Embed Size (px)
Citation preview

SYNTHESIS OF AN ANTI-CANCER COMPOUND,
SARKOMYCIN, AND SOME ANALOGS
by
GEVORK MINASKANIAN, B.S., M.S.
A DISSERTATION
IN
CHEMISTRY
Submitted to the Graduate Faculty of Texas Tech University in
Partial Fulfillment of the Requirements for
the Degree of
DOCTOR OF PHILOSOPHY
Approved
August, ^ f 9

mi
ACKNOWLEDGMENTS
I am deeply indebted to Dr. John Marx for his guidance and
encouragement throughout the course of this synthetic project. His
understanding of chemical concepts and of the student-advisor re
lationship was instrumental in the development of my ability to
carry out chemical research.
In addition, I wish to thank Drs. Richard Bartsch, John Kice,
Richard Redington and Jerry Mills for their helpful suggestions.
This project was supported by the Robert A. Welch Foundation.
I am deeply grateful for their financial assistance.
11

TABLE OF CONTENTS
Page
ACKJNOWLEDGEMENTS 11
LIST OF SCHEMES v11
LIST OF FIGURES viii
CHAPTER I. INTRODUCTION 1
Characteristics of Sarkomycin 1
Early Structural Studies of Sarkomycin ... 2
Decomposition of Sarkomycin 6
The Absolute Configurat ion of Sarkomycin 8
Previous Syntheses of Sarkomycin and
Related Compounds 9
Scope and Purpose of the Present Work . . . 19
CHAPTER I I . RESULTS AND DISCUSSION 20
Or ig inal Synthetic Plan for Sarkomycin . . . 20
The F i r s t Attempts to Produce 2-Carbo-niethoxy-Cyclopent-2-enone (27) in Quantity 23 Improved Synthesis of 2-Carbomethoxy-cyclopent-2-enone (27) 27
2-Carbomethoxy-3-Cyanocyclopentanone (28) . 30
The Phenomenon of Enol-Keto Tautomerlzation for 2-Carbomethoxy-3-Cyanocyclopentanone (28) 33
Protect ion of Ketone. Formation of Ethylene Ketal of 2-Carbomethoxy-3-Cyanocyclopentanone (29) 36
Attempts on Select ive Hydrolysis of N i t r i l e in Ethylene Ketal of Cyano Ester (29) . . . 38
111

Page
CHAPTER I I I .
CHAPTER IV.
Select ive Reduction. Formation of the Ethylene Ketal of 2-Hydroxymethylene-3-Cyanocyclopentanone (32) 41
The Cyano Der ivat ive of Sarkomycin; 2-Methylene-3-Cyanocyclopentanone (47) . . . . 47
Neighboring Group Par t i c ipa t ion In the Hydrolysis of the N i t r i l e Group in (32). Formation of the Ketal Lactone (49) 48
Lactone Derivat ive of Sarkomycin (50) . . . . 49
Sarkomycin From Keto Lactone (50) 49
El iminat ion of Anchimeric Assistance by Using 2,3-Dlhydropyran as the Hydroxy1 Protect ing Group 53
Amide Der ivat ive of Sarkomycin; 2-Methylene-3-Amidocyclopentanone (54) 55
Sarkomycin ( 1 ) , From 2-Methylene(0-Tetrahydro-pyranyl )-3-Carboxyl1c Acid (53) 56
Looking fo r Bio logical A c t i v i t y 57
CONCLUSION 61
EXPERIMENTAL 64
Modi f icat ion of the Procedure for the Synthesis of 2-Carbomethoxycyclopentanone-Cyclopentadlene Diels-Alder Adduct (34) . . . 65
2-Carbomethoxycyclopent-2-enone (27) from Pyrolysis of Adduct (34) 65
Preparation of Sodium Salt of 2-Carbomethoxycyclopentanone (38) 66
Preparation of Benzeneselenenyl Chloride (35) 67
Preparation of 2-Carbomethoxy-2-Benzene-selenenylcyclopentanone (36) 67
Preparation of 2-Carbomethoxycyclopent-2-enone (27) 68
Preparation of 2-Carbomethoxy-3-Cyanocyclo-pentanone (28) 69
iv

Page
Preparation of the Ethylene Ketal of 2-Carbomethoxy-3-Cyanocyclopentanone (29) 71
An Attempt to Iso la te Stereoisomers of Ethylene Ketal of 2-Carbomethoxy-3-Cyanocyclopentanone (29) 72
Preparation of the Ethylene Ketal of 2-Hydroxymethylene-3-Cyanocyclopen-tanone (32) 73
Preparation of 2-Methylene-3-Cyano-cyclopentanone (47) 74
Preparation of Ketal Lactone (49) . . . . 75
Preparation of Keto Lactone (50) 76
Preparation of Sarkomycin; 3-Carboxy-2-methylenecyclopentanone (1) From Keto Lactone (50) 76
Preparation of Ethylene Ketal of 2-Methylene(0-Tetrahydropyranyl)-3-Cyanocyclopentanone (51) 77
Preparation of the Ethylene Ketal of 2-Methylene(0-Tetrahydropyranyl)-Amidocyclopentanone (52) 78
Preparation of the Ethylene Ketal of 2-Carbomethoxy-3-Am1docyclopentanone (30) . 79
Preparation of 2-Methylene(0-Tetrahydro-pyranyl )-3-Carboxylic Acid (53) 80
Preparation of Amide Derivat ive of Sarkomcyin; 2-Methylene-3-Amidocyclo-pentanone (54) 80
Preparation of Sarkomycin (1) From 2-Methylene-(0-Tetrahydropyranyl)-3-Carboxylic Acid (53) 81
LIST OF REFERENCES 83
APPENDIX A: Schematic Diagram of Pyrolysis Apparatus . . . . 87

Page
APPENDIX B: Infrared and Nuclear Magnetic Resonance Spectra 89
APPENDIX C: Publication Arising From This Work 122
VI

LIST OF SCHEMES
Page
SCHEME I 4
SCHEME II 7
SCHEME III IQ
SCHEME IV IT
SCHEME V 12
SCHEME VI -13
SCHEME VII 14
SCHEME VIII -15
SCHEME IX 16
SCHEME X -18
SCHEME XI 22
SCHEME XII 26
SCHEME XIII 27
SCHEME XIV 32
SCHEME XV 50
SCHEME XVI 52
SCHEME XVII 62
VI 1

LIST OF FIGURES
Figure Page
1 Schematic Diagram of Pyrolysis Apparatus 88
2 IR spectrum of 2-Carbomethoxy-2-Benzeneselenenylcyclo-pentanone (36) 90
3 NMR spectrum (60 MHz) of 2-Carbomethoxy-2-Benzeneselenenylcyclopentanone (36) 91
4 IR spectrum of 2-Carbomethoxy-3-Cyanocyclopentanone (28) 92
5 NMR spectrum of (60 MHz) of 2-Carbomethoxy-3-Cyanocyclopentanone (28) 93
6 Dependance of enol-keto tautomerlzation with Temperature In 1800-1600 cm"^ region of the IR spectrum 94
7 Dependance of enol-keto tautomerlzation upon dilution with Acetone-d^ (a-c), Benzene (d-f), and CDCU(g-l), as monitored by NMR In 6 3.8-4 p.p.m. region . . . . . 95
8 IR spectrum of Ethylene Ketal of 2-Carbomethoxy-3-Cyanocyclopentanone (29) 96
9 NMR spectrum (100 MHz) of 2-Carbomethoxy-3-Cyanocyclopentanone (29) 97
10 IR spectrum of cis-2-Carbomethoxy-3-Cyano-cyclopentanone (29) 98
11 NMR spectrum (100 MHz) of cis-2-Carbomethoxy-3-Cyanocyclopentanone (29) 99
12 IR spectrum of Ethylene Ketal of 2-hydroxymethylene-3-Cyanocyclopentanone (32) 100
13 NMR spectrum (100 MHz) of Ethylene Ketal of 2-hydroxy-methylene-3-Cyanocyclopentanone (32) 101
14 Reduction of Ethylene Ketal of 2-Carbomethoxy-3-cyanocyclopentanone (29) with L1BH, a) with added NaOH (compound 32) b) without addgd NaOH (compound 45) 102
15 IR spectrum of 2-Methylene-3-Cyanocyclopentanone (47) a) heat b) In chloroform 103
16 a) NMR spectrum (60 MHz) of 2-Methylene-3-Cyano-cyclopentanone (47) after distillation b) NMR spectrum (100 MHz) of (47) before distillation . . . . 104
17 IR spectrum of Ketal Lactone (49) 105
viii

Figure Page
18a NMR spectrum (100 MHz) of Ketal Lactone (49) 106
18b Expansion of 6 4.2-4-7 region of Figure 18a showing
coupling constant assignments 107
19 IR spectrum of Keto Lactone (50) 108
20 NMR spectrum (100 MHz) Keto Lactone (50) 109
21 IR spectrum of Sarkomycin prepared from Keto Lactone (50) 110
22 NMR spectrum (100 MHz) of Sarkomycin prepared from Keto Lactone (50) Ill
23 IR spectrum of Ethylene Ketal of 2-Methylene (0-Tetrahydropyranyl)-3-Am1docyclopentanone (52) . . . 112
24 NMR spectrum (100 MHz) of Ethylene Ketal of 2-Methylene -(0-Tetrahydropyranyl)-3-Am1docyclopentanone (52). . . 113
25 IR spectrum of Ethylene Ketal of 2-Carbomethoxy-3-Amldocyclopentanone (30) 114
26 NMR spectrum (60 MHz) of Ethylene Ketal of 2-Carbo-methoxy-3-Am1docyclopentanone (30) 115
27 IR spectrum of 2-Methylene(0-Tetrahydropyranyl)-3-Carboxylic acid (53) a) In CHCI3 b) neat 116
28 NMR spectrum (100 MHz) of 2-Methylene(0-Tetra-hydropyranyl)-3-Carboxylic acid (53) 117
29 IR spectrum of 2-Methylene-3-Am1docyclopentanone (54) 118
30 NMR spectrum (100 MHz) of 2-Methylene-3-Am1docycl opentanone (54) a) in Acetone-d^ DCl-D^O solution b) in CDCI3 119
31 IR spectrum of Sarkomycin (1) prepared from 2-Methylene (0-Tetrahydropyranyl)-3-Carboxyl ic acid (53) 120
32 NMR spectrum (100 MHz) of Sarkomycin (1) prepared from 2-Methylene(0-Tetrahydropyranyl)-3-Carboxyl1c acid (53) a) before bicarbonate ext ract ion b) a f t e r ex t rac t ion 121
IX

X
CHAPTER I
INTRODUCTION
Studies of antibiotics have indicated that the Streptomyces
microorganisms are a rich source of a variety of compounds. About 200
soil Streptomyces strains were examined In Japan in the 1950's by
Yamamoto's anti-cancer screening method, and the existence of sub
stances exhibiting selective toxicities to the cells of Yoshida sar
coma of the rat was confirmed. Among those anti-cancer substances
obtained, one named sarkomycin was found to be the lowest In toxicity
and the most effective on Ehrlich cancer of mice. A brief description
2 "? of sarkomycin was first reported by Umezawa ' In 1953. One year
later, the production, extraction, toxicity, and effects of this drug 4
were recorded by Umezawa in detail.
Characteristics of Sarkomycin
The strain No. W-115-C was isolated from the soil at Kamakura
4 3
by S. Umezawa and T. Okuda. When Okami and Umezawa examined the
strains Isolated by the above authors, strain No. W-115-C was found
to be a rare species. Therefore, this strain was included in the
group tested for anti-cancer properties. In this first test, the
broth filtrate of this strain prolonged the survival period of rats
bearing sarcoma. The active principle in the broth filtrate was named
sarkomycin.
Sarkomycin was extracted as an oil by a solvent or adsorption
process. Little change in its properties was observed on purification.
Active material did not sublime from acid oils at 1mm Hg pressure and 1

100°C Sarkomycin showed acidic characteristics and exhibited weak
antibacterial effects. The free acid of sarkomycin was soluble in
water, methanol, ethanol, butanol, ethyl acetate, and was sparingly 3
soluble in petroleum ether.
Sarkomycin exhibited a curious toxicity. It was more toxic by
subcutaneous injection than by intravenous injection. No latent tox
icity was noted.
Sarkomycin was Injected daily in a monkey Intravenously for 20
days. Daily injection of 100 mg/kg produced no evidence of toxicity.
When 200 mg/kg was injected, the monkey showed signs of nausea, but
survived, and tolerated further daily Injections of 100-150 mg/kg.
The injection of 100 mg/kg repeated 20 times caused no change In the 3
white or red blood cell count.
Sarkomycin obtained by the solvent process has a potency of
1-3 g/ml. The LD was 800-1,600 mg/kg by Intravenous injection,
400-800 mg/kg by subcutaneous injection, and 4800-6400 mg/kg by oral 3
administration.
Early Structural Studies of Sarkomycin 5
In 1955, Hooper, ejt aj_. reported evidence which Indicated that
the structure of sarkomycin Is 3-carboxy-2-methylenecyclopentanone (1).
Sarkomycin (1)

The structural proof was based on chemical analysis and Infrared
spectroscopy, which in turn proved of value in following the purifi
cation of sarkomycin. Analysis of crude materials showed broad bands
at 2920, 1720, 1550, 1460, 1405, 1380, 1200, 950, 855, and 820 cm"^
A small band or shoulder at 1640 cm" was found in all active samples.
With increasing purity, the relative intensity of this band increased.
It was assigned to a carbon-carbon double bond and correlated well
with antibacterial activity. The only other assignments that could be
made on crude sarkomycin acid or sodium salt were the probable presence
of a carboxyl carbonyl and at least one additional carbonyl. The
broad band in the 1250 cm' region was suggestive of a five or six
membered ring system. With increasing purity, these bands were more
sharply defined and tended to confirm the original assignments.
Like many other structural studies, structural determination of 5
sarkomycin was carried out by Hooper, et_ aj_. by a series of reactions,
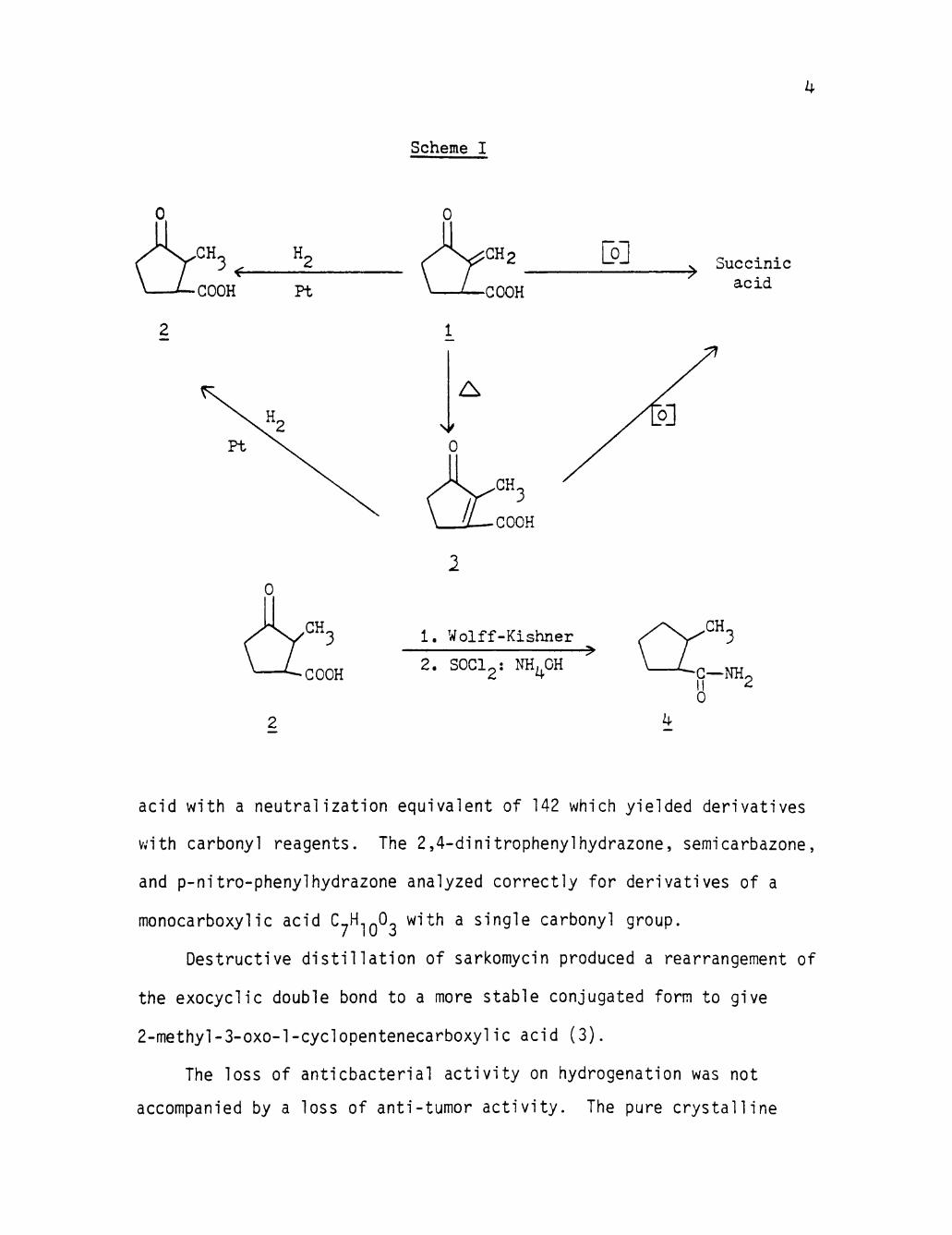
as Illustrated in Scheme I (page 4).
Hydrogenation of sarkomycin in neutral aqueous solution with
platinum or Raney nickel catalyst and 1 to 3 atmosphere of hydrogen
destroyed the antibacterial activity. Acidification and solvent
extraction yielded a dense oil, which unklike sarkomycin preparations,
was partly crystalline. By extraction with petroleum ether or carbon
disulfide, a crystalline product was Isolated in amounts of 26 to 60
per cent of the weight of crude hydrogenation product.
The hydrogenation product was purified by repeated recrystalli-
zation and sublimation to give a white, crystalline product (2) melting
sharply at 99-99.5°C and analyzing for C^H.|Q03. The material was an
Scheme I
COOH
H,
Pt
Co] • >
Succinic acid
Pt
H,
A
0
(V COOH 1. Wolff-Kishner
2. SOCI2: NH^OH CY II 0
acid with a neutralization equivalent of 142 which yielded derivatives
with carbonyl reagents. The 2,4-dinltrophenylhydrazone, semicarbazone,
and p-nitro-phenylhydrazone analyzed correctly for derivatives of a
monocarboxylic acid C^H 0 with a single carbonyl group.
Destructive distillation of sarkomycin produced a rearrangement of
the exocyclic double bond to a more stable conjugated form to give
2-methyl-3-0X0-1-cyclopentenecarboxylic acid (3).
The loss of anticbacterial activity on hydrogenation was not
accompanied by a loss of anti-tumor activity. The pure crystalline

/
hydrogenation product was claimed to be fully as active in preventing
excessive weight gain and prolonging life of mice bearing the Ehrlich
ascites tumors as the most highly purified preparation of sarkomycin.
Further work cast doubt on this early conclusion. The rearrangement
product was, however, devoid of both anti-bacterial and anti-tumor
activity. All experimental evidence was in complete accord with the
proposed structure of sarkomycin (1).
In a later paper, Maeda and Kondo studied the structures of some
decomposition products formed after freeze-drying of the sodium salt
of sarkomycin in aqueous solution, those formed after storage of
sarkomycin in aqueous solution, those formed after storage of sarko
mycin in acid solution, or those formed after storage as a neutral
amorphous powder. The results are briefly described below.
The purified syrup of sarkomycin was colorless. It was dissolved
in water and shaken with chloroform, then a main part of the activity
was transferred into the solvent, but a part of that remained In the
aqueous layer. The active fraction transferred into chloroform was
named sarkomycin A and another active fraction left in the aqueous
layer was named sarkomycin A'.
Then, sarkomycin A was separated into two fractions, the one was
a main fraction and another was minor. The latter was named sarkomycin
B. Sarkomycin B was confirmed to be a decomposition product of
sarkomycin A. The different solubility in the chloroform extraction
was the main difference between A and A'. But, it was found that
sarkomycin A', after it had been kept in the neutral aqueous solution
in the ice box, changed its transferability into chloroform, and

became identical with A. Therefore, the existence of A' as a compound
different from A seems conclusive. In all sarkomycins, the presence
of a carbonyl group was confirmed by the 2,4-d1n1trophenyl hydrazone
test and by the infrared absorption. The optical rotations of dried
20 syrups of the sarkomycins were determined. Thus, [a] -. of sarkomycin
A was -45° and that of A' -57° and that of B -33°. 5
Hooper et_ . presented the structure of sarkomycin A as
3-carboxy-2-methylenecyclopentanone (1). No structures were assigned
to sarkomycins A' and B.
Decomposition of Sarkomycin
When sarkomycin was dissolved in commercially available ether and
evaporated, It turned to an inactive and water-insoluble powder. It
was soluble in alkaline water and turned to gel with acid. This o
polymerized product has been studied by Yamada and others and poly
merizat ion of sarkomycins was confirmed to be catalyzed by peroxides.
However, the addi t ion of polymerization i n h i b i t o r s , such as hydro-
quinone, d id not s t a b i l i z e sarkomycin in ethanol , ethyl acetate or
water.
As l a t e r described by Maeda, sarkomycins A, A' and B changed to
a polymer ca l led the Z-crysta l by the react ion with formic ac id .
Also, the Z-crysta l was the main decomposition product of sarkomycins
A and B during the storage of the sarkomycin acid so lu t i on , and t h i s
decomposition product would be produced from sarkomycin A through
sarkomycin B. The Z-crysta l was produced quan t i t a t i ve l y from a l l
sarkomycins by the act ion of formic ac id . Treatment of the Z-crysta l

Scheme II
Decomposition of Sarkomycin
s : •H o >» a o
f
I 0)
o e o •g
o
o ^ O
0) > bO S cd o H •!*: .H TJ O ^ TS s
-P cti O O CO w w p
0)
• ^
^
-p to
o I
!S3
O
-o^ 'o~"o
'o:zP
« «
( I I I i )
I I 1 1
CVi (Ni
Cvi
"OHQ

8
with 5% HCl at 100° converted i t into a new polymer, called the
M-crystal.
Decomposition products of sarkomycin are summarized in Scheme I I .
The above data and the sarkomycin structure, proposed by Hooper
and others, suggested the structures of the Z-crystal and the M-crystal,
as shown in Scheme I I , though there is no proof for these structures.
The Absolute Configuration of Sarkomycin g
I t was shown by Tatsuoka, et_ a l . , that the natural sarkomycin
is in the levorotatory form [ct] ^^ -32.5° (C=l, MeOH).
In 1963, Sato, ejt aj_., determined the absolute configuration
of cyclopentanone-3-carboxyl1c acid (5) , the start ing material for
Toki's synthesis of natural (-)-sarkomycln, and consequently as
signed the (S) configuration to sarkomycin ( l a ) .
COOH
(-)-i
CH2^NR2
COOC^H^
0
^/y-3 t^GOOH H
( - ) - l a
12 However, af ter careful invest igat ion, H i l l et_ a]_.
showed clearly that Mannich condensation of (5) gives a mixture of
positional Isomers, predominantly the 5-, not the 2-subst1tuted pro
duct, and that consequently Toki's synthetic sarkomycin was extremely
impure

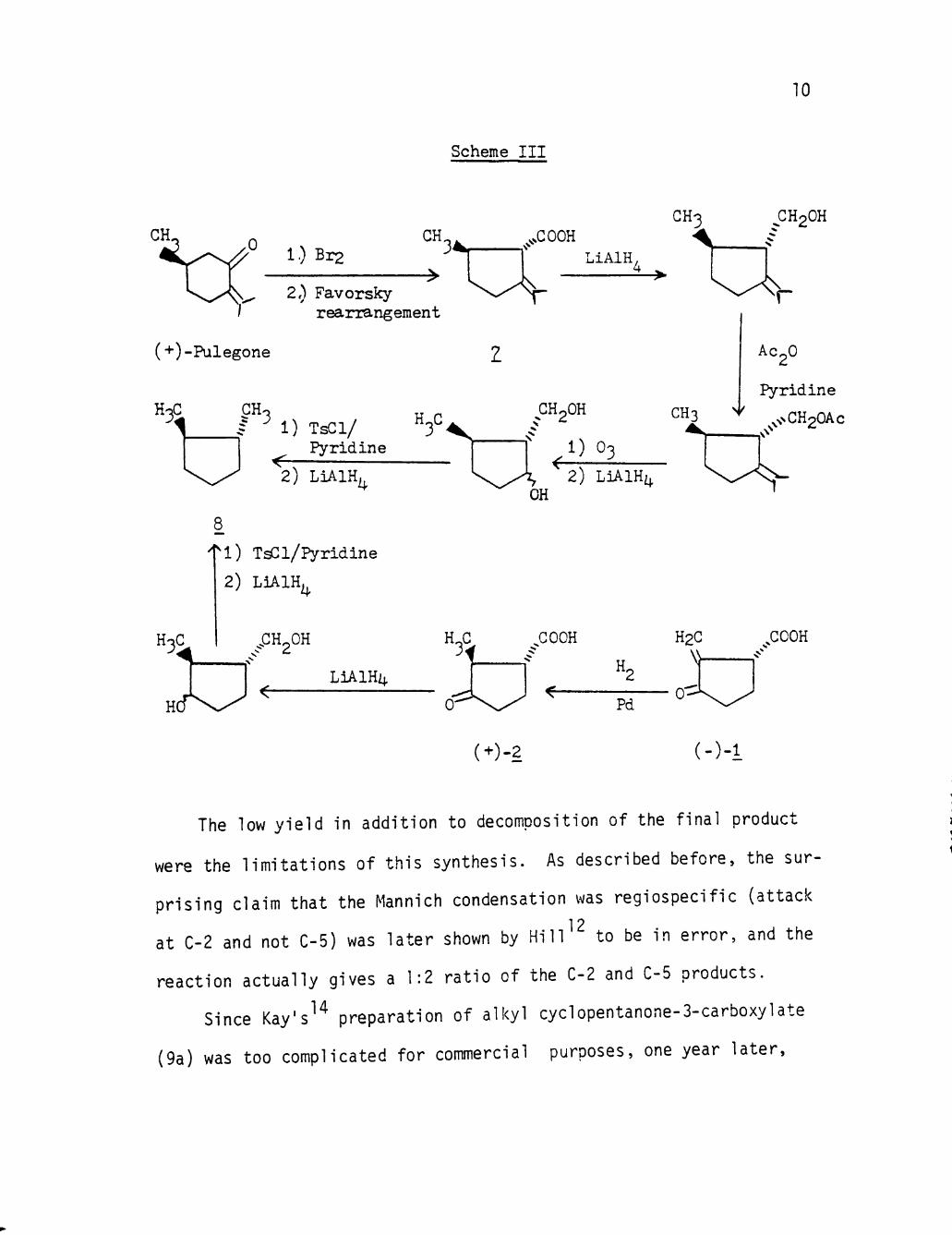
1 ? Since the absolute conf igurat ion of pulegone is known, there-
12 f o r e , H i l l appl ied pulegenic acid (7) as an unambiguous standard
fo r conf igurat ional cor re la t ions by the reactions shown in the Scheme
I I I (page 10).
These cor re la t ions demonstrate that (-)- l ,2-d1methylcyclopentane
( 8 ) , derived from pulegenic acid ( 7 ) , has the ( IR, 2R) conf igurat ion
shown, and that (+)-dihydrosarkomyc1n (2) has the ( IR, 2S) conf igur
a t i on . Natural (-)-sarkomycln consequently has the (R) con f igura t ion ,
in contrast to the e a r l i e r published conclusion.
Previous Syntheses of Sarkomycin and Related Compounds
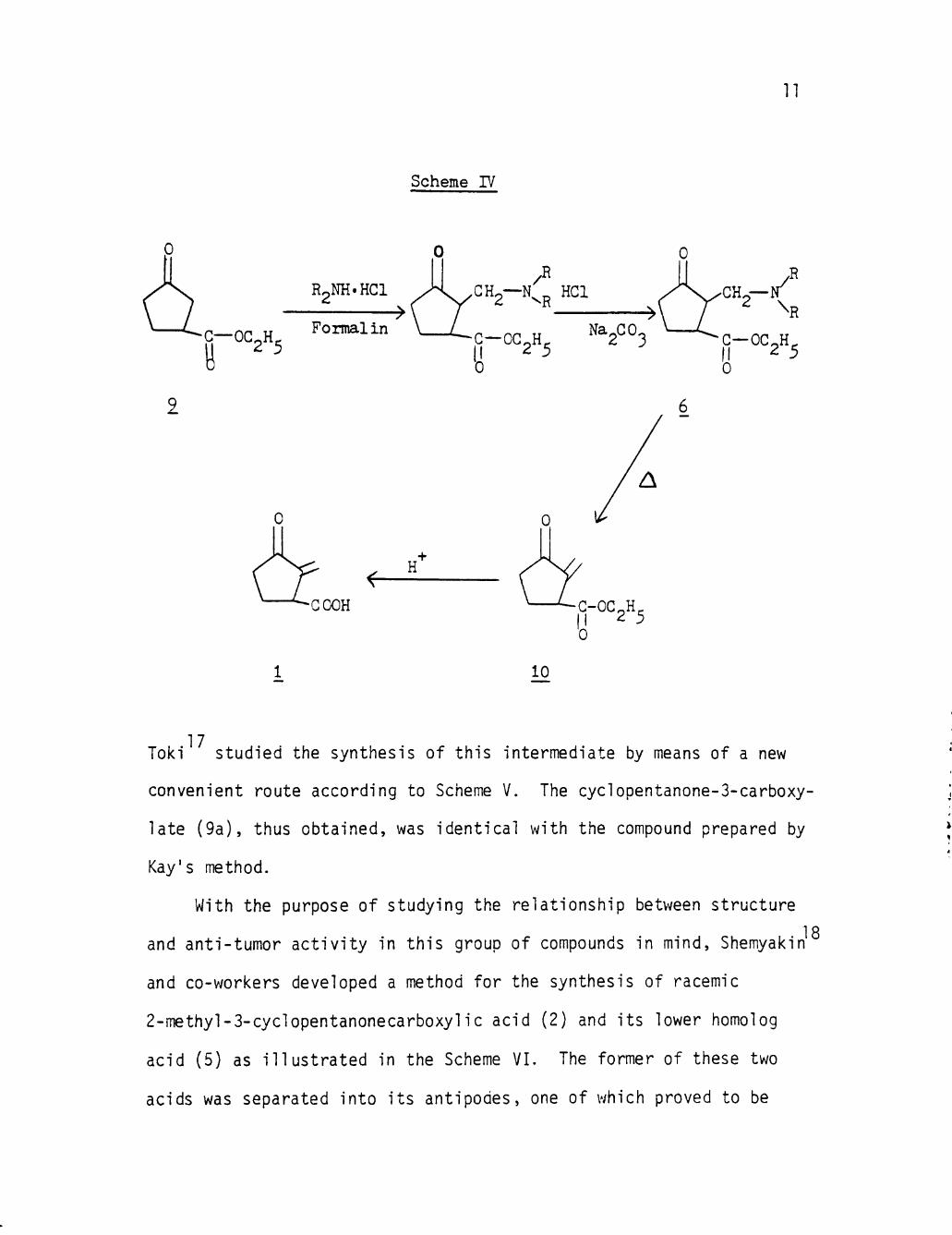
The synthesis of rac-sarkomycin was f i r s t achieved by Toki
who obtained the ul t imate product by a four step process (Scheme IV)
s t a r t i ng from ethyl cyclopentanone-3-carboxylate (9 ) , prepared by
14 Kay's method.
15 As claimed by Tok i , the Mannich react ion was successful ly
appl ied to compound (9) to Introduce a methylene group by treatment
wi th hydrochloride of a secondary amine and fo rmal in , fol lowed by
thermal e l im ina t ion of the amine. However, hydrolysis of the ethyl
ester (10) to the free ac id , dl-sarkomycin, was accompanied by de
composition of the product under severe condit ions while a large
amount (80-90%) of the ester remained in tac t under mi ld condi t ions.
1 fi One year l a t e r , the same author published a paper applying the
four-stage process (Scheme IV ) , using o p t i c a l l y act ive cyclopen-
tanone-3-carboxyl1c acid (5) as s ta r t i ng material to synthesize d-
and 1-sarkomycins.
Scheme I I I
10
1.) Bra CH,
2,) Favorsky rear rangement
(+ ) -Pu legone
H-tiC
HoC
1) TsCl / P y r i d i n e
2) LIMH^
8
^ 1 ) TsGl /Pyr id ine
2) LiAlH^
.CH OH
LiMHi^
H^C
,COOH CH3
LiAlH,
CHoOH CH3
^ 1) O3
OH 2) LiAlHi^
COOH
H,
Pd
(-^)-2
H2C
J : H 2 0 H
Ac^O
( - ) - l
P y r i d i n e
^NNCH20AC
COOH
The low yield in addition to decomposition of the final product
were the limitations of this synthesis. As described before, the sur
prising claim that the Mannich condensation was regiospecific (attack
at C-2 and not C-5) was later shown by Hill^^ to be in error, and the
reaction actually gives a 1:2 ratio of the C-2 and C-5 products.
Since Kay's^^ preparation of alkyl cyclopentanone-3-carboxylate
(9a) was too complicated for commercial purposes, one year later.
C—OC^H-
Scheme IV
RgNH.HCl
Formalin
<r
COOH
H
G H 2 ~ N 4 HCl
C—OC^H-II 2 5
10
Na2C03
C-OC„H-jl 2 5 0
6
11
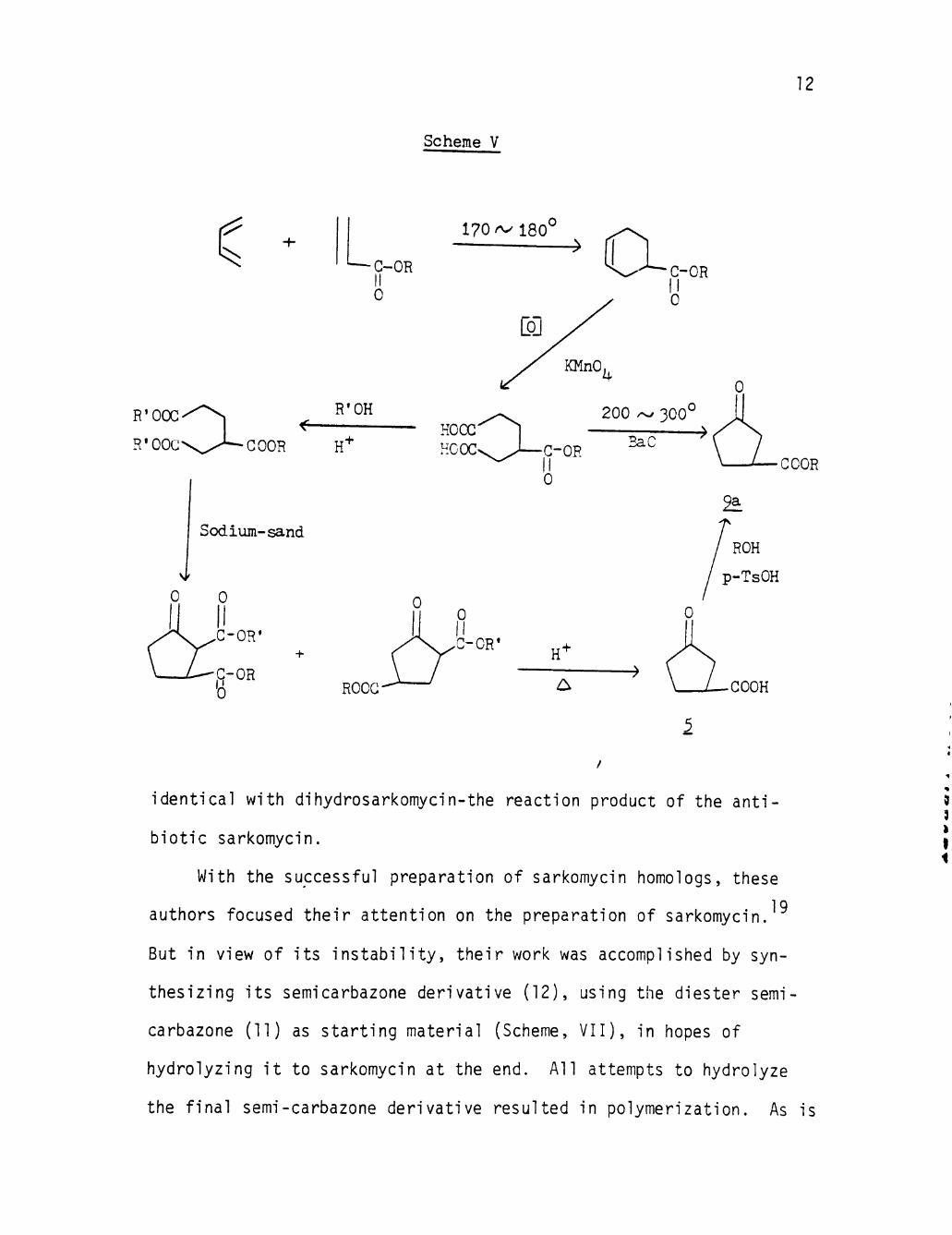
Toki studied the synthesis of this intermediate by means of a new
convenient route according to Scheme V. The cyclopentanone-3-carboxy-
late (9a), thus obtained, was identical with the compound prepared by
Kay's method.
With the purpose of studying the relationship between structure
and anti-tumor act ivi ty in this group of compounds in mind, Shemyakin
and co-v/orkers developed a method for the synthesis of racemic
2-methyl-3-cyclopentanonecarboxylic acid (2) and i t s lower homolog
acid (5) as i l lus t ra ted in the Scheme VI. The former of these two
acids was separated into i t s antipodes, one of which proved to be
18
Scheme V
12
R'OOC
R'OOG GOOR
Sodium-sand
C-OR*
•C-OR II 0
R'OH
H"
ROOG
170^180'
HOCC HCOG
^
0
C-CR'
- ^
G-CR II 0
200 ^ 300
H"
GOOR
-GOOH
19
Identical with dihydrosarkomycin-the reaction product of the an t i
b io t ic sarkomycin.
With the successful preparation of sarkomycin homologs, these
authors focused thei r attention on the preparation of sarkomycin
But in view of i t s i n s t a b i l i t y , the i r work was accomplished by syn
thesizing i t s semicarbazone derivative (12), using the diester semi
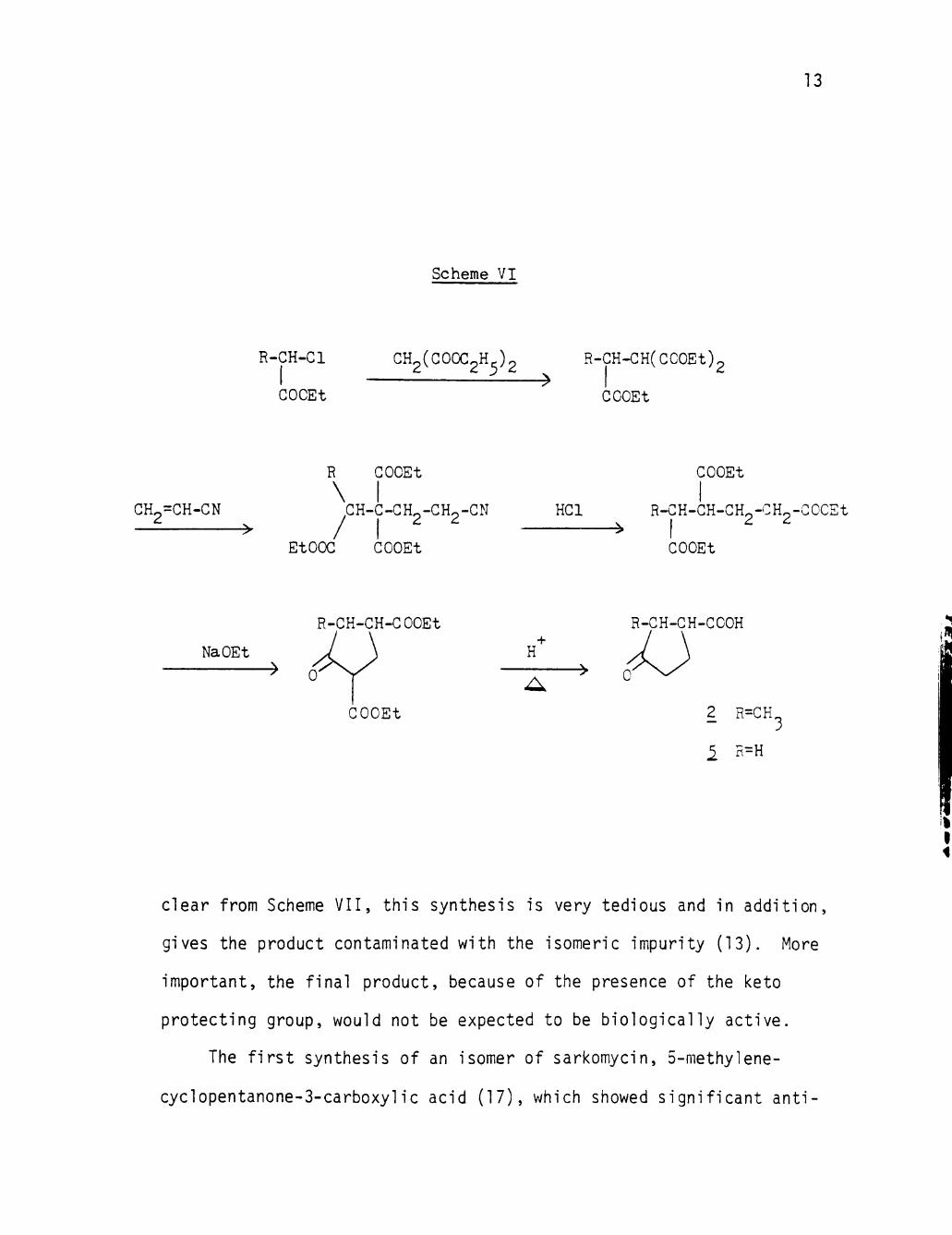
carbazone (11) as star t ing material (Scheme, V I I ) , In hopes of
hydrolyzing i t to sarkomycin at the end. Al l attempts to hydrolyze
the f ina l semi-carbazone derivative resulted in polymerization. As is
Scheme VI
13
R-GH-Gl
GOCEt
GH2(G00C2H.)2 ^
R-GH-GH(GCOEt).
COOEt
GH2=CH-GN
R GOOEt
\ I GH-G-GH^-GH^-GN / I 2 2
EtOOG GOOEt
HGl
GOOEt
R-CH-GH-GH^-CH^-GOCSt
GOOEt
NaOEt • ^
R-GH-GH-GOOEt
GOOEt
H • »
R-GH-GH-GOOH
0
2 R=GH,
^ E=H
clear from Scheme VII, this synthesis is very tedious and In addition,
gives the product contaminated with the isomeric impurity (13). More
Important, the final product, because of the presence of the keto
protecting group, would not be expected to be biologically active.
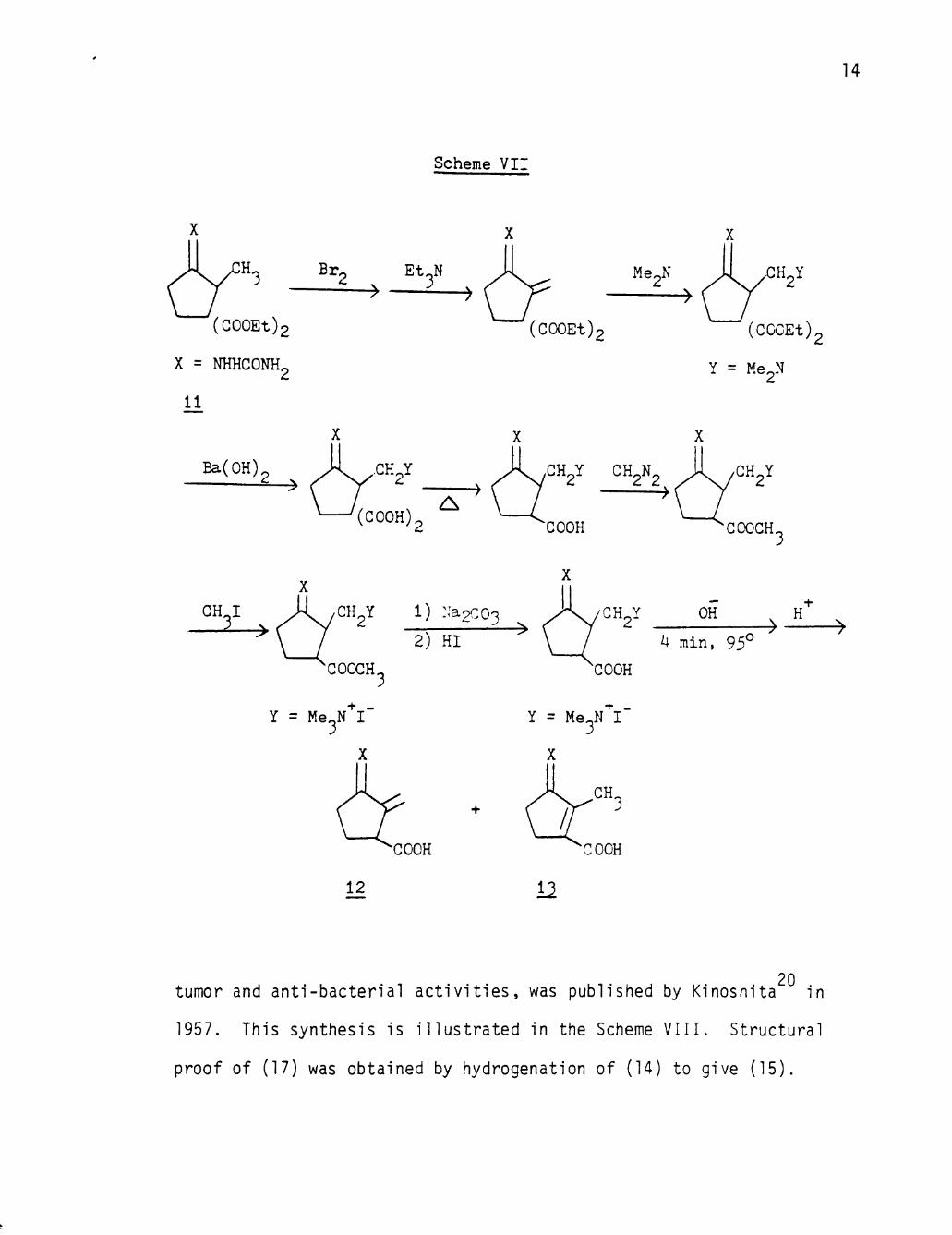
The f irst synthesis of an isomer of sarkomycin, 5-methylene-
cyclopentanone-3-carboxyl1c acid (17), which showed significant anti-
Scheme VII
14
(G00Et)2
X = NHHCONH,
11
Ba(OH) ..GH^Y 2_ /V'"2 w. (GOOH), A
(GOOEt).
Ct
Me^N
GH^Y GH^N^
GOOH
(GOOEt),
Y = Me^N
GH I A
GH^Y
GOOGH.
Y = Me^N I
1) Na2C03
2) HI
GOOH
12
• >
yV/CH^Y u GOOH
Y = Me^N'^l"
n
OH 4
H
4 min, 95° • ^
20 tumor and anti-bacterial a c t i v i t i e s , was published by Kinoshita in
1957. This synthesis is i l lus t ra ted in the Scheme VIII. Structural
proof of (17) was obtained by hydrogenation of (14) to give (15).
Scheme VIII
15
15
I^xaformaldehyde YH
COOH Me^N.HGl
• >
GH^OH YH G ^ V ^
^COOH
Y = Me2N«HGl
-OCH.
° / ^ 2 / 1^
Pt
"''Y_) -GOOGH,
11
H
g-0CH3 GOOH
17
0
^ ^GOCH
16
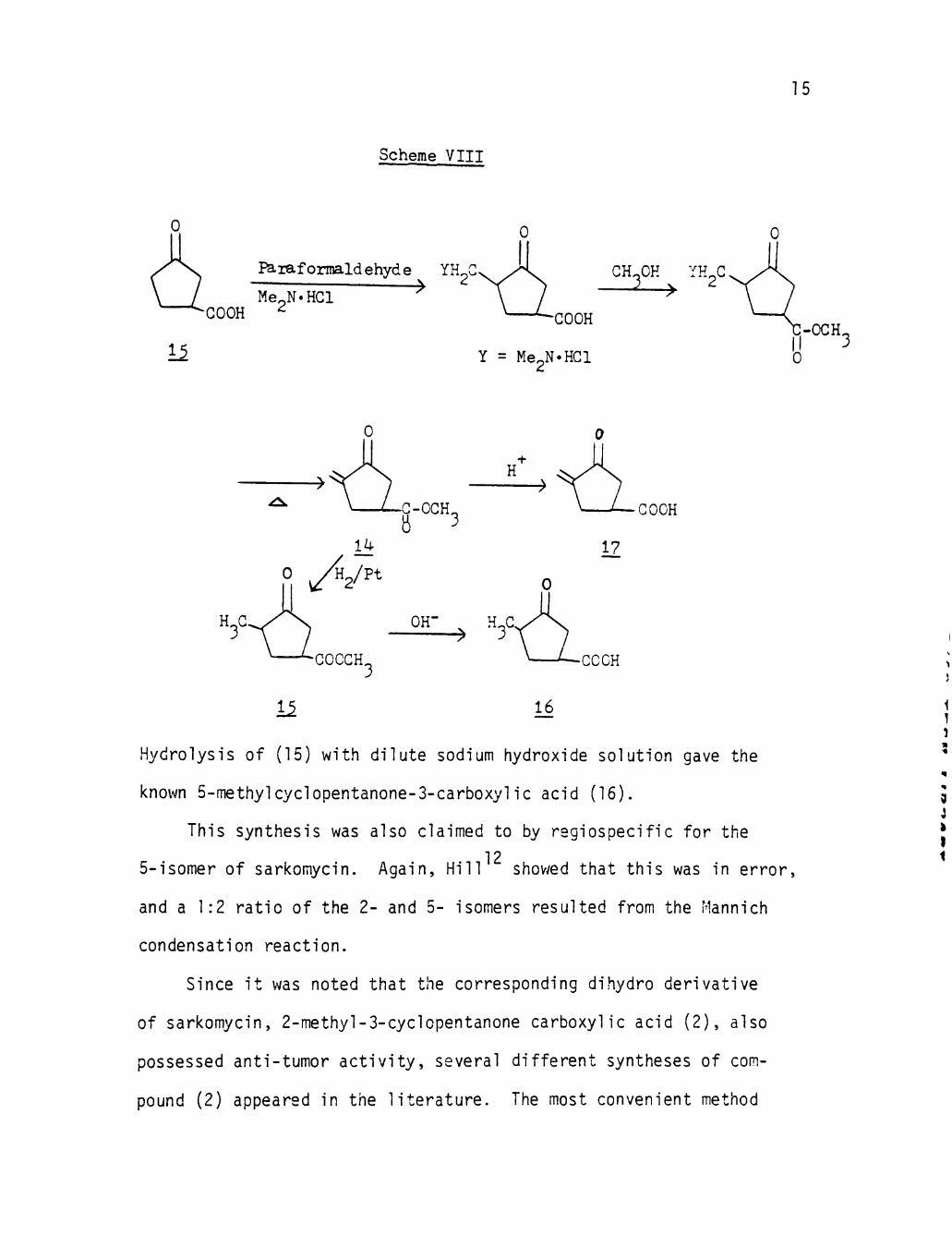
Hydrolysis of (15) wi th d i l u te sodium hydroxide solut ion gave the
known 5-methylcyclopentanone-3-carboxylic acid (16).
This synthesis was also claimed to by reg iospec i f ic fo r the
12 5-1somer of sarkomycin. Again, H i l l shovyed that th i s was In e r ro r ,
and a 1:2 r a t i o of the 2- and 5- isomers resulted from the Mannich
condensation react ion .
Since i t was noted that the corresponding dihydro der ivat ive
of sarkomycin, 2-methyl-3-cyclopentanone carboxyl ic acid ( 2 ) , also
possessed ant i - tumor a c t i v i t y , several d i f f e ren t syntheses of com
pound (2) appeared In the l i t e r a t u r e . The most convenient method

16
21 was reported by N. Smirnova and co-workers, which can be expressed
by the Scheme IX.
Scheme IX
R-GH-COOCpH I
H G2OOG-GH-COOC2H
Ethyl aciylate •^
R
H G2OOG ^ C H - G ( G 0 0 G 2 H - ) 2
GH2-GH2-GCCG2H^
NaOEt . / ^ N / ' ° ' ' ' 2 « 5
(GOOG2H )2
0
H3G200G^>< R
H , GH-OH • ^
Reflux 1 hr
0
a' (G00G2H^)2
2 ?=CK^ GOOH
i
i
i 1
9
J 1
As described above, the early syntheses of sarkomycin left much
to be desired in terms of se lec t iv i ty , yield, and elegance. A major
diff iculty in any synthesis l ies in the presence of the exocyclic
methylene group conjugated with keto and carboxylic functionalities
on a five membered ring system, which makes the system too reactive 22 tov^ard polymerization and other reactions. Jankowski in 1971 came

17
up with the idea of making rac-homosarkomycin (25) in order to study
the effect of ring size on stability and biological properties. His
synthesis is represented in Scheme X (page 18). He claimed his method
can well be applied to make further sarkomycin analogs also. As
illustrated in the Scheme X, the starting material, the keto-ester
(18), was quantitatively ketalyzed to the ketal-ester (19). This
product was reduced with LIAIH^ to the ketal-alcohol (20). Treatment
of product (20) with thionyl chloride gave the chloro-ketal (21).
The dehydrochlorination of compound (21) (DBU, DMSO) resulted in a
mixture of endo and exo olefins (22) (55%). After applying different
methods for the isomerlzation of the double bond, this author claimed
that the treatment of the crude mixture with isopropanol in the
presence of traces of sodium isopropoxide resulted in the exo migration
of the double bond (80-90%). The exo product (22) then was treated *1
with NBS (CCl,, reflux 12h). A mixture of four products was obtained I
and the major product was reported to be 3-bromo-methylene-ketal (23) ,
in 33% yield. The crude product mixture (23) was stirred with "
Cu„(CN)« and the mixture of exo and endo 3-cyano-methylene-ketal (24) c Z ••
was isomerized to obtain 70% of the exo product (24). The acid from 9
hydrolysis of the latter compound (2 h reflux, 5% HCl) gave a mixture }
of the exo and endo homosarkomycin (25) (40%).
It is very obvious from the experimental procedure that the
author tried to synthesize a molecule named homosarkomycin (25) with
out considering the impractibility of the intermediate steps and
impurity of samples due to the different isomeric products. Since
it is not possible to separate the isomers in any step mentioned above;
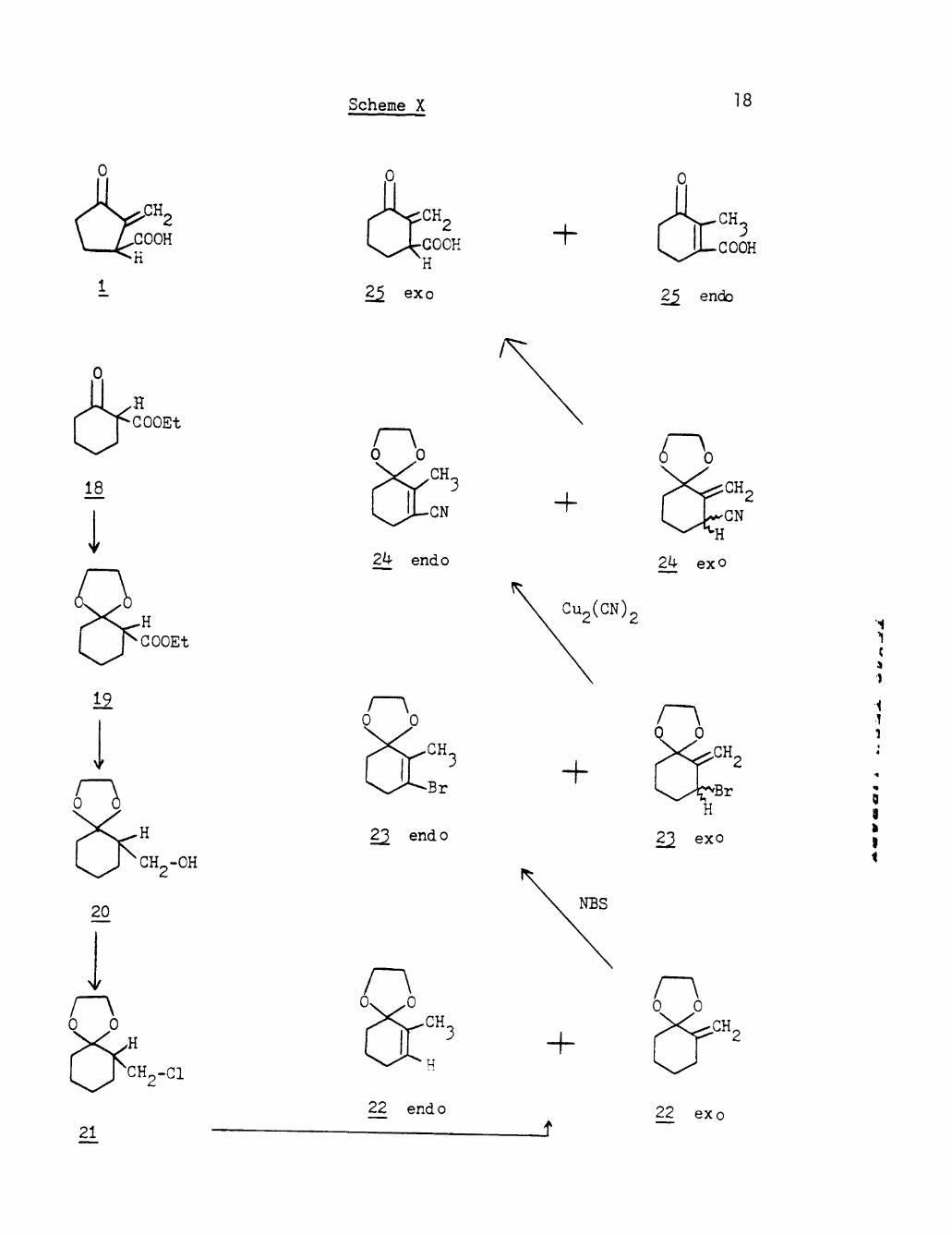
Scheme X 18
0
\ ACOOH COOH H
25 exo
-h
0
l^^^^^COOH
25 endo
18
I
H GOOEt
1 J ^ COOEt
11
5' GH2-0H
20
0 0 H
GH2-GI
0^0 CH,
ON
2^ endo
/ \ 0 0
23 endo
22 end o
+ Zk exo
CU2(CN)2
r^ H-
CH,
'Br H
2; exo
- f -
0 0
22 exo
Q I \ I 1
21

19
therefore, this procedure is by no means reasonable to use for a sarko
mycin synthesis.
Scope and Purpose of the Present Work
Considering previous procedures of sarkomycin synthesis, it Is
clear that, in spite of the simplicity of the structure of sarkomycin,
the synthesis of the antibiotic is connected with specific difficult
ies because of the great reactivity of the compound and its great
tendency to polymerize and isomerize. Therefore, many methods which
require the use of high temperatures for the reaction, or energetic
reagents (especially alkaline or acid media) cannot be used for pro
ducing sarkomycin. Its formation is possible only with mild reagents
in a limited range of pH values and at moderate temperatures.
The primary goal of this research was to develop a rational regio-
specific synthesis of sarkomycin and to synthesize some molecules that ^
are closely related to sarkomycin which might provide compounds with '
enhanced stability and possibly important biological properties, and i
to submit them for screening as possible anti-cancer agents. Moreover,
sarkomycin still stands as a synthetic challenge because of the easily
polymerizable enone functionality. Because of the low stability of
sarkomycin, the synthetic plan was to make a suitable penultimate
precursor of sarkomycin which possesses good stability in storage and
handling. Then, to develop a good procedure to convert this latter
compound to sarkomycin. Also, some of the intermediates in the pro
jected route could be converted into sarkomycin analogs.
1 «
i

CHAPTER I I
RESULTS AND DISCUSSION
Original Synthetic Plan for Sarkomycin
In order to plan a synthesis of sarkomycin, one has to consider
the known properties and chemistry of a-methylene ketones. The
a-methylene ketone structural unit of sarkomycin is also found in
many natural ly occurring sesquiterpenes and ant ib io t ics , such as the
fol lowing examples:
H
GOOH
Sarkomycin 1_
/ ^
Ghiloscyphone 23
'GOOH
. 24 Methylenomycin
Moreover, the potential utility of the highly reactive 0=C-C=CH2
functional group has been demonstrated repeatedly throughout the liter
ature.
a-methylene ketones are widely used in organic synthesis, especially
25 for their ability to function as excellent Michael acceptors.
The most common ways to introduce a methylene group a to a ketone
include the following:
-the base catalyzed condensation of formaldehyde with a ketone. 26
-the i n i t i a l Mannich a-am1noalkylation of C-H acidic compounds followed by quaternization and 3-elimination of the dialkylamino group.
-several indirect solutions recently developed to eliminate the serious synthetic restr ict ions which accompany attempts of
20
1 ^
1
i
1 3
J 9 %
i

21
28 a-methylenation of ketones.
None of these valuable methods affords a simple, direct solution to
this problem, however.
The first successful direct a-methylenation of ketones was re-
29
ported by Gras in 1978, by a method which promises to be of consider
able importance. The approach is analogous to the classical Mannich
reaction but, by suitable choice of substrates, the a-methylene ketone
is the direct product isolated from the process.
GH^, GF^GOO'H^N ^ ^
-^•\^ C\^0 TKF or dioxane
0
• >
•i
However, these procedures are not applicable to unsymmetrical <
ketones, since condensation may occur on either side of the carbonyl
group, and mixtures of products result. So, in our work, an entirely \ different approach was considered, in which the carbon atom to become
the methylene group is already present in the molecule in a higher ' 9
oxidation state. I 1
Based on the considerations described above, the original syn
thetic plan was proposed for the synthesis of sarkomycin according to
the Scheme XI which illustrates the key intermediates.
This scheme (XI) makes use of 2-carbomethoxycyclopentenone (27)
as the key starting material. This highly reactive compound was 30 originally synthesized in this laboratory by Marx, Cox, and Norman

Scheme XI
22
26
a 28
0 II G-OGH,
GN
• ^
G-OCH,
^G=N
GH2OH
G=N
32
-§-NH2
20
0 0
• ^
GH20H
GOOH
il
0 0 0 W G-OGH
GOOH
3
31
GOOH
and the desire to utilize it as a synthon for natural products was
an important factor in choosing it as the starting material for the
present work.
The high rate of polymerization of compound (27) with many re-
30 agents makes It difficult to Introduce the last carbon to form the
sarkomycin skeleton. Therefore, addition of HON presented in the
Scheme XI is the key step to success in the synthesis. If this could
be carried out successfully, all that would remain to be done is to
(.
i 1
9 9 »
9 1

23
transform the ester and cyano groups into methylene and carboxylic
groups, respectively. These transformations, however, are not routine
or trivial, since the ester and cyano groups react very similarly with
most reagents. It was expected that much experimentation would be
necessary to find suitable conditions for these selective transformat
ions. If such conditions could be found, it was anticipated that they
might have wider chemical applications for carrying out chemical diff
erentiations between the ester and cyano groups.
A further advantage of the proposed synthesis was that, with a
few modifications at various steps, other functional groups closely
related to those present in sarkomycin can be Introduced in order to
make new analogs of sarkomycin. Such compounds were planned for clin
ical testing. Some analogs of sarkomycin, as described in earlier
sections, do show anti-tumor activity. i
i
The First Attempts to Produce 2-Carbomethoxy- ) Cyclopent-2-enone (27) in Quantity ^
The f i r s t synthesis of 2-carbomethoxycyclopent-2-enone (27) was J
30 reported in 1972 by Marx, Cox, and Norman. This compound was ob-
tained in 50% y i e l d by oxidat ion of 2-carbomethoxycyclopentanone (25) 9
wi th selenium dioxide in re f lux ing dioxane. The resu l t i ng compound (27)
is f a i r l y stable in dioxane so l u t i on , but a l l attempted p u r i f i c a t i o n
techniques resul ted in rapid polymerization of the product. However,
the compound was trapped by adding dienes to the reaction mixture.
The trapping reagent of choice, cyclopentadiene, reacted smoothly
at 25°C wi th (27) to give a 1:1 mixture of the endo and exo adducts (34),
which were separated by s i l i c a gel chromatography. Pyrolysis of e i ther
Isomer or the mixture of (34) at 438°C, and trapping at -10°C gave a

24
° P GOOCH^ M 0 1) SeO^ A \ ^ AJ' ^ °="3 ' , ^^ty-Y° ''"' - (V°™^
26 ^ 2 7
pure sample of 2-carbomethoxycyclopent-2-enone (27). However, the com
pound was not produced in quantity and the procedure used was tedious.
Since this highly reactive molecule has potential value for natural
product synthesis and is also a potential synthon prostaglandins, the
early work of this project was concerned with scaling up the reaction
sequence, increasing the yield, and working out conditions to make the
procedure much more convenient.
Our preparation of adduct (34) was similar to the previously pub- 'i
lished procedure. However, a careful study of the reaction condit- «
ions resulted in some improvement for this process. In the original ^
procedure, the SeO, oxidation was carried out by refluxing 25 min. in "J
dioxane, the excess cyclopentadiene was added at 25° and the mixture
stirred overnight. We found that optimum oxidation period was 5 min., 9
and that if the cyclopentadiene was added at reflux, the entire reaction J
was over in an hour.
The original cracking of the adduct (34) used a very small pyroly
sis apparatus which could produce only about 250 mg of (27) in three
days of reaction time. This slow process was caused because a slow
stream of nitrogen was used to carry adduct (34) (heated to 60-65°) into
a small pyrolysis chamber.

25
A new large-scale pyrolysis apparatus (see Appendix A for schematic
diagram) was devised. This Included vertical instead of horizontal
mounting and made it possible to scale up this reaction to large
quantities. However, as described in the experimental part, the success
of this reaction was fully dependent on the purity of the adduct (34).
Adduct (34) had to be purified from selenium containing impurities.
Column chromatography was used for this, and it proved to be very
tedious to scale up, so somewhat contaminated material was used. It
was possible to produce large quantities of 2-carbomethoxycyclopentenone
(27) in a short time in a run. However, the yield was still low and
some selenium impurities were present in the product (see experimental
details, page 65). Therefore, even with modification this general
method was still judged to be not very satisfactory for practical
reasons.
. , 32
i 33 ,, . 1
1
After we had started our work, Reich^'^ reported on improved syn- J
thesis of compound (27), as Illustrated in Scheme XII.
This procedure was based on the discovery by Whitehouse"" that m
selenoxides undergo clean syn elimination to form olefins at or below
room temperature. This procedure was considered a very convenient ^ method to convert the ketone to the enone by using the phenylselenenyl J derivative (36).
As is shown in the Scheme XII, the f i r s t step of the synthesis was
the one-pot preparation of a-phenylselenoketone (36) by m.aking the
sodium enolate followed by rapid addition of benzeneselenenyl chloride
(35) to the THF-enolate solution. Without further purification, the
crude selenide (36) was oxidized with 15% H O in CH^Cl - It was
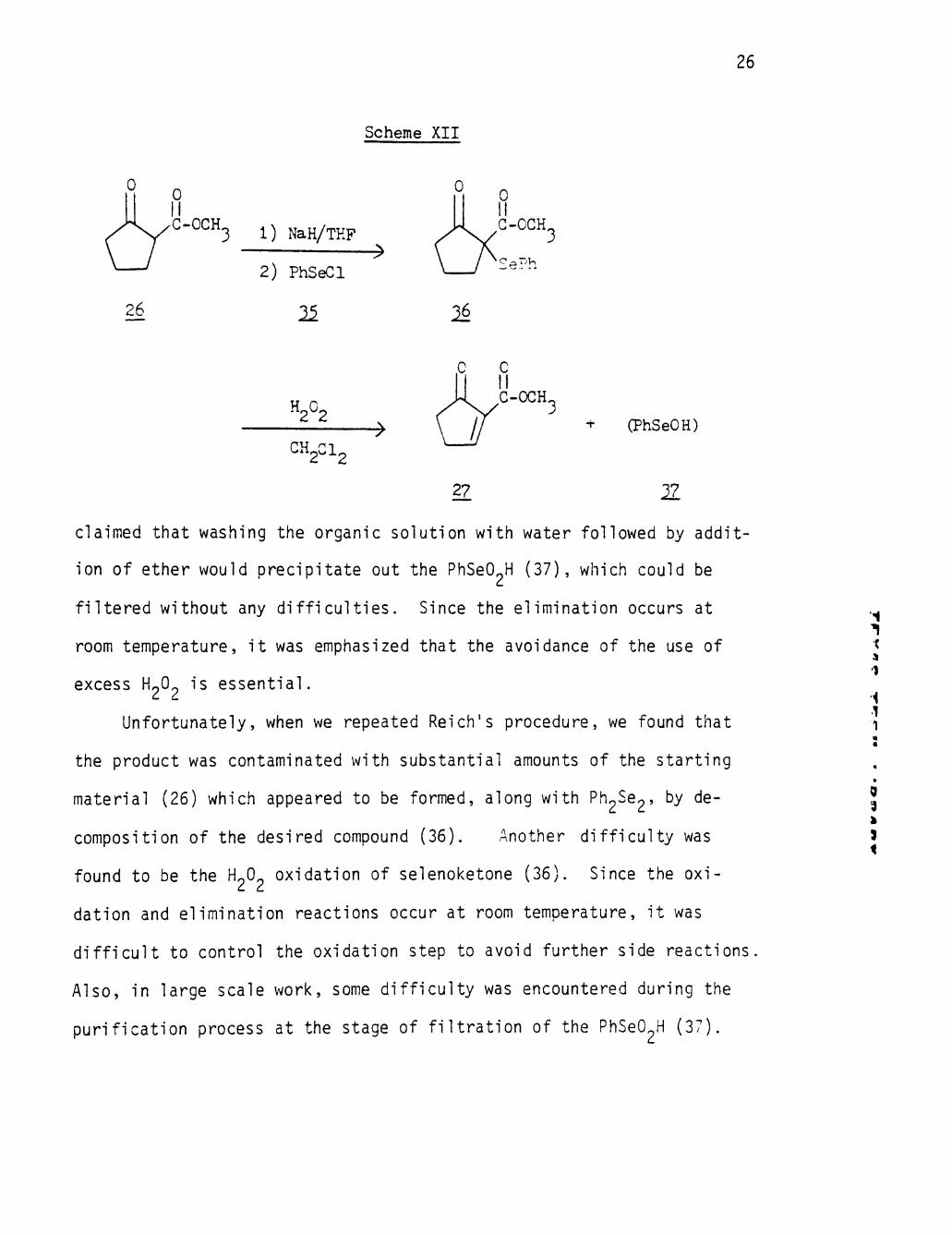
Scheme XII
26
^ 0 II
J<^G-OCH
26
3 1) NaH/TKF
2) PhSeCl
11
• ^
^
H2O2 • ^
CH2CI2
27
+ (PhSeOH)
IL
claimed that washing the organic solution with water followed by addit
ion of ether would precipitate out the PhSeO^H (37), which could be
f i l tered without any d i f f icu l t ies . Since the elimination occurs at
room temperature, i t was emphasized that the avoidance of the use of
excess H O is essent ia l .
Unfortunately, when we repeated Reich's procedure, we found that
the product was contaminated with substantial amounts of the start ing
material (26) which appeared to be formed, along with Ph2Se2, by de
composition of the desired compound (36). Another difficulty was
found to be the HO oxidation of selenoketone (36). Since the oxi
dation and elimination reactions occur at room temperature, i t was
d i f f icul t to control the oxidation step to avoid further side reactions
Also, in large scale work, some difficulty was encountered during the
purification process at the stage of f i l t ra t ion of the PhSeO^H (37).
1
i
i 1
Q 9 » 9 i
27
In view of these major problems, i t was necessary to Improve the
procedure substantially, if we were going to use the compound as the
starting material for a long synthesis.
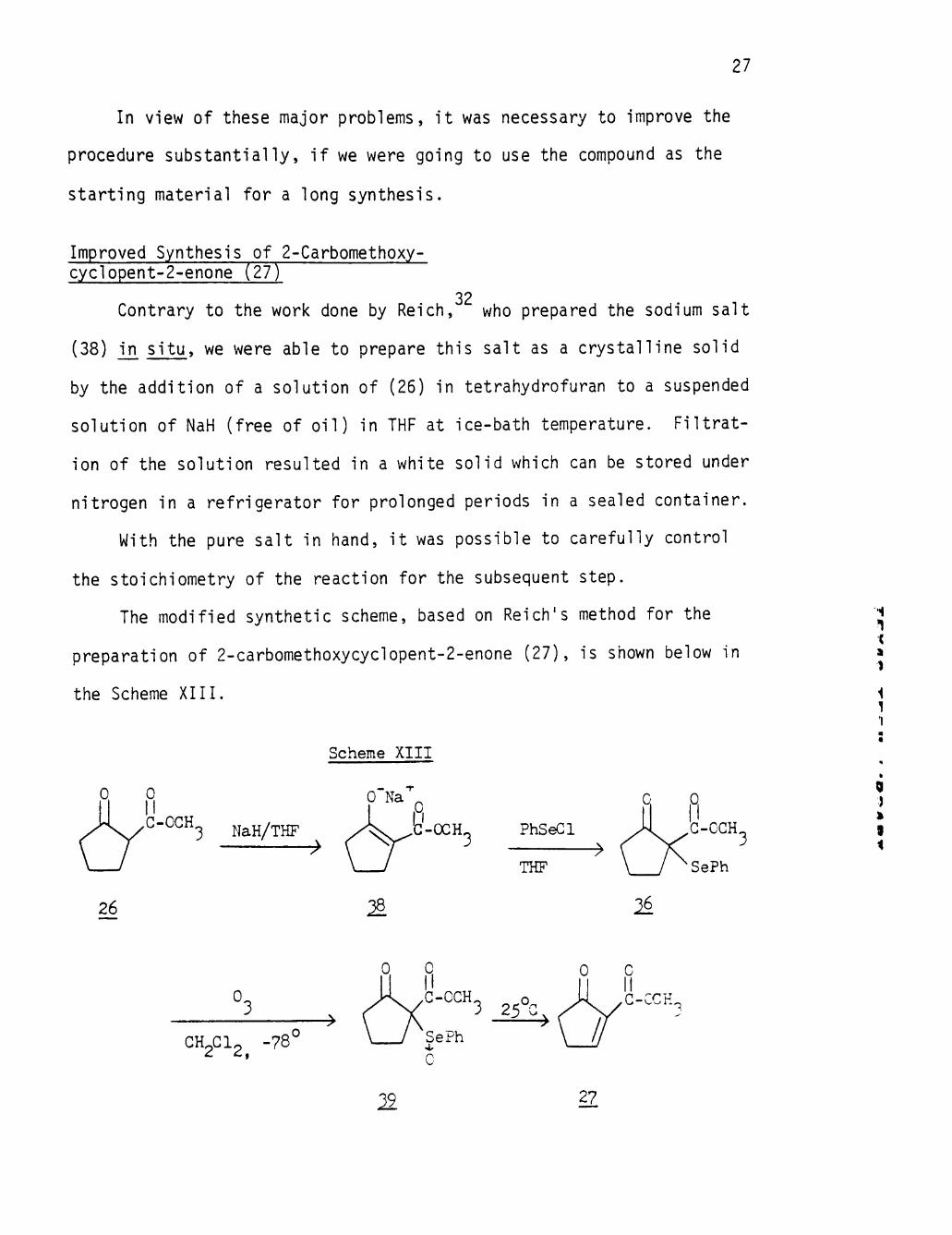
Improved Synthesis of 2-Carbomethoxy-cyclopent-2-enone (27)
32 Contrary to the work done by Reich, who prepared the sodium salt
(38) in s i tu , we were able to prepare this salt as a crystalline solid
by the addition of a solution of (26) in tetrahydrofuran to a suspended
solution of NaH (free of oil) in THF at ice-bath temperature. Fil trat
ion of the solution resulted in a white solid which can be stored under
nitrogen in a refrigerator for prolonged periods in a sealed container.
With the pure salt in hand, i t was possible to carefully control
the stoichiometry of the reaction for the subsequent step.
The modified synthetic scheme, based on Reich's method for the
preparation of 2-carbomethoxycyclopent-2-enone (27), is shown below in
the Scheme XIII.
Scheme XIII
0 Na
NaH/THF • ^
& ' •
PhSeGl • ^
THF
26 ^ ^
t
1
\ 1 1
0 J
9 i
0.
CH2CI2 -78
3 25^
^ 27

28
The reagent, benzeneselenenyl chloride (35), was prepared by the
32 method described by Reich. This procedure involves passing chlorine
gas through a solution of Ph^Se^ (commercially available) in dry hexane.
The resulting orange crystals of PhSeCl (35) were filtered and dried by
a vacuum pump which gave a pure compound with m.p. of 62-64°C. This
compound can be stored in a sealed container under nitrogen atmosphere.
While this work was in progress, the pure PhSeCl became commercially
avallable.
Addition of a solution of PhSeCl in THF to a suspended solution of
the sodium salt (38) was conducted under nitrogen atmosphere. The re
action was very fast, but an additional 15 minutes of stirring of the
mixture assured completion of the reaction. Extraction with CH^Cl^
gave the phenylselenenyl derivative (36) in 91% yield. The advantage
of this procedure over the original published method is that by using Jj
dry THF and a 1:1 stoichiometric ratio of the starting materials there J
is no sign of any starting material (26) remaining based on spectral i
analysis. Also, the compound was not heated sufficiently to cause de- «
composition. Several attempts were made to purify this compound, !
but the experiments showed that the crude compound is very sensitive to J
deselenation by either heat or basic media. However, the crude com- i
pound was used for the next step without any difficulty, and any selen
ium containing impurities could easily be removed in the following
stages.
With the a-selenenylated carbonyl compound in hand, the next
stage involved oxidation followed by fragmentation of the resulting
selenoxide. A variety of methods is available for this process.

29
Sodium periodate, peracids (usually m-chloroperbenzoic acid) , ozone^^
and hydrogen peroxide are the most common reagents for the oxidat ion.
The choice of the reagent usually depends on the other funct ional i t ies
present in the molecule.
Hydrogen peroxide ( typ ica l ly 30-50% W/W), one of the most frequent--DC
ly used oxidants, was employed by Reich for the oxidation of compound
(36). However, on scaling up the reaction, some solubility and sep
aration problems were encountered, as mentioned previously.
Our experiments showed that selenoxide elimination would not occur
at -78°C. Therefore, It was possible to employ ozone as the oxidant
in a suitable solvent cooled by a dry ice/acetone bath. The blue
color of the ozonized solution of (39) in CH«CK was used as a good
indication of the end point of the reaction. The excess ozone was
swept away at -78°C by passing oxygen through the clear solution. The
solution was warmed slowly to room temperature, and a precipitate
formed. This precipitate was a mixture, formed from the elimination 37
product PhSeOH by the following disproportionation:
Drying and weighing of this yellow-white powder gave a good approxi
mation of the yield of this reaction (ca . 50%). Drying and removal of
the solvent of the clear filtrate left compound (27) (yellow oil),
which was still contaminated with some selenium-containing materials
(NMR absorption in the aromatic region). This procedure was used to
prepare 2-carbomethoxycyclopentenone (27) conveniently on a 50 g scale-
3 PhSeOH < — » PhSe-SePh + PhSe(0)0H + H^O Q

30
All spectral data on compound (27) were in accord with the assigned
structure. The vinyl proton signal in the NMR spectrum appears as a
triplet (J=2.5 Hz) at 6 8.38 p.p.m. This is a very deshielded value
for a vinyl proton and reflects the great polarity in the enone chromo-
phore of the compound.
2-Carbomethoxy-3-Cyanocyclopentanone (28)
With a practical method available for preparing large quantities
of (27), the next step in the synthesis vyas the Introduction of a
functional group in the C-3 position of the cyclopentenone (27).
Hydrocyanation is the formal addition of hydrogen cyanide to a
double bond. Since this nitrile functionality can then be readily
converted to a carboxylic acid or amide, hydrocyanation is an effective
tool for introducing these groups at specific points in an organic
1 T 38 molecule.
Diethylaluminum cyanide, (C2Hc)2AlCN, is an efficient hydro
cyanation reagent with applications in the chemistry of natural pro-
39 ducts and steroids. This reagent effects hydrocyanation more
efficiently than hydrogen cyanide alone or even a solution of hydrogen 'J 9
cyanide and triethylaluminum In THF. For example, the reaction of t
Et AlCN with cholestenone in toluene is about 10,000 times as fast as
the reaction of the hydrogen cyanide-triethylaluminum-THF solution 39 reagent.
The mechanism of this method of hydrocyanation of enones has been
40 studied in detail. The monomeric molecule of Et2AlCN was assumed
to act as both the enone-activating (via Lewis acid properties of
0

31
the Al) and the cyanating agent. An in i t i a l concerted activation and
41 cyanation of the carbonyl function with Et AlCN has been proposed,
since i t was observed that formation of the 1,2-addltion product (such
as 27a) is extremely rapid. The 1,4-product (such as 27b) was produced
slowly via the reconverted enone (27), because all the steps are revers
ib le . This mechanism also explains the recommended usage of excess
reagent for all hydrocyanation methods using Et^AlCN. This reagent 42
can also be used to form cyanohydrins from ketones very easi ly.
Et
Et2AlGN
27
Et >
1-0 GN 0 G-OGH
3
27a
i
"I
i 1 1 •«
«
27h 9
Taking into consideration all these facts , in addition to ease
of handling and commercial avai labi l i ty of Et^AlCN, i t was reasonable
to think of this reagent as a f i r s t choice for the hydrocyanation
reaction. But, since the use of excess Et AlCN is necessary, gener
ation of some cyanohydrin (1,2-addition) is unavoidable.
32
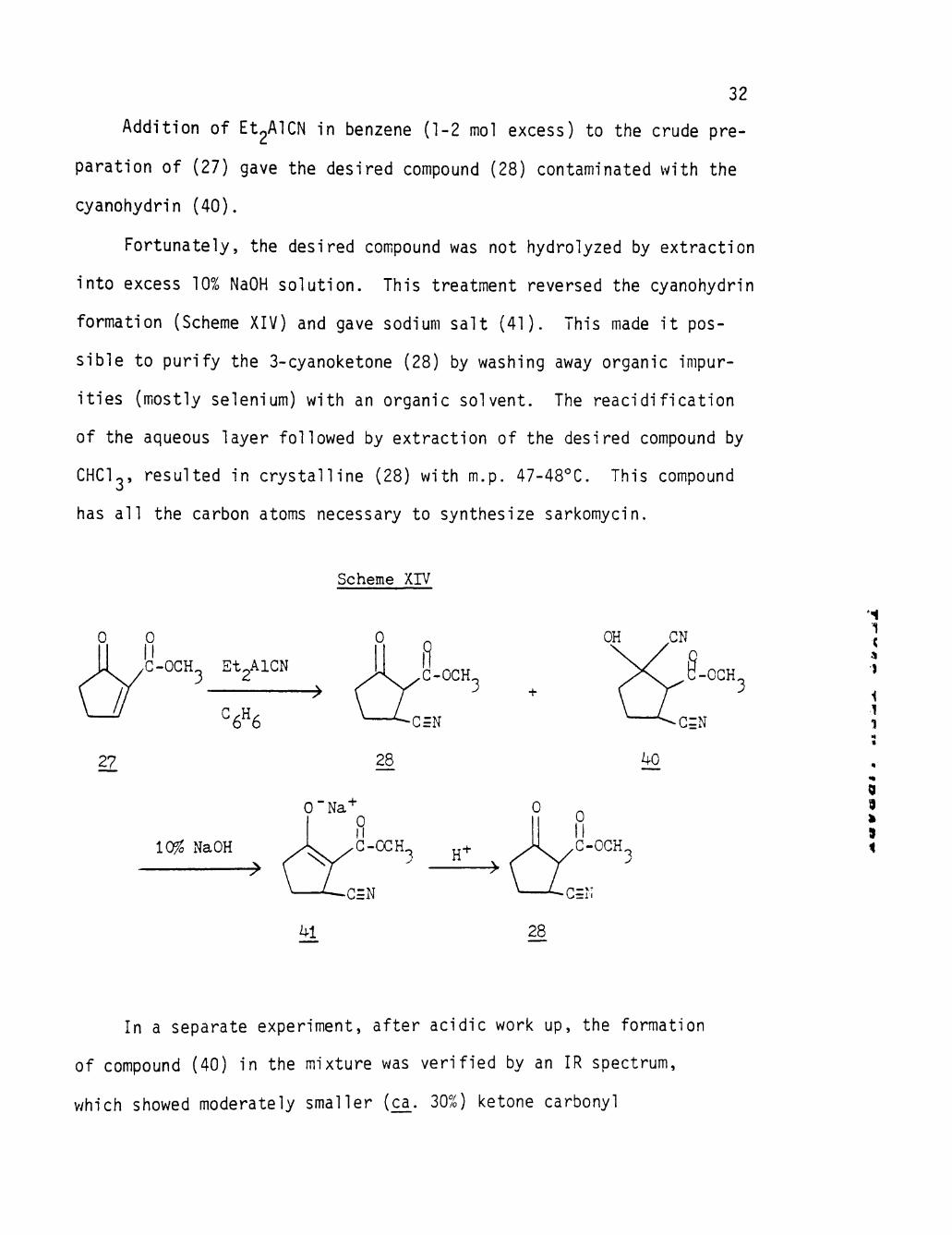
Addition of Et2AlCN in benzene (1-2 mol excess) to the crude pre
paration of (27) gave the desired compound (28) contaminated with the
cyanohydrin (40).
Fortunately, the desired compound was not hydrolyzed by extraction
into excess 10% NaOH solution. This treatment reversed the cyanohydrin
formation (Scheme XIV) and gave sodium salt (41). This made it pos
sible to purify the 3-cyanoketone (28) by washing away organic impur
ities (mostly selenium) with an organic solvent. The reacidification
of the aqueous layer followed by extraction of the desired compound by
CHCU, resulted in crystalline (28) with m.p. 47-48°C. This compound
has all the carbon atoms necessary to synthesize sarkomycin.
Scheme XIV
27
0 II G-OGH Et2AlGN
'=6«6
10^ NaOH ->
• ^
G-OGH 3
^G=N
28
— G = N
^1
H'
OGH,
G=N
kO
-G=N
28
1
)
i 1
Q 9 9 1
In a separate experiment, after acidic work up, the formation
of compound (40) in the mixture was verified by an IR spectrum,
which showed moderately smaller (ca_. 30%) ketone carbonyl

33
absorpt ion due to the presence of some cyanohydrin, in comparison with
the i n f ra red spectrum of pure (28).
I t i s worthwhile to mention here that up to th is point the pro
cedures developed allow us to prepare the pure compound (28) in large
scale in 35-40% y i e l d from the s ta r t i ng material (26) , without any
requirements fo r chromatographic techniques.
The Phenomenon of Enol-Keto Tautomerlzation fo r 2-Carbomethoxy-3-Cyanocyclopentanone (28)
The cyanoketoester (28) showed some In te res t ing tautomeric behavior,
which was Invest igated in some d e t a i l .
The term tautomerism is usual ly defined as "s t ruc tu ra l isomerism
wi th a low ba r r i e r to interconversion between the isomers." Since the
actual range of values can only be a r b i t r a r i l y def ined, the "phenom
enon" of tautomerism simply "fades" in to ordinary isomerism for high i
ba r r i e r s . Tautomeric e q u i l i b r i a cover an important area of organic -i
chemistry, and NMR and IR spectroscopy have been widely applied to i
t h e i r study.
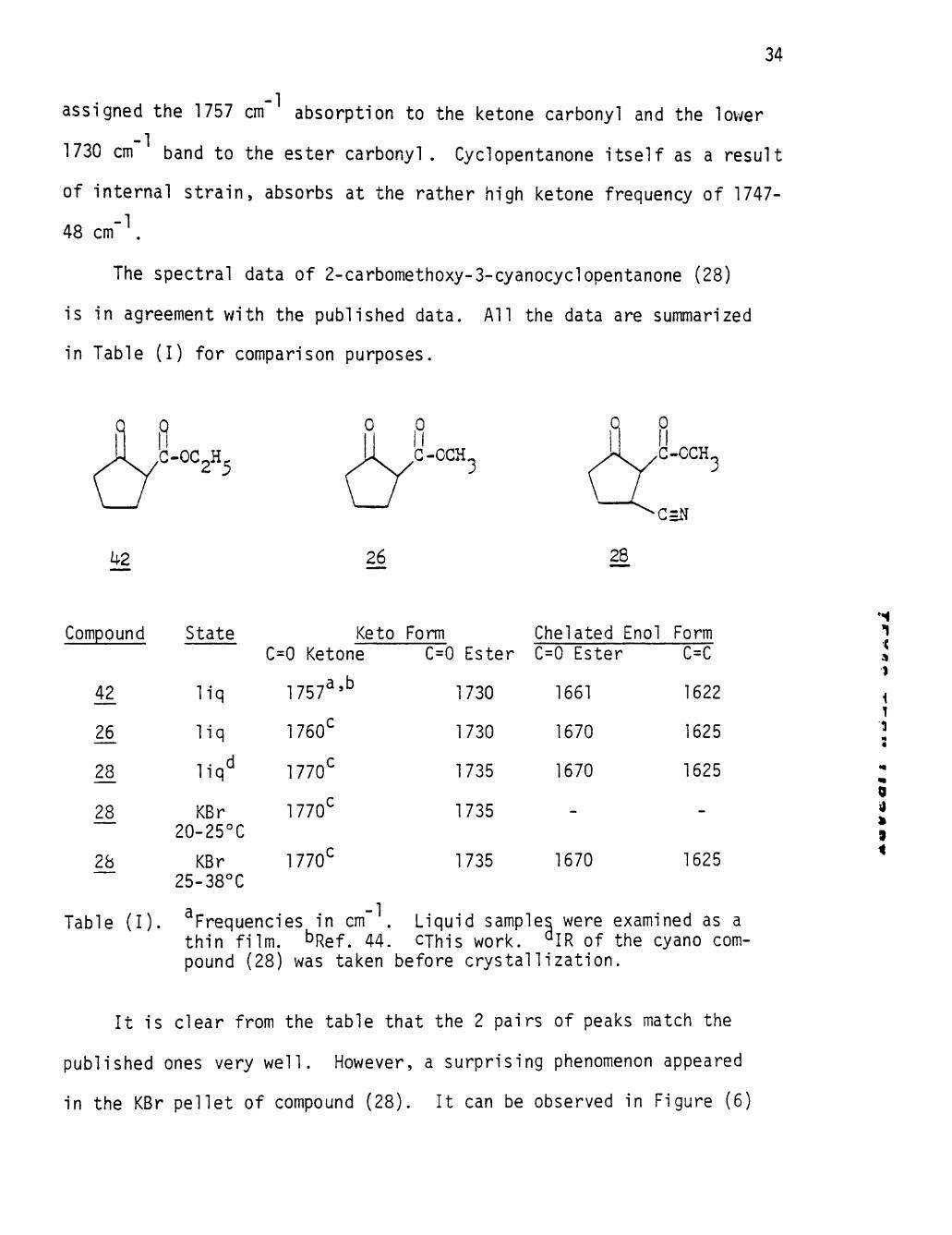
In the most extensive examination of 3-ketoesters reported,
43 Leonard et_ a_l_. invest igated the IR spectra of a number of 3-keto-
esters derived from six and five-membered cyc l i c systems. Later re-
44 inves t iga t ion of t h i s work by G i lber t and co-workers confirmed and
extended the general izat ions out l ined by Leonard et_ aj_. For enol izable
3-ketoesters, four bands appear in the double bond s t re tch ing region,
a pa i r of higher frequency (1760-1710 cm' ) associated wi th the keto
tautomer and a lower frequency pa i r (1665-1600 cm ) due to the chelated
enol . In the case of 2-carbethoxycyclopentanone (42) , G i lber t ejt aj_.
i
21
1
34
assigned the 1757 cm absorption to the ketone carbonyl and the lower
1730 cm band to the ester carbonyl . Cyclopentanone i t s e l f as a resul t
of in terna l s t r a i n , absorbs at the rather high ketone frequency of 1747-
48 c m " \
The spectral data of 2-carbomethoxy-3-cyanocyclopentanone (28)
is in agreement with the published data. A l l the data are summarized
in Table ( I ) for comparison purposes.
Q 0 /-y=-°=2«5
1 ^ ^ G-OCH
VJ a n >I,C.0CH3
•C=N
il2 26 28
Compound State
42
28
28.
2b
l i q
l i q
l i q
KBr 20-25°C
KBr 25-38°C
Keto Form Chelated Enol Form C=0 Ketone
1757^'^
1760^
1770^
1770
1770'
-1
C=0 Ester 0=0 Ester
1730 1661
1730
1735
1735
1670
1670
.,
1735 1670
C=C
1622
1625
1625
1625
Table (I). Frequencies in cm thin film. ^Rgf. 44,
Liquid samples were examined as a CThis work. ^IR of the cyano com
pound (28) was taken before crystallization.
i
i 1
0
9
It is clear from the table that the 2 pairs of peaks match the
published ones very well. However, a surprising phenomenon appeared
in the KBr pellet of compound (28). It can be observed in Figure (6)

35
(page 94) that the IR spectrum changed with the temperature of the KBr
pellet. At or below room temperature (spectra 6a-b), the compound in
the pellet exists only in the keto form. Upon prolonged exposure (10-
20 min.) to the IR beam (spectra 6c-f), 2 new peaks appear due to the
chelated enol tautomer. In cooling down the same pellet (spectra 6g-i),
the new peak disappears again and reappears with heating (spectra 6j-l).
These data illustrate very well the low energy barrier between the two
isomers, in addition to the preferred stability of the keto form in
the solid state.
The classical examples of tautomerism in 3-dicarbonyl compounds
are also well-suited for investigation by NMR spectroscopy because
the interconversion rates between the tautomers are generally slow,
thus permitting direct determination of the proportions of the keto
and enol tautomers by integration of NMR spectra. Many systematic 'J
investigations, often dealing with the effects of structure and solvent '>
on the position of the tautomeric equilibrium, may be found in the \ T ^ ^ 45,46 I literature.
attributed to the difference in polarity of the keto form and the
47 chelated enol. It is noteworthy that in all the cases examined,
an enhancement of enol content is observed with decreasing solvent
polarity, such as from ethanol to cyclohexane.
Different chemical shifts were observed for the two methoxy
signals of the tautomeric forms, keto and enol, in compound (28).
To the best of our knowledge, this is the first example of a tauto-
The effect of solvent polarity on the position of equilibrium in 3
tautomeric systems has been recognized for many years and has been *

36
merle mixture which has been studied by observing the methoxy signals
instead of the broad acidic OH proton signals for which makes i t more
d i f f i c u l t to d i f ferent ia te between the two forms quant i tat ively. There
fore, th is might be a useful compound for studying the effect of sol
vents on the posit ion of the tautomeric equil ibrium in addition to the
dissociation of intramolecular and intermolecular hydrogen bonds on
d i lu t ion. Some experimental data are reproduced in Figure (7)(page 95 )
which show an equil ibrium sh i f t to the keto tautomer in acetone-d^ D
solution (spectra 7a-c). However, the enol tautomer is enhanced in
benzene (spectra 7d-f) and chloroform solution (spectra 7g-i).
It should be mentioned that the acidic proton in the enol tautomer
can be observed at about 6 10.2 p.p.m. and is variable with solvent.
This is comparable to the literature values for acidic protons for
dicarbonyl compounds.
Protection of Ketone. Formation of Ethylene Ketal of 2-Carbomethoxy-3-Cyanocyclopentanone (29)
With a supply of the cyano keto ester (28) assured, we turned to
ways to convert it to sarkomycin and derivatives. The problem presented
by this compound was to find a way to selectively hydrolyze the nitrile
or to reduce the ester without disturbing the other functional group.
A number of approaches were investigated.
In order to do any further transformations on (28), it was
necessary to protect the ketone group. The ethylene ketal, one of the
most common protecting groups for ketones, was investigated first and
proved quite satisfactory for the purpose.
i
1
9
3 1

37
Refluxing a benzene solution of (28) with a three-fold excess of
ethylene glycol and a catalytic amount of p-toluenesulfonic acid into
a Dean-Stark trap to remove the water gave the desired ethylene ketal
(29) in a moderately good yield. Pure crystalline compound (29) was
obtained upon column chromatography followed by two recrystallizations
from ether and pet ether and melted at 47-48°C.
0
G-OGH^ HOGH2GH2OH
0 0 0
^ > \ /
H-", G^H^ -//,,.
XyL'-OGH, ^
G=N ' 6 6 ''G=N "^^C=N
28 major (trans) minor (cis) 29
Initially, it was assumed that this compound was the pure trans
sample gave a complex absorption pattern for overlapping chemical shifts
of C-2 and C-3 hydrogens which, in turn, made it impossible to assign
the stereochemistry or determine if it was a single stereoisomer. A
very careful investigation of this sample was carried out with H NMR
(100-MHz) at different temperatures from -60 to 80°C. A double reson
ance technique and use of the Eu(fod)^ NMR shift reagent was attempted.
but all experiments failed to give any meaningful data on the stereo
chemistry. However, investigation of various fractions from column
chromatography of the material showed a slight change in peak intensity
A
< "I
1
isomer (29) (the thermodynamically more stable product) based on its 1
sharp melting point. However, the NMR spectrum of the crystalline s
0 9
i 1

38
in the 6 3.1-3.2 p.p.m. region which in turn suggested that the sample
might be a mixture of the cis and trans isomers. Therefore, a much
more careful chromatographic separation was carried out. The major
fractions were found to contain mainly a single component, which \vas
later assigned as trans, but this was never obtained completely pure.
However, the last fractions contained a material which was judged to be
the pure C2S_ Isomer m.p. 73-74°C. An NMR signal for the C-2 and C-3
protons at 6 3.1 was found to be diagnostic for this isomer, and it was
judged by this method that the original trans:cis ratio was ca . 10:1.
However, for our purposes, there was no need to separate the Isomers
(vide infra) and the original crystalline mixture was used in subsequent
steps.
Attempts on Selective Hydrolysis of Nitrile J in Ethylene Ketal of Cyano Ester (29) '5
The conversion of cyanides and amides to carboxylic acids is con- ^ 1
sidered a routine procedure, but in practice it is not always straight- i 49
forward. Often vigorous and strong catalysts, such as concentrated «•
0
acids or strong alkali hydroxides, are needed to effect the hydrolysis. j
In general, the yields of these reactions are fair to good, but J
occasionally the severe reaction conditions cause decomposition of
the desired acid or hydrolyze other functional groups in the molecule.
Since the hydrolysis of the ester group definitely would happen much
faster than the nitrile moiety under vigorous conditions, a more
selective way was needed for this transformation. 50
Cook ejt aj_. have shown that cyanides may be hydrolyzed to amides in good yields by solid-phase catalysis at room temperature. The pro-

39
cedure is to s t i r a methylene chloride solution of the cyanide with
manganese dioxide. The water necessary for this reaction is the
moisture held by the surface of the catalyst . This procedure was
applied to the ethylene ketal of 2-carbomethoxy-3-cyanocyclopentanone
(29). Several attempts were conducted by s t i r r ing a mixture of com
pound (29) and manganese dioxide in CH2CI2 cit room temperature for
different intervals of time. But, there was no evidence of hydrolysis
even after several days of reaction time. Even after refluxing the
mixture overnight, only a trace amount of amide was isolated based on
the IR spectra. The rest of the compound could not be recovered by
any solvent extraction from the mixture.
An early report on the hydrolysis of n i t r i l e s in alkaline solution,
51 catalyzed by hydrogen peroxide, has been published by 01iveri-Mandala.
Our f i r s t attempt to accomplish this base catalyzed hydrolysis of the
n i t r i l e to the amide (30) with H O in THF or acetone solution was very
successful. This reaction was done by s t i r r ing a solution of compound
(29) in THF with 30% HO followed by dropwise addition of 10% solution
of sodium carbonate to keep the pH between 7 and 8. Stirr ing for one
hour at room temperature, followed by work up, resulted in crystal l ine
ethylene ketal of 2-carbomethoxy-3-amidocyclopentanone (30) which melts
at 114-n5°C.
The above transformation simplified the problem since amides are
usually easier to hydrolyze than nitriles. However, the problem was
not yet solved, since a selective hydrolysis of the amide was still re
quired. Amides which resist hydrolysis with alkalines or acids can
i 1
Q 9
5 1
be converted to the corresponding acids by treating with nitrous acid. 52

40
G-OGH.
G=N 2^2
29
CH-
20
Therefore, after successful preparation of amide (30), several attempts
were conducted on hydrolysis of amide (30) with nitrous acid in
acidic solution (pH 2-3). Unfortunately, the results were not satis-
53 factory since the high concentration of acid necessary to complete
the deamination reaction caused hydrolysis of the ketal protecting
54
group.
It is also known^^ that the hydrolysis of moderately hindered
amides will proceed with nitrous acid in strong sulfuric acid solution.
Unfortunately, the presence of the acid sensitive ketal protecting
group made it difficult to investigate these types of reactions. Sev
eral attempts to hydrolyze amide (30) to the carboxylic acid using the
strongly acidic reagents resulted in the hydrolysis of ketal and/or
ester functionality.
55
In 1965, Olah reported the hydrolysis of amides to the corres
ponding acids by nitrosonium tetrafluoroborate (NO BF^-) at low temp
eratures and in oragnic solvent systems. We repeated this experiment
according to the published procedure. However, the resulting material
1
i 1 1
9 1

41
showed an extensive amount of ketal hydrolysis. It is entirely pos
sible that one could find conditions to selectively hydrolyze the amide
group in (30), but these discouraging results prompted us to investi
gate a different approach.
Selective Reduction. Formation of the Ethylene Ketal of 2-Hydroxymethylene-3-Cyanocyclopentanone (32)
The alternative approach to use of amide (30) as an Intermediate
in a synthesis of sarkomycin is to reduce the ester group first,
leaving the amide intact. A reagent considered for this purpose was
LiBH^ (LBH), but the result was the reduction of both carbonyl
functionalities. With this procedure blocked, we directed our atten
tion to alternate modes of reduction of the ester functionality as
described below.
A search of the literature ' ^ revealed that LiBH. (LBH), in a
functional groups. It was expected that there should be no hydrolysis
are maintained.
However, when the reaction was run, a major unexpected difficulty
arose. Although the ester group was reduced and the cyano was mainly
not attacked, the ketal group was destroyed. This conclusion was simply
based on the disappearance of the ketal singlet at 5 3.98 p.p.m. in the
NMR spectrum of the reduced compound in Figure (14b) (page 102).
I
suitable solvent (THF), was the reagent of choice for transformation of i
the ester to a primary alcohol in the presence of the nitrile and ketal J
i
of the ketal group whatsoever since strongly basic reaction conditions ' 9 1

42
The initial exploratory work on studying this reaction started by
using super dry solvent (refluxing THF over NaK for several days before
distillation) under an inert atmosphere, and monitoring each run in
different intervals of time at room and reflux temperatures. Different
solvents, contamination of LBH, and different experimental procedures
were considered, but all attempts failed to keep the ketal group intact.
Since the destruction of the protecting group was the problem of
this step, therefore, it seemed reasonable to consider another protect
ing group, such as the ketal of 2,2-dimethyl-l,3-propanediol instead of
ethylene glycol. This new reagent generates a more stable, six membered
ring, protecting group which was assumed to be much more stable toward
LBH.
This reaction was first studied on a model compound, and the ketal .^
was formed by the routine ketalization procedure. ^
The ketal ester (43), after distillation (61% yield), showed a '
sharp peak in the infrared spectrum at 1740 cm' for the ester carbonyl. i
The NMR spectrum (CDCU) showed two singlets, one at 6 0.83 and another ^ n
at 1.1 p.p.m., due to the two unequivalent methyl groups and a sharp ) 9
singlet at 6 3.75 was assigned to methoxy protons. J
In a similar reaction, this protecting group was introduced into
compound (28) and after careful chromatography (13% ether in pet ether)
and two recrystallizations from ether and pet ether, the resulting
white crystals of compound (44) melted at 95-97°C.
The neat infrared spectrum (before crystallization) showed ester
carbonyl absorption at 1740 in addition to cyanide absorption at

43
r 0
><^G-OGH
26
28
3 OH OH
H , G^H^
OH OH
H , G^H^ • ^
0 0
OGH,
G=N
hh
2225 cm" . However, the major peaks in the NMR spectrum were almost
identical to those for compound (43).
Unfortunately, this protecting group also did not remain intact
under several sets of reaction conditions investigated. After careful
study of the reaction, the infrared spectrum revealed that there was
no carbonyl absorption, even when using less than half the theoretical
amount of the LBH. This meant that the disappearance of the ketal group
59
was not just a simple ketal hydrolysis process.
The system of NaBH. and AICK in diglyme reported by Brown^ and
co-workers gives good yields in the reduction of esters to alcohols at
room temperature. This reaction was applied to compound (29) but in
addition to reduction of the ester, the ketal group also was destroyed
A 1
0 '.J SI 9 1

44
In reviewing the literature on these seemingly trivial and re
lated reactions, some examples of reduction of acetals and ketals to
ethers in high yield by means of lithium aluminum hydride-aluminum
fin chloride were found. These reactions remove the ambiguity of the
absence of the ketone carbonyl group, in addition to explaining the
additional methylene peaks in the NMR spectra.
0 -
C -
LUlHzi
AlGl 3
OGH2GH2CH
LiAlH/
AlGl 3
GH20GH^
Therefore, in our case, it was reasonable to consider that the
generation of some decomposition products (acting like a Lewis acid)
from the LiBH. catalyzed the reductive opening of the protecting
group to give the ether-alcohol, whose probable structure is (45).
Keeping in mind the above findings, our attention was directed
mainly towards reductions which could take place under mild condit
ions in basic media.
It is known that the reduction of simple aliphatic esters with
sodium borohydride is extremely slow and therefore not practical for
industrial processes. Thus, we needed a reducing agent stronger fii
than NaBH^, and weaker than LiBH.. However, Brown has shown that
1
"I
i ^ \
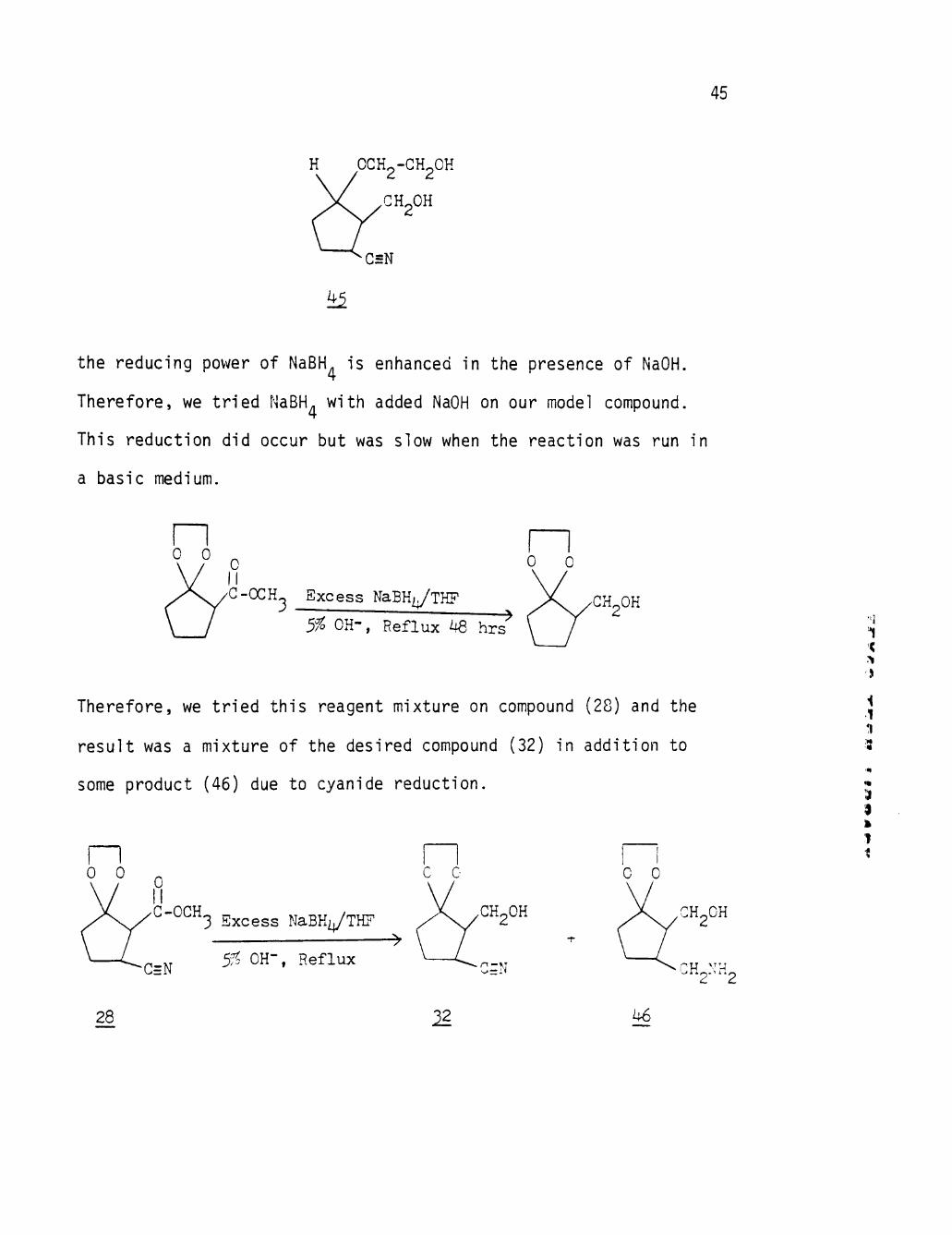
45
H OCH2-GH2OH
GH2OH
G5N
^
the reducing power of NaBH is enhanced in the presence of NaOH.
Therefore, we tried NaBH^ with added NaOH on our model compound.
This reduction did occur but was slow when the reaction was run in
a basic medium.
0 0 0
C-OCH^ Excess NaBH^THF - ^
5% 0H-, Reflux ^ hrs
GH2OH
Therefore, we tried this reagent mixture on compound (28) and the
result was a mixture of the desired compound (32) in addition to
some product (46) due to cyanide reduction.
0 0 0 c 0 0
3 Excess NaBHi/THF • ^
G=N % OH", Reflux
GH2OH
-iN
GH2OH
1
J
1 'I
9
1
28 ^ i^

46
This reaction was very encouraging, since there was no sign of
ketal hydrolysis. Therefore, we turned our attention again to our
original plan of using LBH with the new modification of adding NaOH,
P K . , ^ G - O G H Excess LiBH^THF X\y^\^^
\ ^C=N 5^ 0H-. Reflux ^ C C = N
5 minutes
28 ^2
ue
This latter reaction, as hoped, was very successful. No ketal
hydrolysis and very little cyanide reduction was observed. It was
assumed that the addition of £a. 2 eq. of H 0-NaOH per mole of i
LIBH gives a reagent mixture which presumably contains LiB(OH) H J
as the major species and carries out the desired transformation J
cleanly. 2
Even with the LiBH /NaOH-H 0 system, some reduction of the
cyanide group was observed in the course of the reaction. Therefore,
(32) by investigation of the conditions of the reaction. It turned
out that the best result, with minimal reduction of the C=N group,
was accomplished by stirring of the basic solution of LBH in THF
solution for 24 hours at room temperature. For comparison purposes
NMR spectra of the resulting compounds from LBH reduction in the
absence and presence of the NaOH are reproduced in Figure (14)
the first effort was directed toward improving the overall yield ''

47
(page 102). One can see the dramatic effect the addition of aqueous
NaOH has on the course of the reduction leaving the ketal group
intact and reducing only the ester.
The Cyano Derivative of Sarkomycin; 2-Methylene-3-Cyanocyclopentanone (47)
After developing this key step of the synthesis to produce the
protected 2-hydroxymethylene-3-cyanocyclopentanone (32) cleanly,
our attention was then focused on converting this into the cyano
derivative of sarkomycin (47). This transformation requires re
moval of the ketal protecting group, followed by a dehydration
process.
Various mild acidic conditions for the ketal hydrolysis were
investigated, monitoring the reaction by NMR. It was found, using 1 K "I
several combinations of time and HCl concentration in acetone i
solution, that the ketal hydrolysis was followed by subsequent ^
dehydration to give the desired cyano derivative of sarkomycin (47) • «
directly. This method revealed that the best conditions involved •; ))
treatment of neat compound (32) with a 1:1 mixture of 1 N HCl- * f
acetone solution and stirring at room temperature for 12 hours.
The result was a mixture of 2-methylene-3-cyanoketone (47) and
2-hydroxymethylene-3-cyanocyclopentanone (48).
It proved possible to carry out molecular distillation of the
crude compound (0.25 mm Hg,50 to 80°C oil bath) without any major
polymerization. However, the distilled compound was contaminated
with the alcohol (48). The distilled material was stable for at

48
0 0
^ ^ r = N Rooni Temp . 1 2 h r
^
,GH2CH 5% H C l - A c e t o n e
0
a_ bl
GH2OH
G=N
kQ
least several weeks in an organic solvent (CHCK) when stored in a
refr igerator . The d i s t i l l a t ion was only carried out once on a
very small scale due to lack of material. Presumably (47) and (48)
could be separated by a more careful d i s t i l l a t i on .
Neighboring Group Participation in the Hydrolysis of the Nitr i le Group in (32). Formation of the Ketal Lactone (49T
Considering the ethylene ketal of 2-hydroxymethylene-3-cyano-
cyclopentanone (32) to be a mixture of cis and trans isomers (ca.
10:1 r a t i o ) , our general synthetic plan was to hydrolyze this
mixture and separate the acid derivative (trans isomer) from the
lactone (cis isomer) by means of sodium bicarbonate solution.
• 1
^
» 1 1
0 ,0 0 0
.GH2OH Q^-
G=N
GH2OH .
''^^'^COOVi
t rans - 32 c is - ^2 ^9 7 T

49
Surprisingly, this was not the case. After 12 hours of reflux
ing in 10% sodium hydroxide-methanol solution, the only recovered
product was ketal lactone (49). After careful examination of the
reaction procedure, it became clear that this reaction could be
conducted in a surprisingly short period of time (1 hour). This unusual
ease of hydrolysis and the recovery of a single product suggests
that epimerization at C-3, followed by neighboring groups assistance
by the hydroxy1 group is occuring in the hydrolysis reaction. The
proposed mechanism is illustrated in Scheme XV (page 50). This
result meant that the stereochemistry of the precursors is not
important for the synthesis of the lactone (49).
Lactone Derivative of Sarkomycin (50) ^
Since we had been directed to the synthesis of the ketal lactone J
(49), our next step was to hydrolyze the ketal protecting group
and to study the resulting compound more carefully. Our first i «
attempt indicated that the ketal protecting group was not hydrolyzed
by 25% HCl-acetone solution in two minutes as occurs in similar j
30 *
published examples. However, smooth hydrolysis of the protecting \
group of the ketal lactone (49) was accomplished by stirring a 1:1 mix
ture of acetone and 0.5 N HCl solution for 48 hours at room temper
ature. The resulting crystalline compound (50) after several re
crystallizations melted at 45-46°C.
Sarkomycin From Keto Lactone (50)
Having the lactone analog of sarkomycin in hand, and consider
ing our major goal, synthesis of sarkomycin, it seemed to be

Scheme XV
50
GH2OH
32
OH" I 1 C 0
6- GH.,0"
1^ (^
^ = N
slow -^
OH-
0 C
0 0
^6-' ' '////,
GH,
10 ^ ^ G ;
N
GH2OH
OH-
0 0
HN^ •OH
0 0
2NP -0
• ^
0 0
^
0
desirable but challenging to transform this molecule into sarko-
myc1n.
fi? fi? During the course of literature investigation ' several
attractive routes for nucleophilic cleavage of lactones via a
nonbasic nucleophile (e.g. sulfur or selenium anion) were encount
ered. If such nucleophilic opening could be accomplished, pre
sumably a subsequent transformation (e.g. oxidation) would yield
. )
\
1 •

51
sarkomycin under mild conditions as illustrated in the examples
below:
(™2?n ° •\_J
0
^ ^ ( C H ^ ) ^ OH
V • \
SePh
Lc"]
^ GH SLi
'^O —^ ^
-.(^Vn °
CCOH
Several attempts were conducted in transforming the keto-
lactone (50) to sarkomycin using PhSe" and CH.S' as nucleophiles
fi? fi*^ for cleavage processes as recommended. ' Unfortunately, all
attempts to convert the keto lactone into sarkomycin via published
procedures met with failure. Either starting material or decomp
osition products were obtained.
Unexpectedly, during hydrolysis of the ketal group of (49), it
was found that prolonged treatment of keto lactone (50) with 0.5-
1 N HCl in acetone-HpO solution gave a slow eliminative opening
of the lactone to produce (±)-sarkomycin directly. This reaction
was studied more carefully using several combinations of time and
acid-acetone concentrations.
The hydrolysis of the ketal (49) to give keto lactone (50) was
completed by stirring a 1:1 solution of acetone 0.5 il HCl overnight
•4
\ 1 1
I
I I
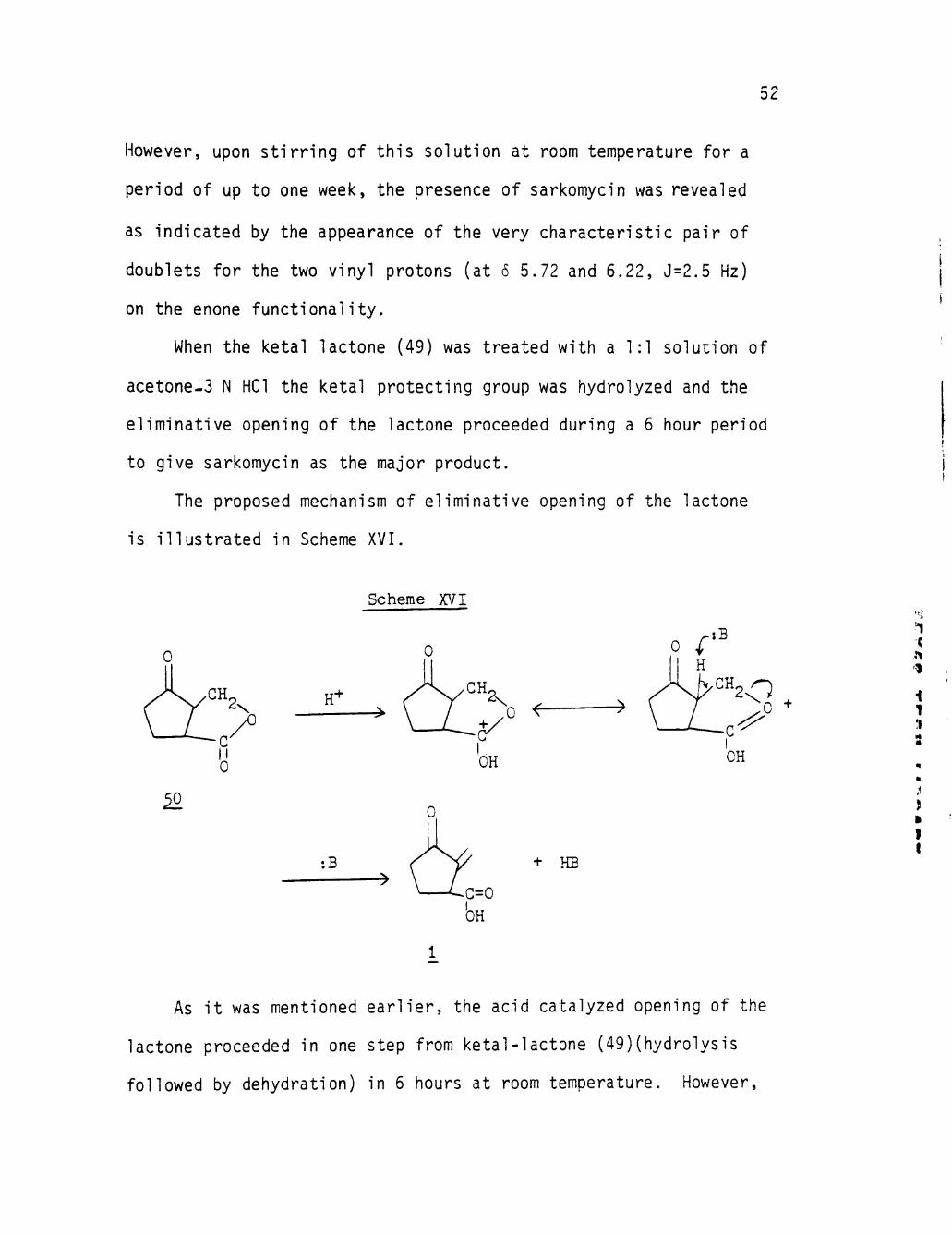
52
However, upon stirring of this solution at room temperature for a
period of up to one week, the presence of sarkomycin was revealed
as Indicated by the appearance of the very characteristic pair of
doublets for the two vinyl protons (at 6 5.72 and 6.22, J=2.5 Hz)
on the enone functionality.
When the ketal lactone (49) was treated with a 1:1 solution of
acetone-3 N HCl the ketal protecting group was hydrolyzed and the
eliminative opening of the lactone proceeded during a 6 hour period
to give sarkomycin as the major product.
The proposed mechanism of eliminative opening of the lactone
is illustrated In Scheme XVI.
io
H'
:B
Scheme XVI
0
* cr:> OH
• ^
+ HB
1
i 1 n
J »
I I
As it was mentioned earlier, the acid catalyzed opening of the
lactone proceeded in one step from ketal-lactone (49)(hydrolysis
followed by dehydration) in 6 hours at room temperature. However,

53
such a strong acidic solution (3 N)is not a recommended media for
sarkomycin preparation because after additional stirring of this
solution vinyl protons disappeared due to polymerization. There
fore, a longer reaction time with a 0.5 N HCl solution and subsequent
extraction of sarkomycin with NaHCO^ after 4 or 5 days is the
recommended procedure among the conditions studied.
Elimination of Anchimeric Assistance by Using 2,3-Dihydropyran as the Hydroxy1 Protecting Group
Since the slow opening of the lactone in acidic media caused
slow polymerization of sarkomycin, the synthesis described above
was not considered as a method of choice.
Due to the fact that epimerization, followed by neighboring 1
group p a r t i c i p a t i o n of the hydroxyl group, resulted in the lactone a
der iva t i ve instead of the acid-alcohol (33) , i t seemed reasonable i
to th ink of using a protect ing group fo r the alcohol moiety in ]
order to e l iminate anchimeric assistance.
The formation of tetrahydropyranyl (THP) ethers by the react ion J
of 2,3-dihydropyran wi th alcohols under mild acid cata lys is is a t
very useful method fo r protect ing alcohols from strong basic media.
I t appeared that ^ - to luenesu l fon ic acid would be a sa t i s fac to ry
ca ta lys t fo r te t rahydropyrany lat ion. Since the removal of the THP
pro tec t ing group can be done under mi ld aqueous acid ic condi t ions,
th i s procedure was considered as the method of choice for our pur
poses. The react ion proceeded very w e l l , though some se l f conden
sat ion of dihydro-4H-pyran occured a lso. However, th is did not
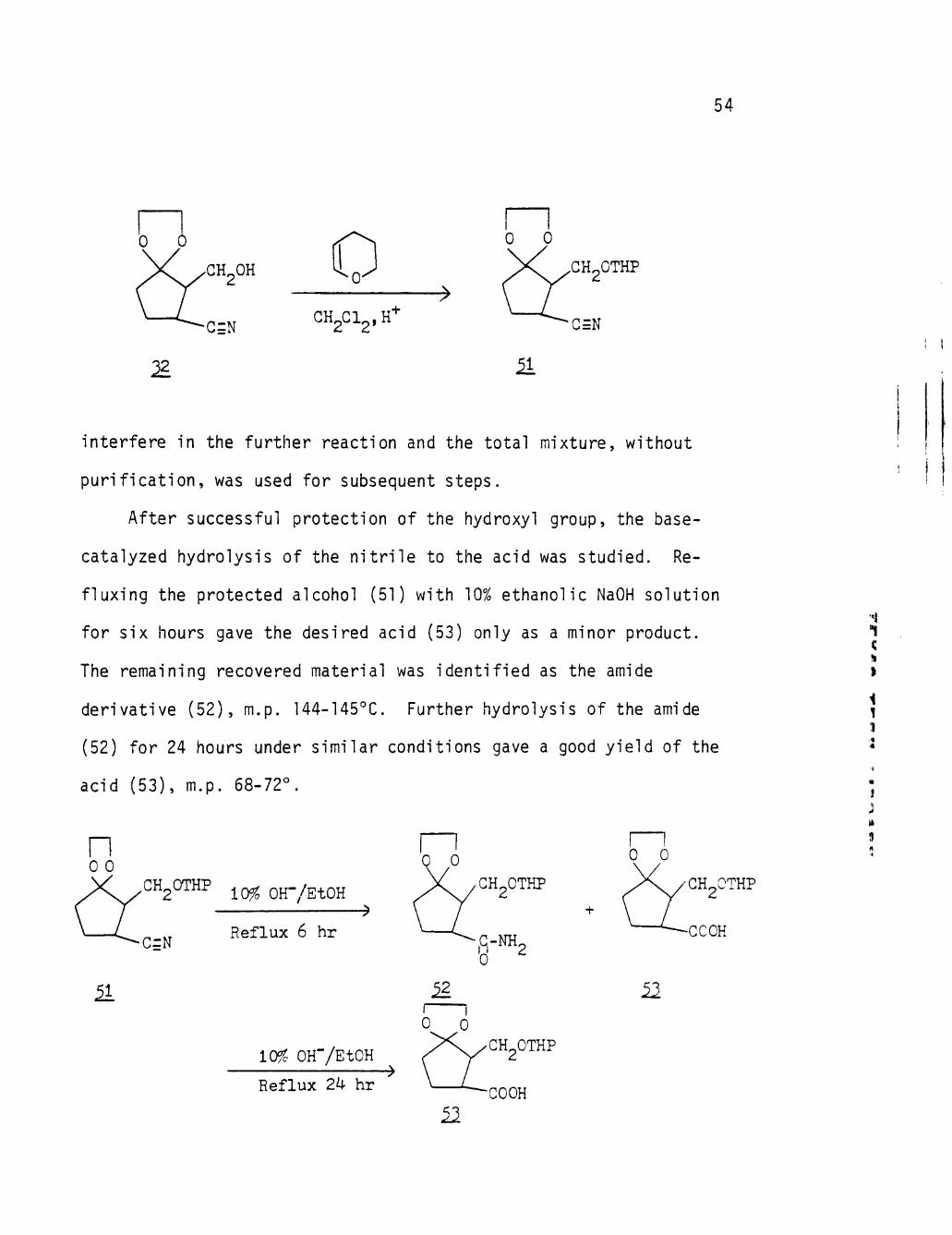
54
0 0
&
GH2OH
^
GH2Cl2,H"*" • ^
0 0
6:.. GHgOTHP
Ik
interfere in the further reaction and the total mixture, without
purification, was used for subsequent steps.
After successful protection of the hydroxyl group, the base-
catalyzed hydrolysis of the nitrile to the acid was studied. Re
fluxing the protected alcohol (51) with 10% ethanolic NaOH solution
for six hours gave the desired acid (53) only as a minor product.
The remaining recovered material was identified as the amide
derivative (52), m.p. 144-145°C. Further hydrolysis of the amide
(52) for 24 hours under similar conditions gave a good yield of the
acid (53), m.p. 68-72°.
GH OTHP
GrN
10^ OH"/EtOH
Reflux 6 hr
^
5 11
0 0
GH2OTHP
g-NH^
GH2CTHP
GOOH
^
0 0
10^ OH'/EtOH
Reflux 2^ h r
><,^GH20THP
-GOOH
>
1 I .« 4
n

55
These resu l ts confirm the idea tha t anchimeric assistance was
a id ing i n the n i t r i l e hydrolysis i n the cyano alcohol (32) , since
pro tec t ion of the OH group as a THP ether prolonged the react ion
time dramat ica l ly (one hour vs. 24 hours) and led to the trans Isomer
(53) Instead of the c i j^ lactone (49) (vide i n f r a ) .
Amide Der ivat ive of Sarkomycin; 2-Methylene-3-Amidocyclopentanone (54)
With the pure c r y s t a l l i n e amide (52) in hand, several attempts
were made to prepare the amide der iva t ive of sarkomycin (54) by an
acid-catalyzed hydrolysis-dehydrat ion sequence. However, due to the
high water s o l u b i l i t y of the amide de r i va t i ve , i so la t i on problems
were encountered in the course of t h i s react ion. The f i r s t successful
•i attempt of the dehydration process was carried out by monitoring i
c the reaction in an NMR tube. A solution of amide (52) in acetone-dg J
and 0.5 N DCl-D^O af ter standing at room temperature for 5 hours ^
showed complete hydrolysis of both protecting groups. After 24 hours a
at room temperature, 60-70% of the mixture was the amide derivative
of sarkomycin (54), as revealed by the NMR spectrum (Figure 30,
Appendix B).
0 6 ,0, .GH OTHP 5% DGl-D^O
^ ^
D6-Acetone ^p ^ .G-NH2 i f 2
0
i2 ^
:i

56
Further attempts to separate amide (54) from an aqueous solution
by means of organic solvents were made but were Inefficient due to
the high water solubility of the amide (54). However, a small portion
which was extracted by excess CHC1-, showed the characteristic pair
of doublets for the two vinyl protons (at 6 5.58 and 6 6.24 p.p.m.)
on the enone functionality.
Sarkomycin (1), From 2-Methylene (0-Tetrahydropyranyl )-3-Carboxy1 ic Acid (53)
With successful preparation of hydroxy protected cyanoketal (51)
and hydrolysis of the cyanide moiety to carboxylic acid (53), what
was left to accomplish was the removal of protecting groups followed
by a dehydration process to prepare sarkomycin. The removal of pro
tecting groups and dehydration took place under very mild acidic -l
conditions (a 1:1 mixture of 0.5 N HCl-acetone) and stirring for 12 J
hours at room temperature. ^
The resulting NMR spectrum of the crude mixture showed two pairs ;«
of doublets at 5 5.70and 5 6.24p.p.m. due to the vinyl protons of
sarkomycin as a major product (our reported values for vinyl protons i
of sarkomycin generated from keto lactone (50) was 5.72 and 5.22). |
The product was purified by sodium bicarbonate extraction of the mix
ture, followed by reacidification and work up which resulted in almost
pure sarkomycin, based on spectral analysis. The successful synthesis
of sarkomycin from the keto lactone (50) and from the protected
hydroxy acid (53) confirms the structures and stereochemistry to all
intermediates in this synthesis. The route from the keto lactone

57
gave no hydroxy acid and the route from the protected hydroxy acid
gave no detectable amount of lactone.
Since the keto lactone (50) has to have a cis_ ring junction (the
trans isomer would be highly strained), the groups in the protected
hydroxy acid (53) must be trans. Since isomerlzation can occur in
the nitrile hydrolysis reaction on either (32) or (51), anchimeric ass
istance is postulated during the hydrolysis of compound (32), which
isomerizes all the compound to the £1^ form, whereas when the
hydroxy group is protected as in (51) the isomerlzation gives the
more stable trans arrangement of the functional groups. Thus all
the stereochemical assignments are consistent.
0 0 0 0
GH20THP OH" X>^ H20THP
' 'G=N
51
' ' 'GOOH GOOH
Looking for Biological Activity
Dr. Robert B. Ing at "Division of Cancer Treatment", of the
National Cancer Institute in Silver Spring, Maryland, responded to
our inquiries by supplying information for the procedures which are
used at NCI for biological screening of compounds for possible anti
cancer activity. These procedures are summarized below.
Before submission of any sample at the institute, a computer
search is made to determine if any previous testing has been done on
I

58
it. If the preliminary search suggests that there is interest in
testing the compound, it can be submitted. An NSC number is assigned
to the sample and a computer print out of the molecule is sent to
the supplier.
The extent of testing that is carried out depends upon the
amount of material available and the toxicity of the compound. The
more toxic a substance, the smaller the dose and, therefore, the
greater number of tests per given amount of sample which can be
performed. Conversely, the less toxic the material, the more sam
ple needed per test. The usual requirement is 1.5 gram of a sub
stance. However, a limited amount of testing can be done with 100
milligrams of compound.
From our work, three samples were mailed, the cyano, amide and n^
lactone derivatives of sarkomycin. 1
i 1 I «
0 GH,
/ ^ '
\—/-^c^c ^ — ^ c = r ^ '—^Vr^^"2 i ^cc ^ ^G=N ^ ^G-NH,
^ ^ ^
250 mg (crystalline) ^00 mg (in GHGl^) 200 mg ( in GHCl^)
The computer search revealed that there was no previous evidence
of biological screening. Therefore, the NSC number of 317614 was
assigned to keto lactone (50) and 318815 to cyano derivative (47).
The number for amide (54) will be available soon.
As described in the text, sarkomycin (1) is active against the 65
ascites type of tumors, but according to one study is inactive

59
against solid tumors. However, one can speculate that the kato-
lactone (50), which can be considered as a "protected" form of
sarkomycin, may be able to penetrate cell walls better than sarko
mycin.
fifi According to recent literature, Ranieri and Calton reported
the isolation and structure elucidation of a new anti-neoplastic
agent quadrone (55) obtained from extracts of the broth from
A. terreus. The crude broth exhibited significant inhibitory
activity in vitro against human epidermoid carcinoma and in vivo
activity against P 388 lymphocytic leukemia in mice.
In a private communication. Dr. Amos Smith suggested that
possibly the active agent is not quadrone (55) itself, but rather
the open form (56) as the active ingredient. Since, as described ^
in the text, keto lactone (50) undergoes eliminative opening to J
sarkomycin (1) in dilute acid, we hope an analogous reaction may
the structural similarity between quadrone and our keto lactone 0)
is obvious.
mycin, since they already contain the methylene ketone functional
group. They are more stable than sarkomycin itself, and we have
been able to store them for extended periods of time in organic
solvents below zero degrees centigrade. These samples were sent
for biological screening in CHCl^ solution to minimize the possi
bility of decomposition on storage. It will likely be several
months from this writing before any testing results are available
1
occur in vivo, by analogy to Smith's speculation. In any event, :
f I »
Compounds (47) and (54) are closer structural analogs of sarko- '
60
GOOH
i6
H"
7 ^GOOH
19.
1 K •I
i 1 1

CHAPTER III
CONCLUSION
The purpose of this research was to synthesize sarkomycin and
some of its analogs for biological screening. The total regio
specific synthesis of sarkomycin and three analogs (cyanide, amide
and lactone), as developed in this work, is represented in Scheme
XVII.
In the course of the synthesis of these easily polymerizable
molecules, several new steps were developed as described below.
On the course of the reduction of ethylene ketal of 2-carbo-
methoxy-3-cyanocyclopentanone (29) with LBH, we encountered the
problem of losing the ketal protecting group. This phenomenon was
rationalized by the LBH or its decomposition products causing ,«
reductive opening of the ketal protecting group. This problem was ••
solved by addition of ca_. 2 eq. H20-NaOH per mole of LiBH^ to carry I
out the desired transformation cleanly . J
Very mild conditions were studied for the final step of the i
synthesis, which introduced the a-methylene ketone functionality '
by an acid-catalyzed dehydration process.
Successful acid-catalyzed opening of keto lactone (50) to
sarkomycin (1) suggests that the lactone may also open in vivo,
which gives it a high potential value for biological activity.
In addition to the synthetic work, a careful spectral analysis
of 2-carbomethoxy-3-cyanocyclopentanone (28) indicated this compound
is a good model for studying the enol-keto tautomerlzation phen-
61
M1111111111111111111
I M I I I I M H I M H I I I
l l l l l l l l l l l l l l l l l l l t
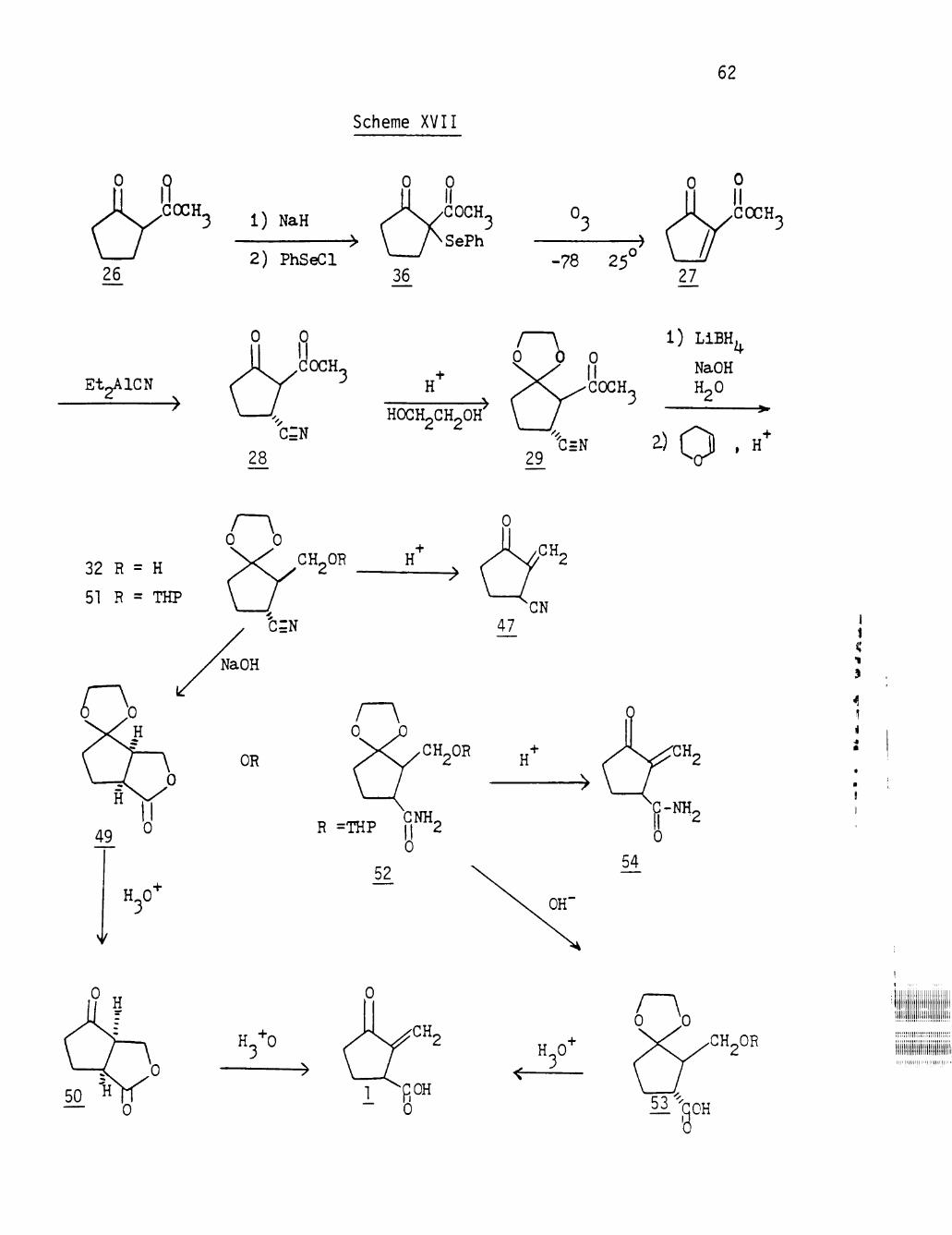
62
Scheme XVII
>k COCK
26
3 1) NaH • »
2) PhSeCl
0 II COCK,
SePh'
36 -78 25
0 II COCH-
27
Et2A ICN s OCH,
H
C=N HOGH2CH2OH
D 0 0
> ^ ^ \ ^ C O C H ,
1) LiBH^
NaOH H2O
28 29 1:=N Z) Q'»'
32 R = H
51 R = THP
r-\ 0 0
t=N
0
CH2OR H • ^
GH,
GN 47
/~A GH2OR
R =THP n ^ 2 0
52
H ^
GH,
0
54
OH'
i
H " 0
• ^
HO
0 )o
& 53 "cOH
CHgOR M t t t I I t t t l l

63
omenon. To the best of our knowledge, this is the first example
of a tautomeric mixture which could be studied by observing the
methoxy signals instead of the broad acidic OH proton signals, which
makes it easier to differentiate between the two forms quantitative
ly.
i 1
I
n
I I f 4

CHAPTER IV
EXPERIMENTAL
General
All routine NMR spectra were run on a Varian A-60 or XL-100
spectrometer in CDCK solutions unless otherwise stated. All NMR
chemical shifts are reported in 6 units downfield from internal ref
erence TMS. Infrared spectra were obtained with a Perkin-Elmer 457 as
a thin film or in CHCU solution (wavelength calibrator: Beckman
polystyrene film vs. air 1603.0 cm' ). The NMR and IR spectra are re
produced in Appendix B. All melting points are uncorrected and were
determined after at least one recrystallization and drying at 0.1
mm Hg. Melting points were obtained in open capillaries on a Labor
atory Device's "Mel-Temp." The ozone generator was model 03V5-0 from J
"Ozone Research and Equipment Corp.," Phoenix, Arizona. A multiple ^
unit furnace from "Hev-duty Heating Equipment Co." was used as a j
pyrolysis chamber. i
Silica gel, 60-200 mesh, high purity, from Sargent-Welsh, was
used for all column chromatography. Columns were packed as a slurry
using the first eluting solvent as the packing solvent.
Elemental analyses were done commercially by Chemalytics, Inc.
Tempe, Arizona.
Unless otherwise stated, when reactions were worked up using an
organic-aqueous separation, the organic layer, after extraction with
appropriate aqueous solutions, was dried over anhydrous MgSO^ for a
few minutes, then filtered and the organic sol vent(s) removed with a
Buchi Rotavapor-R with v^ater aspirator vacuum of 20-30 mm Hg. 64

65
Benzene, chloroform, acetone, ether, and petroleum ether were
purified according to standard procedures and fractionally distilled.
Tetrahydrofuran (THF) was dried over sodium-potassium alloy (NaK) for
24 hours and distilled before use.
Modification of the Procedure for the Synthesis of 2-Carbomethoxycyclopentenone-Cyclopentadiene Diels-Alder Adduct (34)
To a solution of 14.2 g (0.10 mol) of 2-carbomethoxycyclopent
anone (26) in 25 ml of reagent grade dioxane was added 12.2 g (0.11
mol) of Se02 and the mixture was refluxed for five minutes. A quick
addition of 66 g of freshly prepared cyclopentadiene (by dropping
funnel) was followed by refluxing the solution for one hour, com
pleting the reaction.
Removal of all volatile materials by a water aspirator was fol- •'
lowed by absorption of the residue on 25 g of silica gel and separation »
by chromatography on 600 g of silica gel packed in petroleum ether 1
(b.p. 40-60°C). After elution with 20% ether in petroleum ether, a .'
1:1 mixture of the two stereoisomeric adducts (34) was obtained
(6.2 g, 36%).
30 The NMR spectrum of this mixture matched the published spectrum.
2-Carbomethoxycyclopent-2-enone (27) from Pyrolysis of Adduct (34)
A 1:1 mixture of the two isomeric adducts (34) was placed in a
dropping funnel and that was connected to a slow stream of nitrogen.
The pyrolysis chamber was heated to 400°C in a vertical position as
illustrated in Appendix A (Figure 1). I i '
I Mil
I!

66
A liquid film (3 mm) of adduct (34) flowed down due to gravity,
with the aid of nitrogen gas, to the pyrolysis chamber. After passing
through the heated zone, the exit gases were condensed in a cold trap
(dry ice/acetone). The desired compound (27) was collected in the cold
trap, in addition to selenium impurities. This procedure can be re
produced in moderately large quantities in a short period of time.
The best results are obtained if the adduct (34) is well purified from
selenium impurities before pyrolysis. The reaction should be done
under a ventilation hood.
Preparation of Sodium Salt of 2-Carbomethoxycyclopentanone (38)
The apparatus used consisted of a 2000 ml three-necked flask,
equipped with magnetic stirrer, which was fitted to a pressure equaliz
ing dropping funnel and syringe Inlets for nitrogen. The flask con
tained a stirred suspension of 25 g (1.04 mol) of NaH (57% dispersion
in mineral oil; washed free of oil with 3x100 ml of dry pet ether) in
600 ml of dry THF under nitrogen at 0°C. To this solution was added,
dropwise, a solution of 100 g (0.7 mol) of commercially available
2-carbomethoxycyclopentanone (26) in 200 ml of THF over a period of
one hour. After stirring the solution at room temperature for an
additional 15 minutes, the solution was cooled and a v/hite precipitate
was filtered out. The filter cake was washed several times with dry
pet ether. After drying it at 25° at 0.1 mmHg for 2 hours, 105 g (90%)
of compound (38), as a white powder was obtained.
* ,»
1
' i 1 1 1 1 1 1 1 1 1 1 1 1
I ; ; I
I t M t M

67
This compound can be kept for extended periods of time under
nitrogen in a sealed container in a refrigerator. The solubility of
the salt is 4 g/100 ml in THF at room temperature.
Preparation of Benzeneselenenyl Chloride (35)
The following procedure is similar to the method described by 32
Reich. Chlorine gas was passed into a magnetically stirred solution
of 25.0 g (80 mmol) of Ph2Se2 in 200 ml of dry hexane. The appearance
of the white precipitate of PhSeCl-, which can be removed by the
addition of another small quantity of Ph2Se2, indicates the end point
of the reaction. The solution was heated to dissolve any crystallized
PhSeCl (if the solution is cloudy at this point, it should be filtered),
and then the clear solution was allowed to crystallize in the refrig
erator very slowly. A quick filtration of the large orange crystals,
'li followed by drying the crystals by a vacuum pump (0.1 mm for 2 hours) i
resulted in 26 g of PhSeCl (85%), m.p. 62-64°C. Some unreacted Ph2Se2 I
present in the filtrate can be recovered by cooling the solution and j
filtering the precipitate. :
Benzeneselenenyl chloride (35) is now available for purchase from '
chemical companies which will minimize the possible toxic hazards in '
making the compound.
Preparation of 2-Carbomethoxy-2-Benzene-selenenylcyclopentanone (36)
32 The following is an improvement of the method described by Reich.
A solution of 50 g (0.26 mol) of benzeneselenenyl chloride (35) in
100 ml of dry THF was added rapidly to a suspended solution of 43 g
(0.26 mol) of the sodium salt of 2-carbomethoxycyclopentanone (38) in

68
500 ml THF under nitrogen. An additional 15 minutes stirring took
place in order to complete the reaction. To decompose any unreacted
PhSeCl which may have been present, 100 ml water was added, followed by
stirring the solution for 5 to 10 minutes. Extraction of the organic
compounds was done with CH2Cl2» followed by washing the organic layer
with excess water to remove the salts generated in the reaction.
After drying the organic layer, the solvent was evaporated by a water
aspirator at room temperature and then by a vacuum pump. In order to
minimize any decomposition of the product.
The resulting dark yellow oil (71 g; 91%) shows bands in the
infrared (film) spectrum (Figure 2) at 3060 (C-H aromatic), 1760-1710
(0=0 and COO), 1575 (0=0 aromatic), 725 and 675 cm'^ (C-H aromatic).
This spectrum is a good indication of the absence of any 2-carbo
methoxycyclopentanone (26). The nuclear magnetic resonance spectrum , i
(Figure 3) shows peaks at 6 1.7-2.8(6 H, mul t , CH^), 3.75(3 H, s , ]
COOMe), and 7.2-7.9 p.p.m. (complex mul t , Ph). However, in tegra t ion , I
also shows some possible Ph2Se2 impur i ty . ]
Preparation of 2-Carbomethoxycyclopent-2-enone (27)
Without any fu r ther p u r i f i c a t i o n , a solut ion of 71 g crude
2-carbomethoxy-2-benzeneselenenylcyclopentanone (36) in 350 ml dry
OH 01 was added to a 500 ml round bottom reaction f l ask . Ozonized
oxygen (about 0.4 mmol O3 per min) was bubbled through the so lut ion
fo r 12 to 14 hours at -78°C. The end point can be determined by the
appearance of the l igh t -green to blue color of excess O3 in the
s o l u t i o n . The excess ozone was swept away by passing O2 through the
react ion f l a sk . Bringing the clear yel low so lu t ion back to room temp-

69
erature resulted in a white prec ip i ta te . Fi l t ra t ion followed by a i r
drying of the solid material gave 21 g of a white and yellow powder
which is a mixture of PhSe02H and Ph2Se2,^^ resulting from decompo
si t ion of PhSeOH. The clear f i l t r a t e was dried over anhydrous mag
nesium sulfate and the solvent was removed with a rotatory evaporator
which resulted in a yellow o i l . The overall yield calculated on the
amount of the elimination product (PhSeOH) formed was 17.5 g(48-50%).
The structural identification of the compound, 2-carbomethoxy-
cyclopent-2-enone (27), was based on spectral analysis.^° The infra
red spectrum of the neat compound shows bands at 3060 (aromatic Im
pur i ty) , 1750 (0=0 ketone), 1725 (C=0 e s t e r ) , 1620 (C=C), and 1580 cm"
(aromatic impurity). The nuclear magnetic resonance spectrum in CDCl
shows peaks at 6 2.1-2.9 (4 H, mult, CH2), 3.92 (3 H, s, COOMe),
5.35 (reference solvent CH2CI2), 7.2-8.2 (m, aromatic impurity), and
8.38 p.p.m. (1 H, t , J=2.5 Hz, vinyl H). Pure samples of this corn-on
pound have been prepared previously in this lab, but the material
was judged to be of sufficient purity to be used in the subsequent
step. This compound polymerizes after standing in a refrigerator for
several days.
Preparation of 2-Carbomethoxy-3-Cyanocyclo-pentanone (28)
Into a 500 ml three-necked flask, equipped with magnetic s t i r r e r ,
syringe in le ts for nitrogen, and also connected to oil bubbler, was
placed the total crude 2-carbomethoxycyclopent-2-enone (27) from the
previous step in 250 ml of dry benzene in an atmosphere of nitrogen.
The mixture was cooled to 0°C and 150 ml of 1.69 M commercially avail-

10
able (Alfa) Et2AlCN (3 portions of 50 ml each) were added by syringe
over a few minutes time. The solution was s t i r red overnight at room
temperature. The work up started with cooling the solution to ice-bath
temperature followed by the addition of ice-cold water and then acid
i f i ca t i on with 5% HCl solut ion. The organic layer was extracted by
CHCl-j and washed several times with excess water. Any cyanohydrin
present in the solution due to the addition of excess Et2AlCN was
reversed by shaking the solution with 10% NaOH which in turn generates
the sodium sa l t of the cyano keto ester (28). Impurities were washed
away by an excess of CHCl followed by reacid i f icat ion of the aqueous
layer with 5% cold HCl. This resulted in the desired compound; a dark-
orange o i l which was crysta l l ized by standing at room temperature for
a few days. Two recrystal l izat ions of the product from ether and pet
ether gave 16 g (38% based on sodium salt of 2-carbomethoxycyclopen- i 4 .1*
tanone) of cyano keto ester (28) as white crystals, m.p. 47-48°C. »
The infrared spectrum of (28) (liquid film before crystallization) | f
(Figure 4) shows bands at 2225 (-C=N), 1770 (0=0, ketone), 1735 (0=0 \
ester), 1670 (0=0 conj ester), and 1625 cm"^ (conj C=C). The NMR
spectrum (Figure 5) shows two singlets at 6 3.86 and 3.88 p.p.m.
(3 H total) due to two methoxy signals corresponding to enol and keto
tautomers. In addition, this spectrum shows absorptions at 6 3.5-3.7
(1.5 H, m, methine protons), 2.1-3 (4 H, complex absorption, CH2), and
10.3 p.p.m. (£a. 0.5 H, s, broad, -OH enol). Anal. Calcd. for CgHgO^N:
C, 57.46; H, 5.42. Found: C, 57.55; H, 5.62.
The phenomenon of enol-keto tautomerlzation is well observed from
the Infrared spectra (KBr pellet) in Figure (6). These spectra show

71
two bands in the carbonyl region fo r the pure keto Isomer (Spectra
6a-b) : 1770 (0=0, ketone) and 1735 cm'^ (0=0, es te r ) . However, upon
prolonged exposure to the IR beam, two new peaks appeared due to the
chelated enol tautomer at lower frequency (Spectra 6 c - f ) : 1670
(0=0, conj es ter ) and 1625 cm" (conj C=C). Upon cool ing the pe l l e t
to 20-25°C, the new peaks disappeared again as i l l u s t r a t e d in Figure
(6)(Spectra 6h-1) .
Dependance of enol-keto tautomerlzat ion upon d i l u t i o n wi th d i f
fe rent solvents is demonstrated in Figure (7) by the ra t i o of the
methoxy signals ( fo r more d e t a i l s , see the discussion page 33 ).
Preparation of the Ethylene Ketal of 2-Carbomethoxy-3-Cyanocyclopentanone (29)
A mixture of 15.25 g (91 mmol) of 3-cyanoketone (28) , 250 ml of
reagent benzene, and about 30 mg of p-toluene sul fonic acid was t reated i
w i th 34 g of ethylene glycol and ref luxed fo r 8 hours into a Dean- J
Stark t r a p , then 4 hours in to a large Soxhlet ext ractor f i l l e d with |
anhydrous MgSO . The so lu t ion was then cooled, 100 ml of CHCl^ was
added, and then the so lu t ion was washed with saturated br ine. Addi t
ional washing wi th 10% NaOH so lu t ion was done to ext ract any unreacted
s t a r t i n g mate r ia l . Drying, fol lowed by solvent removal gave 16.4 g
(82%) of crude compound. The product was absorbed on s i l i c a gel and
chromatographed on a s i l i c a gel column which had been packed in petro
leum ether (40-60°) . Some selenium impur i t ies were washed away by
using pet ether. The pure compound, m.p. 40-40.5°C, was co l lec ted by
e lu t i on o f the column wi th 15% ether in pet ether.

72
The desired compound shows a mixture of two stereoisomers based
on spectral data. The neat Infrared spectrum before crysta l l izat ion
(Figure 8) shows bands at 2225 (C=N), and 1740 cm" (COOCH3). The
NMR spectrum (Figure 9) in CDCI3 shows peaks at 5 1.7-2.5 (4 H,
complex mult, CH2), 3.14-3.62 (2 H, mult, Cii-C00CH3 and CH-C=N), 3.78
(3 H, s, COOCH3), and 3.85-4.2 p.p.m. (4 H, mult, -OCH2-CH2O-). Anal.
Calcd. on the mixture of two Isomers for C^QH,30«N: C, 56.86; H,
6.20. Found: C, 56.55; H, 6.28.
A s l igh t change in peak intensity of the column chromatography
fractions at 6 3.16 p.p.m. implied that compound (29) was a mixture of
two stereoisomers which could possibly be separated by careful chroma
tography.
An Attempt to Isolate Stereoisomers of Ethylene Ketal of 2-Carbomethoxy-3-Cyanocyclopentancne (29) j
if
Further elution of the column from the previous experiment with »
pure ether resulted in one stereoisomer (minor product; almost pure)
which is believed to be the cis isomer (the thermodynamically less
stable compound). The Infrared spectrum (Figure 10) is almost iden
tical (except for a few peaks in the finger print region) to Figure
(8), which is a mixture of two isomers. However, after crystallization
in ether, the pure compound with m.p. 73-74°C, showed NMR (CDCI3)
(Figure 11) peaks at 6 1.82-2.52 (4 H, mult, CH2) 3.04-3.18 (2 H,
mult, CH-COOCH3 and C H - C E N ) , 3.78 (3 H, s, COOCH3), and 3.98 p.p.m.
(4 H, s, -OCH2-CH2O-). Subsequent attempts to separate the pure trans
isomer by chromatography and recrystallization techniques failed.
J J J J J J!

73
In order to assign the peaks fo r the r i g h t Isomers, the mixture
was studied w i th ^H NMR (100-MHz) between 6 3.1-3.6 and 1.7-2.5
p.p.m. at d i f f e r e n t temperatures from -60 to 80°C, then a double
resonance technique was attempted but a l l experiments f a i l e d , because
of overlap chemical s h i f t s of C-2 and C-3 hydrogens. The mixture was
used in subsequent steps.
Preparation of the Ethylene Ketal of 2-Hydroxymethylene-3-Cyanocyclopentanone (32)
To a so lu t ion of 0.52 g (2.4 mmol) of the ethylene ketal of 2-
carbomethoxy-3-cyanocyclopentanone (29) in 5 ml dry THF, under ni trogen
atmosphere, was added 0.2 ml of 5% NaOH so lu t i on . Following the
add i t ion of 10 ml of 0.8M so lu t ion of LiBH^ (LBH) in THF, the mixture
was s t i r r e d at room temperature for 24 hours. Af ter dropwise addi t ion
of water to decompose excess LBH, the oraanic materials were extracted
wi th CHC1-, and washed several times wi th excess water. Drying, I
fo l lowed by solvent e x t r a c t i o n , resul ted in 0.33 g (78%) of a yel low
o i l . A l l attempts to c r y s t a l l i z e th i s compound f a i l e d .
Molecular d i s t i l l a t i o n of the crude compound in an 80-90°C o i l
bath (0.25 mm Hg)resul ted in an almost pure compound. However, i t is
possible to pu r i f y the compound by column chromatography on s i l i c a gel
by using pure ether as the eluent a f te r washing away the im.purities by
20% ether in pet ether.
The neat i n f ra red spectrum (Figure 12) shows a broad band at 3500
due to the -OH moiety and a band at 2220 cm"^ (C=N). However, the
NMR (00013) spectrum (Figure 13) shows peaks at 6 1.6-2.7 (5 H,

74
complex mult, CH2), 3.06 (1 H, mult, CFi-C=N), 3.80 (1 H, d, J=8 Hz,
CH2OH), 3.84 (1 H, d, J=8 Hz, CH2OH) and 3.98 p.p.m. (4 H, s, -OCH2-
CH2O-).
A separate experiment was conducted under the same conditions
described above, except that the NaOH was left out of the reaction.
After work up, there was no ketal peak detectable by NMR (Figure
14-b). For comparison purposes, both NMR spectra are reproduced
in Appendix B (Figure 14)(see discussion for more detail, page 41 ).
Preparation of 2-Methylene-3-Cyanocyclopentanone (47)
To 0.88 g (4.80 mm.ol) of cyano ketal alcohol (32) was added a
1:1 mixture of 1 N HCl and reagent acetone, and the homogenous solution
was stirred for 12 hours at room temperature. The organic compounds
were extracted twice with excess CHOI3 and the combined extracts were
dried and the solvent was removed at 25°C under reduced pressure. The
resulting 0.47 g crude oil (81%) was a mixture of compound (47) and
2-hydroxy-methylene-3-cyanocyclopentanone (48). Molecular distillation
of the crude product was carried out during two hours at 0.25 mm Hg
and 80-90° bath temperature. The result is an almost pure sample of
2-methylene-3-cyanoketone (47) based on spectral analysis.
The infrared spectrum of the neat compound Figure (15-a) and in
chloroform solution Figure (15-b) show bands at 3450 (broad, -OH),
2220 (C=N), 1750 (C=0, carbonyl), and 1645 cm" for carbon-carbon
double bond moiety. The NMR spectrum (Figure 16-a, after distil
lation, and Figure 16-b, before distillation) in CDCI3 shows two
doublets, one at 6 5.78 (J= 2.5 Hz) due to vinyl proton trans to keto
group and the second at 6 6.28 p.p.m. (J= 2.5 Hz) due to the other
! i • 1

75
v iny l proton which is ci_s, to the carbonyl group. ' The NMR
spectrum also shows peaks at 6 3.5-4.2 p.p.m. (mul t , -01^2^*^) ^^^ ^° ^^^
undehydrated compound, 2-hydroxymethylene-3-cyanocyclopentanone (48).
This mixture i s stable in CHOI3 so lu t ion fo r at least several weeks i f
i t i s kept in a r e f r i g e r a t o r .
Preparation of Ketal Lactone (49)
To a so lu t ion of 0.50 g (2.7 mmol) of cyano ketal alcohol (32)
in 5 ml of 95% ethyl alcohol was added 5 ml of 5% NaOH so lu t i on . The
mixture was allowed to re f l ux fo r one hour. Af ter coo l ing , the basic
so lu t ion was washed several times wi th excess CHOI3 in order to remove
a l l organic impur i t i es . Then, the water layer was ca re fu l l y a c i d i f i e d
w i th 5% HCl so lu t i on . Extract ion of the compound with CHOI3 was
fol lowed by drying and removal of the solvent which resul ted in 0.32
g (64%) of ketal lactone (49). Af ter several r ec rys ta l l i za t i ons from
an e ther -pet ether s o l u t i o n , the white crys ta ls melted at 67-67.5°C.
A separate experiment showed that in 15 minutes re f lux ing tim.e, about
ha l f of the s t a r t i n g material was consumed.
The i n f ra red spectrum of the pure compound in CHOI3 (Figure 17)
shows a band at 1765 cm"^ (0=0 lactone) . The NMR spectrum (Figure
18a) in CDCI3 shows peaks at 5 1.46-2.34 (4 H, mul t , CH2), 2.7-3.26
(2 H, mul t , two methinyl protons) , 3.96 (4 H, s , -OCH2-CH2O-), 4.32
(1 H, dd, J=8 ,10Hz) , and 4.46 p.p.m. (1 H, dd, J=4, 10 Hz). This is
consistent wi th the formulat ion of the compound as ketal lactone (49)
(see Figure 08b) fo r s p l i t t i n g assignments). Anal. Calcd. fo r
C H^pO.: C, 58.68; H, 6.56. Found: 0 , 58.76; H, 6.24.
I I I
J J J J

76
Preparation of Keto Lactone (50)
Hydrolysis of the protecting group of 0.23 g (1.25 mmol) of ketal
lactone (49) took place by stirring a 1:1 mixture of acetone and 3 N
HCl solution for six hours at room temperature. Recovery of the organic
compounds took place by CHCI3 extraction and was followed by drying
and evaporation of the solvent. The resulting crystalline material,
0.16 g (91%), showed a sharp melting point of 45-46°C after several
recrystallizations from CHCl3-ether solution.
The Infrared spectrum (Figure 19) of the compound in CHOI3
solution shows bands at 1780 (0=0 lactone) and 1750 cm"^ (0=0 ketone).
The NMR spectrum (Figure 20) in 00013 ^^ ° ^ ^ ^^^^^ °^ 1.96-2.74
(4 H, mult, CH2), 2.84-3.22 and 3.28-3.56 (2 H total, mult, methinyl),
4.44 (1 H, d, J=5.5 Hz) and 4.45 p.p.m. (1 H, d, J=4.0 Hz, CH.2-0-C=0).
Analysis Calcd. for C-^Hg03: 0, 59.99; H, 5.75. Found: C, 59.86;
H, 5.75.
Spectral analysis of the crude material (before crystallization)
showed some eliminative opening of the lactone to give sarkomycin
3-carboxy-2-methylenecyclopentanone (1). This could be avoided by
weakening the acid strength, but this Increased the hydrolysis time
to impractical length. (See the discussion part, page 49 ).
Preparation of Sarkomycin; 3-Carboxy-2-methyl-enecyclopentanone (1) From Keto Lactone (50)
The final step leading to sarkomycin, from keto-lactone (50) as a
substrate, is the eliminative opening of the lactone ring. This react
ion was achieved by stirring (0.32 g, 2.2 mmol) a 1:1 mixture of 0.5 g
(3.5 mmol) keto lactone (50) in 3 N HCl and acetone solution for 3

77
hours at room temperature. Organic compounds were extracted three
times wi th CHOI3 and the combined extracts were dr ied and the solvent
was removed at 25°C under reduced pressure. The resu l t ing material
was i d e n t i f i e d as a mixture of sarkomycin (35%) and lactone Isomer
(50) , based on spectral analysis (0.2 g, 80%).
The resu l t i ng IR spectrum, In CHCI3 (Figure 21) , of sarkomycin
shows bands at 1730-1720 (0=0) and 1640 cm"^ (C=C). The nuclear
magnetic resonance spectrum (Figure 22) shows peaks at 2.2-2.7
(complex mul t , CH2), 6 5.72 (1 H, d, J=2.5 Hz, v inyl proton, trans
to 0=0 ketone), and 6.22 (1 H, d , J=2.5 Hz, v iny l proton, c is to
0=0 ketone).
A neat sample of sarkomycin polymerizes a f te r standing at room
temperature f o r several days. However, i t is possible to slow down the
polymerizat ion process wi th storage of the compound in a sui table
solvent (CHOI3) at temperatures below zero.
Preparation of Ethylene Ketal of 2-Methylene (0-Tetrahydropyranyl)-3-Cyanocyclopentanone (51)
To a magnetical ly s t i r r e d so lu t ion of 0.81 g (4.42 mmol) of cyano
ketal alcohol (32) and reagent grade p-toluenesulfonic acid (0.05 g,
0.26 mmol) in 10 ml of anhydrous methylene chlor ide at room temper
ature was added 10 ml of 2,3-dihydropyran. The solut ion was s t i r r e d
fo r one hour at room temperature. Addit ion of saturated brine solut ion
was fol lowed by the ex t rac t ion of the organic compounds with CHCI3.
Drying and removal of the solvent gave (2.18 g) the desired compound
(51) which was contaminated wi th self-condensation products from the
2,3-dihydropyran.

78
Structural proof was based on spectral analysis. The IR (film):
2225 cm" (C=N) and the NMR (CDCI3) showed peaks at 5 3.9 (4 H, s,
-OCH2-CH2O-) and 4.5 p.p.m. (1 H, broad, s, methinyl proton on tetra-
hydropyran ring).
The crude compound was used for the next stage without further
purification.
Preparation of the Ethylene Ketal of 2-Methylene (0-Tetrahydropyranyl)-Amidocyclopentanone (52)
To the total crude sample of protected hydroxy cyano ketal (51)
(from the previous experiment) was added a solution of 1:1 5% NaOH
and 95% ethyl alcohol and the mixture was refluxed for 6 hours. After
cooling and acidifying the solution, organic materials were extracted
by CHCI3. Drying and removal of the solvent gave a yellow oil, which
upon dissolving in ether, precipitated out (0.24 g) the crystalline
amide derivative (52). After two recrystallizations from ether, the
pure white crystals melted at 144-145°C. By considering the amount
of amide which can be recovered as the acid derivative (53) in the
next step, the yield for the two step reaction from cyano ketal
alcohol (32)—^(52) is 60%.
The infrared spectrum of the pure crystalline material (Figure 23)
in CHOI 3 solution shows bands at 3500 and 3350 (N-H amide, stretching),
1680 (0=0 amide) and 1600 cm"^ (N-H amide, bending). The NMR
spectrum (Figure 24) of crystals in CDCI3 solution shows absorptions at
6 1.3-2.94 (12 H, mult, CH2 and CH), 3.2-3.72 (4 H, mult, Chl20),
3.92 (4 H, s,-0CH2-CH20-), 4.66 (1 H, broad, s, methinyl proton on

79
tetrahydropyran ring), 5.76 and 6.68 p.p.m. (1 H each, broad, s,
amide). Anal. Calcd. for CT.H-.,O^N: C, 58.93; H, 8.12. Found: C,
14 ^J 0
58.39; H, 8.19.
Preparation of the Ethylene Ketal of 2-Carbomethoxy-3-Amidocyclopentanone (30)
To a magnetically stirred solution of 0.77 g (3.6 mmol) of the
ethylene ketal of 2-carbomethoxy-3-cyanocyclopentanone (29) at room
temperature was added 5 ml of 30% hydrogen peroxide. After dropwise
addition of 3 ml of 10% sodium carbonate solution (keeping pH between
7 and 8), the mixture was allowed to stir magnetically at room temp
erature for one hour. The organic compounds were extracted by CHOI3
and washed with cold water and dried. Upon dissolving the crude yel
low oil in ether, white crystals (0.27 g; 32%) precipitated out, which
upon recrystallization from ether and pet ether, melted at 114-115°C.
The infrared spectrum of pure crystals of (30) in CHCI3 (Figure
25) shows bands at 3520 and 3420 (N-H amide, stretching), 1735 (0=0
ester), 1690 (C=0 amide), and 1600 cm" (N-H amide, bending). The
NMR spectrum (Figure 26) in CDCI3 shows peaks at 6 1.8-2.2 (4 H,
broad, s, CH^), 3.2-3.5 (2 H, broad, s, methinyl protons), 3.75 (3 H,
s, COOCH3), 3.9-4.1 (4 H, mult, -OCH2-CH2O-) and 6.0-6.7 p.p.m. (2 H,
broad, s, NII2). Anal. Calcd. for C^QH.,50^N: C, 52.39; H, 6.59.
Found: C, 52.52; H, 6.50. All the experimental data mentioned above
are consistent with the formulation of the compound as ketal amide
(30). In addition the spectral data are closely related to those
from the ketal amide (52).
J J J jj

80
Preparation of 2-Methylene((3-Tetrahydro-pyranyl)-3-Carboxyl1c Acid (53)
A mixture of o.58 g (2.0 mmol) of ketal-amide (52) and 10 ml of a
1:1 mixture of 10% NaOH and 95% ethyl alcohol were ref luxed for 24
hours. The cooled react ion mixture was ca re fu l l y a c i d i f i e d wi th 5%
HCl so lu t ion and the oragnic compounds were extracted twice with
excess CHCI3. The combined organic solut ions were dr ied over MgSO^
and the solvent ex t rac ted. The resu l t i ng material (0.51 g; 87%)
c r y s t a l l i z e d from ether and pet ether so lu t ion and the pure crysta ls
melted at 69-72°C.
The IR spectrum of the pure crys ta ls (Figure 27-a) in CHOI3
shows bands at 1750 (C=0 dimer), and 1710 cm" (C=- carboxyl ic ac id ) .
However, the neat spectrum of the compound before c r y s t a l l i z a t i o n
(Figure 27-b) shows a broad peak at 3000 cm' charac ter is t i c of acid
f u n c t i o n a l i t y , 1730 (0=0 dimer) and 1705 cm" (C=0 carboxyl ic ac id ) .
The NMR spectrum in CDCI3 (Figure 28) shows peaks at 6 1.36-2.28
(10 H, complex, CH^), 2.42-3.0 (2 H, broad, mul t , methinyl protons),
3.24-3.86 (4 H, mul t , Cli2-0), 3.92 (4 H, s , -OCH2-CH2O-), 4.68 (1 H,
broad, s , methinyl proton on tetrahydropyran r i n g ) , and 9.85 p.p.m.
(1 H, broad, s , ac id ic proton). Anal. Calcd. for ^^^^^Z^s' ^ '
58.72; H, 7.74. Found: 0, 58.28; H, 7.68.
Preparation of Amide Der ivat ive of Sarkomycin; 2-Methylene-3-Amidocyclopentanone (54)"
In a l i k e manner as described prev ious ly , 0.1 g (0.35 mmol) of
hydroxy protected ketal amide (52) was hydrolyzed for 12 hours with a
1:1 mixture of 0.5 N HCl-acetone so lu t i on . Af ter several extract ions
of organic compounds wi th excess CHCI3, 0.03 g (57%) of desired

81
compound (54) was obtained. This reaction was also conducted very
successfully in an NMR tube in acetone-d^ and DCl-DoO solution. 0 I
The neat Infrared spectrum of the crude extract (Figure 29) shows
bands at 3350 (broad, OH and NH), 1730 (0=0 ketone), 1670 (C=0 amide)
and shoulder at 1620-1640 cm"^ (0=0 and NH amide, bending). Both
the NMR spectra of compound (54) in acetone-dg and DCI-D2O solution
(Figure 30a) and in CDCI3 (Figure 30b) shows two doublets, one at
6 5.58 (J=2.5 Hz) and second at 6 6.24 p.p.m. (J=2.5 Hz). The NMR
spectrum also shows peaks at 6 3.2-4.2 p.p.m. (mult, -CH^^OH) due to
the undehydrated compound In addition to some undesired complex
absorptions in the crude mixture.
Preparation of Sarkomycin (1) From 2-Methylene-(O-Tetrahydropyranyl)-3-Carboxyl1c Acid (53)
To a 5 ml solution of 0.38 g (1.32 mmol) of compound (53) was
added a 0.5 N HCl solution and the mixture was stirred overnight at
room temperature. Extraction of the organic compounds with excess
CHCK followed by drying and evaporating the solvent resulted in
0.06 g (33%) crude oil containing sarkomycin as its major compound.
Structural proof was based on spectral analysis. The neat IR
spectrum (Figure 31) of pure sarkomycin after extraction with sodium
bicarbonate shows bands at 6 1740-1730 (0=0 carbonyl) and 1640 cm"
(C=C). This spectrum matches the published spectrum. ' The NMR
spectrum of the crude mixture in 00013 (Figure 32a) shows the pres
ence of polymeric materials. However, after bicarbonate extraction
of sarkomycin, the resulting spectrum (Figure 32b) shows two pairs
of doublets at 6 5.7 (1 H, J=2.4 Hz) and 5 6.23 p.p.m. (1 H, 0=2.5 Hz)

82
for vinyl protons. This spectrum also shows complex absorption at
5 2.2-2.7 p.p.m. for methylene protons. Vinyl protons from this
experiment are in well accord with those generated from keto lactone
(50).

LIST OF REFERENCES
1. T. Yamamoto, and T. Yamaoka, J. Antibiot. Ser A, 7_» 1 (1954).
2. H. Umezawa, T. Takeuchi, and K. Nitta, J. Antibiot. Ser A , 6, 100 (1953). =:
3. H. Umezawa, T. Yamamoto, T. Takeuchi, S. Yamaoka, K. Nitta and Y. Okami, J. Antibiot. Ser A, 6, 147 (1953).
4. H. Umezawa, Y. Yamamoto, T. Takeuchi, T. Osato, Y. Okami, S. Yamaoka, T. Okuda, K. Nitta, K. Yagishita, R. Utahara, and S. Umezawa, Antibiotics and Chemotherapy, 4, 514 (1954).
5. I. R. Hooper, L. 0. Cheney, M. J. Cron, 0. B. Fardig, D. A. Johnson, D. L. Johnson, F. M. Patermiti, H. Schmity, and W. B. Wheatley, Antibiotics and Chemotherapy, 5_, 585 (1955).
6. L. R. Hooper, Private Communication to J. N. Marx (1976).
7. Kenji Maeda, and Shinichi Kondo, J. Antibiot., Jj[, 37 (1958).
8. T. Hara, Y. Yamada, and E. Akita, J. Antibiot. Ser A, 10, 70 (1957). —
9. S. Tatsuoka, A. Miyake, M. Inoue, S. Wada, H. Iwasaki, K. Ogata, J. Antibiotics Japan Ser B, 9, 157 (1956).
10- Yasuhiko Sato, Shiro Nishioka, Osamu Yonemitsu, and Yoshio Ban, Chem. Pharm. Bull., 21, 829 (1963).
11. K. Toki, Bull. Chem. Soc. Japan, 3£, 450 (1957).
12. R. H. Hill, P. J. Foley, and C. A. Gardella, J. Org. Chem., 32 , 2330 (1967). —
13. E. J. Eisenbraun, and S. M. McElvain, J. Am. Chem. Soc., 77_, 3383 (1955). —
14. Kay, et aj_., J. Chem. S o c , 89 , 1646 (1906).
15. Mannich, et a^., Arch. Pharm., 575 (1936).
16. K. Toki, Bull. Chem. Soc. Japan, 31, 333 (1958).
17. K. Toki, Bull. Chem. Soc. Japan, 3^, 233 (1959).
18. M. M. Shemyakin, et_ ., Zhur. Obs. Khim., 27_ 742 (1957).
83

84
19. M. M. Shemyakin, G. A. Ravdel, E. S. Chaman, Yu. B. Shvetsov, and E. I. Vinogradova, Chem. and Ind., 1320 (1957).
20. S. 'Jmezawa, and Mitsuhiro Kinoshita, Bull. Chem. Soc. Japan, 3£, 267 (1957).
21. N. V. Smirnova, ejt al., Zhur. Obs. Khim., 2J_, 1312 (1957).
22. K. Jankowski, Tetrahedron Lett., 1733 (1971).
23. S. Hayashi, A. Matsuo, Tetrahedron Lett., 1289 (1970).
24. T. Naneishi, N. Kitahara, Y. Takiguchi, M.Arai,S. Sugawara, J. Antibiot., 27 , 386 (1974). For a total syrrthesis of methylenomycin A, see:
R. M. Scarborough, Jr., A. B. Smith III, J. Am. Chem. Soc., 99 , 7085 (1977).
25. a. J. Hooz, R. B. Layton, J. Am. Chem. Soc., 9^, 7320 (1971) and references therein. —
b. G. Stork, J. D'Angelo, J. Am. Chem. Soc., 96_, 7114 (1974).
26. A. T. Nielsen, W. J. Houlihan, Org. React., 16, 1 (1968). E. J. Corey, D. E. Cane, J. Org. Chem. , 36, 3tr70 (1971).
27. a. For a recent review, see M. Tramontini, Synthesis, 703 (1973), b. Y. Jasor, M. Gaudry, M. J. Luche, A. Marquet, Tetrahedron,
3^, 295 (1977).
28. A. J. Manson, D. Wood, J. Org. Chem. , 32_, 3434 (1967). R. B. Miller, B. F. Smith, Tetrahedron-Jrett., 5039 (1973). G. M. Ksander, J. E. McMurry, M. Johnson, J. Org. Chem., 42 ,
1180 (1977). — F. Huet, M. Pellet, J. M. Conia, Tetrahedron Lett., 3505 (1977).
29. Jean-Louis Gras, Tetrahedron Lett., 2111 (1978).
30. J.N. Marx, James H. Cox, and Lewis R. Norman, J. Org. Chem., 37_, 4489 (1972). —
31. Early developmental work on this reaction by Brian Walker and Clint Melton (unpublished) is acknowledged.
32. H. J. Reich, J. M. Renga, and I. L. Reich, J. Am. Chem. Soc., 97 , 5434 (1975).
33. D. N. Jones, D. Mundy, and R. D. Whitehouse, J. Chem. Soc. Chem. Comm., 86 (1970).
34. G. Ayrey, D. Barnard, and D. T. Woodbridge, J. Chem. Soc. , 2089 (1962).

85
35. a. D. L. J. Olive, Tetrahedron, 34, 1049 (1978). b. Chem. Abstr., 88, No. 3, (197ff.
36. K. B. Sharpless, M. W. Young, and R. F. Lauer, Tetrahedron Lett., 1979 (1973).
37. T. Hori and K. B. Sharpless, J. Org. Chem., 43, 1689 (1978).
38. W. Nagata and M. Yoshioka, Yuki Gosei Kagaku Kyakai Shi, 26, 2-22 (1968); Chem. Abstracts, 68, 77327Y (1968). —
39. L. F. Fieser and M. Fieser, Reagents for Organic Synthesis, Vol. 1, John Wiley and Sons, 1967, p. 244.
40. R. V. Stevens, C. G. Christensen, W. L. Edmondson, M. Kaplan, E. B. Reid, and M. P. Wentland, J. Am. Chem. Soc, 93, 6629 (1971). = '
41. W. Nagata, M. Yoshioka, and S. Hirai, J. Am. Chem. Soc., 94, 4635 (1972). =
42. H. Gerlach, Helv. Chem. Acta., 55, 2962 (1972); Chem. Abstr., 78, 42894k (19721: = =
43. N. J. Leonard, H. S. Gutowsky, W. J. Middleton and E. M. Peterson, J. Am. Chem. Soc., £4, 4070 (1952).
44. Sara Jane Rhoads, J. C. Gilbert, A. W. Decora, T. R. Garland, R. J. Spangler, and Mary Jane Urbigkit, Tetrahedron, 19, 1625 (1963). ~
45. J. L. Burdett and M. T. Rogers, J. Am. Chem. Soc., 86 , 2105 (1964). ""
46. M. T. Rodgers and Jane L. Burdett, Can. J. Chem., 4|, 1516 (1965).
47. G. W. Wheland, Advanced Organic Chemistry, 3rd Edition, John Wiley, New York, 1960, p. 680.
48. G. Allen and R. A. Dwek, J. Chem. Soc.,(B), 161 (1966).
49. B. 0. Chains and J. A. Challis, The Chemistry of Amides, J. Zabicky, Ed. Interscience Publishers, New York, 1970, p. 816.
50. M. J. Cook, E. J. Forbes, and G. M. Khan, J. Chem. Soc. Chem. Commun., 121, (1966).
51. E. Oliveri-Mandala, Gazz. Chim. Ital ., 52 , 107 (1922); Chem. Abstr., 16, 2112 (1922]^ ~"

86
52. L. Bouveault, Bull. Soc. Chim. France, 9, 368 (1892).
53. R. Sandler and W. Karo, Organic Functional Group Preparations, Academic Press, New York, 1972, p. 320.
54. L. Tsai, T. Miwa, and M. S. Newman, J. Am. Chem. S o c , 79, 2530,(1957). =
55. G. A. Olah and J. A. Olah, J. Org. Chem., 30, 2386 (1965).
56. R. F. Nystrom, S. W. Chaiken, and W. G. Brown, J. Am. Chem. Soc., Zl, 3245 (1949).
57. H. C. Brown and K. Ichikawa, J. Am. Chem. Soc, 83, 4372 (1961).
58. H. 0. Brown, Hydroboration, W. A. Benjamin, Inc., New York, 1962, p. 245.
59. H. C. Brown and B. 0. Subba Rao, J. Am. Chem. Soc, 78^,2582 (1956)
60. E. L. Eliel, V. G. Badding, and M. N. Rerick, J. Am. Chem. Soc., 84, 2371 (1962).
61. H. 0. Brown and P. Geoghegan, J. Am. Chem. Soc., 1522 (1967).
62. R. M. Scarborough, Jr. and Amos B. Smith, III, Tetrahedron Lett., 4361 (1977).
63. D. Liotta and H. Santiesteban, Tetrahedron Lett., 4369 (1977).
64. H. J. E. Loewnethal, Tetrahedron, 6, 303 (1959).
65. G. B. Magill, et. al., Cane Res., ] | , 960 (1956).
66. R. L. Ranieri and G. J. Calton, Tetrahedron Lett. , 499 (1978).
67. Dr. Amos B. Smith, III, University of Pennsylvania, Philadelphia.
68. T. Hori and K. B. Sharpless, J. Org. Chem., 43 , 1689 (1978).
69. R. Kaiser and D. L. Hopper, Mol. Phys., 8, 403 (1964).
70. G. Klose, Mol. Phys., i, 585 (1963).

APPENDIX A
Schematic Diagram of Pyrolysis Apparatus
87
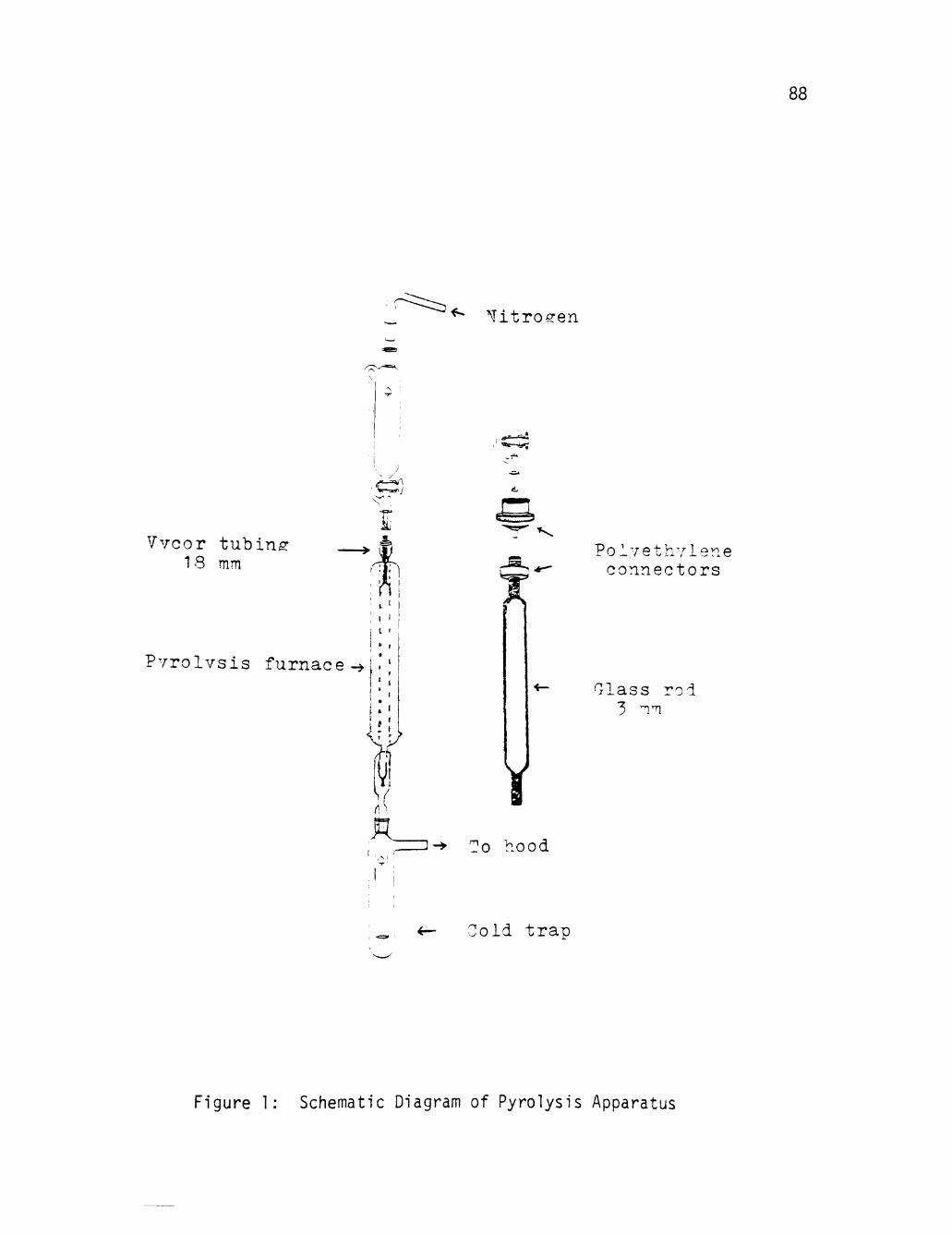
^ ^Titro^en
88
Vycor tubinsr 1 8 mm
\
Pyrolvsis furnace-»L'
; '
/I V
^-^ To hood
P o l y e t h y l e n e c o n n e c t o r s
Glass rod 3 nm
!old t r a p
Figure 1: Schematic Diagram of Pyrolysis Apparatus

APPENDIX B
Infrared and Nuclear Magnetic Resonance Spectra
89
90
KO CO
01 c o ta
c (U
o u >> o
*>» sr <u c <u
Hj CO (U
c N c
CQ t
C\J I
>» o
4->
£ o .a ^ ta o
I c\j
o
3
O cu Q. to
11 11 11 11 11 11 11 11 11 11 11 11 11 11 11 I I
I I I I I I I I I I I I I I I I I I I I I I I I I I I I I I I I I I

91
. 0 1
01 1
9 00 '
o
o •d"
a.
o
12 a.
1-
o I " '
1
•6
0
o
^q r
O "00
U3
o e ta + j c O) Q. O
O
(V c tu
cu CO QJ c a;
O) CO
I CM
I >> X o
o s_ fO
I C\J
o
N
o U3
3 s_
-(-> (J cu a. C/)
c>o
OJ
CD

92
C30 CVJ
CU e o c: fa + j
<u Q -O CJ
u o c: (a >5
c_j I
I > X o O)
o -Q S -<tJ I
C\J
f -o
3
4->
u CD Q . to
OJ i> 3
cn

93 ^ 1 A^
I-
— o
a.
. _ 9
J
l5
-
o
s^.
0 0
OJ c o c +J c <u a. o >> u o ta
O I
CO I
>> X o
4-> <U E O
& . ra o
I C\J
^-o
N
o U3
o
3 & -O <U Q . to
^
j 9
. .
cu s -3 CD
I
4
o 00

94
-M •r—
3 i -
S 4J
C o
• r --f-> 03 N
•r—
s. sa £ o
a cu a . to
cn 1—t
O) -c: + j
+J ^f-3 fO 4->
o 4-> CU
1
o c cu
4 -
o c o
• r -CJ1 cu s-
1 E CJ
o O O
CU u
vo r—
1 c o ta o
-a cxD c: cu a. CU a
. . VO
CU ^ 3
r ^
C •r—
CU L. 3
4-> IT3 S.. CU a.
a> £ • r~ cu
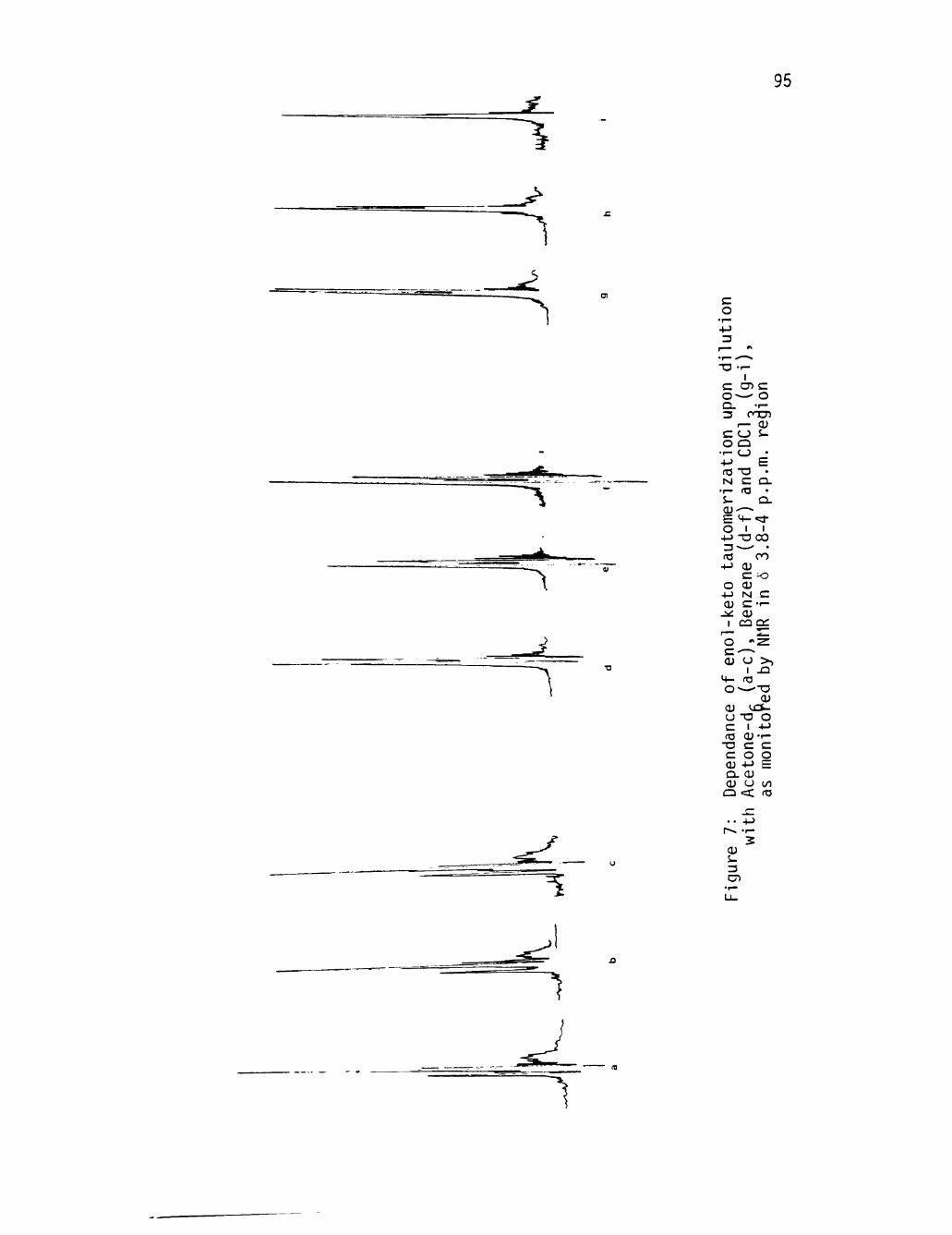
95
>
^
^
_ ^ ,
1
4-> 3
•r— --o •
c o-Q . 3
n
' • -
O) c o
' r -
c^ncn
C O O Q
" F " 1
+ J o
fo "a M
• r—
s-<u-
c (O
cu S-
. E
. Q .
. £2-
E M-«^ O 1 1
+ J T 3 C a 3 -rtJ
4->
o 4-> CU
.:<i 1
1
O C ' cu
M -O
CU
1
cu •
CO
C K3 (U N
c cu
c • r -
CQC2::
A
CJ
ta
.^— Z
J 3
-a cu
U 3 -
u -o c ta -a c: CU
1
cu c o 4->
Q . CU CU u
Q <
r>.
cu S-3
^ + J
i C7)
o 4-> •r—
c o E (/I ta
1
1

96
CVJ
2S u
CD
z D Z
g<
CU sz o e (T3 +J £= (U o. o CJ
u o e fO
I
ro >•>
X o
. e +-> cu E O
J 3 03
CJ I
CXI
03
CU
CU E CU
LU
< + -O
3 i -+J U CU O L to
QJ
CJ)
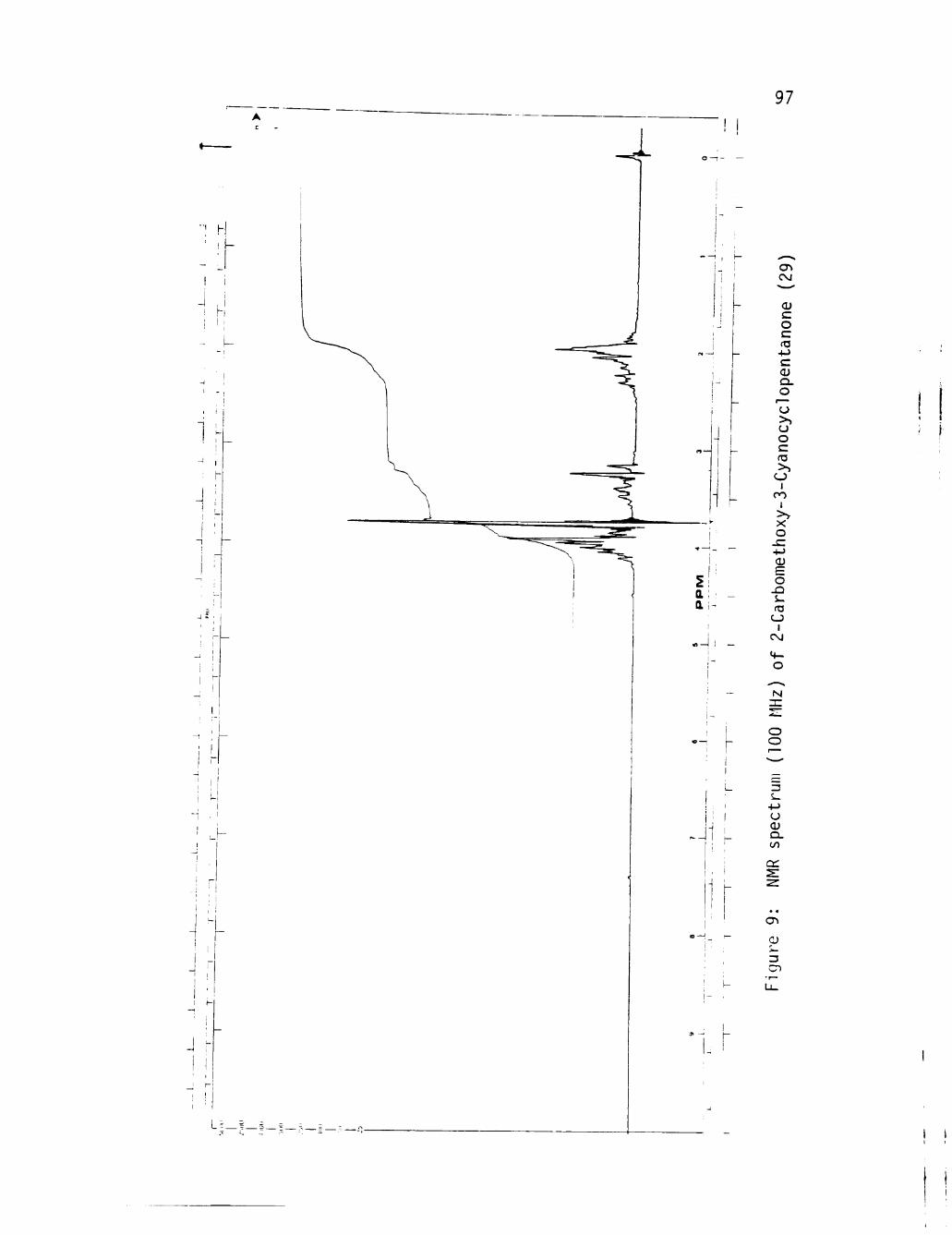
97
hi
J
i 1
^ 1
CM '
( ) j 1
n —
i cn C\J
cu
o c 03 4-> e cu CL O
_ 1 —
r CJ >> u o
1
-Cya
n
c^ >v X
1 ^ O ! ^
' - " - - 4-) ! • I "
- ' £ 1 o ^ - •£ a i- 03
C_)
M — 1 1 1—
**-O
N
r o o
h
3 i_ 4-> CJ O) Q. to
cn
J r
r
^
cn a 3 cn
r

98
•2. U
UJ IS 2 3 Z Ul
>
t
C\J
cu c o
ta
cu Q .
o CJ
O
03
C ^ I
CO I
o
i t l i t I t t t t
I I
!
I
1 1 1
if
1 1 1 : 1 1 I: 1 1 1 : 1 f' !' ' ' f' !' ' ' f' !' ' ' !' i i I I i i I I i i I I i
o JZi s.. 03
CJ I
eg I
toj « j |
«f-o
3 i-4-> U cu Q . to
cn
cu 3
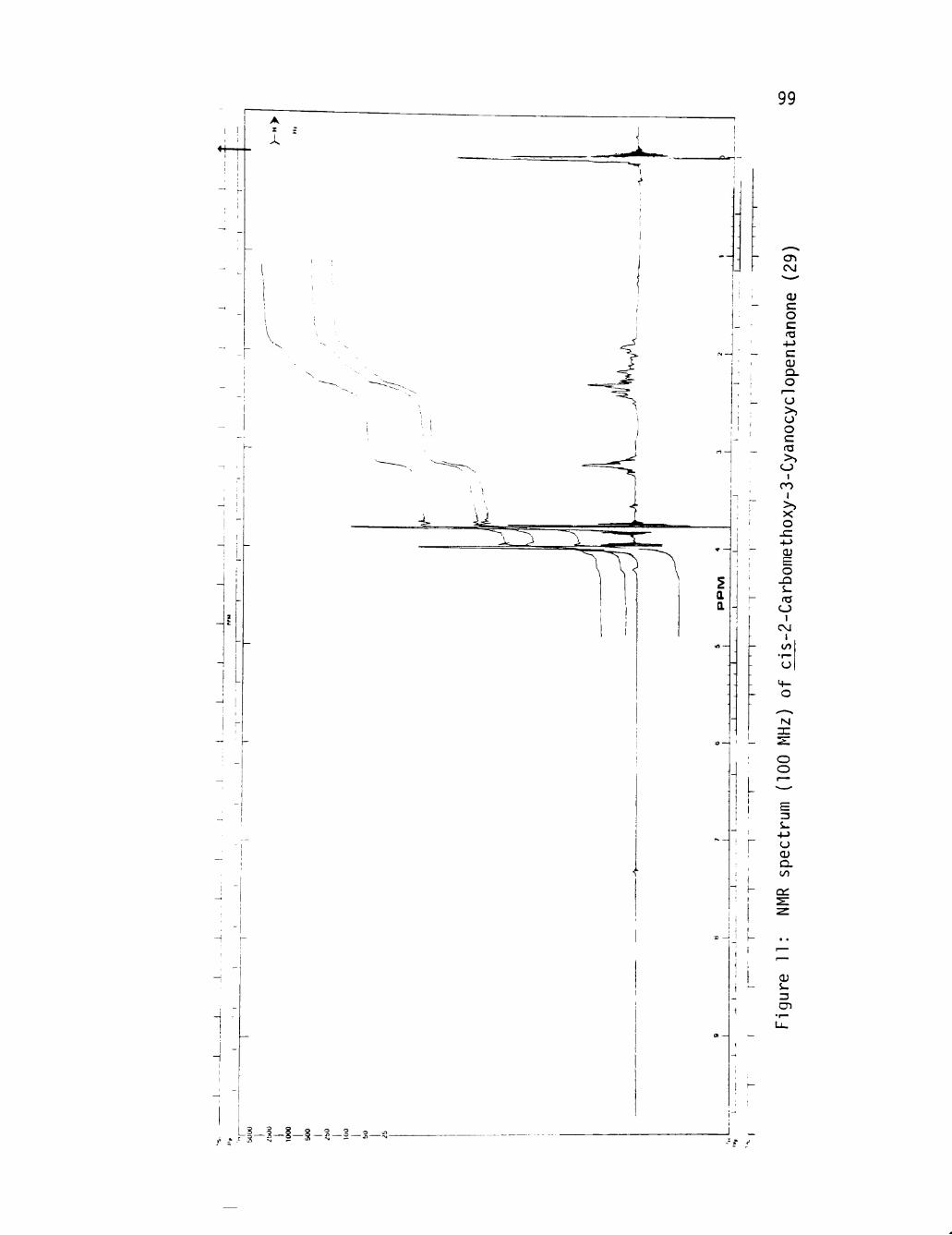
X
99
=i=
r
h
cn CM
cu c o 03 +-> e cu Q. O
U > a o c 03 >>
CJ I
CO
I
X
o (U E o
JZi i -03
C_J I
CM I
to
CJ
4 -O
N
O O
E 3 i-
4-> CJ
cu to
CU i -3 CD
-I
•5 5 S ^ : i — Jl y -

100
! 2
01
z
S < 3
CM CO
CU c o c 03
c CU O L O
CJ > CJ o c 03 >»
CJ I
CO I
cu cu
4-> cu E >^ X o i -
>> I
CM
M -O
03 +-) cu
cz cu
o
3
+-) u cu to
CM
CU S-3 CD
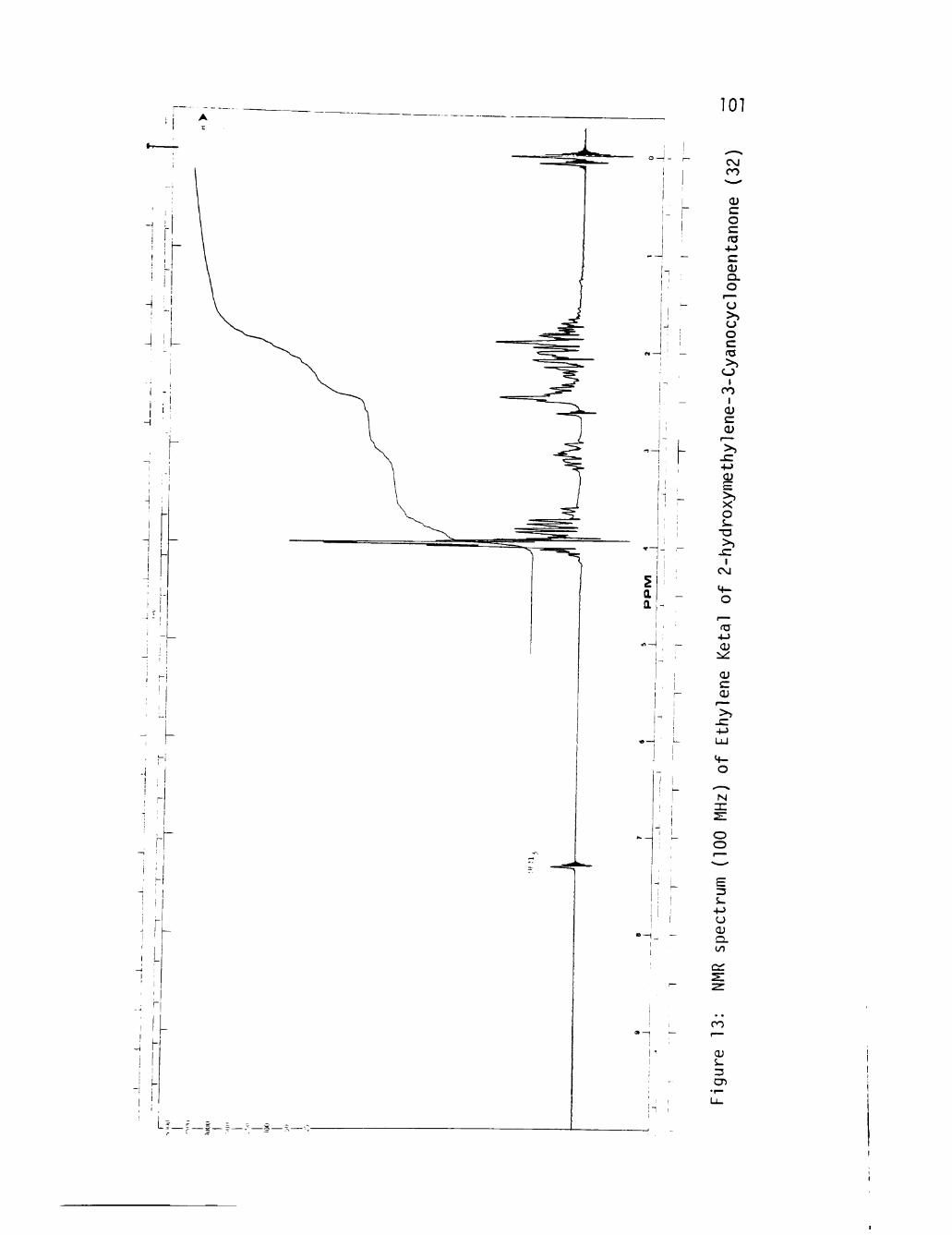
101
CM
cn
cu c o 03
-M e cu CL
o CJ
> u o c 03
C_)
CO I
0)
cu
+-> a; E > X o S-. -a >
. E I
CM M -O
03 +J <U
i ^
OJ
<u
O
N
O O
CJ <u Q. to
cn
cn
3 cn

102
(Q
CM LD
CU X 3
c o c 03
3 O CX
+-> E c o cu a a.—' o r— zc CJ o > , 03 CJ S o 03 (U >^"a
CJ t 3 I 03
CO I +J
>^ =5 X o O - C
(U 2 E 0 - —
JD JO s_ 03
CJ CM CM
CO 4 -O
03
<U
-a e 3 o Q. E O u
CU ZIZ f— o > , 03
4-> UJ T3
<U
<+- -a O T3
03 O JZ
•r- 4-> +J -r-U S 3
-a-—> <U 03
•• zc ^ CQ
CU
3 -(-) CD'i -

103
u a: UJ oo 2 3 Z
<u c o c ta -M c: cu o. o u >» o o c ts E
CJ o I ^ -
CO o I s>
<u o C I —
cu .c r— CJ >»
^ c 4-> - 1 -(U I JZi
CM O 03 _ <U
•M 03 U CU Q . to
CO
cu 3 C7>

104
o • r -
03
to
" ^
CU 4-> <+-03
I _<^
_ ^
(0
c cu o 0 +-> C 03 03 .—
4-> f— E •!— CU +-> Q . to
°^ ^ i -
I-CJ "-^
CO ^ cu E <+-cu o
.E M +-> n: cu 2:
2 : 1 o
CM O <+- C I . o _
' - ^ 3 M S-
nz +-> 2: u
OJ O Q . CO (/)
cc: E 2: 3 2 : S-
+-) -—V CJ J3 (U Q.
03
CO
cu S-3 CT)

105
cn
CU E O
CJ 03
OJ 4-> CU
i^
«+-o E 3 S--M CJ CU CL to
cn
<v 3 cn

106
I L
1 1
1
A
r:
i I
k-
cn
cu E O +J
u 03
03 +.> CU
^-o
N
o o
o <u to
1 1 I 03
CO
Lh cu s -3 cn
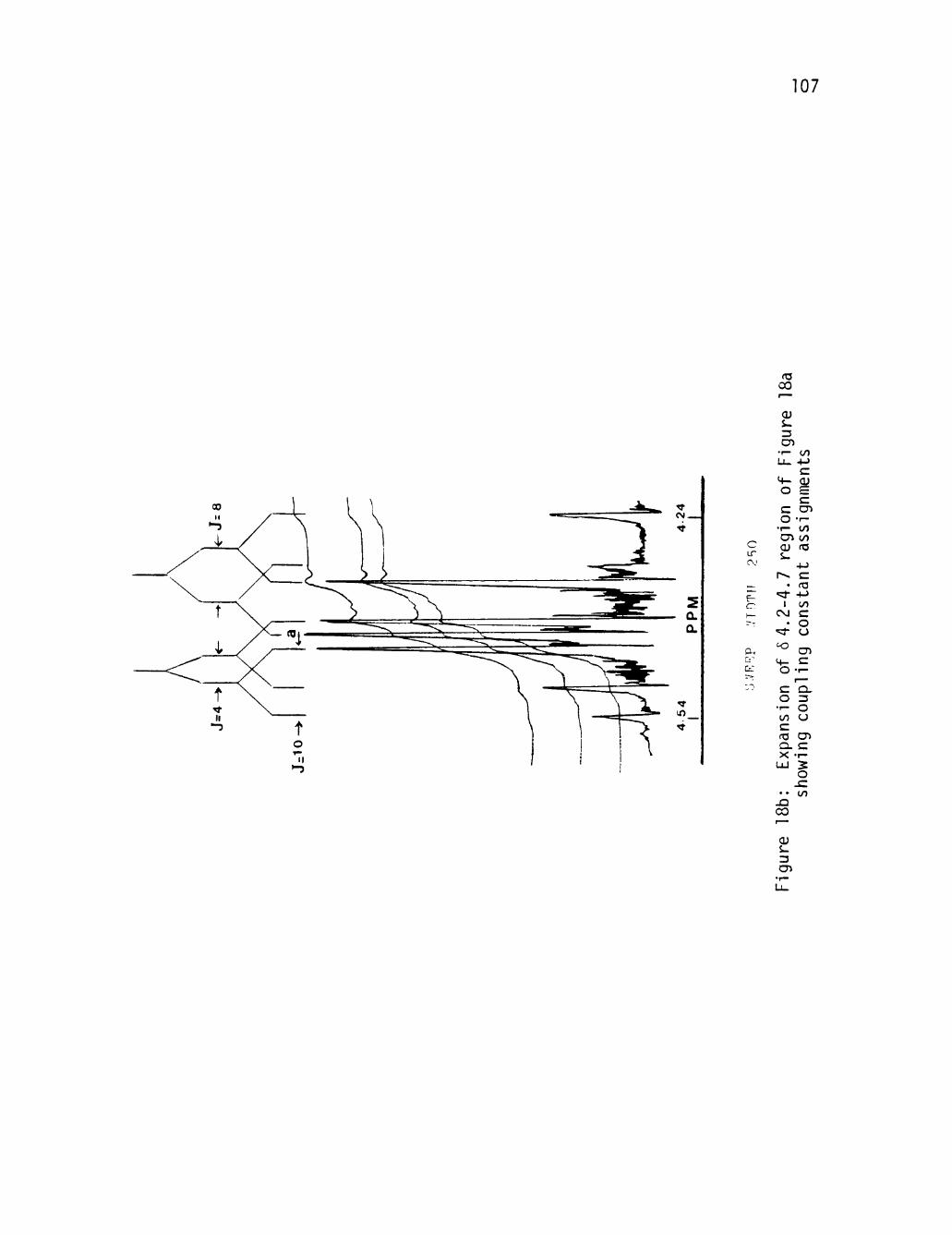
107
<„' LT cv
, f^ r-1
r-
P-. t~ U-
-_~!
ta 00 '—
cu s-3 as
•1— to U_ +J
E ^— gj O £
E E cn o -r-
•1— to cn to cu 03 i .
+ j r«» E
• 03 « ^ -t->
1 to CM E
• O ' ^ O
' ^ C7i H- C O - 1 -
^ " E CX O 3
•r- O I/) CJ E 03 CJ) Q . E X • ! -
LU 2 o x:
• * to JZl CO '— cu s_ 3 cn

108
o
<u E o
4-> CJ Ol
o +J <u
t f -o
3
4J U CU Q. to
cr>
CD
s -3 CT>

I
109
- \\- J
o LO
CU E O 4-> CJ 03
O +-> CU
N
o o
£ 3 S_ 4-) CJ OJ Q . to
o CM cu i -3 CJ)

no
o LO
<U E O
4-> CJ 03
Z
O 4-> (U
O
• a cu
03 Q . CU i .
CJ
o .^ S-03
CyO
4 -O
3
4->
u O) Q . to
cn
CNJ
<u S-3
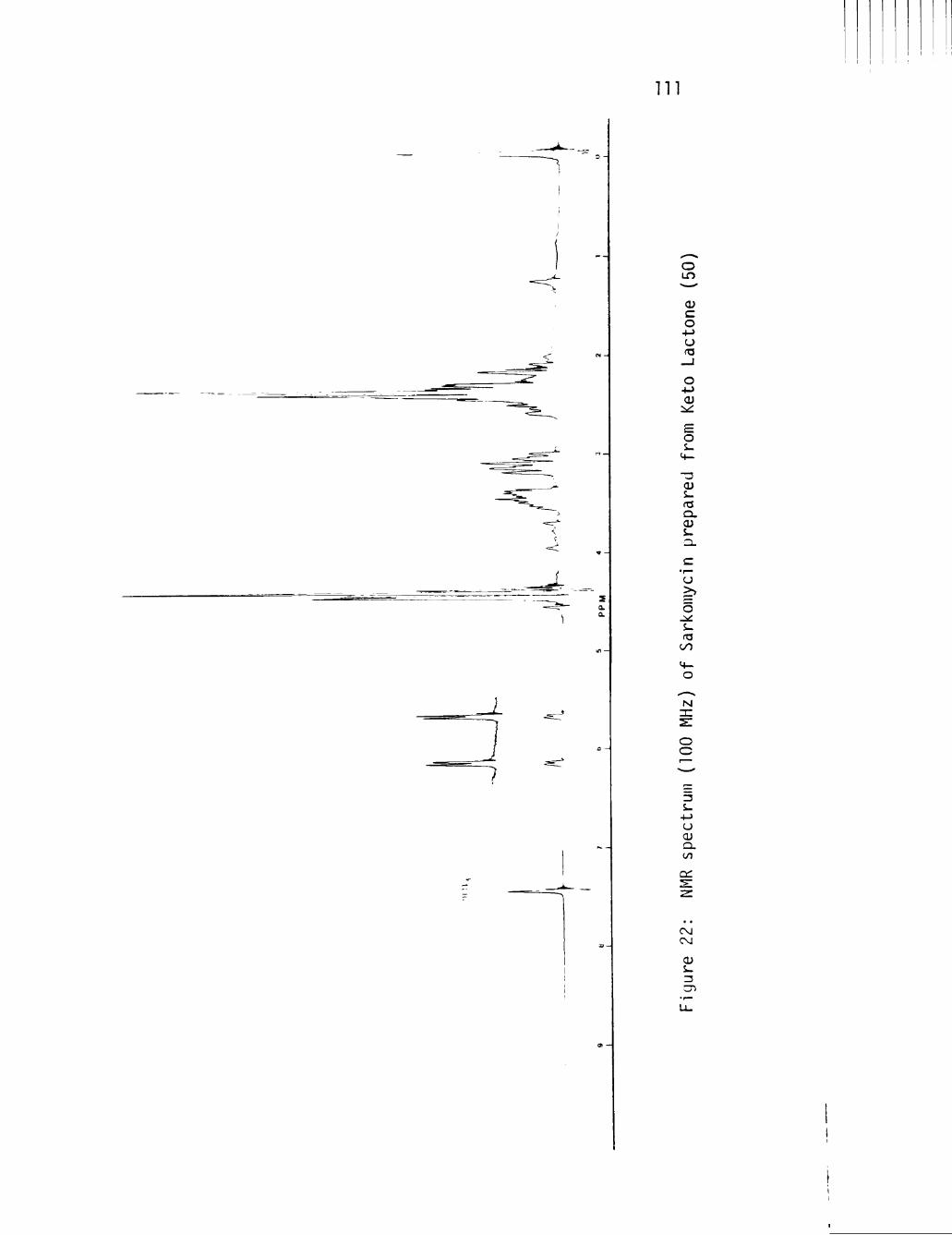
Ill
_JL
~1
o LO
OJ E
o u 03
O
CU
o
cu s -03 Q . CU s -
CJ
>> o i -03
C>0 t ( -
o
N
O c
3 S-+J CJ cu Q . to
QC
CM CM
CU

112
CO I
03
Q . O
T3 >^
- C 03 i_
-M CU
f— I
o cu E CU >,CM
JE LO + J V — CU
s: (u I E
CM O E
M— 03 O 4->
E I— <u 03 d.
-M O CU I—
>>^ CJ
>^ cu u E O <U T3
r— •!— >, E -c: < 4->
<4-O
E 3 L.
CJ (U Q .
CO CM
cu s -3 CJ)

113
'-1i -
I CO
03
i . CL o t . -o >>
JE 03 4J CU
I— I
o I
cu E <u
+-> cu
CM O J LO
^ ' o
cu I— E 03 O
• M E cu 03
i^ 4-> E
OJ (U E CL CU O
'>)'o - C >> 4-> O LU O
• a
O E <c
o o
CJ
cu
a. to 1 L cn
CM
cu s . 3 cn

114
2 2 o
5 z UJ
> ° 5
O CO
cu E o E OJ
+J E CU Q . O O
u o -a
<: I
CO I
>^ X
o - E +-> CU
o JZt i-ta
CJ I
CM • + -O
03 +J CU
cu E (U
<4-O
i_ +-> CJ cu CL to
LO CM
CU
3
cn

115
! J 1
. - I
o
q 00 '
o
9 9
— o J
O CO
<v c o E 03
CU Q. O
CJ
U O
13
I CO
I >^ X o
JE 4-> CU £ O
J2 S.-03
CJ I
CM <^-O
o •6'
.9 03 4-> CU
1 ^
a.
9 i n •
.a. a.
9 " in
CU E
CU
'>) -E
t ( -O
9 fi'
o
N
9 o
J _ O
o CO
3 S_
CJ O) Q . to
CO CM
OJ i . 3 CD
9 9 00

116
2 2 u
2' 3 Z UJ
CO LO
u OJ
u
u
(0
o -^ o -
a: 33
5 3
z LU ^ > s< S5
"
o r
3 3 3
3
>
> i X o
JZi ^ 03
CJ 1
fV 1
^ i ^ ^ i ^
r—
>> E 03 i . >) Q . O 4-) S- 03
T3 CU >> E
- E rT3 - ^ S- J3 +-> CU
1— 1 CO
O 1— — - C J cu n : E CJ cu
>— E >>-^
<U 03 2:
I CM
S-+-> u cu CL to
cn
CM
<u
3 cn

117 "1 I
I I
u
1 a.
r
r
U S -
CO LO
u 03
a
X o J 2
03 C J
I CO
I
E 03 &->^ Q. O
" 3 >
- E 03 S .
- M CU
h-I
O
CU E CU
'>> - E 4-> (U
; T -
1 CM
O
M
O O
3 S-
+-> CJ CU CI.
us cn
CO CM
cu
3 cn

118
LO
CU E O E 03 4-> E CU Q . O
O > u o -a
I C O
I cu E cu
I CM
<+-O
i -
CJ <u a. to
cn
as CM
OJ 3
cn

119
LO
cu E O E 03
+J E CU £3-O
I— CO U I— >>CJ CJ Q O O
• o •r- E £ -r-
<c CO J 2
I CU E E CU O
CU
s CM
*+-o
^—^ M
2 :
o o r—
• •
E 3 S-
+ J CJ (U CD. to
cn
o to
O CM Q
1 r— C J Q
CD • o
1 CL) E O 4-> O) CJ
< E
• r -
03
o CO
cu S-3 CJl

120
I CO
I
221 o
2
i!
E 03
£1. O
-o >» 03 s> 4 J cu
I — I
o cu E CU
+ j cu I
CM
S- CO <+- LO
- a cu "O S- - r -03 CJ a . 03 cu S- CJ CL-r-
-—- o JZl
E i -•r- 03 O CJ
i o .^ S-03
<>0
<+-o
3 i -+J u cu Q . to
cc
cn O) i -3 cn
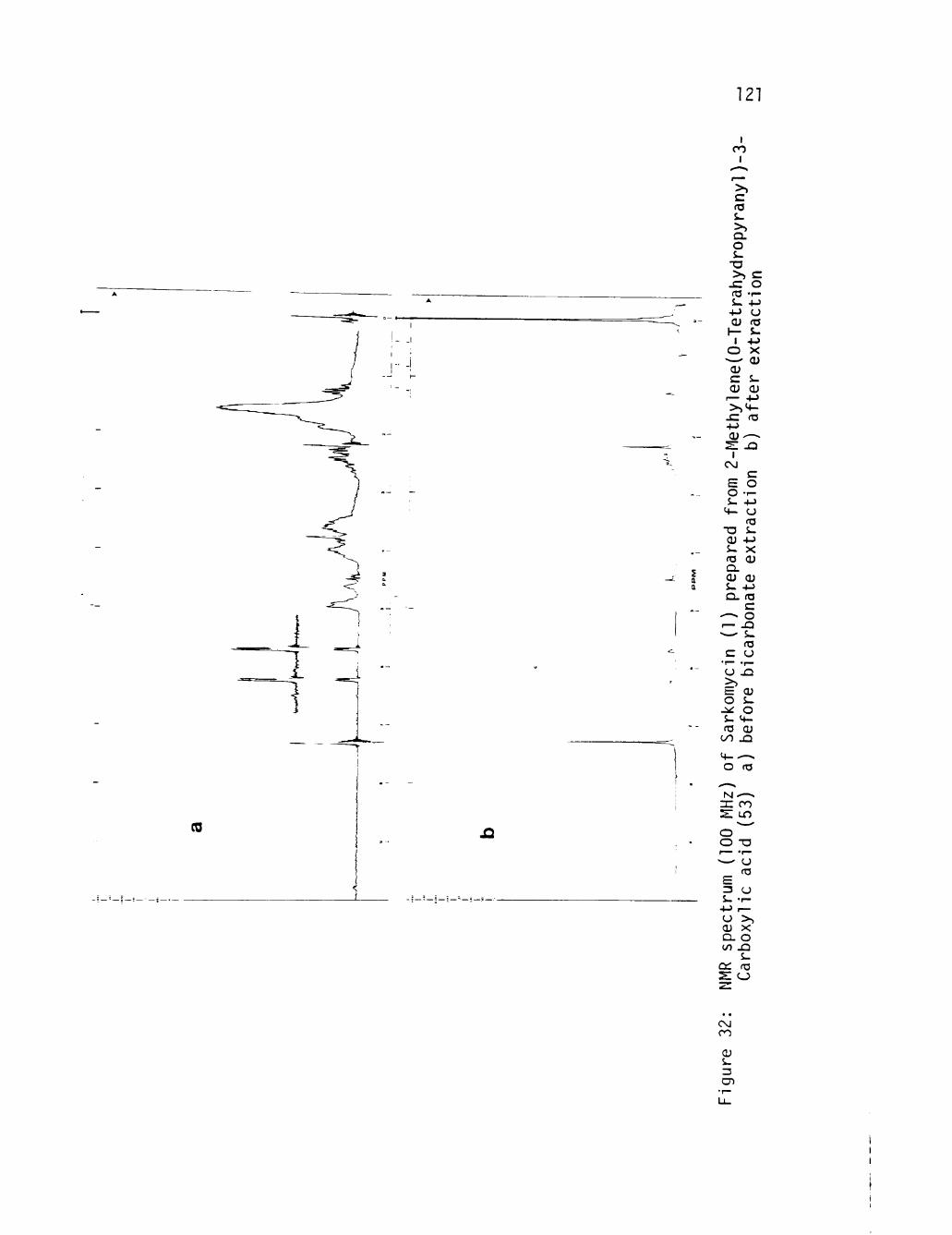
121
I CO
I
X
03
>> a. o s~
• o >» E
Jd O 03 - r -i - +J
4-> CJ CU 03
h - S-I 4J
O X v - ' O)
<u E S-<u CU
r— +J > ^ ^
x : 03 -t-> < u - ^
s : JO I
E £ O O • . -i - -M
M- O 03
• a i -cu +-) i - X 03 CU CL CU cu 1 - -M
E - - ^ O <— JZi ^-^ s -
03 E CJ
CJ J2 >> fc CU O i -
Ji^ o
03 (U CyO JD
4 - . O 03
: r CO 2 : LO
o o -a —' u
03
CU X CL O to J3
S-ci: 03 2 : CJ
CM CO
CU
as

APPENDIX C
Publication arising from this work
Submitted to Tetrahedron Letters May 25, 1979 Accepted for publication July 20, 1979
122

123
2-Carbomechoxycyciopencenone as a Synthon. Synthesis of Sark.onycin~
John N. M.ar-x* and Gevork Minaskanian
Departnenc oi Chemistry, Te/.as Tech '.niversicy, Lubbock, Texas 79409
'•.D ser act: A new synthesis of sarkomycin (II), a lompound -active igamst dscities-type tumors,
.3 reported. "treatment of 2-carbomethoxycyclopentenone (I) with St^AlC^; generated zne carbon
skeleton, selective functional ^roup manipulations then gave the Keto Lactone X, and acid-
ritalyzed eliminative opening of this gave sarkomycin.
Several years ago, we reported" the first synthesis of I-c3rbomethoxycyclopent-2-enone
vD 3 highly reactive molecule whicn is a potential synthon for prostaglandins and other
natural products. Ve now report its use in the first regiospec
'IL'>. an antibiotic which is also active against ascites tumors
atural products. Ve now report its use in the first regiospecific syr.tr.esis of sarkomycin 3, ~
0 0
(V""' &; Previouslv, sarkomycin has been synthesited by a :-'.ana'.ch condensation route' or. 3-car-
ioxvcvciooentanone. The reaction gives a 1:2 -.ixture of cr.e . jr.a :• isomers, in spite JC I' 5 . . . . ' i
the original clai.-s of regiospecir icity. The otner reported svntnesis is an mvoivea one
bv Shemvakin,' which gave sarkomycin semicarbazone, -..nich cculd -ot be v.-dro L;-ted vitr.cut .;
p o 1 yme r i za 11 r r .
Our orisinai route' to I-carbomettioxycyclopentenone CI) involved SeO, oxication o: 2- ]
^-.arbomethoxycyclopentancne (III), followed by trapoing tne I for^'.ec is its cvcioDent ic .ene \\
adduct, purification, and the pyrolvsis to give the nure compound. Reicn"' 'as s'.nce retortec |
m improved rvnthesis of I, ?asea on the ^neny Iseienuinvl C'^rivativ- I'., :it m ::r -cncs, :.e

124
product formed by the reported procedure is contaminated with substantial anouncs of the
starting compound (III) which appears to rie formed, nlong •-•ich PhSeSeP'^, by decomposition of
the desired compound IV. '.v'e have therefore improved the procedure as follows.
Treatment of 2-carbometnoxycyclopentanone (II) vith N'aH in THF gave the solid Ma salt
of H I . This was suspended in fresh dry THF under M^ and 1 eq. of PhSeCl in THF -as added at
-^ .Atter L5 min., the solvent was removed in vacuo below 25' and replaced witn CH-,C^-, tnen
excess 0 was passed in until saturated at -78°, followed by 0, to remove the 0 , tnen the
clear solution was allowed to warm to 25°, filtered, and the solvent removed to give almost
jjure I, which was used directly in the next reaction.
Although 2-carboraechoxycyclopentenone (I) pol\'merizes readilv with -r.any reagents," it
reacted smoothly with a slight excess of Et,AlC" in benzene. Purification by extraction of
linj product into aqueous MaOH to reverse some cyanohydrin formation and then careful reaciai-9
rication gave the cyano compound V, mp -7-<i8', (35-"0''< yield from III). The ketone group in
V -jas protected as its ketal, which was a £3_. 10:1 :Tiixture of two isomers, mp Jij-ao.S". Silica
gel chromatography allowed separation, in the last fractions, of the minor isomer, mp 72-7-^',
w;-'.ich was assumed to be cis, though N'MR did not allow assignments (cnemical shifts of C-2 and
C-3 H's overlap). The mixture was used in subsequent steps.
.Among a number of methods investigated to reduce the ester or -ivarolyze the nitrile m VI,
Li3H did reduce the ester selectively, but the ketal group was not stable to this reagent,
•'ovever, addition of . 2 eq. of H^O-MaOH per mole of LiSH, lave a reagent mixture, presum
ably containing Li3(0H)-,H^ as the major species, which effected ti-.c desired transformation
cl.janly in THF at room temp, to give the cyano alcohol VII.T [IR : 3500 br, 222'): :;::R
'TDCi ) : 1.0-2.7, mult, 5 H; 3.06, mult, 1 H; 3.80, d, J = 3, 1 H; 3.34, d, J = 5, 1 ti; 3.'-S,
, ^Hj.
Hvdroivsis of the cyano group in Vila with 5", aqueous N'aCH w,;S c-mpLete with:.-. I '••r ::
reflux. Aciaification gave tne protected keto lactone VIII, mp h --^^.-^ :s ;;ie onLv jroduct.
The hvdrolysis andoubtedly involves epimerization it 'J-3 ma ncig-ooring ^riup part icioati:-
•v/ the CH group, since tne tetrahydropyranyl ether derivative (Vllh) required 0 hr reflu:< vi::;
''••. N'.iCH to give tne amide UX) , mn 144-l-.3% and a rurchpr 24 hr refia/. to ;i/e :':e ic-.d X.
np 09-72'-
Removal of the i ctal group of VIII by brief treatment witn 5'. nCl gave trie eto .a.tone
>:. mp 45-46" [IR ('CHCl ): 1730, 1750 cm""; NMR (CDCl^): L.95-2.:'3, m, - H; ..i5, m, 1 H;
j.44, m, 1 H; 4.44, d, J = 5.5; 4.45, d, J = 4.0], This compound can be considered as a
"protected" and more lipophilic form of sarkomycin. Surprisingly, mild treatment of this
•.eto-lactone with 5-10% rfCl in ac£tone-H,0 gave slow eliminative onening to prcdace •-;
s.irkomycin (II),*"' [IR matcnes the published spectrum; N'MR " 1.5-2.5, m; o.nS, c, ; = 2.J,
H; 0.21, d, J = 2.5, 1 H].
On the other hand, acid treatment of the protected <eto hyaroxv icii (I ^ve r=movni -^z
':ie protecting groups and elimination o: H.O t.: give iarkomvcLn. T-.e co-.T .r.: iW > give--
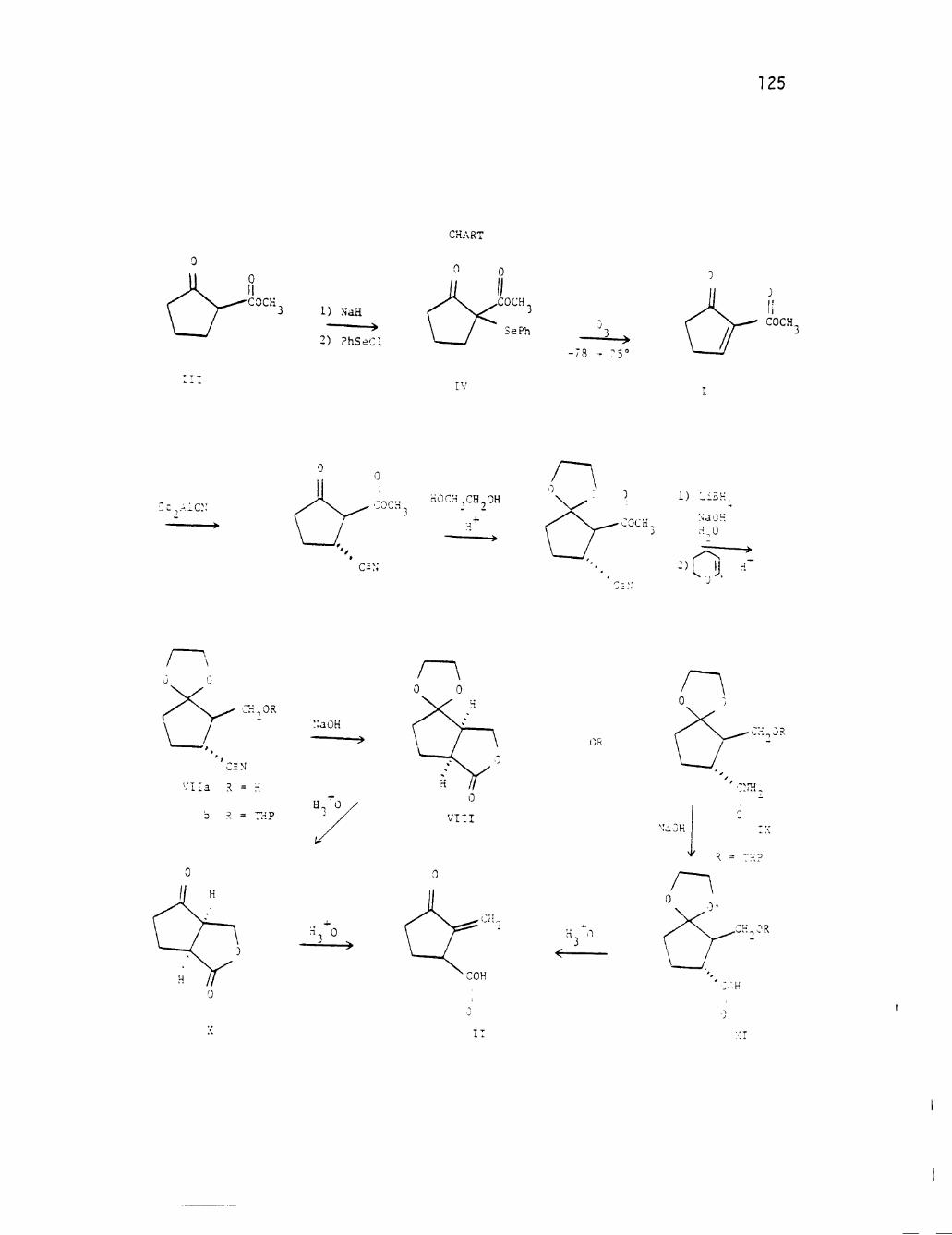
CHART
125
0 II COCH 3 1) N'aH
2) PhSeCJ &
COCH.
SePh
"8 - 2;
: i i IV
.-^ic:: HOCH^CH^QH
4.
\ ^
:ocH,
1) l i S H ,
NaOK H,0
2) o.
CH.OR
b R = THP
:;aOH
a, 0
S 0
COH
OR
H j O
r~\ rn -OS
;aOH
^ = T:-;?
r~\ :K,OR
I I

126
sarkomycin much faster and cleaner than does the keto-lactone X, and no trace of X could be
detected. This behavior confirms the stereochemical assignments to all intermediates in the
s-.-n thesis.
Modifications of the above scheme are being carried out to prepare several compounds with
structural similarity to sarkomycin.
Acknowledgement. Assistance in early stages of the project by Arthur w. Carnrick. Brian
•»aiker, and Clinc Melton is gracefully acknowledged, as is financial assistance by the .Robert
A. '.jelch Foundation.
References.
•3.
?res<:;nted at the .ACS-CSJ Chemical Congress, Honolulu, April 2-6, 1979.
J. .N'. M.arx, J. H. lox, and L. R. Norman, J. Org. Chem., }]_, -*c)9 (1972).
H. L'mezava, e_t . , Antibiotics and Chemotherapy, -_, 514 (19541; I. R. Hooper, et al. ,
loid, 2' 535 (1955) .
3. 3. Magill, et al., Cancer Research, 16. 960 (1965); for reviews, see 3. Goto and H.
.-jnnno, Chirvo, 38, 72 (1956); S.-C. Sung, Antibiotics, L^. .56 (1967); S. \ondo.
Antibiotics Jaoan Ser. 3., 15, 210 (1963).
N. Toki, 3ull • Chem. Soc., Japan, 30, ^50 (1957); 3. Umezawa and M. Kinoshita, 3ull.
Chem. Soc. , Jaoan, 30, 267 (1957).
R. H. Hill, ?,. J. Toley, and C. A. Gardella, J. Or^. Chem., 21- ^^30 (1967).
M. -M. Shemyakin, L. A. Shchukina, Z. I. Vinogradova, M. M. rColosov, R. G. Raduel, "j 3.
Shretosov, £. M. 3amadas, E. S. Chanan, -C. M. Ermolaev, and S. ?. SemKin, Zhur- Cbshcnei
Nhim., 27, 7 2 (1957).
H. J. Reich, J. M. Reuga, and I. L. Reich, J. .Am. Chem. Soc. , 97, 5424 ('1975).
Comoound III exists in th.e keto form in the crystalline state: IR.._ : 2225, I7~0, 1";0 xbr
cm~", but IS a ca. 1:1 -:ato-enol mixture m the melt or solution, new IR peaks at 1670
and lb25 cm"''"; NMR (,CDC1 ) I 2.1-3.0, 4 ;-:, mult; 3.5-3.7, mult. ^ . 1.5 -l; 3.36 anc 3.3i,
singlets, 3 H total; 10.3, broad s, C3_. 0.5 H.
-0. See discu.isions on iifficu3.ties encountered m a su.iilar reaction m ene syntnesis ::
Me tnvleno mycin .A: R- M. Scarborough, Jr., and A. 3. Smith III, JACS, i"^, I'85 i, I - ) .
11. Wit.l 5" HCl, opening required at least a week it 25'; witn 10% HCl, atout :ne c y was
required. Slow decomposition occurred under the reaction conditions, and a maximum
estimated vield of 50-60% of sarkomycin was produced under tne conditions investigati..






























