Embed Size (px)
Citation preview

PHYSICAL REVIEW B VOLUME 35, NUMBER 3 15 JANUARY 1987-II
Surface segregation in dilute binary fcc transition-metal alloys
M. C. DesjonqueresService de Physique des Atomes et des Surfaces, Centre d'Etudes Nucleaires de Saclay, 91191Gif sur Yve-tte Cedex, France
D. SpanjaardLaboratoire de Physique des Solides, Universite de Paris —Sud, Batiment 510,
Centre Universitaire d'Orsay, 91405 Orsay Cedex, France(Received 19 June 1986)
We present a tight-binding study of the surface segregation energy of transition-metal impuritiesin fcc transition metals. Various approaches to the self-consistent determination of the impurity po-tentials are discussed and their results are compared. When size effects can be neglected, the trends
followed by the segregation energy are qualitatively given by the difference of surface energies be-
tween the solute and the solvent species if electronic correlation effects are not too large. These size
and electronic correlation effects are finally discussed.
I. INTRODUCTION
It is well known that the surface composition of abinary alloy may differ from the average composition ofthe bulk. This surface segregation is a phenomenon ofgreat importance in metallurgy, chemisorption, andheterogeneous catalysis, for example. However, its studyis difficult both experimentally and theoretically. Indeed,it needs surface sensitive techniques (Auger spectroscopy,field ionization microscopy, ultraviolet photoemissionspectroscopy, etc. ) and moreover it is sometimes difficultto reach the thermodynamic equilibrium. Finally thesamples must be free of undesirable dissolved impuritiesand the experiment should be performed in goodultrahigh-vacuum conditions since the presence of impuri-ties or adsorbed gazes can modify drastically the surfacecomposition. Nevertheless, many experimental studies'have been devoted to this subject but they led sometimesto controversial results. Furthermore, it is not obviousthat the segregating species should be the same for thewhole range of composition of the alloy. For example, ithas been recently shown that in the case of a Cu Nilalloy, the surface is enriched in copper for 0&x&0.84and in nickel for x) 0.84.
On the other hand, various theories have been set upbased on phenomenological interface energies or pair po-tentials' ' on macroscopic effects (elasticity)' ' ' and onmicroscopic electronic approaches. " In this latter case,the study of very dilute alloys (isolated impurities) is avery interesting starting point since one does not need toaverage over configurations and the only quantity of in-terest is the solution energy of the impurity as a functionof its distance to the surface. ' However, this problem isnot as simple as it seems since one must cope with thequestions of charge transfers and self-consistency whichactually exist also for concentrated alloys.
In this paper, we will adopt the electronic approachstarting from the tight-binding approximation. We wi11
first briefly review the various assumptions which can bedone to deal with the problem of self-consistency in the
calculation of the solution energy of a transition impurityin a transition metal, discussing their range of validity. Inparticular, we will illustrate, on the case of a linear chain,the consequences of these various assumptions. We willthen consider the segregation of a transition impurity at(100) and (111) faces of a fcc transition metal. Even in avery dilute alloy the concentration of impurities at thesurface can be finite. In order to investigate the role ofthe concentration of surface impurities we will also con-sider the limit in which the surface plane is made only ofimpurities [monolayer of transition metal atoms on the(100) face of a fcc transition metal]. Finally, we will dis-cuss the influence of electronic correlations and size ef-fects.
II. MODELS
The segregation energy of a single impurity can be writ-ten as
Esegr =Edis, surf Edis, bulk
where Ed;, ,„,f (b„lk) is the variation of total energy when asurface (bulk) host atom is replaced by an impurity atom.Let us neglect any 1attice relaxation around the impuritysite and first consider the one-electron contribution at 0K. Actually the replacement of a host atom by an impur-ity produces a change in the one-electron total energysince the potentials of both atoms are interchanged and anadditional perturbation potential, different at the surfaceand in the bulk, is induced in the vicinity of the impurityin order to screen the difference of charge between it andthe host metal. Since here we will only study the case oftransition impurities in a transition metal, we will neglectthe effect of s and p electrons and describe the valence dband in the tight-binding approximation. In fact, the con-tribution to the segregation energy of the s-p electronsshould be small due to their delocalized nature. In thetight-binding framework the total perturbation potentialdue to the impurity leads to a modification of the effec-tive atomic levels and possibly of the hopping integrals.
35 952 1987 The American Physical Society

35 SURFACE SEGREGATION IN DILUTE BINARY fcc. . . 953
This last effect will be neglected in the main part of thispaper but will be discussed in Sec. V B. Such an approxi-mation is valid when the extension of the orbitals of theimpurity is comparable to that of bulk atom (i.e., for com-parable bandwidths of the corresponding metals, in thesame crystallographic structure). Furthermore, we as-sume that the perturbations of the atomic levels due to theimpurity do not extend beyond the first shell of neighbors.Under these conditions the expression of E„g, becomes
EE„s,——f E 5n (E)dE 5Ec,„—l, (2)
where 5n(E) is the variation of the total d density ofstates between the systems in which the impurity is at thesurface and in the bulk. 5EC,„& is the variation of theCoulomb energy between these two systems which hasbeen counted twice in the integral and is given by the ex-pression'
5EC,„I——g ZI5 Vl + , 5ZI5—VI,I
(3)
where the I indices are a given orbital on a given atom, ZIis the corresponding filling, 5V~ is the variation of the cor-responding effective orbital energy, and 5Z~ is the varia-tion of charge due to 5VI. If we assume that 6VI is in-dependent of the symmetry of the considered orbitals, theexpression above becomes
5ECoul ZIB( VIS VIB)+ 2 (ZIS ZIB)( VIS VIB)
+ZMS( VMB VMS )
+ T(ZMB MS )( VMB VMS )+5 Coul rois (4)
E[n (E, VI ) no(E)]dE =Z—I —ZM, (5)
where n (E, VI) and no(E) are, respectively, the total den-sities of states of the matrix with one impurity (with po-tential VI) and with no impurity. ZI and Z~ are thenumber of d electrons of the impurity and the host.
This variation of density of states is related to the totalphase shift rI(E) by'
where Z» (rs) and V» (rs) are the charge and the potentialof the impurity in the bulk (surface). Similarly, ZMB lMsland VMB ~Ms~ are the charge and the potential of an atomof the solvent in the bulk (surface). 5Ec,„l „„,is the varia-tion of the Coulomb energy of the neighbors around thebulk and the surface sites successively occupied by the im-purity. Although it can be derived straightforwardlyfrom Eq. (3), its expression is lengthy. We will give it ex-plicitly in the following when we will assume the localneutrality.
The self-consistent determination of the potentialaround an impurity in a metallic matrix is a very oldproblem. ' ' In a tight-binding scheme, several approxi-mations can be done. In the most common one, the per-turbing potential is localized on the impurity site only andit is determined in order to satisfy the Friedel' sum rule(model I). This ensures the global neutrality of the systemwhich can be written as
n (E, VI ) —no(E) =—1 dr)(E)
with
rI(E) = —arg det(1 —Go V),
where gq[z] is the total phase shift when the impurity is inthe surface (bulk) and the global neutrality condition [Eq.(5)] can be written as
1 rl(EF—)=Zr —ZM .'ij
(7b)
This model has been used for the calculation of segrega-tion energies by Dreysse and Riedinger and by Masuda-Jindo. ' However, in their expression of E„g, theCoulomb term 5EC,„& is missing. According to our calcu-lations this latter term is very important and can play theleading role as we will discuss later (Sec. IV). This model,which has the advantage of being extremely simple and toallow Friedel charge oscillations, sets some problems.Indeed these charge oscillations should induce potentialoscillations which are neglected. Therefore, as we will seebelow, the local variations of charge are overestimated,especially when ZI —Z~ is large.
Since it is known that the charge transfers in metals arerather small' (less than 0.1 e/at. ), another possible ap-proximation can be made. First one calculates the impur-ity potential by assuming a local neutrality of this impuri-ty, then the contribution of the substrate to the segrega-tion energy is computed in a non-self-consistent way byallowing a sma11 variation of the Fermi level in order toensure the global neutrality (model II). This kind ofmodel has already been used, for example, by Simonet al. ' to evaluate the interaction energy between an im-purity and a stacking fault in transition metals. In thisscheme one writes the segregation energy as a sum of twocontributions. The first is due to the perturbation of thelocal density of states at the impurity site at the surfaceand in the bulk and can be written as
EFE.'.„=f E[nls(E, Vls) —.MS(E VMS)]dE
E
f E[IIIB(E,VIB) nMB(E, VMB )ldE
—ZI [( Vls —VMs) —( VIB —VMB)]
—(ZI —ZM)( VMS —VMB) .
The second is due to the perturbation of the remainingpart of the system and has the form
EFE„s,——f (E EF ) 5nII (E)dE . — (9)
In these formulas n &s (»] and n Ms [MB j are, respectively,the local densities of states of the impurity and the matrixat a surface (bulk) site. 5nII is the variation of the totaldensity of states of the remaining sites between the sys-
where Go is the unperturbed Green operator and V is theperturbation potential. The first integral term in Eq. (2)becomes
E E
f E 5n (E)dE = ——f [r)s(E) rIB(—E)]dE, (7a)

954 M. C. DESJONQUERES AND D. SPANJAARD 35
tems in which the impurity is at the surface and in thebulk. It can be calculated with the phase-shift method[Eq. (6)]. However, the accuracy of this model may be-come questionable when Zz —ZM is large since the varia-tion of the Fermi level necessary to ensure the global neu-trality can no more be considered as small.
Finally one can adopt another point of view. Since thescreening length of an impurity in a metal is rather short,it is well justified to neglect the perturbation of the local
densities of states on sites beyond the first shell of neigh-bors. If we now assume, as previously, that the chargetransfers are small, one can calculate the perturbing po-tentials at the impurity site and its neighbors by a localcharge neutrality condition (model III). With these as-sumptions the expression of E g is unchanged save forthe fact that now n&s and n&B are also a function of theperturbing potential on the neighboring sites. E„g, be-comes
IJEsegr =
1: nearest neighbors
of the surface impurity
Eon'(E, Vts Vi's . . )dE —ZM Vis
J E one's(E, VtB, . . . , VJ g, . . . )dE ZM VI—BI: nearest neighbors
of the bulk impurity
(10)
We have already shown that a similar model can ex-plain successfully the general trends observed in chem-isorption on transition metals. ' Although it neglects theFriedel charge oscillations, it has the advantage to allowfor potential oscillations. We will see in the followingthat the neglect of the potential oscillations (model I, forexample) leads to unphysical charge transfers (0.5e toseveral electrons), whereas the potential oscillations need-ed to obtain physical charge transfers would be close tothe ones derived from a strict local charge neutrality con-dition. Therefore this approximation is expected to bebetter for larger values of Zz —Z~ than the previous ones.
To illustrate this model let us assume that all the localdensities of states are constant on an energy range 8' andvanish everywhere else. This bandwidth 8' is determinedso that the exact and approximate local densities of stateshave the same centered second moment p2 (pz NP, ——where A' is the number of neighbors and P a transfer in-tegral). In this very simple case it can be shown straight-forwardly that
8g —Rs~MS 10
(ZM —5)
8'gVIB (ZM ZI ) ~
10
~s+IS ~MS + ~IB8'g
VMB ——0 is the origin of the energies and 8'z[s] is the bulk(surface) bandwidth. Let us compare the perturbing po-tentials due to the impurity at the surface and in the bulk
~rs —&Ms
~IB +MB
This ratio is smaller than 1. We will see in the next fewsections that this property will remain true in all con-sidered cases.
Since E„z,——0, E„z, is obtained directly from Eq. (8) inwhich the potentials are given by Eqs. (11). One finds
~a- ~sE„s,= (Zr —ZM )(10 Zr —ZM ), —
which can be rewritten in the form
Wg —8'sE„s,= [Z~(10 Zs ) —Z—M(10 —ZM )] (12a)
=&s(Zr) rs(ZM—), (12b)
where rs(Z) is the surface energy of a metal with a bandfilling Z calculated within the same approximations. Ac-cording to this formula, the impurity I will segregate ifthe surface tension of the corresponding metal is smallerthan the host one. This criterion is widely used in theliterature. However it assumes that the densities of statesare rigidly displaced by the perturbing potentials. Asshown in our chemisorption studies, ' this is not the casesince the band part of the densities of states must have thesame limits. This assumption of "rigid bands" is ratherwell justified when Zz —ZM is small but breaks down oth-erwise. On the other hand, let us notice that in the partic-ular case Zz ——ZM+1, the segregation energy is equal tothe core level shift between surface and bulk atoms' seenin photoemission which, within the thermodynamical ap-proach of Johansson and Martensson is precisely givenby Eqs. (12) when some terms are neglected.
III. LINEAR CHAIN
The perturbing potential of the impurity must satisfyEq. (7b). Then the segregation energy is given by Eq. (7a).
To check the sensitivity of the final results to the vari-ous approximations, discussed in the preceding section, wewill now calculate the segregation energy on a linearchain. Although this study is of little practical interest, itis an ideal case to check the consequences of each approx-imation. Indeed one needs no further assumptions sinceall the local densities of states can be calculated exactly(see the Appendix for the expressions of the Green func-tions). Moreover the strong dissimilarity between the"surface" and "bulk" densities of states enhances the ef-fects. The comparison between the various models is il-lustrated on the particular case of a filling of the substrateband equal to 0.8.
A. Model I
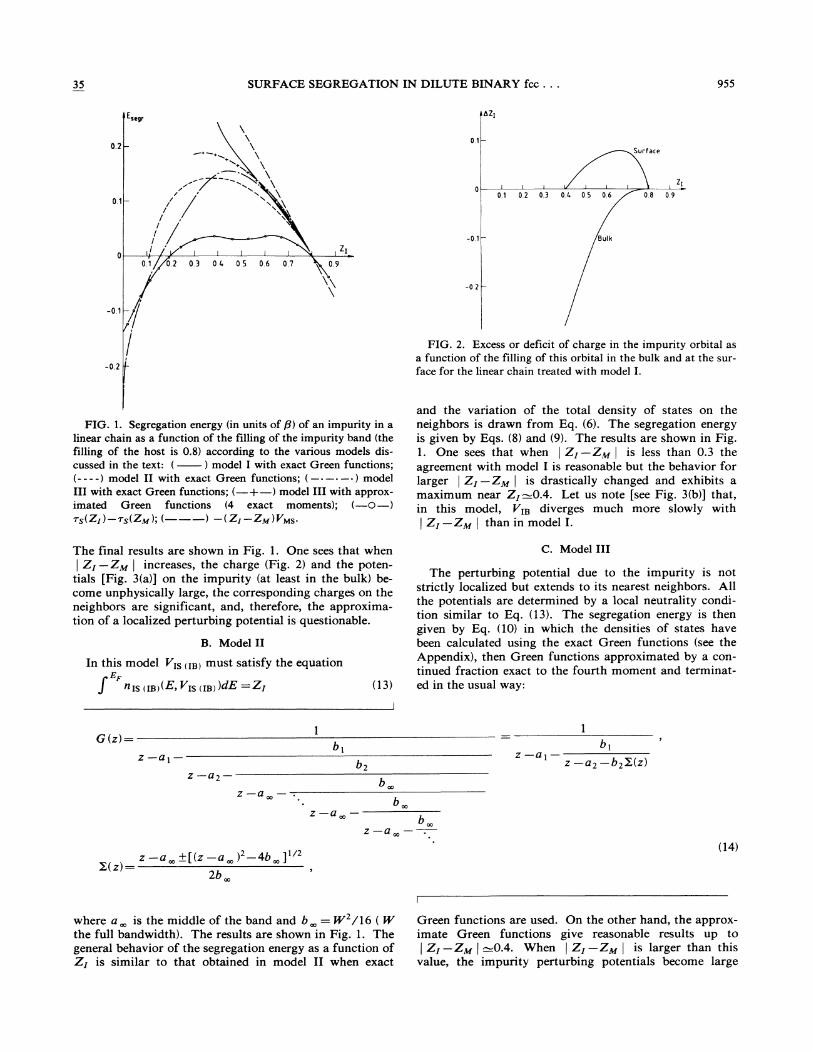
35 SURFACE SEGREGATION IN DILUTE BINARY fcc. . . 955
E$tgf(}UZI
0.2— 0.1—
0.1—
/)I'
/I
l H0.1 0.2 0.3 0.4 0.5 0.6 0.7
Zi
0.9
-0.1—
I I I
0.1 0.2 0.3 0
ZI
8 0.9
-0.2—
-0.2
FIG. 2. Excess or deficit of charge in the impurity orbital asa function of the filling of this orbital in the bulk and at the sur-face for the linear chain treated with model I ~
FICx. 1. Segregation energy (in units of P}of an impurity in alinear chain as a function of the filling of the impurity band (thefilling of the host is 0.8) according to the various models dis-cussed in the text: ( ) model I with exact Green functions;(- - - -) model II with exact Green functions; ( ———) modelIII with exact Green functions; (—+—) model III with approx-imated Green functions (4 exact moments); (—0—)
rs(ZI ) —&s(Zw); (———) —(Zz —ZM) VMs.
and the variation of the total density of states on theneighbors is drawn from Eq. (6). The segregation energyis given by Eqs. (8) and (9). The results are shown in Fig.1. One sees that when
~ZI —ZM
~
is less than 0.3 theagreement with model I is reasonable but the behavior forlarger
~ZI —ZM
~
is drastically changed and exhibits amaximum near ZI-0.4. Let us note [see Fig. 3(b)] that,in this model, V&& diverges much more slowly with
~ZI —ZM
~
than in model I.
In this model V&s [&B] must satisfy the equation
fEFnIS (IB)(Ei ~IS (IB) )dE ZI (13)
The final results are shown in Fig. 1. One sees that when
~ZI —ZM
~
increases, the charge (Fig. 2) and the poten-tials [Fig. 3(a)] on the impurity (at least in the bulk) be-come unphysically large, the corresponding charges on theneighbors are significant, and, therefore, the approxima-tion of a localized perturbing potential is questionable.
B. Model II
C. Model III
The perturbing potential due to the impurity is notstrictly localized but extends to its nearest neighbors. Allthe potentials are determined by a local neutrality condi-tion similar to Eq. (13). The segregation energy is thengiven by Eq. (10) in which the densities of states havebeen calculated using the exact Green functions (see theAppendix), then Green functions approximated by a con-tinued fraction exact to the fourth moment and terminat-ed in the usual way:
G(z) =z —a&—
z —a2—z —a
OO
z —aOO
z —aOO
z —a&—z —op —b~X(z)
z —a + [(z —a ) 4b„]'~—&(z)=
2b
(14)
where a is the middle of the band and b = 8' /16 ( Wthe full bandwidth). The results are shown in Fig. 1. Thegeneral behavior of the segregation energy as a function ofZI is similar to that obtained in model II when exact
Green functions are used. On the other hand, the approx-imate Careen functions give reasonable results up to
~ZI —ZM
~
=0.4. When~
ZI —ZM~
is larger than thisvalue, the impurity perturbing potentials become large

956 M. C. DESJONQUERES AND D. SPANJAARD 35
and have a noticeable effect on the coefficients of the con-tinued fraction beyond the second level and therefore set-ting a„=a, b„=b when n~2 is a poor approxima-tion.
The trends followed by the segregation energy can beunderstood using simple arguments. When
~ZI —Z~
~
issmall, a straightforward application of the Hellman-Feynman theorem ' shows that
E. s = (ZI ——ZM)I Ms .
Notice that this formula gives E„g, with a good accuracywhen
~ZM —ZI
~~0.2. On the other hand when ZI van-
ishes, the potential on the impurity goes to infinity andone has to solve a vacancy problem. It is clear that thecreation of a vacancy needs an energy smaller when thevacancy is at the end of the chain than in the bulk of itsince in this latter case one creates two "surfaces, " in ad-dition to the removal of one atom in both cases. There-fore when the valence shell occupancy of the impurity de-creases strongly, the segregation energy should becomenegative. Consequently, one should reach a maximum be-tween Zr ——0 and Zr ——ZM.
In Fig. 1 we have also plotted the values of the segrega-tion energy deduced from Eq. (12b). In the calculation ofthe surface energies we have used exact Green functionsand we have limited the extension of the surface perturba-tion to the first two atoms of the semi-infinite chain. Theperturbing potentials on these two atoms have been deter-mined by a local neutrality condition. The overall trendfollowed by the segregation energy is similar to the previ-ous results, however the quantitative agreement withmodels II and III is rather poor. Let us recall that the ex-pression of the segregation energy given by Eqs. (12) as-sumes that the perturbing potentia1 due to the impurityrigidly shifts the corresponding density of states. This isnot at all the case for a bulk atom in a linear chain sinceeven an infinitesimal perturbing potential produces abound state. This latter effect being typical of a linearchain, we expect that the segregation energy will be betterapproximated by Eq. (12) in the three-dimensional case.
In conclusion, when~ZI —ZM
~
&0.3 the models I, II,and III agree within an accuracy of 20%. When
~Zl —ZM
~
is larger, model I leads to unphysical local
(b) '
charges and becomes inadequate. In the same limit, themodels II and III agree qualitatively but the actual nu-merical values becomes strongly sensitive to the approxi-mation made both for self-consistency and for Greenfunctions. Finally the perturbation potential due to theimpurity is smaller at the surface than in the bulk in allmodels as predicted in the preceding section.
IU. SEGREGATION OF A TRANSITION-METALIMPURITY ON fcc TRANSITION-METAL SURFACES
We first study an isolated impurity and calculate itssegregation energy, using the models developed in thepreceding section, for a (100) face of a fcc transition met-al. We investigate also the (111)face of the same metal toprecise the influence of the surface crystallography. Inthis latter case we only use model III which is more tract-able since it does not need off-diagonal Green functions.Then we also compare the energy of a monolayer of im-purities on the (100) surface to the energy of a system inwhich all these impurities are solved in the bulk. Finally,we will also consider the case where the impurity is a va-cancy.
A. Model I
The d band is described in the tight-binding approxi-mation using a basis set composed of the d atomic orbi-tals
~i,k ) centered on all sites i; A, = 1,2, . . . , 5 corre-
sponds respectively to (xy, yz, zx, x y, 3z r—) with—(iA
~jp)=5,J5». Since we assume that the perturbing
potential is localized on the impurity site, the phase shiftcan be expressed in terms of the intra-atomic Green func-tions g;;" of the pure host on this site only. Let us recallthat for symmetry reasons, g;;" vanishes when k&p for asemi-infinite (100) crystal. Furthermore in the bulkg"=g =g and g =g and at the surface g =gAll the elements of the Green functions have been ex-pressed as continued fractions exact up to the thirteenthlevel and terminated in the usual way. They have beencalculated using tight-binding parameters (given in TableI) suited to the 3d series (bandwidth of -4.4 eV). Sincethe densities of states of all the fcc transition metals havevery similar shapes, the segregation energies are propor-tional to the host bandwidth and therefore their values for4d or Sd impurities and hosts can be derived from the re-sults on the 3d series by a proper scaling. Let us noticethat the surface potential VMs (necessary to satisfy theFriedel sum rule in the semi-infinite pure crystal) pro-duces a band of surface states which extends in energybeyond the top of the bulk band. This should be taken
I I I
0.2 0.4 0-6 0 ~ 8 0.2 0.4 0.6 0.8 0. 2 0.4 0.6 0.8 TABLE I. Hopping integrals (in eV) for the d series.
FIG. 3. Impurity potentials in units of P ( V,a in the bulk,
V&s —VMs at the surface) according to the three models for alinear chain. In model III, the solid curve has been calculatedwith exact Green functions, and the dashed curve with approxi-mated Green functions (4 exact moments).
dd0dd 7T
dd5
3d series
—0.5670.256
—0.032
4d and Sd series
—0.9660.436
—0.055
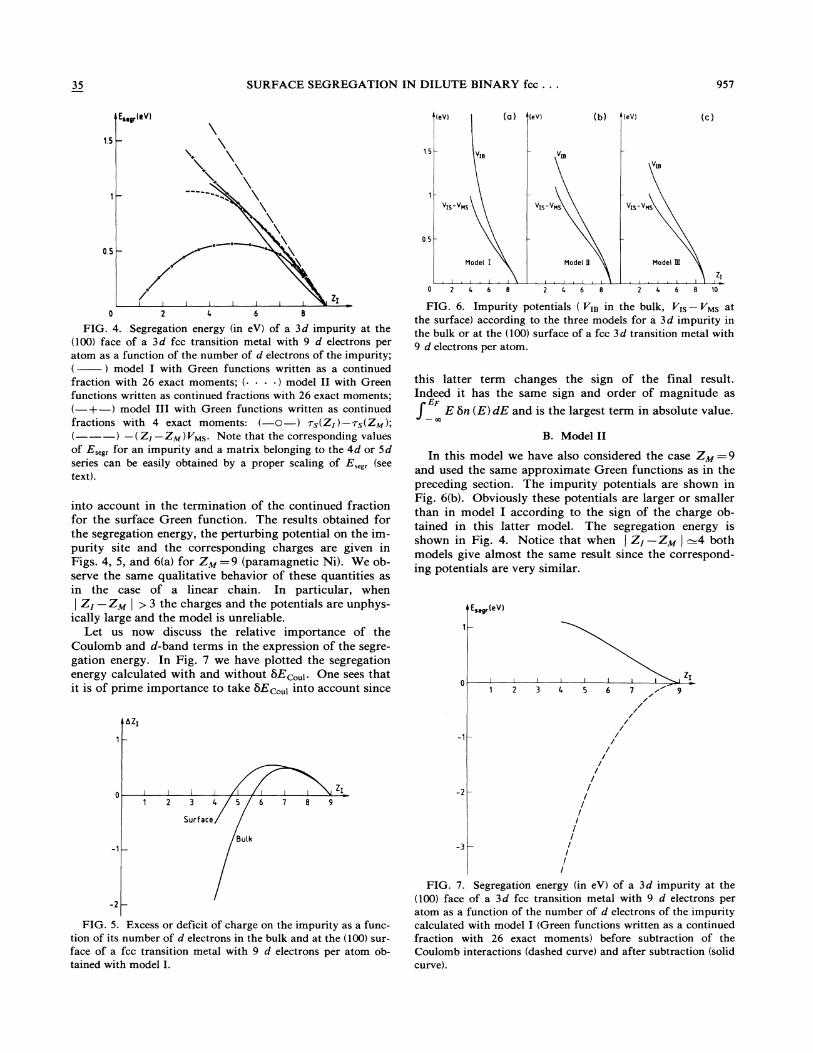
35 SURFACE SEGREGATION IN DILUTE BINARY fcc. . . 957
'j{ey) {G) )~{ev) {b) "(ey) (c)
1.5—
IS MS VIS-V VIS- V
0.5—0.5—
0 2 0 & 8 2 C 6 8
ZII I
2 0 6 8 10
6 B
Zy
into account in the termination of the continued fractionfor the surface Green function. The results obtained forthe segregation energy, the perturbing potential on the im-purity site and the corresponding charges are given inFigs. 4, 5, and 6(a) for Z~ ——9 (paramagnetic Ni). We ob-serve the same qualitative behavior of these quantities asin the case of a linear chain. In particular, when
! ZI —ZM ! & 3 the charges and the potentials are unphys-ically large and the model is unreliable.
Let us now discuss the relative importance of theCoulomb and d-band terms in the expression of the segre-gation energy. In Fig. 7 we have plotted the segregationenergy calculated with and without 6EC,„~. One sees thatit is of prime importance to take 5EC,„& into account since
)az,
Surface
8 9
FIG. 5. Excess or deficit of charge on the impurity as a func-tion of its number of d electrons in the bulk and at the (100) sur-face of a fcc transition metal with 9 d electrons per atom ob-tained with model I.
FIG. 4. Segregation energy (in eV) of a 3d impurity at the(100) face of a 3d fcc transition metal with 9 d electrons peratom as a function of the number of d electrons of the impurity;( ) model I with Green functions written as a continuedfraction with 26 exact moments; (. ~ ~ ~ ) model II with Greenfunctions written as continued fractions with 26 exact moments;(—+—) model III with Green functions written as continuedfractions with 4 exact moments: (—O—) 7g(ZI ) —7q(ZM );(———) —(ZI —ZM) VMs. Note that the corresponding valuesof E„g, for an impurity and a matrix belonging to the 4d or Sdseries can be easily obtained by a proper scaling of E„g, (seetext).
FIG. 6. Impurity potentials ( VqB in the bulk, VIs —VMs atthe surface) according to the three models for a 3d impurity inthe bulk or at the (100) surface of a fcc 3d transition metal with9 d electrons per atom.
this latter term changes the sign of the final result.Indeed it has the same sign and order of magnitude as
fEFE 5n (E)dE and is the largest term in absolute value.
I(E ~{eV)
I
3
Zr
5 6 7 ~ 9J'
//
//
//
//
//
//
//
//
//
//
I/
FIG. 7. Segregation energy (in eV) of a 3d impurity at the(100) face of a 3d fcc transition metal with 9 d electrons peratom as a function of the number of d electrons of the impuritycalculated with model I (Green functions written as a continuedfraction with 26 exact moments) before subtraction of theCoulomb interactions (dashed curve) and after subtraction (solidcurve).
B. Model II
In this model we have also considered the case Z~ ——9and used the same approximate Green functions as in thepreceding section. The impurity potentials are shown inFig. 6(b). Obviously these potentials are larger or smallerthan in model I according to the sign of the charge ob-tained in this latter model. The segregation energy isshown in Fig. 4. Notice that when ! ZI —Z~! =4 bothmodels give almost the same result since the correspond-ing potentials are very similar.

958 M. C. DESJONQUERES AND D. SPANJAARD 35
C. Model III E$09f (eV)
1.5-II (e V) (b)
For the sake of simplicity and to avoid large computingtimes we have used densities of states exact up to thefourth moment and given by a continued fraction ter-minated in the usual way [Eqs. (14)]. This approximationshould be reasonable for the unperturbed Green functionssince for the pure host density of states, the coefficient b2is rather different from b „,whereas higher-order coeffi-cients are much closer to this value.
Moreover it is easy to derive analytical expressions forthe corrections to the first two pairs of coefficients of thecontinued fraction when the intraatomic potentials Vi aretaken into account. ' Therefore, these coefficients arecomputed numerically once only with all VI set to zerothen the corresponding analytic expressions are used.This saves a large amount of computing time. On theother hand, we have seen in the case of the linear chain,that Green functions with four exact moments lead tovery reasonable results up to
~Zi —ZM
~
=4.The results obtained for the segregation energy and im-
purity potentials are exhibited in Figs. 4 and 6(c) forZ~ ——9, and for the (100) face. They closely follow the re-sults derived from model II up to
~ZI —ZM
~
=4. Forlarger values of
~ZI —Z~
~
the results may become pro-gressively unreliable as discussed above in the case of alinear chain.
It is interesting to compare the segregation energy ob-tained in this model with the result derived from Eq.(12b), in which the surface energies have been calculatedwith the same assumptions (densities of states with fourexact moments, two surface potentials satisfying localcharge neutrality). When
~ZI —ZM
~
=1, this result al-most coincides with the previous ones (models II and III),but deviates progressively when
~ZI —ZM
~
increasessince the assumption of a rigid density of states is less andless accurate although the overall behavior remains physi-cally reasonable. Note, however, that Eq. (12b) gives abetter approximation to the segregation energy than in theone-dimensional case although in both cases it is underes-timated. Finally, the evaluation of the segregation energygiven by Eq. (15) is very reasonable when
~ZI —ZM
~
(2and overestimated otherwise (see Fig. 4).
We have also studied the (111) face of a fcc metal withthe same band filling (Z~ ——9) since this allows to precisethe influence of the number of bonds broken by the sur-face. The segregation energy and the perturbing poten-tials are given in Figs. 8(a) and 8(b) with the correspond-ing results for the (100) face. One sees that the surfaceimpurity potential ( Vis —VMs) is closer to the bulk im-purity potential ( ViB) for the (111)than for the (100) face.This is expected since the (111) face is more close packedthan the (100) face. In fact, if one uses rectangular densi-ties of states one can easily show from Eqs. (11) that theperturbation of the impurity potential when going fromthe bulk to the surface [ ViB —(Vis —VMs)] is roughlyproportional to the number of broken bonds at the sur-face. As a consequence, the segregation energy has thesame sign but is smaller on the (111) than on the (100)face for all elements. This rather intuitive behavior (infact followed experimentally in CuNi alloys ) is actually
VIS-VgS ()t))
VIS-VgS (100
0.5- (10
FIG. 8. (a) Comparison of the segregation energies (in eV) ofa 3d impurity at the (111)and (100) faces of a 3d fcc transitionmetal with 9 d electrons per atom as a function of the numberof d electrons of the impurity. Solid curves: model III withGreen functions written as continued fractions with 4 exact mo-ments; dashed-dotted curves: ~q(ZI) —w~(Z~); dashed curves:—(Zi —Z~)VMs. (b) Comparison of the impurity potentialsV» in the bulk, and V&s —VMs at the (111) and (100) surfacescorresponding to the results shown in (a) (model III).
predicted by Eqs. (12) but could not be obtained if theCoulomb terms are omitted. Also plotted in Fig. 8(a) arethe evaluations of the segregation energies derived fromEq. (12b) which are underestimated, and from Eq. (15)which are in rather good agreement with the results of
ZM~
&3.In order to investigate the influence of the band filling
of the matrix ZM we have also calculated the segregationenergies of transition impurities in a (100) fcc metal withZM ——8 and ZM ——7. They are shown in Fig. 9. Actuallywhen
~ZI —ZM
~
increases around ZI ——ZM, the segrega-tion energy varies less and less rapidly as expected fromthe estimations given by Eqs. (12) and (15). Furthermore,as stated above, within the so-called "equivalent core ap-proximation" the segregation energy of a Z + 1 atom in aZ metal is equal to the surface core level shift measured
i(E ~~(eV)
1.5- (o)
Zq
0.5'- Zq
2
I
2
ZI
-0.5-
FIG. 9. (a) Comparison of the segregation energies (in eV) ofa 3d impurity at the (100) face of a 3d fcc transition metal with9, 8, and 7 d electrons per atom as a function of the number ofd electrons of the impurity. Solid curves: model III; dashed-dotted curves: ~z(ZI )—~q(Z~ ); dashed curves:—(ZI —Z~) VMs. (b) Comparison of the segregation energies (ineV) per impurity atom for a rnonolayer of impurities (solidcurves) and a single impurity (dashed curves) according to modelIII for a d impurity at the (100) face of a fcc transition metal asa function of the number of d electrons of the impurity. Thed-band fil?ing of the host is 9, 8, and 7 d electrons.

35 SURFACE SEGREGATION IN DILUTE BINARY fcc . . ~ 959
in a photoemission experiment. In this special case, ac-cording to Eqs. (15) and (12), an approximate value of thesegregation energy is given by —VMs and rs(Z+1) r—s(Z) I.n Figs. 10(a) [(100) face] and 10(b) [(111)face] we have plotted these estimated values with thesegregation energy derived from model III as a functionof the host band filling. The general behavior of thesethree evaluations are very similar: they have the same or-der of magnitude and they change sign around the middleof the series. However, quantitatively one observes adiscrepancy between the estimated values and E„z, calcu-lated with model III which can be as large as -0.08 eVwhen ZM —6. In practice, transition metals do not havethe fcc structure for a band filling around this value.Therefore to have a precise comparison between thesethree evaluations in realistic cases it would be highlydesirable to consider the real crystallographic structurecorresponding to each band filling.
Let us now consider the value of the variation of energyper solute atom when a complete monolayer of impuritiesis formed at the surface arising from the segregation ofdilute (without interactions) impurities in the bulk. Thecomputation has been performed using model III for the(100) face and the results are given in Fig. 9. When com-paring to the previous results, one sees that the trends foror against segregation is reinforced.
In Figs. 4, 8, and 9 one remarks that the calculatedvalues of E„s, increase continuously with
~ZI —ZM ~.
However for obvious reasons and as already seen in thecase of the linear chain, this quantity should reach a max-imum and then decrease to reach the segregation energyof a vacancy at ZI ——0. This energy is negative since thenumber of bonds which has to be removed to create thevacancy is smaller at the surface than in the bulk. Similarto the case of the linear chain, one must use more andmore accurate Green functions (i.e., more and more exactlevels of the continued fraction) to obtain the decreasingpart of E„z,. This is feasible but costly. Moreover, theoutput of such a calculation is not expected to comparewell with experiments since the impurity and the host ele-ment are, in this case, far apart in the Periodic Table andconsequently the size effects are certainly important anddifficult to estimate in a microscopic model as we will dis-cuss in Sec. V. Nevertheless the calculation of the limit-
i)E [eVj
ZM
8
FIG. 11. Electronic contribution to the formation (in thebulk, dashed curve) and segregation [at the (100) surface, solidcurve] energies of a vacancy in a fcc transition metal of thesecond or third series as a function of the d-band filling.
ing case ZI =0 (i.e., vacancy) can be performed with agood accuracy using model III. In this calculation theperturbation of the intraatomic potentials due to the va-cancy is assumed to extend on1y on the first nearest neigh-bors of the vacancy site. The results are shown in Fig. 11and exhibit the expected parabolic behavior as a functionof the d-band filling.
Finally let us note that in Eq. (1) there appears to be adifference of dissolution energy of an impurity at the sur-face and in the bulk. From this latter quantity (Ed;, b„ik)one can get the alloy formation energy in the dilute limit.We have checked that, in model III, these energies havethe right order of magnitude (i.e., most of the time &0.1
eV). If the Coulomb term in Ed;, b„,k is omitted one getsmuch too large values ( —1 eV). Similarly one getsreasonable values for the formation energy of a vacancy inthe bulk (a few eV). Notice that for a detailed compar-ison with experiments one should take into account arepulsive contribution (see Sec. V B) which decreasessomewhat this formation energy without changing its or-der of magnitude. If one omits the Coulomb term one ob-tains completely unphysical results except, of course whenthis term vanishes (ZM —5).
Esegr teV
0.3—
0.2—
0.1—
(ap
~' ~segrteVI(b)
,ZM
V. INFLUENCE OF ELECTRONIC CORRELATIONSAND OTHER CONTRIBUTIONS
TO THE SEGREGATION ENERGY
A. Electronic correlations
FIG. 10. Segregation energies of a 3d impurity with ZM+1d electrons in a 3d fcc transition metal with ZM d electrons as afunction of ZM. Solid curves: model III; dashed-dotted curves:7 g(ZM+ 1 ) —'Tg(ZM ); dashed curve: —( ZI —ZM ) VMs. (a) (100)surface; (b) (111)surface.
The electronic correlation term can be estimated from asimplified Hubbard Hamiltonian
correl g i ia ia' ~
I&, a, aa&a'
M. C. DESJONQUERES AND D. SPANJAARD 35
where U; is the average Coulomb integral within the dband and n; the occupation number of the spin orbital aat site i .For a small value of U/8' ( W is the d band-width) a perturbation theory up to second order is likelyto be valid. The first-order term in U produces a shift of
the center of gravity of the local density of state n;(E).This term is implicitly taken into account by choosing thecorrecting potentials such that all atoms remain neutral.The second-order term is given by
E E n; (E, )n;(E2)n;(E3)n;(E4)Eco„e)———45 U; dE3 dE4 dE ) dE2 (17)
Let us discuss qualitatively the influence of this term onthe segregation energy. A good order of magnitude ofE,'„„&is given by
45U; Z;Elcorrel
eff
2Z1—10
2
(18)
where Z; is the number of d electrons at site i and 8' ff isproportional to the exact bandwidth ( 8',rr-8' for a rec-tangular density of states of width W'). If we assume thatthe Coulomb integral is the same for an impurity or ahost atom the contribution of electronic correlations tothe segregation energy becomes
Ecorre~ 45 U2segr 8'S
ZI10
2ZI1—10
2
r
ZM
10
2ZM1—10
2
(19)
This expression is the difference between the correlationcontribution to the surface energy of metal I and of thehost M. Therefore Eq. (12b) is still valid when correla-tion effects are taken into account. E'„'g,"' gives a negativecontribution to the segregation energy when
~
ZI —5~
iscloser to zero than
~ZM —5
~
and positive otherwise andtherefore has a sign opposite to that of the one electronterm [see Eq. (12)].
Quantitatively one can have significant deviations fromEq. (18) when EF approaches a peak of n;(E). In someparticular cases, the use of the exact second-order pertur-bation equation [Eq. (17)) may even change qualitativelythe correlation contribution to the segregation energy aswe will see below. Indeed, in this scheme one should alsotake into account the corresponding contribution of theneighbors which vanishes in the simplified model sincethe presence of the impurity does not change the local ef-fective bandwidth. Therefore we have computed E,",g,
"'using the exact equation for E,'„„~. The results for thesegregation energies of an impurity I in a metal M withZM ——9 as a function of ZI are given in Fig. 12. We havechosen UM ——2 eV for the host atom and UI ——1.5 eV forthe impurity (ZI &9) in the first transition series since itis known that U increases along a d series. These valuesare derived from photoemission and Auger experimentsusing the same perturbation treatment of the Hubbard
model. In the other series we have taken UM ——2.5 eV andUI ——l.'7 eV (Ref. 29) (ZI &9). Let us notice all theseCoulomb integrals are not known with a good accuracyand can be varied slightly around these values. One seesthat the trends discussed just above are followed: the de-crease of the segregation energy is the more pronouncedas the filling of the d valence shell of the impurity is near5. In the first series the ratio U/8 is larger than in theother series and the contribution of electronic correlationsis stronger and may become of the same order of magni-tude as the one electron contribution. (In this case the va-lidity of a second order perturbation theory may be ques-tionable. ) Therefore if we ignore size effects which areindeed small if the solute and the solvent atoms belong tothe second half of the same series (see Sec. V B), it is diffi-cult to draw any definite conclusion concerning the oc-currence of segregation in the first series since it dependscrucially on the values chosen for the Coulomb integrals.Actually in this case, our results lead to small segregationenergies and this is consistent with experimental datawhich do not show any systematic trend and are some-
) Esegr(eV)
(a)() Esegr [e~]
(b)
1.5—Impurity
solated xmono[ayer
impurity&-
TS0.5—
z,
FIG. 12. (a) Influence of Coulomb correlations on the segre-gation energy per atom of isolated 3d impurities, of a monolayerof 3d impurities [within model III and from Eqs. (12)] at a (100)surface of a 3d fcc transition metal with 9 d electrons per atom.Solid curve: U~ ——2 eV, UI ——1 5 eV; dashed curve:UM ——UI ——0. (b) Influence of Coulomb correlations on thesegregation energy per atom of isolated 4d or 5d impurities[within model III and from Eqs. (12)] at a (100) surface of a 4dor 5d fcc transition metal with 9 d electrons per atom. Solidcurve U~. 2.5 eV, UI ——1.7 eV; dashed curve: U~ ——UI ——0.

35 SURFACE SEGREGATION IN DILUTE BINARY fcc. . . 961
times controversial [for instance for Cr (Ref. 30) and Fe(Ref. 31) in Ni].
If we now consider a monolayer of impurities [see Fig.12(a)] the effect of electronic correlations on the segrega-tion energy is of the same order of magnitude as for iso-lated impurities. Therefore the segregation energy per im-purity atom remains larger for a monolayer than for anisolated impurity. Consequently, for a given range ofvalues of U segregation occurs at dilute concentrationsbut stops for a finite concentration of impurities at thesurface. In the second or third series the electronic corre-lation effects decrease the segregation energy but the signof the latter seems well established since one would needmuch larger values of U to reverse it [Fig. 12(b)].
Let us notice that the presence of electronic correlationeffects when treated with the exact second-order perturba-tion expression may lead sometimes to amazing effects.Indeed according to the simplified expression (12) thesegregation energy of element B in metal A has a sign op-posite to that of element A in metal B since the elementwith the smallest surface energy will segregate to the sur-face. If we apply this simple rule to Cu Ni alloys, oneshould find always an enrichment of the surface in Cu.However Sakurai et al. have recently reported that Nisegregates to the surface when the bulk Ni concentrationis in the range 0—16 at. %. We do think that correlationeffects can help to understand this effect when treatedwith the exact second-order perturbation expression [Eq.(17)]. To illustrate this point, we have considered the caseof elements A and B with a filling of the d band equal,respectively, to Zz ——9.0 and Zz ——9.8 with U=2 eV andthe bandwidth suited to the 3d series ( W=4.4 eV). Whenone considers an impurity B in meta1 A, one finds thatelectronic correlation effects enhance the segregation of Bat the surface of A: the segregation energy changes from—0.11 to —0.17 eV. This effect is surprising since thesimplified formula [Eq. (18)] would predict the oppositetrend; indeed we replace a host atom by an impuritywhich has less holes in its d shell and therefore less corre-lation energy is lost when the impurity is in the bulk thanwhen it is in the surface. When the exact second-order ex-pression is used this trend is still obeyed on the impuritysite, but is counterbalanced by the correlation contributionof the neighbors due to the modification of the shape oftheir local densities of states which brings more statesnear the Fermi level when the impurity is at the surface.
Correlatively, when one considers an impurity A in ametal B, the segregation energy of A in B which is posi-tive when U=O (E„s,——0.20 eV) becomes slightly nega-tive (E„s,———0.02 eV) when electronic correlation effectsare taken into account. This agrees with the intuitive ar-gument stated above that correlation effects favor general-ly the segregation at the surface of the element closest tothe middle of the series.
In conclusion, one finds that A segregates in a pure Bmetal and B segregates in a pure A metal. We do thinkthat a similar effect might be responsible for the behaviorof CuNi alloys, corresponding roughly to the filling of thed band chosen in our calculation. (We have left out themagnetic effects in Ni-rich alloys. ) Let us note that thesegregation energies of A in B and B in A are opposite
)) E„g„(eV)0.6—
liEsegr{eV)
0.2—
2
-0.2—
FIG. 13. Influence of Coulomb correlations on the segrega-tion energy of a d impurity with ZM+1 d electrons at a (100)face of fcc transition metals of the same series with ZM d elec-trons as a function of ZM. Solid curve: model III withCoulomb correlations included; dashed-dotted curve:~g(ZM+1) —7g(ZM ) with Coulomb correlations included. Forcomparison we have also drawn the segregation energy (modelIII) without Coulomb correlations (dashed curve) and —VMs(dotted curve). (a) 3d series, UM ——UI ——1.5 eV; (b) 4d or 5dseries, UM ——UI ——1.7 eV.
only in oversimplified models. In more realistic studies,the deformation of the local densities of states breaks thisperfect antisymmetry.
Finally we have calculated the effect of electroniccorrelations on the segregation energy of a Z + 1 impurityin a Z metal. We have taken U=1.5 eV and U=1.7 eV,respectively, for the first and second or third transitionseries whatever the considered element. As expected, thiseffect is dramatic in the first series and weaker in thesecond or third series (Fig. 13). In this latter case the gen-eral behavior of E
gis not changed.
B. Other contributions
Until now we have neglected the size difference betweenan impurity and a host atom. These size effects have beenwidely discussed in the literature and several analytical ex-pressions' ' ' have been given within macroscopicmodels based on continuous elasticity theory. Since wehave started with a microscopic point of view, we cannotuse straightforwardly these models. As in our previousstudies' ' let us decompose the energy per atom in thefollowing way
E(r) =E„~(r)+Eb,„d(r) . (20)
E p is a repulsive energy which will be assumed to bepairwise and described by a potential of the Born-Mayertype which decreases with distance as Ae ~' (the range ofthis potential is limited to first nearest neighbors). Eb,„dis the one electron contribution previously calculated. Itis proportional to the hopping integrals which are knownto vary roughly as e q'. These exponential laws are de-rived from a fit to microscopic calculations or to experi-mental results which leads to 3 (p/q (5. Here, as in Ref.32 we adopt p /q =3. At equilibrium one can thenstraightforwardly show that the repulsive part of the

962 M. C. DESJONQUERES AND D. SPANJAARD 35
cohesive energy E„h of a pure metal is the half of the to-tal energy in absolute value. Consequently, the repulsiveenergy of a pair of nearest neighbors c~M in a fcc metalM as a function of r is given by
eMM(r) = Ecch(M) p(r ——R )
12(21)
where RM is the nearest-neighbor equilibrium distance.When one considers an I impurity, two simple assump-tions can be made on HAMI(r)
EMI(r) 2 [eMM(r)+EII(r)] (22a)
or
HAMI ( r) [EMM ( &)&II ( &)] (22b)
In both cases eMI(r) is intermediate between EMM(r) andeII(r) which is physically reasonable. The correspondingrepulsive contributions to the segregation energy of a sin-gle impurity for a (100) face of a fcc metal are, respective-ly,
E, s,c=2[EMM(RM ) EII(RM )]-,Esc~sr=4[&MM(RM)l' [[&MM(RM)]' —[&II(RM)]'
(23a)
(23b)
From Eqs. (21) and (23) one can get a rough estimationof E,",g, . Taking experimental values of E„h and p valuesfrom Ref. 32, the results are given in Table II for diluteimpurities of the first, second, and third transition-metalseries in Ni, Pd, and Pt, respectively. One sees clearlythat E,",g, is only important for impurities belonging tothe first half of the series which are strongly oversizedwhen compared to bulk atoms.
Notice that when Eq. (23a) is used, E,",sp, can be writtenin the following way:
Escs'c =6 (
I Ecch(~)I
—I&coh(l)
I)
—p, (R+ co(1 I M I
6(24)
[(X~—I )P +(P+EI3) ]' (NIIP )' =bPI(NI—I)'I
( N~, coordination number of the neighbor). It is clearthat the impurity energy gain is larger in the bulk than inthe surface since Nl is larger in the bulk. Let us nowcompare the contribution of the nearest neighbors in thebulk and at a (100) fcc surface, for example. In the sur-
The first term of E,';sp, is in fact the repulsive contribu-tion to the difference of surface energy of the metal Mand the metal I: rs(ZI ) rs(Z—M ). The second termtends to expel from the interior an impurity larger than ahost atom ( RI )RM ) and vice versa to keep in the interioran impurity such that RI &RM. This effect is antisym-metric for small deviations in size, but when
~
(RI —RM)/RI~
increases () 10%), the absolute valueof the segregation energy becomes quickly much largerfor oversized impurities.
Let us now discuss the size effects on the band termEh,„d(r). When going from the beginning to the end of ad series, the extension of the d orbitals decreases. There-fore if in a Ni, Pd, or Pt matrix we replace an atom by animpurity at the left of the same series, the hopping in-tegral P between this impurity and its nearest neighborsincreases by hP. This obviously leads to a gain in bandenergy of the impurity, roughly proportional to(XI)'I hp, (1VI, coordination number of the impurity)and of each nearest neighbor roughly proportional to
TABLE II ~ Contribution (in eV) of the repulsive term to the segregation energy.
Impurityin Ni
CoFeCrVT1
(RN; —Rr )/Rr
—4x 10-'4x 10-'0
—53x10—'—138x 10-'
prR
9.128.869.608.408.18
E,';gc„[Eq. 23(a)]
000
—0.6—1.8
E,",~P, [Eq. 23(b)]
000
—0.6—1,2
Impurityin Pd
RuMoNbZr
Rpd —Rr )/R
57 x 10-'11x10-'
—38 x 10-'—132 x 10-
prRr
10.3710.058.317.69
E,",g', [Eq. 23(a)]
—0.4—1.0—2.2
E,";g", [Eq. 23(b)]
0—0.4—0.8—1.4
Impurityin Pt
IrReWTaHf
Rpt —Rr )/R
22x 10—'1 1 x 1011x10—'
—31x 10-'—121 x 10
prRr
11.2010.569.778.458.00
E,",gc„[Eq. 23(a)]
—0.2—0.4—0.8—1.8
E,",gP, [Eq. 23(b)]
—0.2—0.3—0.6—1.4

35 SURFACE SEGREGATION IN DILUTE BINARY fcc. . . 963
TABLE III. Contribution to the segregation energy due to the variation of hopping integrals betweenthe impurity and its neighbors (in eV).
Element in Pt
PtIrReWTaHf
( Rp& —Ri )/Rl
022)& 1011 &&
10-'11'10-'
-31X10 '—121 X 10-'
qlR
3.773.803.583.312.862.71
P (eV)
0.510.510.630.680.820.99
ZI
9.58.35.84.63.52.4
rh, PEsegr
000.20.30.40.65
E„~g,— b,p[(V 12—V 8)ZI(10—ZI )
+ ( 12/~12 —4/W8 —4/v 12)
&& ZM(10 —ZM )1 (25)
where, if we take a geometrical average for hopping in-tegrals between two different atoms
~P= [Pl «M )PM(RM )]'" PM(RM ) . — (26)
For a given metal P is given by
P=[ , (dda +2ddn—. +2dd5 )]'~ (27)
where ddo, ddt, and dd5 are the Slater-Koster hoppingintegrals. In fact
~
ddt.~
=~
ddcr~
/2 and dd5=0; there-fore
(28)
The hopping integrals decrease exponentially with dis-tance and consequently
131(RM ) =PI(RI )e (29)
Then taking the d bandwidth 8' as the energy differencebetween points X5 and X3 in the Brillouin zone, we find
~
ddcr~
= W/8 . (30)
(This formula is also valid for bcc metals within a 10%%uo
accuracy. )
To illustrate these effects we have chosen the exampleof 5d impurities in Pt. For reasonable values of the band-width and the band filling one sees in Table III that thiscontribution is only noticeable for impurities in the firsthalf of the series.
If we now superimpose the repulsive effects discussedabove, the total size effects nearly vanish when
face the ener y gain is proportional to4bg/~8+4bP/ 12, while in the volume it is propor-tional to 126,P/~12; therefore larger than in the surface.The final energy balance of both contributions clearlyfavors the bulk impurity. The proportionality factors canbe estimated roughly with a rectangular density of stateswith a width R,ff fitted to give the centered second mo-ment p2 of the exact density of states[ W,ff —2v 3(pz)' ]. One finds that the contribution ofthese terms to the segregation energy is
~(RI —RM)/RM
~
is not too large. Otherwise, when Ribecomes significantly larger than RM (by a few percent),the repulsive term dominates and favors the segregationof the impurity. Moreover, in this case (strongly over-sized impurities), it is very likely that lattice relaxation be-comes important. We expect this effect to favor also asegregation to the surface since the relaxation of the bondlength between the impurity and the host is easier at thesurface.
VI. CONCLUSION
It is clear from this work that the surface segregationenergy of impurities in fcc transition metals can be wellestimated when the solvent and solute elements are nottoo far apart in the Periodic Table, since in this case sizeeffects are not important. In the opposite case the calcu-lation of the segregation energy requires a betterknowledge of the interactions between solute and solventatoms (repulsive energy and hopping integrals). Moreoverone should certainly also take into account the lattice re-laxation and one can wonder if the approximation of astrict local charge neutrality still holds. When
~Zi —ZM
~
&4, the trends obeyed by the segregation en-
ergy follow qualitatively the difference of surface energiesof the two components, apart in systems in which correla-tion effects can be important (3d series). This fact justi-fies empirical expressions of segregation energies based onthis difference. Furthermore, as can be expected, the ab-solute value of the segregation energy increases with thenumber of bonds broken at the surface.
On the other hand we have shown that when theCoulomb interactions which have been counted twice inthe sum over one electron states in the Hartree approxi-mation are not subtracted from the total one-electron en-
ergy, one gets completely unreliable results.Finally, it would be interesting to perform a systematic
study of various alloys at finite concentrations since thefinal aim of surface segregation calculations is the deter-mination of concentration profiles as a function of thedistance to the surface at finite temperature. Indeed, upto now, at least to our knowledge, only Cu-Ni alloys havebeen studied in detail. '
ACKNOWLEDGMENTS
We are grateful to Pr. J. Friedel for having drawn ourattention to this problem and for his comments. Wewould also like to thank Dr. B. Legrand for a criticalreading of the manuscript.

M. C. DESJONQUERES AND D. SPANJAARD 35
APPENDIX
1. Infinite chain
Let us consider an infinite chain in the tight-bindingapproximation with one spherical orbital per site and letus call —P (P & 0) the hopping integral between neighbor-ing sites. The atomic level of this orbital is taken as theorigin of energies. If we replace an atom of this chain byan impurity, the effective atomic level on the correspond-
ing site becomes VqB and on its neighbors becomes VNB.We assume that there is no perturbation of the hoppingintegrals.
In these conditions the local Green functions on the im-purity site is
z —VNB P Goo2 S
Gtt(z) =[(z —VNB —P Goo)(z —Vtn) —2P ]
and on the nearest neighbors
(Z —VIB)(z —VNB —p Goo) —pGNN(z) =
2 S 2 S 2I(z —VNB —P Goo)[(z —VNB —P Goo)(z —VtB) —2P ] I
where Goo is given by
z —(z' —4P')'"Goo(z) =
2 2
with
—(E —4P )' if z =E & —2P(z —4P )' ='t'(4P E)' if—z =E+i 0 and
jE ~&2P
(E 4f3 )' if z—=E &2P .
Goo(z) is actually the local Green function on the atom atthe extremity of a semi-infinite linear chain with all atom-ic levels set to zero.
2. Semi-infinite chain
The impurity atom is located at the extremity of thechain and has an atomic level Vqs while on its neighborthe atomic level becomes VNs.
The corresponding local Green function on the impuri-ty site is
z —VNs —/3 Goo2 S
Gtt(z) =(z —Vts)(z —VN. —n'G~) —n' '
and on the nearest neighbor
z —Vis6NN(Z)
(z —Vts)(z —VNs fJ Goo) P— —
See, for instance, the compilation of experimental results per-formed by F. F. Abraham and C. R. Brundle, J. Vac. Sci.Technol. 18, 506 (1981).
M. P. Seah, J. Catal. 57, 450 (1979);Surf. Sci. 80, 8 (1979).Toshio Sakurai, T. Hashizume, A. Jimbo, A. Sakai, and S. Hy-
odo, Phys. Rev. Lett. 55, 514 (1985).4A. R. Miedema, Z. Metallkd. 69, 287 (1978); D. Kumar, A.
Mookerjee, and V. Kumar, J. Phys. F 6, 725 (1976); V. S.Sundaram and P. Wynblatt, Surf. Sci. 52, 569 (1975); J. L.Moran-Lopez and L. M. Falicov, Phys. Rev. B 18, 2549(1978).
5W. M. H. Sachtler and R. A. Van Santen, Appl. Surf. Sci. 3,121 (1979).
6D. McLean, Grain Boundaries in Metals (Oxford UniversityPress, London, 1957).
7G. Kerker, J. L. Moran-Lopez, and K. H. Bennemann, Phys.Rev. B 15, 638 (1977).
P. Lambin and J. P. Gaspard, J. Phys. F 10, 651 (1980); 10,2413 (1980).
R. Riedinger and H. Dreysse, Phys. Rev. B 27, 2073 (1983); H.Dreysse and R. Riedinger, ibid. 28, 5669 (1983).
~ K. Masuda-Jindo, Phys. Lett. 107A, 185 (1985).J. P. Muscat, J. Phys. C 15, 867 (1982).
2G. Allan, Ann. Phys. (Paris) 5, 169 (1970).J. Friedel, Nuovo Cimento 7, 287 (1958).
~4P. A. Wolff, Phys. Rev. 124, 1030 (1961); A. M. Clogston,
ibid. 125, 439 (1962).~5G. Toulouse, Solid State Commun. 4, 593 (1966).' F. Gautier, G. Moraitis and J. C. Parlebas, J. Phys. F 6, 381
(1976); M. C. Desjonqueres and M. Lavagna, ibid. 9, 1733(1979).J. P. Simon, A. M. Papon, and P. Guyot, Philos. Mag. B 44,69 (1981).
' M. C. Desjonqueres and D. Spanjaard, J. Phys. C 15, 4007(1982); 16, 3389 (1983).
' W. F. Egelhoff, Jr., Phys. Rev. Lett. 50, 587 (1983).OB. Johansson and N. Martensson, Phys. Rev. B 21, 4427
(1980).2tH. Hellman, Einfuhrung in die Quantenchemie (Franz Deu-
ticke, Vienna, 1937); R. P. Feynman, Phys. Rev. 56, 340(1939).M. C. Desjonqueres and F. Cyrot-Lackmann, J. Phys. F 5,1368 (1975).
Y. S. Ng, T. T. Tsong, and S. B. McLane, Jr. , Phys. Rev. Lett.42, 588 (1979); Y. S. Ng, S. P. McLane, Jr., and T. T. Tsong,J. Vac. Sci. Technol. 17, 154 (1980).R. Hultgren, P. Desai, D. Hawkins, M. Gleiser, and K. Kel-ley, Selected Values of the Thermodynamic Properties ofBinary Alloys (American Society for Metals, Cleveland, 1973).H. Wollenberger, in Physical Metallurgy, edited by R. W.Cahn and P. Haasen (North-Holland, New York, 1983), p.1139,and references therein.

35 SURFACE SEGREGATiON IN DILUTE BINARY fcc. . . 965
26F. Kajzar and J. Friedel, J. Phys. (Paris) 39, 397 (1978); G.Treglia, F. Ducastelle, and D. Spanjaard, ibid. 41, 281 (1980).J. Friedel and C. M. Sayers, J. Phys. (Paris) 38, 697 (1977).G. Treglia, F. Ducastelle, and D. Spanjaard, J. Phys. (Paris)43, 341 (1982); G. Treglia, M. C. Desjonqueres, F. Ducastelle,and D. Spanjaard, J. Phys. C 14, 4347 (1981).J. P. Bourdin, M. C. Desjonqueres, D. Spanjaard, and J.Friedel, Surf. Sci. 157, L345 (1985).A. Mosser and J. Werckmann, in Proceedings of the FourthColloquium on Solid Surface Physics and Chemistry (Antibes,
Juan-les-Pins, 1978); in ReUue "Le Vide" (French Vacuum So-ciety, Paris, 1979); A. Zalar, Proceedings of the Fourth International Conference on Solid Surfaces, Cannes, France, 1980,edited by D. A. Degres and M. Costa iSociete Franchise DuVide, Paris, 1980), pp. 201 and 1311.K. Wandelt and G. Ertl, J. Phys. F 6, 1607 (1976); C. R.Brundle, E. Silverman, and R. J. Madix, J. Vac. Sci. Technol.16, 474 (1979).
D. Spanjaard and M. C. Desjonqueres, Phys. Rev. B 30, 4822(1984).





























