).
9. Zhang, T. & Luo, T. High-contrast, reversible thermal conductivity regulation utilizing the phase transition of polyethylene nanofibers. ACS Nano 7, 7592 (2013).
10. Shen, S., Henry, A., Tong, J., Zheng, R. & Chen, G. Polyethylene nanofibres with very high thermal conductivities. Nat. Nanotech. 5, 251–255 (2010).
11. Singh, V. et al. High thermal conductivity of chain-oriented amorphous polythiophene. Nat. Nanotech. 9, 384–390 (2014).
12. Kim, G.-H. et al. High thermal conductivity in amorphous polymer blends by engineered interchain interactions. Nat. Mater. 14, 295–300 (2015).
13. Wang, X., Ho, V., Segalman, R. A. & Cahill, D. G. Thermal conductivity of high-modulus polymer fibers. Macromolecules 46, 4937–4943 (2013).
14. Phan, L. et al. Reconfigurable infrared camouflage coatings from a cephalopod protein. Adv. Mater. 25, 5621–5625 (2013).
15. Sariola, V. et al. Segmented molecular design of self-healing proteinaceous materials. Sci. Rep. 5, 13482 (2015).
16. Vural, M. et al. Programmable molecular composites of tandem proteins with graphene oxide for efficient bimorph actuators. Carbon 118, 404–412 (2017).
17. Demirel, M. C., Cetinkaya, M., Pena-Francesch, A. & Jung, H. Recent advances in nanoscale bioinspired materials. Macromol. Biosci. 15, 300–311 (2015).
18. Pena-Francesch, A. et al. Materials fabrication from native and recombinant thermoplastic squid proteins. Adv. Funct. Mater. 24, 7401–7409 (2014).
19. Jung, H. et al. Molecular tandem repeat strategy for elucidating mechanical properties of high-strength proteins. Proc. Natl Acad. Sci. USA 113, 6478–6483 (2016).
20. George, M. C., Rodriguez, M. A., Kent, M. S., Brennecka, G. L. & Hopkins, P. E. Thermal conductivity of self-assembling symmetric block copolymer thin films of polystyrene-block-poly(methyl methacrylate). J. Heat Transfer 138, 024505 (2016).
21. Losego, M. D., Moh, L., Arpin, K. A., Cahill, D. G. & Braun, P. V. Interfacial thermal conductance in spun-cast polymer films and polymer brushes. Appl. Phys. Lett. 97, 011908 (2010).
22. Foley, B. M. et al. Protein thermal conductivity measured in the solid state reveals anharmonic interactions of vibrations in a fractal structure. J. Phys. Chem. Lett. 5, 1077–1082 (2014).
23. Ghossoub, M. G., Lee, J.-H., Baris, O. T., Cahill, D. G. & Sinha, S. Percolation of thermal conductivity in amorphous fluorocarbons. Phys. Rev. B 82, 195441 (2010).
24. Einstein, A. Elementary observations on thermal molecular motion in solids. Annalen der Physik 35, 679–694 (1911).
25. Cahill, D. G., Watson, S. K. & Pohl, R. O. Lower limit to the thermal conductivity of disordered crystals. Phys. Rev. B 46, 6131–6140 (1992).
26. Rashidi, V., Coyle, E. J., Sebeck, K., Kieffer, J. & Pipe, K. P. Thermal conductance in cross-linked polymers: effects of non-bonding interactions. J. Phys. Chem. B 121, 4600–4609 (2017).
27. Pena-Francesch, A. et al. Pressure sensitive adhesion of an elastomeric protein complex extracted from squid ring teeth. Adv. Funct. Mater. 24, 6227–6233 (2014).
28. Flory, P. J. Principles of Polymer Chemistry (Cornell Univ. Press, Ithaca, NY, 1953).
29. Treloar, L. R. G. The Physics of Rubber Elasticity (Oxford Univ. Press, New York, NY, 1975).
30. Atilgan, A. R. et al. Anisotropy of fluctuation dynamics of proteins with an elastic network model. Biophys. J. 80, 505–515 (2001).
31. Pena-Francesch, A. et al. Mechanical properties of tandem-repeat proteins are governed by network defects. ACS Biomater. Sci. Eng. 4, 884–891 (2018).
32. Sakai, V. G. & Arbe, A. Quasielastic neutron scattering in soft matter. Curr. Opin. Colloid Interface Sci. 14, 381–390 (2009).
33. Zaccai, G. How soft is a protein? A protein dynamics force constant measured by neutron scattering. Science 288, 1604–1607 (2000).
34. Gabel, F. et al. Protein dynamics studied by neutron scattering. Q. Rev. Biophys. 35, 327–367 (2002).
35. Gordon, M. & Taylor, J. S. Ideal copolymers and the second-order transitions of synthetic rubbers. i. Non-crystalline copolymers. J. Appl. Chem. 2, 493–500 (1952).
36. Volino, F. & Dianoux, A. J. Neutron incoherent scattering law for diffusion in a potential of spherical symmetry: general formalism and application to diffusion inside a sphere. Mol. Phys. 41, 271–279 (1980).
37. Allen, P. B. & Feldman, J. L. Thermal conductivity of disordered harmonic solids. Phys. Rev. B 48, 581–588 (1993).
38. Larkin, J. M. & McGaughey, A. J. H. Thermal conductivity accumulation in amorphous silica and amorphous silicon. Phys. Rev. B 89, 144303 (2014).
39. Shenogin, S., Bodapati, A., Keblinski, P. & McGaughey, A. J. H. Predicting the thermal conductivity of inorganic and polymeric glasses: the role of anharmonicity. J. Appl. Phys. 105, 034906 (2009).
40. Allen, P. B., Feldman, J. L., Fabian, J. & Wooten, F. Diffusons, locons and propagons: character of atomic vibrations in amorphous Si. Philos. Mag. B 79, 1715–1731 (1999).
41. Feldman, J. L., Kluge, M. D., Allen, P. B. & Wooten, F. Thermal conductivity and localization in glasses: numerical study of a model of amorphous silicon. Phys. Rev. B 48, 12589 (1993).
42. Feldman, J. L., Allen, P. B. & Bickham, S. R. Numerical study of low-frequency vibrations in amorphous silicon. Phys. Rev. B 59, 3551 (1999).
AcknowledgementsJ.A.T. and P.E.H. acknowledge support from the Office of Naval Research (grant no. N00014-15-12769). M.C.D., B.D.A., A.P.-F. and H.J. were supported by the Army Research Office (grant no. W911NF-16-1-0019) and the Materials Research Institute of Pennsylvania State University. Access to the HFBS was provided by the Center for High Resolution Neutron Scattering, a partnership between the National Science Foundation and NIST under agreement no. DMR-1508249. Certain commercial material suppliers are identified in this paper to foster understanding. Such identification does not imply recommendation or endorsement by the NIST, nor does it imply that the materials or equipment identified are necessarily the best available for the purpose.
Author contributionsJ.A.T. and A.P.-F. contributed equally to this work. P.E.H. and M.C.D. conceived the idea and supervised the research. J.A.T. performed the TDTR and TDBS measurements/analysis. J.A.T performed the thermal analysis. A.P.-F. fabricated the protein films and performed the rheology, structural and mechanical analysis and temperature-modulated differential scanning calorimetry measurements. A.P.-F. performed the neutron scattering measurements in collaboration with M.T. H.J. worked on the cloning, recombinant expression and purification of proteins under the supervision of B.D.A. All authors contributed to writing and revising the manuscript, and agreed on the final content of the manuscript.
Competing interestsThe Penn State Research Foundation and the University of Virginia Patent Foundation have applied for a US provisional patent, application no. 62/711,010, filed 27 July 2018, related to the structural protein-based thermal switch produced in this work.
Additional informationSupplementary information is available for this paper at https://doi.org/10.1038/s41565-018-0227-7.
Reprints and permissions information is available at www.nature.com/reprints.
Correspondence and requests for materials should be addressed to M.C.D. or P.E.H.
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
NATurE NANoTECHNology | VOL 13 | OCTOBER 2018 | 959–964 | www.nature.com/naturenanotechnology964

ArticlesNATure NANoTecHNology
MethodsTDTR. As described in detail in the Supplementary Information, TDTR is an optical pump–probe technique that allows for measurement of the thermal conductivity of thin films and other nanomaterials. As our TR films are solvent-cast on top of an optically thick, 80 nm Al film, we performed TDTR measurements through the supporting SiO2 substrate to avoid non-thermal responses, as shown in Fig. 1a. As this sandwiched metal film acts as a thermal transducer (that is, we are probing the temperature of the Al film), we used a bidirectional model to determine the thermal conductivity of the biopolymer film.
Time-domain Brillouin scattering. Also described in detail in the Supplementary Information, our time-domain Brillouin scattering (TDBS) measurements made use of a similar apparatus as our TDTR measurements, other than that our pump and probe beams passed through the TR film and subsequently interacted with the metal transducer. The pump–metal interaction led to a temperature rise at the TR–metal interface, causing the formation of an acoustic pressure front that traversed through the biopolymer film. As our probe pulse was at normal incidence to this acoustic wave, it underwent a partial reflection due to the change in density (that is, change in refractive index) and the remainder reflected off the metal film. At integer values of the probe beam’s wavelength, it underwent self-interference due to these partial reflections, allowing for determination of the velocity of this coherent wavefront.
Rheology. Rheological measurements of TR proteins were performed on protein disks (diameter, 2 mm; thickness, 1 mm) in a Rheometric Scientific ARES rheometer using a parallel plate geometry (diameter, 3 mm) with a custom-made liquid reservoir bottom plate. Protein disks were adhered to the plates with Click Bond CB200 acrylic adhesive. Oscillatory shear measurements were performed from 0.001 to 100 rad s−1 with 2% strain.
Dynamic mechanical analysis. Dynamic mechanical analysis measurements were performed in a TA 800Q dynamic mechanical analysis instrument on protein film samples with dimensions of 15 mm × 2.5 mm × 0.2 mm (thickness). Film-tension dynamic measurements were performed from 0.1 to 200 Hz, with an amplitude of 5 µ m and a preload of 0.01 N.
Temperature-modulated differential scanning calorimetry. A TA Instruments Q200 differential scanning calorimeter was used for the specific heat measurements of TR proteins. The instrument was calibrated for specific heat Cp measurements using Tzero aluminium pans and sapphire standard materials, including cell resistance and capacitance, cell constant and temperature calibration. TR protein samples were annealed on a hot plate at 100 °C for 30 min to remove water before the experiment. Hydrated proteins were immersed in deionized water for 1 h, excess water was removed, and samples were sealed in hermetic pans. Ambient samples were heated at 2 °C min−1 from 25 to 210 °C (beginning of thermal degradation) with a modulation period of 60 s and a temperature amplitude of 0.318 °C. Hydrated samples were run from 5 to 50 °C with the same rate, modulation period and amplitude parameters. The measured heat capacity values in both ambient and hydrated states are provided in the Supplementary Information.
Neutron scattering. QENS experiments were performed at the NIST Center for Neutron Research (NCNR), Gaithersburg, MD. Experiments on TR proteins under ambient conditions were performed on the high-flux backscattering spectrometer (HFBS)43, with an energy resolution of 1 µ eV, dynamic range of ± 15 µ eV and Q range of 0.25–1.8 Å−1, at 295 K. The resolution function was measured at 4 K (the signal is completely elastic). Quasi-elastic experiments on D2O-hydrated SRT TR polypeptides were performed on a disk chopper time-of-flight spectrometer (DCS)44, with an energy resolution of 64 µ eV (wavelength λ = 6 Å), dynamic range of ± 0.5 meV and Q range of 0.1–2 Å−1, at 295 K. Vanadium was used as the resolution function in DCS measurements.
Data availability. The data that supports the plots within this paper and other findings of this study are available from the corresponding authors upon reasonable request.
references 43. Meyer, A., Dimeo, R. M., Gehring, P. M. & Neumann, D. A. The high-flux
backscattering spectrometer at the NIST Center for Neutron Research. Rev. Sci. Instrum. 74, 2759–2777 (2003).
44. Copley, J. R. D. & Cook, J. C. The Disk Chopper Spectrometer at NIST: a new instrument for quasielastic neutron scattering studies. Chem. Phys. 292, 477–485 (2003).
NATurE NANoTECHNology | www.nature.com/naturenanotechnology