Embed Size (px)
Citation preview

2 7 8 ACETALS AND KETALS Ch . 8
94McElvain and Stevens. J . Am . Chem . Soc .. 68. 1917 (1946); X~cElvain and Venerable. ibid., 72. 1661 ( 1950) .
'=Avy. Bull . soc . chim . France . (4) 49. 12 (1931) . "Brand and Schreber. Ber., 75. 156 (1942) . 97 Palornaa and Kantola. Ber.. 65. 1593 (1932) . 98 Uey. / . Chem . Soc.. 1057 (1937) . Q9 Rotbart. 4nn . chim., (11) 1. 458 (1934) .
lDO Witzemann e t al., Org . Synlheses. Col1 . Vol . 11. 307 (1943) . l'' Shriner and Kleiderer. Org . Syntheses. Col1 . Vol . 11. 538 (1943) . 'O' Allen and Clark. Org . Syntheses. 24. 3 (1944); Woodward and doer in^,
] . ..l m. Chem . Soc.. 67. B68 (1945) . '03 Johnson e t al., J . Am . Chem . Soc., 69. 2364 (1947) . '04Albers. Kallischnigg. and Schmidt. Ber., 77. 623 (1944) . 'O'McElvain and Clarke. J . Am . Chem . Soc., 69. 2657 (1947) . 106ivlcElvain and Clarke. / . Am . Chem . Soc., 69. 2661 (1947) . 'O7Uhle and Jacobs. J . Org. Chem., 10. B1 (1945); Hartung and Adkins. J . Am .
C k m . Soc., 69. 1535 (1947) . lo8Wihut and Beets. Rec . Lrav . chim., 59. 653 (1940) . lo9Scheibler and Depner. Ber .. 68. 2151 (1935) . "O Bergel. klorrison. and Rinderknecht. J . Chem . Soc., 265 (1944) . "l Magnani and .dcElvain. J . Am . Chem . Soc .. 60. 2210 (1930) . l'' Feazel and Berl. J . Am . Chem . Soc., 72. 2278 (1950) . aslow low and Lawton. J . Am . Chem . Soc .. 72. 1723 (1950) . l14Vogel and Schinz. Helv . Chim . Acta . 33. 127 (1950) . " ' ~ c C u s k e r and Vogt. J . Am . Chem . Soc .. 59. 1307 (1937) . l16hlcElvain. Chem . Revs., 45. 453 (1949) . "'Price and Pappalatdo. ] . Am . Chem . Soc., 72. 2613 (1950) . l18Lunt and Sondheimer. J . Chem . Soc., 3361 (1950) . "9 Icke et al., Org . Synlheses. 29. 6 (1949) . lZ0 ~ a n e t z k y . Verkade. and Meerburg. Rec . trav . chim., 66. 321 (1947) . "'Deno. J . Am . Chem . Soc., 69. 2233 (1947) . 1 aa Kraus. Ann . chim.. (12) 4 . 817 (1949) . "l Willimann and Schinz. Iielv . Chim . 4 c l a . 32. 2 151 (1949) .
Aldehydes
CONTENTS METHOD
140 . Formylation with Carbon Monoxide (Gattermann-Koch) ................... 141 . Formylation with Cyano Compounds (Gattermann) ............................ 142 . Formylation with N-Methylformanilide ................................................ 143 . Formylation of Phenols with Chloroform (ReimerTiemann) ............ 144 . Formylation of Phenols (or Amines) with Hexamine (Duff) ............ 145 . Hydroformylation o f Unsaturated Co~npounds .................................... 146 . Formylation o f Ketones with Formic Es t e r s ...................................... 147 . Interaction of Halomethyl Compounds and Hexamine (Sommelet) .. 148 . Interaction of Benzyl Halides and Sodium 2-Propanenitronate ...... 149 . Decomposition of Arylsulfonohydrazides .......................................... 150 . Cleavage o f Schiff B a s e s .................................................................... 151 . Hydrolysis of gemDihal ides ................................................................
........ . 152 Interaction of Pyridinium Sal ts and p-Nitrosodimethylaniline 153 . Hydrolysis o f 2-Alkoxy-3.4-dihydro-1.2-pyrans ................................ 154 . Hydrolysis of Aldehyde Derivatives .................................................. 155 . Oxidation of Aromatic Side Chains .................................................... 156 . Oxidation o f Olefinic Compounds ........................................................
.......................... . 157 Oxidation o f Methyl Ketones by Selenium Dioxide 158 . Oxidation o f Primary Alcohols ............................................................ 159 . Dehydrogenation of Primary Alcohols ................................................ 160 . Oxidative Cleavage o f Glycols ............................................................ 161 . Selective Reduction o f Olefinic Aldehydes ...................................... 162 . Reduction o f Acyl Chlorides (Rosenmund) ........................................ 163 . Reduction o f Thiol Esters ............. : ...................................................... 164 . Reduction o f Nitriles (Stephen) ..........................................................
.............. . 165 Interaction of Grignard Reagents and Orthoformic Es t e r s ...... . 166 Interaction of Grignard Reagents and Ethoxymethyleneaniline
167 . Decomposition o f Glycol Monoalkyl Ethers ...................................... 168 . Thermal Uecomposition of Acids ........................................................ 169 . Decomposition of a-Hy&oxy Acids .................................................... 170 . Decarborylation of a-Keto Acids ........................................................ 171 . Decarboxylation o f Glycidic Acids .................................................... 172 . Hydrolysis o f Olefin Dibromides ........................................................ 173 . Degradation o f Acid Amides and Azides ............................................ 174 . Acid Treatment of Primary Acznitroparaffins .................................... 175 . Isomerization o f Unsaturated Alcohols ..............................................
.................... . 176 Condensation of Aromatic Hydrocarbons with Chloral 177 . Fomylation o f Acetylenes ..................................................................
PAGE
280 280 281 281 282 282 282 282
283 283 284 285 285 286 286 287 288 288 289 290 290 291 291 292 292 293 293 294 294 294 295 295 296 236 296 296 297 297

280 ALDEHYDES ch. 9
CONTENTS (continued) METHOD
...................................................................................... Table 25. Aldehydes .................................................................................. Table 26. Dialdehydes
Table 27. Olefinic Aldehydes ...................................................................... Table 28. Acetylenic Aldehydes .................................................................. Table 29. Halo Aldehydes ............................................................................
...................................................................... Table 30. Hydroxy Aldehydes .................................................................................. Table 31. Aldo Ethers
...................................................................................................... References
PAGE 298 303 303 30 5 305 306 307
309
A large number of methods exis t for the preparation of aldehydes, many of which are very limited in their scope. The more general methods are given here. An excellent review on the synthesis of aromatic aldehydes has been p~bl ished."~
140. Formylation with Carbon Monoxide (Gattermann-Koch)
AICl+2uCb ArH + CO + HCl -------A ArCHO
Aromatic aldehydes are prepared by passing carbon monoxide and dry hydrogen chloride through an ether or nitrobenzene solution of a n aro- matic hydrocarbon in the presence of a catalyst, commonly aluminum chloride with cuprous chloride a s a carrier. The process i s illustrated by the synthesis of p-tolualdehyde (51%):' A convenient procedure for obtaining an equinolar mixture of anhydrous hydrogen chloride and carbon monoxide cons is ts in dropping chlorosulfonic acid on formic acid,"' viz.,
In most reactions a t atmospheric pressure the yields are about 30-50%, whereas a t a high pressure of carbon monoxide the yields are 80-90%." This method is particularly suitable for the reaction of mono- and poly- alkylbenzenes. It i s not applicable to phenols and aromatic ethers. The reaction has been considered in detail.'4"
141. Formylation with Cyano Compounds (Gattermann)
ArH + HCN + HCl ZnC1,\ ArCH = NH HCI 5 ArCHO
A mixture of hydrogen cyanide and hydrogen chloride in the presence of zinc chloride reacts with an aromatic compound to form an aldimine hydrochloride which on hydrolysis produces the corresponding aldehyde.
METHODS 141-143 28 1
The reaction can be carried out more conveniently and in equally good yields by substituting zinc cyanide for the hydrogen cyanide (70-90%).7''7' Potassium chloride impurity in this catalyst i s necessary?' Sodium cyanide has a l s o been used?9 With these modifications, phenols7' and ethers " a s well a s hydrocarbons 74' '" react (cf. method 140).
T X 142. Formylation with N-Methylformanilide
ArH + C6H, N(CH,)CHO 5 ArCHO + C& NHCH,
This synthesis is applicable to many aromatic compounds, including alkoxyl or N,N-dimethylamino derivatives of benzene'" and naphthalene,"' naphthols,'06 indole,'" and certain reactive hydrocarbons, namely, anthra- cene,"' 1,2-benzanthracene,"' 3 ,4-benzpyrene ,"' and pyrene!04 The high-melting polynuclear hydrocarbons react best in the presence of a solvent, such a s o-dichlorobenzene. For example, a solution of anthra- cene, methyl formanilide, and phosphorus oxychloride in o-dichlorobenzene i s heated 1 hour a t 90-95O; then an aqueous solution of sodium acetate is added, and the solvent and N-methylaniline are removed by steam dis- tillation. The solid residue is readily purified to yield Panthraldehyde (84%);'' With liquid or low-melting compounds a solvent is not required.
The conversion of thiophene and i ts derivatives to the corresponding aldehydes by this procedure has been extensively studied, the yield of 2-thiophenealdehyde being 76%?60
unsym-Diarylethylenes react in a similar manner to yield unsaturated aldehydes, Ar,C= CHCHO."'
Other ca ta lys ts and reagents have been used. In the presence of aluminum chloride, 2-naphthol reacts with formamide to give 2-naphthol- l-aldehyde (45%):''
143. Formylation of Phenols with Chloroform (Reimer-Tiemann)
NaOH; C,& OH + CHCl, + o- and p-HOC6H4CH0
HCI
Substituted phenols react with chloroform and alkali in alcohol solution t o yield o- and p-hydroxybenzaldehydes. The yields are often l e s s than
I 50%, the para- isomer predominating." The procedure involves heating an alkaline ethanolic solution of the reactants for several hours, followed by acidification and isolation of the product by steam distillation or crystallization. An example i s the synthes is of 2-hydroxy-l-naphthalde- hyde (48%):'

2 8 2 ALDEHYDES ch. 9
144. Formylation of Phenols (or Amines) with Hexamine (Duff)
This reaction is readily accomplished by heating the phenolic com- pound at 150-160' for 10 to 30 minutes with a mixture of glycerol, boric acid, and hexamine. The phenolic aldehyde is liberated by acidification and steam distillation. By this general procedure, sixteen phenolic alde- hydes have been prepared. Although the yields are only 15-20%, the method requires little time and furnishes a reasonably pure product which is the ortho isomerg6 (cf. method 143).
The method has been extended to the formation of pdialkylamino- benzaldehydes in 3 5 4 5 % yields?g
145. Hydroformylation of Unsaturated Compounds
Pressure RCH= CH, + CO + H, - RCH,CH,CHO Catalyst
Addition of carbon monoxide and hydrogen to an alkene linkage in the presence of cobalt catalysts gives aldehydes in an average yield of
The reactions may be carried out in the usual hydrogenation apparatus. The poisonous properties of carbon monoxide and cobalt carbonyls call for considerable care. Compounds made by hydroformyla- tion include cyclopentanealdehyde from cyclopentene (65%), P-carbethoxy- propionaldehyde from ethyl acrylate (74%), and ethyl P-formylbutyrate from ethyl crotonate (71%).
146. Formylation of Ketones with Formic Esters
Acylation of ketones having reactive methylene groups by higher esters has been shown to be an excellent method for preparing Pdiketones (method 203). If the acylating ester i s an alkyl formate, then a keto aldehyde is formed (50-80%).'71-174 The formylation is simply brought about by adding sodium metal to a mixture of the ketone and ester in anhydrous ether. Oftentimes, the product is isolated a s the sodium sal t of the hydroxymethylene form. The point of attack is unpredictable in unsymmetrical ketones, CH3COCH,R.'73s'74
147. Interaction of Halomethyl Compounds and Hexamine (Sommelet)
(C% bN4 ArCH,X - [A~CH, (C& &N,]+c~- H'9 ArCHO
METHODS 147-149 2 8 3
Substituted benzyl halides react with hexamine in boiling alcohol to form addition compounds which decompose on heating with water to give aldehydes:5'" An excellent discussion of the reaction has been pre- sented, and improvements in the conditions have been made?4' Aqueous acetic acid ( l : l ) i s recommended a s solvent for the entire process, and there is no need to isolate the intermediate salt . The procedure is il- lustrated by the synthesis of l-naphthaldehyde (82%):'' In other in- stances, the addition compound is first prepared in chloroform solution, isolated, and then decomposed with water or dilute acetic acid, as in the synthesis of 2-thiophenaldehyde (53%):'
The reaction is applicable to the formation of m- and p-dialdehydes, but not the ortho isomer, from the bis-(chl~rometh~l>benzenes,"~ a s well a s aldehyde esters, e.g., p-carbomethoxybenzaldehyde~' and halo alde- hydes, e.g., l-bromo-2-na~hthaldeh~de:~
A somewhat similar reaction is the conversion of substituted benzyl- amines to the corresponding benzaldehydes by treating their formaldehyde condensation product with hexamine .g7
148. Interaction of Benzyl Halides and Sodium 2-Propanenitronate
ArCH,Br + [(cH,),cNO,]- ~ a + + ArCHO + (CH,XC= NOH + NaBr
A general procedure for the conversion of p-substituted benzyl halides to the corresponding benzaldehydes consists in treating the halide with sodium 2-propanenitronate suspended in absolute ethanol. The resulting instable nitronic ester breaks down into acetoxime and the carbonyl com- pound. The yields are in the range 68-77% for benzaldehydes having a methyl, bromo, carbomethoxyl, cyano, or trifluoromethyl group in the para position. However, p-nitrobenzyl chloride undergoes C-alkylat ion to fur- nish the stable substituted nitropropane, ~-No,C~H,C&C(CH,XNO,?~' The reaction has been extended to the synthesis of o-tolualdehyde (73%
149. Decomposition of Arylsulfonohydrazides
C;I~SO cl Ne2COs ArCONHNH, 2 ArCONHNHS02C6HS + ArCHO
Aromatic and heterocyclic aldehydes have been prepared from hydra- zides, via the arylsulfonyl derivative, in 50-65%'23 and 20-40% yields,'24 respectively; the method fails in the aliphatic series. The hydrazide is treated with benzenesulfonyl chloride in pyridine, and the subsequent product i s isolated by precipitation with water and decomposed by heat- ing with sodium carbonate in ethylene glycol or glycerol a t 160'.

2 8 4 ALDEHYDES ch. 9
Benzhydrazides in small quantities have been oxidized t o the alde- hydes with potassium ferricyanide in excess ammonium hydroxide (30- 60%).'27
150. Cleavage of Schiff Bases
ArCH=NR + H,O -+ ArCHO + RNH,
Several preparations of aldehydes have been developed that involve the formation and cleavage of Schiff bases. The condensation of ani- lines or phenols with formaldehyde and p-nitrosodimethylaniline leads to such intermediates. These substances can be isolated and converted by an exchange reaction with formaldehyde in acetic acid to the cor- responding aldehydes. p-Dimethylaminobenzaldehyde is made in this manner in 5% yield.'"
When a methyl group on an aromatic nucleus is activated by a nitro group in the ortho or para position, condensation with nitrosobenzenes can occur to give a Schiff base; subsequent hydrolysis furnishes the aldehyde. An example is the synthesis of 2,4dinitrobenzaldehyde (32%).'86
(NO, XC6H3CH0
Condensation of diethylaniline and formaldehyde in the presence of sulfanilic acid gives the structure
which can be isolated and oxidized with potassium dichromate to the benzylidene compound; the latter on alkaline hydrolysis gives p-diethyl- aminobenzaldehyde in 50% yield.'88
h i n o chlorides, which are readily prepared by the action of phosphorus pentachloride on anilides, are reduced by anhydrous stannous chloride to imino intermediates which on hydrolysis yield aromatic aldehydes (50- 7%); applications in the aliphatic series are poorly d e ~ c r i b e d . " ~ " ~
METHODS 150-1 52
C1 H
h most cases , the crude imino chloride i s treated directly by adding it to a solution of stannous chloride saturated with dry hydrogen chloride; the aldehyde is then liberated by steam distillation. The procedure i s illustrated by the synthesis of o-tolualdehyde (70%).'" h i n o chlorides have a lso been prepared by treatment of ketoximes with phosphorus penta- chloride, viz., RR'C= NOH -+ RCCI= NR', in preparations of benzalde- hyde and p-chlorobenzaldehyde (70-85%):' As in the Stephen reaction (method 164), groups ortho t o the irnino chloride group hinder the reaction.
Schiff bases from other sources furnish aldehydes (methods 166 and 170).
151. Hydrolysis of gem-Dihalides - ArCHO ArCH, 5 ArCHX, H'0
Toluenes substituted with chloro, bromo, fluoro, or cyano groups can be dichlorinated or dibrominated and the resulting benzal halides hydro- lyzed directly to the corresponding aldehydes in the presence of calcium carbonate or sulfuric acid (50-70%).'35v'36 o- and p-Xylene have been converted to the corresponding dialdehydes.''9"40 In the halogenation of certain cresols, the carbonate or acetate esters are used in order to pre- vent nuclear ha l~~ena t ion . '~"
Aliphatic gem-dihalides require more vigorous conditions for hydroly- s i s than do the benzal halides. Examples are found in the treatment of certain l ,l-dichloroalkanes , like 1 ,ldichloro-3-methylbutane and 1,l- dichloro-3,3dimethylbutane, with water and, in some cases , magnesium oxide for 4 hours a t 200-300'. The aldehydes are formed in 6 0 4 % yields (cf. method 222).
152. Interaction of Pyridinium Salts and pNitrosodimethylaniline
P yridine ArCf&CKH,Cl - [A~CH,COCH,~,H,IB; I (CHlhNC6bNO
H+ ArCH,COCHO t-- ArCH,COCM = E!OC6H4N(CH, 1,
H, 0
Compounds containing reactive halogens (ArCH-CHCf&X or ArCOCH,X) readily form pyridinium salts. Rearrangement of these prod-

286 ALDEHYDES ch. 9
ucts with p-nitrosodimethylaniline to a nitrone followed by hydrolysis with acid gives a,P-unsaturated aldehydes or substituted g l y o ~ a l s . ' ~ ~ Substituted benzyl halides, ArCH'X, undergo the ser ies of reactions to give the corresponding aldehydes, ArCHO. Terephthaldehyde i s made in this way in a 70% over-all yield.''P
153. Hydrolysis of 2-Alkoxy-3,4dihydro-1-2-pyrans
Hydrolysis of 2-alkoxy-3,4-dihydro-1,2-pyrans with dilute hydrochloric acid furnishes a convenient synthesis of glutaraldehyde (R= H) and other 1,5-dicarbonyl compounds. The starting materials are obtained by the 1,4addit ion of vinyl ethers t o a,P-unsaturated carbonyl compounds. The wide selection of diene systems includes acrolein, crotonaldehyde, meth- acrolein, cinnamaldehyde, P-furylacrolein, methyl vinyl ketone, benzal- acetone, and benzalacetophenone. Ethyl vinyl ether is preferred a s the dienophile. The yields in the cyc lka t ion s t ep are in the range of 2 5 4 7 % and in the subsequent hydrolysis s tep , 57-85%?6s
154. Hydrolysis of Aldehyde Derivatives
RCH-NOH + C,H5CH0 -4 RCHO + C,H,CH= NOH
Oftentimes, aldehydes are isolated and purified a s their derivatives, and their regeneration i s then of importance (cf. method 195). The fission of the oxime, semicarbatone, hydrazone, etc., may be accomplished by acid hydrolysis or by an exchange of the nitrogenous moiety with another carbonyl compound, such a s benzaldehyde, for which i t has a greater affinity.
Semicarbazones of volatile aldehydes may be hydrolyzed by steam dis- tillation in the presence of phthalic anhydride.''' A synthetic route for aromatic aldehydes involves the hydrolysis of semicarbazones which have been prepared by the interaction of dithio ac ids and semicarbazide hydro- chloride in pyridine solution."* "
ArCSSH - %NNHCONH2 ArCH = NNHCONH, % ArCHO
METHODS 154-1 55 287
The hydrolysis of succinaldehyde dioxime must be carried out with care because of the instability of the dialdehyde. This s t ep has been accomplished in 60% yield by treating the dioxime with ethyl nitrite in dioxane or with sodium nitrite in dilute sulfuric acid."'
The adducts formed from amine bisulfites and aldehydes are readily purified by crystallization from organic solvents and, like the sodium bi- sulfite addition products, are readily decomposed by the action of dilute acids .'"
Acetals are readily hydrolyzed by dilute mineral acids; however, the yields are not always satisfactory. These subs tances are not affected by alkaline reagents. The sensit ive dl-glyceraldehyde aceta l i s con- verted to i t s aldehyde in 80% yield by the action of dilute sulfuric acid under mild conditions?" Other procedures are illustrated by the treat- ment of acetals which are formed by the interaction of Grignard reagents and orthoformic es ters (method 165).
Olefinic aldehydes have been prepared by bromination of the diethyl- aceml derivatives followed by dehydrobromination (cf. Acetals and Ketals); the unsaturated aldehydes are readily liberated by mild acid treatment of their a ~ e t a l s . ~ Alkoxy aldehydes have a l s o been synthesized through aceta l intermediates, which in turn are prepared from sodium alkoxides and bromoacetals."'
a-Hydmxy aldehydes have been prepared by hydrolysis of the oximes resulting from the action of Grignard reagents on certain isonitroso ketones ."'
155. Oxidation o f Aromatic Side Chains
Cr03 ArCH, - ArCH(OCOCH3), % ArCHO
(CH,COhO %"
Oxidation of the methyl group in substituted toluenes with chromium trioxide in ace t ic anhydride forms crystalline diacetates, which are stable t o further oxidation. These compounds are readily hydrolyzed in acid solution t o the corresponding aldehydes (40-50% over-all).'*' "' The procedure i s generally applicable t o the preparation of benzaldehydes carrying nitro, halo, and cyano substituents.
Other oxidative procedures have been described. The heterogeneous liquid-phase oxidation of toluene with manganese dioxide in 65% sulfuric acid is important in the production of benzaldehyde and salicylaldehyde. An example of i t s application in the laboratory i s found in the preparation of 3,5-dimethylbenzaldehyde (48%) from m e ~ i t ~ l e n e . " ~ In a comparison

288 ALDEHYDES Ch. 9
of other oxidants, chromyl chloride i s outstanding; however, it must be employed with care. The hydrocarbon is added slowly to a chloroform solution of this reagent, and the addition complex i s carefully decomposed with dilute sulfurous acid to give the aldehyde. Yields range up to 80% (Etard reaction)?" The internal oxidation-reduction of nitrotoluenes can be brought about by refluxing with alkaline sodium polysulfide, e.g., p- aminobenzaldehyde from p-nitrotoluene (75%).'56
Benzyl halides have been oxidized directly with selenium dioxide9' or copper nitrate?'
156. Oxidation of Olefinic Compounds
RCH=CH, -% RCHO + CH'O 4 0
Aldehydes result from the decomposition of certain ozonides. The technique is similar to that used for the preparation of ketones (method 182). High yields are obtained by catalytic hydrogenation of the o ~ o n i d e s . " ~ This s t ep coupled with Grignard and dehydration reactions has been used a s a procedure for the degradation of an aldehyde to i ts next lower homolog, viz.,
C6YMa' - 4 0 RCH'CHO ----a RCH,CHOHC,H, + RCH= CHC6H, 2 RCHO
Dialdehydes result when cyclic olefins are ozonized!'~mproved directions for the ozonolysis of unsaturated esters in glacial acetic acid to yield aldehyde esters have been given!'6 The same procedure i s ap- plied to the preparation of aliphatic aldehydes containing halo,"7 hydroxyl:18 and ether groups.'''
Oxidation of olefinic side chains with ozone to form aromatic aldehydes gives erratic results and therefore other oxidants are employed!'0 For this purpose, the most widely used oxidant i s nitrobenzene in dilute alkali; the mixture is allowed to react a t moderate temperatures for several hours. Thus, hydroxy benzaldehydes may be obtained from propenyl- phenols, which in turn are readily prepared by the Claisen rearrangement of Oalkyl ethers (method 100). Sodium dichromate in the presence of sulfanilic acid, which removes the aldehyde a s i t is formed, gives yields a s high a s 86% in the oxidation of isoeugenol and i s ~ s a f r o l e . ' ~ ~
157. Oxidation of Methyl Ketones by Selenium Dioxide
METHODS 157-1 58 289
The preparation of certain substituted benzils by treatment of aryl benzyl ketones with selenium dioxide is discussed later (method 183). If a methyl ketone is treated under these conditions, the methyl group i s oxidized t o an aldehyde group.'76 The reaction is carried out by reflwing a mixture of selenium dioxide and ketone in dioxane or alcohol for several hours. Preparative details are found in the procedures for phenylglyoxal (72%)'77 and glyoxal (74%)t7' the latter i s isolated a s its bisulfite derivative.
4-Methylquinoline and l-methylisoquinoline, which have reactive methyl groups, are converted to quinoline-4aldehyde (61%) and isoquinaldehyde (42%), respectively, by means of this ~ e a ~ e n t ! ' ~ " ~
158. Oxidation of Primary Alcohols
R C h O H 'O'- RCHO
Controlled oxidation of a primary alcohol with a mixture of sulfuric and chromic acids gives the corresponding aldehyde. In the preparation of low-molecular-weight aldehydes, an aqueous medium i s used and the product is removed by steam distillation, thus preventing further oxida- tion. This procedure is well illustrated by the preparation of propion- aldehyde (49%)' and isovaleraldehyde (60%): Certain benzyl alcohols are dissolved in aqueous acetic acid for chromic acid ~ x i d a t i o n . ~ OIe- /inic aldehydes are produced by a rapid low-temperature (5-20') oxidative procedure, a s illustrated by the preparation of 2-heptenal (75%) from 2- heptenol!O Aldehyde etbers such a s methoxyacetaldehyde and ethoxy- acetaldehyde have been prepared by the chromic acid oxidation of the corresponding alcohols in 17% and 10% yields, respectively."
Aldehydes have been formed from alcohols by the use of other oxidizing agents. Dihydroxyacetone has been oxidized with excess cupric acetate to hydroxypyruvic aldehyde in 87% yield.'' p-Cyanobenzyl alcohol treated a t 0' with a chloroform solution of nitrogen tetroxide gives practically pure p-cyanobenzaldehyde Aromatic alcohols containing nitro groups have been oxidized to the corresponding nitro aldehydes with con- centrated nitric acid, e.g., o- and p-nitrobenzaldehydes (80-85%)!4 m-Nitrobenzenesulfonic acid in basic media has been used for the oxida- tion of substituted benzyl alcohols, most satisfactorily for the water- soluble phenolic benzyl alcohol^?'^ Selenium dioxide, or less effectively tellurium dioxide, oxidizes benzyl alcohol slowly to benzaldehyde."'
The Oppenauer reaction has been applied in the conversion of aliphatic and aromatic alcohol^.'^^ The alcohol, a high-boiling aldehyde (such a s cinnamaldehyde), and aluminum alkoxide catalyst are heated, and the volatile aldehyde is removed a s i t is formed.

ALDEHYDES Ch. 9
Alumlnum RCH,OH + R'CHO ---+ RCHOT + R'CH,OH
alkoxlde
In this manner, benzaldehyde and n-butyraldehyde have been obtained in 95% and 72% yields, r e s p e c t i ~ e l y . ' ~ This procedure is employed more extensively in the preparation of ketones (method 180).
159. Dehydrogenation of Primary Alcohols
Catalyat RCH,OH - RCHO + H,
Catalytic dehydrogenation of primary alcohols in the vapor phase has been studied in detail?'6 Formerly, a copper catalyst '2 was used; how- ever, it has been found that this catalyst i s eas i ly poisoned.3s A copper chromite catalyst a t 300-34r0 and atmospheric pressure gives improved and consistent yields (50-70%) and retains its activity over long periods .m. 34. '8 Side reactions, such a s dehydration, condensation, and
es ter formation, do.not occur appreciably under these conditions.'' Preparation of the catalyst and the apparatus have been de~c r ibed . '~ '~ ' "
Catalytic dehydrogenation of alcohols has been conducted with yields a s high a s 90% by passing the vapor mixed with air over si lver or copper- sdve r catalysts.'11 lsS1 "' A three-step synthesis of DL-glyceraldehyde from glycerol cons is ts in protecting two of the hydroxyl groups by ketal formation with acetone, followed by air oxidation over a si lver catalyst and then hydrolysis of the ketal (53% ove ra l l yield)?" Methacrolein, H,C=C(CH,)CHO, is made by the air oxidation of methallyl alcohol (95%):" A laboratory-scale model for the air oxidation of teuahydro- furfuryl alcohol over a silver gauze catalyst has been described."'
Liquid-phase dehydrogenation is carried out under a pressure of ethyl- ene, which serves a s a hydrogen acceptor."
Ethoxyacetaldeh yde, an aldehyde ether, is readily prepared in 35% yield from Cellosolve by the vapor-phase dehydrogenation technique.'6
Similar techniques are employed for the catalytic dehydrogenation of secondary alcohols (method 181).
160. Oxidative Cleavage of Glycols
HI0 or R C H O H C H O H R ' . ~ RCHO + R'CHO
P ~ ( O O C C H S ) ~
Certain P-amino alcohols and glycols and their dehydroderivatives, i.e., a-ketols, a-ketals , and diketones, are readily oxidized with periodic acid or lead tetraacetate to aldehydes. A review of the method has been made ."'
METHODS 160-162 29 1
The reactions are usually carried out at a moderate temperature, using water as the solvent for periodic acid and organic solvents for lead tetra- acetate; however, both reagents can b e used in aqueous solvents. Addi- tion of the oxidizing reagent t o the glycol instead of the reverse gives an improved yield.'60 The yields are high, and the method has found exten- s ive application in both analytical and preparative procedures. It has been applied in the preparation of aldehydes containing a double bond or hydroxyl, carboxyl, ester , or ether groups.'47"4"16s Oxidation of 1,2- cyclohexanediols with lead tetraacetate leads t o substituted adipic alde- hydes in 68% yields?4s
Several small-scale synthetic routes for obtaining intermediates for cleavage to aldehydes by lead tetraacetate have been proposed.'4''146
CHl- CHCYBr (a) R M ~ X - RCH,CH= CH, BIZ;. RCH,CHOHCH,OH 9
80% KOAc
CH& HOAc H + ( b ) RCOCI h RCOCHN, ----+ RCOCH,OAc -+
9Wo 90% 80%
RCOCH'OH 2 RCHO
161. Selective Reduction of Olefinic Aldehydes
RCH= CHCHO RC&CH,CHO Catalyst
i
Aldehydes may be prepared by se.lective hydrogenation of substituted acroleins in much the same manner a s the selective reduction of unsatu- rated ketones (method 196); however, there are few examples adequately described.g'-9sv "W 236
162. Reduction of Acyl Chlorides (Rosenmund)
RCOCl H' RCHO + HCI Catalyst
Selective catalytic hydrogenation of an acyl chloride to an aldehyde can be accomplished with varying yields; the method has been reviewed." The preferred catalyst i s palladium suspended on bariwn sulfate. The reaction may be carried out in the liquid phase by bubbling hydrogen. through a hot solution of the acyl chloride in xylene or teual in in which

292 ALDEHYDES (3.9
the catalyst is suspended, or in the vapor phase by passing the acyl chloride over palladinized asbes tos a t about 200°.64 In the former pro- cedure, the reduction has been arrested a t the aldehyde s tage by careful control of the temperature6' (lowest point a t which hydrogen chloride i s evolved) or by use of a catalyst "regulator" which inactivates the cata- lyst for reduction of the aldehyde. Typical reductions with and without catalyst poisons are found in the preparation of P -naph tha~deh~de (81%)s6 and 2,4,6-uimethylbenzaldehyde (80%):' respectively. The reaction i s applicable to acyl chlorides carrying halogen, nitro, or e s t e r groups,6s' 67' "' and even a double bond although this may migrate during the reacti0n.6~ Hydroxyl groups should be protected by acetylation.
Phosphorus- or sulfur-containing compounds formed in the preparation of the acyl chlorides hinder the reaction and therefore must be removed?"
163. Reduction of Thiol Es ters
The reduction of a carboxyl group to an aldehyde group can be effected by a reductive desulfurization of the thiol es ter with Raney nickel. The thiol esters are prepared by the reaction of the acyl chloride with an ex- c e s s of ethyl mercaptan in pyridine or by reaction with lead mercaptide in dry ether. The hydrogenolysis i s then carried out by refluxing an ethanolic solution of the thiol es ter with Raney nickel for 6 hours. By this new synthesis, propionaldehyde and benzaldehyde have been prepared in 73% and 62% yields, respectively .'60
164. Reduction of Nitriles (Stephen)
HCI SnCll RCN - RC(Cl)== NH. HCI 4 (RCH=NHXSnCl, 3 RCHO
HC I
Nitriles may be converted to their imino chloride sa l t s by the action of dry hydrogen chloride in ether. These intermediates are reduced by an- hydrous stannous chloride to stannic aldimoniurn chlorides, which on hydrolysis yield aldehydes. Chloroform may be added to facilitate the solution of the nitrile. The quality of the stannous chloride catalyst is important; the preparation of an active and dependable form has been described." The yields are usually high for many aromatic nitriles, a s in the preparation of P-naphthaldehyde (95%).'9 The reaction has a l so beer. employed in the heterocyclic ser ies , a s in the synthesis of 4-methyl- thiazole-5aldehyde (40%):' The reduction of the cyano group in the
METHODS 164-166 293
presence of an ester group leads to a n aldehyde ester, e.g., methyl cyano- benzoate t o methyl p-formylbenzoate (90%)."
However, it has been shown that the method may not be a s general a s originally supposed, especial ly in the preparation of the aliphatic alde- hydes:01S2's' Also, groups orfho to the nitrile group hinder the reaction.
Instead of reducing the imino chloride with stannous chloride, a s indi- cated above, sodium amalgam may be used in the presence of phenyl- hydrazine. The resulting phenylhydrazone is then hydrolyzed.'4
165. Interaction of Grignard Reagents and Orthoformic Es ters
The reaction of ethyl orthoformate and Grignard reagents gives ace ta ls which are hydrolyzed readily by dilute acid to aldehydes. This method has been employed extensively for the preparation of aliphatic and aro- matic aldehydes. A study of the optimum conditions has been made, using the conversion of bromobenzene to benzaldehyde a s a model synthesis (90%)."*" Comparative studies of various aldehyde syntheses that em- ploy Grignard reagents (methods 154, 166, and 167) show that this one is the most practicalf6*" however, the possibility of a sudden exothermic reaction limits the s i ze of the run. Longer reaction times a t room or re- flux temperature help overcome this d i f f i c ~ l t ~ ! ~ " ~ Examples of the better preparative procedures are found in those for n-hexaldehyde (5 0%)," p-tolualdehyde (7%),'=and phenanthrene-Faldehyde (42%)?24
N,N-Dialkylf~rmamide'~"~ or ethyl formatea0 and Grignard reagents have been used with some success ; however, the former reaction is complicated and frequently produces tertiary amines a s the chief product, and the lat- teh forms secondary alcohols by further reaction of the aldehyde. Sub- stituted benzaldehydes have been prepared from aryllithium compounds and h'-methylformanilide in good yields!aa
166. Interaction of Grignard Reagents and Ethoxymethyleneaniline
Aromatic Grignard reagents react smoothly with ethoxymethyleneaniline to give imines which are easi ly hydrolyzed to aldehydes. The reaction is e a s y to cary out, i s adaptable to large-scale preparations, and gives high yields (65-82%)." Its use i s limited by the availability of the ethoxymethyleneaniline, which may be prepared in a pure condition from the dry silver s a l t of formanilide and ethyl iodide.

294 ALDEHYDES
167. Decomposition of Glycol Monoalkyl Ethers
A large number of symmetrical diaryl- or dialkyl-acetaldehydes, diffi- cult to obtain by other means, have been prepared by the reaction of ethyl ethoxyacetate, or ethyl phenoxyacetate, with Grignard reagents followed by treatment of the resulting glycol monoalkyl ether with anhydrous oxalic acid or dilute sulfuric acid.'9 The yield in the first s tep is 40-80%, and the yield in the subsequent transformation is 5G80%.
Unsymmetrical dialkylacetaldehydes may be obtained by s tar t ing with an a-keto ether?'
By this procedure, 2-(a-naphthy1)-propionaldehyde has been obtained in a 74% yield?'
The method has been further studied in i t s application for the syn- thesis of ketones (method 202).
168. Thermal Decomposition of Acids
RCOOH + HCOOH % RCHO + CO2 + H20
The old method of heating the calcium sa l t s of formic and a second carboxylic acid for aldehyde formation has been modified by the use of a catalytic decomposition technique. By this scheme, the acid vapors are passed over thorium oxide, titanium oxide, or magnesium oxide a t 300°;2'3 or the acids are heated under pressure a t 260' in the presence of titanium dioxide?I4 In the latter procedure, non-volatile ac ids can b e used. With aliphatic acids over titanium oxide, reaction occurs only when more than seven carbon atoms are present, the yields increasing with increase in the molecular weight (78-90%). Aromatic ac ids having halo and phenolic groups are converted in high yields to aldehydes, e.g., salicylaldehyde (92%) and p-chlorobenzaldehyde (83%). Preparation of a thorium oxide catalyst has been describeda6' (cf. method 186).
169. Decomposition of a-Hydroxy Acids
RCHOHC0,H RCHO + CO + HaO
METHODS 169-171 ' 295
High-molecularweight aliphatic aldehydes have been made by the dis- tillation of a-hydroxy acids, which are prepared by the hydrolysis of the corresponding a-bromo acids. The reaction is carried out under dimin- ished pressure or in an atmosphere of carbon dioxide. Details for the procedure are found in the preparation of octanal (57%)43 and undecanal
Preparation of the a-bromo acid and i t s subsequent hydrolysis are a l s o described. A later modification has been the distillation of the a-methoxy acid in the presence of copper. This procedure gives an almost quantitative yield in the preparation of hep tade~ana l .~ '
Aldehydes have a l s o been prepared from a-hydroxy acids by oxidation with lead tetraacetate in glacial acetic acid, e .g ., tridecanal (55%) and pentadecanal (58%)46 (cf. method 160).
170. Decarboxylation of a-Keto Acids
C6Y NH R C K O O H 3 RCH==NC6H, 3 RCHO Heat
a-Keto ac ids are readily decomposed to aldehydes and carbon dioxide. The decarboxylation may be brought about by heating the a-keto acid or i t s arylimino derivative. By the latter procedure, a solution of the keto acid in aniline i s boiled, which causes the formation of water, carbon dioxide, and a Schiff base, RCH=NC6H,; hydrolysis of this product gives the aldehyde.164 Oftentimes, decarboxylation is accomplished in higher yields by heating the glyoxylic acid in N,N-dimethyl-p-toluidine at 170°170 or in diphenylamine a t 150-200°.a56
Another modification i s the decomposition of the bisulfite-addition compound of the keto acid a s illustrated by the synthesis of phthalalde- hydic acid (41%).'66
171. Decarboxylation of Glycidic Acids
Aromatic and aliphatic aidehydes have been prepared in good yields by the decarboxylation and isomeritation of the corresponding glycidic acids. Es ters of the latter are obtained by treating a ketone with ethyl chloro- acetate in the presence of sodium amide (method 127). The glycidic es ters a re first converted to the sodium sa l t s with sodium ethoxide and then treated with aqueous hydrochloric acid under gentle reflux. By this procedure, a-phenylpropionaldehyde has been prepared from acetophenone in an over-all yield of 3 8 % y 7 Other detai ls have been d i s c ~ s s e d . ' ~ '

2 9 6 ' ALDEHYDES ch. 9
A similar route i s the formation and isomerization of substituted ethyl- ene oxides."' This synthesis has been carried out without isolating the intermediates 1'
R 'MgX RCOCH,CI - RR'COHCH,Cl % HC? RR'CHCHO
172. Hydrolysis of Olefin Dibromides "'
(CH,hCOH BI'- (CH,XCBrCH,Br 2 (CH,XCHCHO
Over-ell yield 75%
173. Degradation of Acid Amides and Azides
(a) U-Brorno AzidesUa (cf. method 220).
Heat H 0 RCHBrCON, 4 RCHBrNCO 2 (RCHBrNH,) RCHO
(b) Monosubstituted Malonyl Azides
C a b OH RCH,CH(CON,)-, - RCH,CH(NHGO,C,HS X H'q RCH,CHO
NeOCl RCH= CHCONH, + RCH= CHNHCO,CH, H'O- RCH,CHO
CH30H
174. Acid Treatment of Primary Acinitroparaffinsl"
NaOH H + RCH,NO, 4 RCH= NONa - RCHO
3. """"" 0
R=methyl, ethyl, isopropyl, and n-butyl.
175. Isomerization of Unsaturated Alcohols
METHODS 176-177
176. Condensation of Aromatic Hydrocarbons with C h l ~ r a l " ~ " ~ '
ArCH, + C1,CCfIO 5 ArCH,CHOHCCI, (% A r c h C H 0
177. Formylation of Acetylenes 'lb "'
(a C,H,C=CNa + HC0,R - C 6 H S C ~ C -CH0 18%
-zoo (b) CH,(CH,);C~ C N ~ + HCO,R ---+ CH,(CH, hC=C:-CfiO
24%
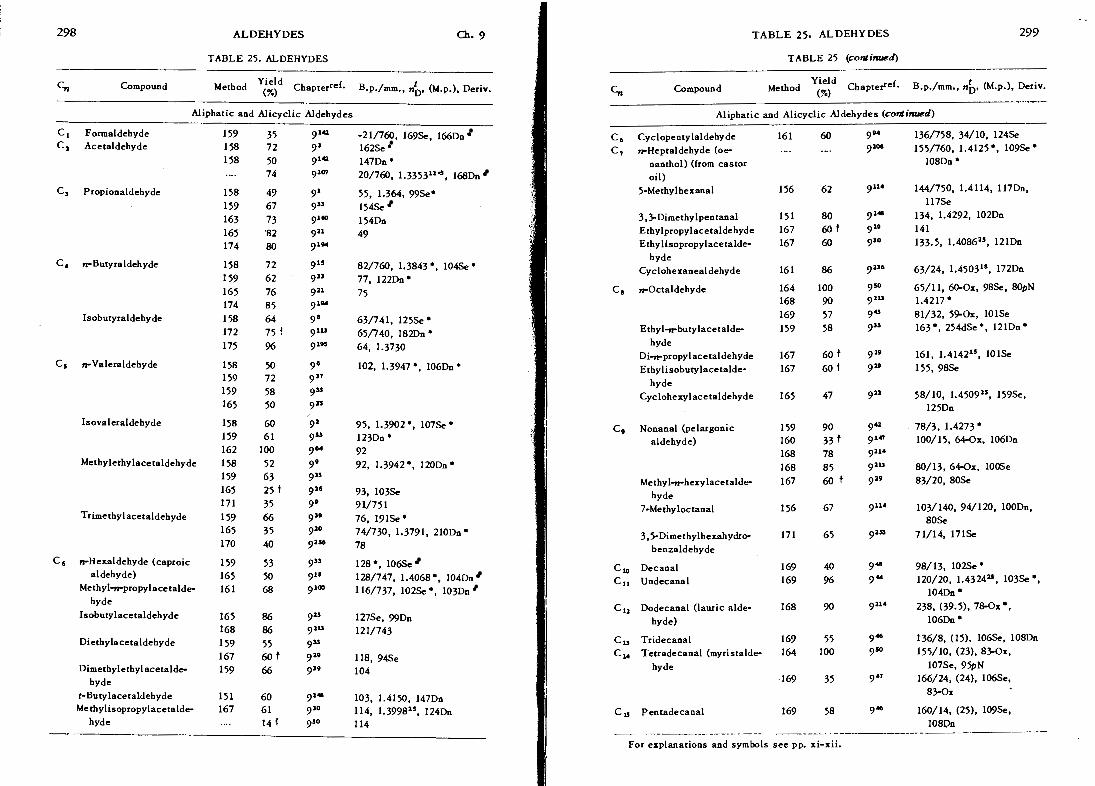
I 298 ALDEHYDES fi. 9
TABLE 25. ALDEHYDES
c, Compound Method chapterref. B.p./mm., nb, (h4.p.). Deriv.
Aliphatic and Alicyclic Aldehydes
C 1 Formaldehyde 159 35 914 -21/760, 169%. 166Dn Cl Acetaldehyde 158 72 9' 162Se
158 50 914 147Dna .... 74 914 20/760, 1.335311", l68Dn
C, Propionaldehyde 158 49 9I 55, 1.364, 99Se0
159 67 9" 154% 163 73 9lW 154Dn 165 '82 911 49 174 80 9l"
Isobutyraldehy de
C5 n-Valeraldehyde
Isovaleraldehyde 158 60 9'
159 61 9- 162 100 9"
Methylethylacetaldehyde 158 52 9,
159 63 913 165 25 i 916 171 35 9,
Trimeth~lacetaldeh~de 159 66 9=
165 35 9'O 170 40 gas
C, n-Hexaldehyde (caproic 159 53 9" 128 *, 106Se aldehyde) 165 50 91, 128/747, 1.4068 *, 104Dn
Methyl-n-propylacetalde 161 68 91m 116/737, 102% *, 103Dn hyde
Isobutylacetaldehyde 165 86 9= 127Se, 99Dn 168 86 9lU 121/743
Diethylacetaldehyde 159 55 933 167 60 t 919 118, 94Se
Dimethylethylacetaldc 159 66 939 104 hyde
t-Butylacetaldehyde 151 60 9" 103, 1.4150, 147Dn Methylisopropylacetalde 167 61 9'O 114, 1.3998". 124Dn
hyde . .. . 141 9'O 114
TABLE 25. ALDEHYDES
TABLE 25 (continued)
Yield c, Compound Method
chaptefief. B.p./mm., nb, (MP.). Deriv. (%)
Aliphatic and Alicyclic Aldehydes (continued)
C6 Cyclopentylaldehyde 161 60 9" C, ~Heptaldehyde ( o e . .. . . .. . 9lW
nanthol) (from castor oil)
5-Methylhexanal 156 62 9114
3,3-Dimethylpentanal 151 80 gra Ethylpropylacetaldehyde 167 60 t 919 Ethylisopropylacetalde- 167 60 9'O
hyde Cyclohexanealdehyde 161 86 9136
C, n-Octaldehyde 164 100 9" 168 90 9lU 169 57 9U
Ethyl-trbutylacetalde 159 58 9- hyde
Di-trpropylacetaldehyde 167 60 t 919 Ethylisobutylacetalde- 167 60 t 9"
hyde Cyclohexylacetaldehyde 165 47 9
C, Nonanal (pelargonic 159 90 9 ' = aldehyde) 160 33 t 9147
168 78 9114 168 85 91U
Methylrrhexylacetalde 167 60 ? 919 hyde
7-Methyloctanal 156 67 g114
3,FDimethylhexahydro- 171 65 9'" benzaldehyde
C, Decanal C,, Undecanal
C,, Dodecanal (lauric alde- 168 90 9214 hyde)
C, Tridecanal 169 55 9& C14 Tetradecanal (rnyristalde 164 100 950
hyde -169 35 9 47
C, Pentadecanal 169 58 9&
-- - - For explanations and symbols see pp. xi-xii.

T A B L E 25. A L D E H Y D E S
TABLE 25 ( C m f i ? ~ ~ e d )
300 ALDEHYDES Ch. 9
TABLE 25 (continued)
c, Compound Yield
Method ( %)
chapterref. B.p./mm., nb. (M.p.), Deriv.
Aliphatic and Alicyclic Aldehydes (continued)
C Hexadecanal (palmitalde 164 100 9" (34). 8 8 0 x , 107Se. 97pN hyde 169 47 9 47 202/29, (34). 107Se. &Ox
C,, Heptadecanal (margaric 160 80 9l' (63) aldehyde) 169 52 947 204/26, (36), 108Se. 90-Ox
C, Octadecanal ( s t ea ra lde 164 100 9" (381, 89-Ox, 109Se. lOlpN hyde
Aromatic Aldehydes
C, Benzaldehyde 147 70 9 148 73 9'" 64/13, 1.5446, 235Dn 149 73 91U 222Se 150 85 9- 88/40,158Phe 151 70 9Ia 179 155 44 9 a15 158 95 9 lS 162 96 9" 163 62 9'- 235Dn 165 89 9 168 93 9 a14
.... 97 9"
C, Phenylacetaldehyde 160 72, 9'- 84/14, 9 7 - 0 ~ 162 80 ' \ 9 = 156Se 164 33 9" 165 58 9'l 195. 99-Ox 171 50 9'" 95/22, 121Dne 173 75 9'" 82/12, 58Phe 147 70 9" 88/19, l l l P h 148 73 9'" 72/6, 1.5430a5, 193Dn8 150 70 9 93/19, 101Ph 155 65 9115
165 73 9" 166 8 1 917 155 60 9'" 84Phe 164 50 9-O 198/756, 2 12Dn 140 51 9" 205 140 65 9 74 114Ph 148 70 9" 72/6, 1.5420, 234Se 149 60 9"' 198pN 155 80 gals
164 77 9" 106/10, 2OOpN 165 74 9l' 166 82 9"
C, a-Phenylpropionalde 171 38 t 9'" 93/10, 76/4, 135Dn hyde
c, Yield
Compound Method (%l chapterref. B.p./mm., ntD. (M.p.), Deriv.
Aromatic Aldehydes (contirered)
C, P - ~ h e n ~ l ~ r o ~ i o n a l d e hyde
2,GDimethylbenzalde- hy de
3,5-Dimethylbenzalde hyde
C, 3-Phenyl-2-methylpropanal prrPropylbenzaldehyde p-Isopropylbenzaldehyde 2,3,GTrimethylbenzalde-
hyde 2,4,5-Trimethylbenzalde-
hyde 2,4,GTrimethylbenzalde-
hyde
1,2,3,4-Tetrahyd~o-2- naphthaldehyde
C 1, p-~Butylbenzaldehyde 2,3,5,GTetramethyl-
benzaldehyde a-Naphchaldehyde
C 1, 2,4,6-Triethylbenzalde- hyde
pPhenylbenzaldehyde crPhenylbenzaldehyde 2-(a-Naphthyl)-propion-
aldehyde l- Acenaphthaldehyde
C, Diphenylacetaldehyde 9-Formylfluorene
C, a$ -Diphenylpropion- aldehyde
P h t h r a l d e h y d e l-Phenanthraldehyde
For explanations and symbols s e e pp. xi-xii.

302 ALDEHYDES Ch. 9
TABLE 25 (continued)
c, Yield
Compound Method (X) chapterref. B.p./mm., nb, (M.P.), Deriv.
Aromatic Aldehydes (cordimped)
C U 2-Phenanthraldehyde 150 85 913" (59)*, 195-Ox* 162 70 9 m (59.5), 282%.
3-Phenanthraldehyde 150 85 9- 275Seb 162 90 9" (80), 145-Ox
9Phenanthraldehyde 150 90 9"' (101), 223Se 162 90 3" (101) 165 4 2 f 9aa4 (101)
1,2,3,CTetrahydrophe- 150 68 9- (129) nanthrene-+aldehyde
C, 2,4,GTriisopropylbenz- 140 65 9 78 126/4 aldehyde
Cl, Pyrene-3- aldehyde 142 53 9" ((126) C, 1,2-Benzanthracene-l@ 142 64 91m (148)
aldehyde C,, 3,4-Benzpyrenc5-aldehyde 142 70 9Im (203)
Heterocyclic Aldehydes
C S Furfural %Furaldehyde Tetrahydrofurfuraldehyde 2-Thiophenealdehyde
%Thenaldehyde a-Pyrrole aldehyde CMethylthiazole-5-
aldehyde
3-Methyl-2-thiophenealde-
hyde 5-Methyl-2-thiophenealdc
hyde Nicotinaldehyde
C, P-Furylpropionaldehyde C, Thianaphthene-3- aldehyde
Coumarin-3- aldehyde
TABLE 27. OLEFINIC ALDEHYDES 303
TABLE 25 ( c W i - 4
c, Yield
Compound Method (%) chapterref. B.p./mm.. nb, (M.p.), Deriv.
Heterocyclic Aldehydes (contimped)
C m Quinolinc2-aldehyde 176 50 9'- (69) QuinolincCaldehyde 157 6 1 91M (84.5). 1 8 2 - 0 ~
176 36 * 9'" 123/4, (51). 179Pi lsoquinaldaldehyde 157 42 9'" (55.5), 197Se
C, Dibenzofuran-2-aldehyde 140 81 9 77 (68), 162Ph
For explanations and symbols see pp. xi-xii.
TABLE 26. DlALDEHYDES
$I Compound Method (%) chapterref. B.p./mm., nb , (MP.), Deriv.
Cl Glyoxal C, Malonaldehyde C, Succinaldehyde
CS Glutaraldehyde C, Adipic dialdehyde
C, Phthaldehyde lsophthaldehyde Terephthalaldehyde
For explanations and symbols see pp. xi-xii.
TABLE 27. OLEFlNlC ALDEHYDES
c, Compound Method chapterref. B.p./mm., nb. (M.p.). Deriv. (%l
Aliphatic and Alicyclic Olefinic Aldehydes
C, Acrolein .... 48 9 191
.... 85 9 191
C, Methacrolein (2-Methyl-2- 159 95 9.217 propenal) 159 90 919s
C5 2-Pentenal 158 50 9 154 70 9
2-Methyl-2-butenal 36 30 231s
P -Methylcrotonaldehyde 19 40 2 a9 ---p .- . -- . - - - .- - --
For explanations and symbols see pp. xi-xii.

ALDEHYDES
TABLE 27 (continued)
Ch. 7
c, Compound Method (%) chapterref. B.p./mm., nh, (M.p.1, Deriv.
Aliphatic and Alicyclic Olefinic Aldehydes (confimred)
2-Hexenal
3-Hexenal Hexadienal a-Isopropylactolein
l-Cyclopentenylfotmalde-
hyde
2-Heptenal
l-Cyclohexenealdehyde
2-Cyclopentenylacetalde-
hyde
4-Octenal Octatrienal 2-Ethyl-2-hexenal 2-Ethyl-3-hexenal 3,GDihydrwtolualdehyde
2-Nonenal
l l-Undecenal
Aromatic and Heterocyclic Olefinic Aldehydes
C, P -Furylacrolein 36 54 2'U 95/9, (52) C, p-Formylstyrene (p-Vinyl- 27 52 2"" 93/14, 1.59601s. 131Ph
benzaldehyde C, a-Methylcinnamaldehyde 36 67 P4 124/14, 2 0 8 s a
For explanations and symbols see pp. xi-xii.
T A B L E 29. HALO ALDEHYDES 30 5
TABLE 28. ACETYLENIC ALDEHYDES
t c, Compound chapterref. B.p./mm., n ~ , Deriv. Method (7%)
C, Propargyl aldehyde 158 46 9"O 55 C 4 2-Butynal 177 28 t 9"' 105-110/755, 1.446"
136Dn
C , 2-Heptynal 177 24 9 54/13, 1.452117, 74Dn C, Phenylpropargyl aldehyde 43 70 t 3- 116/17, 1.6032's, 108-Ox
154 81 9U8 117/17,1.6032's
For explanations and symbols see pp. xi-xii.
TABLE 29. HALO ALDEHYDES
c, Compound Method chapterref. B.p./mm., ntD. (M.p.1, Deriv. ( %)
Aliphatic and Alicyclic Halo Aldehydes
Cl Ttifluotoacetaldehyde Tribromoacetaldehyde
(bromal) C, -Chloroptopionaldehyde
p ,P ,P-Ttifluoropropion- aldehyde
C 4 a-Bromoisobutyraldehyde
C, a-Bromo-n-valeraldehyde 2,f Dibromo-2-methyl-
butanal C, Btomoparacetaldehyde
2-Methyl-2,3-dichloro- pentanal
C, a-Bromoheptaldehyde l-Bromocyclohexanealde-
hyde C. '+Chlorononaldehy de
Aromatic Halo Aldehydes
For explanations and symbols s e e pp. xi-xii.

AL1)lillY I)ES
TABLE 29 (continued)
Ch. 9
p-
% Compound Method chapter 'ef - L3.p./mm.. &, (t4.p.). Deriv. (%) -
Aromatic l l a lo Aldehydes (continued) - --P
C , p-Chlorobenzaldehyde 151 6 0 9 111/25. (47) 168 89 9214
p-Bromobenzaldehyde 148 75 9'" (57). 229Se I 5 1 6 9 9*' (57) 155 51 1 914 (57) 164 6 2 9-O (57). 257Dn
p-lodobenzaldehyde 56 100 4I3O (77), 121Ph* 164 56 9I3O (77), 2 5 7 I h
C , p-Trifluoromethylbenz- 148 77 9"' 67/13, 1.4630 a ldehyde
C* a-Bromobenzylaceta lde 6 6 9 0 4 (82) hyde hydrate
C l-Bromo-2-naphthaldc 147 40 9 (118) hyde
p----
For explanat ions and symbols s e e pp. xi-xii.
TABLE 30. HYDROXY ALDEHYDES
t =,I Compound chapterref . H.p./mm., n ~ , (MP.). Deriv. Method (%)
Aliphatic Iiydroxy Aldehydes --
C, Glycolaldehyde 156 25 911' (76), 162Ph .... 25 9''' (87) . .. . 9 6 911,
C , a- t lydroxypropionalde 9 6 35 5'" 114/9, 127pN hyde
dl-Glyceraldehyde 154 80 9-' 139 159 59 1 9"' (133)
Hydroxypyruvic aldehyde 158 87 9 l2 (160). 135-OX
C , 4Hydroxybutanal 160 42 9Im 60/8. 1.4403, 1 1 8 t h
C S 5-Hydroxypentanal 99 79 5""5/3, 1.4514"
Methylethylglycolic 154 50 917s a ldehyde
>Methyl-3hydroxy- l 5 6 75 9'" 6;/13, 142pN butanal
a , a - ~ i m e t h ~ l - / 3 - h ~ d r o x ~ - 102 8 0 5Im 85/15, (97) propionaldehyde
C 6 2-Methyl-3-hydroxy- 102 8 6 5'- 86/12, 1.4373 pentanal
2-Isopropyl-3hydroxy- 102 52 5'01 84/10, 1.4603, 1261)n
T A B L E 31. A L D O E T f l E R S
TABLE 30 (continued)
c, Compound t
vethod y:;y Chapterref. B.p./mm., nD, (M.~.) ,
Aliphatic Hydroxy Aldehydes (conlinued)
C , ~e th~1-n -bu ty lg lyco l i c 89 15 t 5'- 87/35, 143Se a ldehyde 154 50 917' 88/35. 143%
C, 2,2,4-Trimethyl-3 102 .... 5Im 110/13, 1.4443 hydroxypentanal
C, PHydroxynonanal 160 23 t 9 120/0.1, (54)
Aromatic Hydroxy Aldehydes
C , Salicylaldehyde
m-Hydroxybenzaldehyde Resorcyl a ldehyde 3,4DihydroxybenzaIde-
hyde
C* Benzylglycolic aldehyde Methylphenylglycolic
a ldehyde 2-Ethyl-4-hydroxybenz-
a ldehyde
C , Ethylphenylglycolic a ldehyde
C l-Naphthol-2-aldehyde 2-Naphthol- l-aldehyde
C Diphenylglycolic a l d c
hy
For explanat ions and symbols s e e pp. xi-xii.
TABLE 31. ALDO ETHERS
Compound Yield t
Method (%) Chapter'ef B.p./mm.. nD, (M.p.), Deriv.
Aliphatic Aldo E the r s
C, Methoxyacetaldehyde 158 17 9" 92, 125Dn 160 51 9" 89,124Dn
- - ..................................... -~ .-
For explanations'and symbols s e e pp. xi-xii.

308 ALDEHYDES ch. 9
TABLE 31 (coniinued)
Aliphatic &do Ethers (corrtinucd)
C4 y - Metho~~propionaldc 121 63 6Ie hyde
Ethoxyacetaldehyde 158 10 91r 106, 117Dn 159 35 g'6 106/760 *. 1.3956 160 40 9" 91, 116Dn
C, /?-~ethoxyisobut~ralde- 121 51 6Ie 129. 1.4030aT, 102Dn hyde
rrPropoxyacetaldehyde 160 28 9 68/100. 119/748. 86Da
Cb 5-Methoxyvaleraldehyde 1% 78 9-l 59/14.5 a-Methyl-7-methoxy- 171 59 9'" 66/55, 1.4280a', 88Dn
butyraldehyde C8 2-Methyl-2,3-dimethoxy 115 85 6= 67/12. 1.419619
pentanal
Aromatic Aldo Ethers
C, Phenoxyacetaldehyde
C, crEthoxybenzaldehyde 3,bDimethoxybenzalde-
hyde (veratraldehyde)
C, 2-Ethyl-bmethoxybenz- aldehyde
3-Ethoxy4methoxy benzaldehyde
3-Methoxy4ethoxybenz- aldehyde
3,4,5-Trimethoxybenz- aldehyde
Cll 3,bDiethoxybenzaldc hyde
C, ~Phenoxybenzaldehyde 2-Ethory- haphthaldc
hyde Cu nrBenzyloxybenzaldc
I hyde
For explanations and symbols see pp. xi-xii.
REFERENCES FOR CHAPTER 9 30 9
REFERENCES FOR CHAPTER 9
'Hurd and Meinert, Org. Syntheses , Coll. Vol. 11, 541 (1943). a BouveauIt and Rousest, Bull. SOC. c h i n France, ( 3 ) 11, 300 (1894); Wey-
gand, Organic Preparations, Interscience Publishers, New York, 1945, p. 143. 'Wertheim, J. Am. Chem. Soc., 44, 2658 (1922). 4Smith e t al., J. Org. Chem., 4, 323 (1939).
West, J. A m C h e m Soc., 42, 1663 (1920). 'Kiihn and Grundmann, Ber., 70, 1897 (1937). Iv . Braun, Ber., 56, 2272 (1923). 'Fossek, Montush., 2, 614 (1881); 4, 660 (1883).
Neustzdter, Monatsh., 27, 882 (1906). l0Delaby and Guillot-~llt?gre, Bull. SOC. chim. France, 53, 308 (1933); Martin,
Schepartz, and Dauberc, J. Am. Chem. Soc., 70, 2601 (1948). 'l Hurd and Abernethy, J . Am. Chem. Soc., 63, 1966 (1941). " Evans, Can , and Krantz, I . Am. Chem. Soc., 60, 1628 (1938). '=Ashley e t al., J. Chem. Soc., 115 (1942). I4Helferich, Streech, and Giinther, J . prakt. Chem., 151, 251 (1938). I5Davies and Hodgson, J . Soc. Chem. Ind. (London), 62, 109 (1943); Lauchen-
auer and Schinz, Helv. Chim. Acta, 32, 1265 (1949). "Smith and Bayliss, J. Org. Chem., 6, 437 (1941). "Smith and Nichols, J. Org. Chem., 6, 489 (1941). I8Bachman, Org. Syntheses, Coll. Vol. II, 323 (1943). l9 Maxim and Mavrodineanu, Bull. SOC. chim. France, (5) 2, 591 (1935). 'OCampbell, 1. Am. Chem. Soc., 59, 1980 (1937). "Wood and Cornley, J. Soc. C h e m Ind. (London), 42, 429T (1923). 'l Fried and Elderfield, J. Org. Chem., 6, 574 (1941); cf. ref. 21. " Brunner and Farmer, J. Chem. Soc., 1044 (1937). " Cohen, J. C h e m Soc.. 432 (1935). " Letch and Linstead, J . Chem. Soc., 450 (1932); cf. ref. 21. "Linstead and Mann, J. C h e m Soc.. 2069 (1930). lTSah, Rec . trav. c h i m , 59, 1024 (1940). " Wuyts, Berman, and Lacourt, Bull. SOC. chim. Be lg . , 40, 665 (1931). " Behal and Sommeler, Bull. SOC. c h i m France, (3) 31, 300 (1904); (4) 1, 401
(1907); Stoermer, Ber., 39, 2288 (1906). "Barnes and Budde, J. Am. C h e m Soc., 68, 2339 (1946). " Fieser, Joshel, and Seligman, J. Am. Chem. Soc., 61, 2136 (1939). 'l Bouveault, Bull. SOC. c h i n France, (3) 11, 300 (1894); (4) 3, 119 (1908). U Dunbar and Arnold, J. Org. C h e m , 10, 501 (1945); I n d . Eng. C h e m , Anal.
Ed., 16, 441 (1944). '4Dunbar, J. Org. Chem., 3, 242 (1938). 35Lazier and Arnold, Org. Syntheses . Coll. Vol. 11, 142 (1943). 3bRedemann and Icke, J. Org. C h e m , 8 , 160 (1943).
Kraft and Herbst, J. Org. Chem., 10, 492 (1945); cf. ref. 36. '8Adkins e t al., J. Am. Chem. Soc.. 55, 2992 (1933). 39 Conant, Webb, and Mendum, J. Am. Chem. Soc., 51, 1246 (1929). "Reeve and Adkins, J. Am. Chem. Soc., 62, 2874 (1940).
Davies and Hodgson, J. C h e m Soc., 282 (1943). U Weygand, Organic Preparations, Interscience Publishers, New York, 1945, p.
145. UBlaise , Bull. SOC. c h i m France, (3) 31, 483 (1904).

3 10 ALDEHYDES Ch. 9
*Davies and Hodgson, I. Soc. Chem. Ind (London), 62, 128 (1943). &Darzens and Levy, Compt. rend., l%, 348 (1933).
Laurer, Gensler, and Miller, I. Am. Chem Soc., 63, 1153 (1941); c f . ref. 47. " Le Sueur, I. Chem. Soc., 87, 1888 (1905); 85, 827 (1904). "Pickard and Kenyon, J . Chem Soc., 103, 1947 (1913). " Williams, Org. Syntheses, 23, 63 (1943). s ~ t e p h e n , I . Chem Soc.. 1874 (1925). "Harington and Moggridge, I . Chem. Soc., 445 (1939).
Wi l l iam, J . Am. Chem Soc., 61, 2248 (1939). "Slotta and Kethur, Bet., 71, 335 (1938). UHenle, Bet., 35, 3039 (1902); 38, 1362 (1905). 55Lieber, I . A m Chem Soc., 71, 2862 (1949). "Hershberg and Cason, Org, Syntheses, 21, 84 (1941). " Barnes, Org. Syntheses, 21, 110 (1941). "Mosettig and Mozingo in Organic Reactions, Vo l . 4 , John Wiley & Sons, New
York, 1948, p. 362. Fieser and Hershberg, J . Am. Chem. Soc., 62, 52 (1940); c f . re f . 58.
" Mosettig and van de Karnp, I. A m Chem. Soc., 55, 2995 (1933); c f . ref. 58. ' H a y e s , I. Am. Chem Soc., 71 , 2581 (1949). " Boehrn, Schurnann, and Hansen, Arch. Pharm., 271, 490 (1933). " L o c k and Schrnidt, I. prakt. Chem, 140, 231 (1934).
Weygand and Meusel, Bet., 76, 503 (1943); FrEschl and Danoff, I . prakt., Chem, ( 2 ) 144, 217 (1936); Rosenmund, Ber., 51, 585 (1918). " Rosenmund and Zetzsche, Ber., 54, 425 (1921). 66English, Jr., and Vel ick , I . Am. Chem. Soc., 67, 1413 (1945). 6'Shoesmith e t al., J . C h a Soc., 2760 (1926). "Harris e t al., I. Am. Chem Soc., 67, 2098 (1945). *Glattfeld and Sua i t i f f , I. A m Chem Soc., 60, 1386 (1938). r°Colernan and G a i g , Org. Syntheses, Coll. Vol . U, 583 (1943).
Adams and Levine, J . Am. Chem. Soc., 45, 2373 (1923). " Adams and Montgomery, I. A m Chem Soc., 46, 151 8 (1924). "Holloway and Krase, Ind. Eng. Chem, 25, 497 (1933). "Hinkel, Ayling, and Morgan, I. Chem. Soc., 2793 (1932). "Arnold and Sprung, J . Am. Chem. Soc., 60, 1699 (1938). "Baker, I. A m Chem. Soc., 65, 1576 (1943). "Hinkel. Ayling, and Beynon. I. Chem. Soc., 778 (1937). . ~ - .
"Fuson e t al., I. Am. Chem. ~ b c . , 64, 30 (1942); Org. Syntheses, 23, 57 (1943).
* ~ i e d z i e l s k i and Nord, I . Org. Chem., 8 , 147 (1943). * Russell and Lockhart, Org. Syntheses, 22, 63 (1942). 'l Hodgson and Jenkinson, I . Chem. Soc., 469 (1929). '' Boyd and Robson, Biochem. l., 29, 555 (1935). " Weygand, Organic Preparations, Interscience Publishers, New York, 1945, p.
403. "Dunn, Waugh, and Ditmer, I . Am. Chem. Soc., 68, 2118 (1946); Wiberg, Org.
Syntheses, 29, 87 (1949). Fuson and Cooke, Jr., J . Am. Chem. Soc., 62, 1180 (1940).
'6Campaigne and LeSuer, I . Am. Chem. Soc., 70 , 1557 (1948). " Hewett, I. Chem. Soc., 297 (1940). "Coles and Dodds, I . A m Chem. Soc., 60 , 853 (1938); Mayer and Stieglitz,
Ber., 55, 1846 (1922); Rupe and Brentano, Helv. Chim. Acta, 19, 586 (1936); Ruggli and Bruckhardt, t b i d , 23, 443 (1940).
REFERENCES FOR CHAPTER 9 31 1
"Weygand, Organic Preparations, Interscience Publishers, New York, 1945, p. 156; Grammaticakis, Bull. SOC. chim. France, (5 ) 7 , 537 (1940).
' O ~ a d g e r , I . Chem Soc., 536 (1941). "Fisher , I. A m Chem. Soc., 56, 2056 (1934). " Baker, Nathan, and Shoppee, j. Chem. Soc., 1848 (1 935).
l "'Palfray, Bull. SOC. ch im France, ( 5 ) 7 , 414 (1940). " ~ r i o n , Ann. ch im, <11) 1, 43 (1934).
1 '' Burdick and Adkins, I . A m Chem Soc., 56, 438 (1934). " Duff , J . Chem Soc., 547 (1941 ). " Graymore and Davies, I . Chem Soc., 293 (1945). g '~ommelet , Compt. rend., 157, 852 (1913).
Duf f , I . Chem. Soc., 276 (1945). 'ODskita, Ber., 48, 1491 (1915). '" Fieser, Hartwell, and Jones, Org. Syntheses, 20, 11 (1940). '"Fieser and Hershberg, I. A m Chem Soc., 60, 2547, 2558 (1938). 'OS ~ i l s m e i e r and Haack, Bet., 60, 119 (1 927). '04 ~ o l l m a n n e t al., A n n , 531, 108 (1937).
I 'OaShabica e t al., J . Am. Chem Soc., 68, 1156 (1946).
l l W ~ u g g l i and Burckhardt, Helv. Chim A c t 4 23, 447 (1940). lmTiemann, Bet., 33, 3721 (1900). "'Keagle and Hartung, I. Am. Chem Soc., 68, 1609 (1946); Mannich and
Budde, Arch. Pharm, 270, 283 (1932); and Weygand, Organic Preparations, Inter- science Publishers, New York, 1945, p. 161.
'00 Gattermann and Wieland, Laboratory Methods of Organic Chemistry, The Macmillan Co., New York, 1938, p. 235.
" O ~ o s e s , Bet., 33,2624 (1 900). lUDey , I. Chem. Toc., 1059 (1937); Rotbart, A n n chin'., (11) 1, 439 (1934).
l '"Newman, I. A m Chem Soc., 57, 732 (1935). "'Whitmore e t al., I. A m Chem. Soc., 55. 1136 (1933).
I "'Henne and Hill, J . A m Chem Soc., 65, 752 (1943); Henne and Perilstein, 1 ibid. 65, 2183 (1343).
per is her and- ~oewenberg , Ber., 66, 666 (1933); Gattermam and Wieland, Lab- oratory Methods of Organic Chemistry, The Macmillan Co., New York, 1938, p. l 384.
Noller and Adams, I. Am. Chem Soc., 48, 1074 (1926). "'Noller and Bannerot, I. A m Chem Soc., 56, 1563 (1934). "'Fisher and Feldmam, Bet., 62, 856 (1929). " 'Fisher, Bet., 76, 734 (1943). "O Ferguson, Chem Revs., 38, 227 (1946). '"Pumrnerer and sch&amsgruber, Bet., 72, 1840 (1939). '"Adams and Lipscomb, I . Am. Chem. Soc., 71, 519 (1949). '"McFadyen and Stevens, I . Chem. Soc., 584 (1936). '" Buchman and Richardson, J . A m Chem. Soc., 61, 892 (1939). ' " ~ i e m a n n , Lewis, and Hays, I . Am. Chem. Soc., 64, 1678 (1942); Panizzon,
Helv. Chim. Acta. 24, 24E (1941). 126 Natelson and Gottfried, I . Am. Chem Soc., 63, 487 (1941); 64, 2962 (1942).
' " ~ a l b and Gross, Bet., 59, 727 (1926). '" Williams, Witten, and Krynitsky, Org. Syntheses, 26, 97 (1946). '" Button and Shoppee, I . Chem Soc., 548 (1937). "'Bachrnann and Kloetzel, I . Am. Chem. Soc., 59, 2209 (1937). "'Shoppee, J . Chem. Soc., 40 (1933).


3 14 ALDEHYDES Ch. 9
"'Spense and Wild, 1. Chem. Soc., 338 (1935); Walker, 1. Am. Chem. Soc., 55, 2821 (1933).
Moureu and Delange, Compt. rend. 133, 105 (1901); Chem. Zentt., ii, 461 (1901).
alaMilas, Sussman, and Mason, 1. Am. Chem Soc., 61, 1844 (1939). '"Sabatier and Mailhe, Compt. tend., 154, 561 (1912); 158, 986 (1914). "4Davies and Hodgson, 1. Chem. Soc., 84 (1943). "' Law and Perkin, 1. Chem. Soc., 259 (1907).
Segesser and Calvin, 1. Am. Chem. Soc., 64, 825 (1942). Hanus, J. prakt. Chem, 158, 254 (1941).
al"Astin, Newman, and Riley, J . Chem. Soc., 391 (1933); Fisher and Eisner, ibid., 6, 169 (1941); Weygand, Kinkel, and Tietjen, Chem. Ber., 83, 394 (1950).
t Jacobson, J. Am. Chem Soc., 72, 1491 (1950). "'Wille and Saffer, Ann., 568, 34 (1950). "l Gresham and Gr igsb~ , 1. Org. Chem., 14, 1103 (1949). "'Heme, Pelley, and Alm, 1. Am, Chem Soc., 72, 3370 (1950). "'Emerson and Patrick, 1. Org. Chem., 14. '790 (1949). "4Dornfeld and Coleman, Otg. Syntheses, 28. 83 (1948). 'l5 Lunt and Sondheimer, 1. Chem. Soc., 3361 (1950). ""6arek and Hahn, Catalytic Oxidation of Organic Compounds in the Vapot
Phase, Chemical Catalog Co., New York, 1932, pp. 37-99. Church and Lynn, Ind. Eng. Chem., 42, 768 (1950).
"' Bremner e t al., J. Chem. Soc., S25 (1950). '*Uoureu, Chovin, and Brunet, Bull. soc. chim France, (5) 15, 96 (1948). '" Bowen andwilkinson, 1. Chem. Soc., 750 (1950). "'Newman and Mangham, J . Am Chem. Soc., 71, 3342 (1949). a'aKumler, Saait, and Alpen, J . Am Chem. Soc., 72, 1463 (1950). "'Eliel and Burgstahler, J . Am. Chem. Soc., 71, 2251 (1949). " '~etweiler and Amstutz, J. Am. Chem. Soc., 72, 2882 (1950). "'Elliot and Harington. J . Chem Soc., 1377 (1949). U6Heilbron e t al., J. Chem. Soc., 737 (1949). U7Witzemann e t al., Org. Syntheses, Coll. Vol. 11, 304 (1943). "'Allen and Edens, Org. Syntheses, 25, 92 (1945). '"Alexander, J . Am. Chem Soc., 70, 2592 (1948). "Curtius, 1. prakt. Chem, 94, 273 (1917); Smith in Organic Reactions, Vol. 3,
John Wiley & Sons, New York, 1946, p. 384. 14' Cook e t al., 1. Chem Soc., 142 (1950). U' McCoubrey and Mathieson, 1. Chem. Soc., 701 (1949). "'Crounse in Organic Reactions, Vol. 5, John Wiley & Sons, New York, 1949,
pp. 290-300. '*~ngyal e t al., 1. Chem. Soc., 2700, 2704 (1949); 2141 (1950). "'Angyal, Tetaz, and Wilson, Org. Syntheses, 30, 67 (1950); cf. Gaylord and
Becker, 1. Org. Chem., 15, 312 (1950). '"Wood e t al., 1. Am. Chem. Soc., 72, 2992 (1950). '"King and Nord, J. Otg. Chem., 13, 635 (1948). '4'Schmerling, 1. Am. Chem. Soc., 68, 1653 (1946).
English and Barber, 1. Am. Chem. Soc., 71, 3310 (1949). 'soPaul and Tchelitcheff, Bull. soc. chim France, (5) 15, 200 (1948). ''' Bergmann and Pinchas, J. Org. Chem., 15, 1184 (1950). UaHorrom and Zaugg, J . Am Chem. Soc., 72, 723 (1950). U'Horning, Homing, and Platt, 1. Am. Chem. Soc., 71, 1771 (1949).
REFERENCES FOR CHAPTER 9 3 15
's4Elderfield, Pitt, and Wempen, 1. Am. Chem. Soc., 72, 1334 (1950). "'Ecary, Ann. chim., (12) 3, 450 (1948).
Trister and Hibbert, Can. 1. Research, 14B, 421 (1936). '57Cagniant, Bull. SOC. chim. France, (5) 16, 849 (1949). '" Kipnis and Omfelt, 1. Am. Chem. Soc., 70, 3948 (1948). '" Johnson and Shelberg, 1. Am. Chem. Soc., 67, 1745 (1945). moU/eston and Michaels, 1. Am. Chem. Soc., 72, 1422 (1950) Org. Syntheses,
31, 108 (1951); cf. refs. 223 and 247. '"Ilass and Bender, J . Am. Chem. Soc., 71, 1767 (1949). "'Hass and Bender, Org. Syntheses, 30, 99 (1950). m' Ruzicka e t al., Helv. Chim. Acta, 31, 433 (1948). m4 Buck and Zimrnermann, Org. Syntheses, Coll. Vol. 11, 549 (1943). m' Longley and Emerson, 1. Am. Chem. Soc.. 72, 3079 (1950). '" Bert, Compt. rend., 221, 77 (1945). "' Davies and Hodgson, 1. Soc. Chem. Ind. (London), 62, 90 (1943). ='Herbst and Manske, Org. Syntheses, Coll. Vol. 11, 389 (1943).
Djerassi in Organic Reactions, Vol. 6, John Wiley & Sons, New York, 1951, p. 207.

10
Ketones
CONTENTS
METHOD PAGE
178 . Acylation of Hydrocarbons (Friedel-Crafts) .............................................. 317 179 . Oxidation of Secondary Alcohols .................................................................. 3 23 180 . Oxidation of Alcohols by Ketones (Oppenauer) ........................................ 324 181 . Catalytic Dehydrogenation of Secondary Alcohols .................................... 325 182 . Oxidation of Olefinic Compounds (Ozonolysis) ........................................ 325 183 . Oxidation of Methylene Groups .................................................................... 326 184 . Cleavage of P-Keto E s t e r s ........................................................................ 327
...................................................... . 185 D e ~ a r b o x ~ l a t i o n of Acylmalonic Acids 330 .............................................................. 186 . Thermal Decarboxylation of Acids 331
............................................ 187 . Interaction of Giignard Reagents and Nitri les 332 188 . Interaction of Organometallic Reagents and Anhydrides .......................... 333 189 . Interaction of Organometallic Reagents and Acyl Chlorides .................... 333 190 . Interaction of Grignard Reagents and Amides ............................................ 335 191 . Interaction of Grignard Reagents and q P 4 l e f i n i c Ketones .................... 335 192 . Interaction of Grignard Reagents and Halo Ketones ................................ 335 193 . Interaction of Organometallic Reagents and Es t e r s .................................. 336 194 . Interaction of Organometallic Reagents and Sa l t s of Carboxylic Acids .. 336 195 . Hydrolysis of Ketone Derivatives ................................................................ 336 196 . Selective Reduction of a. &Olefinic Ketones ............................................ 337 197 . Par t ia l Reduction of Pheno l s ................................................................... 338 198 . Alkylation of Ketones ............................................................................. 339 133 . Interaction of Diazomethane and Carbonyl Compounds ............................ 340 200 . Catalytic Hydration of Acetylenic Compounds .......................................... 340 201 . Dehydration and Rearrangement of a-Diols ................................................ 341 202 . Decomposition of Glycol Monoalkyl Ethers ................................................ 341 203 . &Diketones by Acylation of Ketones .......................................................... 342 204 . a. p 4 l e f i n i c Ketones from Acetylenic Carbinols ...................................... 343 205 . y . 8 4 l e f i n i c Ketones from Alkenyl Es t e r s of P-Keto Acids .................. 343 206 . Cyclopentenones from Lactones .................................................................. 343 207 . &Halo Ketones from Acyl Chlorides and Olefins .................................... 343 208 . &Halo Ketones from Alkenyl Es t e r s ....................................................... 344 209 . Hydroxy Ketones from Phenolic Es t e r s (Fr ies) ........................................ 344 210 . a-Keto Acids from Azlactones ...................................................................... 344 211 . P-Keto Es t e r s by Condensation of Es t e r s .................................................. 345
.................... . 212 p-Keto Es t e r s by Selec t ive Cleavage of %a-Diacyl E s t e r s 346 2 13 . ,&&to Es t e r s by Alkylation of P-Keto E s t e r s .......................................... 346
............................. 214 . P-Keto Es t e r s from Ethyl t-Butyl Acylmalonic Es t e r s 347 215 . P-Keto Es t e r s by Acylation of Es ter Enola tes .......................................... 347
METHOD 178
CONTENTS (continued)
METHOD
.................................................... . 216 /3-Keto Nitri les by Acylation of Nitr i les .................................................................. . 217 Hydrogenolysis of 1. f Diketones
............................................................ 218 . Acid Treatment of Aciniuoparaffins ........................................................................ . 219 Pyrolysis of Glycidic Acids
.............................................................. . 220 Rearrangement of a - B r m o Azides ............................................ . 221 Degradation of Disubsti tuted Glycolic Acids
........................................................................ . 222 Hydrolysis of gem-Dihalides .................................................................. 223 . Isomerization of Vinyl Carbinols
224 . Condensation of Furans with Unsaturated Ketones .................................. ........................................................................ . 225 Condensation of Anhydrides
226 . Acylation of Certain Heterocyclic ~ b m ~ o u n d s .......................................... 227 . Addition of Aldehydes t o Olefins ................................................................ 228 . Interaction of Hydriodic Acid and Diazo Ketones .................................... 229 . 8-Diketones from Substi tuted Furans .......................................................... 230 . a-Diketones by Oxidation of Aryl Acetylenes .......................................... 231 . Y-Diketones from Ketones ............................................................................ 232 . Olefinic Ketones from Hydrocarbons a n d Carbon Monoxide ...................... 233 . a , P - ~ l e f i n i c Ketones from Diketene and Aldehydes ................................ 234 . p-Keto Es t e r s by the Reformatsky Reaction .............................................. 235 . Hydrolysis of p-Iminonitriles ........................................................................ Table 32 . Monoketones .......................................................................................... Table 33 . Diketones ................................................................................................ Table 34 . Olefinic Ketones .................................................................................. Table 35 . Acetylenic Ketones .............................................................................. Table 36 . Halo Ketones ........................................................................................ Table 37 . Hydroxy Ketones .................................................................................. Table 38 . Keto Ethers ............................................................................................ Table 39 . Keto Aldehydes ...................................................................................... References ................................................................................................................
3 1 7
PAGE
178 . Acylation of Hydrocarbons (Friedel-Crafts)
Catalyst ArH + RCOCl ...---, ArCOR + HCI
Many organic compounds react with carboxylic acids. acyl halides. or anhydrides in the presence of certain metallic halides. metallic oxides. -
&dine. or inorganic ac ids to form carbonyl compounds . The reaction is generally applicable to aromatic hydrocarbons . Benzene. alkylbenzenes. biphenyl. fluorene. naphthalene. anthracene. acenaphthene. phenanthrene. higher aromatic ring systems. and many derivatives undergo the reaction . In addition. olefinic and heterocyclic compounds have been converted to ketonic compounds . Therefore. a large number of ketones have been pre- pared by this reaction . Excellent reviews are available.1°
Benzene is usually acylated by the addition of anhydrous aluminum chloride to a benzene or carbon disulfide-benzene solution of the aliphatic

3 18 KETONES Ch. 10
or aromatic acyl halide, a s in the preparation of phenyl benzyl ketone (83%),' benzophenone (go%),' and stearoylbenzene (65%).'
The mono- and poly-alkylated benzenes are treated using modifications of the above procedure. Monoalkylbenzenes are added to a preformed com- plex of acyl halides and aluminum chloride in carbon tetrachloride4 (Per- rier modification). In th is manner, the manipulation is easier, no tars are encountered, and the yields are improved (85-90%). The procedure shows no advantage, however, in the acylation of alkoxy- or chloro-aromatic compounds. The addition of benzoyl chloride to p-alkylbenzenes in the presence of aluminum chloride in cold carbon disulfide is a good procedure for making p-alkylbenzophenones (67-87%).5 The condensation of homologs of benzene with oxalyl chloride under similar conditions yields pop'-&- alkylbenzophenones (30-55%).17 Polyalkylbenzenes have been acylated with acetic anhydride and aluminum chloride (2.1: 1 molar ratio) in carbon disulfide in 54-80% yields.47 Ferric chloride catalyst has been used under similar conditions.' Acetylation of p-cymene with acetyl chloride and aluminum chloride in carbon disulfide yields 2-methyl-5-isopropylaceto- phenone (55%L9
Studies on the conditions of the reaction have been made using simple compounds a s model substances. A comparison of thirty-nine metallic chlorides shows alurninum chloride to be the most effective in the prep- aration of p-methylacetophenone." Optimum yields resul* when the molar ratios of aluminum chloride to anhydride, acyl chloride, and acid are 3.3, 1.0, and 2.5, respectively. Halogen and oxyhalogen carriers are not help- ful." Inconsistent yields in the Friedel-Crafts reaction have been attrib- uted to the presence of ferric chloride or moisture in the aluminum chloride catalyst." Prolonged heating causes condensation of the ketone product. It has been shown that cessation of hydrogen chloride evolution may not be a satisfactory criterion for judging completeness or optimum period of re- action.I4 For the most part, the success of the reaction depends on the use of mild conditions and pure reagents1""
Other aromatic compounds have been acylated by varying procedures. A.general procedure for the preparation of alkyl biphenyl ketones has been described whereby the acyl halide is added to a mixture of biphenyl, aluminum chloride, and carbon disulfide (62-90%).18 Nitrobenzene or carbon disulfide i s used a s the solvent in the preparation of 2-acetyl- fluorene (83%)'9s'1 and the isomeric 2- and ~a~~lphenanthrenes.~'~~~ A convenient method for obtaining pure 2-acylphenanthrene is the acyla- tion of 9,lO-dihydrophenanthrene followed by sulfur dehydrogenation. In this case, only the 2-position is attacked; the over-all yield i s about 48%.la Anthracene i s acylated in the (60%)." The isomeric acetylacenaphthenes have been prepared from the hydrocarbon and acetic
METHOD 178 319
acid, using hydrogen fluoride a s c a t a l y ~ t . ~ ' ~ ~ ~ Substituted tetralins have been prepared by the FriedeI-Crafts reaction under mild conditions. Thus, tetralin or i t s 7-alkylated derivative reacts with acid anhydrides in the presence of aluminum chloride and nitrobenzene solvent at 0' (60-80%).15 Naphthalene is acetylated or benzoylated almost exclusively in the alpha position by the action of an acyl chloride and aluminum chloride in methyl- ene or ethylene chloride solution.'' Also, on treatment with benzoyl chlo- ride in the presence of iodine, i t i s converted predominantly to the a- benzoyl isomer (52%).16 Aroyl halides respond better than anhydrides to this treatment.
Heterocyclic ketones derived from furan or thiophene have been pre- pared similarly using an iodine catalyst. Short reaction time and low tem- perature are used. Thus, thiophene and acetic anhydride heated for 1 hour with a small quantity of iodine a t about 100' yields 2-acetylthiophene (86%); similarly, furan yields 2-acetylfuran (75%).59-Bther catalysts for the acylation of furan and thiophene have been used, namely, zinc chlo- ride:' silica-metal oxides,61 stannic ~h lor ide ,~ ' aluminum ~hlor ide ,~ ' boron t r i f l u ~ r i d e , ~ ~ * ~ ~ ' ~ ' and orthophosphoric acid.66 The last-named catalyst has been employed for the preparation of eleven compounds including 2- acetylthiophene (94%), 2-benzoylthiophene (99%), and 2-acetyl-5-methyl- thiophene (91%). Other oxygenated acids have been studied, but ortho- phosphoric acid is the most effective and produces the fewest side reactions. In general, the acid anhydride a s acylating agent i s preferred over the acyl halide. In introducing large acyl groups, i t i s convenient to use merely the organic acid and phosphorus pentoxide. Yields of acylated thiophene range from 45% with acetic acid to 97% with oleic acid.M
y-Aryl-substituted acids, Ar(CH,),COOH, or their halides undergo an internal Friedel-Crafts reaction to give l-tetralones.15 The acids 'may be cyclized directly with 85-95% sulfuric acid a s in the preparation of 4- methyl-l-tetralone (74%).0° However, sulfonation by-products may occur. Thus, l-tetralone from y-phenylbutyric and sulfuric acid mixrure i s obtained in 49% yield, whereas i t i s prepared from the acyl chloride and aluminum chloride in 92% yield.79 A better catalyst for direct cyclization i s hydro- fluoric acid. The organic acid i s simply treated at room temperature with 10 parts hydrofluoric acid for several hours. In this manner, l-tetralone (92%), l-hydrindone (73%), 1,2-bent-10-anthrone (75%), and other difficultly obtained anthrones have been preparedz4 I n preparing acyl chlorides with thionyl chloride for the Friedel-Crafts reaction, care must be taken to re- move this reagent completely since i t may lead to side reactions. Better results have been obtained by employing phosphorus pentachloride for formation of the acyl halide, but again the harmful phosphorus oxychloride

320 KETON ES Ch. 10
must be removed. This is readily accomplished by codistillation with benzene. The acyl chloride may be cyclized without further purification. A solution in benzene, nitrobenzene, or chlorobenzene is added to alumi- num chloride below 250.17 Polyphosphoric acid has also been applied in the synthesis of cyclic k e t ~ n e s . ~ '
Ring closure of this type has been brought about by the reaction of a lactone, namely, y,y-dimethylbutyrolactone, with benzene and aluminum chloride to give 4,4-dimethyl-l-tetralone (70%).e6 Tetralones containing halogen atomse7 or alkoxyl groups17*'' have been prepared. Also, P- haloalkyl ketones of the type ArCOCH,CH,CI undergo intramolecular con- densation to furnish l-iadanones14
Diketones have been prepared by the Friedel-Crafts method. Both acyl chloride groups in adipyl chloride react with benzene in the presence of aluminum chloride to form the diketo compound, 1,4dibenzoylbutane (81%).'~ When diketene is treated with benzene under the conditions of the Friedel-Crafts reaction, benzoylacetone, C,H, COCH,COCH,, is formed (73%L90
This synthesis of 1,3-diketones may be extended by the use of other available diketenes.
Olefinic ketones have been obtained from the reaction of acyl chlorides oranhydrides with olefins using the conditions of the Friedel-Crafts re- action. The intermediate chloro ketones are oftentimes stable and must be treated with sodium bicarbonate or dimethylaniline to complete the de- hydrohalogenation. In this manner, l-acetyl- l-cyclohexene (62%) 91*103
and l-butyryl-l-cyclohexene are prepared.
COR COR
More recently, i t has been shown that acetylation of cyclohexene with acetic anhydride in the presence of stannic chloride is l ess troublesome and does not necessitate dehydrohalogenation."'
The reaction has been investigated in detail using diisobutylene and acetic anhydride whereby methyl octenyl ketones are formed in yields a s high a s 60%. Studies of catalysts show zinc chloride to be the most effective. It is used in relatively small concentrations compared with
METHOD 178 32 1
the catalyst requirements for aromatic hydrocarbons. A low temperature (40°) is maintained to prevent polymerization of the olefin. On a small scale, a preformed complex of the anhydride and zinc chloride i s prepared and treated with the 01ef in .~~ '~ '
Under these conditions, the addition of acyl chlorides to acetylene leads to P-chlorovinyl ketones (62-80%).~~
AlCl RCOCl + HC= CH a RCOCH =CHCl
Ketones containing a double bond have also been prepared by the re- action of unsaturated acyl halides with aromatic hydrocarbons 96 in the usual Friedel-Crafts manner. Acylation of benzene and i ts homologs with P,P-dimethylacroyl chloride leads to dimethylvinyl aryl ketones, (CH,),C=CHCOAr (75-70%)."'~ The latter compounds are stable and do not undergo intramolecular condensation.
Three types of halo ketones, differing in the position of the halogen atom, have been prepared by the Friedel-Crafts reaction: ( l ) a halogenated acyl chloride and an aromatic hydrocarbon give a haloalkyl aryl ketone, e.g., P-bromopropiophenone, C,H, COCH,CH,Br, (93%)"' from benzene and P-bromoacetyl chloride; (2) an aryl halide upon acylation' gives a halo- aryl alkyl ketone, e.g., p-fluoroacetophenone (74%) from fluorobenzene and a preformed acetic anhydride-aluminurn chloride complex 11° or p-bromo- acetophenone (79%)") from bromobenzene and acetic anhydride; and (3) an aryl-substituted alkyl halide on acylation gives an aryl alkyl ketone con- taining a halogenated side chain, e.g., P-(p-acetylphenyl)-ethyl bromide, p-CH3COC6H,CH2CH,Br (83%),11' from P-phenylethyl bromide and acetyl chloride. In general, the reactions are carried out in carbon disulfide with aluminum chloride catalyst.
Phenolic ketones have been prepared by modifications of the Friedel- Crafts reaction. In preparing acyl derivatives of phenol, a preformed complex of phenol and aluminum chloride is treated with an acyl chloride. Ortho and para isomers are formed with the latter predominating.1a3 On the other hand, in preparing acyl derivatives of the polyhydric phenols and naphthols, a preheated solution of zinc chloride and acylating acid is treated with the hydroxy compound (Nencki reaction).12r126 This pro- cedure gives poor yields when applied to the monohydroxy phenols.1a7 Phloroglucinol, sym-C6H,(OH),, condenses with acetonitrile in the pres- ence of zinc chloride and hydrochloric acid to give phloroacetophenone (87%) (Hoesch-Houben r e a ~ t i o n ) . ' ~ ~ " ~ An imino chloride is probably formed, viz., CH3CN + HCI 4 CH,C(CI) =NH, which reacts with the phenol to give an intermediate ketimine hydrochloride.

322 KETONES Ch. 1 0
Acylation of aromatic ethers yields the corresponding keto ether^.'^' Typical examples are found in the conversion of anisole with aluminum chloride and appropriate acyl halide to p-methoxybutyrophenone (85%)13' and p-methoxyphenyl benzyl ketone (84%).13' Mild catalysts like iodinez6 and phosphorus pentoxidea9 are also effective.
Aryl-substituted y-keto acids are readily obtained by acylation of aro- matic compounds with succinic anhydride, e.g., P-benzoylpropionic acid (85%)."'
Phenol,136 bromobenzene," t - b ~ t ~ l b e n z e n e , " ~ and acenaphthene "' give keto acids in good yields. The reaction is applicable to other aliphatic dibasic acid anhydrides like glutaric anhydride,ls9 adipic polyanhydride,'40 and maleic anh~dride, '~ ' furnishing w -aroyl acids. An excellent dkcussion including experimental conditions and procedures has been given.'4a
Optimum conditions for the reaction of naphthalene,670 b i ~ h e n ~ l , ' ~ ~ and c h l ~ r o b e n z e n e ' ~ ~ with phthalic anhydride have been determined. The corresponding keto acids are obtained in 90-98% yields. In this type of condensation, nitrobenzene i s stated to be far superior to other solvents with respect to solvent power and ability to slow side reactions.'*
Another variation consists in the reaction between an aromatic nucleus and the ester-acyl chloride of a dibasic acid followed by hydrolysis of the resulting keto ester. This synthesis affords w-aroyl aliphatic acids in 8 5 4 5 % yields and i s applicable to benzene, i t s alkyl, halo, alkoxy, and alkylalkoxy derivatives a s well a s to thiophene and naphthalene.'39*'47
When the interaction of an ester-acyl chloride and an aromatic nucleus is employed for the synthesis of a keto ester, then a reesterification step i s r e~ommended . '~~ Certain a-keto esters have been prepared by using ethyl oxalyl chloride, COaCICOaCaH,, a s the acylating agent, e.g., ethyl a-thienyl glyoxylate (50%),IQ ethyl a-naphthylglyoxylate (46%),15' and ethyl p-biphenylylglyoxylate (70%).'5a An example of acylation of an aromatic ester i s found in the preparation of the para and meta isomers of ethyl acetylphenylacetate (8O%).ls3
Nitro- and amino-aromatic compounds do not respond favorably. How- ever, acylations of acetanilide with acetic anhydride using iodine
METHODS 178-179 323
catalysta6 or with acetyl chloride and aluminum chloride ~ a t a l y s t " ~ have been reported. o-Nitrophenyl 2-thienyl ketone has been prepared."'
Use of a-cyanopropionyl chloride results in a cyano ketone, e.g., a- cyanopropiomesitylene, C6Ha(CH3),COCHCNCH3 (2O%).lS6
179. Oxidation of Secondary Alcohols
Oxidation of secondary alcohols to ketones with sulfuric-chromic acid mixture proceeds readily. In general, the reaction i s carried out in an aqueous medium keeping the temperature a t 20-40°. Occasionally, the reaction temperature i s elevated to 50-80° for additional period^.^'^*^'" Vigorous stirring i s required for slightly soluble alcohols. The yields vary from 60% to 80% for the C,-C,, aliphatic ketones. Isopropyl S-butyl ketone i s prepared by carrying out the oxidation of the alcohol a t 40' for 36 hours (68%).ls9 Substituted cyclohexanones have been prepared in good yields (70-93%) with widely varying reaction times and tempera- tUres.16"72,675 Oxidation of insoluble aromatic carbinols i s carried out with acetic acid a s the solvent. Thus, m-biphenylmethylcarbinol and 2- phenylcyclohexanol are oxidized a t 45-50' to the corresponding ketones in 80% yield.173147 Concentrated nitric acid at reflux temperature for 20 minutes has been used for the preparation of hexamethylacetone (81%).17' The mechanism of chromic acid oxidation of alcohols has been dis- CUSSed.16b 175.186
Among the diketones prepared by oxidation of an alcohol group are the the benzils from the corresponding benzoins and aliphatic a-diketones from the acyloins. The oxidation of the former i s accomplished with copper sulfate in pyridine, e.g., benzoin to benzil (86%),l9O and the latter with cupric acetate in 70% acetic acid, e.g., 4-hydroxy-3-hexanone to dipropionyl (70%).19' Ferric chloride in a boiling ether-water mixture i s also used a s an ~xidant . '~ ' Certain alicyclic 1,2-diketones are prepared by oxidation of the acyloins with chromic anhydride in glacial acetic acid, e.g., 3,3,6,Gtetramethyl-1,2-cyclohexanedione (64%).'01 Improve- ments in carrying out oxidations of benzoins and in processing the re- action mixtures have been de~cr ibed.""~~ In one oxidation procedure, a catalytic quantity of cupric acetate is employed, which i s continuously regenerated by the action of ammonium nitrate. The reduction product of the latter is ammonium nitrite, which i s decomposed simultaneously to nitrogen snd water.194 Benzoins carrying halo,lg5 m e t h ~ x y l , ' ~ ~ ~ ' ' and di- alkylamino'99 groups have been oxidized.
Secondary acetylenic alcohols, prepared in good yields from acetylenic Grignard reagents and aldehydes, are oxidized to acetylenic ketones

3 24 KETONES a. 10
(40-80%).2'3~104 The oxidation is carried out at 0-20' by means of chromic acid with acetone a s solvent. An acetone layer of the unsaturated ketone separates, preventing further oxidation.
Preparations of halo ketones, such a s a,a'-dichloroacetone (75%)"' and l-chloro-4-phenyl-2-butanone (82%),14' and keto ethers, such a s 4 methoxycyclohexanone (65%)'" and sym-dialkoxyacetones (40-70%),"" have been carried out by the oxidation of the corresponding alcohols with chromic-sulfuric acid mixture. Methyl esters of certain a-hydroxy acids can be oxidized to the a-keto esters with lead tetraacetate in boiling benzene a s in the preparation of methyl phenylglyoxylate (84%).2'3 Also, esters of lactic acid, CH,CHOHCO,R, have been converted to pyruvic esters by the action of potassium permanganate.2'"~692 This same reagent has been employed for changing mandelic acid to the a-keto acid, ben- zoylformic acid (72%).'14
A general synthesis for y-keto acids involves the oxidation of y- lactones with bromine in the presence of magnesium The lactones are readily obtained by interaction of oxides and sodio- malonic esters with subsequent hydrolysis and decarboxylation (method 323). The over-all yields are excellent.
Nitro alcohols from the condensation of aromatic aldehydes with sodium salts of nitroparaffins are oxidized to a-nitro ketones with chromic- acetic acids, a s illustrated by the preparation of a-nitroacetophenone, C6H5 COCH,NO, (80%)."~
180. Oxidation of Alcohols by Ketones (Oppenauer)
hZetal
R,CHOH + R',CO X2C0 + R:CHOH alkoxide
Oxidation of alcohols by ketones in the presence of a metallic alkoxide has proved especially valuable in the steroid The literature to 1951 has been re~ iewed .~" An extensive investigation of experimental conditions using aluminum t-butoxide has been carried out."3 The merits of various ketones a s hydrogen acceptors have been considered. In general, methyl ethyl ketone and cyclohexanone are best for high-mole- cular-weight alcohols. The condensation products from these ketones may be removed by steam distillation. Benzil i s recommended for preparing aldehydes and ketones capable of being distilled from the reaction mix- ture below 100'. Benzil or quinone may be used for ketone products boiling from 100' to 200°, especially if they are likely to condense. The optimum temperature, duration of reaction, and concentration of re- actants may vary for the alcohol oxidized. In general, 0.5 mole of alkox- ide per mole of alcohol gives good results; however, an additional 0.5
METHODS 180-182 325
mole of alkoxide is recommended to remove any water present in the re- action mixture. A high ratio of 40 to 80 moles of ketone for 1 mole of a steroid is desirable. For simpler alcohols, 20 moles of acetone or methyl ethyl ketone, 3-10 moles of cyclohexanone, or 1-3 moles of quinone or benzil are satisfactory. The equilibrium is displaced by the large excess of the ketone reactant to give the desired product. It i s preferable to carry out the oxidation at 55-60'. The use of an inert diluent, such a s benzene, toluene, or dioxane, minimizes ketone condensation products.
The reaction has been extended to nitrogen-containing csmpounds by the use of an alkali alkoxide, such a s potassium t - b ~ t o x i d e . ~ ~ ~
181. Catalytic Dehydrogenation of Secondary Alcohols
-H l R,CHOH R2C0
Catalyst
Ketones are formed in good yields by vapor-phase dehydrogenation of secondary alcohols over copper chromite catalyst. An example is the conversion of cyclohexanol to cyclohexanone (60%)."~ A liquid-phase dehydrogenation using Raney nickel catalyst at 170' has proved success- ful for preparing C,-C, aliphatic ketones (73,95%).226 The catalyst can be reused. The procedure has been modified by employing a hydrogen acceptor, such a s cyclohexanone. The mixture of catalyst, hydrogen acceptor, alcohol, and toluene is merely refluxed for short periods.22'
The reaction may also be performed over a mixed-oxide catalyst at 280' and 100 atm. of ethylene, which serves as the hydrogen a~ceptor , '~ ' a s illustrated by the preparation of P-tetralone from 1,2,3,4-tetrahydro-
l
l 2-naphth01.~"~ By the same procedure, diisobutyryl, a diketone, has been prepared from the acyloin (27%).'"
Dehydrogenation of 1,Cpentanediol over a copper chromite catalyst in the liquid phase yields the corresponding hydroxy ketone, 5-hydroxy-2- pentanone (30%)."~
182. Oxidation of Olefinic Compounds (Ozonolysis)
R2C =CHR % R,CO + RCHO H20
Ozonolysis of olefins has found little application in the preparation of ketones for synthetic purposes. Since the ozonides may be explosive, the method has been limited to the reaction of small quantities of olefins, mostly for degradation studies and location of double bonds.
Improved conditions for the oxidation of olefins with ozone to ketones (60-70%) have been des~ribed.''"~" The use of Dry Ice temperature and methylene chloride a s solvent lessens the loss of volatile olefins in the

326 KETONES c h . 10
oxygen stream. The ozonides are decomposed by zinc and water in the presence of acetic acid or by catalytic hydrogenation with 1% palladium- calcium carbonate catalyst. Ozonides also react with Raney nickel to give aldehydes and ketones.lS4 A new ozonizer has been described.'"
Several olefinic compounds have been oxidized with potassium per- manganate or chromic acid to furnish ketones. An example i s the oxidation of diisobutylene to methyl neopentyl ketone (56%).'"
Methylenecyclobutane has been converted to cyclobutanone by oxidation to the corresponding glycol with perfonnic acid and subsequent cleavage of the glycol with lead tetraacetate (75% ~ v e r - a l l ) . ' ~ ~
183. Oxidation of Methylene Groups
Se02 ArCOCH,Ar ArCOCOAr
or Nitrogen oxldes
Compounds containing reactive methylene groups are readily converted by suitable oxidizing agents to carbonyl derivatives. Reviews of the re- action employing selenium dioxide or nitrogen oxides ''' are given.
Selenium dioxide i s commonly applied to a methylene group activated by a carbonyl group, although an adjacent double bond, aromatic ring, or heterocyclic ring may also subject i t to attack. The conversion of alde- hydes and methyl ketones leads to glyoxals (method 157). Best results are obtained when only one methylene group i s present. For example, aryl benzyl ketones have been oxidized almost quantitatively to substi- tuted benzils by treatment with selenium dioxide and acetic anhydride a t 140-150° for 3 to 4 hours.S66 Dioxane has been used a s solvent with this oxidizing agent. The products are purified by activated-charcoal treatment Other experimental details are illustrated in the preparation of methyl phenyl diketone (60%)567 and 2,4,Gtrimethylbenzil (83%).56'
Cyclic ketones like cyclohexanone 569 and cycloheptanone S70 yield the corresponding a-diketones in 35% and 90% yields, respectively.
Compounds having methylene groups situated between two activating groups-ketone, acid, or ester-are readily oxidized with selenium di- oxide to furnish t r i k e t o n e ~ , ~ ~ ' keto d i e ~ t e r s , ~ ~ ' a,D-diketo or a-keto acids.574
Another procedure utilizes oxides of nitrogen. An example i s the oxidation of diethyl malonate to diethyl oxomalonate, CO(C02C2H5),, with nitrous anhydride (76%).575 Synthesis of alkyl aryl a-diketones has been accomplished under similar conditions (30-40%).5'6
A benzyl side chain is changed to a benzoyl group by vigorous oxida- tion. For example, 4,4'-dia~etylaminodi~hen~lmethane'~' i s converted with chromic acid to the benzophenone in 70% yield. Also, 2-benzoyl-
METHODS 183-184 3 27
pyridine i s made from 2-benzylpyridine in 86% yield by the action of potassium ~ermangana te . '~~
Oxidation of cyclohexene with chromic anhydride in acetic acid gives a 37% yield of 2-cyclohexenone; likewise, l-methylcyclohexene goes to 3-methyl-2-cy clohexen-l-one (20%).~'
Certain aromatic compounds containing alkyl groups have been con- verted to carbonyl derivatives by liquid-phase oxidation of these groups with air in the presence of chromium oxide catalysts.
0 2 ArCH2CH3 - ArCOCH,
Catalyst
By the simple procedure of passing dispersed air through a suspension of m-diethylbenzene, 1% chromia, and 4% calcium carbonate at 130' for 40 hours, a 50% yield of m-ethylacetophenone i s obtained."' Likewise, aromatic ester^,'^^*'^^*'^^ a ~ e t o ~ h e n o n e s , ~ and halogenated benzenesz4' containing alkyl groups yield the corresponding keto esters, diketones, and halo ketones, respectively. Manganese dioxide catalyst has also been used.24" Tetralin can be oxidized to a-tetralone with dispersed air in the absence of a catalyst (56%).241
184. Cleavage of p - ~ e t o Esters The formation of 0-keto esters and their cleavage represents an im-
portant synthesis for many types of ketones. The methods of synthesis of various 0-keto esters are considered under methods 211 to 215 and have been reviewed.'I4 Quite often the intermediate 0-keto esters are not isolated but are cleaved directly to ketones. With few exceptions (methods 266 and 308), the cleavage always results in the formation of a ketone. Syntheses involving these cleavages are considered here.
Monoalkylation of ethyl acetoacetate and subsequent ketonic hydroly- s i s gives methyl ketones of the type CH3COCH2R (acetoacetic ester s ~ n t h e s i S).
NaOC2H5 ; H + CH3COCH2C02C2Hs CH3COCHRC02C2H5 -+ CH3COCH2R
R X 1 NaOC 2H5 ;R'X H +
CH3COCHRR8.+ CO2 + C2Hs OH +- CH3COCRR8C02C2H5
The over-all yields resulting from the use of primary alkyl bromides are 50- 70%. The method i s illustrated by the preparation of methyl n-amyl ketone. (61%)."' Monoalkylation with secondary alkyl bromides is l e s s complete, and the over-all yields are lower (20-30%).
Dialkylation followed by hydrolysis gives methyl ketones of the type CH3COCHRR8 The over-all yields are 30-40%, often depending on which

1 3 28 KETONES Ch. 10 l rC
alkyl group is introduced fir'st when R and R'are different.as0*a51 For 1 example, in the preparation of methyl methylisopropylacetoacetate, better
results are obtained if the methyl group i s substituted first (60% es ter yield)"' (cf. method 213). Hydrolysis of disubstituted acetoacetic esters, CH,COCRR'CO,C,H,, in which R and R' a re methyl or ethyl groups usually gives ketones in 60 to 80% yield^.'^'^^^^ When R i s a n-butyl group and R' i s either a n- or S-butyl group, the ketones are formed in low yields, ester formation being favored (cf. method 308); however, these particular ketones are available in good yields by cleaving the corresponding t- butyl ace to acetate^.^^^
SulfuricaS6 or ac ids are used for the ketonic hydrolysis, a s in the preparation of methyl n-amyl ketone. Also, the hydrolysis i s brought about by boiling with acetic-sulfuric acid mixture,"' hot 5% potassium hydroxide s ~ l u t i o n , ' ~ ~ or hydriodic acid i f the hydrolysis is especially d i f f i~ul t .~" Benzylacetone, C6H, CH,CH,COCH,, i s formed by hydrolysis of the corresponding p-keto ester with water a t 150-250' and 200 atm. Dialkylated p-keto esters are stable to th is treatment; therefore, a single ketone can be obtained from a mixture of mono- and di-alkylated p-keto esters.253
Difunctional compounds have been prepared by th is ser ies of reactions. Alkylation with unsaturated h a l i d e ~ ' ~ ~ " ' ~ or alkylation of unsaturated p-keto esters262 leads to olefinic ketones. Halogenation of a substituted acetoacetate followed by acetic-sulfuric acid hydrolysis gives a-halo ketones. An example of this transformation i s the chlorination of ethyl benzylacetoacetate with sulfuryl chloride (69%) followed by hydrolysis and decarboxylation to give a-benzyl-a-chloroacetone (84%)."' If alkoxy halides are used, keto ethers result. In th is manner, 6-ethoxybutyl methyl ketone (35% o ~ e r - a l l ) ~ ~ ~ and 6-phenoxybutyl methyl ketone (61%)a9a have been prepared. Similarly, alkylation using dialkylamino halides yields dialkylamino ketones in about 60% over-all yield,306 a s illustrated by the conversion of y-diethylaminopropyl chloride and ethyl sodioacetoacetate to l-diethylamino-5-hexanone (60%).'07 An example of the reaction of a
halogenated ester leading to a keto a c i d is found in the preparation of 8-ketononoic acid (68%).297 ~ K e t o - a - a l k y l ac ids have been prepared by a one-step hydrolysis and decarboxylation of certain cyanoacetoacetic esters.296
RCHCNCH(COCH,)CO,C,H, 2 RCH(COOH)CH,COCH, + CO, + C,H5 OH
a-Keto acids have a lso been obtained by treating a-oxalyl es ters with boiling dilute sulfuric acid for 6 hours (8-94%).'" These starting materials are prepared by condensation of ethyl oxalate and a second es ter (method 2 1 1).
METHOD 184
NaOC2H, (CO,C,H, ), + RCH,CO,C,H, - CaH, O,CCOCHRCO,C,H,
l H+ RCH,COCO,H + CO, + GH, OH
P - ~ e t o es ters prepared by additional methods (methods 21 1-215) are cleaved to give other types of ketones. (1) Acylation of the sodium eno- laces of disubstituted acetic esters followed by hydrolysis and decarboxy- lation g ives ketones of the type R'COCHR,.
H+ J R'COCHR, + CO, + C,H, OH c R'COCR,CO,C,H,
The over-all yield from ester and acid chloride is 38-58%.a62 (2) Self- condensation of high-molecular-weight es ters and hydrolysis of the re- sulting p-keto es ters gives symmetrical ketones of the type RCH,COCH,R.
The over-all yields (R equals n-C,-C,, n-C,,, and n-C,,) from the e s t e r s vary from 55% to 78%.as9 Certain heterocyclic ketones, namely, 8-acetyl- quinoline and p-acetylpyridine, have been prepared through a mixed es ter
(3) If acetoacetic es ter i s acylated in the form of i t s sodium enolate and carefully hydrolyzed, a new p-keto ester i s formed. Alkylation of this keto es ter followed by hydrolysis gives ketones of the type RCOC\Rt.
NaOCaHS ; CH,COCH,CO,C,H, 9 CH,COCH(COR)CO,C,H,
RCOCl l NH,
The over-all yields are stated to be 13-2073 from the acid ~hlor ide ; '~ ' however, the directions are not ~ l e a r . ' ~ ~ ~ ' ~ ~ If the chloride of a dibasic acid is used, a diketone results. Thus, terephthalic acid chloride gives p-diacetylbenzene (15% over-all).'" o-Chloroacetophenone, a halo ketone, has been prepared from ethyl acetoacetate and o-chlorobenzoyl chloride ( 54%). a90

l 3 30 KETONES Ch. 10
Aminomethyl ketones have been prepared by the a-oximination of P-keto es ters followed by reduction and
HCl RCOCH(NHAc)CO,C,H, ----+ RCOCH'NH, HCI
Symmetrical ketones are sometimes prepared from acyl chlorides by way of diketenes and 0-keto acid^.^'"
2RCKaCOCI (C'H5)3N% RCH,COC(R) =C =O 2 (RCH, ),CO + CO,
The addition of ethyl sodiomalonate to olefinic ketones followed by ring closure and P-keto es ter cleavage leads to 1,3-cyclohexanediones. The reaction has been applied to the formation of 2-alkyl-5-phenyl- l, 3- cyclohexanediones and, is typified by the preparation of 5,5-dimethyl- 1,3-cyclohexanedione (B)%)."~ Other cycli tat ions for formation of four- and five-membered rings have been described.sssss86
185. Decarboxylation of Acylmalonic Acids
A convenient method for preparing al icyclic or aromatic methyl ketones consists in the acylation of the ethoxymagnesium derivative of diethyl malonate with the appropriate acyl chloride, followed by acid hydrolysis and decarbosylation of the resulting P k e t o d i e ~ t e r . ~ " " ~ The l a s t s tep is carried out l ike the ketonic cleavage of P-keto es tersa6 ' The over-all yields are 60-85%.
The method is especially valuable for the preparation of certain sub- stituted acetophenones, namely, o- and p-nitroacetophenone and o-chloro- a c e t ~ ~ h e n o n e . " ~ Methods involving Grignard, Friedel-Crafts, or nitration reactions are apparently not applicable for the preparation of these nitro compounds, and the Friedel-Crafts reaction is not applicable to the prep- aration of o-chloroacetophenone. Although the acetoacetic ester synthesis has been used for the preparation of these and other substituted aceto- phenones, it may be complicated by 0-acylation and a l so by cleavage a t either acyl group (cf. method 212).
High-molecular-weight aliphatic ketones of the type RCOCH,R8 are made by acylation of substituted dibentyl es ters of malonic acid followed by hydrogenolysis and d e c a r b ~ s y l a t i o n . ~ ~ ~
METHODS 185-186 331
Decomposition of acylated malonic e s t e r s over aromatic sulfonic ac ids leads to 6-keto es ters (method 214).
186. Thermal Decarboxylation of Acids
Catalyst RCOaH - R,CO + CO,
Heat
Symmetrical ketones (R equals ethyl, propyl, isopropyl, n-butyl, iso- butyl, and n-heptyl) have been prepared in nearly "quantitative" yields by passing the acid vapors over thoria a t high temperatures. Studies have been made of metallic oxide ca ta lys ts and temperature of reaction. In
general, a thoria catalyst h a s been favored a t temperatures of 400- 500' The apparatus and catalyst preparation have been described.31g
More recently, it has been shown that a thoria "aerogel" catalyst is superior to the thoria-hydrogel and thoria-on-pumice catalysts. High yields a t a lower temperature !310°) and high flow rates are ~ b t a i n e d . ~ "
The distillation of lauric acid (or other high-boiling acids) over the catalyst bed i s difficult and gives poor yields; however, when the lower- boiling methyl es ter i s used, laurone is obtained in a 93% yield.3" Similarly, the ethyl es ter of 7-undecenoic acid gives undecylenone (86%).
A large number of unsymmetrical ketones have been prepared by the thermal decarboxylation however, the yields are not re- corded. In general, by using a large excess of the short-chain acid (which minimizes formation of the long-chain symmetrical ketone) over thoria a t 400°, yields of about 50% are ~ b t a i n e d . ~ ' * ~ ~ ~ Methyl benzyl ketone and other alkyl aryl ketones have been synthesized in th is manner (65%).3'9 The u s e of manganese oxide catalyst a t 400' gives about the same
The thermal decarbosylation of a mixture of barium sa l t s has been used to prepare unsymmetrical ketones; the yields are not stated."' The earlier procedure has been modified by carrying out the reaction in v a ~ u o ' ~ ~ in an iron flask. Glass reaction vesse ls are inferior. In th is manner, a large number of the high-molecular-weight methyl ketones, C,, C,,, C,'-C,,, and C,,, are p~epa red in 54-6774 yields. Cyclopentanone has been synthesized in 80% yield by distillation of adipic acid from barium hydroxide a t 275°.3'7 In a study of metallic oxides and carbonates, magnesium oxide i s preferred for the liquid-phase ketonitation of stearic acid a t 330-360' (95%).3'8 A convenient method for the preparation of dibentyl ketone i s the reaction of p h e n ~ l a c e t i c acid, acetic anhydride,

332 KETONES Ch. 10
and fused potassium acetate at 150' (41%).~~' Several pyridyl ketones have been made in a similar
Acids which have no a-hydrogen atoms may yield unsymmetrical ketones on decarboxylation instead of the anticipated symmetrical compounds."'
187. Interaction of Grignard Reagents and Nitriles
R'MgX + RCN 4 RR'C=NMgX % RCOR'
Grignard reagents react with nitriles to form ketimine sal ts which on hydrolysis give ketones. For the most part, the procedure is successful only for high-molecular-weight aliphatic and aromatic nitriles, although the lower-membered aliphatic niuiles respond favorably with aromatic Grignard reagents.3s3~a54*3" P oor results have been associated with a competing reaction of the Grignard reagent a t the a-hydrogen of the nitrile to form a hydrocarbon and a magnesium derivative which may react further a t the nitrile group, viz., RCH,CN + R'MgX + R'H + (RCHCN)M~X.~~'
Alkyl a-naphthyl ketones from a-cyanonaphthalene and XMgX are pre- pared when R is methyl through n-hexyl, cyclohexyl, or phenyl (35-60%)?46 The Grignard reagents are treated with the cyanide for 5 hours in boiling toluene or benzene-ether mixture. The intermediate ketimine sal t i s then hydrolyzed with aqueous ammonium chloride. Acylated aromatic com- pounds can be prepared readily in this way, avoiding isomeric mixtures encountered by the Friedel-Crafts method. Thus, the pure acetyl-, propionyl-, and benzoyl-phenanthrenes have been synthesized (50- 87%).21~347.34"
Ketones from fatty acid nitriles and high-molecular-weight Grignard reagents are often contaminated with hy d r o c a r b ~ n s . ~ ~ ~ * " ~ This difficulty can be avoided by discarding the ethereal solution containing the hydro- carbon products before the hydrolysis of the ketimine salt.351 The ketone-hydrocarbon mixture has been separated by dissolving the ketone in warm concentrated sulfuric acid, removing the insoluble hydrocarbons, and then reclaiming the ketone by diluting the acid solution with at er.^''
The reaction of olefinic Grignard reagents with nitriles to give olefinic ketones i s not common. An example is the preparation of 4-hexen-+one from allylmagnesium bromide and propionitrile (25%)."'
Nitriles carrying relatively unreactive halogen atoms have been used to prepare halo ketones. Thus, 4-chloro-2-ethoxybutyronitrile, C&ClCH,CH(OC,H,)CN, has been converted to 3-chloro-l-ethoxypropyl alkyl ketones in 40-75% yields.369 Reaction of methyl Grignard reagent and o-bromophenyl cyanide gives o-bromoacetophenone (80%).~~'
Diphenylacetoin, a hydroxy ketone, has been prepared in 45-60% yield by the action of benzyl Grignard reagent on phenylacetaldehyde cyano- hydrin.37'
METHODS 187-189 333
An important method for the preparation of keto ethers is the reaction of cyano ethers with Grignard reagents. In this manner, a large number of a-alkoxy aliphatic ketones have been made ( 3 0 - 7 0 % ) . ~ ~ ' " ~ ~ " ~ ~ Like- wise, phenoxymethyl alkyl ketones have been prepared (20-64%).380 When the Grignard reagent contains an U -alkoxy group, w -alkoxy ketones are formed.379
Ethyl ,&oxovalerate, a P-keto ester, is prepared from ethyl Grignard reagent and ethyl cyanoacetate (58%).'"~~" Amino ketones are conven- iently made by the action of aromatic Grignard reagents on y-diethyl- aminobutyronitrile, (C,H,),NCH,CH,CH,CN, in 80-90% yields.3n'
188. Interaction of Organometallic Reagents and Anhydrides
R ' M ~ X (RCO),O - RC - OCOR % RCOR'
\
A large number of ketones have been prepared by treating anhydrides with Grignard reagents. It has been shown that the yields are best at low temperatures (-75'). Primary, secondary, and tertiary aliphatic or aromatic Grignard reagents give high yields when treated with acetic, propionic, or butyric anhydrides.389'3Q'
A variety of ketones may be made using cadmium alkyls (50-70%). In the preparation of alkyl aryl ketones, reaction of the aliphatic rather than the aromatic anhydride i s preferred.3Q2 Keto acids result when phthalic anhydride3Q2-394 or dimethylsuccinic anhydride (60-70%)395 is used.
Acetylemc ketones of the type RF= CCOCH, are prepared by the re- action of acetic anhydride and acetylenic Grignard reagents. The latter compounds are readily made from acetylenic hydrocarbons and ethyl- magnesium chloride, and are added slowly to the anhydride at a low temperature. This procedure prevents a secondary reaction of the de- sired product with a second molecule of Grignard reagent. In this man- ner, Soctyn-bone (58%) and 3-nonyn-2-one (55%) are prepared.3Q6 Sodium phenylacetylide has been treated with various anhydrides, including acetic, benzoic, cinnamic, and crotonic, to give the corresponding phenyl- acetylenic k e t ~ n e s . " ~
189. Interaction of Organornetallic Reagents and Acyl Chlorides
Addition of cadmium alkyls to acyl chlorides yields the corresponding ketones. The method has been reviewed,400 and the experimental condi-

3 34 KETONES Ch. 10
tions have been studied.*' The cadmium reagents are readily prepared by adding anhydrous cadmium chloride to Grignard reagents. It i s advisable t o check the completeness of the cadmium alkyl formation by the standard Gilman tes t for Grignard reagent. The u s e of an alkyl bromide for forma- tion of the cadmium reagent and of benzene as solvent during the coupling reaction has improved the yield. A variety of ketones have been prepared, and yields of 50-80% may be expected i f highly reactive ketones are not formed and if the cadmium alkyl i s not secondary or tertiary.40'*4Da In the preparation of methyl n-butyl ketone (74%), n-propyl-n-heptadecyl ketone (65%), acetophenone (85%), and ethyl a-fury1 ketone (61%), the smaller alkyl fragment comes from the cadmium alkyl. Either the aryl or alkyl cadmium compound i s satisfactory for formation of alkyl aryl ketones.
The r-action h a s been extended to the formation of difunctional com- pounds. High-molecular-weight keto e s t e r s and halo ketones are formed by using carbalkoxy acyl chlorides and halogenated acyl chlorides, re- spectively. Methyl 4-keto-7-methyloctanoate (75%) and l-chloro-2- hexanone (51%) have been prepared in th is way.40'*403 Also, P-aroyl- propionic es ters are made by the reaction of diarylcadmium reagents with P-carbomethoxypropionyl chlorideO6" In the preparation of a carbalkoxy acyl chloride having a branched carbon skeleton, an ester interchange may occur to give a mixture of the two possible carbalkoxy acyl chloride^.^" Alkoxy acyl chlorides react with cadmium alkyls to give keto ethers, as in the preparation of 7-phenoxypropyl methyl ketone (78%) from y-phenoxy- butyryl chloride and methylcadmiumaDa and of certain 2-alkoxyethyl phenyl ketones from P-alkoxypropionyl chloride and d iphenyl~admium.~"~
A large number of methyl and ethyl ketones have been prepared in about 70% yields by employing zinc alkyls; however, full directions are not given.40s Reaction of zinc alkyls and unsaturated acyl chlorides in the presence of a zinc-copper couple gives olefinic ketones in 75-90% yields. By th is procedure, 5-ethyl-4-hepten-+one (74%) and 3,4-diethyl-4-hexen-2- one (83%) are made.406 High-molecular-weight keto ac ids (C,,-C,,) have been prepared in good yields (77-92%) by adding ethereal Grignard reagents to anhydrous zinc chloride, replacing the ether with benzene a s solvent, and then treating with carbethoxy acyl chlorides under re flu^.^^'*^^'
In general, the cadmium reagent i s preferred to the zinc reagent be- cause i t i s more readily prepared and i s l e s s reactive toward the car- bony1 group.
Grignard reagents have been used directly in mono- 409*4'0*539 and di- ketone4I5 formation. More recently, i t h a s been found that a catalytic quantity of cuprous chloride greatly increases the An ex- ample i s the formation of hexamethylacetone in 70-80% yield from t-
butylmagnesium chloride and trimethylacetyl chloride. Diketones have
METHODS 183-192 335
also been prepared by coupling magnesium enolates of certain ketones with high-molecular-weight acyl chlorides.539
19. Interaction of Grignard Reagents and Amides
~ R ' M ~ X RCONH,
YHMgX , RC-OMgX % RCOR' \
This reaction has been used extensively for the preparation of neo- pentyl and t-butyl ketones from n-alkyl Grignard reagents and t-butyl- acetamide and trimethylacetamide, respectively, (52-78%).'""a8 In addition, a large number of halo ketones have been prepared by the re- action of aromatic Grignard reagents with chloro-substituted aromatic amides (60-80%).~~"-~~ ' For example, benzyl Grignard reagent and m-chloro- phenylacetamide react to give benzyl m-chlorophenyl ketone (80%). In a similar manner, the u s e of mandelamide or p-methoxyphenylacetamide leads to hydroxy ketones or keto ethers, r e s p e ~ t i v e l y . ~ ' ~
191. Interaction of Grignard Reagents and a,D-Ol efinic Ketones
Aliphatic and aromatic ketones have been prepared by th is method. The Grignard reagent adds 1 : 4 to the conjugated ketone system. This i s
illustrated by the addition of ethyl Grignard reagent to ethylideneacetone, CH,CH=CHCOCH,, to give a 75% yield of 4-methyl-2-hexanone.437 Highly branched ketones have been prepared in small yields.438*43g The amount of 1 : 4 addition varies considerably with the Grignard (cf.
method 89). Certain methoxy-substituted chalcones, h C H = C H C O h , have been treated suc~essful ly . '~ '
192. Interaction of Grignard Reagents and Halo Ketones
The most successful application of th is method has been the synthesis of 2-substituted cyclohexanones by the action of either aliphatic'" or aromatic445 Grignard reagents on 2-chlorocyclohexanone. An example i s the formation of 2-phenylcyclohexanone (R = C,H,) in 60% yield.443 The

3 36 KETON ES Ch. 10
aromatic moiety may a lso be substituted with alkyl or alkoxyl groups. The method has been extended to the preparation of 2-phenylcyclopentanone (50%).'w
173. Interaction of Organometallic Reagents and
(cf. method 71)
RCOaC,Hs + R'MgX 4 RC -0MgX % RCOR' \
R'
194. Interaction of Organometallic Reagents and W t s of Carboxylic Acids"% 451
/ RC0,Na + R h g X 4 RC -0MgX H'O- RCOR'
\ R'
175. Hydrolysis of Ketone Derivatives
Oximes, which are produced by several synthetic routes (cf. Chapter 27), are readily hydrolyzed to carbonyl compounds. Thus, the acetylbenzoyl monoxime, prepared by the nitrosation of propiophenone, has been con- verted to the diketone by hydrolysis with dilute sulfuric acid.'"
In another instance, the action of aliphatic Grignard reagents on methyl a-nitrosoethyl ketone with subsequent acid hydrolysis furnishes a-hydroxy ketones of the type CH3(R)COHCOCH3.'56 The oxime of l-methylcyclo- penten-5-one is hydrolyzed by dilute sulfuric acid (54%). It i s prepared by the action of nitrosyl chloride on l-methylcyclopentene with subsequent dehydrohalogenation with pyridine.598
METHODS 195-196 337
A method for hydrolyzing p-quinone oximes with the aid of cuprous chlo- ride has been described; the yields are excellent.'s9
Aliphatic ketones have been prepared by a five-step synthesis from nitroparaffins.
RCHO R'CKNO, ----+ RcHOHCHR'NO,
1
The nitroparaffins are condensed with aldehydes to yield nitro alcohols (70-80%), which on acetylation and treatment with an aqueous methanolic solution of sodium bicarbonate are converted to nitroGlefins (80434%). These compounds are reduced to the corresponding ketoximes by zinc and acetic acid (50-60%)."~ Reduction with iron and dilute hydrochloric acid gives good yields of either ketones or ketoximes, depending upon the amount of hydrochloric acid used.679 The ketoximes can be hydrolyzed to ketones by refluxing with dilute sulfuric acid in the presence of forma- lin, which ac t s a s a hydroxylamine acceptor (80%). The over-all yields from the nitroGlefins are 40-60%. In this manner, certain otherwise dif- ficultly obtainable ketones are prepared. Semicarbazones have been con- verted to ketones by treatment with sodium nitrite in glacial acetic acid,454 with aqueous oxalic acid,'" or with phthalic anhydride.'90
a-Keto acids or es ters may be prepared by the hydrolysis of the cor- responding oximino esters with 85% formic acid and nitrosylsulfuric acid a t o ~ . ' ~ ' Although a-oximino acids can be obtained in excellent yield from a-halo ac ids or substituted acetoacetic or malonic ester^,'^' their hydrolysis may proceed poorly.29s
Elimination of carbon dioxide from a carboxylic acid in the presence of a diazonium sa l t leads to an aryl hydrazone (Japp-Klingemann). S u b sequent hydrolysis in the presence of pyruvic acid furnishes the carbonyl compound, a s illustrated by the preparation of 2-n-butyrylpyridine (81%)."'
196. Selective Reduction of a ,POle f in ic Ketones
RCH = CHCOR H' , RCH,CH,COR Catalyat
Selective hydrogenation of a , b o l e f i n i c ketones to saturated ketones can be accomplished through careful control of the temperature, duration of reaction, and use of a catalyst active enough to permit low-temperamre hydrogenation.'6' Thus, mesityl oxide, benzalacetone, and benzalaceto- phenone have been reduced in 70-100% yields to the corresponding satu- rated ket~nes . '~ ' Preparations of nickel catalysts used in these reductions are d e ~ c r i b e d . ' ~ ~ " ~ ~

338 KETONES Ch. 10
Other olefinic ketones have been reduced selectively a t room tempera- ture and atmospheric pressure over a platinum or palladium catalyst to give good yields of the ketones, namely, 5-methyl-3-heptanone (94%)F7*468 diisobutyl ketone and a-benzylacetophenone (81-95%).6'8 Selective hydrogenations of some 3alkyl-2-cyclohexenones have been carried out over palladinized charcoal in essentially quantitative Preparation of platinum catalyst has been d e ~ c r i b e d * ' ~ Many olefinic ketones prepared by the aldol condensation or by acylation of olefins have been hydrogenated; however, the yields are not always stated"' Benzalacetone, C6H5CH=CHCOC;H3, is selectively reduced to b e n y l - acetone in a 63% yield by the action of sodium amalgam in acetic acid- alcohol solution.476
Unsaturated keto e s t e r s obtained by the Knoevenagel condensation have been selectively hydrogenated in good yields with Raney nickel catalyst a t room temperature and 45 atm. to saturated keto esters, e.g., ethyl a-heptylacetoacetate (97%) from ethyl a-heptylideneacet~acetate.~~~
197. Part ial Reduction of Phenols
ON a ONa OH 0 I I
Phenols can be partially hydrogenated in the presence of alkali to cyclohexanones. An example is the synthesis of dihydroresorcinol, or 1,3cyclohexanedione, by hydrogenation of resorcinol in the presence of Raney nickel and an equimolar quantity of sodium hydroxide (95%).'01 Under these same conditions, pyrogallol furnishes a stable enedi~lone. ' '~
OH OH 0 00. $aoH -soH
/ ONa I ONa OH
Hydrogenation of 2-naphthol in the presence of palladium and an organic base like N-ethylmorpholine gives 2-tetralone (40%);'03 other conditions for its reduction lead to other products.484ea5 By means of Raney nickel and alkali, 1,Gdihydroxynaphthalene h a s been partially reduced to G hydroxy-l-tetralone.'"
Reductions of th is type may a lso be carried out by the action of sodium and ammonia, sodium and alcohol,a6 or Raney nickel-aluminum alloy and alkali.'"
METHOD 198
198. Alkylation of Ketones
Many highly branched ketones have been prepared by the alkylation o f simpler ketones, sodium amide or sodium alkoxides generally being used t o form the enolate ion. For example, ketones of the type RCOR', where R and R' represent many combinations of methyl (Me), ethyl (Et), n-propyl (Pr), isopropyl, n-butyl, S-butyl, t-butyl, isoamyl, Et,CH-,
'
Et$ -, n-Pr,CH -, n-PrMeCH -, isoPrCH, -, and n-PrMe,C -, have
been prepared; however, the yields are not always reported.'" Alkylation of alicyclic ketones like cyclopentanone and cyclohexanone has also been studied. In these reactions a l l available a-hydrogens may be re- placed, disubstitution on one s ide of the carbonyl group occurring f irst.489-493 Alkyl aryl ketones of the types ArCOCH,R, ArCOCHR'R", and ArCOCR'R'Y' are made by alkylating dcetophenone and its deriv- at ives with ally1 or benzyl ha l ide~. '~ ' In general, the reactivity of the alkyl halide decreases with increasing carbon content and complexity. Oftentimes, an alkyl sulfate is employed a s the alkylating agent. A re- view of the earlier work has been presented.494 The method is illustrated by the conversion of diisopropyl ketone to hexamethylacetone in the presence of sodium amide (52%).16'
Methyl y-chloropropyl ketone, CH,CO(CH,),CI, undergoes intramolecular cyclization to methyl cyclopropyl ketone under the influence of 50% aqueous sodium hydroxide.694
The effect of the basic reagent has been studied in the methylation of phenylacetone. hionomethylation proceeds better with sodium isopropoxide than with sodium ethoxide. Introduction of a second alkyl group is accom- plished best with potassium t-butoxide. Sodium t-amylate al lows many alkylations that fail or give poor results when carried out with sodium amide.493 1,l-Disubstituted 2-tetralones are conveniently prepared by alkylation in the presence of sodium hydride, no monosubstituted products being formed with th is reagent.496
The temperature of the reaction has been shown to be important. For example, in the alkylation of 2-methylcyclopentyl phenyl ketone, the reaction carried out a t the temperature of the refluxing benzene solution gives the desired product; however, the u s e of boiling xylene leads to 0-alkylated products, and boiling toluene g ives mixtures.66"
Diketones have been alkylated by a modified procedure."00'50' The monosodio derivative is prepared in ether by treating the diketone with powdered sodium. It is then. allowed to react with the alkyl iodide in acetone or dioxane solution. This scheme has been applied in the prep-

340 KETONES Ch. 10
aration of n-butylbenzoylacetone, PhCOCH(n-Bu)COCH,, ethylacetylace- tone, CH3COCH(C2H,)COCH3, and other high-molecular-weight compounds. In a similar manner, acyloin enolates are alkylated with primary halides in ethyl ether or toluene to furnish a,a-dialkyl- a'-hydroxy k e t o n e ~ . ' ~ ~
Alkylation with ally1 bromide leads to olefimc ketones, e.g., 2-allyl- cyclohexanone (62%) and a-allylethyl ethyl ketone (56%) from the corre- sponding k e t o n e ~ . " ~ ' ~ ~ ~ Desoxybenzoin, C6H,C&COC6Hs, and ,&ðyl- aminoethyl chloride, (C,H,),NC&CH,CI, combine to form the correspond- ing amino ketone.'04
19. Interaction of Diazomethane and Carbonyl Compounds
RCHO + CH,N, 4 RCOCH, + N,
Diazomethane reacts with carbonyl compounds to introduce methylene In the case of aldehydes, nitrogen is lost and the corre-
sponding epoxide, methyl ketone, or higher homolog of the starting alde- hyde i s formed, depending on the nature of the R group and catalytic influences. Similarly, ketones yield epoxides and homologous ketones. The latter may react further with additional diazomethane. For these reasons, the reaction may be complicated.
Cyclic ketones react to form the higher homologs; for example, c y c l e hexanone i s converted to cycloheptanone (63%).'06
An extension of this reaction has been the use of other aliphatic di- azoalkanes. Benzaldehyde and the appropriate diazo compound give propiophenone, butyrophenone, and valerophenone in almost quantitative yield.507 Furylaldehyde also reacts to form fury1 alkyl k e t o n e ~ ~ ~ ~
200. Catalytic Hydration of Acetylenic Compounds
Catalyst RC= CH + H,O + RCOCH,
This method finds commercial application in the production of acetalde- hyde from acetylene. Mercuric sa l ts in the presence of dilute sulfuric acid act a s the catalyst. The reaction has been extended to higher alkylacetylenes, which are obtained in about 60% yield from sodium acetylide and alkyl halides. These compounds are readily hydrated in aqueous solutions of acetone, methanol, or acetic acid to give 80-90% yields of the corresponding methyl ketones, for example, methyl butyl, methyl amyl, and methyl hexyl ket~nes . '~ ' Hydration has been accom- plished by passing the acetylenic hydrocarbon and steam over a phos- phoric acid catalyst at 150-204' and atmospheric pressure.509
Acetylenic carbinols (from sodium acetylide and a ketone) are readily hydrated in the presence of mercuric sulfate to give the corresponding
METHODS 200-202 341
hydroxy ketones in high P-Keto acids have been prepared by hydration of acetylenic acids.512 a-Acyloxy ketones, R,C(OCOCH,)COCH,, are made by the action of carboxylic acids on acetylenic ~ a r b i n o l s . ~ ~ ~
201. Dehydration and Rearrangement of a-Diols
The classical example of this method is the rearrangement of pinacol to pinacolone (72%).'13 The reaction is usually brought about by dilute sulfuric acid. A second procedure is the passage of a mixture of the pinacol and steam over silica-phosphoric acid at 275-300'; the yield of pinacolone is 94%.'14 Benzopinacol, (C6H,)2COHCOH(C6H,),, is dehy- drated and rearranged by iodine in acetic acid (96%).'15 Under the same conditions, diphenyl-(l-hydroxy-l-cyclopentyl>carbinol undergoes re- auangement accompanied by ring expansion to form 2,2-diphenylcycle hexanone (98%).'16
The reaction has been extended to other pinacols; however, their preparation may involve lengthy procedure^.^'^ Certain benzoins on re- duction with metals and acids yield diols which are then converted to desoxybenzoins."'"20 These conversions involve the migration of a hydrogen atom rather than an alkyl group. Similarly, aromatic keto ethers and amino ketones have been prepared.520ss21
A modification of this reaction is the hydrolysis and rearrangement of olefin d i b r ~ m i d e s . ' ~ V h e most successful of these conversions is the preparation of methylisopropyl ketone (59%) from trimethylethylene dibr~mide."~
202. Decomposition of Glycol Monoalkyl Ethers
H+ +
RCOCHR', c---- RC(OC,H,)- CR: 6&96%
Ketones of the type RCOCHR:, where R represents methyl, ethyl, iso- propyl, n-butyl, n-hexyl, or phenyl, and R' represents ethyl, n-propyl, n-butyl, or phenyl, have been prepared by a series of reactions similar to that used in the preparation of aldehydes (method 167).'"
In an analogous manner, the monoethyl ether of dihydroresorcinol reacts with alkylmagnesium halides to form ~alkyl-2-cyclohe~enones.~~~

KETONES Ch. 10
Q d Q o0 0 R OMgX R
203. PDike tones by Acylation of Ketones
R'CO,C,H L R'COCHRCOCH, (Type 11) B a n e
The acylation of ketones having reactive methylene groups by es ters or anhydrides 54a*543 is a common and convenient method for preparing P-diketones. An ester is used in the presence of a base, and an anhydride with boron trifluoride. From an unsymmetrical ketone two types of ketones result, depending on which a-hydrogen atom reacts. In general, the boron trifluoride method leads to the formation of type I ketones, R'COCH,COCH,R, whereas the basic reagent method favors type I1 ketones, R'COCHRCOCH,. Either sodium amide544*549 or sodium hydide 545.549 is preferred a s the basic reagent. Unsymmetrical ketones having only one reactive s ide (such a s acetophenone) respond the same by either method.54a Also, symmetrical ketones take the same course by both methods, e.g., acetone to a c e t y l a ~ e t o n e . ~ ~ ~ ' ~ ~ ' Many representative ketones-methyl ethyl, methyl isopropyl, methyl isobutyl, methyl t-butyl, diisobutyl, methyl n-amyl, cyclohexanone, and acetophenone-have been converted to diketones. The acylating agents are varied and include ethyl esters or anhydrides of acetic, propionic, wbutyric, isobutyric, n-valeric, n-caproic, benzoic, anisic, phenylacetic, lauric, and nicotinic acids. Thus, a large number of P-diketones have been prepared in vary- ing yields, mostly in the range of 30-60%.
P - ~ i k e t o n e s are a l so formed by acylation of the enol es ters of ketones with anhydrides in the presence of boron tr if l~oride.~"
If the acylating es ter is diethyl oxalate, then an a,y-diketo ester, or a substituted glyoxalate, is formed555'ss7 These substances are important intermediates in the synthesis of certain P-keto e s t e r s (method 307).
NaOC,H5 RCOCH, + (CO,C,H,), + RCOCH,COCO,C,H,
METHODS 204-207
204. a, P - ~ l e f i n i c Ketones from Acetylenic Carbinols
RCH2COH(R')C~ CH HCO,H
+ RCH = C(R')COCH,
Ethynyl carbinols on heating with formic acid are isomerized to a,/?- olefinic ketones; for example, isohexylmethylethynylcarbinol is taken to 3,7-dimethyl-3-octen-2-one (48%)'84 and 1-ethynyl-1-cyclohexanol to 1- acetyl-l-cyclohexene (70%). '~~ Small amounts of unsaturated aldehydes may contaminate the product.
2 0 5 Y, 6-Olefinic Ketones from Alkenyl Esters of P - ~ e t o Acids
170-250' CH,COCH,CO,CH,CH==CH, - CH,COCH,CH,CH= CH, + CO,
Acetoacetates or benzoylacetates of P,y-unsaturated alcohols-methal- lyl alcohol, crotyl alcohol, methylvinylcarbinol, cinnamyl alcohol, etc.- on heating a t 170-250 ' evolve carbon dioxide and produce y, 6 -01efinic ketones (23-88%).'95 The unsaturated acetoacetates are readily prepared by the action of diketene on the corresponding unsaturated alcohols.
206. Cyclopentenones from Lactones
CH.
y-Methyl-y-lactones having a methylene group adjacent to the y-carbon are converted conveniently to 2-alkyl-3-methyl-2-cyclopentenones (30-50%). The method is not applicable, however, to the preparation of 2-cyclo- pentenone and 3-methyl-2-cyclopentenone. The lactone is simply warmed over phosphorus pentoxide, and the product is dist i l led from the reaction m i ~ m e . ' ~ ~
207. p - ~ a l o Ketones from Acyl Chlorides and Olefins
Catalynt R'COCI + RCH=CHR - RCHCICHRCOR'
Addition of acyl halides to olefins in the presence of catalytic amounts of aluminurn chloride, stannic chloride, or zinc chloride gives P-halo k e t o n e ~ . ' ~ ~ An example is the addition of propionyl chloride to ethylene

3 4 4 KETONES Ch. 10
to form ethyl /khloroethyl ketone (45%).98 Sometimes the addition prod- ucts are very unstable and undergo spontaneous dehydrohalogenation to olefinic ketones"' (cf. methods 20 and 178).
208. a-Halo Ketones from Alkenyl Esters
RC0,CR'-CH, RCO,CR'BrCH,Br --r RCOBr + R'COC%Br
The dibromide derivatives of alkenyl esters spontaneously cleave in the cold to form a-bromo ketones and acyl halides. In this manner, 1- bromo-2-hexanone (67%) and l-bromo-bheptanone (80%) are prepared. The alkenyl esters are prepared by the catalytic addition of organic acids to alkylacetylenes (30-35%).60'
209. Hydroxy Ketones from Phenolic Esters (Fries)
OCOR 0 6 or ~ C O R
/ / COR
An ester of a phenol may be converted to the isomeric o J r p-hydrory ketone, or a mixture of both, by treatment with aluminum chloride. Crit- ical discussions of the reaction have been presented6" with respect to the influence of temperature, solvents, ester-reagent ratio, and the struc- ture of the acyls0' and phenory By varying the first three factors, i t is often possible to prepare predominantly either of the iso- meric ketones. The reaction is exemplified in the preparation of o= and p-propiophenol (35% and 40%, respectively) and 2-hydroxy-4,Gdimethyl- acetophenone (80%).606
210. a-Keto Acids from Azlactones ArCH=C-CO
I 1 NaOH; N O -
Hydrolysis of certain unsaturated azlactones with aqueous sodium hydroxide followed by treatment with dilute hydrochloric acid yields
METHODS 210-211
a-keto acids. The azlactones are .readily prepared from substituted benzaldehydes and hippuric In this manner, phenylpyruvic acid (72% over-all)610 and m-chlorophenylpyruvic acid (52% over-all)6" have been prepared Other applications have been d e ~ c r i b e d . ~ ~ ' ~ ~ ' ~ * ~ ' ~
211. P - ~ e t o Esters by Condensation of Esters
The acetoacetic ester condensation consists of a base-catalyzed re- action of two esters (at leas t one having an a-hydrogen atom) to form a P-keto ester. The scope, limitations, experimental procedures, and applications have been r e v i e ~ e d . ~ ' ~ ) ~ " * ~ ~ '
Variations of the reaction include condensation of the same ester, a mixed ester condensation, and ester cyclizations. Improvement in yield of the self-condensation reaction is obtained by removing the alcohol produced, the reaction being forced to completion. In this manner, methyl esterssi6 catalyzed by sodium methoxide and ethyl esters14' catalyzed by sodium ethoxide are self-condensed (50-85%). Ethyl isobutyrate and ethyl isovalerate do not respond to sodium alkoxide catalysis; however, these compounds are readily self-condensed with the aid of diisopropyl- aminomagnesium bromide.626 Another promising reagent is sodium hy- d~ide. '~ ' Mixed ester condensations in which only one ester has an a- hydrogen atom are satisfactory. These are l e s s complicated than a con- densation of two different esters each having reactive a-hydrogens. Thus methyl benzoate condensed under "forcing" conditions with methyl acetate, propionate, or butyrate forms the a-alkylbenzoylacetates, C6H,COCHRC0,CH,, in 45%, 61%, and 41% yields, r e spec t i~e ly . "~ Similarly, condensation between ethyl oxalate and these esters produces a-ethoxalyl ester^.'^'^^"
An example is the synthesis of ethyl a-ethoxalylpropionate (R = CH,) in 70% yield.6" Ethyl oxalate and ethyl succinate form ethyl a-ethoxalyl- succinate (83%).624 In a mixed ester condensation, the use of a more reactive ester, such a s the phenyl or biphenyl ester, helps to prevent side Simple heterocyclic esters, namely, ethyl nicotinate and ethyl 8-quinolinecarboxylate, undergo the mixed ester condensation in good yields.a'0*a8'p6"' The internal condensation of ethyl adipate to give 2-carbethoxycyclopentanone (Dieckmann reaction) is an example of c yclization (8

346 KETONES Ch. 10 I 212. P-Keto Esters by Selective Cleavage of a,a-Diacyl Esters I
NaOCaH,;
RCOCl
The acylation of simple 0-keto es ters with acyl chlorides to form di- acylacetic es ters proceeds readily; however, the subsequent cleavage for removing the smaller acyl group is complicated in that the original keto es ter may be regenerated. The optimum conditions for the conversion of bentoylacetoacetic es ter to benzoylacetic es ter with ammonium chloride and ammonium hydroxide have been studied.63' The over-all synthesis of this es ter has been described (57%).6'a An improved procedure for the ammonolysis of ethyl a-acetyl-&oxocaproate using gaseous ammonia has been des~r ibed . "~ By a similar process, a ser ies of al icyclic 0- keto es ters has been prepared in over-all yields of 20-40%.6"
Variations of the above procedures are sometimes employed. D-Keto es ters may be obtained by akoholys is of the intermediate diacyl esters by sodium methoxide in methanol,634 a s in the preparation of methyl 0- oxocaprylate (88%).635 The starting P-keto es ter can be converted to the new P-keto es ter in a single step. Thus, in the synthesis of ethyl ben- ' toylacetate (55%), ethyl acetoacetate and ethyl benzoate are converted directly to th is keto es ter by distilling the lower-boiling product, ethyl acetate, thereby forcing the reaction to completion.636
Finally, the sodium enolate of the new D-keto es ter may be alkylated directly to give 0-keto esters of the type RCOCHRCO,C,H,.~~~
213. P - ~ e t o Esters by Alkylation of P K e t o Esters
This reaction has been considered above (method 184) with respect to e a s e of mono- and di-alkylation. A large number of condensing agents have been compared, including sodium and potassium ethoxide, sodium in dioxane or toluene, sodium hydride, sodium amide, and sodium or potassium t - b ~ t o x i d e . ~ ~ ' In general, sodium ethoxide i s recommended in the alkylation of acetoacetic ester with primary halides (73%); potassium ethoxide with branched halides, such a s isobutyl and S-butyl halides
METHODS 213- 215 347
(61% and 55%); and potassium t-butoxide for introducing a second alkyl group in a-substi tuted acetoacetic es ters (60-80%). The other reagents a r e successful in certain cases. Alkylation of 2-carbethoxycyclo entanone with methyl, ethyl, or isopropyl iodides gives the corresponding i k e t o es ters in 82%, 74%, and 59% yields, Other examples are found in the preparation of ethyl monomethyl- (71%) and dimethyl- acetoacetic es ters (54%)644 and ethyl n-butylacetoacetate (72%).645 Alkylations by ethyl b e n ~ e n e s u l f o n a t e , ~ ~ ' isopropyl ace ta te or isopropyl alcohol in the presence of boron t r i f l ~ o r i d e , ~ ~ ~ ~ ~ ~ ~ and dimethyl ~ u l f a t e ~ ~ ' have proved more successful than those by the corresponding alkyl halides.
P - ~ e t o es ters containing a double b~nd,"""~ an alkoxyl 29z
or an amino group"0307 are formed by alkylating acetoacetic ester with a substituted alkyl halide.
214. P-Keto Esters from Ethyl t-Butyl Acylmalonic Esters
RCOCl C,H,0MgCH(C0,C,II,)COaC(CH,)3 --+ RCOCH(CO,C,H,)CO,C(CH,),
+ RCOCII,CO,C,H, + CH,=C(CH,), + CO,
Olefin elimination and decarboxylation of ethyl t-butyl acylmalonates proceeds easi ly on treatment with toluenesulfonic acid to form D-keto esters of the type RCOC&C0,CaH,.654 By th is procedure, acyl
acetates where R is ethyl (63%), cyclohexyl (65%), 2-fury1 (70%), benzyl (46%). or propenyl (35%) have been prepared. The limiting factor in this excellent method is the availability of ethyl t-butyl malonate; i t s syn- thesis h a s been d e s ~ r i b e d . ~ ~ '
A reaction similar to the above involves the acylation of malonic ester through its magnesium enolate. Thus, the reaction of propionyl chloride with the es ter enolate leads to diethyl propionylmalonate. Thermal de- composition of th is compound with P-naphthalenesulfonic ac id yields ethyl propionylacetate (57%). Th i s modification appears to be general in that it has been extended to the u s e of aliphatic, aromatic, and car- balkoxy acyl chlorides.65a
215. p-Keto Esters by Acylation of Ester Enolates
RCOCl + Na+ [CR'2C0,C,H,]- --+ RCOCR',CO,C,H, + NaCl
The acylation of the sodium enolates of es ters (prepared by sodium triphenylmethide) with acyl chlorides gives the corresponding a,a-di- substituted D-keto esters, RCOCR'ACO,C,H,. The synthesis i s direct, and the product is free from monoalkylation products usually encountered

348 KETONES Ch. 10
by the dialkylation of P-keto esters. By this procedure, ethyl dimethyl- acetoacetate (51%), ethyl n-butyryldimethylacetate (58%), and ethyl benzoyldimethylacetate (65%) have been prepared.s23p6s' In a similar manner, the acylation of malonic ester i s performed through its magnesium enOlate.65~s6SB.6SS
216. P-Keto Nitriles by Acylation of Nitriles
RCHICN + R'CO,C,H, a R'COCHRCN + CIHSOH
In the presence of sodium ethoxide, nitriles having reactive a-methylene groups may be acylated with es ters to form P-keto nitriles. The method i s general and i s illustrated by the reaction of alkyl cyanides, where R i s C, to n-C,, with ethyl benzoate to form the corresponding alkylbenzoyl- acetonitriles in 53-60% yield.6s9 Aliphatic e s t e r s a l so react; for example, phenylacetonitrile with ethyl acetate gives a-phenylacetoacetonitrile, C6HsCH(CN)COCH, (64%).660 In the c a s e of the higher-boiling nitriles, the alcohol product i s removed by distillation, thereby increasing the yield and decreasing the reaction time.16'
The method h a s been extended to the preparation of numerous acyl- acetonitriles in the benzene, naphthalene, furan, and the thiophene series. Modifications of the procedure including the substitution of commercial sodium methoxide for sodium ethoxide and the use of an inert solvent to facil i tate stirring have been employed.66'
If the acylating es ter i s capable of undergoing self-condensation in the presence of sodium ethoxide, sodium triphenylmethide i s substituted for the latter. An example i s the reaction of acetonitrile with ethyl n-butyrate t o give n-butyrylacetonitrile (52%).""
217. Hydrogenolysis of 1, +Diketones *07
CH,COCH,COCH,CH(CH,), H,, C a t e l y s t
% CH3CH,CH,COCH,CH(CH,)l 4 2%
218. .Acid Treatment of Acinitroparaffins
NaOH H + R,CHNO, + R,C=N-ONa - R,CO 4 80-85%
0
219. Pyrolysis of Glycidic acid^^*^*'*^*'"^
METI-IODS 220-226
220. Rearrangement of a-Bromo Azides8'*'*"'**
Where R equals ethyl, n-butyl, or cyclopentyl, over-all yields of 35%, 77%, and 60%, respectively, have been obtained.
221. Degradation of Disubstituted Glycolic Acidss4"
222. Hydrolysis of gem-Dihalides46*46' (cf. method 151)
223. lsomerization of Vinyl Carbinolss"
RMgX C u CH, =CHCHO CH, = CHCHOHR -----* CH,CII,COR
30-50% 50-707.
224. Condensation of Furans with Unsaturated Ketones
H C Z H ,+ HC-CH 1) 11 + H,C=CHCOCH,R -4 I( 11
FH CH,C CCH,CH,COCH,R
0 \ / 0
Furans and unsaturated ketones undergo a condensation similar to the Diels-Alder type (cf. method 34) to give furyl-substituted ketones; for example, a-methylfuran and methyl vinyl ketone react under mild acidic conditions to yield 5-methylfurfurylacetone (65%).s29
225. Condensation of Anhydrides ""
226. Acylation of Certain Heterocyclic Compoundss'*

350 KETONES Ch. 10 I
1 , 227. Addition of Aldehydes to Olefins "' l
Diacetyl RCHO + R'CH =CH, - RCOCH,CH,R' peroxide
Typical compounds prepared include 4-decanone (41%), 4-dodecanone (57%), and 7-pmtadecanone (75%).
228. Interaction of Hydriodic Acid and Diazo Ketones s'7~5'B
RCOCHN, + HI -4 RCOCH, + NI + I,
229. y-Diketones from Substituted Furans 589'sv'
230. U-Diketones by Oxidation of l y l Acetylenes'"
Cr0,-CH,CO,H ArC =CAr + ArCOCOAr
231. YDiketones from KetonesSv3
Diacetyl peroxide RR'CCOCHRR' 2RR'CHCOCHRR'- I
RR'CCOCHRR'
232. Olefinic Ketones from Hydrocarbons and Carbon M o n ~ r i d e ~ ~ ~
3CH3CH,CH, + CO AlCl,, 125 atm., 12 hr. (CH,),CH = CHCH,COCH(CH,),
23%
233. a,/%Olefinic Ketones from Diketene and Aldehydes
R C H 0 CH,=C-CH, +CH,=C-C-CHR CH3COC-CHR -C01
I I 0-CO
I I 0-CO
I C0,H
CH,COCH - CHR
l METHODS 234-235
234. P - ~ e t o Es ters by the Reformatsky R e a c t i ~ n ~ ~ ' * ~ ~ ~
Zn C6H5C0,C,H5 + (CH,),CBrCO,C,H, --+ C6H5COC(CH3),C0,C,H,
(5 27.1
235. Hydrolysis of ~ - 1 m i n o n i t r i l e ~ ' ~ ~
ArCN + CH,CN NaNH, + ArC( = NH)CH,CN , ArCOCH,CN 40-609. 60-707.

35 2 KETONES Ch. 10
TABLE 32. MONOKETONES
C, Compound Method chapterref. B.p./mm., n b , (M+). Deriv. (R
C , Acetone (purification only)
C, Methyl ethyl ketone
C, Methyl n-propyl ketone
Methyl isopropyl ketone
Diethyl ketone
C, Methyl n-butyl ketone
Methyl isobutyl ketone
Methyl sbutyl ketone
Methyl I-butyl ketone
Ethyl n-propyl ketone
C7 Methyl n-amyl ketone
Methyl isoamyl ketone
4-Methyl-2-hexanone
Aliphatic Ketones
TABLE 32. MONOKETONES 353
TABLE 32 (mntimed)
c, Compound Method ~ h a ~ t e r ~ ~ ~ . B.p./mm., nb . (MP.), Deriv. (S.)
Aliphatic Ketones (contirered)
C 7 Methyl neopentyl ketone 182 56 10"' 125/760, 1.4018" 222 96 1 0 ~ 122, lOODn
Methyl t-amyl ketone 179 36 10'" 130/733, 1.4100, 112Dn 3,4-Dimethyl-%pentanone 184 36f loan 138, 1.4094*, 113Seb Ethyl n-butyl ketone 179 70 10'~' 148/756, 103Se
181 89 10"' 148, lOlSe 186 46 10" 14/767, 1.4092*
. 195 48t 104s Ethyl isobutyl ketone 189 70 10- 135/735, 1.407*, 152Seb
195 48t 10- Ethyl S-butyl ketone 179 63 10" 78Dn
184 78 10'" 136/760, 1.402*, 137Seb Ethyl t-butyl kemne 190 78 loa7 125/729, 1.4052, 144Dn Di-n-propyl ketone 179 70 10'" 144/7%, 132Se
186 50 10"' 145/767, 1.4069, 134Se 225 60 10s' 145
n-Propyl isopropyl Le- 184 79 10'" 136/760, 1.4075, 119Se tone 189 60 10" 132, 119Se
Diisopropyl ketone 179 74 10'" 125/742, 1.4001, 98Dn 184 78 10'" 125/7GO. lGOSe 187 58 1 125, 1GOSe
C' Methyl n-hexyl ketone 179 % 10'" 173, 1.4154 181 95 10'" 172, 121Se 184 70 10'" 172, 122Seb 200 91 1 0 170
Methyl isohexyl ketone 184 47t 10'" 171, 1.4146 184 77 1 0 ' ~ 164/74, 154Se . . . . . . . . 10'" 164/757, 1.414419, 77Dn
f Methyl-2-heptanone 179 68 10"' 162/760, 1.415, 82Se 3,4-Dimethyl-2-hexanone 191 20 loU9 158, 120Se
196 80 1 0 ' ~ 155, 1 1 S e 1% 90 1 0 158, 126Se
4-Ethyl-lhexanone 195 48t loU3 3-Methyl-3-ethyl-2- 189 48 10'" 79/20, 1.420Gb, 7 4 h
pentanone Ethyl isoamyl ketone 183 40 loaa 163, 132Se
1% 92 10dn 160. 132Se )-Methyl- f hcptanone 1% 94 10" 161 Ethyl neopentyl ketone 189 51 loU' 92/150, 1.410*, 136Dn n-Propyl n-butyl ketone 201 25 lonT 170. 96% n-Propyl isobutyl ketone 217 42 10"' 150/750, 124Se n-Propyl L-butyl ketone 179 41 10'" 1 2 4 h
190 67 1oU7 145/738, 1.4107, 116Dn Isopropyl sbutyl ketone 179 68 1 0 ' ~ 65/50, 1.4080, 71Dn
189 70 1ou4 145, 1.4059
For explanations and symbols see pp. xi-xii.

TABLE 32. MONOKETONES 35 5 354 KETONES Ch. 10
TABLE 32 (continued) TABLE 32 (cvntinued)
c, Compound Method ( X ) Chapterref. B.p./mm.. n h , (up.), Deriv. c, Compound Method c%) ~ h a p t e ~ e f . B.p./mm.. n b , (MP.). Deriv.
Aliphatic Ketones (continued) Aliphatic Ketones (continued) --
C,, Di-n-undecyl ketone 184 98 10'" (69). *Ox* (l awon e) 184 55' 10" (69)
186 93 10"' (69)
C m Di-n-uidecyl ketone 184 97 10" (79). 51-Ox* (myristone)
C,s Di-n-heptadecyl ketone 186 95 loUS (89h 63- Or* (stearone)
lsopropyl I-hutyl ketone 190 l98
Methyl n-heptyl ketone 181
185 186
4-Methyl-2-octanone 182 3-Methyl-3-ethyl-2- 189
hexanone Ethyl n-hexyl ketone 1%
19 5 5-Ethyl-fheptanone 187
195 Dilrbutyl ketone 184
186 n-Butyl isobutyl ketone 188 n-Butyl I-butyl ketone 190
198 Diisobutyl ketone 1% Isobutyl sbutyl ketone 184
188 Isobutyl I-hutyl ketone 198 Isopropyl neopentyl 193
ketone Isopropyl I-amyl ketone 189 Di-t-butyl ketone 179
185 189 198
Methyl n-octyl ketone 196 sym-Teuaethylacetone 225 Di-n-amyl ketone 225
184 184 186
Methyl n-decyl ketone 185 Di-n-hexyl ketone 184 Methyl n-undecyl ketone 185
Di-n-heptyl ketone 184 Di-n-octyl ketone 184 Methyl n-he~tadecyl 185
ketone Di-n-nonyl ketone 184
Di-n-decyl ketone 184
Alicyclic Ketones
C Cyclobutanone C S Methyl cyclopropyl
ketone Cyclopentanone
C 6 Methyl cydohutyl ketone
C, Methyl cyclopentyl 179 54t loin ketone
3,fDimethyl- l-cycle 186 30 1oSs4 pentanone
l-Ethylcyclopentanone 184 64 10'" 2-Methylcpclohexanone 179 85 1 0 ' ~ f Methylcyclohexanone 179 90 1 0 ' ~
179 78 1 0 ' ~ 179 88 10"'~ 196 100 104"
4-Methylcyclohexanone 179 74 10ln 179 70 10" 179 70 loin
Cycloheptanone 186 40 10'" 199 63 1 0 ' ~
pmtanone 2-Methyl-5-ethylcyclo- 184 88 loam
pentanone . Methyl cyclohexyl ketone 179 85 10'"
185 66 t 10'" 2-Ethylcyclohexanone 179 86 10""
184 74 10'" 192 41 10'" 198 43 10-
For explanations and symbols see pp. xi-xii.

KETONES Ch. 10
TABLE 3 2 (continued) --
c n Compound Method chapterref. &~./mm., n b , (Up.), Deriv. (7-3 --.--p
Alicyclic Ketones (continued)
3,+Dimethylcyclohex- anon l
3,>Dimethylcyclohex- anone
C 9 a-Methyl-a-cyclop~ltyl- acetone
* 2,2,5.>Tetramethylcy- clopentanone
2-n-Propylcycloher anone
3-~~Propylcyclohex- anone
3-Isopropylcyclohex- anone
4-n-Propylcyclohex- anone
CIsopropylcyclohex- anone
3-Methyl->ethylcyclo- hexanone
2,2,GTrimethylcyclohex- anone
c10 2,2.G6Tetramethylcyclo- hexanone
cis-Decalone 2-Decalone
C 1, Dicyclopentyl ketone l-Methyl-2-decalone
C La 4-Cyclohexylcyclohex- anone
---
Aromatic Ketones
C8 Acetophenone 178 83 10' 88/16, (20) 178 86 10" (19), 60-Ox* 183 6 3 10'* 187 70 loss 205/760. 1.541, 199Se
T A B L E 32. MONOKETONES 357
TABLE 32 (cvntirmed)
'42 Compound Yield Chapterref. B.p./mm., n b , (M.P.), Deriv.
(S.)
Aromatic Ketones (continued)
C8 Acetophenone (CO*
timed)
Cg Methyl benzyl ketone
Phenyl ethyl ketone
bhdanone
C, Phenyl n-propyl ketone
Phenyl impropy1 ketone
Ethyl b e n ~ l ketone Ben zylacerone
For explanations and symbols see pp. xi-xii.

K E T O N E S
TABLE 32 (cvnfimed)
Ch. 1 0
G2 Compound Method (%l Chapterref. B.p./mm, n 6 , (up.) . Deriv.
Aromatic Ketones (continued)
2,ODimethylacetw phenone
2,?Dimethylaceto- phenone
3,ltDimethylaceto- phenone
3,SDimethylaceto- phenone
a - T e t r d one
Cll Phenyl n-butyl ketone
3 Phenyl-2-pentanone 4 Phenyl->pentanone 5-Phenyl-2-pentanone Phenyl isobutyl ketone Phenyl scbutyl ketone Phenyl f-butyl ketone
FPhenyl-f pentanone Pi valophenon e f Methyl-3-phenyl-2
butanone fMethyl-&phenyl-l
butanone 2,4,9Trimethylaceto-
phenone 2,4,GTrimethylaceto-
phenone 2-Phenylcyclopmtanone
T A B L E 32. MONOKETONES
TABLE 32 (continued)
Compound Method C.hapterref. B.p./mm., n b , (M.P.), Deriv. (7.1
Aromatic Ketones (continued)
1Methyl- l - teual one
f Methyl- l-tetralone
4Methyl- l-tetralone 7-Methyl-l-tetralone
Phenyl neopentyl ketone mPropylpropiophenone Mesirylacetone
p-n- Butylacetophenone p-Isobutylacetophenone p-S-Butylacetophenone p-f- Butylacetophenone Ehletbyl-5-isopropylact
tophenone Acetodurene
Acetoisodurene Acetoprehni tene 2Phenylcyclohexanone
lt Phenylcyclohexanone Methyl clrnaphthyl ketone
Methyl &naphthyl ketone 6-Acetyltetralin
Benzylpinacolone p+Amylacetophenone pIsoamyl acetophenone p-* Amylacetophenone p-l- Amylacetophenone Acetopmtamethyl-
benzene Ben zoph en one
For explanations and symbols see pp. xi-xii.

KETONES
TABLE 32 (continued)
Ch. 10 T A B L E 32. MONOKETONES
TABLE 32 (continued)
Compound Merhod Chapterref. B.p./mm., n b , (Up.), Deriv. (so)
Aromatic Ketones ( m n t i w d )
C ,, Benzophenone (mm 1 s 87 t inued) 187 57
222 89 Ethyl a-naphthyl ketone 187 37
G Propionyl teualin Fluorenone
Cl, Phenyl benzyl ketcue (desoxybenzoin)
p-Methylbenwphenone 4-Phenylhexahydroacc
tophenone p-Cyclohexylaceto-
phenone PAcecyl biphenyl f Acetyl biphenyl
C l, Benzylacetophenone Di benzyl ketone
a,a-Diphenylacetone Di-c-tolyl ketone 0-Ethylbenzophenone ~ E t h y l benwphenone 0.P'-Dimerhylbenro-
phenone Ethyl Cbiphenylyl
ke tme ZAcetylfluorcne
C16 p-mPropylbenzo- phenone
Compound Merhod (%) ~ h a ~ t e ~ ~ ~ . B.p./mm., n b , (Up.), Deriv.
Aromatic Ketones (continued)
C ,, pIsopropy1 benzo- 178 phencne 187
Mcsityl phenyl ketone 183 l- Acetylphenanthrene 187 EAcetylphenanthrene 178
l78 f Acetylphenanthrene 178 pAcetylphenanthrene 184
l87 P Acetylanthracene 178
C ,, p-+Butylbenzaphenone 178 p - s Butyl benzaphenone 178
187 p-t-Butyl benmphenone 178 Benwyli sodurene 178 Phenyl a-naphthyl ketone 178
l78 ZPropionylphenan- 178
thren 178 l87
f Propionylphenan- 178 threne 187
PPropicnylph man- 187 threne
pPropionylanthracene 178
C, Laurophenone 187 p - s Amy l benzoph enone 178 2,ZDiphenylcycloher 201
anone
C Dimesityl ketone 189 Phenyl fbiphcnylyl 187
ketone Phenyl kbiphenylyl 178
ketone 1-Benzoylacenaphthcne 190 f Benzoylacenaphthene 178
c a1
/3,~Diphcnylpropiw 17 8 phencue 191
Di-a-naphrh~l ketone 187 1-Benzoylphenanthrene 178 PBenzoylphenanthrene 187 f Benzoylphenanthrene 178
l87
For explanations and symbols see pp.
5 5 40 83 85 15 53t 64 83 t 5 9 60
69 88 50 74 78 5 2 86 2 3 45t 77 23 22 86
1 1
90 60 98
5 6 46
75
75 70
85 70 75 8 85 2 0 GO -- xi-xii.

362 KETONES Ch. 10
TABLE 32 (continued)
c, Compound Method ~ h a p t e r ~ ~ ~ . B.p./mm., n b , (M.P.). Deriv. ( %)
Aromatic Ketones (continued) --
C ,l 9Benzoylphenanthrene 187 65 10" (m) 9Anthraphenone 178 65 10'' (148) 2,f Diphenyl-l-indenone 19 71 2'' 238/6, (151)
C +( Sttaroylbenzene 178 65 10' (65) C M Phenyl uiphenylmethyl 201 96 loUS (180)
ketone
C symTeuaphmylacetone 189 52 lo4' (134)
189 36 10'" . .. 39 10'" (134)
C 33 Pentaphenylacetone 189 70 lo4' (181)
Heterocyclic Ketones
C 4 3-Thiophanone 560 22 39' 85/24, 19% C6 1Acetylfucan 178 6 6 10" 48/5, (32), 150%.
178 48 lo6' 90/43, 1.5015'O, (32) 178 77 1 0 " ~ 48/5 178 76 10" 48/5, 22ODn 189 28 1ou1 58/3 199 75 1oP6 169173, 148Se
lAcetylthiophme 178 70 10" 88/8, 1.5666
178 8 3 106' 91/9, 1.566 178 79 1 0 90/10. (10.5). 1.5662 178 73 10" 81/7 178 86 loBP 78/4, 1.5666
C7 ~Fury l ace tone 195 40 loam 180 Ethyl 2-fucyl ketone 178 52 10" 77/17, (28). 189%
178 81 lo6* 6316 189 6 1 10" 82/15, 189% 199 100 1oU6 183, (30), 189Se
1Acetyl->methylfuran 178 42 1 0 73/8, 191Se G Thienylacetone 219 87 10'~' 106/12, 1.5366'~. 195Se Ethyl 2thienyl ketone 178 79 10 '~ 89/6 2-Acetyl->methyl- 178 91 1 0 83/2, 1.5622, 217%
thiophene Methyl 2-pyridyl ketone 184 50 loam 190. 121-Ox* Mechyl f pyridyl ketone 184 81 loam 218, 137Ph*
184 96 10'" 92/5, (14), 177HC1 186 36 1 0 ' ~ 108/23 187 50 10'" 220, 11301
Mehyl kpyridyl ketone 184 80 1 0 ' ~ 212, 1 4 l h *
C O n-Propyl 2-fury1 kemne 178 93 10" 78/7 1-(a- Fury1)-2butanone 195 70 106* 76/12, 1.4680" l-(a-Teuahydrofury1)-3 1% 73 10" 81/2, 1.4459'~
butanone
I TABLE 33. DIKETONES 36 3
TABLE 32 (continued)
cm Compound 1
Method (%) Chapterref. B.p./mm., nD, (M.p.). Deriv.
Heterocyclic Ketones (cuntirmbd)
C. >Methyl-2propiofuran 199 100 1 0 " ~ 96/14, 164% n-Propyl l thienyl ketone 178 89 1oa4 96/4 n-Propy1 fpyridyl ketone 189 30 10'" 98/3, 1.5128, 104Pi f Pyridylacetone 186 40 10'" 123/1, 185Se
C9 >Fury1 lchienyl ketone 178 66 10" 136/3. 1.669414 2-Fury1 2-pyrryl ketone 189 42 10* 144/1.5, (70) rrPropyl f pyridyl ketone 187 40 10'" 98/3, 1.5136. 130Ph 2n-Butyrylpyridine 195 81 10"' 217.1.5078,75Pi
C, Methyl 2bmzofuryl 178 37 10'' 119/5, (72), 207% kemne 570 80 3sM 136/11, (W), 154Ph
f Acetylthianaphth m e 178 70 10" 137/3, 250%
Cll 2-Benzoylfucan 178 70 1 0 150/3, (44), 122-0s Phenyl l thienyl ketone 178 90 10" 209/40, (56), 9 f O x 1Acetylquinoline 201 62 10''' (46), 54Ph f acetylquinoline 184 95 10'" (98.5) 8-Acetylquinoline 184 52 1 0 1 16/0.7, (43.5). 253Dn
C 1Acetyldibazofnran 178 57 10" 220/18 2-Acetyldibenzochicr 178 25 lon (112), 235%
phene
For explanations and symbols s ee pp. xi-xii.
TABLE 33. DIKETONES
c, 1
Compound ~ ~ & ~ d chapterref. B.p./mm., n D. (MP.), Deriv. (%)
Aliphatic Diketones
CS Acetylacetone 203 45 10'" 136 203 54 1oBU 141/758 203 85 1oBa 136, 150-Ox*
C, Dipropionyl 179 70 10ln 35/10, 18>0x* Propionylacetone 203 35 loUD 157
203 46 1 0 ' ~ 157/754, 193Cu 203 60 loS4 158, 198Cu
Acewnylacetone 229 M loBR 79/15, 89/25 Methyldiacetylmechane 203 32 loS4 79/30
For explanations and symbols see pp. xi-xii.

KETONES
TABLE 33 (continued)
Ch. 10
c72 Compound "'ld (%) chapterref. B.p./mm.. 4. (Up.), Deriv.
Aliphatic Diketoner (continued)
C7 n-Bucyrylacetone 203 45 10- 203 48 lose
Isobutyrylacetone 203 30 1oS4 203 41 low m 3 54 iom
f Methyl-2,4-hexane 203 31 low dione 203 45 10-
203 60 10- 3 Methyl-2,Fhexanc 184 83 loam
&one Diacetylerhylmethane 198 30 10-
C B bvaluylacetone 203 62 10- Propionyl-m butyryl- 203 70 1osU
methane f Methyl-2,4heptanedione 203 44 lose
203 47 10- Isovalerylacetone 203 64 10- Pivaloylacetone 203 43 1oSU Diisobutyryl 181 27 10"' Isopropyldiacetyl- 198 35 1 0 ' ~
methane
C9 Caproylacetone 203 54 1 0 ' ~ 203 61 10IU
Di-n-butyrylmechane 203 76 1 0 ' ~ Merhylpropionylbucytyl- 203 46 lose
methane Propionyl-isovaleryl- 203 75 10'"
methane Diirobutyrylmethane 203 28 lose ~Bucyldiacetylmechane 198 38 1oSa
203 53 loS* 203 67 lobn
Diaceryldiethyhethane 198 32 loSm
Cm Dipivaloyl 179 36 10'- 179 50 10la
C,, 2,5-Undecandione 229 86 10" (33) Diisovalewlmechane 203 76 loS4 116/20. 1.4565"
Alicyclic Diketones
CS Cyclopentan- 1,2-dione 184 67 10'" 97/20 C &Methyl-cyclopentan- 184 65 10'" 98/17
TABLE 33. DIKETONES 365
TABLE 33 (continued)
Gz Compound Method '"ld (%) ~hapter'ef- B.p./mm.. n:. (Up.), Deriv.
Alicydic Diketones (continued)
C6 1,f Cyclohaanedione 1,4-Cyclohexanedione
C, l,>Cycloheptanedione C' Tetramethyl-l , fcyclo
butanedione 2-Acetylcyclohexanone
5,5-Dimerhyl-1.3 cyclo- hexanedione
Cg 5-Iropropyl-1,fcyclo hexanedione
2-Propionylcyclohexanone
clo ~ ~ t h ~ l - ~ b p r o p ~ l - l , + c y - clopentanedione
Aromatic Diketones
CQ Acetylbenzoyl
Ninhydrin (uiketohy- drindene)
C l-Phenyl- 1.2-butanedione Benzoylacetone
C,, ~ P r o p i o n y l a c c tophenone
For explanations and symbols see pp. xi-rii.

KETONES
TABLE 33 (conlinued)
Ch. 10 TABLE 34. OLEFINIC KETONES 367
TABLE 34 (continued)
c, CO mpound Method chapterref. B.p./mm., ntD, (Mp.), Deriv. ( %) c, Yield
Compound Method (X) ~ h a p t e ~ e f - B.p./mm., ntD, (MP.). Deriv.
Aromatic Diketones (continued) - -
Aliphatic Olefinic Ketone6 (continued)
C Methyl isopropmyl 36 80 2lR 58/200. 1.4232 ketone (continued) 200 91 1O9O
Cl, Dibenzoylmethane 203 71 1oSP 202 80 loU' 222 5 9 t 1om 183 75 10- 179 83 10'"
1% 76 10- 179 47 10'" 178 45 10" 178 81 loop
Heterocyclic Diketones
Diphmyl triketone 4-Methyl benzil Meeityl t- bury1 ketone
184 4 8 t 205 31 188 42 187 25 .... 15 20 65 36 8 7 36 90 36 80 36 loo
C,, 1,2-Dibenzoylethane p-Tolil P,P'-~iaceryl biphenyl
4 H e x m - 3 one 1.2-Diacerylerhylene >Methyl- l -p rn tm-3 m e f Methyl-f pmten-Zone
4-Methyl-f pmten-Zone (meeityl oxide)
C7 tr-3 Heptrn-lone FHepteo-2-me (crotyl-
acetone) f Methyl- l-hexen- F o n e ?Methyl-thexen-f one
Tetrahydrohroylacetone Aceryl-2-thenoylmethane
?Methyl--5hexen- 2-one (methallylacetone)
3.4-Dimethyl-f peaten- C ,, Di-2-thenoylmethane
2- Furoyl-lthenoyl- methane Z m e
3,4-Dimethyl-3pmten- l o n e
3,4-Dimethyl-4-pmte~ Zone
3 ,~Dimethyl -Cpmtm- l o n e
4,4-Dimethyl- l-peaten-
For explanations and symbols eee pp. xi-xii. I
TABLE 34. OLEFINIC KETONES f one
C O 3 Methyl-f heptm->me f Methyl-f heptm-)-me 4-Methyl-bheptm-3 one 2-Methyl-2,Fheptadien-
t o n e f Ethyl-Fhexen-Zme %Ethyl- l-hexen-3 m e 3,4-Dimethyl-f h e m -
>,one 5,FDimethyl-f hexen-
l o n e 4,FDimethyl-4-hercn-
3-one 4,FDimethyl-Fhexeo-
f one
Aliphatic Olefinic Ketonee
C, Methyl vinyl ketone 26 8 1 t 2"' 81/734 36 1 5 t 2m 81, 1.4095"
15 10"' 81,1.4095",140%* 181 6 3 10""
C, Methyl propenyl ketone 36 42 2" l l P 1 2 5 Ethyl vinyl ketone 178 22 lo9' 102/740, 1.4192, 129Dn Methyl isopropenyl 24 9 8 2- 38/85, 1.4235, 173% 1 8 1 h
ketone 26 92 24n 971734
For explanations and symbols see pp. xi-xii.

KETONES
TABLE 34 (continued)
c, Yield
Compouud Method (%) ~ h a p t e r ~ ~ ~ . B.p./mm.. n b , (Up.). Deriv. P
Aliphatic Olefinic Ketones (continued)
C9 FMethyl-5-octen-&one 36 45 2'* 86/25. 1.4413 5- Ethyl-4hepten-3-one 189 74 10- 1 19/740. 10% 2.3 Dimethyl-bheptar 184 86 10"' 76/13, 163Se
Gone 3-Propyl-f hexen-tone 19 68 2" 7 U 9 , 142% 2,4,FTrimethyl-4- 178 40t 10'~' 174/755
hexen-f one
Alicyclic Olefinic Ketones
10'" 53/12. 220% 10"' 161/760. 1.4771. 127-01 279 68/22. 172%. 163Dn
1 0 ~ 67/25. 1.4879. 168Se. 117Dn
10'" 74/12. 211% C 7 l-Aceryl- l-cyclopen- tene
2 .3 Dimethyl-tcyclopm- tenone
f Methyl-kyclohexen- l-one
C. l-Cyclopentenylacetone a -Pr~p~l idenecyc lopm-
tanone 2.2, f Trimethyl-4-cyclo-
pentenone f Ethyl-2-cyclohexenone 3 ,SDimethyl -kyc lo
hexen-l-one I-Ac etyl- 1- cyclohexen e
C 9 fMerhyl-2.n-propyl-l- cyclopentenone
l-Propionyl- l -cycle hexene
2-Allylcycloheranone
fn-Propyl-bcycloher e n m e
31so~ropyl-Z cyc loher enone
f ~ e t h ~ l - F e t h y l - 2 - c y c l e hexm-l-one
T A B L E 34. OLEFINIC KETONES 369
l TABLE 34 (calinuedl
I C, Compound eth hod (S chapterref. ~.p./mm., n b . wp.) . ~ c r i v . Yield
Alicydic Olefinic Ketmes (continued)
C m 2,EDimeQyl-1-acetyl-l 204 5 6 1 0 ~ ~ 118/49. 1.4810". 2OlSe cyclohexene
Cl, ~Cyclohe.yl idenecyclw 36 70 zUO 150/22. 1.5084". 188% hersnone
Cp Phenyl vinyl k e t m e C m Phenyl propenyl ketone
Benzalacetone a-Methylacrylophenone
Cll Isopropylideneacc tophenone
C,, l-Phenyl-l-hexcn-)-one l-Phenyl-4-hexm- l-one +Phenyl- l-hexen-Fone Phenyl >methyl-+
butenyl ketone o-Methylstyryl ethyl
L a m e
Cl, Benzalpinacolme l-Benwyl- l-cyclohexene
C Benzalacetophenone (chalcme)
C16 bmwDibenwylechylene 2.4-Diphenyl-2-buten-
t o n e C17 Di benzalacetme
P
Aromatic Olefinic Ketones
20 78 2'" 178 6 1 1 0 ~ ~ 95/2 36 78 2'4 128/8. (42) 26 70 2=' 60/3. 1.5354
178 35 10- 106/5. 1 . 5 5 7 9 ~ 178 40 10 lm
194 40 10- 121/4, 1.559819, 168PN 205 88 loD' 99/0.30, 1.5458". 132Se 205 83 10'~' 97/1. 1.5270", 130Se 205 74 10'~' 86/1.1.5193". 103Dn 205 76 10'~' 100/2.1, 1.5223". 177%
Heterocyclic Olefinic Ketones
C, Furfiualacetme 36 6 6 2m 116/10, (38) C Furfuralacetofuran 36 89 2" (90) C Furfuralacetophenone 36 90 2- 179/7,(26)
>'Ihenalacetophenone 36 96 zUI (59)
For explanations and symbols see pp. xi-xii.

KETONES Ch. 10
TABLE 35. ACETYLENIC KETONES
c, Yield
Compound Method (%) Chapterref- B.p./mm., n;, (Up.), Deriv.
C, Methyl ethynyl ketone 179 40 loam 86. 181Dn, 143PN C S + Prntyn-lone 179 6 7 loa4 74/95, 1.4380m, 149Dn
C 6 n-Propyl ethynyl ketone 179 70 loam 66/100, 1 3 7 h
C8 3Octyn-lone 179 80 loam 76/15, 88Dn, 109% 188 5 8 t 10'% 76/15, 1.4446"
C9 + N o n y e l o n e 188 55t 1 0 ' ~ 87/13, 1.4463" Wenyl ethynyl ketone 179 80 loam (51). 214Dn
Cl, 4-Phenyl-+ b u t y e l o n e 188 45t 10'- 102/3, 1.5735" 188 55 10'" 125/14
Cl, Phenyl phenylethynyl 189 74 10414 (55) ketone 193 85 lou4 (66)
For explanations and symbols s e e pp. xi-xii.
TABLE 36. HALO KETONES
c, Compound Method Chapterref. B.p./mm., n b (h4.p.). Deriv. (S.)
Niphatic and Alicydic Halo Ketanes
C , Chloroacetone 66 72 449s 120 184 10-
Bromacetone 66 44 4"' 42/13 apt-Lli bromoacetone 66 60 46'4 98/22, (26.5) a ,y-Dichloroacetone 179 75 10'" 175, (45). a ,a ,at-~ribromoacetone 66 60 4'Y 116/14, (29) Hexafluoroacetone 182 60 1 0 57/93, 1.3288
hydrate
C Methyl a-chloroethyl 66 62 44" 113, 1.4171 ketone
Methyl a-bromoethyl 66 50 4& 34/12, 1.4571 ketone
Methyl Pchloroethyl 73 67 4Ia4 50/15 ketone 207 40 1 0 ' ~ 48/15
Chloromethyl ethyl 66 21 44% 138, 1.4372 ketone
Ekomomethyl ethyl 57 55 4''9 155, 1.4670 ketone 66 17 4a 50/12, 1.4670
Chloromethyl P c h l o r w 207 45 1 0 81/2.5 ethyl ke tme
Chloromethyl Piodoethyl 57 84 (55) ketone
=.a'-Dibromodiacetyl 66 71 44es (117)
T A B L E 36. HALO KETONES
TABLE 36 (cunfinued)
c, hl thod Yield
Compound (K) Chapterref. B.p./mm., n b . (Up.), Deriv.
Niphatic and N i c y d i c Halo Ketmes (continued)
C S Methyl a-chloro-rrpropyl ke tme
Methyl y-chlorcm- propyl kemne
Methyl a-bromo-n- propyl ketone
Chloromethyl n-propyl ketone
Bromomethyl n-propyl ketone
Methy l a-chloroisopropyl ketone
Methyl a-bromoi sopropyl ketone
Bromomethyl i sopropyl ketone
l-Bromo-5.chlor-l pentanone
Ethyl ,&chloroethyl ketone
a-Chloroethyl Pchloro- ethyl ketone
Di-Pchloroethyl ketone Bromoethyl Pbromoethyl
ketone 2,+Dibromo-3-rnethyl-
2-butanone 1,5-Di bromoacetyl-
acetone Acetyltriiluoroacetme
Bromomethyl imbutyl ketone
2-hlthyl-l-chloro-+ pentanone
2-Chlor-%methyl-4- pentanone
2,fDibromo-+ methyl-l pentanone
l-Chloro-3,+dimethyl- l h t a n o n e
l-Bromo-3,+dimethyl-1 butanone
For explanations and symbols see pp. xi-xii.

KETON E S
TABLE 36 (mntinue4
Ch. 10
C, Compound Method Yield
(%) Chapterref- B.p./mm., ntD, (Up.), Deriv.
Aliphatic and Alicyclic Halo Ketones (cwrlimrcd)
1 Bromocyclohexanone
2-Chloro- f heptanone 1- Bromo-Gheptanone 3 Methyl+ bromo- 2-
h exanon e 3.4-Dimethyl-Cchlom
2-pmtanone
C., Chlommethyl n-hexyl ketone
f Bromo-3-methyl-4- heptanone
E Ethyl- l-chlom-3 hexanone
4,SDimethyl-Schloro- f hexanone
Methyl a- bmmocyclo- hexyl ketone
Bromomethyl cyclo- hexyl ketone
l-Acetyl- 1.2-dibromo- cyclohexane
l-(Dibromoacetyl) l- bromocyclohexane
C l-Bromo-stridecanone
Aromatic Halo Ketones
C., w-Fluoroacetophenone w-Bromoacetophenone w-Dichloroacetophenoae w -Di bromoacetophenone w-Trifluoroacetophmone w-Trichloroacetophenone
m Bromophenacyl bromide p-Bromphenacyl bromide o-Chloroacetophenone
I T A B L E 36. HALO K E T O N E S 373
I TABLE 36 (continued)
l Yield c, Compound Method chapterref. B.p./mm., nfD. (MP.), Derir.
(%) -
Aromatic Halo Ketones (continued)
C n o-Bromoacetophenone 56 80 4"' 112/10, 177Se 187 80 1 0 189Dn 212 65 1OW 117/12, 175Sea
m-Chloroacetophmone 56 83 4'" 113/11. 1.5494. 183 76 1 0 ' ~ 92'3, 232Sea
mBromoacetophenone 56 56 4'" 132'17. 1 . 5 7 5 5 . 2 3 s ~ mIodoacetophenone 56 53 4"' 117/4, 1.6220 p-F1 uoroacetophenone 178 74 10'" 79/10, 1.5081"
178 76 loll1 196. 219% p-(aloroacetophenone 178 78 10LU 126/24
178 83 10" (12), 204Se' p-Bromoacetophenone 178 79 10'" 117/7, (50.5), 129-Ox* p-Iodoacetophenone 56 52 4'" 140/9, (84)
178 95 10"' (85)
acetone a-Bromo-a-phenyl- 66 69 4- 127/7
acetone Chloromethyl benzyl 57 85 4'"O 135/19, %/l
ketone Bromomethyl b m y l 57 62 4'19 10U0.2, 1 .559319"
ketone a-Clloropropiophenone 178 66 1 0 133/26 a-Btomopropiophenone 66 42t 4643 139/20,1.5686" P-Chloropropiophenone 178 6 5 10lm (50)
178 85 10'" (48) p- Bromopropiophenone 178 9 3 10"' (59) a.a-Dikomopropio- 66 83 4 180/64,(30.5)
phenme a , P D i bromopropio- 178 98 1 0 ' ~ (56)
phenone o-(lhlorobenzyl methyl 189 60 1oW9 130/15, 120-01
ketone p-Chlorobenzyl methyl 178 16 10"' 86/1
ketone o-Chloropropiophenone 56 85 4'U 106/12, 173Se o-Bromopropiophenone 56 77 4'U 118/11, 179Se mChl oropropiophenone 56 73 4 333 (46),180%
m Bromopropiophenone 56 44 4"' (40), 183% p-Clloropropiophmone 56 76 4"' 118/2, (35), 177%
p-Bromopropioph mone 56 58 4 140/2, (G) , 171Se
p-Methylphenacyl bromide 6 6 94 4=' (50) p-Acetobenzyl bromide 54 46 4 lW 136/5 m Trifluoromethylacc 187 50 10'" 202
tophenone 189 91 1 0 3 202
For explanations and symbols s e e pp. xi-xii.

KETONES Ch. 1 0
TABLE 36 (continued)
Compound Method Cbaptedef- B.p./mm.. n b , (Kp.). Deriv. (%)
Aromatic halo Ketones (continued)
Clo a-Bromolrpropyl 66 98 4'* 154/23 phenyl ketone
Chloromethyl ,&phenyl- 57 85 4'w (40). 1 4 6 h ethyl ke tme 179 82 10'" 111/5, (41). 1 4 7 h
4-Phenyl-f chloro-2- 184 60 f 1 0 99/4. 1.5268, 139Dn but anon l
4-Phenyl-+ bromo-2 66 81 4- 155-160/30 bu tanone
Benzalacetone &chloride 74 34 4"O (93) Benzalacetone dibromide 74 57 4 0 9 (125) 1,f bis-Chloroacetyl- 57 83 4'= (98)
benzene
C ,, a- Bromoisobutyl phenyl 66 80 4- 145- 155/20, (52) ketone
C a-Bromoacetylnaph- 6 6 80 46s 215/15 thalene
C,, a-Bromoisobutyryl- 178 70 l 170/24 mesitylene
o-Chlorobenzophenone 178 8 6 t 0 180/15,(44) o-Bromobenzophenone 178 52 10" 153/0.05, 13f Ox*
178 80 loU1 190/14 p-C3lorobenzophenone 178 82 10"' (78). 106Phe. 185Dne
C,, Phenyl a-chlorobenzyl 53 79 4'" (67) ketone 62 65 4Y" (68)
o-Chlorobcnzyl phenyl 190 73 10- (71), 8 6 0 . ketone
m-Chlorobenzyl phenyl 190 42 10"' (43), 102-Ox ketone
P-Chlorobcnzyl phenyl 190 70 1 0 ~ ' (138), 9 6 0 ~ ketone
o-Chlorophenyl benzyl 190 71 1 0 ' ~ ~ 178/5, 1 3 2 0 . ketone
mChloropheny1 bcnzyl 190 72 loU9 (62), 120-Ox* ketone
p-Chlorophenyl benzyi 190 77 10"' (108), 12fOx ketone
CCblorobenzil 183 93 105" (73) 4-Bromobcnzil 183 94 0 (87) 2.2'-~ichlorobenzil 179 3 9 t 10'~' (129)
C,, a-Chlorodi benzyl 66 80 4"' 195/12, (68.5) ketone
a-Bromodibenzyl ketone 66 99 4 (49) Benzalacetophenone di- 74 96 4"' (113)
chloride
I TABLE 37. HYDROXY KETONES
TABLE 36 (continued) A-
I
c, Yield
Compound Method (%) ~hapter'ef. B.p./mm., n b (M.P.), Deriv.
Aromatic Halo Ketones (cat inzed)
C a- Bromo-4-propionyl- 66 75 4=' (79) biphenyl
C ,, p o - B r o m o a c e t y l a ~ 66 50 4- (107) thracene
Heterocyclic Halo Ketones
thiophene 2Bromoacetyl- 66 80 4'w 98/1.5. 1.6258
thiophene
furan Cl, 4-@holy1 chloromerhyl 57 50 4'= (101)
ketone
For explanations and symbols see pp. xi-xii.
TABLE 37. HYDROXY KETONES
c, Yield
Compound t Method (%) Chapterref. B.p./mm.. n D , (M.p.), Deriv.
Aliphatic and Alicydic Hydroxy Ketones
C, Acetol (l-hydroxy-2 95 58 5 52.3 42/12 propanone)
C, l-Hydroxpf butanone 84 44t 5 w 74/13, 1.4302" 102 28 71/12, 1.435'~
but anm l Dimethylacetylcarbinol 89 26t 5" 140, 87-0.. 165% 2&droxycyclopentanone 104 16 5 74/10, 1.4701"
(propionoin) >Hydroxy3-hexanme 79 51 158 76/12, 1.4280'' f Methyl-f hydroxy-2 200 60 1 0 = ~ 73/50. 1.4200, 150%
pentanone
For explanations and symbols see pp. xi-xii.

3 76 KETONES Ch. 1 0
TABLE 37 (continued)
c, Compound Method Yield (%) chapteflef. B.p./mm., nb (Up.), Deriv.
Aliphatic and Alicydic Hydroxy Ketones (continued)
C, f Methyl-4-hydroxy-l penranone
4-Methyl-4-hydroxy-1 pentanone (diacerone alcohol)
2-Merhyl- l-hydroxy-f pentanone
fEthyl-4-hydrox)~> tutanone
2-Hydroxycyclohexanone
C7 OHydroxy- 2-hepranone
2-Hy droxy-4-heptanone +Methyl-4-hydroxy2-
hexanone 2-Methyl-Fhydroxy-f
hexanone 2-Hydroxymethyl- l -cydw
hexanone
C8 2-Hydroxy-4-octanone 5-Hydroxy-4occanone
(buryroin) +Methyl-f hydroxy-l
hepranone +Methyl-4-hydroxy-2-
hepranone >~eth;l-Fhydroxy-3-
heptanone 5-Methy14-hydroxy4-
heptanone 6-Methyl-lhydroxy-4-
heptanone 4- Ethyl-4-hydroxy- f
hexanone 2,2-Dimethyl-Fhydrory-
fhexanone 2,FDimethyl-4-hydrow
f hexaaone (iso- buryroin)
%(a-Hy droxy-n-propyl) cyclopenranone
C9 +Methyl-4-hydroxy-l octanone
TABLE 37. HYDROXY KETONES 377
TABLE 37 (continued)
Compound Method c h a ~ t e f l e f . B.p./m.. n;. (M.P.), Deriv. ( %)
Aliphatic and Alicyclic Hydroxy Ketones (continued)
Clo 2,2,5,FTetramethyl-4- 104 GO 5 6s6 85- 95/ 12 hydrory-fhexanone (pivdoin)
2-(l'-Hydroxycycb 102 40 5 105 99/3, ( 3 0 , 7&0x
Aromatic Hydroxy Ketones
C8 mHydroxyacetophenone 93 48 5" (95) 2,4-Dihydroxyacetw 178 65 1 0 " ~ (1 44)
phenone 2,FDihydroxyacetw 209 77 10m (203)
phenone 2,3,4-Trihydroxyact 178 57 1 0 " ~ (172)
tophenone 2,4,GTrihydmxyact 178 87 101a9 (219)
tophenone
C9 Acetylphenylcarbin01 95 72 s5* 123/13, l lfOx, 126Dn 1 9 50 lou5 137/24, 194Se, 170Dn
Methyl benzoylcarbinol 95 87 5'* 123/14, 134-Ox
a,PDihydroxypmpiw 98 90 5619 (82)
phenone o-Propioph enol 209 ' 35 10"' 115/6 p-Propiophenol 178 82 10'- (149), 170Se
209 50 10"' (148)
C,, Phenyluimethylact 105 49 5* (47) tylcartinol
C,, Benzoin 79 93 5156 (134) 79 97 5 lS7
104 92 56" (1 29) 105 30 5 m (133)
0.0'-Dichlorobenzoin 104 40 56a (57) mm'-~ichlorobenzoin 104 22 5 M (76) pop'-~ichlorobenzoin 104 88 56" (88) 4,4 ' -~ih~droxy b e n d 97 89 55U (235)
(benzaai win) --
For explanations and symbols see pp. xi-xii.

378 KETONES Ch. 10
1 TABLE 37 (continued)
c, Compound
--p
Aromatic Hydroxy Ketones (continued)
C Diphenylacetoin 187 45 10"' (52). 169 Se. 84NBz 0.p'-~imethoxybenzoin 104 73 56U (113)
(ani soin)
C17 2',4';d- rim ethyl benzoin 105 63 5w (103) C, PNaphthoin 104 78 56U (126), 172-Ox
Heterocyclic Hydroxy Ketones
C, 2-Hydroxyacetylfuran 114 74 5'" (82) Cl,, a-Furoin 104 38 5M (135)
2.2'- he no in 104 30 5m (109)
For explanations and symbols see pp. xi-xii.
TABLE 3'8. KETO ETHERS
Compound Yield
Method (%) chapterref. B.p./mm., n b , (M.p.), Deriv.
Aliphatic and Alicyclic Keto Ethers
C, Mexthoxymethyl methyl 179 29 1 0 ' ~ 115/756, 1.3982, 111PN, 1 6 3 h ketone 187 48 10'" 1 14/746,1.3980,159h*, 109pN*
C, l-Methoxyethyl methyl 187 37 10"' 116/739, 1.3936, 141Se ketone
CMethoxylbutanone 121 73 6" 66/50, 138/745. 1.4050 195 75 10ns 140/745
Methoxymethyl ethyl 187 49 10"' 133/757, 1.4063 ketone 187 59 1 0 " ~ 132, 198Dn*
symDimethoxyacetone 187 45 1 78/18, 1.4174, l2OSe Ethoxyacetone 187 65 10'" 36/28, 1.4000, 96Se*
C6 l-Methoxypropyl methyl 187 29 10"' 71/95, 1.4015~'. 147Se ketone
Methoxymethyl wpropyl 187 51 1 0 ' ~ 153/745, 1.4119 ketone
Methoxymethyl isopropyl 187 30 1 0 " ~ 144, 163Dn ketone 187 44 10'" 145/748, 1.4078
l-Methoxyethyl erhyl 187 22 10"' 136/750. 1.4019, 120% ketone
4-Ethoxy.2-butanone 121 77 6iii 150/764, 74/50 Ethoxymethyl ethyl 187 84 loan 147/752, 1.4068
ketone wPropoxymethyl methyl 187 52 10"~ 49/6, 1.4052
ketone
I TABLE 38. KETO ETHERS 379
TABLE 38 (continued)
c, Yield
Compound Method (S.)
chapterref. B.p./mm.. n h , (MP.). Deriv.
Aliphatic and Alicyclic Keto Ethers (continued)
C 6 Isopropoxymethyl methyl 187 48 10''~ 35/10, 1.4004, 144Dn ketone 187 53 10"' 142 .142h
C7 l-Methoxy-)-hexanone 187 23 10"~ 67/8, 1.4180", 70Dn Methoxymethyl wbutyl 187 34 10"' 169/744, 1.4173
ketone Methoxymethyl isobutyl 187 30 10"' 164/751, 1.4140
ketone
hlethoxymethyl sbu ty l 187 32 10"' 164/757, 1.4162 ketone
Methoxymethyl t-butyl 187 19 10'" 159/743, 1.4193 ketone
l-Methoxyethyl n-propyl 187 33 1 0 ' 155/746, 1.4091, 169% ke ton 187 73 10'" 93/100, 170Se
l-Methoxyethyl isopropyl 187 13 10"' 58/31, 1.4092, 146Se ketone
l-Methoxypropyl ethyl 187 79 10'" 63/40, 1.4080''. 145Se ketone
a-Methoxypinacolone 124 59 6"' 83/4,189Dn
wPropoxymethy1 ethyl 187 46 10"~ 56/4, 1.4122 ke ton l
Isopropoxymethyl ethyl 187 41 10'" 47/11, 1.4082, 103Dn ketone
symDiethoxyacemne 187 67 10lm 105/35, 1.4202. 91Se 1hkthoxycyclohexanone 179 46 loam 59/8, 1.4519" 4-Methoxycyclohexanone 179 65 loam 85/14, 1.4560, 178Se, l5ODn
Cs Methoxymethyl wamyl 187 46 10'" 191/753, 1.4220 ketone
Methoxymethyl isoamyl 187 71 10"' 186/752, 1.4210 ketone
l-Methoxyethyl wbutyl 187 63 10"' 82/36, 1.4160, 154Se ketone
l-Methoxyethyl isobutyl 187 21 10"' 52/9, 1.4128, 145Se ketone
l-Methoxyethyl sbuty l 187 43 10"' 77/36. 1.4158, 127Se ketone
l-Methoxyethyl I-butyl 187 14 10"' 64/34, 1.4130, 121% ketone
l-Methoxypropyl wpropyl 187 69 10"' 86/42, 1.4131". 157Se ketone
l-Methoxypropyl iso- 187 44 10'" 66/23, 1.4159, 136% propyl ketone
GEthoxy-lhexanone 184 60t 10"' 92/13. 64Dn Ethoxymethyl sbu ty l 187 29 10"' 173/743, 1.4158
ketone
For explanations and symbols see pp. xi-xii.

380 KETONES Ch. 10
TABLE 38 (continued)
c, Compound Method Yield
(%) C h a p t e P f * B.p./mm.. n;, (M.P.), Deriv.
Aliphatic and Alicydic Keto dthers (continued
C s l-Isopropoxy-3-methyl- 187 17 lom4 160, 88Dn l butanone
Methyl a - ( s tu toxy> 187 69 10'" 163/750, 1.4080, 118Se ethyl ketone
Merhoxymethyl c y c l e 187 22 1 0 " ~ 87/14, 1.4486". 129Dn pentyl ketone
C9 3-Methyl-Gethoxy-l hexanone
Methoxymethyl c y c l e hexyl ketone
Aromatic Keto Ethers
C9 Phenoxyacetone
Clo Phenoxymethyl ethyl 187 62 10'" 100/5, 1.5201, lO2Se ketone
a-Methoxypropiophenone 124 60 G 173 89-95/4, 160Dn ,&hlethoxyethyl phenyl 189 30 10"" 1.5250, 176Dn
ketone a-Ethoxyacetophenone 124 81 6'" 127/11, 128Se
187 68 10"" 122/15, 1.5250 p-Methoxypropiophenone l l 6 88 G% 152/19
178 87 10, . 125/4 p-Ethoxyacetophenone 178 77 1019 147/16, 1.5429 " 2 , F Dimethoxyacr 178 71 loU4 160/15
tophenone 3, FDimethoxyacc 190 57 10" (43)
tophenone
Cl , y-Phenoxypropyl methyl 189 78 10'~' 121/2, (50). llODn kemne
Phenoxymethyl n-propyl 187 64 10'" 112/4, 1.5148, 108Se ketone
P E h o x y e t h y l phenyl 189 82 10"" 1.5190, l6lDn ketone
n-Propoxymethyl phenyl 187 37 1 118/6, 1.5150 ketone
C,, S-~henoxybutyl methyl 184 6 1 t 10'~' 130/2, 1.507laS, lOlDn ketone
P-n-Propoxyethyl phenyl 189 82 10"" 1.5193, 1 5 8 h ketone
TABLE 30. KETO ALDEHYDES 38 l
TABLE 38 (contitwed
Aromatic Keto Ethers (continued)
Cl' ,&Isopropoxyerhyl 189 69 loa4 1.5083, 175Dn phenyl kemne
C ,&Naphthoxyacemne l 1 5 85 6" (77) C ,, Phenoxymethyl phenyl 187 45 low 187/8, (74), 187Se
ketone mMerhoxybenwphmone 179 2 5 t 10"' (38)
187 77 1 0 185/4, (40) p-Merhoxybenzophenone 178 89 10" (62.5), 180Dn p-Phenoxyacemphenone 178 6 8 10"' 154/2, (49)
16 Cl, P-Methoxyphenyl benzyl 130 74 1oU9 (77), 118-Ox
ketone 2-Methoxy bmzil 179 6 0 t 10"' (72) 4-Methoxy benzil 179 30 10'~' (63), 124-Ox
Cl, 2-Ethoxybenzil 173 6 0 t loaU (102) 4-Ethoxybenzil 173 6 0 t 10"' (71) Desoxyanisoin 221 98 10'" (1 12) 2,2'-~imethoxyben zi1 179 4 0 t 10"' (129) 3,3'-~imethoxybenzil 179 6 0 t 10"' (83) 4,4'-~imethoxybenzil 179 5 2 t 10"' (133)
(anisil) 179 97 1 0 ' ~ ~ (132), 255Se*
For explanations and symbols s e e pp. xi-xii.
TABLE 39. KETO ALDEHYDES
Yield 'n compound
t Method (%) chapterref. B.p./mm., n D, (M.p.), Deriv.
C 7 Pivaloylacetaldehyde 146 50 9 171 45/13, l 2 K u Ilydroxymethylenr 146 80 9 17s
D methyl isotutyl ketone i a- Formylcyclohexanone 146 60 9 ~ 7 4 88/14, 1.5130
! C 8 Cydohexyldyoxd 157 59 9181 72/17 J l-Methyl-3-hydroxp 146 45 9 '" 87/12 2 methylenr2-cyclo- F hexanone
? Phenylglyoxd 152 8 7 t 9'" 9 l77
(73) m 157 72 97/25
C9 p-Acetylbenzaldehyde 162 43 9r34 130Ph, 181-Ox
For explanations and symbols see pp. xi-xii.

KETONES ca. l 0
TABLE 39 fcont~nued)
l 1
I c, Compound Method Yield (%) chapterref. B.p./-.. nb. (M.p.). Deriv.
l C ,, Mesitylglyoxal 157 83 9 l" 106/4, 1.5520'~ 2-Hydroxymethylene- l- 146 94 9'" 180/28
tetralone
C,, PNaphthylglyoxal 152 30 9 189 (109) C,, p-Xenylglyoxal 152 90 9l" (121)
For explanations and symbols see pp. xi-rii.
REFERENCES FOR CHAPTER 10 383
REFERENCES FOR CHAPTER 10
'Bergmann, bchapiro, and Eschinazi, J . Am. Chem Soc., 64, 559 (1942); Al- len and Barker, Org. Syntheses, Coll. Vol. 11, 156 (1943).
'Grummitt and Case, J. Am. Chem Soc., 64, 880 (1942). 'Seidel and Engelfried, Ber., 69B, 2578 (1936). 4Mowry, Renoll, and Huber, J. Am. Chem. Soc., 68, l105 (1946). ' Bachmann, Carlson, and Moran, J. &g. Chem., 13, 916 (1948). 6N011er and Adams, J. Am. Chem Soc., 46, 1889 (1924). 'Smith and Guss, J. Am. Chem Soc., 59, 805 (1937). 'Marvel, Saunders, and Overberger, J. Am. Chem Soc., 68, 1086 (1946).
Allen, Org. Syntheses, Coll. Vol. 11, 3 (1943). 'OGroggins, Unit Processes in Organic Synthesis, McGraw-Hill Book Co.,
New York, 1947, pp. 759-770; Thomas, Anhydrous Aluminum Chloride in Organic Chemistry, Reinhold Publishing Corp., New York, 1941, pp. 205-393.
"Dermer e t al., J. Am. Chem. Soc., 63, 2881 (1941). l' Groggins and Nagel, Ind. Eng. Chem, 26, 1313 (1934). "Riddell and Noller, J. Am Chem. Soc., 52, 4365 (1930). '4Calloway and Green, J. Am. Chem. Soc., 59, 809 (1937). " Johnson in Organic Reactions, Vol. 2, John Wiley & Sons, New York, 1944,
p. 114. l6 Calloway, Chem. Revs., 17, 327 (1935). l' Johnson and Glenn, J. Am. Chem Soc., 71, 1092 (1949). "Long and Henze, J. Am. Chem. Soc., 63, 1939 (1941). l9 Bachmann and Sheehan, J . Am. Chem Soc., 62, 2688 (1940). 'OMosettig and van de Kamp, J . Am Chem Soc., 52, 3707 (1930). "Bachmann and Struve, J. Am Chem Soc., 58, 1660 (1936). " Riegel, Gold, and Kubico, J . Am. Chem. Soc., 64, 2221 (1942). "Fieser and Cason, J . Am. Chem. Soc., 61, 1742 (1939).
Fieser and Hershberg, J . Am. Chem Soc., 61, 1272 (1939). "Smith and Lo, J . Am. Chem. Soc., 70, 2209 (1948).
Chodroff and Klein, J . Am. Chem Soc., 70, 1647 (1948). " Fahim, J. Chem. Soc., 520 (1949). * Pines, Strehlau, and Ipatieff, J . Am. Chem. Soc., 71, 3536 (1949). "Kosolapoff, J . Am Chem Soc., 69, 1651 (1947). 'O Baddeley, J . Chem. Soc., S99 (1949). "Ray and Rieveschl, Org. Syntheses, 28, 3 (1948); cf. ref. 4. "Merritt and Braun, Org. Syntheses, 30, 1 (1950). "Mason and Terry, J. Am. Chem. Soc., 62, 1622 (1940). ',Read, 1. Am. Chem. Soc., 44, 1751 (1922). "Vogel, J. Chem. Soc., 612 (1948).
Fourneau and Barrelet, Bull. soc. chim. France, 47, 77 (1930). " Maxwell and Adams, J. Am. Chem. Soc., 52, 2959 (1930). "Weygand and Mensdorf, Ber., 68B, 1831 (1935).
Hennion and UcLease, J . Am. Chem. Soc., 64, 2421 (1942). "Kadesch and Weller, J. Am. Chem. Soc., 63, 1311 (1941). 41Kloetzel and Herwg, J. Am. Chem. Soc., 72, 1991 (1950). 4'Vi11iams and Osborn, J. Am Cbem. Soc., 61, 3438 (1939). 4'Immediata and Day, J. Org. Chem., 5, 516 (1940). ,,Sachanen and Caesar, Ind. Eng. Chem., 38, 45 (1946).
Johnson and Offenhauer, J. Am Chem. Soc., 67, 1046 (1945).

KETONES Ch. 10
I 1 " Hu&es, Ingold, and Taher, I . Chem. SOC., 953 (1940). 4 7 H u k r et al., I . Am. Chem. Soc., 68, 1109 (1946). -Drake and Bronitsky, I . Am. Chem SOC., 52, 3718 (1930). * Fieser and Kilmer, I . Am. Chem. Soc., 62, 1357 (1940). 'O~teyemark and Gardner, I . A m Chem. Soc., 5 5 4886 (1930). "Meyer, Org. Syntheses, Coll. Vol. I , 60 (1941). ' 'Bergmam, I . Org. Chem., 4, 4 , (1939). "Van and Wagnet, I . Org. C h e m , 9 , 163 (1944). ' 4 F u ~ ~ n e t al., 1. Am. Chem Soc., 66, 682 (1944). "May and Mosettig, I . Am. Chem. Soc., 70, 686, 688 (1948). "shildneck, Org. Syntheses, Coll. Vol. 11, 236 (1943). "Cook, I . Chem Soc., 1284 (1926).
Bachmam, I . A m Chem Soc., 57, 555 (1935). "Hartough and Kosak, I . A m hem. Soc., 68, 2639 (1946). 60 Hartough and Kosak, I . Am. Chem. Soc., 69, 1012 (1947).
"Hartough, Kosak, and Sardella, I . Am. Chem. Soc., 69, 1014 (1947). "Johnson and May, Org. Syntheses. Coll. Vol. 11, 8 (1943); c f . re f . 4. 63 Mimis , Org. Syntheses, Coll. Vol . 11, 520 (1943); re fs . 59 and 60.
-Levine e t al., I . Org. Chem.. 13, 409 (1948); 1. A m Chem. Soc., 71, 1207 (1949).
6'Hartough and Kosak, I . Am. Chem. Soc., 70, 867 (1948). "Hartough et al., 1. Am. Chem Soc.. 69, 3093, 3096, 3098 (1947); Kosak and
Hartough, Org. Syntheses, 28, 1 (1948). 67 Emling, Beatty, and Stevens, I . Am. Chem Soc., 71, 703 (1949).
" Farrar and Levine, I . Am. Chem. Soc., 72, 3695 (1950). 69 Gilman, Rowe, and Dickey, Rec. trav. ch im, 52, 396 (1933).
"Hansch and Lindwall, I . Org. Chem., 10, 383 (1945); c f . ref. 71. 71 Farrar and Levine, I . Am. Chem Soc., 72, 4433 (1950).
"Burger and Bryant, I . Org. Chem, 4, 119 (1939). " Buu-Hoi and Royer, Rec. trav. chim., 69, 866 (1950). 74Hart and Tebbe, I . A m Chem. Soc., 72, 3286 (1950). "Snyder and Werber, I . Am. Chem Soc., 72, 2965 (1950). "Lev in , Graham, and Kol lof f , I . Org. C h e m , 9 , 384 (1944). 77 Cope and Field, I . Am. Chem. Soc., 71, 1591 (1949). 18 Pacaud and Allen, Otg. Syntheses, Coll. Vol. 11, 336 (1943).
79 Martin and Fieser, Org. Syntheses, Coll. Vol. 11, 569 (1943). 80Kloetzel, I . A m Chem. Soc., 62, 1708 (1940). "Alexander and Mudrak, I . Am. Chem. Soc., 72, 3194 (1950). "Adkins and Davis, I . A m Chem. Soc., 71, 2955 (1949). " Weygand and SchGder, Bet., 74B, 1847 ( 1941). 8 4 B a c h m a ~ and Struve, I . Am. Chem. Soc., 62, 1618 (1940). 8%ewman, I . Am. Chem. Soc., 62, 1685 (1940).
Amold, Buckley, and Richter, I . Am. Chem. Soc., 69, 2323 (1947). Fieser and Seligman, I . Am. Chem. Soc., 60, 170 (1938).
"Thomas and Nathan, 1. Am. Chem. Soc., 70, 331 (1948). 89 Fuson and Walker, Org. Syntheses, Coll. Vol. 11, 169 (1943). 90 Boese, Ind Eng. Chem., 32, 16 (1940). 91 Lutz, Org. Syntheses, 20, 29 (1940). 9'Christ and Fuson, I . Am. Chem. Soc., 59, 893 (1937). 9' Colonge and Duroux, Bull. SOC. chim. Ftance, (5e ) 7 , 459 (1940). 94Colonge and Mostafavi, Bull. SOC. chzm. Ftance, ( 5 ) 5, 1478 (1938).
REFERENCES FOR CHAPTER 10
95 Byrans and Doumani, Ind. Eng. Chem., 35, 349 (1943). 96 Fuson, Christ, and Whitman, I . Am. Chem Soc., 58, 2451 (1936). 97 Royals and Hendry, I . Org. Chem., 15, 1147 (1950). 98McMahon e t al., I . Am. Chem. Soc., 70, 2971 (1948). 99 Price and Pappalardo, I . Am. Chem Soc., 72, 2613 (1950).
' * ~ a r z e n s , Compt. rend., 211, 435 (1940). 'OIColonge and Mostafavi, Bull. SOC. chim. France, ( 5 ) 6 , 341-354 (1939). 'mIlawthorne and Robinson, I . C k m . Soc., 763 (1936). "Ruz icka , Koolhaas, and Wind, Helv. Chim. Acta. 14, 1157 (1931). '04 Jones and Ramage, I . Chem. Soc., l856 (1938). ""ruce and Sack, I . A m Chem. Soc., 70, 3959 (1948). 'MCampbell, LaForge, and Campbell, I. Org. Chem., 14, 348 (1949). '07Conant and Kirner, I . Am. Chem Soc., 46, 239 (1924).
Cohen, Wolosinski, and Scheuer, I . A m Chem. Soc., 71, 3440 (1949). '* Baker and Barkenbus, I . Am. Chem. Soc., 58, 263 (1936). " O Renoll, I . A m Chem. Soc., 68, l l60 (1946). "'Fosdick and Campaigne, I . A m Chem. Soc., 63, 974 (1941); Buu-Hoi, Hoan,
and Jacquignon, Rec. trav. chim., 68, 784 (1949). "' Foreman and McElvain, I . Am. Chem. Soc., 62, 1436 (1940). "' Adams and Noller, Org. Syntheses, Coll. Vol. I , 109 (1941). "4Kimura, Ber., 67, 395 (1934). ""Houben and Fischer, I . prakt. Chem., 123, 313 (1929). "6Davis , I . Am. Chem Soc., 63, 1677 (1941). "'Patrick, McBee, and Hass, I . A m Chem. Soc., 68, 1135 (1946). "'Berliner, I . Am. C k m . Soc., 66, 534 (1944). ' l9 Newton and Groggins, Ind. Eng. Chem., 27, 1397 (1935). '"Bergmann, I . Org. Chem., 4 , 9 (1939). 'l' Borsche and Scriba, Ann., 540, 90 (1939). "'Fisher, Oakwood, and Fuson, I . Am. Chem. Soc., 52, 5038 (1930). "'Sandulesco and Girard, Bull. soc. chim. France, ( 4 ) 47, 1300 (1930); c f .
Close, T i f fany , and Spielman, I . Am. Chem. Soc., 71 , 1265 (1949). ' l4 Cooper, Org. Syntheses, 21, 103 (1941). "' Badhwar and Venkataraman, Org. Syntheses, Coll. Vol. 11, 304 (1943). 'l6 Brewster and Vatters, I . Am. Chem. Soc., 64, 2578 (1942). "' Coulthard, Marshall, and Pyman, I . Chem. Soc., 280 (1930). "8Spoerri and Dubois in Organic Reactions, Vol. 5, John 'Viley & Sons, New
Y O K ~ , i949, p. 387. "9 Gulati. Seth, and Venkataraman, Org. Syntheses, Coll. Vol. 11, 522 (1943). "Ov. Auwers, Potz, and Noll, Ann., 535, 228 (1938). '"Ungnade, Chem Revs., 38, 419 (1946). "'Stuart and Tallman, I . Am. Chem. Soc., 65, 1579 (1943); Skraup and Nieten,
Ber., 57, 1294 (1924). '"Buck and Ide, I . Am. Chem. Soc., 54, 3012 (1932). " 4 ~ i l l a n i and Lang, I . Am. Chem. Soc., 72, 2301 (1950). "%omerville and Allen, Org. Syntheses, Coll. Vol . 11, 81 (1943).
Fieser, Gates, and Kilmer, I . Am. Chem. Soc., 62, 2968 (1940). '" Fieser and Price, I . Am. Chem. Soc., 58, 1841 (1936). '"Fieser and Peters, I. A m Chem. Soc., 54, 4347 (1932). IrPFieser et al., I . Am. Chem. Soc., 70, 3197 (1948). lmHill, I . A m Chem. Soc., 54, 4105 (1932). I 4 l Papa e t al., I . A m Chem. Soc., 70, 3.356 (1948).

KETONES Ch. 10
14' Berliner in Organic Reactzons, Vol. 5 , John Wiley & Sons, New York, 1949, p. 229.
14'Henze and Holder, 1. A m Chem Soc., 63, 1943 (1941). Groggins, lnd. Eng. Chem., 22, 620 (1930). Groggins and Newton, lnd. Eng. Chem., 21 , 369 (1929).
'& Fieser, Org. Syntheses, 20, 1 (1940). ' 4 7 ~ a p a , Schwenk, and Hankin, 1. Am. Chem. Soc.. 69, 3018 (1947). "McElvain, I . Am. Chem. Soc., 51, 3124 (1929); Roberts and McElvain, ibid.,
59, 2007 (1937). '*Sengupta, I . prakt. Chem., 151, 87 (1938). "OBlicke and Tsao, I . Am. Chem Soc., 66, 1645 (1944). ='Blicke and Feldkamp, 1. A m Chem Soc., 66, 1089 (1944). 1'1 Blicke and Grier, I . A m Chem. Soc., 65, 1726 (1943). " 'papa e t al., I . A m Chem. Soc., 68, 2133 (1946). " 4 Ferber and BGckner, Ber., 72B, 999 (1939). "'Steinkopf and G b t h e r , Ann., 522, 31 (1936).
Fuson, Ullyot, and Gehrt, I . Am. Chem Soc., 60, 1200 (1938). '" Yoke, Louder, and Smith, I . Chem. Education, 10, 374 (1933). " 'Sheaill , I . A m Chem. Soc., 52, 1990 (1930). "9Young and Roberts, I . Am. Chem. Soc., 67, 321 (1945). 16" Bennett and Eldet, I . Chem. Education, 13, 273 (1936).
Grignard and Fluchaire, Ann. chim., (10) 9 , 14 (1928). 16'smith e t al., I . A m Chem. Soc., 61, 3082 (1939). 16'\r/agner and Moore, I . A m C h e m Soc., 72 , 974 (1950). '64~%itmore and Lewis, I . Am. Chem. Soc., 64, 2965 (1942). 16'Whitmcae and Stahly, I . A m Chem. Soc., 55, 4155 (1933). '"Mam and Porter, I . Chem. Soc., 456 (1944). 167Powell, I . Am. Chem. Soc., 46, 2516 (1924). l" Mosher and Langerak, I . Am. Chem. Soc., 71, 286 (1949). '69Signaigo and Cramer, I . Am. Chem Soc., 55, 3329 (1933). ""Macbeth and Mills, $. Chem. Soc., 711 (1945). "'Ungnade and McLaren, I . Org. Chem., 10, 30 (1945). "'Pezold and Shriner, I . Am. Chem. Soc., 54, 4709 (1932). "'Price and Karabinos, I . Am. Chem. Soc., 62, l160 (1940). 174 Bartlett and Schneider, I . Am. Chem Soc., 67, 143 (1945). 17' Westheimer, Chem. Revs.. 45, 419 (1949). 176 Osterburg and Kendall, I . Am. Chem. Soc., 42, 2618 (1920). 17' Gredy, Ann. chim., ( 1 1) 4 , 22 (1935). "'Kornfeld, Jones , and Parke, I . A m Chem. Soc., 71, 150 (1949). ' " ~ a r l i n , I . Am. Chem. Soc., 67, 932 (1945). ""Ungnade and Ludutsky, I . Org. C h e m , 10, 521 (1945). "l Frank, Berry, and Shotwell, I . A m Chem Soc., 71, 3889 (1949). "' Adkins and Hager, I . Am. Chem. Soc., 71, 2965 (1949). "'Adkins and Hager, I . Am. Chem Soc., 71, 2967 (1949). 184Shunk and Wilds, I . Am. Chem. Soc., 71, 3946 (1949). " 'Lock and Schreckeneder, Ber., 7 2 B , 516 (1939). '06Neidig e t al., I . Am. Chem. Soc.. 72 , 4617 (1950). l'' Tsatsas , Ann. chim., (12) 1, 348 (1946). l" Ungnade, I . Org. Chem., 13, 364 (1948). "' Fieser and Cason, I . Am. hem. Soc., 62, 434 (1940). " " ~ l a r k e and Dreger, Org. Syntheses, Coll. Vol . I , 87 (1941).
REFERENCES FOR CHAPTER 10
''l R'uggli and Herzog, Helv. Chim. Acta, 29, 111 (1346). "'Kinney, I . A m Chem. Soc., 51, 1596 (1929). 19'Pearl and Dehn, I . Am. Chem. Soc., 60, 57 (1938). 194Veiss and Appel, I . Am. Chem. Soc., 70, 3666 (1948); Klein, ibid., 63,
1474 (1941). '9'Gilman and Broadbent, I . Am. Chem. Soc., 70, 2619 (1948). 196 Backer, Rec. trav. c h i m , 57, 978 (1938). 19' Fuson and Robertson, I . Org. C h e m , 7 , 469 (1942). 19'Kinney, I . Am. Chem. Soc., 51, 1596 (1929); c f . re f . 190. 199 Jenkins , Buck, and Bigelow, I . A m Chem Soc., 52, 4496 (1930). '''Hartman and Dickey, I . Am. Chem. Soc., 55, 1229 (1933). '01 Leonard and Mader, I . Am. Chem. Soc., 72, 5390 (1950). 1°'Dane, Schmitt, and Rautenstrauch, Am. , 532, 37 (1937). 'O' Bowden, Heilbron, Jones , and Veedon, I . Chem Soc., 39 (1946). ' 0 4 ~ r a u d e e t al., I . Chem. Soc., 612 (1949). ' O S Conant and Quayle, Org. Syntheses, Coll. Vol. I , 211 (1941). '06 Elderfield and Ressler, I . Am. Chem. Soc., 72, 4067 (1950). '07 Marvel and Walton, I . Org. Chem., 7 , 88 (1942). "'Henze and Rodgers, I . A m Chem. Soc., 61, 433 (1939); 62, 1759 (1940). 'wMariella and Leech, I . Am. Chem. Soc., 71, 3558 (1949). "O Adkins e t al., I . Am. Chem. Soc., 71, 3629 (1949). " 'Lea and Robinson, I . Chem Soc., 2354 (1926). "'Lecaard e t al., I . Am. Chem Soc., 71 , 2997 (1949). "' Baer and Kates, I . Am. Chem. Soc., 67, 1482 (1945). "4Hurd and McNamee, Org. Syntheses, Coll. Vol. I , 244 (1941), note 11. "'Dimroth and Resin, Rer., 75B, 322 (1942).
Russell and Vanderwerf, I . A m Chem. Soc., 69 , 11 (1947). "' McRae, Charlesworth, and Alexander, Can. I . Research, 21B, 1 (1943). "'Colonge, Watteau, and Cumet, Bull. SOC. chim. France, ( 5 ) 14, 246 (1947). "'Long and Troutman, I . Am. Chem SOC.,. 71, 2469 (1949); Parkes and Wil-
liams, I . Chem. Soc., 67 (1934). '10 Ford-hloore and Rydon, I . Chem. Soc., 679 (1946). "' Bersin in Newer Methods of Preparative Organic Chemistry, Interscience
Publishers, New York, 1948, p. 143. ''l 0 ppenauer, Org. Syntheses, 21, 18 (1941). "' Adkins and Franklin, $. Am. Chem Soc., 63, 2381 (1941). "4Woodward, ~ e n d l e r , and Brutschy, I . A m Chem. Soc., 67, 1425 (1945);
Lutz, Jordan, and Truett, ibid., 72, 4085 (1950). "'Hurd, Greengard, and Roe, I . Am. Chem. Soc., 61, 3359 (1939).
Paul, Bull. soc. chim. France, ( 5 e ) 8 , 514-518 (1941). '" Kleiderer and Komfeld, $. Org. Chem., 13, 455 (1948). "' Speck and Bost, I . Org. Chem., 11, 788 (1946). '" Kyrides and Zienty, I . Am. Chem. Soc., 68, 1385 (1946).
Kolfenbach et al., lnd. Errg. C h e m , 37, 1178 (1945). '"Henne and Perilstein, I . Am. Chem. Soc., 65, 2183 (1943). "'Church, Whimore, and McGrew, 1. Am. Chem. Soc., 56, 181 (1934). " 'Ifenne and Hill, I . A m Chem. Soc., 65, 752 (1943). "4Cook and Whitmore, I . Am. Chem. Soc., 63, 3540 (1941). "'Moersch and Vhitmore, I . Am. Chem. Soc., 71, 819 (1949); Mosher and COX,
ibid., 72, 3701 (1950). " 6 H e ~ e , Shepard, and Young, 1. Am. Chem. Soc., 7 2 , 3577 (1950).

KETONES REFERENCES FOR CHAPTER 10 389
1 "'Roberts and Sauer, J . A m Chem. Soc., 71, 3928 (1949). ! '" Mowry, J . Am. Chem. Soc., 67, lO5O (1945).
1'9Emers~n e t al., J . Am. Chem. Soc., 68, l666 (1946). 140Senseman and Stubbs, Ind Eng. Chem., 25, 1286 (1933). 14' Thompson, Org. Syntheses, 20, 94 (1940). 14'Huntress, Hershberg, and C l i f f , J . A m Chem. Soc., 53, 2720 (1931). 'U Winkler, Chem. Ber., 81, 256 (1948). 'UHuntress and Walter, J . Am. Chem. Soc., 70, 3704 (1948).
Emerson and Lucas, J . Am. Chem. Soc., 70, 1180 (1948). '46 Emerson e t al., J . Am. Chem. Soc., 68, 674 (1946). 14' Emerson et al., J . Am. Chem. Soc., 69, 1905 (1947). ' ' ~ i v i e r and Farine, Helv. Chim. Acta, 1% 865 (1929). '" Bartlett and Schneider, J . Am. Chem. Soc., 67, 141 (1945). ''O Clarke, 1. Am. Chem. Soc., 34, 679 (1911). "' Clarke, J . Am. Chem. Soc., 33, 529 (1911). '" Willst'%ter and Hatt, Ann., 418, 152 (1919). "'Connor and Adkins, J . Am. Chem. Soc., 54, 3420 (1932).
Renfrow, J . A m Chem. Soc., 66, 144 (1944). '''Renfrow and Walker, J. Am. Chem. Soc., 70, 3957 (1948).
~ o h n s o n and Hager, Org. Syntheses, Coll. Vol. I , 351 (1941). '" Dehn and Jackson, J . Am. Chem. Soc., 55, 4284 (1933). "' Adams, Abramovitch, and Hauser, J . Am. Chem. Soc., 65, 552 (1943)
Briese and McElvain, J . A m Chem Soc., 55, 1697 (1933). "O Leuchs, Heller, and Hoffman, Ber., 6% 875 (1929). '6'Cope, Hofmann, and Hardy, J . Am. Chem. Soc., 63, 1855 (1941). "'Hudson and Hauser, J . Am. Chem. Soc., 63, 3163 (1941). '6'Bouveault and Locquin, Bull. SOC. chim. France, (3) 31, 1153 (1940). le4Price and Meisel, J . Am. Chem. Soc., 69, 1497 (1947). %'Strating and Backer, Rec. trav. c h i m , 55, 904 (1936). m6HGckel and Kindler, C h e m Ber., 80, 203 (1947). %'Shive, Crouch, and Lochte, 1. Am. Chem. Soc., 63, 2983 (1941). %'Case and Reid, I . Am. Chem. Soc., 50, 3064 (1928). H9Cornubert and Borrel, Bull. soc. chim. France, (4) 47, 307 (1930).
Comubert e t al., Bull. soc. chim. France, (5e) 12, 371 (1945). "'King, Barltrop, and Walley, J . Chem. Soc., 279 (1945). "'Rydon, J. C h e m Soc., 1549 (1939). 17' Julian and Oliver, Org. Syntheses, Coll. Vol. 11, 391 (1943).
a74Mercer, Robertson, and Cahn, I . Chem. Soc., 999 (1935). "'Heilbron et al., 1. Chem. Soc., 1338 (1931).
Ramart-Lucas and Labaune, Ann. chim., (10) 16, 295 (1931). "'Mosettig and van de Kamp, I . Am. Chem. Soc., 55, 3445 (1933). "' Dean, Dickinson, >ayle, and Lester, J . A m Chem. Soc.. 72, 1740 (1950). '" Kolloff and Hunter, J . A m Chem. Soc., 63, 492 (1941). nostrong and McElvain, J . lm. Chem. Soc., 55, 816 (1933). '"Campball e t al., J . A m Chem. Soc., 68. 1845 (1946). "'Youtz and Perkins, J . Am. Chem. Soc., 51, 3514 (1929). "'Ruggli and Gassenmeier, Helv. Chim. Acta, 22, 501 (1939). "4Schechter, Green, and LaForge, J . A m Chem Soc., 71, 3165 (1949). "'Ruzicka and Schinz, Helv. Chim. Acta, 23, 964 (1940). '"Cope, Hoyle, and Heyl, 1. Am. Chem. Soc., 63, 1843 (1941). '"' v. Rraun and Rudolph, Ber., 67 , 278 (1934).
'" McPhee and Klingsberg, J . Am. Chem. Soc., 66, 1132 (1944). Becker, Helv. Chim. Acta, 32, 1584 (1949). sharp, Sutherland, and Wilson, J . Chem. Soc., 346 (1943).
"'Anderson, Crawford, and Sherrill, J . A m Chem. Soc.. 68, 1294 (1946). "' Brown and Partridge, I . Am. Chem. Soc. 67, 1423 (1945). w'Finkelstein and Elderfield, J . Org. Chem., 4, 371 (1939). " 4 ~ ~ ~ a r d and Fraser, Org. Syntheses, Coll. Vol. I , 475 (1941). "'Adickes and Andcesen, Am., 555, 41 (1943). s 6 ~ u a n , Bull. soc. chim. France, (5e) 5, 1341 (1938). "'Gaubert, Linstead, and Rydon, J . Chem. Soc., 1977 (1937). "" Ames and Bowman, I . Chem. Soc., 329 (1950). w9 ~ g ~ l , Halberstadt, and Barendregt, Rec. trav. chim., 68, 387 (1949). 'OOOpenshaw and Robinson, J . Chem. Soc., 944 (1937). "' Fieser e t al., I . Am. C h e m Soc., 57, 1852 (1935); 58, 2320 (1936). "' Friedmann, J . prakt. Chem., 146, 159 (1936). "' Oldham and Ubbelohde, J . Chem. Soc., 20 1 ( 1939): s4 Bouveault and Bongert, Bull. soc. chim. France, (3) 27, 1038, 1046, 1088
(1902). " 5 B ~ u v e a u l t and Locquin, Bull. SOC. chim. France, (3) 31, 588, 1153 (1940).
Breslow e t al., J . Am. C h e m Soc., 66, 1921 (1944). "'Elderfield e t al., J . Am. Chem. Soc.. 69, 1258 (1947). ""Mannick and Margotte, Ber., 68B, 278 (1935). IR Elderfield et al., J . Am. Chem. Soc. 68, 1584 (1946). '" Jackman et al., J . Am. Chem. Soc., 70, 2884 (1948). '" Jones e t al., J . A m Chem. Soc., 70, 2846 (1948). '"Walker and Hauser, J . Am. Chem. Soc., 68, 1386 (1946); c f . ref. 313. "' Bowman, J . Chem. Soc., 322 (1950). . '14 Reynolds and Hauser, Org. Syntheses, 30, 70 ( 1950). ' " ~ o n g and Troutman, 1. Am. Chem. Soc.. 71, 2473 (1949). ' l6 Bowman, J . Chem. Soc., 325 (1950). 'l7 Noyce, J . Chem. Education, 26, 275 (1949). '" ~abat ier ' and Mailhe, Compt. rend., 158, 831 (l9 14); Senderens, A n n ch im,
(8) 28, 243-343 (1913). ' l9 Herbst and Manske, Org. Syntheses, Coll. Vol. 11, 389 (1943). 'n,Yistler, Swann, and Appel, Ind. Eng. Chem., 26, 388 (1934). '"Swann, Appel, and Kiscler, Ind Eng. Chem., 26, 1014 (1934). ='Senderens, Bull. SOC. chim. France, (4) 3, 824 (1908). "'perkins and Kenyon, J . Chem. Soc., 99, 57 (1911); 101, 629 (1912); 103,
1936 (1913). 'l4 Cowan, J e f f e r y , and Vogel, J . Chem. Soc., 171 (1940). '" Krafft , Ber., 15, 1712 (1879).
Morgan and Holmes, J . Soc. Chem. Ind. (London), 44, 109T ( 1925). 'YJ Thorpe and Yon, Org. Syntheses, Coll. Vol. I , 192 (1941). 310 Curtis, Dobson, and Hatt, J . Soc. Chem. Ind (London), 66, 402 (1947).
'29Dougherty, J . A m Chem Soc., 50, 571 (1928). '"'Hurd and Thomas, 1. Am. Chem Soc., 58, 1240 (1936). '"Miller, Cook, and Whitmore, J . Am. Chem Soc., 72, 2732 (1950). "' Wibaut e t al., Rec. trm. chim., 58, 362 (1939). " ' ~ o g e l , 1. Chem Soc., 912 (1931). "4Henshall, 1. Soc. Chem Ind (London), 62, 127 (1943). " V o g e l , 1. Chem Soc., 2033 (1928).

KETONES al. 10
=Birch e t al., I . A m Chem Soc., 71, 1362 (1949). "'Winkler, Chem Bet., 81, 258 (1948). '"Maeder, Helu. Chim Acta, 29, 124 (1946). "'\"ebb and Webb, J . A m Chem Soc., 71, 2285 (1949). -Burger and Walter, J . A m Chem Soc., 72, 1988 (1950). '41 Yarnall and Tal l is , J . Org. C h e m , 4 , 270 (1939). '4' Newman and Magerlein in Organic Reactions, Vol. 5, John Viley & Sons,
New York, 1949, p. 421. =v. Braun and Kroper, Ber., 62, 2880 (1929); v. Braun and Teu f f er t , i b i d ,
62, 235 (1929). . '"v. Braun, Ber., 67, 218 (1934); Newman, J . A m Chem. Soc., 57, 732 (1935). Rohrmann, Jones , and Shonle, J. A m Chem. Soc., 66, 1856 (1944).
'"Nunn and Henze, J. Org. Chem., 12, 541 (1947). '47 Bachmann and Boamer, J. A m Chem Soc., 58, 2098 (1936); Callen, Dorn-
feld, and Coleman, Org. Syntheses, 28, 6 (1948). " Bachmann, J. Am Chem Soc., 56, 1365 (1934).
Whitmore e t al., J. Am Chem Soc., 67, 2059 (1945). Sherk, Augur, and Sof fer , J . A m Chem Soc., 67 , 2239 (1945).
"' Blicke, J. Am Chem. Soc., 49, 2847 (1927). '" Gilmaa and Jacoby, J. Org. Chem., 3, 108 (1938). '"Shriner and Turner, J. Am Chem Soc., 52, 1268 (1930). 's4Hauser, Humphlett, and Weiss, J. Am. Chem. Soc., 70, 426 (1948). '"Pearson, J . A m Chem. Soc., 72,4169 (1950).
Hauser and Renfrow, 1. A m Chem. Soc., 59, 1823 (1937). ' s 7 R h i ~ e s m i t h , J. A m Chem Soc., 58, 596 (1936). "'Kumler, Strait, and Alpen, J. A m Chem. Soc., 72, 1463 (1950).
Alpen, Kumler, and Strait, J . A m Cbem Soc., 72, 4560 (1950). Baddeley and Kenner, J. Chem. Soc., 303 (1935).
"' Bachmann, Carlson, and Moran, J. Org. Chem, 13, 920 (1948). "'Whitmore et al., J . A m Chem Soc., 69, 236 (1947). '" Reeve and Adkins, J. Am Chem Soc., 62, 2874 (1940). '
S4LaForge, J. A m Cbem. Soc., 50. 2477 (1928). "' Frank and Weatherbee, J . A m Chem. Soc., 70, 3482 (1948). "'Vogel and Schinz, Helu. Chim. Acta, 33, 123 (1950). '67 Cagniant, Bull. SOC. chim France, ( 5 ) 16, '847 (1949). "'Behrens e t al., J . A m Chem Soc., 70, 2837 (1948). '69 Wilson and Henze, J. A m Chem. Soc., 63, 2112 (1941). '" Borsche and Herbert, Ann., 546, 297 (1941). '"Young, McKinnis, Webb, and Roberts, J . A m Chem Soc., 68, 295 (1946). " ' ~ u ~ g l i and Hegedus, Helv. Chim Act& 25, 1292 (1942); Ruggli and Zeller,
i b i d , 28, 744 (1945). "'Henze and Rigler, J . Am. Chem Soc., 56, 1350 (1934).
Barnes and Budde, 1. Am. Chem. Soc., 68, 2339 (1946). 37'Wallace and Henze, J . A m Chem Soc., 64, 2882 (1942). lT6Henze e t al., J. Am. Chem. Soc., 64, 1222 (1942). "' Rigler and Henze, J. A m Chem Soc., 58, 474 (1936). '78Henze, Benz, and Sutherland, 1. A m Chem. Soc., 7 1 , 2122 (1949).
Elderfield, Pitt , and Vempen, 1. A m Chem Soc., 7 2 , 1334 (1950). - ~ h i t n e y and Henze, 1. A m Chem. Soc., 60, 1148 (1938). 'O' Cross and Henze, J . A m Chem Soc., 61, 2730 (1939). '8'Niemann, Benson, and Mead, 1. Org. Chem, 8 , 397 (1943).
38s Speer and Henze, 1. A m Chem. Soc., 61, 1226 (1939).
"4Rubin, Paist, and Elderfield, J. Org. C h e m , 6 , 260 (1941). "'Bachmann and Ferguson, J . A m Chem Soc., 56, 2082 (1934). '06 Anderson et al., J . Am Chem. Soc., 67, 2197 (1945). 387 Decombe, Ann. c h i m , (10) 18, 92 (1932).
'M Humphlett, Weiss , and Hauser, 1. Am. Chem. Soc., 70, 4020 (1948). " 9 N e m a n and Smith, J . Org. Chem., 13, 592 (1948). ' P O Yewman and Booth, J . A m Chem. Soc., 67 , 154 (1945).
'9'Campaigne and Reid, J. Am. Chem. Soc., 68, 1663 (1946). '"de Benneville, J. Org. Chem, 6 , 462 (1941). '9'Ullyot e t al., J . Org. Chem, 10, 440 (1945). '94 ~ e i z m a n n et al., J. Chem Soc., 1367, 1371 (1935). ' 9 ' ~ i e s e r and Daudt, I . A m Chem Soc., 63, 785 (1941). '"Kroeger and Nieuwland, J. A m Chem. Soc., 58, 1861 (1936). '"Nightingale and Wadsworth, J . Am. Chem Soc., 67, 416 (1945). ' "Hopf f , Nenitzescu, Isacesu, and Cantuniau, Ber., 69, 2249 (1936). 399Hopff e t al., Ber., 69B. 2249 (1936). "'Cason, Chem Revs., 40, 15 (1947). "'Cason, J. Am. Chem Soc., 68, 2078 (1946); c f . ref. 426. 401 Gilman and Nelson, Rec. trau. c h i m , 55, 520 (1936).
"'Bunnett and Tarbell, J . Am Chem Soc., 67, 1944 (1945). "4Les l i e and Henze, J. A m Chem Soc., 71 , 3480 (1949). "'Michael, J. A m Chem Soc., 41, 41 1 (1919). 406Colonge and J o l y , Ann. c h i m , (11) 18, 314 (1943). " ' Jones , J . A m Chem Soc., 69, 2 350 (1947). "'Schmidt and Shirley, J. A m Chem Soc., 71, 3804 (1949).
Badertscher and Whitmore, J . A m Chem Soc., 55, 1564 (1933). 410Thoms, Arch. Pharm, 263, 246 (1925). 41' Whitmore e t al., J . Am Chem. Soc., 60, 2790 (1938). 4!' Baker, J . Chem Soc., 447 (1938). 41'Kadesch, J. A m Chem Soc., 66, 1211 (1944). 4'4Thitmore, Whitmore, and Cook, J. A m Chem Soc., 72, 51 (1950). 41'Hurd and Kelso, J. Am. Chem. Soc., 62, 2184 (1940). 416 Cook and Percival, J . Am Chem. Soc., 71 , 4 141 (1949). 417 Ford, Thompson, and Marvel, J. Am Chem Soc., 57, 2621 (1935). 4'8Kohler and Baltzly, J. Am. Chem Soc., 54, 4023 (1932). 4'9Stehman, Cook, and mitmore, J . Am. Chem. Soc., 71 , 1509 (1949). 4'0Lester and Proffi t t , J. Am Chem. Soc., 71, I877 (1949). 4''Kipnis and Ornfelt, J. Am. Chem Soc., 70 , 3948 (1948). 4"Kantor and Hauser, J . Am. Chem Soc., 7 2 , 3290 (1950).
Pinson and Friess, J . Am. Chem Soc., 72 , 5333 (1950). 4 '4H~rd and Cohen, J . A m Chem Soc., 53, 1071 (1931). 4''McKennis and du Vigneaud, J . Am. Chem. Soc., 68, 832 (1946); c f . ref. 426. 4'6Cason and Prout, Org. Syntheses, 28, 75 (1948). 4'' Whitmore, Noll, and Meunier, J. Am. Chem Soc., 61, 683 (1939). 4'8Whitmore et al., J . Am. Chem. Soc., 60 , 2462 (1938). 4'9 Jenkins , J . Am. Chem. Soc., 55, 703 (1933). UOJenkins, J . A m Chem. Soc.. 55, 1618 (1933). 4'' Jenkins , J. A m Chem Soc., 56, 682 (1934). "' Jenkins , J . A m Chem. Soc., 55, 2896 (1933). U'Montagne, Ann c h i m , (10) 13, 53 (1930).

KETONES Ch. 10
4'4Vhitmore and Sloat. J . A m C h e m Soc., 64, 2969 (1942).
' I 4'SHey, J . G e m . Soc., 1232 (1930). 4'6Adams, Harfenist, and Loewe, I. A m C h e m Soc., 71, 1628 (1949). 4" Kohler, A m Chem. 1.. 38, 51 1 (1907). 43'Colonge, Bull. SOC. c h i m Frmzce, (5) 3, 413 (1936). 439 ~ o l o n g e , Bull. SOC. c h i m France, (5) 2, 754 (1935). ""Smith, Chase , and Rhodes, J . A m Chem. Soc., 66, 1547 (1944).
I 441 Whitmore and Pedlow, J . A m Chem. Soc., 63 , 758 (1941). UIKohler , A m Chem. J., 29. 352 (1902). U'Newman and Farbman, 1. A m Chem. Soc., 69, 1550 (1944); Bachmann e t al.,
72, 1997 (1950). U4Dice, Loveless , and Gates , J . A m C h e m Soc., 71, 3546 (1949). 44'Newman and Booth, J . Org. Chem., 1 2 , 737 (1947); Mueller and May, J . A m
Chem Soc., 71, 3313 (1949). u A r n ~ l d r Buckley, and Dodson, J . A m Chem Soc., 72, 3154 (1950). 44''Whianore and Forster, 1. A m Chem. Soc., 64, 2967 (1942).
Sah, Rec. trrm. chim., 59. 1025 (1940). Braude and Coles , J. Chem Soc., 2012 (1950). Wieland, Chem Ber., 81, 3 14 (1948).
4" Salkind and Beburuschwili, Ber., 42, 4500 (1909). 4"Hartman and Roll, Org. Syntheses, 23, 1 (1943). 45'Nightingale and Janes , J . Am. chem. Soc., 66, 352 (1944). U 4 ~ t . Goldschmidt and Veer , Rec. bav. c h i m , 65, 796 (1946); Hey and ~ o r r i s ,
J. Cbem Soc., 2319 (1948); Keagle and Hartung, J. A m Chem. Soc., 68, 1608 ( 1946).
4" Hey and Morris, J . C h e m Soc., 48 (1948). 456 Freon, A n n c h i m , (11) 11, 465 (1939). U'Locquin, Bull. SOC. chim. France, (3) 31, 1147 (1904).
Barry and Hartung, J. Org. C h e m , 12, 460 (1947). 4'9Sumerford and Dalton, J . Am. Chem. Soc., 66, 1330 (1944). "'Marvel and Sperry, Org. Syntheses, Coll. Vol. I , 95 (1941). "' Bigelow and Hanslick, Org. Syntheses, Coll. Vol . 11, 244 (1943). "'witt ig and Vidal , C h e m Ber., 81, 368 (1948). 46'Schmerling, J. A m Chem. Soc., 68, 1650 (1946). t
"4Adk ins , Reactions of Hydrogen, U. o f Wisconsin Press , 1937, p. 129. "'Covert, Connor, and Adkins, J . Am Chem Soc., 54, 1658 (1932). 4 6 6 C ~ v e r t and Adkins, J. A m Chem. Soc., 54, 4116 (1932). "'Powell andSecoy, J. A m Chem Soc., 53, 765 (1931). "'Colonge, Bull. SOC. chim. France, (4) 49, 444 (1931). " ' ~ a v o n , Ann. c h i m , (9) 1, 144, 196 ( 1914); Skita, Ber., 4 3 3393 (1910). 4'0 Adams, Voorhees, and Sh iner , Org. Syntheses, Coll. vo l . I , 463 (1941). 4 ' 'C~ longe , Bull. SOC. c h i m France, (4) 49, 441 (1931); (4) 41, 327 (1927); (5)
3, 416 (1936); (5) 6, 335, 342 (1939); Morgan and Hardy, J. Soc. Chem Ind (Lon- don), 11, 518 (1933); Thoms, Arch. Phann., 263, 241 (1925); Powell, Munay, and Baldwin, J . A m Chem. Soc., 55, 1153 (1933).
4'1 Haeussler and Dijkema, Ber., 77, 601 (1944). 4"Rupe and Willi, Helv. Ch im Acta, 15, 845 (1932). 4'4Hente, Y i l son , and Townley , J . A m Chem Soc., 65, 964 (1943). 4's\100ds e t al., J . Am. Chem Soc., 71, 2028 (1949).
Dippy and Lewis , Rec. trav. chim., 56, 1000 (1937). 4"Raiford and Peterson, J . Org. Chem., 1, 549 (1937).
REFERENCES FOR CHAPTER 10 393
4'8 Berliner and Berliner, J . A m Chem. Soc., 72, 222 (1950). Metayer, Ann. c h i m , (12) 4, 201 (1949).
4'0 Bailey and Lutz , J . A m Chem Soc., 70, 2412 (1948). Q'Thompson, Org. Syntheses, 27, 21 (1947).
. *'Pecherer, Jampolsky, and Wuest, J . A m Chem Soc., 70, 2587 (1948). *'Stork and Foreman, J . A m C k m . Soc., 68, 2 173 (1946). 4'4Papa, Schwenk, and Breiger, J . Org. C h e m , 14, 366 (1949); Papa, J. A m
Chem Soc., 71, 3246 (1949). *' Adkins, Rossow, and Carnahan, J . A m Chem. Soc., 70, 4247 (1948). 4-96 Cornforth, Cornforth, and Robinson, J. Chem Soc., 690 (1942).
*'Sprague and Adkins, J . Am. Chem. Soc., 56, 2670 ( 1934). 4"Haller and Bauer, A n n c h i m , (8) 29, 313 (1913); Nasarow, Ber., 70, 594
(1937). Cornubert e t al., Bull. SOC. chim. France, (4) 49, 1260, 1498-1 528 (193 1);
(5) 5 195 (1935). 490Chanley, J . Am. Chem Soc., 70, 244 (1948); Fischer and Wunderlich, Ber.,
74, 1546 (1941). 4 9 1 ~ o b o t k a and Chanley, J. Am. Chem Soc., 71, 4136 (1949). 491 Colonge, Bull. SOC. chim France. (5e) 5, 99 (1938); c f . ref. 491. 49 1 Conia, Bull. SOC. ch im France, (5) 17, 537 (1950). 494Haller and Bauer, Ann. chim., (8) 28, 373 (1913). 49'Haller, Bull. soc. c h i m France, (4) 31, 1073-1144 (1922). " 'Sof fer e t al., J. A m Chem Soc., 72, 3704 (1950). 49'Vhianore and Laughlin, J . Am. Chem Soc., 55, 3732 (1933). 49'Haller, A n n c h i m , (8) 29, 313 (1913). "'Suter and Weston, I. A m Chem. Soc., 64, 534 (1942). m o ~ p r a g u e , Beckman, and Adkins, J. A m Chem. Soc., 56, 2665 (1934). 'O' Adkins, Kutz, and Cof fman, J. A m Chem. Soc., 52, 3218 (1930). '01 Speck and Bost, J . Org. C h e m , 11, 788 (1946). 50s Vanderwerf and Lemmerman, Org. Syntheses, 28, 8 (1948). 504 Eis leb , Ber., 74, 1437 (1941).
' O S Eistert i n Newer Methods o f Preparative Organic Chemistry, Interscience Publishers,.New York, 1948, p. 513.
w6Kohler e t al., J. A m Chem Soc., 6 1 , 1059 (1939). 'O' Adamson and Kenner, J . Chem. Soc., 184 (1939). '00 Thomas, Campbell, and Hemion , J. A m C h e m Soc., 60, 719 (1938). '09Schaad and Ipat ie f f , J. A m C h e m Soc., 62, 178 (1940). " 'Leers , Bull. SOC. c h i m France, 39, 423 (1926). ' " H e ~ i o n , Uavis , and Maloney, J . A m Chem. Soc., 71, 2813 (1949). "'Moureu and Delange, Bull. SOC. c h i m France, (3) 29, 666 (1903). "'Hill and Flosdorf, Org. Syntheses, Coll. Vol . I , 462 (1941). "4Emerson, J. A m Chem Soc., 69, 1212 (1947). "'Bachmann, Org. Syntheses, Coll. Vol . 11, 73 (1943).
Burger and Bennet, J . A m Chem Soc., 72, 5414 (1950). "' Bouveault and Locquin, Bull. SOC. chim. France, (3) 35, 629-649 (1906);
Leers , i b i d . (4) 35, 597 (1924); (4) 39, 421, 423, 433, 655 (1926); Parry, J. Chem. Soc., 107, 108 (1915); Meerwein, Ann., 396, 201, 250 (1913). '" Kohler and Vygaard, J . A m Chem Soc., 52, 4133 (1930).
Ballard and Dehn, J . Am. Chem. Soc., 54, 3969 (1932). " O J enk ins , Buck, and Bigelow, J. Am. Chem Soc., 52, 4495, 5198 (1930). " ' v . Wacek, Ber., 77B. 85 (1944).

KETONES
i' " 'Evers e t al., J . Am. C h a Soc., 55, 1136 (1933).
1: ''aWhianore, Evers , and Rothrock, &g. Syntheses, Coll . Vo l . U, 408 (1943). ' l4 Brown, J. C h e m Soc., 2577 (1949).
I "'Bardan, Bull. SOC. c h i m France, (4) 49, 1426, 1551, 1875 (1931); (5) 1, 141 368, 370 (1934); Elphimoff-Felkin, i b i d . (5) 17, 497 (1950).
' '6Ramonczai and Vargha, J . A m C h e m Soc., 72, 2737 (1950). I Bouveault, Bull. soc. c h t m France, (3) 35, 629-649 (1906).
'"Delaby and Dumoulin, Bull. SOC. chim. France, (4) 33, 602 (1923); (4) 39, 1583 (1923).
Alder and Schmidt, Ber., 76, 192 (1943). "'Nenitzescu and Gavat, A m , 519, 260 (1935). ' "Ru le and 'Ihompson, 1. C h a Soc., 1762 (1937). "'Goldschmidt and Veer , Rec. t rm. c h i m , 67, 504 (1948). "'Man and Hauser, 1. A m C h e m Soc., 72, 3294 (1950). '" Weis s and Hauser, J . A m Chem. Soc., 71, 2023 (1949). '" Frank and Phi l l ips , J . A m G e m . Soc., 71, 2804 (1949). '"Kharasch, Urry, and Kuderna, J . Org. Chem., 14, 248 (1949).
Wolfrom e t al., J. A m C h a Soc., 65, l516 (1943); 67, 1793 (1945); 71, 3509 ( 1949).
' sT ru i t t e t al., J . A m C h e m Soc., 71, 3511 (1949). 'a91hitmore and Randall, J . A m G e m Soc., 64, 1244 (1942).
Johnson and Degering, J . Org. C h e m , 8, 10 (1943). ' 41Lev ine , Conmy , Adams, and Hauser, J . Am. C h e m Soc., 67, l5 l0 (1945). '4 'Adams and Hauser, J . A m C h e m Soc., 67, 284 (1945); 66, 345 (1944). ' 4aYeerwein and V o s s e n , 1. prakt. Chem., 141, 157 (1934). 'U Adams and Hauser, J . A m C h e m Soc., 66, 1220 (1944). 'USwamer and Hauser, 1. A m C h e m Soc., 72, 1352 (1950). '"Denoon, Org. Syntheses, 20, 6 (1940). '" Adkin S and Rainey, Org. Syntheses , 20, 7 (1940). '"Smith and King, J . A m C h e m Soc., 65, 442 (1943). '"Green and LaForge, 1. Am. Chem. Soc., 70, 2287 (1948); c f . ref . 545. "'Smith and Engelhardt, J. A m C h e m Soc., 7 1 , 2671 (1949). " 'Al len , Abell, and Normington, Org. Syntheses , Coll. Vo l . I , 205 (1941). "'Magnani and McElvain, Org. Syntheses, 20, 32 (1940). '"Harris and Levine , J . A m Chem. Soc., 71, 1120 (1949).
Harris and Levine , J . Am. Chem. Soc., 70, 3360 (1948). "' Royals , J . Am. C h e m Soc., 67, 1508 (1945).
Dessert and Halverstadt, J. A m C h e m Soc., 70, 2595 (1948). Snyder, Brooks, and Shapiro, Org. Syntheses, Coll . Vol . 11, 531 (1943).
'" Jackman, Bergman, and Archer, J. Am. C h e m Soc., 70, 499 (1948). '" Elks , Ell iott , and Hems, J . C h e m Soc., 629 (1944). MOReid and Calvin, J . A m C h e m Soc., 72, 2948 (1950). ' 61Hesse and B;cking, Ann., 563, 31 (1949). ='Hesse and B k k m a n n , Ann., 563, 37 (1949). " a Frank and V a r l d , Org. Syntheses, 27, 91 (1947); Uowry and Ringwald, J .
Am. C h e m Soc., 72, 2037 (1950). '"Waitkins and Clark, Chem. Revs., 36, 235 (1945); Rabjohn i n Organic Reac-
t ions, Vol . 5, John Wiley & Sons, N e w York , 1949, p. 331. Riebsomer, Chem. R e v s , 36, 157 (1945). Hatt, Pilgrim, and Human, J . C h e m Soc., 93 (1936). Wegmann and Dahn, Helv. Chim. Acta, 29, 1248 (1946).
I REFERENCES FOR C H A P T E R 10 395
%'keins tock and Fuson, J. A m C h e m Soc.. 58, 1233 (1936); Fuson and Hoch, ibid.. 71, 1585 (1949).
'69 Rauh, Smith, Banks , and Diehle, J. Org. C h e m , 10, 199 (1945). S70Harr, Voter , and Banks, J . Org. C h e m , 14, 836 (1949). 171 Tee t e r s and Shriner, J. Am. Chem. Soc., 55, 3026 (1933).
57'.4stin, Newman, and Ri ley , J. C h e m Soc., 391 (1933); re f . 573. s7 'Mi l ler , Ber., 66, 1668 (1933). '74Chakravarti and Swaminathan, J. Indian C h e m Soc., 11, 715 (1934); C. A.,
29. 1080 (1935). '7'Dox, Org. Syntheses, Coll. Vol . 1, 266 (1941). ' 76Co le s , Vanske , and Johnson, J . A m C h e m Soc., 51, 2269 (1929). ' 77Postowsky and Lugowkin, Ber., 68, 852 (1935). "" Killian, Hennion, and Nieuwland, J. A m Chem. Soc., 56, 1786 (1934). S79 Vuller and Krauss, Monatsh., 61, 206 (1932). '"' Franke, Kroupa, and Schmid, Monatsh., 66, 412 (1935). '" 'Cason, J. Org. C h e m . 13, 227 (1948). ' " 'Yale , J. A m Chem. Soc., 69, 1547 (1947). '" 'Mattar, Hastings, and Walker, J . C h e m Soc., 2455 (1930). '"4Shriner and Todd, Org. Syntheses, Coll . Vol . 11, 200 (1943). '" Erickson and Ki tchens , J . A m C h e m Soc., 68, 492 (1946).
Voodward and Blout, J . A m C h e m Soc., 65, 562 (1943). 5 " 7 ~ i n c e n t , Thompson, and Smith, J . Org. C h e m , 3, 606 (1939). 581 Frank and Hall, J . A m C h e m Soc., 7 2, 1645 (1950).
' " 9 L ~ t z and Wilder, J. A m C h e m Soc., 56, 978 (1934). "' Yunsdiecker, Ber., 75B. 452 (1942). IP1Y0ung and Allen, Org. Syntheses, Coll . Vol . 11, 219 (1943); note 2. '9' Huggli and Zaes l in , Helv. C h i m A c t 4 18, 848 (1935). IP3 Kharasch, MC Bay, and Urry, J. A m C h e m Soc., 7 0 , 1269 (1948). 594 Saunders, Org. Syntheses, 29, 1 (1949).
5 ~ ' Kimel and Cope, J. A m C h e m Soc., 65, 1992 (1943). '96Frank, Armstrong, Kwiatek, and Price, J . A m Chem. Soc., 70, 1379 (1948).
p ines and Ipa t i e f f , J . A m C h e m Soc., 69, 1337 (1947). '9" Gaddis and Butz , J . A m Chem. Soc., 69, 1203 (1947). ' 99 c a t c h e t al., J. C h e m Soc., 278 (1948). 6mCardwell and McQuillin, J . Chem. Soc., 714 (1949); c f . ref . 599. 601Slanina, Hemion , and Nieuwland, J . A m C h e m Soc., 58, 891 (1936). 60' Blatt i n Organic Reactions, V o l . 1, John Wiley & Sons, New York , Chapter
11; Blatt, C h e m Revs., 27, 413 (1940). 60'Tarbell and Fanta, J . A m C h e m Soc., 65, 2169 (1943). 604 Ralston, McCorkle, and Baurer, J . Org. G e m . , 5, 645 (1940).
605Miller and Hartung, Org. Syntheses, Coll . Vo l . 11, 543 (1943). '06 Smith and Opie, J . Org. C h e m , 6, 427 (1941). 607 Amin and Shah, Org. Syntheses, 28, 42 (1948). 600 Carter i n Organic Reactions, Vo l . 3, John Wiley & Sons , New York , 1946, p.
198. '09 Buck and Ide, Org. Syntheses, Coll. V o l . 11, 55 (1943). 6 ' 0 H e r b ~ t and Shemin, Org. Syntheses, Coll . Vol . 11, 1, 519 (1943). 6'1Buck and Ide, J . A m C h e m Soc., 54, 3307 (1932). 6 1 ' B ~ ~ k , B a l t d y , and Ide, J . A m C h e m Soc., 60, 1789 (1938).
Johnson i n Organic Reactions, Vol . 1, John Wiley & Sons , New York , 1942, p. 253.

KETONES
614Hauser and Hudson i n Organic Reactions, V o l . 1, John Wiley & Sons, New York , 1942, p. 266.
l 1 61 'Sh i ve r~ , Dillon, and Hauser, I . A m C h e m Soc., 69, 119 (1947); Ilamell and Levine , J. Org. C h e m , 15, 162 (1950).
' l6 Royals, I . A m C h e m Soc., 70, 489 (1948). '" ~ l o ~ d and Miller, I . A m C h e m Soc., 69, 2354 (1947).
l "' COX and McElvain, Org. Syntheses, Coll. Vo l . 11, 272 (1943); Steck, Hallock, and Holland, J. A m Chem. Soc., 68, 131 (1946).
' l9 Shivers, Hudson, and Hauser, I . Am. Chem. Soc., 45, 2051 (1943). 6 '0Abram~vi tch and Hauser, I . Am. Chem. Soc., 64, 2271 (1942). '"1nglis and Roberts, Org. Syntheses, Coll. Vo l . I , 235 (1941). '" Hauser and Walker, I . A m C h e m Soc., 69 , 295 (1947). '"Hudson and Hauser, I . A m C h e m Soc., 63, 3156 (1941). ' l4 Friedman and Kosower, Org. Syntheses, 26, 42 (1946). 6 2 ' ~ l b e r t s o n , J. A m C h e m Soc., 70, 669 (1948). 6 a 6 F r ~ ~ t i c k and Hauser, I . A m C h e m Soc., 71, 1350 (1949). " 'Pinkney , Org. Syntheses, Coil. V o l . 11, 116 (1943). 6"'Zanetti and Beckmann, I . A m C h e m Soc., 50. 1438 (1928).
Sarger, Robinson, and Smith, J. C h e m Soc., 721 (1937). "Oi lman and Broadbent, I . A m C h e m Soc., 70, 2755 (1948); Bloom, Breslpw,
and Hauser, ibid., 67, 2206 (1945). ' " Sh iner and Schmidt, I . A m Chem. Soc., 51, 3636 (1923). ''l Shriner, Schmidt, and Roll , Org. Syntheses, Coll . V o l . 11, 266 (1943). '" J ackman, Bergman, and Archer, I . A m Chem. Soc., 70, 499 ( 1948).
~ u n s d i e c k e r , Ber., 75B, 447, 455, 460 (1942). 6'sSoloway and LaForge, J. A m Chem. Soc., 69 , 2677 (1947). 6 ' 6 ~ c ~ l v a i n and Weber, Org. Syntheses, 23, 35 (1943). "' Bouveault and Locquin, Bull. soc. c h i m France, (5) 31, 3813 (1940). 618 S passow, Org. Syntheses, 21, 46 (1941). 639 Coulson, J. Chem. Soc.. 1409 (1934).
'"cardwell , I. C h e m Soc., 719 (1949). 6411\Tewman and Balborsky, I. A m C h e m Soc., 72, 4296 (1950). 6 4 a ~ e n f r o w and Renfrow, I. A m C h e m Soc., 68, 1801 (1946). 643 Bartlett and Bavley, I . Am. C h e m Soc., 60, 2416 (1938).
Folkers and Adkins , I. A m C h e m Soc., 53, 1416 (1931). '-Marvel and Hager, Org. Syntheses, Coll. Vo l . I , 248 (1941). '" Breslow and Hauser, J. A m C h e m Soc., 62, 261 1 ( 1940). 6 4 ' A d a m ~ , Lev ine , and Hauser, Org. Syntheses, 27, 35 (1947). '" Bowden, I . A m C h e m S o d , 60, 131 (1938). 649 Wallingford, Thorpe, and Homeyer, I . Am. C h e m Soc., 64, 580 (1942).
\filler, Dessert , and Anderson, J. A m C h e m Soc., 70, 500 (1948). '" Breslow, Baumgarten, and Hauser, I . A m Chem. Soc., 66 , 1286 (1944). ''l Riegel and Li l ien fe ld , I. Am. Chem Soc., 67, 1273 (1945). 6'aHauser and Renfrew, Org. Syntheses, Coll. Vo l . 11, 268 (1943). '"Lund, Ber., 67, 935 (1934). '"Hunter and Hogg, I. A m C h e m Soc., 71, 1924 (1949). 6'6von Dwring and Wiberg, I . Am. C h e m Soc., 72, 2608 (1950). 6"Fr~n ing and H e m i o n , J. A m C h e m Soc., 62, 653 (1940). ''' Bloom and Hauser, I . A m C h e m Soc.,66, 152 (1944).
Dorsch and McElvain, I . A m C h e m Soc., 54, 2960 (1932). Jul ian e t al., Org. Syntheses, Coll. Vo l . 11, 487 (1943).
I R E F E R E N C E S F O R C H A P T E R I O
"'Wiley and Adkins, I . A m C h e m Soc., 60, 914 (1938). 661 Long, I . A m Chem. Soc., 69 , 990 (1947).
66'AAbcamovitch and Hauser, I . A m C h e m Soc., 64, 2720 (1942). 6 6 4 K r ~ e k e r and McElvain, I . A m C h e m Soc., 56, 1172 (1934). "'Milas e t al., I. A m C h e m Soc., 70, 1602 (1948).
Shriner i n Organic Reactions, Vo l . 1, John Wiley & Sons, New York , 1942, p. 9.
" 'L iv ings ton , I . A m C h e m Soc., 69, 1220 (1947). "'Wash, Shive, and Lochte, I . A m C h e m Soc., 63 , 2975 (1941). "9 Johns and Burch, I. A m C h e m Soc., 60, 919 (1938).
Groggins and Newton, Ind Eng. G e m . , 22, 157 (1930). '" Adams, Ch i l e s , and Rassweiler, Org. Syntheses , Coll. Vol. I , 10 (1941). '" Winterfeld and Rgnsberg, Arch. Phann., 274, 40 ( 1936). '"Young e t al., I. A m C h e m Soc., 72, 3635 (1950).
Bergmann and Sulzbacher, I. Org. C h e m , 15, 918 (1950). ' "N icke l s and Heintzelman, I . Org. C h e m , 15, 1142 (1950). '"Shapiro, I . Org. Chem., 15, 1030 (1950). '" Hauser and Humphlett, J. Org. C h e m , 15, 359 (1950). "'Dauben and T i l l e s , I . Org. C h e m , 15, 785 (1950). 6T9Hass , Sus i e , and Heider, I . Org. Cljem., 15, 10 (1950).
Schultz and Mickey, Org. Syntheses , 29, 38 ( 1949). '"Colonge and Pichat , Bull. soc. chim. France, (5) 16, 177, 853 (1949). 681 Dornow, Kuhlcke, and Baxmann, C h e m Ber., 82, 254 (1949).
6''Kaufmann and Adams, J . A m Chem. Soc., 45, 3043 (1923). '04 Helberger, Ann., 529, 214 ( 1937). 6'sKloetzel and S i ldman, I . Org. C h e m , 11, 391 (1946). "'Hatt, Pilgrim, and Stephenson, I . Chem. Soc., 481 (1941). '" Cook, I . C h e m Soc., 1091 (1930).
Adams, Kern, and Shriner, Org. Syntheses, Coll . Vo l . I , 101 (1941); c f . re f . 46 5.
6'9Leonard, Herbrandson, and V a n Heyningen, I. Am. C h e m Soc., 68, 1281 ( 1946).
Kuick and Adkins, J. A m C h e m Soc., 57, 143 (1935). 691Sauer, Org. Syntheses, 31, 68 (1951) including note 5. 69' Cornforth, Org. Syntheses, 31, 59 ( 1951). 6 9 ' D j e r a ~ ~ i i n Organic Reactions, Vol . 6, John Wiley & Sons, New York , 1951,
p. 207. 694 Cannon, E l l i s , and Leal , Org. Syntheses, 31, 74 (1951).

METHOD
11
Quinones
CONTENTS PAGE
236. Oxidation o f Aromatic Hydrocarbons ...................................................... 398 .................. 237. Oxidation of Phenols , Aminophenols, and Aryl Diamines 399
238. Oxidation of 2-Hydroxy-1,4aaphthoquinones ........................................ 399 239. Alkylation of Quinones .......................................................................... 400 240, Quinones by Ring Closure ........................................................................ 400
Table 40 . Quinones ............................................................................................ 402 References ........................................................................................................ 403
236. Oxidation of Aromatic Hydrocarbons
Polycyc l ic quinones a r e prepared by careful oxidation of the corre- sponding hydrocarbons with chromic-sulfuric a c i d mixture in a c e t i c a c i d solut ion or a s a n agi tated aqueous suspens ion , e.g., 2,3dimethyl-l,4- naphthoquinone (80%),' 9,lO-phenanthroquinone (80%): and pcenaphthene- quinone (60%): A laboratory reactor h a s been described inlwhich a n a c e t i c a c i d solut ion of chromic a c i d and another solut ion of hydrocarbon a r e mixed a s a film a t 90°. T h e react ion mixture i s then fed into water t o prevent further oxidation. B y th i s procedure, the yield of 2-methyl- l,4-naphthoquinone h a s been raised from 29% by the usua l p rocess t o 45%:''
Other oxidizing agents have been used. Sodium chlorate with vanadium pentoxide c a t a l y s t a t t a c k s anthracene readi ly but i s not powerful errough for the conversion of hydrocarbons of the naphthalene and phenanthrene series."' An a c e t i c ac id solut ion of 30% hydrogen peroxide h a s a l s o been used.9' l0
METHODS 237-238
237. Oxidation of Phenols , Aminophenols, a n d Aryl Diamines
Derivat ives of phenol or an i l ine c a n b e oxidized t o quinones, the yield and e a s e of oxidation depending on t h e subs t i tuen ts . If a n amino or hy- droxyl group is in the para posi t ion, the react ion proceeds readily, a s il- lustrated by the s y n t h e s i s of quinone from hydroquinone b y oxidation with a sodium chlorate-vanadium pentoxide mix ture (96%)' or with chromic- sulfur ic a c i d mixture (92%):) A para halogen atom usual ly h a s a favor- a b l e effect. Any group in the para posi t ion i s e l iminated or oxidized. o-Quinones a r e usual ly prepared,from the corresponding ca techols . A survey of procedures for the s y n t h e s i s of benzoquinones by oxidation h a s been made.)'
Polymethylquinones and cer tain polycyclic quinones a r e prepared by the oxidation of aminophenols and their polycycl ic analogs. T h e lat ter s u b s t a n c e s a r e readi ly obtained by coupling the corresponding phenolic compound with diazot ized su l fan i l i c a c i d followed by a reduct ive c leavage of the a z o compound.
Oxida o n of t h e crude aminophenol is carried out with chromic or manganese dioxide." T h e over-all y ie lds a r e good (50-90%). For the preparation of 1,2-naphthoquinone, ferr ic chloride i s a milder and a bet ter oxidant than chromic a c i d (94%)." Similarly, diamines a r e oxidized with ferr ic chloride, a s in the s y n t h e s i s of duroquinone (9m).'0
238. Oxidation of 2-Hydroxy-1,Cnaphthoquinones
T h e conversion of 2-hydroxy-3-alkyl-l,4-naphthoquinones by the ac t ion of a lka l ine permanganate into t h e next lower homolog h a s b e e n exten- s i v e l y studied.)' A modified procedure involves the treatment of t h e naphthoquinone with hydrogen peroxide in dioxane-soda solut ion followed

400 QUINONES Ch. 11
by the action of copper sulfate and alkali on a n intermediate acid (93% over-all). It has been established that the hydroxyl and alkyl groups change places in the course of the oxidation. The method has been found valuable in the synthesis of certain homologs difficult t o obtain by direct alkylation (method 239);'
239. Alkylation of Quinones
Diacyl peroxides are good agents for the alkylation of p-bento- and 1,4-naphthoquinones having a free position in the quinoid ring, particu- larly when the normal- or iso-alkyl chains are desired (30-6%)."'" The
method has been widely applied in the synthesis of 2-hydroxy-l,4-naphtho- quinones substituted in the 3-position. The procedure cons is t s in adding slowly a solution of the diacyl peroxide in ether to a solution of the quinone in acetic acid a t 90-95'.
Alkyl groups in the low-molecular-weight range are a l s o introduced by heating the quinone with the corresponding acid, excess red lead, and a promoter, which i s a compound containing a n ac t ive hydrogen, such a s malonic ester or acetoacetic ester."
240. Quinones by Ring Closure
The intramolecular condensation of oaroylbenzoic ac ids in the pres- ence of concentrated sulfuric ac id g ives subst i tuted anthraquinones. The ac id strength, reaction temperature, and period of heating are care- fully controlled to insure optimum yields and to avoid sulfonation prod- u c t ~ . " ~ ' ~ Boric acid has been added a s a sulfonation inhibitor." Sub- stitution in the para position of the aroyl group leads to 2alkyl-:3 2-chloro-," and 2-bromoanthraquinones .'6
A number of anthraquinones have been synthesized by adding dienes to aroylacrylic acids, dehydrogenating the adducts in the form of the es ters , and cyclizing a s before."
METHOD 240
R = H, CH3, R'= H, CH,; R''= H, CH,, OCH,
The diene synthesis ''l with quinones i s valuable in providing hydro- aromatic sys tems which a r e readily dehydrogenated, a s illustrated by the synthes is of 23-dimethylanthraquinone (90% ~ v e r a l l ) . ' ~
CH, KOH
8 o
The synthes is has been adapted to the preparation of 1,2-naphthoquinone and i t s derivatives by a n improved procedure."

40 2 QUINONES Ch. 11
TABLE 40. QUINONES
c, Compound Method ( X ) ~ha@terref . (Wp.)
Benzoquinone
Chlorobenzoquinone Bromobenzoquinone
Methylbenzoquinone crxyloquinone nrXyloquinone p-Xyloquinone
Trimethylbenzoquinone 4,7-Hydrindenequinone
Duroquinone
Acenaphthenequinone
For explanations and symbols see pp. xi-xii.
I REFERENCES FOR CHAPTER 11
REFERENCES FOR CHAPTER 1 1
l Smith and Webster, I. Am. Chem. Soc., 57, 662 (1737). 'L ins tead e t al., I . Am. Chem. Soc., 64, 1778. 2023 (1742); Moore and Hunt-
r e s s , ibid. , 49, 1328 (1727); Steck and Day, ibid. , 65, 454 (1743). 'Crawford and Nelson, I . Am. Chem. Soc., 68, 134 (1746).
Maxwell and Allen, Orp. Syntheses. 24. 1 (1744). 5 F i e s e r e t al., I . Am. &eh. Soc., 61, 3218 (1737); Sah, Rec. trav. chim., 59,
1027 (1740). Veldsaa and Wiardi, Rec. trav. chim., 62, 82 (1743).
7Underwood and Walsh, Org. Syntheses, Coll. Vol. 11, 553 (1743). 'Underwood and Walsh, I . Am. Chem. Soc., 58, 646 (1736); cf. ref. 7.
Arnold and Larson, I . Org. Chem., 5, 250 (1740). 1°Greenspan, Ind. Eng. Chem., 39, 848 (1747). l1 F ie se r and Oxford, I . Am. Chem. Soc., 64, 2060 (1742). l' F i e se r and Chang, I . Am. Chem. Soc., 64, 2043 (1742). 13Vliet, Org. Syntheses, Coll. Vol. I, 482 (1741). 2
14Smith e t al., I . Org. Chem., 4, 318 (1737). l5 Fieser , I . Am. Chem. Soc., 51, 1700 (1727). 16Fieser, Org. Syntheses, Coll. Vol. I, 383 (1741). "Amold and Zaugg, I . Am. Chem. Soc., 63, 1317 (1741); Emerson and Smith,
ibid. , 62, 141 ( 1740). "James, Snell, and Weissberger, I . Am. Chem. Soc., 60, 2084 (1738). 19Smith and Nichols, I . Am. Chem. Soc., 65, 1742 (1743); cf. ref. 14. 'OSmith, Org. Syntheses, Coll. Vol. 11, 254 (1743); Smith and Denyes, I . Am.
Chem. Soc., 58, 304 (1736). "Fieser , Org. Syntheses. Coll. Vol. 11, 430 (1743). " Groggins and Newton, Ind. Eng. Chem., 22, 157 (1730). "Fieser , Org. Syntheses, Coll. Vol. I , 353 (1741). '4Peters and Rowe, I . Chem. Soc., 181 (1745). "Groggins, Stirton, and Newton, Ind. Eng. Chem., 23, 873 (1731). '6Groggins and Newton, Ind. Eng. Chem., 21, 367 (1727).
l "Fieser and Fieser , I . A m Chem. Soc., 57, 1677 (1735). " Butz and Rytina in Organic Reactions, Vol. 5, John Wiley & Sons, New York,
1747. p. 136. l9 Allen and Bell, Org. Syntheses, 22, 37 (1742). 'O Fieser, I . Am. Chem. Soc., 70, 3165 (1748). 'l F i e se r and Martin, Org. Syntheses, 21, 56 (1741). " Fiese r e t al., I . Am. Chem. Soc., 70, 3174 (1748). " Fiese r and Fieser , I . Am. Chem. Soc., 70, 3215 (1748). ' 4 S ~ o t t and Allen, Org. Syntheses, Coll. Vol. 11, 128 (1743). '5Cason in Organic Reactions, Vol. 4, John Wiley & Sons, New York, 1748,
pp. 305-361. Groggins, Stirton, and Newton, Ind. Eng. Chem., 23, 873 (1731).





























