Embed Size (px)
Citation preview

pubs.acs.org/MacromoleculesPublished on Web 10/22/2010r 2010 American Chemical Society
Macromolecules 2010, 43, 9227–9229 9227
DOI: 10.1021/ma101971z
Synthesis of Star Polymers Using ARGET ATRP
Joanna Burdy�nska, Hong Y. Cho, Laura Mueller, and
Krzysztof Matyjaszewski*
Center for Macromolecular Engineering, Department ofChemistry, Carnegie Mellon University, 4400 Fifth Avenue,Pittsburgh, Pennsylvania 15213, United States
Received August 25, 2010Revised Manuscript Received October 13, 2010
Introduction.Starpolymers arebranchedmolecules contain-ing multiple arms connected at a central core that have uniqueproperties compared to their linear analogues.1-3 Star poly-mers typically have lower viscosities than linear polymers andcan also containmany end group functionalities within a singlemolecule,which is useful for various applications.The synthesisof star polymers has been successful by controlled radicalpolymerization (CRP).4-6 Atom transfer radical polymeriza-tion (ATRP)7-9 one of themost powerful CRP techniques, canprovide star polymers by either polymerizing a monomer froma core,10-14 attaching arms to a preformed multifunctionalcore,15,16 or linking preformed arms together.5,17-24 To synthe-size stars by the latter method, often called the arm-firstmethod, a macroinitiator or macromonomer is cross-linked inthe presence of a di- or multivinyl cross-linking agent. Macro-initiators synthesized by ATRP have been successfully cross-linked under normal ATRP conditions. During normal ATRP,an alkyl halide initiator is activated through a reversible redoxprocess by a transitionmetal catalyst in its lower oxidation state(most commonly CuI) to generate radicals and higher oxidationstate catalyst (CuII).25-27 The radical species continues pro-pagation with monomer until it is again deactivated by CuII.Although normal ATRP is useful for synthesis of star polymers,it may lead to a relatively high amount of star-star terminationreactionswhich limits arm conversion andbroadens finalmolec-ular weight distribution.28 This is due to a relatively high radicalconcentration during the polymerization process. In normalATRP, the radical concentration can be controlled by control-ling the ratio of CuI/CuII catalyst and the amount of alkyl halideinitiator in the system. The alkyl halide concentration is fixed inthe macroinitiator, and can only be diluted by solvent. A lowCuI/CuII ratio could be employed, however the reaction wouldbe much slower and would have difficulties in reaching higherconversions. Moreover, a high concentration of radicals insidethe core favors termination and consumesCuI speciesmore thanunder comparable homogeneous conditions.
A solution to these problems was developed for a normalATRP process in that a macromonomer can be used in placeof a macroinitiator. With this technique, the radical concen-tration can be independently controlled by using a smallmolecule alkyl halide initiator in conjunction with macro-monomer arm.29 A star polymer with improved molecularweight distribution could be obtained with this process.However, the reactions typically required repeated injectionsof alkyl halide initiator together with extra cross-linker topush toward higher arm conversion. Additionally, the reac-tion time was typically between 50 and 100 h and an extrasynthetic step is typically required to convert a macroinitiator
to macromonomer. Therefore, star synthesis by ATRP couldstill be improved.
Recently, a new ATRP initiation system was developedwhich allows for a significant decrease in the required Cuconcentration. This system, known as activators regeneratedby electron transfer (ARGET) ATRP utilizes a reducingagent to continuously regenerate the activeCuI species whichis otherwise lost through radical termination reactions.30-36
During ARGETATRP, the concentration of reducing agentdetermines the ratio of CuI/CuII catalyst, which in turndetermines the radical concentration during polymerization.This technique for star synthesis is attractive because it ispossible to control radical concentration during an ARGETATRP. In controlled radical polymerization, radical con-centration is highest at the beginning of the reaction andgradually decreases toward the end, due to the persistentradical effect.37,38 If the radical concentration is too lowduring polymerization, the rate would be very slow. How-ever, if it is too high, more termination reactions occur.Therefore, the use of ARGET ATRP can greatly improvestar synthesis through the ability to adjust radical concen-tration during the polymerization process. At the beginningof the reaction, radical concentration can be kept low byusing a lower concentration of reducing agent to preventstar-star termination. Radical concentration will steadilyincrease as reducing agent is introduced to the system. Atlater stages of the reaction, the star polymers become lesssusceptible toward coupling as they have grown in size andsteric hindrance acts to prevent termination. Consequently,as the reaction progresses, more reducing agent can be fedinto the system resulting in a faster polymerization ratewithout much termination. In this way, the synthesis ofmoreuniform star (co)polymers can be achieved in a relativelyshort reaction time. In this communication, the synthesis ofstar polymers by normal and ARGETATRP are compared.
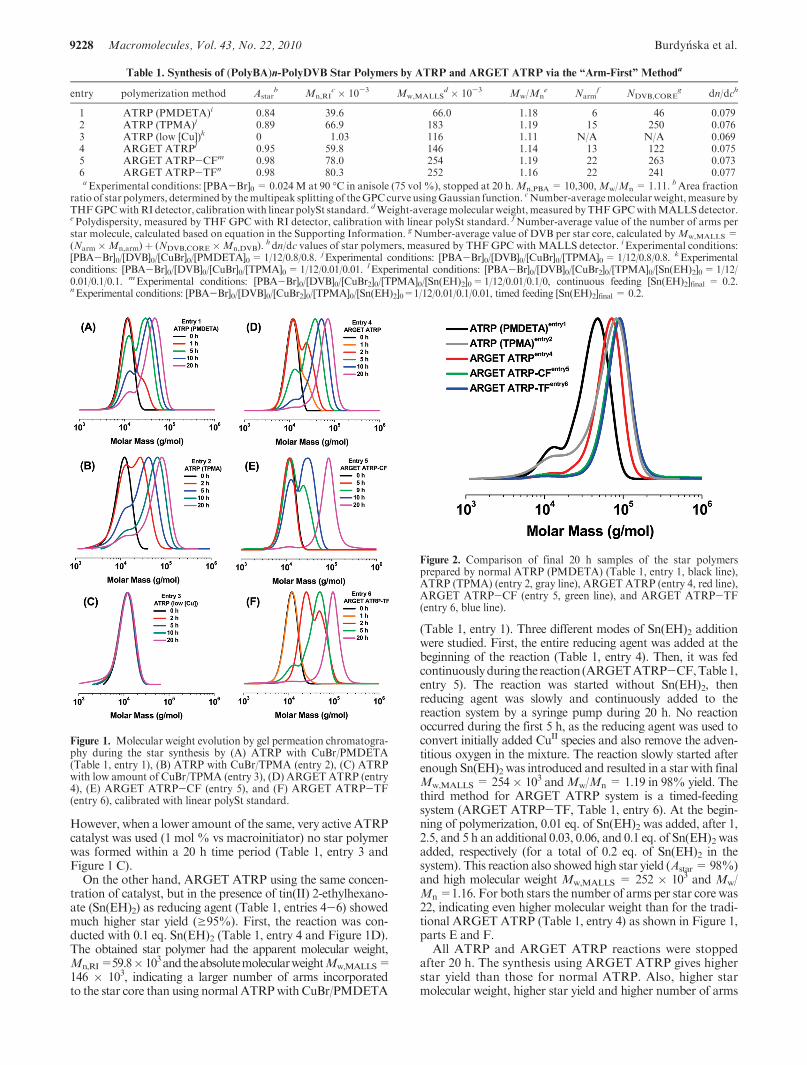
Results and Discussion.Apoly(n-butyl acrylate)-Brmacro-initiator arm (PBA-Br) was synthesized usingARGETATRPconditions to give a product withMn = 10300 andMw/Mn =1.11. This PBA-Br was used in the arm-first star synthesisunder normal ATRP conditions (different ligands and differentcatalyst concentration) and under ARGET ATRP conditions(different reducing agent addition method) (Table 1). Fornormal ATRP star synthesis, CuBr/N,N,N0,N0,N00-penta-methyldiethylenetriamine (PMDETA) and CuBr/tris(2-pyri-dylmethyl)amine (TPMA) were used (Table 1, entry 1-3).When CuBr/PMDETA was used as a catalyst (entry 1), 99%of the divinylbenzene (DVB) was consumed after 20 h. TheGPC curves in Figure 1 A illustrate decreasing RI signalcorresponding to the unreacted macroinitiators and increasingpeak at higher molecular weight region corresponding to starpolymers.Molecular weight (Mn,RI) was 39.6� 103 determinedby GPC with linear polySt standards. Area fraction of starpolymers (Astar) inGPC curve was 84%at 20 h as calculated bydeconvolutionusingGaussian function.Theabsolutemolecularweight measured by multiangle laser light scattering (MALLS)detector (Mw,MALLS) was 66.0 � 103. On the basis of DVBconversion, the dn/dc values in THF at 35 �C (a detailedcalculation is described in the Supporting Information), andMw,MALLS, the calculated number of arms per star core was 6.Similar results were observed when TPMA used as a ligand(Table 1, entry 2).Astar was 89%andMw,MALLS was 183� 103.*Corresponding author. E-mail: [email protected].
9228 Macromolecules, Vol. 43, No. 22, 2010 Burdy�nska et al.
However, when a lower amount of the same, very active ATRPcatalyst was used (1 mol % vs macroinitiator) no star polymerwas formed within a 20 h time period (Table 1, entry 3 andFigure 1 C).
On the other hand, ARGET ATRP using the same concen-tration of catalyst, but in the presence of tin(II) 2-ethylhexano-ate (Sn(EH)2) as reducing agent (Table 1, entries 4-6) showedmuch higher star yield (g95%). First, the reaction was con-ducted with 0.1 eq. Sn(EH)2 (Table 1, entry 4 and Figure 1D).The obtained star polymer had the apparent molecular weight,Mn,RI=59.8� 103andtheabsolutemolecularweightMw,MALLS=146 � 103, indicating a larger number of arms incorporatedto the star core than using normal ATRPwithCuBr/PMDETA
(Table 1, entry 1). Three different modes of Sn(EH)2 additionwere studied. First, the entire reducing agent was added at thebeginning of the reaction (Table 1, entry 4). Then, it was fedcontinuouslyduring thereaction (ARGETATRP-CF,Table1,entry 5). The reaction was started without Sn(EH)2, thenreducing agent was slowly and continuously added to thereaction system by a syringe pump during 20 h. No reactionoccurred during the first 5 h, as the reducing agent was used toconvert initially added CuII species and also remove the adven-titious oxygen in the mixture. The reaction slowly started afterenough Sn(EH)2 was introduced and resulted in a star with finalMw,MALLS= 254� 103 andMw/Mn = 1.19 in 98% yield. Thethird method for ARGET ATRP system is a timed-feedingsystem (ARGET ATRP-TF, Table 1, entry 6). At the begin-ning of polymerization, 0.01 eq. of Sn(EH)2 was added, after 1,2.5, and 5 h an additional 0.03, 0.06, and 0.1 eq. of Sn(EH)2 wasadded, respectively (for a total of 0.2 eq. of Sn(EH)2 in thesystem). This reaction also showed high star yield (Astar=98%)and high molecular weight Mw,MALLS = 252 � 103 and Mw/Mn=1.16. For both stars the number of arms per star core was22, indicating even higher molecular weight than for the tradi-tional ARGET ATRP (Table 1, entry 4) as shown in Figure 1,parts E and F.
All ATRP and ARGET ATRP reactions were stoppedafter 20 h. The synthesis using ARGET ATRP gives higherstar yield than those for normal ATRP. Also, higher starmolecular weight, higher star yield and higher number of arms
Table 1. Synthesis of (PolyBA)n-PolyDVB Star Polymers by ATRP and ARGET ATRP via the “Arm-First” Methoda
entry polymerization method Astarb Mn,RI
c � 10-3 Mw,MALLSd � 10-3 Mw/Mn
e Narmf NDVB,CORE
g dn/dch
1 ATRP (PMDETA)i 0.84 39.6 66.0 1.18 6 46 0.0792 ATRP (TPMA)j 0.89 66.9 183 1.19 15 250 0.0763 ATRP (low [Cu])k 0 1.03 116 1.11 N/A N/A 0.0694 ARGET ATRPl 0.95 59.8 146 1.14 13 122 0.0755 ARGET ATRP-CFm 0.98 78.0 254 1.19 22 263 0.0736 ARGET ATRP-TFn 0.98 80.3 252 1.16 22 241 0.077aExperimental conditions: [PBA-Br]0 = 0.024M at 90 �C in anisole (75 vol %), stopped at 20 h.Mn,PBA= 10,300,Mw/Mn= 1.11. bArea fraction
ratio of star polymers, determinedby themultipeak splitting of theGPCcurve usingGaussian function. cNumber-averagemolecularweight,measure byTHFGPCwithRI detector, calibrationwith linear polySt standard. dWeight-averagemolecularweight,measuredbyTHFGPCwithMALLSdetector.ePolydispersity, measured by THF GPC with RI detector, calibration with linear polySt standard. fNumber-average value of the number of arms perstar molecule, calculated based on equation in the Supporting Information. gNumber-average value of DVB per star core, calculated byMw,MALLS =(Narm �Mn,arm)þ (NDVB,CORE �Mn,DVB).
h dn/dc values of star polymers, measured by THFGPC with MALLS detector. iExperimental conditions:[PBA-Br]0/[DVB]0/[CuBr]0/[PMDETA]0=1/12/0.8/0.8. jExperimental conditions: [PBA-Br]0/[DVB]0/[CuBr]0/[TPMA]0=1/12/0.8/0.8. kExperimentalconditions: [PBA-Br]0/[DVB]0/[CuBr]0/[TPMA]0=1/12/0.01/0.01. lExperimental conditions: [PBA-Br]0/[DVB]0/[CuBr2]0/[TPMA]0/[Sn(EH)2]0=1/12/0.01/0.1/0.1. mExperimental conditions: [PBA-Br]0/[DVB]0/[CuBr2]0/[TPMA]0/[Sn(EH)2]0=1/12/0.01/0.1/0, continuous feeding [Sn(EH)2]final = 0.2.nExperimental conditions: [PBA-Br]0/[DVB]0/[CuBr2]0/[TPMA]0/[Sn(EH)2]0=1/12/0.01/0.1/0.01, timed feeding [Sn(EH)2]final = 0.2.
Figure 1. Molecular weight evolution by gel permeation chromatogra-phy during the star synthesis by (A) ATRP with CuBr/PMDETA(Table 1, entry 1), (B) ATRP with CuBr/TPMA (entry 2), (C) ATRPwith low amount of CuBr/TPMA (entry 3), (D) ARGETATRP (entry4), (E) ARGET ATRP-CF (entry 5), and (F) ARGET ATRP-TF(entry 6), calibrated with linear polySt standard.
Figure 2. Comparison of final 20 h samples of the star polymersprepared by normal ATRP (PMDETA) (Table 1, entry 1, black line),ATRP (TPMA) (entry 2, gray line), ARGET ATRP (entry 4, red line),ARGET ATRP-CF (entry 5, green line), and ARGET ATRP-TF(entry 6, blue line).

Communication Macromolecules, Vol. 43, No. 22, 2010 9229
per star core were obtained with ARGET ATRP as com-pared to normal ATRP. The polymerizations with feedingof reducing agent were quite slow at the beginning, butaccelerated at the later stages. This observation confirms thebenefits of using ARGET ATRP for star synthesis, in thatpolymerization rate (radical concentration) can be preciselycontrolled during polymerization. A comparison of the MWDof all star polymers prepared by ARGET ATRP and normalATRP after 20 h is shown in Figure 2.
Conclusions. The arm-first star synthesis using a macro-initiator synthesized by ATRP could be significantly im-proved by using ARGET ATRP as compared to normalATRP. Not only was less catalyst used, but stars were alsoprepared in essentially quantitative yield (98%) and withhigher MW. Additional improvements can be obtained byapplying slow feeding of the reducing agent to the reactionmixture. Additional studies are currently underway to furtheroptimize the conditions for synthesis of various starsbyARGETATRP.
Acknowledgment. The authors thank the National ScienceFoundation (DMR-10-26060) and acknowledge the GraduateResearch Fellowship for L.M. and thank the members of theCRP Consortium at Carnegie Mellon University for their finan-cial support.
Supporting Information Available: Text giving materials,characterization, and experimental procedures. This material isavailable free of charge via the Internet at http://pubs.acs.org.
References and Notes
(1) Hadjichristidis, N. J. Polym. Sci., Part A: Polym. Chem. 1999, 37,857–871.
(2) Hadjichristidis, N.; Iatrou, H.; Pitsikalis, M.; Mays, J. Prog.Polym. Sci. 2006, 31, 1068–1132.
(3) Hadjichristidis, N.; Pitsikalis,M.; Pispas, S.; Iatrou,H.Chem. Rev.2001, 101, 3747–3792.
(4) Helms, B.; Guillaudeu, S. J.; Xie, Y.; McMurdo, M.; Hawker,C. J.; Frechet, J. M. J. Angew. Chem., Int. Ed. 2005, 44, 6384–6387.
(5) Gao, H.; Matyjaszewski, K. Prog. Polym. Sci. 2009, 34, 317–350.(6) (a) Goh, T. K.; Tan, J. F.; Guntari, S. N.; Satoh, K.; Blencowe, A.;
Kamigaito,M.; Qiao,G.G.Angew. Chem., Int. Ed. 2009, 48, 8707–8711. (b) Pyun, J.; Matyjaszewski, K. Macromolecules 2000, 33,217–220.
(7) Wang, J.-S.;Matyjaszewski,K. J. Am.Chem. Soc. 1995, 117, 5614–15.(8) Matyjaszewski, K.; Xia, J. Chem. Rev. 2001, 101, 2921–2990.
(9) Kamigaito, M.; Ando, T.; Sawamoto, M. Chem. Rev. 2001, 101,3689–3745.
(10) Matyjaszewski, K.; Miller, P. J.; Fossum, E.; Nakagawa, Y. Appl.Organomet. Chem. 1998, 12, 667–673.
(11) Matyjaszewski,K.;Miller, P. J.; Pyun, J.; Kickelbick,G.;Diamanti, S.Macromolecules 1999, 32, 6526–6535.
(12) Ueda, J.; Matsuyama, M.; Kamigaito, M.; Sawamoto, M.Macro-molecules 1998, 31, 557–562.
(13) Angot, S.; Murthy, K. S.; Taton, D.; Gnanou, Y.Macromolecules1998, 31, 7218–7225.
(14) Matyjaszewski, K. Polym. Int. 2003, 52, 1559–1565.(15) Gao, H.; Matyjaszewski, K.Macromolecules 2006, 39, 4960–4965.(16) Whittaker, M. R.; Urbani, C. N.; Monteiro, M. J. J. Am. Chem.
Soc. 2006, 128, 11360–11361.(17) Abrol, S.; Kambouris, P. A.; Looney, M. G.; Solomon, D. H.
Macromol. Rapid Commun. 1997, 18, 755–760.(18) Xia, J.; Zhang, X.; Matyjaszewski, K. Macromolecules 1999, 32,
4482–4484.(19) Zhang, X.; Xia, J.; Matyjaszewski, K. Macromolecules 2000, 33,
2340–2345.(20) Du, J.; Chen, Y. Macromolecules 2004, 37, 3588–3594.(21) Gao, H.; Matyjaszewski, K.Macromolecules 2006, 39, 7216–7223.(22) Gao,H.;Ohno, S.;Matyjaszewski, K. J. Am.Chem. Soc. 2006, 128,
15111–15113.(23) Gao, H.; Tsarevsky, N. V.; Matyjaszewski, K. Macromolecules
2005, 38, 5995–6004.(24) Helms, B.;Guillaudeu, S. J.; Xie, Y.;McMurdo,M.;Hawker, C. J.;
Frechet, J. M. J. Angew. Chem., Int. Ed. 2005, 44, 6384–6387.(25) Tsarevsky, N. V.; Matyjaszewski, K. Chem. Rev. 2007, 107, 2270–
2299.(26) Wang, J.-S.;Matyjaszewski, K.Macromolecules 1995, 28, 7901–10.(27) Matyjaszewski, K.; Tsarevsky, N. V.Nat. Chem. 2009, 1, 276–288.(28) Gao, H.; Matyjaszewski, K.Macromolecules 2006, 39, 3154–3160.(29) Gao, H.; Matyjaszewski, K. Macromolecules 2007, 40, 399–401.(30) Jakubowski, W.; Matyjaszewski, K. Angew. Chem., Int. Ed. 2006,
45, 4482–4486.(31) Jakubowski, W.; Min, K.; Matyjaszewski, K. Macromolecules
2006, 39, 39–45.(32) Matyjaszewski,K.; Jakubowski,W.;Min,K.; Tang,W.;Huang, J.;
Braunecker, W. A.; Tsarevsky, N. V. P. Natl. Acad. Sci. 2006, 103,15309–15314.
(33) di Lena, F.; Matyjaszewski, K. Prog. Polym. Sci. 2010, 35, 959–1021.
(34) Matyjaszewski, K.; Dong, H.; Jakubowski, W.; Pietrasik, J.;Kusumo, A. Langmuir 2007, 23, 4528–4531.
(35) Min, K.; Gao, H.; Matyjaszewski, K. Macromolecules 2007, 40,1789–1791.
(36) Pietrasik, J.; Dong, H.; Matyjaszewski, K. Macromolecules 2006,39, 6384–6390.
(37) Fischer, H. Chem. Rev. 2001, 101, 3581–3610.(38) Tang, W.; Tsarevsky, N. V.; Matyjaszewski, K. J. Am. Chem. Soc.
2006, 128, 1598–1604.



















![Cours Ch Hl[3] _ Atrp](https://img.pdfslide.us/doc/110x75/577d34471a28ab3a6b8d6df9/cours-ch-hl3-atrp.jpg)









