Embed Size (px)
Citation preview

Surface Modification of Mg and Mg Alloys
(Oberflächenmodifizierung von Mg und Mg-Legierungen)
Der Technischen Fakultät
der Universität Erlangen-Nürnberg
zur Erlangung des Grades
D O K T O R - I N G E N I E U R
Vorgelegt von
Can Metehan Turhan
Erlangen 2012

Als Dissertation genehmigt von
der Technischen Fakultät der
Universität Erlangen-Nürnberg
Tag der Einreichung: 25.11.2011
Tag der Promotion: 30.01.2012
Dekan: Prof. Dr.-Ing. habil. Marion Merklein Berichterstatter: Prof. Dr. Sannakaisa Virtanen
Prof. Dr. Gözen Bereket

Acknowledgements:
“Science isn’t about why, it’s about why not.” In this context, first and
foremost, I offer my sincerest gratitude to my supervisor, Prof. Dr. Sannakaisa
Virtanen, who has continuously supported my new ideas with her patience and
knowledge and shared her professional experiences in my own way.
I would like to express my sincere thanks to my former supervisor, Prof. Dr.
Gözen Bereket, who always trusted and encouraged me to push my skills forward
since I was an undergraduate student!
I am also very thankful to my colleagues and dear friends; Dr. Robert Lynch,
Martin Kolacyak, Funda Ezginer, for always being there and ready to share their
experiences during my PhD period.
I would also like to thank Prof. Dr. Patrik Schmuki, Manuela S. Killian, Dr.
Himendra Jha, Dr. Qianqian Li, Florian Seuss, Dagmar Rückle, Ferdinand Singer
Tobias Ruff, Robert Hahn, Martin Weiser, Robin Kirchgeorg and Christian Lehmann
for their help, collaboration and willingness to offer advices during my PhD.
I am very thankful to my friends, Can Yavuz, Ihsan Varol, Kurtulus Erdogan,
Gönül Özcan Yüksel, Rahime Ilkaya, Selma Atsüren, Mehmet Mücahit and Hakan
Kayi, since many years, for their always seeing me as their brother.
Finally, I express my thanks to my parents for their understanding, optimism,
increasing support and especially to my loving wife Ilkay, for her trust and belief in
me.
To all those for lending their helping hands whenever I was in need; thank
you!
Can Metehan Turhan

Dedicated to my mother and father…

iii
Table of Contents
Acknowledgements: .............................................................................................. iii
Table of Contents .................................................................................................. iii
Abstract: ................................................................................................................ vi Zusammenfessung:.............................................................................................. viii
1 INTRODUCTION: CORROSION OF MAGNESIUM AND MAGNESIUM ALLOYS................................................................................................................. 1
1.1 Abstract......................................................................................................... 1
1.2 Corrosion of magnesium .............................................................................. 2 1.3 Corrosion rate measurements ...................................................................... 4 1.4 Anodic hydrogen evolution: negative difference effect ............................... 6
1.5 Improving corrosion resistance of magnesium............................................ 8
2 EXPERIMENTAL METHODS.........................................................................11
2.1 Electrochemical setup..................................................................................12
2.2 Electrochemical methods and corrosion tests.............................................14
2.2.1 Cyclic and linear sweep voltammetry......................................................14
2.2.2 Electrochemical Impedance Spectroscopy (EIS)......................................14
2.2.3 Corrosion tests........................................................................................16
2.3 Surface analytic methods ............................................................................17 2.3.1 Scanning Electron Microscopy................................................................17 2.3.2 Auger Electron Spectroscopy (AES).......................................................19
2.3.3 Energy Dispersive X-ray Spectroscopy...................................................20
2.3.4 Focused Ion Beam..................................................................................20
2.3.5 X-Ray Photoelectron Spectroscopy.........................................................21 2.3.6 Time of Flight Secondary Ion Mass Spectroscopy (ToF-SIMS)................21 2.3.7 Fourier Transform Infrared Spectroscopy...............................................22
3 EFFECT OF ACIDIC ETCHING AND FLUORIDE PRE-TREATMENT ON CORROSION PERFORMANCE OF MAGNESIUM ALLOY AZ91D .............23
3.1 Abstract........................................................................................................23
3.2 Introduction .................................................................................................24
3.3 Experimental route......................................................................................26
3.4 Results and Discussion ................................................................................27
3.4.1 Etching Behaviour..................................................................................27
3.4.2 Fluoride treatment optimization..............................................................32 3.4.3 Chemistry of fluoride layer.....................................................................36
3.5 Conclusions ..................................................................................................41
4 ELECTROCHEMICAL POLYMERIZATION OF POLYPYYROLE ON MG ALLOY AZ91D PART 1: CHARACTERIZATION AND OPTIMIZATION OF ELECTROPOLYMERIZATION PARAMETERS ............................................43
4.1 Abstract........................................................................................................43
4.2 Introduction .................................................................................................44
4.2.1 Synthetic metals and doping....................................................................44 4.2.2 Pyrrole and electropolymerization of conductive polymers.....................46 4.2.3 Conducting polymers on magnesium.......................................................48
4.3 Experimental route......................................................................................49
4.4 Results and discussion .................................................................................50

iv
4.4.1 Electrochemical polymerization and characterization of pyrrole.............50 5.4.2. FTIR-ATR, XPS, ToF-SIMS measurements and SEM analyses...............55 4.4.3 Optimization of electrochemical parameters...........................................58
4.4.3.a Optimization of salicylate concentration and scan rate....................58 4.4.3.b Optimization of potential region.......................................................60
4.4.3.c Influence of electrochemical parameters on adhesion and conductivity of the coatings63
4.4.4 Long Term Corrosion performance of PPy/Mg AZ91D ...........................68 4.5 Conclusions ..................................................................................................69
5 ELECTROCHEMICAL POLYMERIZATION OF POLYPYYROLE ON MG ALLOY AZ91D PART 2: CORROSION BEHAVIOUR OF PPY/AZ91D IN SIMULATED BODY FLUID SOLUTIONS AND FURTHER FUNCTIONILIZATION WITH ALBUMIN MONOLAYERS FOR DRUG DELIVERY APPLICATIONS .............................................................................71
5.1 Abstract........................................................................................................71
5.2 Introduction...................................................................................................72 5.2.1 Corrosion control with ICPs...................................................................72
5.2.1.a Displacement of the electrochemical interface..................................74
5.2.1.b Ennobling the metal by polymer in its oxidized state.........................75 5.2.1.c Self-healing effect by release of dopant anions.................................77
5.2.1.d Barrier effect of the polymer............................................................78 5.2.2 PPy in biomedical research....................................................................78 5.2.3 Mg and its alloys in biomedical research................................................80
5.3 Experimental route......................................................................................81
5.4 Results and Discussion ................................................................................82
5.4.1 Optimization of mechanical stability and adhesion: Effect of different cycles....82
5.4.2 Corrosion behaviour of PPy/AZ91D system in simulated body fluids......86 5.4.3 Modification of PPy layers with Albumin monolayers.............................91
5.4.4 Corrosion behaviour of Alb/PPy/AZ91D system in simulated body fluids94
5.4.5 Polarization curves of PPy/AZ91D and Alb/PPy/AZ91D electrodes......100 6.4.6 Surface characterization of PPy/AZ91D and Alb/PPy/AZ91D electrodes...103
5.5 Conclusions ................................................................................................103
6 ANODIC GROWTH OF SELF-ORDERED NANOPOROUS/TUBULAR LAYERS ON MAGNESIUM ALLOY WE43....................................................105
6.1 Abstract......................................................................................................105
6.3 Experimental route....................................................................................110
6.4 Results and Discussion ..............................................................................111
6.4.1 Optimization of anodisation potential...................................................111
6.4.2 Effect of anodisation time......................................................................117 6.4.3 Initiation of porous layer......................................................................118
6.5 Conclusions ................................................................................................119
7 CORROSION BEHAVIOUR OF CARBON NANOTUBE MODIFIED MAGNESIUM AND MAGNESIUM ALLOY AZ91D ......................................120
7.1 Abstract......................................................................................................120
7.2 Introduction ...............................................................................................121
7.3 Experimental .............................................................................................123
7.4 Results and Discussion ..............................................................................124
7.4.1 Corrosion behaviour of MWNT reinforced AZ91 composites................124

v
7.4.2 Corrosion behaviour of MWNT reinforced Mg composites....................128 7.4.2.a Influence of MWNT addition on the electrochemical behaviour of Mg128
7.4.2.b Characterization of the corrosion product layers...........................134
7.4.2.c Long term corrosion measurements................................................138
7.5 Conclusions ................................................................................................140
8 CONCLUSIONS AND OUTLOOK ...............................................................141 References:...........................................................................................................145 List of Abbreviations: .........................................................................................165
List of Symbols:...................................................................................................167

vi
Abstract:
Progressively, the well explored and studied mechanical properties of a bulk
metal are compared with the corrosion behaviour obtained from its surface, which
enables promising improvements in desired applications. An example is magnesium
metal: where, by developing new types of surface modifications by understanding its
inconsistent corrosion behaviour, it would be possible to apply this engineering metal
safely as a biocompatible metal, in addition to its widely used application areas such
as the automobile and aerospace industries. Accordingly, many scientists worldwide
have focused their attention on new approaches either to fabricate anti-corrosive
coatings or tailor the corrosion rate of magnesium for industrial and biomedical
applications.
This work covers different types of surface/bulk modification of magnesium
or magnesium alloys with different surface pre-treatments, polymer coatings,
nanostructuring and composite formation. For instance, the first part of this thesis
explores the chemistry of a very common surface pre-treatment technique, fluoride
activation by ammonium fluoride (NH4F) on a dual phase Mg alloy (AZ91D) surface
to enhance its corrosion resistance. NH4 treatment was applied after samples were
etched by H2SO4 and it is found that acidic pickling has a detrimental effect on
coating adhesion and uniformity by removing the non-stable, native oxide layer.
Additionally, acidic pickling time was also found to be a very important parameter
resulting in changes in the different distribution of phases on the surface. Therefore
corrosion protection of such a dual phase alloy can be significantly enhanced by
optimizing acidic etching and F-treatment conditions.
The formation of a conducting polymer coating (polypyrrole –PPy-) on Mg
alloy AZ91D is reported for the first time from aqueous solutions of sodium
salicylate by cyclic voltammetry (CV) method. PPy films are characterized by
surface analytic techniques and electropolymerization conditions such as scan rate-
and potential range, while monomer and salicylate concentration are optimized to
achieve adhesive PPy films on Mg alloy AZ91D. Moreover, corrosion behaviour of
PPy/Mg AZ91D alloy in simulated body fluid (SBF) solutions is also reported.
Results show that the adherent PPy coatings hinder the anodic dissolution of Mg
substrate and enhance the corrosion resistance by releasing dopant anions.
Furthermore, functionalization of PPy layers with Albumin (Alb) monolayers
provides better salicylate release profile over 20 days with zero order kinetics and

vii
shows the most effective corrosion performance. Such salicylate doped PPy layers
have a high potential for tailoring the degradation rate of Mg alloys, and therefore
could be used as biomedical components.
Further on, a small example of potentiostatic anodisation of a biorelevant Mg
alloy WE43 in hydrofluoric acid (HF) containing non-aqueous electrolyte shows
growth of self-ordered nanostructures. The morphology of the surface structures
varies with applied potential and anodization time whereby tubular or porous
structures with different sizes and orientation can be grown. Formation of these
structures was found to be potential- and time-dependent. In view of practical
applications, these nanostructured layers may also have considerable potential to help
formation of further biocompatible coatings (i.e. hydroxyapatite) or enhance cell
adhesion, as nanotubes on Ti have an influence on these effects.
In the end, an example related to bulk modification of Mg metal is given and
corrosion behaviour of carbon nanotube (CNT)/Mg composites is demonstrated. Due
to the coupling effect between cathodic CNTs and the anodic Mg alloy, the corrosion
rate is increased by adding increasing amounts of CNTs. Moreover, surface pre-
treatment and dispersion of CNTs was also found to be critical on the corrosion
performance. Therefore, in order to exploit the outstanding mechanical properties of
these composites, additional corrosion protection methods are required.

viii
Zusammenfessung:
Die gut erforschten mechanischen Eigenschaften eines Bulk-Metalls werden
in zunehmendem Maße mit dem auf seiner Oberfläche basierenden
Korrosionsverhalten kombiniert, was vielversprechende Verbesserungen für viele
Anwendungen ermöglicht.
Ein Beispiel ist Magnesium, welches – sollten durch das Verständnis seines
speziellen Korrosionsverhaltens neue Oberflächenmodifikationen entwickelt werden
- zusätzlich zu seinen klassischen Einsatzgebieten wie Automobilbau und Luftfahrt,
sicher als biokomptabiles Metall verwendet werden könnte. Dementsprechend zollen
viele Wisschenschaftler weltweit neuen Ansätzen große Aufmerksamkeit,
Korrosionsschutzschichten herzustellen oder die Korrosionsrate von Magnesium für
industrielle und biomedizinische Anwendungen maßzuschneidern.
Diese Arbeit umfasst diverse Arten der Material- oder Oberflächenmodifikation von
Magnesium oder Magnesiumlegierungen mit unterschiedlicher
Oberflächenvorbehandlung, Polymerbeschichtung, Nanostrukturierungen und
Kompositbildung. Beispielsweise wird im ersten Teil dieser Arbeit die Chemie
hinter einer allgemein üblichen Oberflächenvorbehandlung untersucht, der
Fluoridaktivierung der Oberfläche einer Zweiphasen-Magnesiumlegierung (AZ91D)
mittels NH4F, um ihren Korrosionswiderstand zu erhöhen. Nachdem die Proben in
H2SO4 geätzt wurden wurde die NH4F-Behandlung angewendet und es wurde
herausgefunden, dass das Beizen in Säure einen schädlichen Einfluss auf das
Haftvermögen und die Homogenität einer Beschichtung hat, indem es die instabile
native Oxidschicht ablöst. Außerdem zeigte sich, dass die Beizdauer einen sehr
wichtigen Parameter darstellt und zu Unterscheiden in der Phasenverteilung an der
Oberfläche führt. Aus diesem Grund kann das Korrosionsverhalten solch einer
Zweiphasen-Legierung durch die Optimierung der Bedingungen der Ätz- und
Fluoridbehandlung signifikant verbessert werden.
Des Weiteren wird erstmalig die Bildung einer leitfähigen Polymerschicht
(Polypyrrol –PPy) aus einer wässrigen Lösung von Natriumsalicylat auf der
Magnesiumlegierung AZ91D mittels zyklischer Voltammetrie (CV) gezeigt. Die
Ppy-Filme werden mit Oberflächenanalysetechniken charakterisiert und
Elektropolimersiationsbedingungen wie Scan-Rate, Spannungsbereich und
Monomer- sowie Salicylatkonzentration werden optimiert, um haftende Ppy-Filme
auf der Magnesiumlegierung AZ91D zu erhalten. Auch wird über das

ix
Korrosionsverhalten der Ppy/Mg AZ91D-Legierung in simulierter Körperflüssigkeit
(simulated body fluid - SBF) berichtet. Die Ergebnisse zeigen, dass die anhaftenden
Ppy-Beschichtungen die anodische Auflösung des Magnesiumsubstrats hemmen und
den Korrosionswiderstand verstärken, indem sie Dotierionen abgeben. Außerdem
weisen mit Albumim (Alb)-Monolagen funktionalisierte PPY-Schichten ein besseres
Salicylat-Abgabe-Profil (über 20 Tage, mit zero-order-kinetics) und zeigen ein sehr
zufriedenstellendes Korrosionsverhalten. Solche salicylatdotierten Ppy-Schichten
sind vielversprechend wenn es darum geht, die Degradationsrate von
Magnesiumlegierungen einzustellen und könnten so für biomedizinische
Anwendungen genutzt werden.
Des Weiteren zeigt das Beispiel einer potentiostatischen Anodisierung der
biorelevanten Magnesiumlegierung WE43 in einem nichtwässrigen Elektrolyt, der
Flusssäure (HF) enthält, Wachstum von selbstorganisierten Nanostrukturen. Die
Morphologie der Oberflächenstrukturen hängt ab von der angelegten Spannung und
der Anodisierungsdauer, wodurch röhrenförmige oder poröse Strukturen
unterschiedlicher Größe und Orientierung hergestellt werden können. In Hinblick auf
konkrete Anwendungen könnten diese Nanostrukturierten Schichten beachtliches
Potential bei der Herstellung weiterer biokompatibler Beschichtungen
(Hydroxylapatit) oder in Fragen der Zellanhaftung entfalten, da Nanoröhren auf Titan
diese Sachverhalte beeinflussen.
Schließlich wird ein Beispiel betreffend die Bulkmaterial-Modifikation von
Magnesium gegeben und das Korrosionsverhalten von Kohlenstoffnanoröhren
(carbon nanotubes – CNTs)/Magnesium-Kompositen wird demonstriert. Wegen der
gemeinsamen Verwendung kathodischer CNTs und der anodischen
Magnesiumlegierung wächst die Korrosionsgeschwindigkeit mit der Zugabe von
CNTs. Außerdem wurde gezeigt, dass die Oberflächenvorbehandlung und die
Dispersion der CNTs entscheidend für das Korrosionsverhalten sind. Deswegen
werden zusätzliche Methoden des Korrosionsschutzes dringend benötigt, um die
herausragenden mechanischen Eigenschaften dieser Komposite nutzen zu können.

1
1 INTRODUCTION:
CORROSION OF MAGNESIUM AND MAGNESIUM ALLOYS
1.1 Abstract
In this chapter, a general overview of the corrosion mechanism of magnesium
and its alloys is reported including summary of recent research achievements during
industrial and academic studies. This chapter also covers “anodic hydrogen
evolution”, corrosion rate estimations with the aim of deepening the understanding of
corrosion and protection of magnesium and its alloys and provides a base for later
chapters.

2
1.2 Corrosion of magnesium
After discovery of elemental Magnesium (Mg) by Sir Humphrey Davy in
1808, his assistant, Michael Faraday achieved the production of metallic Mg by
electrolysis in 1833. This invention was followed by the first attempt of commercial
production of Mg by Robert Bunsen in 1852. Afterwards, magnesium was produced
for use in flash lights in Europe and America and also suggested for use as ligatures
to stop bleeding by the physician Edward C. Huse [1]. These were the first signs of
this metal’s use in promising areas of science and technology. In the 1920’s, the first
magnesium parts were produced for race cars and this commercialization followed
with the release of the Volkswagen Beetle in 1936, which had 20 kg of magnesium
within its powertrain. In the following century, interest in Mg has grown and now
this metal is a strong competitor, along with its alloys, for many applications in many
fields in industry.
Magnesium has been used as a “structural” component in aerospace,
automotive [2-5], electronic [6-7] and other performance based industries such as
powertrain applications [8-14] due to its superior mechanical properties as a light
weight metal. On the other hand, due to its very low electrode potential (ESHE = -2.37
V), it can also be used as a sacrificial anode to protect other metals [15-18]. But,
most of the engineering applications of magnesium are limited due to its corrosion
rate. Therefore a significant and detailed understanding of the reasons behind its
corrosion is needed to minimize this drawback.
Upon all engineering metals used in industry, magnesium is the most active
metal and therefore corrodes very readily in many environments (Figure 1). Despite
this major drawback of magnesium, unprotected magnesium may also be more
resistant to atmospheric corrosion as compared to mild steel. This is mainly because
of air formed magnesium oxide which forms on exposure to the atmosphere at room
temperature.
There are two major reasons for the poor corrosion resistance of magnesium.
Firstly, the very electronegative potential of Mg makes it very prone to galvanic
corrosion that can be initiated by internal coupling components with more noble
potential (e.g. impurities [19] or second phases) or by external coupling with

3
dissimilar metals. Therefore analysis of these secondary phases as well as full
chemical composition is very important.
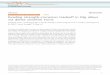
Figure 1: Galvanic series of most used engineering metals
Secondly, the quasi-passive hydroxide [20] film with a formula of Mg(OH)2
is not found to be as stable as other metal oxide films such as aluminum, nickel and
chromium oxide that provide better corrosion properties. Passivation of magnesium
with only this layer shows poor pitting resistance for both pure magnesium and
magnesium alloys. The Pourbaix diagram of magnesium in water is shown in Figure
2. The stability domain of magnesium is always found to be below that of water and
therefore magnesium dissolves in water with hydrogen evolution as Mg+(aq) and
Mg+2(aq) .
Figure 2: Potential-pH (Pourbaix) diagram for pure magnesium in water at 25 °C.

4
The passive film, Mg(OH)2 is found to be slightly soluble in water and does
not provide protection over long periods. However, it also breaks down in the
presence of aggressive ions like chlorides [21-24]. Formation of Mg(OH)2 and
magnesium dissolution in aqueous environments proceeds electrochemically
accordingly to the following reactions;
2H+ + 2e- → H2(g) (cathodic partial reaction)…….………………………………. (1)
2Mg → 2Mg + + 2e- (anodic partial reaction) ………………..………………..… (2)
2Mg+ + 2H2O → 2Mg+2 + 2OH- + H2 (chemical reaction)………………..........… (3)
2Mg + 2H+ + 2H2O → 2 Mg+2 + 2OH- + 2H2 (overall reaction)…..…………...…. (4)
Mg+2 + 2OH- → Mg(OH)2 (product formation)……………………………………(5)
During corrosion of magnesium, there are two important key points; firstly,
hydrogen evolution is observed that (4) is associated with both the cathodic partial
reaction (1) which balances the anodic reaction of magnesium dissolution (2) and the
chemical reaction of Mg+ with water (3). Overall in reaction (4), one mole of
hydrogen gas is generated for one mole of magnesium dissolved. Secondly, in net
reaction (4), H+ is consumed and OH- is formed favouring the formation of Mg(OH)2
and shifting the Mg to its passive domain in Figure 2 by alkalization of the medium
[25].
1.3 Corrosion rate measurements
Side products generated during corrosion of magnesium are helpful for
analyzing corrosion experiments. For instance, in addition to polarisation curves and
other electrochemical techniques, corrosion rate can be estimated more reliably by
collection of H2(g) simply using a burette and funnel as shown in Figure 3. As already
mentioned above, 1 mole of H2(g) is generated when 1 mole of Mg corrodes.
Therefore, by a simple calculation, it can be said that 0.92 ml of H2(g) is produced
from the corrosion of 1 mg of magnesium. Or, if a volume change of 1 ml is achieved
on the burette, this corresponds to corrosion of 1.08 mg of magnesium.

5
But this is not always found to give accurate results because of leaking or the
presence of other dissolved gases in the electrolyte. In these cases, the corrosion rate
can also be calculated from weight loss measurements and both of these results
should be compared for better accuracy.
Figure 3: Setup using for collection of H2(g) during magnesium corrosion.
The corrosion layer of magnesium, namely Mg(OH)2, is slightly soluble in
water. Therefore the removal of the corrosion products before weight loss
measurements is essential. To remove the corrosion products a mixture of 400 g/l
CrO3 + 20 g/l AgNO3 solution at room temperature is used [26]. The corroded
specimen is soaked in the mixture for 5 minutes and the corrosion products were
removed and weight loss values recorded (Figure 4).
Figure 4: Optical image of pure magnesium specimen surface a) before, b) after removal of corrosion products.

6
But in this case, other experimental difficulties occur: if the immersion time
of the specimen is too long, chemical etching of the magnesium substrate result; or if
the soaking time is too short, the complete removal of corrosion products is not
achieved. Therefore an experimental error always exists and should be reported for
corrosion of magnesium (Figure 5).
Figure 5: Comparison of corrosion rates calculated from two different methods for pure magnesium exposed in 3.5% NaCl 1 week.
1.4 Anodic hydrogen evolution: negative difference effect
In principle, corrosion rate of magnesium can be adequately estimated by
using polarisation curves and Tafel extrapolation. But in practice, the corrosion rates
evaluated by this method do not agree with the corrosion rates calculated from
weight loss and hydrogen evolution. Moreover, the deviation between these methods
has been shown to be up to 48% or 96% [27]. The reason behind this is accompanied
by different hydrogen evolution behaviour; more hydrogen is observed at more
anodic potentials or current densities. This kind of behaviour is a common
phenomenon and is called “negative difference effect” (NDE) [28].
For metals, all corrosion reactions take place in anodic and cathodic red-ox
processes. Normally, the cathodic reaction rate is lowered and anodic reactions are
more favoured by increasing the applied potential and therefore the anodic
dissolution rates are lowered. However, for magnesium, the hydrogen evolution

7
behaviour is found to be very different [25] than other metals and this type of
behaviour is very contrary to the fundamentals of electrochemistry theory.
Figure 6: Schematic presentation of negative difference effect (NDE). Expected anodic (Ie,a) and cathodic (Ie,c) current density values are shown on solid black lines. Dashed lines show measured cathodic (Im,c) and anodic (Im,a) current density values.
Figure 6 shows the comparison of NDE with normal polarization curves. The
normal anodic (Ia) and cathodic (Ic) partial reactions are shown by solid black lines
and the system is assumed to follow Tafel kinetics. The rates of reactions are equal to
the intersection point that corresponds to corrosion potential (Ucorr) and corrosion
current (Icorr), as expected. When the potential is increased to a more positive value
(Uapplied), the rate of the anodic reaction rate would be expected to increase to the
value of Ie,a. In parallel, the rate of cathodic reaction would be lower and decreased to
a value of Ie,c. However, for magnesium it is experimentally reported that [25] the
hydrogen evolution reaction, -cathodic reaction- rate increases (blue curve) rather
than decrease when higher potentials are applied. As a result, a significant increase of
the cathodic current value is observed (Im,c). On the other hand, the anodic current
(red curve) values can also increase faster than expected (Im,a). Therefore the
corrosion rates calculated from weight loss or hydrogen gas evolution methods differ

8
from the corrosion rates from polarisation curves at the applied currents by using
Faraday’s Law.
There are four important mechanism proposed to explain NDE [28]. The first
model [23, 29] postulates the breakdown of semi-protective film during anodic
dissolution process. However, the model was criticized because there is no direct
proof present showing this surface film is partially protective and it is not able to
explain the NDE in neutral and acidic solution. In another model [30], unipositive
Mg+ ions are assumed to be produced electrochemically and these intermediate
species can react chemically to evolve hydrogen. However, this mechanism is not
able to explain the drop of the magnesium dissolution at potentials 1000 mV higher
than the Mg+/Mg equilibrium potential. In the third model [31], NDE was explained
by undermining and falling of second phase particles in magnesium alloys. More
undermining would be expected at higher anodic current densities or potentials. But,
this model was also found to be insufficient to explain the NDE phenomena at higher
anodic potentials [32]. Lastly, a further model was postulated to explain the NDE
effect by means of the formation of a MgH2 layer on Mg substrates under certain
conditions. According to this model, reactive MgH2 decomposes into Mg+2 and H2
and therefore a significant amount of H2 can be produced at potentials in the anodic
direction.
Even though many mechanisms are postulated to explain the NDE effect, the
corrosion mechanism of magnesium is still debated and a common mechanism is still
not provided to cover this phenomenon for both magnesium and magnesium alloys
over a wide range of over potential region.
1.5 Improving corrosion resistance of magnesium
In general, mainly two approaches exist to achieve better corrosion resistance
for metals; alloying (e.g. stainless steel, Ni-based super alloys, hard metals) and
surface treatments (e.g. zinc-phosphate coatings, lacquers, paints). It has been
reported that alloying Mg with several metals such as Al, Be, Ca, Cu, Li, Mn, Ni, Re,
Si, Ag, Th, Sn, Zn, Zr and Y changes both the mechanical properties and corrosion
behaviour. These alloying elements either applied alone (binary alloys) or together
with other metals (ternary alloys) are mainly aimed to change desired mechanical
properties. Compared to all these alloying elements, aluminum has the most

9
significant effect on magnesium corrosion. It also improves the strength and hardness
and makes the alloy easier to cast. Commercial Al-Mg alloys with a content between
3-9% Al show the optimum strength and ductility. Zinc, which is next to aluminum,
is always used in conjunction with Al to improve the alloy’s room temperature
strength. For instance, addition of 9% Al together with 1% Zinc gives the well
known magnesium alloy AZ91D. Its combination with rare earth elements also gives
better mechanical properties. Rare earths are usually added as a mixture of Ce, La
and Nd to increase the strength at elevated temperatures.
Due to the fact that this approach is mainly aimed at achieving better
mechanical properties, in general alloying Mg does not provide a sufficient corrosion
protection in aggressive environments [33]. Therefore many industrial surface
treatments are developed to increase its corrosion resistance. Due to its high tendency
to galvanic coupling and its very anodic nature, these treatments have to be pore-free
to avoid aggressive attacks of electronegative anions such as chlorides. One of these
treatments involves coating of magnesium surfaces by other metals such as zinc [34-
36], chromium [37-38], copper[38] and nickel [39-40] metals. Although these
coatings are usually found to enhance corrosion resistance, due to their
electrochemical activity, many challenges are reported for these processes. Therefore
with these types of processes it is usually found to be very difficult to achieve
compact and pore-free coatings with multi-step preparation. Another import
challenge to consider is that the presence of heavy metals reduces the recyclability of
the metal [41].
As an another surface treatment strategy, environmentally friendly ceramic
layers such as phosphate, stannate, silicate and rare earth metal coatings are reported
to increase corrosion behaviour of magnesium, as summerized in a recent review
[42]. Due to their insoluble nature in acidic solutions, phosphate coatings have been
widely investigated since the 90s. During the conversion coating process of
magnesium, magnesium substrate is immersed in a reactive solution that alters the
metal ion concentration as well as the pH at the metal-solution interface. Change in
composition leads to precipitation of the phosphate species onto the magnesium
surface through formation of insoluble magnesium phosphate layers [42]. During
these processes, many factors such as temperature, time, bath stabilizers (Zn, Mn, V,
Ca, Mo, Sn, Co), substrate microstructure and surface state have many influences on
the resulting coating. In addition to phosphate conversion coatings, micro-arc

10
oxidation of magnesium metal has been widely investigated to achieve compact and
pore-free silicate and stannate ceramic layers and this method has become very
popular in the last decade. Proper choice of electrical parameters such as anodisation
potential/current, applied steps, duration and further anodisation in different
electrolyte solutions is very important and therefore this approach very sophisticated
[43].
In this work, in contrast to above mentioned traditional approaches, new types
of surface modifications on the magnesium alloy AZ91D surface is carried out. For
instance, in chapter 3, a very common surface pre-treatment technique, fluoride
activation by ammonium fluoride (NH4) is studied on Mg alloy (AZ91D) surfaces to
enhance corrosion resistance. In chapter 4, a first time formation of conducting
polymer coating (polypyrrole –PPy-) on Mg alloy AZ91D is reported from aqueous
solutions of sodium salicylate by cyclic voltammetry (CV) method. In chapter 5, a
small example of electrochemical formation of self-ordered nanotubular and
nanoporous magnesium oxy-fluoride structures on a magnesium-based alloy are
reported. In the end (chapter 6), influence of carbon nanotube (CNT) addition into
Mg matrix and its effect on the corrosion behaviour is studied.

11
2 EXPERIMENTAL METHODS
In this chapter, a main concept about experimental details is explained with a
common overview. More details and specific explanation concerning how the
experiments are performed will be explained in the experimental part of each chapter.

12
2.1 Electrochemical setup
For electrochemical experiments, three different types of electrochemical
setups were used in order to meet the specific requirements of different works and
reduce the side effects during corrosion tests. Figure 7 shows the electrochemical
setups used during the study. Figure 7a shows a conventional three electrode system
with an o-ring cell sealed to the working electrode (anode). A Cu back-plate was
used to achieve the electrical contact by pressing against the o-ring to expose a
defined electrode area to the electrolyte. Figure 7b shows the same three electrode
system where a simple flask with a total volume of 200 ml replaced the o-ring cell to
anodize bigger surfaces during experiments. A potentiostat/galvanostat Autolab
PGSTAT 30 was used where a Pt foil or stainless steel (SS) sheet served as a counter
electrode and an Ag/AgCl (3 mol dm−3 KCl) with a second capillary filled with the
electrolyte solution used as reference electrode to avoid attack of aggressive Cl- ions.
Results were monitored by NOVA software (Official software of Autolab).
For experiments at higher potentials, a two electrode system was used either
in a flask (Figure 7c) or in the o-ring cell and connected to Heinzinger 350-2 model
DC-Voltage source that interfaced to a computer via a USB-GPIB controller
(National Instruments) combined with a Keithley 188 model digital multimeter. The
anodisation progress and curves were monitored by in-house software programmed
in LabView.

13
Figure 7: Electrochemical setups used during the study; a) A conventional 3-electrode system with an
O-ring cell, b) Same electrochemical 3-electrode system with a simple flask as electrochemical cell, c)
2-electrode system with a power source.

14
2.2 Electrochemical methods and corrosion tests
2.2.1 Cyclic and linear sweep voltammetry
Cyclic voltammetry is a powerful type of potentiodynamic method where the
potential applied to the working electrode can be ramped linearly versus time
like linear sweep voltammetry. Unlike linear sweep voltammetry, that ends when it
reaches a set potential, the potential ramp of working electrode with respect to the
reference electrode is reversed at a set potential where upon it is scanned linearly
back to the initial potential again. This process is called a “cycle” and can be done
multiple times during a single experiment by using a triangle shaped potential
waveform.
The graph where the current measured passing through the working electrode
is plotted versus the potential applied is called a “cyclic voltammogram” and
generally used to study electrochemical analyses of red-ox active couples in an
electrolyte solution. Moreover, this technique is also used for electrochemical
electropolymerization of conductive polymers [44] and characterization of these
materials in monomer free electrolyte solutions to analyze electronic properties of the
polymer films [45-49].
In this work, cyclic voltammetry is used to obtain polypyrrole films on
magnesium substrates and the linear sweep voltammetry for corrosion tests with the
Tafel extrapolation method.
2.2.2 Electrochemical Impedance Spectroscopy (EIS)
Electrochemical impedance spectroscopy is a powerful emerging technique
where the analyses of materials in which ionic conduction is strongly dominated such
as fused salts, conductive polymers, ionically bonded single crystals and metals.
Because of its non-surface destructive background, this technique also helps to
understand the trend of the surface resistance during corrosion tests.
Basically, electrochemical impedance of a system is obtained by applying an
small excitation signal to have a pseudo-linear cell response [50];
Et = Eo sin (ωt)……………………………………………………………(6)

15
In a pseudo linear or linear system, the current response gained from the
sinusoidal excitation potential signal is considered as a sinusoid function at the same
frequency (ω) but shifted in phase (Ф) and shown as following;
It = Io sin (ωt +Ф)……………………………………………………..…. (7)
An expression analogous to Ohm's Law allows calculation of the impedance
of the system by dividing equations 1 and 2.
)sin(
)sin(
)sin(
)sin(0
0 φωω
φωω
+=
+==
t
tZ
tI
tE
I
EZ o
t
t …………………….……(8)
Total impedance of a system is then presented as a complex number by using
Euler’s relationship as following [50];
)sin(cos)( 0 φφω jZI
EZ +== …………………………………………….…(9)
A typical presentation of impedance data, Nyquist plot, is obtained where the real
part is plotted versus the imaginary part in reaction (4) as shown in Figure 8. For
magnesium and magnesium alloys, polarization resistance (Rp: the difference along
the real axis between the resistance at the highest and lowest frequencies) generally is
not equal to the charge transfer resistance (Rct or Rt: the diameter of the Nyquist
curve along the real axis) due to its pseudo-inductive nature in corrosive media (see
chapter 3). Therefore the impedance spectra of magnesium and its alloys can not be
explained easily as fundamental systems with one time constants where the corrosion
process is lying with combination of a single capacitor and a resistor (i.e. corrosion of
carbon steel).
Therefore in this study, further analysis of EIS data with equivalent circuit
modelling is not studied. Instead, the direct relationships are carried out. Even though
equivalent circuits in many cases are very valuable and helpful, the circuit elements
do not have straightforward and simple physical explanations in the case of this
work, as the electrochemical interface is quite complex and dynamic (e.g., corrosion
of Mg leads to an increase of the pH value in the surroundings, and this can influence
both the coating and the Mg substrate behaviour).

16
During EIS experiments, a sinusoidal perturbation of ±10 mV for 10 points
per decade in a frequency range between 100 kHz and 10 mHz used.
Figure 8: Nyquist plot of a magnesium alloy in 3.5% NaCl solution after 20 minutes of immersion.
2.2.3 Corrosion tests
Corrosion tests of magnesium specimens were performed by two methods;
a) A conventional gas evolution setup
b) Electrochemical polarization curves with Tafel Analyses
To determine the corrosion rates from hydrogen gas evolution, surfaces of the
magnesium samples were sealed to expose a defined electrode surface to the
electrolyte and placed in a beaker that is connected to a burette. The change of the
electrolyte level on the burette was recorded every day. To validate the corrosion
rates obtained from gas evolution setup, weight loss measurements were also
performed. Samples were removed from the test medium and immersed into 400 g/l

17
CrO3 + 20 g/l AgNO3 solution at room temperature for 5 minutes and the corrosion
products were removed and weight loss values were recorded [26].
Linear polarization curves were measured between -2.0 and -1.0 V by using a
three electrode system (Figure 7a) with a scan rate of 1 mV/s and the Tafel analysis
of the data was performed by Nova software. Tafel Plots can provide a direct
measure of the corrosion current, which can be related to corrosion rate. Additionally
with this technique, it is possible to measure extremely low corrosion rates, and it can
be also used for continuous monitoring of the corrosion rate of a mixed electrode
system.
2.3 Surface analytic methods
2.3.1 Scanning Electron Microscopy
In electron microscopy, electrons of the specimen surface are excited to high
energy levels [51]. During this process, electrons absorb more energy than their work
function and are ejected from the atomic shell. The energy of the emitted electrons
changes by only changing frequency or energy of the incoming beam. If the
excitation energy is too low, electrons can not escape. Interaction of the beam with
specimen surface produces secondary, backscattered, Auger electrons as well as X-
ray radiation and light (Figure 9).

18
Figure 9: Some atom-high energy electron interactions. The inner electron shells of atoms are lebelled according to standart notation (K, L etc.). a)Electrons pass to next layer of atoms with little loss of energy - Low-angle scattering, b) Back-scattering, c)Characteristic X-Ray emission for secondary electron and d) Emision of a second electron and an Auger electron. These different electrons ejected from the specimen surface are collected by different
detectors to form images. The interaction volume of signals is shown in Figure 10.
In SEM, secondary (SE) and backscattered electrons (BSE) are usually used to
produce images. SEs are produced by inelastic interactions with an energy loss and
produced by the interactions between valence electrons of insulators and
semiconductors or weak conduction-band electrons in metals. These electrons usually
escapes from energy levels below 50 eV and this provides highest spatial resolution
images.

19
Figure 10: Electron interaction volume within a sample. (Figure is not to scale)
BSEs are produced by elastic interactions of beam electrons with nuclei
(Figure 9b). The energy loss is usually less than 1 eV and gives slightly less spatial
resolution than SEs since they come from deeper parts of the specimen (Figure 10).
These electrons generally provide compositional and crystallographic information
due to electron channeling. For elements with higher atomic numbers, more
backscattered electrons are bounced back that makes the image brighter for atoms of
higher atomic number [52].
During this work, a field-emission scanning electron microscope, Hitachi FE-
SEM S4800, was used to investigate morphological characterization of the samples.
2.3.2 Auger Electron Spectroscopy (AES)
As shown in Figure 9d, Auger electrons are emitted from very close to the
surface and gives information about surface chemistry. Different than secondary or
backscattered electrons, Auger electrons are second ejected electrons. After the core
electron is removed by leaving a vacancy, another electron fulfils by releasing
energy. Sometimes this energy is transferred to a second electron and result in

20
ejection. This second ejected electrons are called Auger electrons [53]. Auger
electrons have energy between 100 eV to 2 KeV and mainly absorbed by the
specimen. Therefore, only Auger electron from the surface can be measured, making
Auger Electron Spectroscopy (AES) a separate surface method.
Auger electron spectroscopy (AES) was performed in a PHI Model 670
instrument with primary electron beam voltage of 10 kV. Spectra were detected by
using the manufacturer software and sensitivity factors.
2.3.3 Energy Dispersive X-ray Spectroscopy
When a high energy beam interacts and ejects an inner shell electron (Figure
9c), an outer shell electron moves into the empty orbit with a progression from
higher to lower energy states with the excess energy emanating from the material as
an X-ray of specific energy till all electron states are refilled. The specific energies of
these X-rays are detected by an energy- or wavelength-spectrometer adapted in the
SEM instrument. It usually provides rapid qualitative analyses [54]. On the other
hand where it is the wavelengths of the X-rays that is detected this is referred to as
wavelength dispersive X-ray spectroscopy (WDS), which provides greater elemental
resolution but with a considerably longer analysis time.
In this work, EDAX Genesis, fitted in the same SEM chamber Hitachi FE-
SEM S4800 was used for chemical analysis of the surface compositions.
2.3.4 Focused Ion Beam
The Focused Ion Beam (FIB) is an SEM technique that uses a Ga+ ion beam
in place of an electron gun in SEM. Generated secondary electrons or ions are used to
produce images of surface morphology as previously explained. Differently than
conventional SEM, Ga+ ions allow selective milling or peeling-off of a desired area
on the surface. It is usually used for cross sectional imaging, as well as modification
of semiconductor devices and failure analyses. Additionally, it is also used for
sample preparation for TEM analyses [55].
In this work, cross-sections of the samples were cut using a Zeiss brand
focused ion beam (FIB), Cross-Beam 1540, at the chair of “Allgemeine
Werkstoffeigenschaften” (WW1-Prof. Dr. Mathias Göken). Coarse millings of the

21
cross-sections and the final polishing processes were both executed at an acceleration
voltage of 5 kV. An additional platinum layer was deposited to protect the surfaces
from rounding-off effects.
2.3.5 X-Ray Photoelectron Spectroscopy
XPS, also known as ESCA –electron spectroscopy for chemical analyses-, is
a widely used surface analytic technique because of its sensitivity and reliability. In
XPS, specimen’s surface is irradiated with mono-energetic x-rays and photoelectrons
(photoelectric effect) with different work functions (binding energy) are ejected.
Binding energy and intensity of a photoelectron peak is characteristic for all elements
and hence, XPS detects elemental composition of the surface (top 1–10 nm usually).
It is widely used in surface science and technology applications including: polymers,
catalysis, corrosion, adhesion, semiconductor and dielectric materials, electronics,
magnetic media, and thin film coatings [56].
In this work, chemical composition was determined by employing X-ray
induced photoelectron spectroscopy (XPS)-Phi 5600, using Al Kα radiation.
2.3.6 Time of Flight Secondary Ion Mass Spectroscopy (ToF-SIMS)
In ToF-SIMS, a pulse of ions is used and a cloud of atoms are sputtered.
Some of the atoms are ionized and these particles of one polarity are accelerated into
a spectrometer. After they travel through a tube, depending on the molecular mass of
the particles, they arrive at an ion detector at different times. However, the lighter
ones arrive at the detection system before the heavier ones. The "Time-of-Flight" of
an ion is proportional to the square root of its mass, hence all the different masses can
be detected individually [57]. The ToF-SIMS technique gives chemical composition
of surfaces in ppm sensitivity. Transport of oxygen in ionic materials, reactions at
interfaces, composition of quantum dots and their interfaces, multilayer structures
and monolayers can be analyzed with this technique.
Positive and negative static time-of-flight secondary-ion-mass-spectroscopy
(ToF-SIMS) measurements were performed with a ToF-SIMS V spectrometer from
ION TOF, Münster). The samples were irradiated with a pulsed 25 keV Bi+ liquid-

22
metal ion beam. Spectra were recorded in the high mass resolution mode (m/∆m>
8000 at 29Si).
2.3.7 Fourier Transform Infrared Spectroscopy
FTIR is a very powerful technique to identify the “fingerprint” of organic and
inorganic chemicals. In principle, molecular bonds vibrate at various frequencies
depending on their bonding energies. According to quantum principles, these
frequencies correspond to lowest and higher frequencies between these two states
(ground state (E0) and the first excited state (E1)); the difference in the energy must
be equal to the energy of the detected light.
l
chEE
.01 =− ………………………………………………………………..(10)
Where; h = Planks constant, c = speed of light, and l = the wavelength of light.
The term Fourier Transform Infrared Spectroscopy (FTIR) refers to a recent
development converting data to a spectrum which makes the technique more
sensitive. FTIR is used to identify chemicals in paints, polymers, coatings and drugs
for example [58].
In this work, functional groups were characterized with a Fourier-Transform
Infrared Spectrophotometer from Bruker Instruments, Germany.

23
3 EFFECT OF ACIDIC ETCHING AND
FLUORIDE PRE-TREATMENT
ON CORROSION PERFORMANCE OF MAGNESIUM ALLOY
AZ91D
3.1 Abstract
Fluoride activation of Mg alloy surfaces by ammonium fluoride pre-treatment
is an important procedure used in industry for the removal of the non-stable, native
oxide layer from Mg alloys that can have a detrimental effect on coating adhesion
and uniformity during industrial coating processes. Furthermore, the corrosion
protection properties of the F-coated AZ91D surface can be significantly enhanced
by optimizing the time of acid etching and F-treatment.
The present chapter explores the effect of a surface pre-treatment by acid
etching in H2SO4 on the properties of such a F-coated Mg alloy AZ91D in 3.5%
NaCl electrolytes.

24
3.2 Introduction
Despite their ultra lightness and high strength to weight ratio with a density
that is two thirds of aluminum and one fourth of iron, application of magnesium and
its alloys in a wide number of industrial fields is restricted by their poor corrosion
resistance on which many reviews have been reported in literature [25, 28]. To
overcome this problem, different types of coatings that enhance the corrosion
resistance of Mg and Mg alloys have been developed. Even though a coating
processes including hexavalent chromium (Cr6+) [59] was widely used for producing
conversion coatings, the use of Cr6+ is reduced in recent years as it is found to be
carcinogenic [60]. Afterwards, other alternative procedures have been used in
producing eco-friendly corrosion protection coatings such as stannate[61-62], cerium
oxide[63], phosphate[64-65] and silicate[66] treatments, and some of these including
other industrial treatments have been summarized in recent reviews [41, 43, 67].
Along with the importance of these treatment methods, it has been also found
that pre-treatment methods are very important for the protection of magnesium
against corrosion. The importance of these pre-treatment methods originates in the
need to remove the passive oxide-layer that forms easily due to the high reactivity of
Mg [41] and has a detrimental effect on coating adhesion and uniformity. Industrial
pre-treatment methods such as alkali cleaning[68], acid [69] or fluoride activation
[70-71] are capable of removing this oxide layer. In the case of fluoride activation it
was previously found that this oxide layer is replaced by magnesium fluoride,
enabling further coatings on Mg or Mg dual-phase alloys.
Such a dual-phase alloy is die-cast AZ91D, which contains 9% Aluminum
and 1% Zinc. Aluminum has the most significant effect on magnesium improveing
its strength and its hardness. Moreover, it widens the freezing range and makes the
alloy easier to cast. However, zinc is often used with aluminum to improve room-
temperature strength. Additionally, it also helps to overcome the harmful corrosive
effect of iron and nickel impurities[72].
Figure 11a shows the phase diagram of the Mg–Al alloys which is generally
characterized by solidification of aluminum in magnesium. These alloys have two
phases: a Mg-rich α phase and an Al-rich γ phase, which is also called β phase.
Figure 11b shows the intermetallic β phase compound located at the grain boundaries
with a stoichiometric composition of Mg17Al 12 (at 43.95 wt.% Al) with an α-Mn–

25
type cubic unit cell model that acts as a cathodic centre for the α phase [25, 73]. The
inset EDAX spectra show two sharp peaks for Mg whereas the Al peak is only
prominent for the spectra from the white regions of the SEM images. The spectra
verify that the white regions in SEM images are the Al rich β phase and the darker
regions are the Mg rich α phase. In Mg-Al alloys α phase is found with a hexagonal
closely-packed, hcp structure with a Young modulus value around 45 GPa, where the
same value for the β-phase is about 80 GPa.
For Al-Zn alloys of magnesium in which the Al to Zn ratio is larger then 3:1,
new phases do not appear with zinc. In this case, the zinc substitutes aluminum in the
β-Mg17Al12 phase, creating a ternary intermetallic compound Mg17Al11.5Zn0.5 or
Mg17(Al, Zn) 12 [74-79].
Figure 11: a) Fragment of Mg–Al phase diagram (adopted from ASM Handbook Committee, 1986)
b) EDAX spectra of α- and β-phases of an AZ91D alloy surface after 5 s of etching in 2.5 wt% H2SO4.

26
Song and co-workers have reported that the β phase located at the grain
boundaries mainly serves as a galvanic cathode, where it couples with the α phase
due to the higher cathodic activity of the β phase [32]. Furthermore, they postulated
that, depending on its volume fraction, the β phase may also act as a surface barrier
against corrosion as opposed to a galvanic cathode. Until now, there are no works
that clearly report this postulated barrier-effect-against-corrosion of Mg in AZ alloys.
The main objective of the present chapter is to explore the effect of acidic
etching of AZ91D Mg alloy on the electrochemical properties of the alloy, as well as
on the nature of the F-coating formed on these etched surfaces during NH4F steeping.
3.3 Experimental route
Magnesium AZ91D coupons with dimensions of 2 cm x 2 cm x 0.5 cm were
mechanically ground with 1200 grit emery paper and then polished with 6-, 3-, and 1-
µm diamond paste using ethanol as surfactant followed by cleaning in 1:1
ethanol:acetone mixture for 10 minutes in an ultrasonic bath.
After polishing, the first step of the pre-treatment process, chemical etching
was employed in 2.5 wt.% H2SO4(aq) and the samples were subsequently sonicated in
deionized (DI) water and in ethyl alcohol prior to SEM imaging. To allow for
progressive imaging of the same surface area, SEM images were taken between
etching steps. After the samples were removed from the SEM, they were etched
again allowing the etching progress of the same position to be observed for
approximately 1, 2, 3, 5 and 10 seconds of etching.
For the second step of the pre-treatment process, steeping in NH4F(aq),
samples of different etching durations were prepared and etched as described above.
Etched samples were chemically treated by steeping in 2.5 mol dm-3 NH4F (aq)
solution (pH: 6.35±0.05, T: 80±5°C)[71]. After steeping, the samples were cooled
down to room temperature with DI water and stored in high purity (99.9%) ethyl
alcohol.
In addition to SEM, EIS analyses and OCP monitoring was carried out in 3-
electrode setup with an O-ring cell as already described in section 3.1 and 3.2.2,
respectively. To study the surface chemistry, F-species rich coatings were analysed
with time-of-flight secondary-ion-mass-spectroscopy (ToF SIMS) to depend on the
underlying phase of the alloy surface.

27
3.4 Results and Discussion
3.4.1 Etching Behaviour
To examine the effect of acid etching time on the electrochemical behaviour
of the sample, open circuit potentials OCPs of samples etched for approximately 1, 2,
3, 5 and 10 seconds were recorded. Figure 12 shows the OCP-time curves of the
samples over 20 minutes. After 20 minutes, there is small difference in OCP values
with a value of −1.535±0.004 V for all samples so that the influence of the acidic
etching onto the electrochemical behaviour was not clearly monitored. But on the
other hand, these potentials are, as expected, between the corrosion potentials of
Mg17Al12 (−1.17 V) and pure Mg (−1.62 V) [25].
Figure 12: OCP measurements of AZ91D alloy samples in 3.5% NaCl after different etching times of
1, 2, 3, 5 and 10 s in 2.5% by weight H2SO4.
To examine the changes after etching, the surface morphology of a sample
was monitored ex situ by SEM on several occasions during etching. Figure 133a-d
show SEM images of 1-, 2-, 3- and 5-s etched AZ91D samples allowing the relative
degree of α (Mg rich: dark) and β (Al rich: bright) phases to be observed. As is
evident from the SEM images, the Mg17Al12 rich structures have irregular shapes and
sizes. The results indicate that during the first five seconds of etching, the
interconnected β−phase structures become more visible.

28
Figure 13: SEM images of the same region of an AZ91D alloy sample after different etching times of
(a) 1 s, (b) 2 s, (c) 3 s and (d) 5 s, respectively in 2.5 wt% H2SO4.
Figure14: SEM images of the same region of an AZ91D alloy sample after different etching times of
(a) 3 s, (b) 5 s and (c) 10 s, respectively, in 2.5 wt% H2SO4.

29
Figure 15: Left: peeling-off and undercutting mechanism for AZ91D alloy. Right: tilted SEM image
of AZ91D alloy (at 45) showing the undercutting of the β phase after 10 s of etching in H2SO4
Figure 14a-c show SEM images of another area after 3, 5 and 10 seconds of
etching, which are representative of the overall trend observed for α and β phase
distribution. It is apparent, from the SEM images, that up until 5 seconds of etching
time, the β phase structures stay attached to the surface. However, during further
etching these structures are separated from the bulk alloy, and since they are only
loosely interconnected with each other, they become disconnected from the sample
surface (as shown in the model on the left of Figure 15) and fall into the solution.
Therefore, due to this mechanical instability, a maximum surface fraction of β phase
is obtained after 5 seconds of etching that reduces to a constant plateau for longer
etching times.
In contrast to the sudden change in β-phase, the Mg rich α phase etches in a
relatively constant manner. At the beginning of etching, dissolution of α phase
results in the formation of cracks in the top layer that then peels off to the edge of the
phase boundary revealing an underlying layer. This can be clearly observed in
Figure 13c-d and Figure 14-a. This peeling-off step is followed by the formation of
another crack on the freshly exposed surface. This crack then propagates beneath the
surface resulting in the peeling off of another layer promoting a continuously
repeating process that results in the formation of a cavity in the α phase and exposes
the β phase so that it extrudes from the surface as shown in the tilted SEM image of

30
Figure 15. Taking into account this kind of α-phase behaviour, the etching process
can be characterized as a “peeling-off” mechanism that creates cavities with stepped
edges. This mechanism has the properties that at the beginning of each stage a crack
forms on the surface due to the etching agent. A layer then peels off via
undercutting. Each step reveals more of the surface of each β phase region till
mechanical instability results in detachment from the surface as shown in schematic
of Figure 15.
After SEM investigations, samples were also investigated by EIS so as to gain
a better electrochemical understanding of the electrolyte-AZ91D interface. Spectra
of 1-, 2-, 3-, 5- and 10-seconds etched surfaces (step 1) were recorded and the
Nyquist plots for these samples are shown in Figure 16.
Figure 16: Nyquist plots of etched AZ91D alloy in 3.5% NaCl after different etching times in 2.5 wt%
H2SO4.
EIS experiments show two capacitive loops in the high and middle frequency
(HF and MF) domain. HF capacitive loops are usually attributed to both charge
transfer and the double layer associated with the interface between the electrolyte and
the alloy surface. Although, the capacitive loop in the MF region of all the curves is

31
small, it is always apparent. For MF capacitive loops, proposals are reported by
Song et al. and Baril et al.: According to Song et al. [80], these loops are attributed
to Mg+ concentration [73] within the broken areas of surface-oxide film; Baril et al.
[81-82] proposed that these loops are the relaxation of mass transport in the growing
solid oxide phase. In Figure 16, a clear trend is observed in the charge transfer
resistance Rt (i.e., the diameter of the Nyquist curve along the real axis). The value
of Rt increases from ~260 Ω for the 1-second etched sample to ~750 Ω for the 5-
second etched sample. Although there is an increase of Rt from the 3- to 5-second
etched sample, a decrease in Rt is observed for 10 seconds of etching time.
The polarisation resistance (i.e., the difference along the real axis between the
resistance at the highest and lowest frequencies) is almost the same for all samples
due to the presence of an inductive loop in the LF domain region of all the curves.
In the case of Mg, inductive loops are commonly reported for Nyquist curves
in LF regions as scattered points [32, 82]. Inductive loops in EIS plots were firstly
reported for corroding or dissolving systems [83] and can be explained by a faradaic
impedance if the dissolution of the metal (Me) to solution (i.e., as Me+) involves an
intermediate transition to, for example, (MeOH)ads. Where such an intermediate
transition exists, the imaginary part of the complex representation of the faradaic
impedance can either be capacitive or self-inductive. The nature of the imaginary
part is dependent on which of the two intermediate reactions is greatest which is
decided by the applied voltage. Inductive behaviour is generally assumed to be due
to the presence of adsorbed surface species such as Mg(OH)+ads, Mg(OH)2 ads, [80-82]
and Mg+ads [32]. For Magnesium and magnesium alloys, at open-circuit potential,
inductive behaviour results at low frequencies and is observed as an inductive loop in
Nyquist Plots [84]. In addition, strong inductive loops are also reported due to the
high concentrations of Mg ions on relatively film-free surface [84-85]. However,
Anik et al. [86] found that low-frequency inductive-loops have a tendency to
disappear when the dissolution rate of the alloy decreases in the presence of a
protective oxide layer.
For our results, the scattered points in the low frequency (LF) domain indicate
that the slow reactions at the interface are not stable enough to allow smooth curves.
In addition, the immersion time was less than 1 hour so the corrosion products did
not completely cover the whole surface. An increase in inductance with etching time
was observed for all samples as a corresponding increase in α-phase surface area was

32
observed in SEM images. Therefore, the increased inductance with increased
duration of etching may be attributed to increased presence of absorbed Mg species
on the electrode surface due to an increase in α-phase surface area.
Up until a duration of 5 seconds of etching, the overall surface area increases
at the same time as the area of β-phase surface increases. This etching also causes a
absolute increase in the area of the α phase. Although the α phase surface area
increases during this etching duration, the exposed surface area of the β phase
increases sufficiently so as to create a barrier effect that increases the overall charge
transfer resistance. In addition, the exposed interconnected β phase structures reach a
maximum linkage and play an important role as active cathodic sites on the surface.
For durations greater than 5 seconds the β-phase structures detach from the surface
due to mechanical instability (Figure 14). Thus, for the 10-seconds etched sample,
there is a decrease in charge transfer resistance but a continued presence of inductive
behaviour.
3.4.2 Fluoride treatment optimization
After acid etching in H2SO4 for the optimum time of 5 seconds the treatment
of samples was completed by steeping in ammonium fluoride solution of pH
6.30±0.05 at 80±5°C. The EIS spectra for a range of different sample steeping
durations from 1 minute to 2 hours are shown in Figure 17. For steeping times of 20
minutes or less no inductance effect is present and the polarisation resistance, Rp (i.e.,
the difference along the real axis between the resistance at the highest and lowest
frequencies), increases with increasing steeping time. Thus, for steeping of 20
minutes or less, the ammonium fluoride treatment forms a passive layer that protects
the surface reducing the flow of current and halting the α-phase dissolution of Mg to
Mg2+ via MgOH. For steeping times greater than 20 minutes the polarisation
resistance diminishes and inductance effects reappear and grow as the steeping
duration is increased. These reductions in the benefits of the treatment method after
extended durations of steeping may be due to deterioration of the passive surface-
layer from mechanical stress or chemical etching due to temperature and bubbling of
the solution or chemistry of NH4F, respectively.

33
Figure 17: Nyquist plots of AZ91D alloy samples in 3.5% NaCl for different fluoride treatment times
after 5 s of etching in 2.5% by weight H2SO4.
Figure 18 shows SEM images of NH4F treated surfaces that underwent the
same treatment as the samples shown in Figure 6 for 20 and 120 minutes of steeping.
In Figure 18a the layer that passivates the surface covers the α and β phases. Contrary
to this, in Figure 18b almost all of the chemically more stable β phase has detached
from the surface and unlike Figure 18a the layer that covers the surface has
developed many cracks and fissures. Together these deteriorations in the surface of
the 120-minute sample may facilitate the lower polarization resistance and the
increased inductivity that are observed in the EIS Nyquist curves (Figure 17) of
greater than 20 minutes steeping.

34
Figure 18: SEM images of an AZ91D alloy after different treatment times of (a) 20 min and (b) 120
min, respectively, in 2.5 mol dm−3 NH4F solution.
To further examine the effect of etching time on fluoride pre-treatment of the
surface, EIS was performed on samples for the optimum steeping duration (step 2)
but for different durations of step 1. The EIS results for the 1-s etched sample show a
low Rt of around 1 kΩ (Figure 19) with a complete circle characteristicly indicating a
large inductance. For the 2 seconds etched sample a larger Rt value with relatively
small inductance is achieved. This trend of decreasing inductance and increasing
resistance was observed to continue for increased etching time in the case of 3-
second and 5-second etched samples where Rt values of approximately 6 kΩ and
9 kΩ, respectively, were achieved. However, for the 10-second etched sample, a
dramatic decrease in Rp was observed. As in the case of samples that were not
steeped after etching (as shown in Figure 16), this decrease may be related to the
mechanically unstable β phase as described in our model (Figure 15-left).

35
Figure 19: Nyquist plots of fluoride treated AZ91D alloys after different etching times in 2.5 wt%
H2SO4.
Figure 20: Nyquist plots of AZ91D alloy in 3.5% NaCl after polishing (blue squares: both in the main
image and magnified plot of the inset); after polishing and 5 s of etching (red circles); and after
polishing, 5 seconds etching and 20 min steeping (black triangles).

36
The optimum conditions, found in our study, for the two steps of the
treatment process (i.e., H2SO4 etching and NH4F steeping) have a progressive effect
on charge transfer resistance and surface protection (Figure 16-17). For the polished
sample the Rt is approximately 40 Ω (inset of Figure 20). By etching the surface for
5 s the Rt resistance increases to almost 1 kΩ (Figure 16). We propose that this
increase is due to the increased surface fraction of β phase which increases the
resistance significantly enough to compensate for the increase in surface area due to
surface roughening. We further propose that since the α phase is still present the
faradaic inductance also increases. Finally, through fluoride activation of the surface
by steeping in NH4F(aq) solution, which leads to the formation of a thin fluoride
passivating layer, the α phase becomes insulated from the solution-surface interface
removing the faradaic inductance (Figure 17). Furthermore, the F- passivating layer
creates a resistance in series with the original surface increasing the charge transfer
resistance to approximately 8 kΩ.
3.4.3 Chemistry of fluoride layer
To analyze the morphology and elemental constituents of the fluoride layer,
SEM and ToF-SIMS analyses were performed. These analyses where carried out on
samples that where etched for various durations from 1 to 10 s and steeped for the
optimum steeping duration of 20 min. To investigate the elemental constituents,
ToF-SIMS spectra of both positively and negatively charged secondary ions were
recorded for the different samples along with mapping of the distribution of the
major spectral peaks. All observed ions are summarized in Table 1 and Figure 21
shows the most prominent peaks along with their corresponding assigned chemical
formulae in order of increasing ion mass. The mass numbers marked with an asterisk
indicate the peaks that were resolved clearly for the 5- and 10-second samples only.
On the 1-s, 2-s and 3-s etched samples, characteristic multiplets of peaks resulting
from the isotopic pattern of Mg can be assigned to a series of species consisting of
MgxF2x-1+
and MgxOF2x-3+ in the positive and MgxF2x+1
- and MgxOF2x-1
- in the
negative ion spectra. The counts per total Bi+ shots for these multiplets vary with
increased etching time. An additional series of signals that can not be ascribed to Mg
containing species (due to disagreement with the isotopic pattern of Mg) are obtained
for the 5- and 10-second etched samples. These signals occur as single spectral

37
peaks and are attributed to Al containing species. They were assigned to AlxF3x-
5(OH)2+ and AlxF3x+1
+ in the positive, and AlxF3x-3(OH)2- and AlxF3x-1
- in the negative,
ion-spectra.
Mass
Fragment (Positive Tof
SIMS)
Origin
Mass
Fragment (Negative Tof SIMS)
Origin
18 NH4+ coating 16 O- substrate
24 Mg+ substrate 17 OH- substrate 43 MgF+ MgxF2x-1 19 F- coating 105 Mg2F3
+ MgxF2x-1 62 MgF2- MgF2
81 MgF3- MgxF2x+1 149 - Phthalate
contamination 102 Mg2OF2- Mg2OF2
167 Mg3F5+ MgxF2x-1 121 Mg2OF3
- MgxOF2x-1 191* Al3F4(OH)2
+ AlxF3x-5(OH)2 143 Mg2F5- MgxF2x+1
207 Mg4OF5+ MgxOF2x-3 183 Mg3OF5
- MgxOF2x-1 229 Mg4F7
+ MgxF2x-1 187* Al 2F7- AlxF3x+1
233* Al 3F8+ AlxF3x-1 205 Mg3F7
- MgxF2x+1 269 Mg5OF7
+ MgxOF2x-3 229* Al3F6(OH)2- AlxF3x-3(OH)2
275* Al4F7(OH)2+ AlxF3x-5(OH)2 245 Mg4OF7
- MgxOF2x-1 291 Mg5F9
+ MgxF2x-1 271* Al 3F10- AlxF3x+1
317* Al 4F11+ AlxF3x-1 267 Mg4F9
- MgxF2x+1 331 Mg6OF9
+ MgxOF2x-3 307 Mg5OF9- MgxOF2x-1
353 Mg6F11+ MgxF2x-1 313* Al4F9(OH)2
- AlxF3x-3(OH)2 359* Al5F10(OH)2
+ AlxF3x-5(OH)2 329 Mg5F11- MgxF2x+1
401* Al 5F14+ AlxF3x-1 355* Al 4F13
- AlxF3x+1 415 Mg7F13
+ MgxF2x-1 379 Mg6OF11- MgxOF2x-1
391 Mg6F13- MgxF2x+1
397* Al5F12(OH)2- AlxF3x-3(OH)2
Table 1: Table containing all relevant species detected in ToF-SIMS analysis.

38
Figure 21: Representative section of Tof-SIMS spectra of fluoride pre-treated AZ91D alloy samples
after different etching times of 1, 2, 3, 5 and 10 s in 2.5 wt% H2SO4.
Figure 21 shows a section of the negative spectra illustrating the variation in
spectral counts for the Mg3F7- multiplet at 205 u (unified atomic mass unit) and the
Al2F7- and Al3F6(OH)2
- single spectral lines at 187 and 229 u, respectively. For the
1-s curve only the Mg associated multiplet is observable. As etching time is
increased, the counts per total number of Bi+ shots decreases for this multiplet until 5
s and then increases slightly for 10 s. Furthermore, for the 5-s etched sample, Figure
21 also shows the maximum spectral-line counts for the masses associated with Al
species. For etching times less than 5 s these peaks are almost indistinguishable from
the background noise, probably due to a combination of the low penetration depth of
the ToF-SIMS technique and a smaller apparent surface fraction. In addition, for
longer etching times the peak counts decrease slightly, in agreement with the
decrease of β phase on the surface as observed in SEM images.

39
Figure 22: Left: Positive ion ToF-SIMS image of AZ91D alloy after fluoride pre-treatment. Right:
ToF-SIMS mapping results for the cavity, indicated by the box in the left image, for (a) Mg, (b) F and
(c) NH4 positive ions, and (d) O, (e) OH and (f) F negative ions.
Smaller cavities were also investigated on the 3-s and 5-s etched samples and
they show comparable results. However, larger cavities are represented.
Figure 23: High-magnification SEM images of the steeped edge of a cavity in a pre-treated (5 s 2.5
wt% H2SO4 etched and 20 min fluoride steeped) AZ91D alloy surface.
Figure 22 shows a ToF-SIMS positive ion image for all ion masses of the
overall surface and several higher magnification mappings for the distribution of

40
selected mass values across a cavity (the location of which is marked by a box on the
overall map). The investigated cavity has a relatively large diameter of 50 µm. The
most pronounced signals originating from the large cavity of the 3-s etched sample of
Figure 22 are Mg+, MgF+, NH4+, O-, OH- and F-. The signal of Mg+ is most abundant
in the positive ion detection while NH4+ yields a comparatively small signal. In
negative ion detection mode, F- has the most prominent signals originating from the
cavity. The reason for lack of Mg observed within the cavity can be due to a higher
formation probability for negative ions of the species in the cavity or due to a
topography effect.
Figure 24: Effect of extraction bias on topography in ToF-SIMS
The applied bias to the extractor in this mode can bend the path of the
incident Bi+ beam, resulting in only few incident ions hitting the actual cavity in the
positive detection mode; In negative detection, the beam path is bent in a way that
many incident ions hit the cavity interior. Rossi et al. [87] also observed less
contrast for ToF-SIMS images of cavities recorded by negative-ion detection.
Figure 23 shows a high-resolution SEM image of the edge of a cavity after 5
seconds of H2SO4 etching and 20 minutes of NH4F steeping. The image shows only
the α phase. A highly structured network can be found on the top layer (on the right
of the image) but this coating becomes less visible on approaching the centre of the
cavity. This indicates that the structured coating formed on the cavity circumference

41
is thicker than that formed in the cavity. According to ToF-SIMS mapping results,
these networks correspond to chemically bonded Mg-F structures.
Progressing from the edge of the cavity to the cavity centre the fluoride
containing species disappear from ToF-SIMS analysis (Figure 22f) and the structured
network reduces in density in SEM images (Figure 23). Figure 22d and Figure 22e
show O- and OH- species positioned in the middle of the figure indicating an unstable
oxide layer is still located mainly at the centre of the cavity. This film may be the
result of the formation of a large degree of Mg-OH species on the α phase at the
cavity bottom during the etching step or, indeed, during the steeping step itself, or
indeed it may be due to a thinner coating at the centre that allows the detection of an
underlying O- or OH- species. Oxygen is also observed by EDX measurements (EDX
results in Figure 11 after etching but oxygen peaks were mainly removed from EDX
measurements after fluoride steeping (EDX spectra not shown here).
In the literature it has been reported that a mixed oxide/hydroxide layer is the
favoured product on magnesium alloy surfaces in solutions containing water [88].
Furthermore, it has been proposed that the fluoride layer may be a fluoride
substituted magnesium hydroxide layer with a formula of Mg(OH)2-xFx or a mixture
of Mg(OH)2 and MgF2 but not of MgxOF2x-3 [89]. Our results support a mechanism
including substitution or exchange of hydroxyl ions with fluoride ions, that takes
place after Mg(OH)2 formation, resulting in magnesium fluoride (MgF2) or
magnesium-oxo-fluorides (Mg-O-F) formation, depending on the nature of the alloy
[71]. Where this formation of Mg(OH)2 is greatest (e.g., in the centre of the cavity),
it may not be completely replaced by a fluoride layer during steeping. Alternatively,
it is also possible that Mg-F species may be formed by anodic dissolution of
magnesium followed by a reaction with the fluoride ions in solution, without the
formation of an intermediate Mg(OH)2 stage [41].
3.5 Conclusions
In this chapter, dependence of the corrosion behaviour of AZ91D alloy on the
β phase was investigated in respect to acid etching time. The etching time was found
to be a very important parameter resulting in changes to the different distribution of α
and β phases on the surface. Where samples are only etched, the distribution of α
and β phases changes with increasing etching time and these changes result in

42
increases of both charge transfer resistance and faradaic inductance. In addition,
SEM investigations showed that the etching proceeded via a peeling-off mechanism
that creates cavities with stepped edges on the freshly exposed surface.
Steeping of samples for up to 20 min in NH4F solution removes inductance in
EIS spectra. However, for longer steeping durations, inductance effects reappear.
This reappearance may be due to exposure to solution of the α phase because of
either mechanical instability of the β phase or surface cracking in the protective
fluoride layer.
SEM and ToF-SIMS were used to give a detailed description of the fluoride
layer and highly structured Mg-F networks were found on the top layer of the α (Mg)
phase. According to mapping results, the centre of the cavity showed a drop in F
intensity and a very strong O-/OH- signal that belongs to a mixed oxide/hydroxide
layer. The SEM and ToF-SIMS characterization indicate that the surface layer
formed in NH4F is non-homogenous in thickness and chemical composition.
In parallel to results obtained for NH4F+ pre-treatment in literature [71], a
systematic investigation of experimental parameters showed that optimum conditions
could be achieved for etching and steeping durations of 5 s and 20 min, respectively.
For industrial applications, combination of two steps can significantly affect the
ultimate durability of any surface treatment as the total corrosion performance is
mostly based on the quality of the pre-treatment. In view of modifying magnesium
surface with a thin layer mixture of magnesium oxides/hydroxides or fluorides, it
would be interesting to explore its effect after a zinc-phosphate coating or hard
anodisation process. It would also be of interest to clarify, if a time limit for the pre-
treatment conditions reported here exist- for instance during rapid growth of silicate
coatings during arc-oxidation process.

43
4 ELECTROCHEMICAL POLYMERIZATION
OF POLYPYYROLE ON MG ALLOY AZ91D
PART 1: CHARACTERIZATION AND OPTIMIZATION OF
ELECTROPOLYMERIZATION PARAMETERS
4.1 Abstract
In this chapter, a first time formation of conducting polymer coating
(polypyrrole –PPy-) on Mg alloy AZ91D is reported from aqueous solutions of
sodium salicylate by cyclic voltammetry (CV) method. The polymeric films of PPy
are directly coated onto AZ91D substrates without any pre-treatment or intermediate
steps. The electrochemically formed PPy films are characterized by surface analytic
techniques. Electropolymerization conditions such as scan rate, potential range,
monomer and salicylate concentration are found to play an important role on the
adhesion and hence the corrosion performance of PPy films.

44
4.2 Introduction 4.2.1 Synthetic metals and doping
Since their discovery in 70s [90] by Shirakawa, Heeger and MacDiarmird,
intrinsically conducting polymers (ICP)s such as polythiophene (PT), polyaniline
(PANI) and polypyrrole (PPy) and their derivatives have been explored to be
promising nominees in the development of semiconductor technologies such as
biosensors [91], actuators [92], fuel or bio-fuel cells [93-94], light emitting diodes
(LED) [95] and supercapacitors [96-98].
Semiconductor materials have electrical properties between conductors and
insulators (Figure 25) because of their free electrons in crystalline patterns. However,
electrical conductivity of semiconducting materials can be further improved by
adding certain impurities to increase the number of free electrons and thereby
conductivity. These doping agents can be donor or acceptor atoms depending on
whether they produce electrons or holes that is so called doping. Doping can be
achieved by two possible ways; for instance addition of Antimony (Sb) into a Silicon
(Si) matrix (Figure a) allows four of the five electrons to bond with silicon atoms
leaving one free electron. In this case, positively charged donor atoms lead to
ejection free electrons when a potential is applied (n-type doping). This could be also
achieved when covalent bonds of the silicon matrix are excited by a sufficient
energy. In the second case in Figure b, if Boron (B) with only three valence electrons
is introduced into the crystal structure, a fourth covalent bond cannot be formed. This
leads to the formation of positively charged carriers known as "holes" in the crystal
structure. When the hole is occupied with an adjoining free electron, the electron
filling the hole leaves another as it moves (p-type doping). Doping of ICP polymer
backbones shows the same properties as metallic doping. For instance, p-type doping
is dominant for semi-conductive polymers such as PPy and electron transfer is
achieved by alternation of double bonds in the π-conjugated polymer backbone
followed by delocalization. Transfer of charged species through the carbon chain
therefore allows for electron transport and gives an electronically conductive
material.

45
Figure 25: a) Comparison of conductivities between conductive polymers and other materials. Schematic presentation of b) n-type and c) p-type doping of Si.
Dopant molecules remove or add electrons play an important role during
doping of ICP molecules. For example, when polyacetylene (Figure 26a) is doped
with iodine (I2), it abstracts an electron from the valence band of the polymer and
forms I3- (Figure 26b). This leads to formation of a hole that does not delocalise
completely and the charge is carried along the chain (Figure 26c-d). But, due to low
mobility of the I3- counter ions as compared to the positive charge carriers, higher
concentration of I3- is required to move the polaron close to counter ions. Therefore
doping concentration is found to be very important in the case of conducting
polymers [99-100].

46
Figure 26: a) trans-polyacetylene, b) Formation of radical cation (”polaron”) by removal of one electron and polaron migration (c → d).
4.2.2 Pyrrole and electropolymerization of conductive polymers
Among the conductive polymers, PPy is found to be one of the most suitable
candidates for corrosion protection because of its relatively easy preparation from
aqueous solution and stability at oxidized state. The electropolymerization
mechanism of ICPs is a complicated subject as there have been many different
mechanisms proposed till now. One of the difficulties is the determination of the
different stages of reaction because of the polymerization. However, the insolubility
of the PPy along with its non-crystalline nature makes the characterization difficult.
As a result, there has not been an agreement among researchers concerning the
electropolymerization mechanism of PPy.
Amongst the many polymerization mechanisms, the most discussed
mechanism was proposed by Diaz et al. [101] and confirmed by Waltman and
Bargon [102-103]. The first step of this mechanism implies the oxidation of
monomer M at the electrode to form cation radical M.+ (Scheme 1).
In the second step, because of a greater unpaired electron density in the α-
position of the radical cation, coupling of two radical cations results in the formation
Scheme 1

47
of the dihydromer-dication (Scheme 2). In the stabilization (third) step of
electropolymerization, loss of two protons forms the aromatic dimer of pyrrole
(Scheme 3)
In the next step of the polymerization reaction, oxidation of the dimer results
in the formation of pyrrole trimer (Scheme 4) and propagation of the polymer chain
takes place (Scheme 5).
Electrochemical polymerization of Py always gives PPy in its oxidized
conducting form (doped) and the resulting polymer chain carries a positive charge
every 3 or 4 repeating pyrrole units which are balanced by counter anions. Studies
showed that the composition of PPy chains consist of 65% polymer and 35% counter
anion in weight [104]. Reasons supporting the relevance of Diaz’s mechanism is
believed to be the best one for number of reasons. Firstly, the mechanism is in
agreement with EPR (Electron Paramagnetic Resonance) results. Moreover, the
elimination of H+ is in agreement with the observed drop in pH of the solution during
polymerization [105].
Scheme 4
Scheme 5
Scheme 3

48
Among all other mechanism that are summerized in a recent review [104],
the one proposed by Diaz which features a combination of several successive
reactions (radical cation formation, radical coupling, and deprotonation) is certainly
the most probable due to explained reasons above.
4.2.3 Conducting polymers on magnesium
Because of its relatively easy preparation from aqueous solutions and stability
at oxidized state, PPy and its derivatives are reported to be one of the most important
candidates for corrosion protection. (Details of corrosion protection by ICPs will be
discussed in next chapter). This biocompatible ICP [106-107] is also the most widely
explored one as a drug delivery material with many advantages as summarized in a
recent review [108].
Although there are many studies on the electrochemical polymerization of
pyrrole on widely used metals such as zinc [109], aluminum [110], iron [111], copper
[112] and these works report that PPy coatings show good mechanical stability and
corrosion protection in corrosive media, only a few attempts were aimed to achieve
ICP coatings on Mg substrates. Guo et al. [113] showed electropolymerization of
aniline on AZ91D alloy surface using an electrochemical pulse method in alkaline
solutions, and Jiang et al. [114] reported electropolymerization of pyrrole on a Cu-Ni
plated Mg AZ91D surface. Even less information is available on direct
electrochemical deposition of PPy on Mg [115] surfaces. This type of surface
modification with ICPs may be promising for many applications of Mg alloys,
including their usage as drug eluting stents or other types of self degradable metallic
implant materials. In these applications, surface modification techniques are of
interests which are able to slow down the dissolution rate of Mg, but not completely
stop corrosion.
Due to its fast dissolution rate and very negative corrosion potential of Mg,
direct polymerization of conducting polymers on the Mg surface is difficult. To
overcome this problem, surface passivation is required [112] prior to initiation and
attachment of conductive polymer coating. In a different approach, Petitjean et al.
[116] and Hermelin et al. [117] recently showed electropolymerization of PPy
coatings from sodium salicylate solutions on zinc without any pretreatment.
However, Cascalheira et al. [118] determined the oxidation peak of salicylate at

49
around 1 V and showed the formation of a Cu(II)-salicylate film is favoured at the
electrode surface and contributes to its passivation.
Taking the advantage of sodium salicylate, film formation occurs in a one-
step process, and does not need any preliminary treatment of the metal. They also
claimed that in aqueous media, sodium salicylate is the most suitable candidate that
allows the formation of an adherent and homogeneous PPy film on active metals
without pre-treatment.
Moreover, sodium salicylate is reported to be a drug in medicine as
replacement of Aspirin (Figure 27) and also reported to have analgesic, antipyretic
and non-steroidal anti-inflammatory (NSAID) properties that lower the level of
apoptosis [119-120] in cancer cells.
Figure 27: Molecular structures of a) Sodium salicylate and b) Aspirin.
4.3 Experimental route
Magnesium AZ91D coupons (2 cm × 2 cm × 0.5 cm) were ground with 1200
grit emery paper and then were cleaned in 1:1 ethanol : acetone mixture for 10 min in
an ultrasonic bath. Electrochemical analyses were performed using an o-ring cell
(See 2.1 Electrochemical setup) with a three-electrode system.
PPy films were synthesized from 0.1 mol/l pyrrole (Py) containing aqueous
solutions of 0.5 mol/l carboxylic acid salts, sodium oxalate (Sigma), sodium
malonate (Sigma) and sodium salicylate (Sigma) (pH=6.50) using cyclic
voltammetry (CV) between -0.5 V and +1.0 V at 20 mV s-1 scan rate for 20 cycles. It
is also possible to use conventional methods for electropolymerization. Electrolytes
of 0.5 M sodium oxalate, sodium salicylate and sodium malonate with addition of 0.1
M Py were used. Formation of a black film (pyrrole black) was obtained only from

50
0.5 M sodium salicylate + 0.1 M Py solution. Where the the other electrolytes were
used, either corrosion of AZ91D resulted or no surface coverage was obtained.
Afterwards, electrochemical characterization of thin PPy films was performed
in monomer-free solutions of the same electrolyte via CVs. EIS measurements were
performed in 0.1 M NaSO4 with a sinusoidal perturbation of ±10 mV for 10 points in
each decade over the frequency range from 100 kHz to 10 mHz. Functional groups
were characterized with a Fourier-Transform Infrared Spectrophotometer (Bruker
Instruments - Germany). The chemical composition was investigated by employing
X-ray induced photoelectron spectroscopy (XPS)-Phi 5600. Also, positive and
negative static time-of-flight secondary-ion-mass-spectrometry (ToF-SIMS)
measurements were performed on a ToF SIMS V spectrometer (ION TOF, Münster).
A field-emission scanning electron microscope (Hitachi FE-SEM S-4800) was used
to investigate surface morphology.
After the characterization of PPy films by several surface analytic techniques,
electropolymerization parameters such as scan rate, dopant concentration and
potential region were optimized. Resulting PPy films were tested by a sticky tape
from Tesa (Germany) that was used during pull-off tests to determine the level of
adhesion of PPy coatings.
The electrical conductivity of the coated samples was measured using a four-
probe D.C. technique; constant current was applied to the sample through connecting
probes and the potential drop was measured by means of a multimeter. Then the
current was and the potential was measured again. From a series of such
measurements, a regression value of the resistance and hence of the electrical
conductivity was obtained.
4.4 Results and discussion
4.4.1 Electrochemical polymerization and characterization of pyrrole
Electropolymerization of pyrrole in the presence of salicylate ions has been
studied previously. Cascalheira et al.[118] determined the oxidation peak of
salicylate at around 1 V and showed the role of salicylate ions in the anodic behavior
of copper where formation of a Cu(II)-salicylate film is favoured at the electrode
surface and contributes to its passivation. In references [116-117] it is claimed that in
aqueous media, sodium salicylate is the most suitable candidate that allows the

51
formation of adherent and homogeneous PPy films without pre-treatment on active
metals.
Figure 28: Cyclic voltammogram of AZ91D in 0.5 M sodium salicylate solution, scan rate 20mVs-1. Inset: First cycle
Figure 28 shows the electrochemical behaviour of a magnesium AZ91D
electrode in 0.5 M sodium salicylate solution. It is clearly seen that the surface of Mg
AZ91D alloy can not be passivated easily due to anodic dissolution of Mg ranging
from -0.5 to 0 V (inset figure). However, a drop of current density around 0 V is
observed suggesting lowering of the anodic dissolution rate. At the end of the
forward scan, a decrease of the current drop is obtained with a corresponding peak in
the reverse scan direction at around 0.9 V. This reversible red-ox couple is attributed
to oxidation-reduction of sodium salicylate. At the beginning of the 2nd cycle, the
current density value does not reach as high a value in the potential range of -0.5 / 0
V as in the 1st cycle, probably indicating partial passivation of the surface, but the
current density values are still high. Repetitive 20 cycles in Figure 28 show a shift of

52
current density to more anodic values, suggesting that reversible red-ox behaviour of
salicylate takes place, and dissolution of the AZ91D surface is dominant.
Figure 29: Cyclic voltammogram of AZ91D in 0.5 M Sodiumsalicylate + 0.1 M Pyrrole, scan rate 20mVs-1. Total Charge; Qdep = 4.995 C Inset: First cycle.
Figure 29 shows electrochemical polymerization of Py on AZ91D surface
from sodium salicylate solution. Oxidation potential of Py - that is reported around
0.8 V - can not be detected due to overlapping with the oxidation peak of sodium
salicylate. However, in contrast to the first cycle (inset figure of Figure 28) of
monomer free solution, the intensity of the oxidation peak around ~0.8 V sharply
decreases at the end of the first cycle in Py containing electrolyte (inset figure of
Figure 29). This strong drop of the peak intensity upon addition of pyrrole is a result
of pyrrole being involved in the passivation process and the current decrease
continues in repetitive cycles in Figure 29. A slight current increase was observed at
the end of the each cycle that is attributed to a monomer oxidation process by step by
step polymer film growth.

53
Figure 30: Cyclic voltammogram of PPy film (deposited from 0.1 M pyrrole in 0.5 M sodium salicylate, at a scan rate of 20mVs−1; Qdep = 4.995 C) in a monomer free solution of 0.5 M sodium salicylate at scan rates of (a)10, (b) 20, (c) 30, (d)40, (e) 50 mV s−1.
Figure 30 shows the scan rate dependence of the PPy films in monomer-free
solution. One broad oxidation peak is observed during anodic scanning that is
associated with overlapping of the PPy and salicylate peaks. On the cathodic part, a
wide reduction peak is observed. Anodic and cathodic peak current densities are
related to the oxidation and reduction of PPy. The electrochemical process is
diffusion controlled, as both the anodic and cathodic peak intensities are increased
with increasing scan rate.

54
Figure 31: Nyquist plots of bare and PPy coated AZ91D alloy samples in 0.1 M Na2SO4 solution. Inset: Open circuit potentials of bare and PPy coated AZ91D alloy samples recorded in 0.1 M Na2SO4 solution during 24 hours.
Figure 31 compares the electrochemical impedance of bare AZ91D alloy with
fresh PPy coatings. Electrochemical impedance spectroscopy of pure magnesium in
sodium sulfate solutions has been previously studied [81, 84]. The Nyquist plots
were characterized by two well-defined capacitive loops followed by an inductive
loop [33, 121] (Detailed information about these loops are found in Chapter 4.4). The
PPy coated AZ91D clearly shows a significant increase of the polarization resistance
(Rp) as compared to the bare sample, indicating a protection with a barrier effect of
PPy [121]. Moreover, the inset figure of Figure 31 shows the open circuit potential
(OCP) recorded during 1 day in the same electrolyte. PPy coated samples did not
show noisy oscillations as compared to bare AZ91D, also suggesting that the PPy
coating decreases the dissolution of Mg in Na2SO4 electrolyte.

55
5.4.2. FTIR-ATR, XPS, ToF-SIMS measurements and SEM analyses
Figure 32: FTIR-ATR spectra of PPy coated Mg AZ91D.
FT-IR spectra of a salicylate copper complex have been reported previously
[122]. Santos et al. [123] also showed FT-IR spectra of PPy molecules in sodium
salicylate medium and determined the characteristic peaks with a shift of wave
numbers due to a copper-salicylate complex. In addition, C=O stretching of aromatic
carboxylic acid was not detected in the 1700–1650 cm-1 region as a strong band.
Instead, characteristic salicylate peaks were detected as a (νs(COO-)) band around
1377 cm-1 and shifted to 1392 cm-1, in the case of copper salicylate by splitting into
two components. Similarly, for PPy/Mg AZ91D system, these characteristic
(νs(COO-)) bands were detected around 1597 and 1556 cm-1 in Figure 32, probably
due to formation of a Mg salicylate complex. In addition, characteristic bands of the
ring stretching are found between 1400 and 1500 cm-1. The characteristic peaks of
PPy were detected around 1300 cm-1, 1185 cm-1 and 1035 cm-1, according to
references [124-127].

56
Figure 33: XPS survey spectra of PPy polymer.
Figure 33 shows an XPS survey spectrum of PPy/AZ91D surface containing
mostly C (63.43%), O (28.00%) and N (3.42%). The presence of nitrogen atoms is a
further indication of PPy coating. Additionally, the surface coverage of PPy is
relatively high as the signals from the substrate (Mg, Al, Zn) are very low. Figure 34-
a and -b show SEM pictures of PPy coated Mg AZ91D alloys with a typical
cauliflower type morphology of PPy grains [128] with a diameter ranging between
500nm to 800nm. As the grind striations are still visible on the surface, the
passivation process leads to a very thin film formation in sodium salicylate medium.

57
Figure 34: SEM pictures of fresh PPy surface; a) Lower magnification image with grinding striations are visible. Inset: Higher magnification image showing usual PPy morphology b) High magnification of cauliflower like PPy structures.
Figure 35: ToF-SIMS spectra of Mg AZ91D alloy before (top) and after (bottom) PPy coating. a) Mg+, b) CN- ions, c) C3H3O2
- and d) C5H4NO+, C6H8N+.
ToF-SIMS investigation revealed the successful formation of a PPy layer.
Figure 35a-d shows characteristic fragments after electrochemical treatment in
sodium salicylate (top) and a solution of PPy in sodium salicylate (bottom). Panel a
shows a decrease of the Mg+ signal, indicating a larger film thickness and better
coverage of the coating containing PPy. Panel c shows the fragment of C3H3O2-,
which is related to salicylate. A relative decrease of the signal upon addition of PPy
is observed. The ratio of C3H3O2- to Mg+ stays approximately constant. This implies
that salicylate also incorporates into the PPy layer. Furthermore, an increase in the
appearance of PPy specific fragments (C6H8N+, CN-) is shown in panels b and d.
Also, the signal of C5H4NO+ (panel d) appears only in the PPy containing sample,
indicating a possible reaction of salicylate and PPy.

58
4.4.3 Optimization of electrochemical parameters
4.4.3.a Optimization of salicylate concentration and scan rate
Figure 36: Determination of optimum sodium salicylate concentration by using EIS technique.
First, the effect of salicylate concentration (0.5M, 1.0 M and 3.0 M) on the
resulting coating properties was studied in 0.1 M Py containing salicylate solutions,
while keeping a fixed scan rate (20 mV s-1) and cycle numbers (20 cycles) in the
potential region between -0.5 V and 1.0 V. Figure 36 shows the EIS spectra
measured after coating AZ91D in solutions of varying salicylate concentration. The
variation of the salicylate concentration did not lead to a significant change of the
polarization resistance (Rp) (i.e., the difference along the real axis between the
resistance at the highest and lowest frequencies) of the coated samples. Nevertheless,
PPy films which are synthesized from 0.5 M salicylate + 0.1 M Py solution show the
best corrosion resistance, without any sign of inductive effect in the impedance
spectra. In the case of Mg and Mg alloys, these inductive loops appearing in LF
regions as scattered points and are commonly reported as a sign of active dissolution
[30-32]. This behaviour is generally assumed to be due to the presence of adsorbed
surface species such as Mg(OH)+ads, Mg(OH)2 ads or Mg+
ads [33,34]. Therefore, even
though only a minor effect on the Rp-values is found for different salicylate

59
concentrations in the PPy synthesis solution, the absence of an inductive loop (or
even any scattered data points in the region of the inductive loops of the spectra) is an
indication for somewhat better protective properties of this coating as compared with
those synthesized with higher salicylate concentrations.
Figure 37: Determination of optimum scan rate by using EIS technique.
Using this optimum salicylate concentration, optimum scan rate was
examined at different scan rates of 10, 20 and 50 mV s-1 in the potential region
between -0.5 V and 1.0 V at a fixed cycle number (20 cycles) from 0.1 M Py
containing 0.5 M sodium salicylate solution. Figure 37 shows the EIS examination to
determine the optimum scan rate for PPy films. It is found that AZ91D surfaces look
brown in color instead of “pyrrole black” after electrodeposition at scan rates greater
than 50 mVs-1. At this scan rate, the coating time is not sufficient to achieve a thick
enough PPy layer on AZ91D surface. Hence, samples do not show good corrosion
behaviour with Rp values very close to the bare AZ91D sample. On the other hand,
when a scan rate of 10 mVs-1 is chosen, the propagation of Py molecules does not
take place and acidic etching of the electrolyte removes freshly formed nucleation
sites and the corrosion occurs instead of electrodeposition of a PPy layer. Compared

60
to others, only PPy films synthesized with a scan rate of 20 mV s-1 lead to corrosion
protection of AZ91D in Na2SO4 solutions with a Rp value ~25 kOhm.
4.4.3.b Optimization of potential region
Figure 38: Cyclic voltammograms of AZ91D in 0.5 M Sodiumsalicylate + 0.1 M Pyrrole, between different initial and end potential ranges (a-f) at scan rate 20mVs-1. Inset figures of a-f shows the first cycles during electropolymerization.

61
In order to investigate the optimum potential range, experiments of different
initial (-0.5 V and 0.0 V) and upper potentials (1.0 V, 1.2 V and 1.5 V) were carried
out, while keeping the above mentioned parameters fixed (a scan rate of 20 mV s-1,
cycle number of 20 cycles in 0.1 M Py containing 0.5 M sodium salicylate). After
that, PPy films were again tested by EIS.
Figure 38 shows electrochemical polymerization of Py on AZ91D with
different potential region variations between -0.5 V and 1.5 V. Cyclic
voltammograms with similar shapes were obtained for each condition. However, due
to overlapping of oxidation peaks of sodium salicylate and of Py around 0.8 V,
separate peaks were not observed. In all inset figures of Figure 38, independent of
initial and end potentials, the oxidation peak around ~0.8 V sharply decreases at the
end of the first cycle. This is a sign of pyrrole being involved in the surface
passivation process without oxidative degradation, and the current decrease continues
in repetitive cycles. A slight current increase was observed after each cycle that is
attributed to the oxidation process of Py by step by step polymer film growth.
Figure 39 shows the EIS results of freshly coated PPy films on AZ91D
electrodes prepared by cycling between the different potential regions. All PPy
coatings provide some corrosion protection to the bare AZ91D electrode. However,
change of initial and end potential values during electropolymerization of PPy did not
lead to a very significant improvement of the polarisation resistance of freshly coated
AZ91D samples. Nevertheless, for all end potential values, an initial potential of -0.5
V was found to provide higher Rp values as compared to the initial potential value of
0.0 V.

62
Figure 39: Nyquist plots of bare and PPy coated (fresh) AZ91D alloy samples in 0.1 M Na2SO4 solution.
After 24 hours in Na2SO4 solutions, EIS tests were repeated. Results in Figure
40 show that for all PPy coated samples, the Rp values were increased after 24h in
solution. The most significant increase of Rp value was found for two coatings: 1) Uin
(Initial potential): -0.5V, Uf (Final potential): 1 V; and 2) Uin: 0.0V, Uf: 1 V. For
these coating parameters, Rp values of ~50 and 60 kOhm were observed after 24 hour
immersion. This increase in the Rp values with immersion time can result from
precipitation of corrosion products in the defects of the coatings, as the initial Rp
values for freshly coated samples clearly indicate only weak barrier properties. The
precipitation of corrosion products leads to additional (weak) corrosion protection. In
addition, for these samples the PPy coatings were well adhered on the surface after
24 hours of immersion, whereas coatings prepared with other potential ranges
resulted to peeling-off of the PPy. This is in contrast to freshly prepared coatings
which all showed very good adhesion. Due to these observations, the adhesion
properties of the coatings were further explored, as described in the next section.

63
Figure 40: Nyquist plots of bare and PPy coated AZ91D alloy samples after 1 day immersion in 0.1 M Na2SO4 solution.
4.4.3.c Influence of electrochemical parameters on adhesion and conductivity
of the coatings
Figure 41 demonstrates the relationship between applied potential and level of
adhesion by pull-off tests. The level of adhesion decreases after 24 hours of
immersion in Na2SO4 (from a-e, or from b-f) when wider ranges are chosen during
electropolymerization of Py. Moreover, an increase of initial potential from -0.5V to
0 V results with more adhesive PPy coatings on Mg AZ91D surface.

64
Figure 41: Mechanical adhesion test of PPy films after 1 day immersion in 0.1 M Na2SO4 solution.
Figure 42: SEM images (top-view) of PPy films (a-f) on AZ91D Mg alloys prepared in different potential regions. Inset pictures shows higher magnification images.

65
Figure 43: Tilted SEM images of PPy films (a-f) on AZ91D Mg alloys prepared in different potential regions showing the change of surface roughness by means of applying different potential regions.
SEM images of the coated samples (Figure 42 and Figure 43) indicate that
change of surface morphology can –at least partially- explain the different adhesion
behaviour. For example, when higher initial and end potential values are chosen
during electropolymerization of Py, the level of coating roughness increases (Figure
42a-c Figure 43a-c and Figure 42d-f and Figure 43d-f) and irregular surface
morphologies (inset Figure 42a-f) are obtained. Similar to the results of adhesion
tests in Figure 41, this type of surface morphology shows worse adhesion behavior
after 1 day of immersion in Na2SO4.
Uin (V)
Uf (V)
-0.5 0.0
1.0 16.834 S cm-1 22.312 S cm-1
1.2 1.980 S cm-1 0.459 S cm-1
1.5 0.096 S cm-1 0.681 S cm-1
Table 2: 4-probe conductivity measurement results (through the film –horizontal direction-) of PPy films synthesized onto Mg AZ91D alloys in between different potential regions (All values are reported with and experimental error of ±10%).

66
Another interesting finding is that the change of initial and end potential
values also affect the conductivity of the resulting PPy. Table 2 shows the
relationship between conductivity of the PPy coating and the chosen potential regions
during electropolymerization. For both initial potential values (0.0V and -0.5V), as
compared to end potentials of 1.2 and 1.5V, highly conductive PPy films are
obtained when an end potential of 1.0 V is chosen. However, initial potential value of
0.0 V is found to give a relatively high conductive PPy film with smoother surface
morphology (Figure 42d). It can be concluded that more adhesive and high
conductive PPy films are synthesized when a relatively short potential range is
chosen during electropolymerization.
Figure 44: Charge values of PPy films on AZ91D Mg alloys in between different potential ranges by cyclic voltammetry. Inset table shows the total charge of corresponding samples.
Interestingly, the coating parameters leading to enhanced adhesion and
reduced coating roughness are found to be related to an increase of total charge. It is
found that the total charge is increased when a smaller potential range is chosen
during the electropolymerization (Figure 44). For all conditions, the observed
decrease of charge as a function of cycle number indicates a surface passivation that
corresponds to current drop of the anodic peak around 0.8V. This, moreover,

67
increases the conductivity of the coatings. The resulting more uniform surface
morphology may therefore originate from an increased number of nucleation sites; as
a result better corrosion protection is achieved.
From all above mentioned results, the relationship between adhesion,
conductivity and corrosion resistance is explained with overoxidation of PPy films.
Kaplin [129] et al. showed that overoxidation of PPy films occurs when the end
potential exceeds 1.0 V. Similar results for 0.1 M Py concentration were reported by
Otero [130] et al. By controlling the magnitude of the electrical field, some effects
are obtained over the morphology of the freshly formed polymer. It is reported that
PPy films prepared at lower anodic potentials are found to be denser and
homogeneous. But, PPy films grown at higher anodic potentials form less regular
surfaces [131-132]. Besides, if the potential range chosen is too high, overoxidation
can occur which reduces the electrochemical activity and conductivity of the polymer
film with decreased adhesion to the substrate and loss of mechanical properties [133-
134]. A similar behaviour of PPy films was also found on Mg AZ91D alloy surface
in our work.
Figure 45: FIB-cut measurements showing the film thickness values varying between ~300 and ~1000 nm.
On the other hand, applying different potential regions did not affect the film
thickness values. Figure 45 shows a cross-sectional focused ion beam (FIB-Cut)
measurement performed on a PPy coating (Uin: 0 V, Uf: 1 V). Cauliflower like
surface structures were found to have cavities within, and the thickness values varied

68
between ~300 and ~1000 nm. These values do not change by changing the potential
range applied.
Depending on above mentioned results and corrosion tests, two PPy/AZ91D
electrodes (coated in potential ranges of 0.5/1.0 and 0.0/1.0V) are chosen for long
term corrosion tests, as these coatings show better corrosion protection (Figure 40),
mechanical adhesion (Figure 41) and higher electrical conductivity (Table 2) as
compared to others.
4.4.4 Long Term Corrosion performance of PPy/Mg AZ91D
Figure 46: Nyquist plots of PPy coated AZ91D Mg alloy in 0.1 M Na2SO4 during 10 days. Potential region: 0.0V/1.0V

69
Figure 47: Nyquist plots of PPy coated AZ91D Mg alloy in 0.1 M Na2SO4 during 10 days. Potential region: -0.5V/1.0V.
Figure 46 and Figure 47 show the Nyquist plots of two PPy coated samples in
Na2SO4 during 10 days. The long term corrosion protection of PPy film prepared in
the shorter potential range (higher initial potential) is found to be more durable as
compared to the one in Figure 47. Even though PPy/Mg AZ91D in Figure 47 shows
slightly better Rp values up to first 5 days, a drastic decrease takes place between 5
and 7 days due to peeling-off of the surface film, and results in strong corrosion of
Mg AZ91D substrate. Moreover, the PPy/Mg AZ91D samples coated between
0.0/1.0 V in Figure 46 do not peel-off and show better protection during 10 days,
with only a slight decrease of Rp values.
4.5 Conclusions
This chapter demonstrates direct growth of a conductive polymer
(polypyrrole) thin film on a magnesium alloy (AZ91D) without any pretreatment for
the first time. Fourier-Transform Infrared Spectroscopy (FT-IR), X-Ray
photoelectron spectroscopy (XPS), time of flight secondary ion mass spectrometry
(ToF-SIMS) and scanning electron microscopy results shows that the magnesium

70
AZ91D surface is covered by a thin PPy film. Electrochemical impedance
spectroscopy results (EIS) demonstrate that this sodium salicylate doped PPy layer
has a corrosion protection property in NaSO4 solutions.
Effect of electrochemical coating parameters such as scan rate, sodium
salicylate concentration and potential range on the corrosion behaviour of the PPy
films was studied with electrochemical impedance spectroscopy (EIS). It is found
that the resulting surface morphology and the conductivity are highly affected by the
potential range chosen to deposit PPy on Mg AZ91D alloy surface. For long term (10
days) immersion periods, PPy coatings electrodeposited in between 0.0/1.0 V were
most effective in reducing corrosion rate of Mg. The PPy coatings prepared under
optimized conditions remained well adhesive over this period; no peeling-off was
observed.
In view of practical applications, for instance in biomedical applications of
Mg and its alloys, such salicylate doped PPy layers on Mg alloys have a high
potential for tailoring the degradation rate, since all components (substrate,
passivating agent, coating) of the resulting system are non-toxic. Therefore, these
coatings may have potential for tailoring the degradation rate of Mg alloys for
biomedical and drug-releasing applications.

71
5 ELECTROCHEMICAL POLYMERIZATION OF POLYPYYROLE
ON MG ALLOY AZ91D
PART 2: CORROSION BEHAVIOUR OF PPY/AZ91D IN
SIMULATED BODY FLUID SOLUTIONS
AND FURTHER FUNCTIONILIZATION WITH ALBUMIN
MONOLAYERS FOR DRUG DELIVERY APPLICATIONS
5.1 Abstract
In this chapter, corrosion behaviour of PPy/Mg AZ91D alloy is studied in
simulated body fluid (SBF) solutions at body temperature, Hydrogen gas collection
measurements, polarisation curves and EIS results showed that the adherent PPy
coatings hinder the anodic dissolution of Mg substrate and enhance the corrosion
resistance by releasing dopant anions. Moreover, further functionalization of PPy
layers with Albumin (Alb) monolayers also provides a better controlled release
profile over 20 days with zero order kinetics.

72
5.2 Introduction
5.2.1 Corrosion control with ICPs
Corrosion protection by ICP coatings has been reported to be not favorable
under practical conditions [135-136], as has been discussed in a recent review [137].
However, a good corrosion protection can be achieved in practical applications if
these coatings are applied together with other coatings in sandwich like structures.
For instance, an effective coating against corrosion consists of several layers: a
primer that enhances the adhesion to the metal (~1µm) followed by a thick polymer
layer (~20 µm) containing anticorrosion pigments. Finally, a thin top-coat is applied
[138]. The most important property of a coating is the barrier effect and the
electrochemical inhibition. However, an electrochemical process of anodic and
cathodic reactions takes place on substrate surface [139];
M M+n + ne- …………………………………………… (11)
As a counter reaction, O2 is reduced, according to either;
O2 + 2H2O + 4e- 4OH- (pH ≥ 7) ...................................... (12)
or
O2 + 4H+ + 4e- 2H2O (pH < 7) ......................................... (13)
Reaction (11) corresponds to dissolution of metal substrate that can be
reduced if either a sacrificial coating (e.g. zinc deposition on steel) is employed or
potential of the metal is shifted into the passivation domain by formation of a metal-
oxide layer. Formation of insoluble compounds attached to the metal surface,
resulting from chemical or electrochemical treatment of the metal [139], contribute to
its passivation. However, hindering the diffusion of O2 and H2O by increasing the
thickness of the coating also increases the protection efficiency. Addition of
corrosion inhibitors such as chromate salts, [140-142] is also found in general to
maintain good corrosion properties in metals. Even though chromate-based
techniques provide excellent passivation of metals, they are now prohibited due to
environmental regulations and replaced with alternative solutions as recently
summerized in several reviews [143-144].

73
The use of ICPs against corrosion of the metals was firstly investigated in the
80’s, after DeDerry [145] showed a polyaniline (PANi) film obtained by
electrochemical oxidation of aniline on stainless steel (SS) and mild steel (MI)
surfaces. This work was followed by electropolymerization of Py in neutral medium
on the same metals. First reports [146-147] of the resulting PPy films were not found
to adhere to metal surface. Because of the oxidation potential of the metals is lower
than the oxidation potential of monomer, hence a considerable amount of metal
dissolution was observed during electropolymerization. But this problem was
overcome by changing the rates of electropolymerization. For instance, using
solvents slightly alkaline of water [148] or pre-treatment of metal surfaces [149]
result in more adhesive PPy films. Additionally, choice of electrolytes including
proper anion groups to form insoluble metal-salt layers [150] is found to be another
important way to obtain better PPy films on active metals as shown in Figure 48.
This metallic salt layer only partially passivates the metal by hindering its
electrochemical activity.
Figure 48: Figure shows effect of metallic salt onto adherence of resulting polymer. Dissolution of the metal is favoured (a) if the anions in electrolyte have no ability to form an insoluble metallic complex. In b), more homogenous films are obtained in the presence of a metallic salt layer (b).
The use of ICPs brings an original concept to the protection of metals.
Although the way the ICPs work to protect metals against corrosion is different than
the mechanisms accepted for polymeric coatings in general, the protection
mechanism is still debated. But till now, four main mechanisms [139] are reported to
explain their working principles during corrosion process:

74
a) Displacement of the electrochemical interface,
b) Ennobling the metal by polymer in its oxidized state,
c) Self-healing properties,
d) Barrier effect.
5.2.1.a Displacement of the electrochemical interface
When a metal is coated with another noble one, for instance when iron is
coated with nickel, the electrochemical interface moves from oxidizable metal (Fe) -
electrolyte interface to noble metal (Ni)-electrolyte interface and prevents the
corrosion of Fe substrate. The underlying metal escapes from the aggressive
environment if the top Ni layer is free from any defect (pinholes or scratches). When
an oxidizable metal is coated by ICPs, the same situation is observed if the ICP
remains in its oxidized state. Therefore the electrochemical interface moves to the
polymer-electrolyte interface and the underlying metal is no longer in contact with
the electrolyte as illustrated in Figure 49.
Figure 49: a) Corrosion of a metal in corrosive media by dissolution of metal. b) Displacement of the electrochemical interface to electrolyte/ICP by reducing of polymer.
This effect was reported by Lacroix et al. [151] for a bilayer structure of
PPy/PANi to show the displacement of the electrochemical interface. Direct
electrochemical deposition of PANi onto zinc fails and large amounts of metal
dissolves. But, if a zinc electrode is firstly coated by a thin layer of PPy from
aqueous electrolytes of salicylate, the metal dissolution is not observed during PANi
coating. The film is found to be very stable in acidic electrolytes indicating that the
underlying metal is fully protected. This confirms the displacement of the

75
electrochemical interface to polymer-electrolyte region from its usual location. The
same concept is also reported by Iroh et al. [152-153] by showing that bilayer
structures also lower the porosity and provide better protection compared to single
layer coatings [154].
Recently, Michalik and Rohwerder [155] showed that oxygen reduction is
very crucial to determine the shift of the electrochemical interface. They showed that
the oxygen reduction process can take place at the metal-electrolyte interface again if
the resulting polymer is fully reduced in the presence of small surface defects.
Therefore O2 and H2O can reach the metal surface through the porous surface of ICP
coating by diffusion. On the other hand, they showed that fast reducing of ICP
coatings is also needed in the case of these surface defects. Because the redox
potential of the polymer is generally above that of the metal and thus it is capable of
oxidizing the metal by reducing itself. In this case, the electrochemical interface is
not displaced, but a barrier effect is obtained. Therefore open circuit potential
(OCP)–time curves are generally used to understand the efficiency of the ICP during
corrosion tests as the kinetics of reduction reactions of O2 and H2O is also depends
onto OCP value of the bare metal.
When a metal is coated with a highly doped ICP, and as long as the ICP
remains in its conductive state, OCP of the ICP/Metal system is always above that of
the bare metal electrode. When the ICP is reduced, the potential value drops to a
lower value. For instance, for PPy, the redox potential of O2/H2O is always more
positive than the PPy and hence the reduction of O2 always takes place at the
polymer surface and produces OH- ions [139]. These OH- species are reported to be
responsible for delamination [151-155] in many cases.
As a conclusion, displacement of the electrochemical interface can only delay
the corrosion process for a certain time by reducing the ICP coating through
reduction of O2 on the polymer surface.
5.2.1.b Ennobling the metal by polymer in its oxidized state
The main idea of ennobling the metal surface by ICPs is that the metal surface
is set to a potential within its passive range if it couples with an ICP at its oxidized
state as shown in Figure 50. In general, an oxidizable metal has 3 different electro
kinetic regions: active, passive and trans-passive. When the sample is in contact with

76
a weak oxidant, a counter electrochemical reaction, namely reduction reaction may
correspond to active region with a high corrosion current value. In this region, the
metal has no ability to form an insoluble isolating layer as it has in a passive region
and hence a corrosion process takes place (Red line in Figure 50).
Figure 50: Schematic representation of ennobling mechanism of metal surface that have a passive region.
On the other hand, if the metal is coupled with strong oxidants (blue curve in
Figure 50) such as ICPs, the corrosion current density values correspond to the
passive region and are relatively lower than the first case [139]. In this case, the
width of the passivation range is very important. Because when a metal is coated by
an ICP at its oxidized state, a mixed OCP value is obtained due to galvanic coupling
between the metal and polymer. However, the resulting current corresponds to the net
reaction from the reduction of the polymer and the oxidation of the metal. Therefore,
the most favorable situation is the fact that the reduction of the polymer falls within
the passive area of the metal.
The ennobling effect by ICPs is widely studied with different ICPs coatings
[111, 156-162] In most cases, good protection was reported with the ennobling
mechanism for PANi coatings [111, 160]. However, for PPy, this mechanism has
also been widely investigated. Recently, Michalik and Rohwerder showed that [155]

77
PPy could protect small surface defects easily. But for large defects, PPy was
considered not to have enough capability to passivate iron and ennobling was not
successful [139].
As compared to displacement of the electrochemical interface, it could be
concluded that the ennobling effect is applicable for systems where small scratches
occur during the corrosion. This effect also would be more expected within the
metals that have large passive potential areas.
In view of non-passivating metals such as magnesium, this mechanism can
not be discussed since magnesium metal does not have a passivation region like other
metals (e.g. steel, Al). Therefore, when magnesium is coated by an ICP at its
oxidized state, the resulting current from the reduction of the polymer can not fall
within any passive area of magnesium.
5.2.1.c Self-healing effect by release of dopant anions
As already mentioned with the mechanisms above, due to galvanic coupling
between the metal and ICP, reduction of the polymer and oxidation of the metal
surface is interlinked. In this special case, reduction of O2 can occur either at the
polymer surface or at a scratch and result in formation of OH- (Figure 51). Depending
on the metal substrate, two mechanisms can be predicted: either the metal initiates
the formation of a metal oxide, or the doping anions are released and act as inhibitors
[139, 163]. However, these molecules can only be released when the corrosion starts
[164-166].
Figure 51: Schematic representation of oxygen reduction on PPy surface in neutral solutions.

78
This property of ICPs is described by Chang and Miller [167] for drug release
applications and further details such as mobility, size and valance of the dopant
anions were reported by Weidlich et al. [168].
The self healing effect of the ICPs is generally assumed as an extension of the
ennobling mechanism. But in this case, doping of the polymer is found to be very
important for drug delivery applications. Therefore, choice of a suitable dopant
molecule is very crucial for use of these materials in drug delivery applications [169].
5.2.1.d Barrier effect of the polymer
As previously explained in chapter 6, investigations including
electrochemical polymerization of conducting polymers on active metals such as iron
[111, 170], copper [150], aluminum [110] and zinc [109, 171] report that these
coatings show good adherence on surface and good performance in corrosive media
[139, 172]. Good adherence of ICPs onto the metal substrates acts as a simple paint
layers in general and shows barrier properties. The barrier effect slows down the
corrosion by hindering diffusion of O2 and H2O to reduce the corrosion at the
interface[173]. However, in the presence of very aggressive anions such as chlorides,
diffusion of these anions must be prevented in the barrier effect.
For ICPs, independently of whether the polymer is conductive or not,
diffusion rates of these species are mainly dependent on the porosity of the polymer.
Therefore decrease of the film porosity [174] and changing the surface
hydrophobicity [154-155] is generally reported to enhance barrier properties.
5.2.2 PPy in biomedical research
Among all other ICPs, Py is reported as a strong candidate for biomedical
applications with a soluble nature in neutral or weakly acidic conditions. However,
its low oxidation potential and environmentally friendly nature as compared to
aniline and thiophene make PPy very promising for a wide range of biomedical
applications including the development of artificial muscles [175], neural recording
[176], sensors [91, 177] , actuators [178] stimulation of nerve regeneration [179] and
controlled drug release [171, 180].

79
Among all of these applications, controlled drug release applications by PPy
is very promising. The release of dopant anions from the PPy backbone has been
reported to promote cell survival and growth for biomedical implants [181].
However, it has also been reported that these systems could be used as suitable
platforms for the delivery of a wide range of biomolecules [182] such as glutamine
[183], ATP [184], salicylate[132] and dopamine [185] in the presence of different
dopant anions. On the other hand, dopant anions themselves can also act as drugs if
they have healing properties in biological environments. For instance, sodium
salicylate is reported as a suitable replacement of Aspirin (Figure 27) because of its
analgesic, antipyretic and non-steroidal anti-inflammatory (NSAID) properties.
These properties are also known to lower the level of apoptosis [119-120] in cancer
cells. Therefore use of salicylate during electropolymerization processes may give an
opportunity afterwards for healing by releasing of the dopant anion. Healing ability
of a drug eluting system is related to its drug elution kinetics as shown in Figure 52.
In most cases, zero order kinetics is preferred with a reservoir system showing a
constant release rate. This is mostly obtained if a constant drug source such as pure
liquid drugs or dispersed particles are provided. If the drug source is a non-constant
source (dissolved drug in solution), it usually gives first order (Fickian) release rates
[186]. In this case, the dose released does not provide a constant healing rate and
therefore is not stabilized as like in the zero order kinetics.
Figure 52: Two patterns of drug delivery; a) Fickian and b) Zero order kinetics.

80
5.2.3 Mg and its alloys in biomedical research
In medical science, one of the most promising areas is the development of
biodegradable implant materials and drug eluting stents (DES). As compared to
traditional implant materials such as Ti and stainless steel, biodegradable materials
do not cause long-term irritation or incompatibility with the biologic environment.
Biodegradable implants are mainly made of polymers, such as poly-lactic acid and
derivatives [187-188]. But, these materials are usually found to have limited
mechanical properties and therefore can not meet with the requirements of the
desired applications [189-190].
On the other hand, Mg and its alloys have been regarded as potential
biodegradable materials. In 1878, Mg wires to stop bleeding vessels were first
introduced by Edward Huse [1]. Since then, many researches have been reported
about these materials being used as biodegradable components [191-196].
Although mechanical properties of Mg are found to be very close to human
bone [195], the use of this metal as bio-degradable orthopedic implants is limited due
to its inhomogeneous corrosion. Additionally, alkalization and H2(g) production
during corrosion of Mg [25] is reported to be critical for the biological environments.
Therefore many attempts including physicochemical surface modifications,
bioactive, biomimetic or self-assembled monolayer coatings have been reported to
tailor the magnesium corrosion [197-199]. For instance, albumin attachment onto Mg
surfaces [200-201] is reported to increase corrosion resistance of magnesium in
simulated body fluid (SBF) solutions. Additionally, it is also shown that the surfaces
modified with this most abundant blood protein has a tendency to have
antithrombogenic properties [202-203]. Therefore, these types of protein coatings
could be helpful for further improvement of the Mg surfaces to tailor the corrosion
rates.
In the previous chapter, the direct electrochemical polymerization of PPy on
Mg alloys AZ91D [204-205] from aqueous electrolytes of sodium salicylate was
presented. Results showed that optimization of coating parameters is found to be
critical to achieve adhesive PPy films and Mg surfaces with good mechanical and
corrosion properties. In this part, corrosion behaviour of PPy/Mg AZ91D alloy is
studied in simulated body fluid (SBF) solutions at body temperature. Corrosion
behaviour of PPy/AZ91D system is studied by hydrogen gas collection

81
measurements, EIS and polarisation curves during 1 week of immersion. It is found
that the adherent PPy coatings (1st step) hinder the anodic dissolution of Mg substrate
and enhance the corrosion resistance by releasing dopant anions. Moreover, further
functionalization of PPy layers with Albumin (Alb) monolayers (2nd step) also
provides a better controlled drug release profile over 20 days with zero order kinetics.
5.3 Experimental route
Magnesium AZ91D coupons (2 cm × 2 cm × 0.5 cm) were ground with 1200
grit emery paper and then were cleaned in 1:1 ethanol : acetone mixture for 10 min in
an ultrasonic bath. Electrochemical analyses were performed using an o-ring cell
(See 2.1 Electrochemical setup with a three electrode system)
PPy films (step 1) were synthesized from 0.1 mol/l pyrrole (Py) containing
aqueous solution of 0.5 mol/l sodium salicylate (Sigma) (pH=6.50) by using cyclic
voltammetry (CV) in the potential region between 0.0 V and +1.0 V at 20 mV s-1
scan rate. Different numbers of cycles of 5, 10, 20, 30, 40 and 50 were performed to
optimize mechanical adhesion. Further surface modifications of PPy layers with
albumin monolayers (second step) were achieved by immersion of PPy/AZ91D
electrodes in 1 mM aqueous solution of Bovine Serum Albumin (Sigma) during 1
day. Characterization of Albumin modified electrodes were performed by positive
and negative static time-of-flight secondary-ion-mass-spectrometry (ToF-SIMS)
(ION TOF, Münster).
H2 gas evolution, polarization and electrochemical impedance spectroscopy
measurements were performed in SBF5 solutions [206] at body temperature. (see
Figure 53 and Table 3).
Concentrations are given in mmol/l.
Table 3: Composition of SBF 5 in comparison with human blood plasma.
EIS measurements were performed with a sinusoidal perturbation of ±10 mV
for 10 points in each decade over the frequency range from 100 kHz to 10 mHz.
Polarization tests were performed between -2.0 and -1.0 V with a scan rate of 1
Ion Na+ K+ Mg2+ Ca2+ Cl- H3CO- HPO42- SO4
2- Plasma 142.0 3.6-5.5 1.0 2.1-2.6 95.0-107.0 27.0 0.6-1.4 1.0
SBF5 142.0 5.0 1.0 2.5 131.0 5.0 1.0 1.0

82
mV/s. A simple flask attached with a funnel was used to investigate the H2(g)
evolution measurements. A field-emission scanning electron microscope (Hitachi
FE-SEM S-4800) was used to investigate surface morphology.
A UV-visible spectrophotometer was used to quantitatively analyze drug
release profiles of the specimens. As a drug release medium, phosphate buffered
saline (PBS) is used. The characteristic salicylate peak at 295nm wavelength is
measured.
Figure 53: Preparation of SBF according to [206]
5.4 Results and Discussion
5.4.1 Optimization of mechanical stability and adhesion: Effect of different
cycles
In order to investigate optimum adherence, experiments of different cycle
numbers (5, 10, 20, 40 and 50) were carried out, while keeping the other
electrochemical polymerization parameters fixed (between 0.0 / 1.0 V scan range
with a scan rate of 20 mV s-1 in 0.1 M Py containing 0.5 M sodium salicylate).
Afterwards, adherence of PPy films was tested by double side tapes.

83
Figure 54: Optical images of mechanical adhesion test of freshly coated PPy films for as coated samples (a, c, e, g and h) and after pull-off tests (b, d, f, h and j). Samples coated during 10 (c-d) and 20 (e-f) cycles shows better mechanical adhesion as compared to 5 (a-b), 40 (g-h) and 50 (i-j) cycles.
Figure 54 demonstrates the relationship between cycle numbers and level of
adhesion by pull-off tests. For 5-, 10- and 20-cycle coated electrodes (a, c and e),
homogenous PPy films were observed. After 20 cycles, adherence of PPy films is
decreased. However, electrodes coated for 5, 40 and 50 cycles also show poor
adherence after peel-off test (b, h and i). On the other hand, samples coated during 10
and 20 cycles are not completely peeled-off from the surfaces (d and f). This could be
explained by increase of total charge during the electropolymerization process.

84
Figure 55: Graph shows the increase of total charge during electropolymerization of PPy on Mg alloy AZ91D. The increase of total charge during electropolymerization is shown in Figure
55. The charge rapidly increases after 20 cycles and the resulting PPy layer peel off
the surface for cycle numbers greater than 20 (Figure 54g-j). During
electropolymerization of PPy, the total charge obtained is a combination of the
charge used to build a PPy layer and simultaneous oxidation of the PPy layer itself
after initial stages of electropolymerization. During the initial stages, most of the total
charge is transferred firstly to build a PPy layer by red-ox process of the Py monomer
and therefore follows a linear increase. The total charge increases especially after 20
cycles because of a systematic doping-dedoping process of PPy polymer with
salicylate counter ions. Therefore, in addition to a simultaneous
electropolymerization process of PPy, PPy/AZ91D system also has a charge limit to
obtain good adherence till 20 cycles of coatings (from Figure 54g, i and Figure 54h,
j). When number of electropolymerization cycles is increased, a rapid increase of
charge also results in poor mechanical adherence.

85
Figure 56: SEM images of the PPy/AZ91D surfaces coated with different cycles of a) 5, b) 10 and c) 20. Inset: High magnification images showing the degree of polymerization with cauliflower like surface morphology.
Figure 56 shows the surface morphology of the PPy coated electrodes up to
20 cycles. For 5 cycles, it is found that the Mg AZ91D surface is not completely
covered by the PPy layer (dashed circles in a) and do not adhere onto surface (Figure
56a). Moreover, a cauliflower surface morphology is not present (inset of Figure 56a)
as already confirmed by mechanical pull-off tests of the corresponding sample where
the PPy layer completely peeled off of the surface (Figure 54a). When the cycle
number is increased to 10, formation of typical PPy morphology is slightly observed
(inset of Figure 56b) but a complete coverage is not observed, too (dashed circles in
Figure 56b). However, when the cycle number is increased to 20, the surface is fully
covered with PPy molecules (Figure 56c) with cauliflower structures (inset figure of
Figure 56c) with a film thickness around 1 µm [204].
On the hand, if the cycle number greater than 20 is chosen, the level of
coating roughness increases and very irregular surface structures are obtained (Figure
57a-c). Similar to the results of adhesion tests in Figure 54, this type of surface
morphology shows decreased adhesion during mechanical pull-off test.

86
Figure 57: SEM images of the PPy/AZ91D surfaces coated with cycles of a) 30 b) 40 and c) 50 cycles showing the change of surface roughness and morphology.
From all of the above mentioned results, the relationship between adhesion
and cycle number is explained with similar results as already explained for
overoxidation of PPy films [129-131, 184, 204]. In this case, similar to the present
results, an increase of the surface roughness is obtained with poor mechanical
properties of polymer.
Due to these observations, AZ91D samples coated during 10 and 20 cycles
are used in the further corrosion tests.
5.4.2 Corrosion behaviour of PPy/AZ91D system in simulated body fluids
Figure 58 shows the corrosion rates of PPy/AZ91D electrodes exposed in
SBF5 during 1 week. Corrosion rates were calculated by H2(g) evolution during
corrosion of PPy/AZ91D electrodes and results were compared with bare AZ91D. It
is found that the formation of a PPy layer on AZ91D decreases the corrosion rate
and, moreover, the rate does not increase rapidly as compared to that of the bare
electrode. On the other hand, daily corrosion rate increases with immersion time for
all electrodes. The increase of the corrosion rate with time is most probably due to

87
defects on the PPy surface leading to an increase of the effective surface area by
dissolution-induced roughening of the Mg surface.
Figure 58: Corrosion rates of AZ91D and PPy/AZ91D electrodes exposed in SBF5 during 1 week.
Additionally, electrochemical impedance spectra of coatings were measured
in SBF5 during 1 week (Figure 59). In the beginning of the corrosion tests, a trend of
increase of the polarization resistance (Rp) is observed for both the 10- and 20-cycles
coated AZ91D electrodes. For instance, for 10 coating cycles, the Rp value increases
up to ~20 kOhm after 1 day. But, a significant decrease is observed during 1 week of
immersion and, in addition, inductive loops are observed after 3 days of immersion.
For 20 cycles of coating, a similar trend and character is observed, too. In this case,
increase of Rp values up to ~17 kOhm is found and decrease of the Rp value is
observed after 3 days. For both samples, in parallel to increase of the corrosion rates
in Figure 58, inductive loops are observed during 1 week which is also typical for
magnesium dissolution [73, 80-82].

88
Figure 59: Nyquist plots in SBF5 during 1 week show long term corrosion tests of PPy coated AZ91D Mg alloys coated with different cycles; a) 10 cycles and b) 20 cycles.
Figure 60 shows the mechanical adhesion and pull-off tests of PPy/AZ91D
electrodes. Results imply that inhomogeneous corrosion of magnesium substrates
during 1 week immersion takes place for both the 10 (Figure 60a) and the 20 (Figure
60b) cycles coated electrodes. Both electrodes show partial and random peeling off
of PPy during corrosion.

89
Additionally, pull-off tests of both electrodes (Figure 60b and d) show that
the mechanical adherence of the PPy films is decreased by means of diffusion of
aggressive anions through the polymer/metal interface which initiate the corrosion.
Figure 60: Mechanical adhesion test of PPy films after 1 week immersion in SBF5 solution; a) 10 cycles, b) 10 cycles after pull-off tests, c) 20 cycles and d) 20 cycles after pull-off tests.
Figure 61 shows the SEM image of corroded surface morphologies for 10 and
20 cycles. For both electrodes, similar types of surface structures were observed. In
general, Figure 61a shows that PPy molecules with typical cauliflower surface
morphology disappear most probably due to reducing of polymer from its oxidized
state. This also results in highly porous surface morphology (Figure 61a) enabling
diffusion of water through polymer/metal interface. After the polymer is reduced, it
only acts as a non-specific barrier and looses its electrochemical protection
mechanism. On the other hand, it is well known that for PPy coatings the reduction
of O2 to give OH- always takes place on the PPy layer. These OH- are also reported to
be suspect for delamination of surface layers and hence increase the corrosion rate
(Figure 58).
On the other hand, Figure 61b and d show formation of nano-whiskers and
hollow structures that are most probably formed during passivation of magnesium
surface in SBF5. This also could be explained by self healing property of ICPs by
releasing dopant counter ions from its backbone.
Figure 62 shows the release profile of salicylate during 3 days. The amount of
salicylate released from a 20 cycle coated electrode is slightly higher than 10 cycles.

90
However, the release kinetics for both electrodes show Fickian distribution after 3
days immersion in PBS. It is also worthwhile to stress that the inductive loops are
observed after 3 days during EIS measurements (Figure 59) when the all salicylate is
released from the PPy/AZ91D electrode. Therefore, self healing effect by release of
dopant ions is provided for a temporary period. After all amount of salicylate is
released, dissolution of magnesium takes place and corrosion rate is increased
Figure 61 : SEM images of PPy/AZ91D electrodes after 1 week immersion in SBF5 solution.
On the other hand, salicylate could be of interest in applications that require
the controlled delivery of drugs. To achieve this, the Fickian type of drug release
profile and inhomogeneous corrosion of Mg substrates could be improved by further
functionalization of PPy layer. As already explained above, ICPs could show better
corrosion protection behaviour if they were applied together with other layers in
sandwich type systems. Therefore the PPy layer could be covered to achieve better
corrosion protection and drug release profile.

91
Figure 62: Drug release profile of PPy/AZ91D electrodes in PBS shows a Fickian distribution.
5.4.3 Modification of PPy layers with Albumin monolayers
Further surface modification of PPy/AZ91D electrodes is done by immersion
of these electrodes in 1 mM Albumin solutions during 24 hours. Afterwards, albumin
layers are characterized by ToF-SIMS that provides information about chemical
structures within the protein and consequently may be a promising tool for surface
protein analysis. The fragment pattern generated by amino acids allows for the
confirmation of the attachment of proteins on the surface, determination of their
orientation, and distinguishing of specific proteins deposited from mixtures as well as
to observation of denaturation of protein coatings [207-223].

92
Figure 63 : ToF-SIMS spectra and ratios of ToF-SIMS signals after the different treatment steps: bare AZ91D - AZ; pyrrole covered AZ91D - AZ-P; AZ91D covered with pyrrole and albumin subsequently - AZ-PA; AZ91D steeped in albumin - AZ-A; a) Mg+ - substrate, b) CNO- - protein backbone, c) C4H3N
+ - pyrrole monomeric unit, d) C4H3N+ / CNO- - pyrrole monomeric unit in ratio to
protein backbone, e) C4H8N+ - valine / proline, f) C4H8N
+ / Mg+ - valine / proline in ratio to the substrate.
In Figure 63a-f characteristic fragments for albumin, the pre-treatment and
substrate are displayed. Upon protein adsorption a variety of amino acid signals is
observed, e.g., m/z 70.07 u: C4H8N+ – proline, valine; m/z 84.04 u: C4H6NO+ –
glutamine, glutamic acid; m/z 84.08 u: C5H10N+ – lysine, leucine and m/z 110 u:
C5H8N3+ – histidine. A systematic assignment of typical fragments of amino acids to
their mass spectral signals can be found elsewhere [210, 217, 222].
From Figure 63a, it can be seen that the amount of magnesium decreases
upon coating with pyrrole and albumin. After albumin adsorption only small amounts
of Mg+ could be detected. CNO-, the signal produced by the peptide bonds of the
protein is only clearly visible after albumin adsorption (cf. Figure 63b). A higher

93
amount of CNO- is present on the pyrrole pre-coated surface, indicating better
coverage of the latter. On the pure AZ91D substrate and the pyrrole pre-coated
sample no peptide fragments could be detected. Figure 63c displays m/z = 65, which
contains the fragments C4NH3+ (monomeric repeat unit of pyrrole, m/z = 65.00) and
C3N2H+ (m/z = 65.01). The pyrrole monomeric unit is dominant on AZ-P and a
signal at the same position occurs on the albumin coated samples AZ-PA and AZ-A.
On these two samples the signal has comparative intensity, indicating that it is
a fragment of the protein (possibly originating from tryptophane) and not the pyrrole
layer showing through. Figure 63d shows the C4NH3+ signal in ratio to the peptide
backbone and reinforces the observations from Figure 63c.
Figure 63e and Figure 63f show that the fragment C4H8N+ produced by the
amino acids valine and proline is not observed on AZ and AZ-P, but increases
significantly upon attachment of albumin (AZ-PA / AZ-A). A better coverage of the
substrate with pre-treatment by pyrrole is obvious.
As it has been a common observation that proteins tend to adsorb in
monolayers, i.e. proteins do not adsorb non-specifically onto their own monolayers
[212, 223] and magnesium signals were still observed in low yields after the reaction
sequence, we expect monolayer (or sub-monolayer) coverage of the AZ91D surface.
Figure 64: ToF-SIMS spectra and ratios of different amino acids for the treatment steps. a) histidine signal, b) histidine in ratio to magnesium, c) glutamic acid /glutamine, lysine/leucine.

94
A further observation is that fragments produced by aromatic amino acids,
e.g. histidine, are more pronounced on the AZ-A sample (cf. Figure 64a; all spectra
normalized to total count rate), whereas other amino acids, e.g. glutamine / glutamic
acid, are only detected in lower yields than on the pyrrole pre-treated sample. This
indicates that the albumin possibly adsorbs via π−π interactions to the pyrrole
coating. Figure 64b confirms that the amount of histidine compared to magnesium
still is higher on the pre-treated surface, even though the extent is smaller than for
valine/proline (cf. Figure 63f).
Figure 65: Mechanical adhesion test of Albumin modified PPy films
Moreover, it is clear from the optical microscope images that the adhesion of
PPy layers does not deteriorated during immersion of electrodes in aqueous solutions
of albumin (Figure 65a and c). However, after pull-off tests (Figure 65),
Alb/PPy/AZ91D electrodes do not peel-off of the surface and a good attachment of
albumin onto PPy is achieved.
5.4.4 Corrosion behaviour of Alb/PPy/AZ91D system in simulated body fluids
Figure 66 shows the corrosion rates of Alb/PPy/AZ91D electrodes in SBF5
during 1 week. It is found that further functionalization of PPy layer significantly
decreases the corrosion rate of AZ91D substrate and, moreover, does not increase
rapidly as compared to bare electrode. However, 10 cycles coated Alb/PPy/AZ91D
electrode shows slightly higher increase of the corrosion rate during the first day
(marked as region I in Figure 66). Afterwards, it stabilizes as with the 20-cycle

95
coated electrode. On the other hand, similar to the electrodes without albumin
modification, daily corrosion rate increases with immersion time for all electrodes
mainly due to diffusion of corrosive specicies through the metal/polymer interface.
Figure 66: Corrosion rates of albumin modified PPy/AZ91D electrodes exposed in SBF5 during 1 week.
Corrosion rates from H2(g) measurements are summerized in Table 4. As compared to
bare AZ91D electrode, corrosion rates decrease when a PPy coating is applied to
AZ91D. Moreover, when the electrodes are further modified with albumin
monolayers, a drastic decrease of corrosion rate is observed for both 10- and 20-cycle
coated electrodes and corrosion rate is reduced by ~70 times that of bare AZ91D.
Additionally, electrochemical impedance spectra of Alb/PPy/AZ91D
electrodes were measured in SBF5 during 1 week (Figure 67). For both samples, an
increase of Rp values is observed. But, for 10 cycles of coating, a rapid increase of
Rp value up to ~12 kOhm is found after the 1st day of immersion. On the next days,
impedance of the system did not change significantly. But a decrease of Rp value
with an inductive loop is observed after the 6th day of immersion. For a 20-cycles
coated electrode, systematic increase of Rp is obtained. This increase continues
during 1 week starting from ~9.0 kOhm to ~17.5 kOhm without inductive loops.

96
Moreover, the second capacitive loops at the middle frequency region start to
disappear, which is also an indication of corrosion inhibition of AZ91D substrate
without adsorbed electroactive species [224]. Therefore, after albumin coating, 20
cycles coated electrode found to show slightly better corrosion performance in SBF5
solutions.
Step 1 (PPy)
Step 2 (Albumin)
Corr Rate* (mg.cm-2.day-1)
Corr Rate** (mg.cm-2.day-1)
- - 2.31 - 10 cycles - 0.79 - 10 cycles Albumin 0.60 0.07 20 cycles - 0.56 - 20 cycles Albumin 0.03 -
Corrosion rate of 10 cycle coated sample with albumin coating was calculated separately from two different regions (I and II) of Figure 66: * from region I and ** from region II. Table 4 : Average corrosion rates calculated from Figures 58 and 66 for bare AZ91D, PPy/AZ91D and Albumin modified PPy/AZ91D electrodes, exposed in SBF5 during 1 week.

97
Figure 67: Nyquist plots in SBF5 during 1 week show long term corrosion tests of PPy coated AZ91D Mg alloys coated with different cycles; a) 10 cycles and b) 20 cycles.
After EIS measurements, surface adhesion of albumin modified samples are
also investigated by pull-off tests again. Figure 68 shows that both of the samples are
not heavily corroded as compared to PPy/AZ91 electrodes in Figure 60. However,
very light corrosion is observed for the 20-cycle coated electrode after albumin
coating. As compared to 10 cycles, this electrode also shows better adherence after
pull-off tests (Figure 68 b and d)

98
Figure 68: Mechanical adhesion test of Albumin modified PPy films after 1 week immersion in SBF5.
Figure 69 shows the surface morphology of albumin modified 20-cycle
electrode. In general, surface structures (whiskers in Figure 61) as obtained for the
same electrode without an albumin layer are not observed (Figure 69a). However,
small surface defects (Figure 69b, marked as I in Figure 69a and d marked as II) are
generally found that are most probably formed during the corrosion process. On the
other hand, on defect free surfaces (Figure 69c, marked as III Figure 69a), PPy is
present with typical cauliflower like surface morphology. This may be an indication
of that reducing of the PPy layer does not take place because of sufficient surface
coverage by the albumin monolayer. Therefore diffusion of water through the
polymer/metal interface does not take place and the polymer stays at its oxidized
state. Moreover, the reduction reaction rate of O2 on PPy layer and corrosion of Mg
substrate to give OH- is significantly reduced.

99
Figure 69: SEM images of PPy/AZ91D electrodes after 1 week immersion in SBF5 solution.
In parallel with the findings as explained above, the protection mechanism is
also explained by the releasing profile of dopant counter ions. Figure 70 shows the
release profile of salicylate during 20 days in PBS solution. Both electrodes show the
same type of drug release profiles in orders of µg which is found to be 100 times
lower than that of PPy/AZ91D electrodes in Figure 62. However, the reason of low
amount of dopant present in Figure 70 is that most of salicylate is already released
during the coating of PPy layers during soaking in albumin solutions.
When the electrodes are coated by an albumin layer, drug release profiles
become more durable and follow zero-order kinetics over 20 days with a constant
release of salicylate. It is also worthwhile to stress that the inductive loops are not
observed during EIS measurements for 20 cycles in Figure 67. This may be due to a
self healing effect through the release of dopant ions that is generally provided when
samples are further functionalized by albumin monolayers.

100
Figure 70: Drug release profile of Albumin modified PPy/AZ91D electrodes in PBS:
5.4.5 Polarization curves of PPy/AZ91D and Alb/PPy/AZ91D electrodes
To understand the corrosion inhibition mechanism and the effect of the
coatings on the kinetics of electrochemical reaction rates, polarization curves of bare
and modified AZ91D electrodes were compared when measured directly (Figure 71a)
and after 1 week (Figure 71b) of immersion in SBF5 solution.
The values of Ecorr, icorr, Tafel constants (βa and βc), corrosion rate (CR)
(calculated by AutoLab software, NOVA©), and polarization resistance Rp
(calculated from the Stearn–Geary equation ) [225] (6) are given in Table 5.
Corrosion rates were obtained from these curves.
A parallel trend to EIS results is observed from polarisation curves. As
compared to bare AZ91D, decrease of corrosion current densities with a positive shift
of Ecorr is observed for all coated samples. Additionally, all samples show small
regions of passivation. The strongest effect is found for the Alb/PPy/AZ91D
electrodes. Moreover, after albumin coating, the 20-cycles coated electrode shows
better corrosion protection as compared to the 10 cycles one. The corrosion

101
protection effect of all coatings is clearly related to inhibition of anodic reactions, as
cathodic reactions easily take place on the coated electrodes.
Figure 71: Polarisation curves of bare AZ91D, PPy/AZ91D and Albumin modified PPyAZ91D electrodes in SBF5.

102
Table 5: Potentiodynamic polarization measurement results of pure AZ91D and modified electrodes in SBF5 solution.
Step 1
Step 2
(Albumin)
Ecorr (V)
icorr
(µA/cm2)
ββββa
(mV)
ββββc
(mV)
Corrosion Rate
(mm/year)
Rp
(kΩΩΩΩ. cm2)
Χ
2
- - -1.63 78 220 312 4.09 0.7 1.44 x 10-10
10 - -1.48 9 181 134 1.87 3.7 2.49 x 10-12
20 - -1.47 6 141 97 1.03 4.3 2.12 x 10-12
10 Alb. -1.41 2 136 95 0.09 12.5 9.86 x 10-16
20 Alb. -1.36 1 127 82 0.08 18.1 7.73 x 10-14

103
6.4.6 Surface characterization of PPy/AZ91D and Alb/PPy/AZ91D electrodes
Py/AZ91D Alb/PPy/AZ91D Element Wt% At% Wt% At%
C K 10.88 16.83 36.15 50.25 N K 1.18 1.57 3.32 3.95 O K 45.21 52.51 16.60 17.33 Zn L 1.36 0.39 1.39 0.36 Mg K 21.87 16.71 32.58 22.37 Al K 9.62 6.62 6.54 4.05 P K 5.40 3.24 2.04 1.10 S K 0.16 0.09 0.16 0.08 Cl K 0.51 0.27 0.18 0.09 Ca K 3.81 1.77 1.04 0.43
Table 6: Table shows distribution of the elements on PPy and Alb modified electrodes after 1 week immersion in SBF5 electrolyte. Additionally, chemical composition of surfaces is determined by EDAX
analysis after 1 week immersion in SBF5 (Table 6). As compared to PPy/AZ91D
electrode, more carbon (C) and nitrogen (N) signal is detected for the albumin
modified electrode. This is most probably due to the albumin layer on the PPy.
Moreover, oxygen (O), calcium (Ca), phosphorous (P), chloride (Cl) and sulphur (S)
signals are also found to be less on the albumin modified electrode indicating that the
level of corrosion and formation of corrosion layers including phosphates, chlorides,
sulphates and oxide layers proceeds slowly as compared to PPy/AZ91D electrodes.
5.5 Conclusions
This chapter demonstrates corrosion behaviour of polypyrrole thin films on
magnesium alloy (AZ91D). Electrochemical impedance spectroscopy (EIS) and
corrosion test results show that sodium salicylate doped PPy layers have a corrosion
protection property in SBF5 solutions.
It is also found that PPy layers can be further functionalized by albumin
layers. The resulting surface layers show the most effective corrosion performance by
release of dopant anions. In both cases, PPy layers act as anodic barrier during
corrosion and the corrosion protection mechanism is achieved by release of dopant
anions. The prepared PPy coatings adhered well throughout the period of testing: i.e.
no peeling-off was observed.

104
Dopant release profiles are further improved in addition to the corrosion
performances after the electrodes are coated by albumin monolayers. In addition,
zero order drug release is achieved.
For biomedical applications of Mg and its alloys, such as salicylate doped
PPy layers on Mg alloys, there is a high potential for tailoring the degradation rate,
since all components (substrate, passivating agent, coating) of the resulting system
are non-toxic. Moreover, these coatings have potential for tailoring the degradation
rate of Mg alloys for biomedical applications.

105
6 ANODIC GROWTH OF SELF-ORDERED
NANOPOROUS/TUBULAR LAYERS ON MAGNESIUM ALLOY
WE43
6.1 Abstract
In the present chapter, electrochemical formation of self-ordered nanotubular
and nanoporous magnesium oxy-fluoride structures on a magnesium-based alloy are
explored. The morphology of the surface structures varies with applied potential and
anodisation time. Tubular and porous structures with different sizes and orientation
can be grown.

106
6.2 Introduction Over the past several decades, a variety of self ordering processes by
electrochemical [226] or photoelectrochemical [227] methods such as rapid
breakdown anodisation (RBA) [228], atomic layer deposition (ALD) [229-230] with
or without templates (sol-gel or hydro/solvothermal) [231-232] have been found to
produce oxide nano-structures. These tubular or porous oxide layer structures (Figure
72) can be obtained by anodization of a suitable metal from aqueous or non-aqueous
fluoride-containing solutions by chemical etching of the metal substrates [233] as
summarized in a recent review [234].
Figure 72: The electrochemical anodization process and possible mechanism: a) metal electropolishing, b) formation of insoluble compact anodic oxides, c) self-ordered oxides (nanotubes or nanopores), d) ordered nanoporous layers (reproduced from [234]).
When metals are exposed to a sufficiently anodic voltage in an electrolyte,
depending on the anodization parameters, an oxidation reaction of metal is initiated
where mainly three possibilities exist. The first possibility is that the Mn+ ions are
either solvatized in the electrolyte or the metal substrate continuously dissolved
resulting in electropolishing of the metal (Figure 72a). The second possibility is
formation of an insoluble metal oxide (MO) layer when metal cations (Mn+) react
with O2 from the H2O in the electrolyte (Figure 72b). In some very special
circumstances a third possibility, competition between solvatization and oxide

107
formation resulting in the formation of porous MO layers by chemical etching
(Figure 72c-d).
Over 5 decades ago, anodic growth of porous Al2O3 (Figure 73a) on
aluminum was first reported and this system was found to have a perfect self
organization of pores in the oxide layer [235]. Since then, anodic treatment of other
metal surfaces were studied by many researchers with a growing interest worldwide
became established in many industrial applications or research areas such as
photocatalysis [236-241], solar cells[242-247], dye sensitized solar cells (DSSC)
[245, 248-249], electrochromic devices[250-253], implant materials (cell interactions
and biomedical coatings) on [254-259], drug delivery systems [260-261] and sensing
[262-265] applications.
Figure 73: Cross-sectional and top view (inset) images of; a) porous alumina [266] and b) TiO2 [267] nanotubes.
Formation mechanisms of these structures and models provide better
understanding for the occurrence of self-organization of these porous networks.
These models are mainly based on stress formation at the metal–oxide interface,
optimization of current-flow conditions and attraction of the electric fields as
summerized in a recent review [234]. For instance, electrochemical anodization of
metals in different electrolytes results in different current-time (i-t) characteristics as
overviewed in Figure 74. These curves differ depending on the F- concentration used
for solvatization and result in different types of MO layers. For a widely

108
investigated metal, Ti (Figure 73b), HF is the most used complexig agent which
forms water-soluble [TiF6]-2 structures and complexation occurs with Ti+4 at
intermediate concentrations. The restriction of porous or tubular MO layer formation
to these concentrations is due to the necessity for competition between solvatization
and oxide formation is provided. Else, if the concentration of HF is very low or very
high, either a compact oxide is formed or no-oxide film formation is observed and
electropolishing of the metal surface results.
Figure 74: Typical I–t transitions for different type of metal oxide surface morphologies[234].
An I–t curve for conditions that lead to nanotube formation has three stages:
I) During the initial stage of formation, the curve usually follows the fluoride-free
case and forms an compact oxide layer. In the intermediate stage, stage II, irregular
pores are formed monitored by an current increase that corresponds to increase of the
effective surface area. In the last step, current drops and a regular nano-pores or -
tubes are formed [234].
In addition to porous alumina and tubular TiO2 [234, 267-269], there are also
many works reported in the literature on the formation of ordered porous oxides of
other metals including valve metals, Fe[270-271], Ta [272], Zr [273-274], Nb [275]
and Hf [276], but only a few attempts can be found based on Mg and Mg alloys.
Since magnesium hardly forms defined oxide films in aqueous solutions, it may be a
promising approach to use non-aqueous solutions for anodisation, particularly with
the aim of reducing chemical dissolution during anodisation.

109
Brunner et al. [277] showed the formation of a black, porous oxide-layer on
Mg from water free methanol or ethanol electrolytes containing nitrate ions. Ono et
al. [278-279] studied anodisation of pure Mg [280] in non-aqueous solutions where
they showed the formation of a barrier layer in a solution consisting of a mixture of
amine, ethylene glycol and water. They also investigated the formation of anodic
oxide-films on magnesium surfaces in alkaline-fluoride solutions [281] and for the
first time, using alkaline aluminate solutions, they reported formation of cylindrical
cellular structures [282].
Such surface modifications might be especially interesting for utilizing of Mg
alloys in biomedical applications, as a biodegradable metallic implant material: a
subject of steadily growing interest [195]. In order to optimize the biological
performance, the corrosion behaviour of the material as well as its interactions with
cells need to be tailored. In view of biomedical applications, it is interesting to
mention that on TiO2 nanotubes adhesion, proliferation and differentiation of cells
has been demonstrated to be controlled by the morphology (tube diameter) of
nanotubular layers [283-286]. For Mg alloys, simple chemical surface treatments
such as chemical passivation or biofunctionalization by soaking in simulated body
fluids were recently shown to drastically influence adhesion and survival of cells on
corroding Mg surfaces [287], but specific surface nanostructuring of Mg alloy
surfaces has not been studied in this context.
Figure 75: Backscattered SEM image of the WE43 magnesium alloy.[288]
The alloy selected (WE43) in this chapter is a wrought Mg alloy including
Yttrium (Y), Zirconium (Zr) and Neodymium (Nd). Yttrium has a high solubility in
magnesium and usually added with other rare earth elements to increase creep

110
resistance at high temperatures (300°C) and also to reduce weld cracking [72]. The
microstructure of WE43 alloy consists of different and irregular precipitates of Mg-
Nd or Mg-Y particles precipitated at grain boundaries and the grain interiors [289-
290] as shown in Figure 75. On the other hand, this alloy is one of the likely
candidates for biodegradable implants [201, 291-293]
6.3 Experimental route
Since HF acts as a weak acid and does not dissociate in non-aqueous solvents
such as EG, electrolyte was aged (see below). During the aging of the solution,
titanium was dissolved and therefore the conductivity was increased by formation of
(TiF6)2- ions [267]. Electrolytes of ethylene glycol (EG) were prepared with addition
of hydrofluoric acid (HF): of 500 ml volume and 0.2 M HF concentration. From this
solution 50 ml was ‘aged’ by anodising of Ti overnight for 12 hr at a constant
potential of 120 V in a two electrode cell with the solution as the electrolyte. The Ti
working electrode was sealed to the cell by an o-ring, to expose 1 cm2 of the
electrode surface to the electrolyte, by pressing a Cu plate, which acted as electrical
contact, to the back of the electrode. A platinum foil was used as the counter
electrode. After aging, the aged solution (50 ml) was stirred together with the rest of
the solution (450 ml, non-aged) for 10 minutes. This mixture was found to have the
optimum conductivity to obtain porous surface layers. WE43 Mg alloy samples – cut
from a rod with a diameter of 25 mm – were anodised in the resulting electrolyte
using the same electrochemical cell and counter electrode as used for electrolyte
aging. Prior to anodising, the samples were ground with 1200 grit emery paper. The
chemical composition of WE43 alloy consists of 3.7-4.3% Y with 2.2-4.4% rare earth
elements (Nd) and 0.4% Zr with the balance being Mg.
To investigate the effect of applied potential on the resulting surface
morphology, potential ramp experiments were performed. The potential ramps were
from the 0 V to a maximum potential, Vmax, (of 20, 40, 70, 120, 160, 200, 240 and
300 V) at a sweep rate of 1 V s-1 after which the potential was held constant at Vmax
for 1 additional hour. In addition, effect of the anodisation duration was investigated
for potential ramp experiments with a Vmax of 70 V by applying the same initial
sweep to Vmax (70 V) and by varying the duration the experiment was held at Vmax.

111
A two-electrode setup (Figure 7c) was used as explained in a previous section
(2.1 Electrochemical setup). A field-emission scanning-electron-microscope (Hitachi
FE-SEM S-4800) equipped with an energy dispersive X-ray analyser (EDAX) was
used to investigate the morphology and composition of the surface layers.
6.4 Results and Discussion
6.4.1 Optimization of anodisation potential
Anodisation of the Mg WE43 samples at different potentials indicates the
formation of a compact layer of magnesium fluorides or magnesium oxy-fluorides
(SEM and EDX results, not shown) with some incomplete passivation for potentials
up to Vmax = 40 V, which is followed by a drop in current containing a shoulder. On
such samples randomly distributed surface features such as macro pitting was
observed, but no nano porous or tubular surface morphology could be detected.
Figure 76 shows the top view of an anodized WE43 magnesium alloy surface. The
surface anodized at 20 V shows two remarkable regions under SEM: highly charging
white and relatively less charging black regions. High magnification images (inset
figures of Figure 76) of these areas do not impart either pore or tube formation on the
surface. Only a compact layer of magnesium fluorides or magnesium oxy fluorides is
detected as suggested by EDAX measurements (Figure 77). When the anodisation
potential is increased to 40 V, the same surface morphology is observed (Figure 78).
However, it was found that the white regions have started to crack (Inset bottom-left,
and inset top-left) and a compact oxide layer has started to form at the edge of these
cracks while the compact film on the black regions is thickening.

112
Figure 76: SEM image of WE43 magnesium alloy anodized at 20 V during 1 hour. Inset top-a-) High magnification SEM image of non-charging (black) areas. Inset bottom-b-): High magnification SEM image of charging (white) areas.
Figure 77: EDX spectra of WE43 alloy surface after anodisation by ramping the potential from 0 to 20 V and then holding it at this potential for 1 h in Ti aged 0.2 M HF ethylene glycol solution. Inset bottom-left: Overlay mapping image of O and F. Inset bottom-right: Overlay mapping image of O, F and Mg. Inset Table: Weight and atomic percentage ratio of the elements on the surface.

113
Figure 78: SEM image of WE43 magnesium alloy anodized at 40 V during 1 hour. Inset: High magnification SEM image of a crack.
The surface morphology for 70 V and 120 V anodisation, however,
drastically differs from anodisation at lower potentials. At 70 V elongated
nanoporous structures (nanopores/nanotubes) on the Mg alloy surface are observed
(Figure 79). At this potential, some large recessed areas are formed on the surface;
focusing in these regions reveals nanostructures of both nanopores and nanotubes on
the surface (Figure 79a-f). For example, Figure 79a shows highly packed nanotubular
structures with an average diameter of 100 nm. Porous structures near these tubes
with an average pore diameter of 70 nm are shown in Figure 79b. Moreover, well
defined and separated nanotubes are also found in these regions having an average
diameter of 70 nm (Figure 79c). Lastly, different types of long columns are also
detected (Figure 79d). Among all these different types of surface structures, tubular
forms are the most frequently occurring structures. EDX measurements indicate that
in this case passivation is possible not only by oxide formation but also by formation
of Mg fluorides or oxy-fluorides (Figure 77).

114
Figure 79: SEM images of a WE43 magnesium alloy surface regions anodised in 0.2 M HF ethylene glycol solution at 70 V (a-d) and 120 V(e, f) during 1 h: a) nanotubular structures; b) nanoporous structures; c) separated tubular structures; d) long hollow column structures; e) nanotubular structures; and f) nanoporous structures.
When the applied potential is increased to 120 V, the resulting surface
morphology displays a lesser presence of elongated porous structures as compared to
surface structures formed at 70 V but still displays tubular (Figure 79e) and pore like
(Figure 79f) features. This may be an indication of excess field supported chemical
etching at higher anodisation potentials, in accordance with the slower decrease of
current with time. This effect becomes even more significant at even higher
potentials. Although under the present conditions structures as ideal as grown for Al
or Ti could not be attained; the experiments at 70 V represent an optimum potential
for obtaining of well defined tubular structures on Mg.
This type of surface morphology with more than one kind of structures
suggest that chemical etching and anodisation potential may act as important

115
parameters in the case of magnesium anodisation. Rather than these parameters, also
the type of chemical, solvent used or water addition may influence the surface
morphology such as has been observed for aluminum or titanium. For example,
increase in potential leads to increase in the pore diameter and interpore distance
[294-295] of titanium or amount of HF [275] may also influence surface
morphology. In addition, water addition has an influence on the thickness of oxide
layers [296] in the case of Nb. In addition, an effect due to temperature is also found
to be critical for transition of porous structures to tubular forms for Fe [270].
Although the morphology of oxide layer on these valve metals can be modified by
such parameters, an apparent change of magnesium surface is not observed by
changing these parameters.
During this work, to obtain more ordered nanostructures, many other
experimental parameters were changed as following;
1) Type of base electrolytes (aged with different conductivities with Ti or Mg,
5µS-10mS).
2) Different chemicals as additives (ionic liquids, aluminates, water, different
fluoride sources, NaF, NH4F).
3) Different solvents (methanol, ethanol, glycerol, water, acetone, acetonitrile,
propylene carbonate or their mixtures).
4) Different anodisation temperatures (between -50 to 150 °C)
5) Different types of surface preparation or pretreatments (ground, etched,
polished surfaces or NH4F, NaOH and polymer pre-treated)
6) Area of working electrode, cathodes, continuous stirring or ultrasonication
during anodisation.
All these parameters were tried either separately or in combination with
others to achieve better self-ordered nanostructures. But, these changes resulted in
irregular etching of surface morphologies with formation of a compact oxide layer
similar to those in Figure 77 and Figure 78. Moreover, addition of different
complexing agents (EDTA, oxalates, CN−), annealing of the samples between 300-
600°C on different Mg substrates such as AZ31, AZ91, WE43 and pure Mg also did
not give any self-ordered porous or tubular surfaces. These nanostructures always
took place and tubular forms are only detected in the large pits formed at 70 V under
the previously explained conditions.

116
The morphology in general is found to be similar to TiO2 nanotubes;
however, it is not entirely clear yet, if indeed a barrier layer is present at the bottom
of the tubes. Although under the present conditions structures as ideal as grown for
Al or Ti could not be attained, the experiments at 70 V represent an optimum
potential for obtaining of well-defined tubular structures on Mg.
Figure 80: Current transient as a function of time recorded during electrochemical anodisation of WE43Mg alloy at different potentials in 0.2 M HF/ EG with a sweep rate of 1 Vs-1.
Figure 80 shows the current density as a function of time for samples
anodized between 20 and 300 V. After the potential is increased to the desired
potential by a sweep rate of 1 V s-1, a drop in current density occurs due to oxide
layer formation. After this drop of current density, the plot has a shoulder shape and
reaches to low current densities. At higher potentials more than 120 V (inset Figure
80), after the initial decrease of current density, it again increases which may be due
too temperature increase by high potential. This results to a highly with highly etched
surface morphology dissolution or degradation of tubular or porous structures (SEM
images of 160, 200, 240 and 300 V are not shown here). Afterwards, current
decreases again, probably due to re-passivation of the surface by fluorides. In
addition, at all potentials the current density values are to be found very similar after
1 hour of anodisation which may indicate that the system is diffusion controlled as in
the titanium case [294].

117
6.4.2 Effect of anodisation time
For valve metals, anodisation time is an important parameter influencing the
nanotube layer morphology (i.e. nanotube length). From SEM results and i-t curves
above, an applied potential around 70 V is found as the optimum anodisation
potential to have well defined tubular structures on WE43 magnesium alloy surface
and hence the effect of anodisation time was investigated at this potential. Figure 81a
shows SEM images of a 30 min anodised WE43 surface showing well-ordered
tubular structures of diameter between 50-80 nm (inset figure of Figure 81a). When
the anodisation time is increased to 1 hour (Figure 79a-d), overall surface appearance
looks almost the same but with porous structures of a diameter between 60-100 nm.
A slight increase of pore diameter is observed when the anodisation potential is
increased but the general appearance of resulting surface is found smoother for
shorter time as compared to the 1 hour anodised surface (Figure 79a-d). An increase
of anodisation time to 2 hours leads to the start of chemical etching of the porous
surface (Figure 81b), and after more extended time of 6h all structures are dissolved
due to excess chemical etching, and granular surface morphology results (Figure
81c).
Figure 81: SEM image of WE43 magnesium alloy anodized at 70 V during top left-a) 30 min, top-right-b) 2 hours bottom-c) 6 hours. High magnification image of figure of 7 (top-left-a).

118
6.4.3 Initiation of porous layer
In order to study the initiation phase of nanotube formation, a potential ramp
to Vmax= 70 V followed by a short anodisation time of 3 minutes at Vmax was carried
out. Figure 82 and the details in its insets show four different structural layers: the
first layer (I) is a compact layer (top), the second layer (II) is porous and the third
layer (III) is the propagation layer, i.e., the initiation of these structures is in
agreement with a sequence frequently observed for self-organizing oxide growth
[233]: I, compact layer formation; II, penetration and pore formation underneath the
compact layer; III, ordered pore growth. The growing pores finally lead to a dimpled
scalloped surface (IV).
Figure 82: SEM image of a WE43 magnesium alloy surface anodised in 0.2 M HF ethylene glycol solution at 70V for 3 min. along with a) an inset figure of surface region II; b) an inset figure of surface region III; and c) an inset figure of surface region IV.
With increasing anodisation time, the surface shows well-ordered tubular
structures with diameters of between 50 and 80 nm (as shown for 1 h anodisation in
Figure 79) Overall, anodisation time increases tube order and abundance but for
durations greater than 2 h an increasing loss of the porous structures is observed. This
can be attributed to dissolution processes that begin to dominate the anodisation
reaction.
The mixed type of surface morphology (tubular and porous) and the loss of
the tubular layers after extended anodisation suggest that oxide formation and

119
chemical etching play important interacting roles in the formation of self-ordered
oxide features. This suggests that the anodisation potential, the type of chemical
solvent used, or the addition of water to solution may influence the surface
morphology in a similar way to what has been observed in the case of aluminum and
titanium anodisation, and the effect of these parameters on Mg anodisation will
require further exploration so as to achieve a greater degree and range of ordering.
6.5 Conclusions
Potentiostatic anodisation of a biorelevant magnesium alloy (WE43) in HF
containing non-aqueous electrolyte, after an initial potential sweep, can lead to
growth of ordered oxy-fluoride nanostructures (nanopores and nanotubes). Optimum
conditions for the formation of porous/tubular structures were found to be potential-
and time-dependent. In particular, it should be noted that an optimum parameter
window exists. When the anodisation time or potential is too high, the resulting
surface morphology is destroyed by excess chemical etching. In view of practical
applications, these nanostructured layers on Mg alloys have a considerable potential
in functionalising the surface, for instance in biomedical applications of Mg and its
alloys.

120
7 CORROSION BEHAVIOUR OF CARBON NANOTUBE
MODIFIED MAGNESIUM AND MAGNESIUM ALLOY AZ91D
7.1 Abstract
Carbon nanotubes (CNTs) have been confirmed as good reinforcements for
the improvements of mechanical properties of light metal matrixes such as
magnesium and magnesium alloys. However, the corrosion behaviour of CNT/Mg
composites has hardly been investigated.
This chapter explores the influence of CNT addition on the corrosion
behaviour of pure Mg and Mg alloy AZ91D composites. The corrosion rate is
drastically increased with increased addition of CNTs to the composite matrix due to
the coupling effect between cathodic CNTs and anodic Mg. Additionally, surface
pre-treatment was also found to be critical on the corrosion performance (especially
in low CNT content composite). Moreover, better dispersion of CNTs leads to
reduced corrosion resistivity.

121
7.2 Introduction
Magnesium and magnesium alloys, with a low density of 1.738 g/cm3, exhibit
good mechanical and physical properties. These properties are of interest for use of
Mg alloys in transportation, automobile, electronics and machine tool applications.
However, application of these materials in a wide number of fields is restricted by
their poor corrosion resistance [25, 80].
However, these materials are found to suffer from moderate and high
temperature creep resistance in applications such as gearbox and engine block
components[297]. For instance, for powertrain applications, an elevated temperature
performance at service conditions of 150-200°C and stresses in the range of 50-70
MPa is required where the commercial AM and AZ type of magnesium alloys do not
possess these properties[10].
In order to further improve high-temperature strength and creep resistance of
these materials, development of new types of alloys is the main scope of many
scientists worldwide. But due to high production costs, complexity and their poor
machinability, reinforcing magnesium with nanoparticles or carbon materials instead
of alloying was found to be an alternative solution while preserving the low density
of the material. For example, magnesium or its alloys reinforced with inorganic
nanoparticles such as SiC [298-300], Al2O3 [301-302], Y2O3 [303-304] and SiO2
[305] are reported to increase both high-temperature performance and mechanical
properties.
To further improve the mechanical properties, carbon reinforced magnesium
composite materials also have been developed [306]. Hall [307] and Bakkar et al.
[308] have studied the effect of carbon fibre addition into the Mg matrix and showed
that even though the mechanical properties are improved, the formation of galvanic
cells between the Mg matrix and the carbon fibres reduces the corrosion resistance of
the composites.
Since their discovery in 1991 [309], another carbon material, carbon
nanotubes have attracted remarkable interest due to their excellent mechanical,
electrical and physical properties. There are mainly two types of nanotubes available;
single walled (SWNT) and multi-walled (MWNTs) nanotubes [310-312]. SWNTs are
made of a single sheet of graphene rolled to form a cylinder and MWNTs consist of
multiple rolled layers of graphene or rolled graphite. Depending on the relationship

122
between rolling direction and the unit vectors of the graphene sheets, CNTs can be
metallic or semi-metallic [313]. Semi-conducting nanotubes have bandgaps ranging
between approximately 1.8 eV and 0.18 eV [314]. On the other hand, metallic carbon
nanotubes are extremely conductive, as their one-dimensional nature allows their
charge carriers to carry very large current densities of up to 100 MA/cm2 [315]. For
semi-conducting nanotubes, carrier mobilities are reported as 105 cm2/V s [316]. It
has also been reported that SWNTs show superconductive properties around
temperatures of 5 K [317].
These properties make them very unique materials with a whole range of
promising applications such as in electron gun components in vacuum
microelectronics [318] (in electron microscopes or in microwave amplifiers), in flat
panel displays [319-320], in gas-discharge tubes in telecom networks [321], and in
energy storage [322-324] as summarized in a recent review [313, 325]. CNTs are
also considered as ideal reinforcements for their high strength, light weight, high
performance composites [313] due to their favorable geometrical (high aspect ratio,
high specific surface area) and excellent mechanical properties (very high strength
modulus, extreme rupture strength). Theoretical simulation [326-327] and
experimental results [328-330] have indicated their extraordinary strength (about 150
GPa) and Young’s modulus (about 1 TPa).
Even though the mechanical properties of nanotube modified light metal
composites are in general found to be enhanced, the corrosion behaviour of these
materials has not yet been explored in detail. Due to the very low standard electrode
potential of -2.36 V, and due to the poor protective properties of Mg oxide/hydroxide
layers in many environments, Mg alloys show a very low corrosion resistance. The
low standard potential of Mg of course makes the material very prone to galvanic
corrosion, when coupled to almost any other technologically interesting conducting
material. The first study on the corrosion behaviour of CNT reinforced Mg AZ91D
alloy composites, recently presented by Endo et al. [331], claimed that CNTs act as
good mechanic fillers in Mg AZ91D alloy and reduce the corrosion rate of Mg. On
the other hand, Fukuda et al. [332] also showed the formation of galvanic cells
between the Mg matrix of a AZ31B alloy and CNTs with a high (-surface – Kelvin
Probe) potential difference of 1.1 V. Similar effects of increased corrosion due to
microgalvanic action between CNTs and the Mg matrix were reported by Aung et al.
[333] for Mg/CNT composites.

123
Even though these first reports on the corrosion behaviour of Mg/CNT
composites indicate that CNTs are deleterious due to internal galvanic corrosion,
these studies are still rather preliminary in nature. In the present chapter, the
corrosion process of CNT-reinforced Mg and Mg alloy AZ91D composites is
explored in more detail and over a large time period, from initiation of corrosion until
extended durations of one week. Using a combination of electrochemical and surface
characterization techniques, the aim of the present chapter is to provide further
understanding of the role of MWNTs during Mg corrosion. Even though similar
galvanic corrosion effects are expected to take place as has been reported for
Mg/carbon fiber composites, the nanoscopic size of the local cathodes may lead to a
different behavior than for larger cathodic sites in the material.
7.3 Experimental
A two-step process is applied to produce the MWNT/Mg composites: first, a
pre-dispersion step was performed on MWNTs to avoid formation of large
agglomerates followed by fabrication of MWNT/Mg composite by melt stirring
technique. By using the pre-dispersion step, a relatively homogenous dispersion of
MWNTs in Mg matrix could be achieved that was expected to affect the corrosion
behaviour. The block copolymer Disperbyk-2150 (BYK Chemie GmbH) was first
dissolved in ethanol, which has been proven to be a good dispersing agent to improve
the dispersion of CNTs [334]. MWNTs (Baytubes® C 150P) were added to the as-
prepared solution. This mixture was put into an ultrasonic bath and stirred 30
minutes. After adding Mg (Chempur) or Mg alloy AZ91D (ECKA) chips, the
suspension was further stirred until the ethanol was evaporated. The MWNT coated
chips were then placed in a cylindrical sample crucible in an oven to heat up to
670°C.
Different percentage of MWNTs (0.1 wt%, 1 wt% and 5 wt%) were used to
produce MWNT/AZ91D and 0.1 wt% of MWNTs was used to produce MWNT/Mg
composites. When the Mg chips were molten, the melt was mechanically stirred at
350 rpm during 30 minutes to further disperse MWNTs. After stirring, the molten
MWNT/Mg (MWNT/Mg –dispersed) composite was poured into a mould. A pure
Mg sample was also produced without MWNT addition by the same technique. In
order to check the influence of the pre-dispersion process, the reference samples

124
(MWNT/Mg and MWNT/AZ91D – non-dispersed) were produced using the same
technique without the pre-dispersion step.
Corrosion tests were performed in 3.5% NaCl solution (Sigma-Aldrich, purity
>99.5) that was refreshed daily (250 ml/day) as described in section 2.2.3 Corrosion
tests.
Further analysis by SEM, FIB and AES were also performed (2.3 Surface
analytic methods).
7.4 Results and Discussion
7.4.1 Corrosion behaviour of MWNT reinforced AZ91 composites
Figure 83 shows the corrosion behaviour of MWNT reinforced AZ91D
composites (1 and 5%) after different surface pretreatments. Inset images in Figure
83 show the corresponding corroded surfaces of the composites after 1 day of
immersion in 3.5 % NaCl. The effect of MWNT additions can be observed from the
images but also visually during the experiment by the hydrogen gas bubbling level. It
is obvious that 5 wt% MWNT/AZ91D were highly corroded compared to 1 wt%
MWNT/AZ91D samples. The corrosion rates concur with these results. Corrosion
rates of the composites were found between 9-18 g/m -2day-1 with 1 wt% MWNTs
and ~24 g/m-2 day-1 with 5 wt% MWNTs.

125
Figure 83: Corrosion rate results of MWNT/AZ91 composites in 3.5 % NaCl for different amount of MWNT additions (1 and 5 wt%) and different surface pre-treatments (ground, polished and H2SO4 etched) after 1 day of immersion in 3.5 % NaCl. Inset images: Corresponding optical microscopy taken after 1 day immersion in 3.5 % NaCl.
Montemor et al. [335] have shown that adding Ce or La-modified CNTs into
an amino silane film on AZ31 induced significant reduction of both anodic and
cathodic activity at the metallic surface. However, until now no study has been
carried out to check whether adding “clean” CNTs (i.e. without any pre-treatment) in
the metal alloys could lead to galvanic coupling, which can increase the corrosion
rate of the Mg phase. In our experiments, a strong gas bubbling was observed
proportional to the MWNT amount in the matrix, which could be explained by the
coupling leading to faster dissolution of the Mg phase.
Additionally, surface preparation was also found to be an important factor. In
1 wt% MWNT/AZ91D composite, the highest corrosion rate was measured on the
chemically etched surfaces. The origin of this effect is not yet clear; however surface
pre-treatment seems a critical parameter for the low MWNT-containing composite.
On the other hand, surface pre-treatment was not critical for 5 wt% MWNT/AZ91D
composites. Weight loss of different samples was also measured for 7 days. Strong

126
hydrogen bubbling was also observed during the experiments, especially for the 5
wt% MWNT/AZ91D composite.
For improved mechanical properties, it is known that a homogenous
dispersion of CNTs in a material matrix is important. Without a homogenous
dispersion of CNTs, the excellent properties of CNTs can not be exploited and CNTs
would act as defects rather than reinforcements in the matrix. In order to understand
the influence of the CNT dispersion on the corrosion behaviour of the CNT
magnesium composite, two different samples were prepared. One sample was
produced by using the two step process [336], which has been confirmed that more
homogeneously dispersed MWNTs in AZ91D composite can be achieved; another
sample was produced without a pre-dispersion process, so MWNTs are mainly in
bundles in the AZ91D matrix. Figure 84a-c shows the optical microscopy on the
surfaces of the different samples after 1 day immersion in 3.5% NaCl. The effect of
CNT dispersion can be easily observed even at low MWNT contents. A bigger and
more evenly dispersed corrosion pattern can be observed in Figure 84a (with pre-
dispersion process) compared to a smaller corrosion corner in Figure 84b (without
pre-dispersion process). With well-dispersed MWNTs in AZ91D, formation of
galvanic couples takes place everywhere on the composite surface, hence strongly
increasing the corrosion rate of the alloy. Without pre-dispersion, MWNTs exist
mainly in bundles in the composites, so the coupling effect only takes place at
MWNT agglomerates (as suggested by Figure 84b). Figure 84c exhibits the pristine
AZ91D alloy corroded surface. Corrosion only takes place on a small area (seen in
the circle) and the corrosion level after 1 day is quite low compared to the
MWNT/AZ91D composites.
Figure 84d shows the weight loss for 5 wt% MWNT/AZ91D composites with
and without pre-dispersion. Clearly, a much higher weight loss of the more
homogeneously dispersed CNT/AZ91D composite (with pre-dispersion process)
compared to the less homogeneously dispersed CNT composite (without pre-
dispersion process) is observed confirming that the better the dispersion, the worse
the corrosion behaviour.

127
Figure 84: Optical microscopy of 0.1 wt% MWNT/AZ91D with a) pre-dispersion process, b) without pre-dispersion and c) pristine AZ91 sample after 1 day immersion in 3.5 % NaCl; d) weight loss results of 5 wt% MWNT/AZ91D in 3.5 % NaCl during 1 week with/without pre-dispersing MWNTs. The circle in c) shows the corrosion takes place only in a small area on the pristine AZ91D surface.
The corrosion product of the 5 wt% MWNT/AZ91D sample in 3.5% NaCl
solution was collected and dried after the corrosion test, and the residue was observed
under SEM as seen in Figure 86. The pattern in the circle looks very different from
Fig. 4b, which presents a typical corroded AZ91D composite surface with short,
sharp and needle-shaped crystals. The materials in the circle rather look like to have
relatively long and cylindrical shapes with a smooth surface, which is more like
pristine MWNTs (Figure 85c) and pre-dispersed MWNTs on AZ91D chips (Figure
85d). Therefore, we assume that they are CNT-like materials. This finding suggests
that the CNTs themselves are not dissolved, supporting the proposal that they act as
local cathodes in the composite. (More detailed explanation of the corrosion products
as well as corrosion mechanism will be presented in next section).

128
Figure 85: SEM images of a) 5 wt% MWNT/AZ91 powder dried after corrosion; b) the typical corrosion pattern on 5 wt% MWNT/AZ91 composite; c) pristine MWNTs and d) MWNTs homogenously dispersed on the surface of AZ91 chips [336]. The circle in a) indicates a CNT-like area.
7.4.2 Corrosion behaviour of MWNT reinforced Mg composites
In this section, the influence of the MWNT addition into the pure Mg matrix
is explained by electrochemical means in more detail. Further characterization of
MgH2/Mg(OH)2 layer is also provided by surface analytic techniques.
7.4.2.a Influence of MWNT addition on the electrochemical behaviour of Mg
The OCP of pure Mg and MWNT/Mg composites was measured over 30
minutes in 3.5% NaCl solutions (inset of Figure 86). In the first 5 minutes, the
potential for all samples shifts towards less negative values very fast, indicating rapid
formation of a partially protective Mg(OH)2 layer on the surface. After 5 minutes, the
formation rate of the Mg(OH)2 oxide layer is lowered and the slope of the OCP/time
curves is decreased. Finally, OCP values reach slightly different steady-state values
around -1.65 V. The OCP values are quite similar for all the samples which is not
surprising considering the small area fraction of the MWNTs on the sample surfaces.

129
After OCP measurements, electrochemical impedance spectra were measured (Figure
86). Addition of (only 0.1 wt-%) MWNTs to the Mg matrix leads to a measurable
and significant increase of the dissolution rate. The corrosion behaviour of pure Mg
has been frequently studied by EIS, and interpretation of the behaviour has been
provided for example by Baril et al. [81-82] and Song et al. [80]. According to Baril
et al., capacitive loops in the EIS spectra are the relaxation of mass transport in the
growing solid oxide phase on the Mg surface. Song et al. proposed that these loops
are attributed to unipositive Mg+ ion concentration within the cracked areas [73] of a
surface oxide layer. Most recently, Swiatowska et al. [337] reported that dissolution
of Mg in both NaCl and Na2SO4 electrolytes takes place with no evidence for the
often invoked Mg+ intermediate. Independent of the detailed interpretation of the
origin of some of the spectral features, the most straightforward observation in our
case is evidenced by the decrease of the charge transfer resistance (Rt) (i.e., the
diameter of the Nyquist curve along the real axis) due to the addition of MWNTs in
Mg.
Figure 86: Nyquist plots of pure Mg and MWNT/Mg composites in 3.5% NaCl after 30 minutes at open circuit potential. Inset: Open Circuit Potential versus time curves of pure Mg and MWNT/Mg composites in 3.5% NaCl.

130
The value of Rt is ~2.0 kOhm, ~2.5 kOhm and ~3.0 kOhm for dispersed
MWNT/Mg, non-dispersed MWNT/Mg and pure Mg, respectively. Clearly, addition
of a small amount of MWNTs into the Mg matrix decreases the corrosion resistance
and the corrosion resistance becomes even smaller, when a pre-dispersion step is
applied during preparation of the composites. This is in line with findings on
MWNT/AZ91D composites in which MWNT dispersion was shown to significantly
increase the dissolution rate as already explained in a previous section. The
polarisation resistance (Rp) values - the difference along the real axis between the
resistance at the highest and lowest frequencies – show the same trend as the Rt
values, with an inductive loop in the low frequency (LF) regions. For Mg and Mg
alloys, inductive loops have been attributed to adsorbed surface species such as
Mg(OH)+ads and Mg(OH)2ads [73, 81-82, 337]. Additionally, these inductive loops are
also attributed to Mg ions on relatively film-free surfaces [84, 338].
The reaction of pure Mg in aqueous solutions is given by the equations below
[28]:
Mg(s) Mg+2 (aq) + 2e- (anodic reaction)…………………… (14)
2H2O + 2e- H2(g) + 2OH- (aq) (cathodic reaction) ………….(15)
Mg+2 (aq) + 2OH- (aq) Mg(OH)2 (corrosion product)…….….(16)
According to these reactions, and as is well-known, dissolution of Mg leads
to pH increase in the surroundings. Figure 87 shows the change of pH values for all
samples during 90 minutes of immersion. A rapid increase of pH is achieved during
the first 5 minutes in 3.5% NaCl. After 5 minutes, pH/time curves of different
samples continue to increase with different slopes, indicating different dissolution
rates. The highest pH value is obtained for dispersed MWNT/Mg composite followed
by the non-dispersed one. The pure Mg shows the relatively slowest rate of pH
increase, indicating less dissolution in this time period. This is in good agreement
with the EIS results, and again confirms that the addition of MWNTs leads to
increased dissolution of Mg. As can be seen from the optical inset images of Figure

131
87, when a pre-dispersion step is applied, rate of corrosion also increases and leads to
a macroscopically more uniform corrosion process.
Figure 87: The pH change of each sample in 50 ml 3.5% NaCl during 90 minutes. Inset: Optical images of specimens after 1 day immersed in 50 ml 3.5% NaCl.
In order to better understand the effect of the CNT addition on the corrosion
rate of Mg, potentiodynamic polarization curves were measured (Figure 88). The
polarization curves demonstrate that an acceleration of the cathodic hydrogen
evolution kinetics is observed when MWNTs are added to Mg, but the kinetics of the
anodic dissolution are not significantly influenced by the presence of carbon
nanotubes in the material. Hence, the influence of the MWNTs on the
electrochemical behaviour of Mg is similar to the well-known effects of impurities,
such as Fe, in accelerating kinetics of the hydrogen evolution (see e.g. [339-340]). It
is noteworthy that the composites contain only 0.1 wt-% MWNTs; hence the
MWNTs can be considered as efficient cathodes, as their influence is well observed
even with this small concentration (n.b. the polarization curves are very
reproducible).

132
Figure 88: Polarisation curves of pure Mg and MWNT/Mg composites recorded immediately after immersion in 3.5% NaCl.
Moreover, polarization curves of pure Mg and the Mg/MWNT composites were
compared when measured directly after immersion, and after 6 h of immersion in
3.5% NaCl solution (see Figure 89) and the results are summarized in Table 7. The
values of Ecorr, icorr, Tafel constants (βa and βc), corrosion rate (CR) (calculated by
AutoLab software, NOVA©), and polarization resistance Rp (calculated from
Stearn–Geary equation )[225] (17) are given in Table 7. Corrosion rates were
obtained from these curves.
)ββ(i 2.3
β .βR
cacorr
cap +
= ……………(6)
In all cases, the cathodic current densities are increased with immersion time,
but the effect is the strongest for the composite with dispersed carbon nanotubes
(Figure 89c). Similar effects of faster hydrogen evolution on pre-corroded surfaces
have been previously observed for pure Mg and some Mg alloys [341]. The higher
cathodic currents after 6 h of immersion can result from a larger cathodically active
area after corrosion, as corrosion leads to roughening of the surface. This may also
lead to exposure of a higher number of CNTs on the surface, as the CNTs are
expected to be chemically and electrochemically stable under these conditions. With

133
the progress of dissolution of the Mg matrix, the remaining CNTs probably become
detached but at the same time, new CNTs can become exposed. Moreover, with time,
the surface becomes increasingly covered by corrosion products layers. Therefore,
the corroding surfaces of the Mg/CNTs composites are highly dynamic. The finding
that this immersion-induced increase of the cathodic currents is the strongest for the
composite with dispersed nanotubes is logical in that a fine distribution of the carbon
nanotubes leads to creation of a large number of local galvanic couples on the
surface; hence, dissolution on the microscopic scale can be expected to be highly
non-uniform and to lead to the largest increase of the electrochemically active surface
area. The anodic parts of the polarization curves suggest a slight decrease of anodic
dissolution with time, which may result from the growth of corrosion product layers
on the surface. Overall, however, for all materials the corrosion rates increase with
time indicating the poor protective properties of the formed corrosion product layers.
Table 7: Potentiodynamic polarization measurement results of pure Mg and MWNT/Mg composites in 3.5% NaCl solution.
Electrode Ecorr
(V)
icorr
(µA/cm2)
ββββa
(mV)
ββββc
(mV)
Corrosion
Rate
(mm/year)
Rp
(ΩΩΩΩ. cm2)
Pure Mg (0h) -1.627 36.477 31.335 186.540 1.668 635.790
Pure Mg (6h) -1.531 28.168 58.245 246.000 1.288 726.100
Non-disp. (0h) -1.607 63.185 39.302 230.970 2.890 465.500
Non-disp.(6h) -1.499 71.965 31.342 304.820 3.292 341.370
Dispersed (0h) -1.618 98.291 49.123 281.290 4.469 368.200
Dispersed (6h) -1.520 280.390 151.430 372.990 12.825 332.830

134
Figure 89: Polarisation curves of pure Mg and MWNT/Mg composites after different immersion durations in 3.5% NaCl.
7.4.2.b Characterization of the corrosion product layers
To observe the initial formation of the corrosion layer on MWNT/Mg, SEM
images of the samples were taken after 90 seconds of immersion in 3.5% NaCl. As
compared to pure Mg (Figure 90a), SEM images of both MWNT/Mg composites
(non-dispersed, figures Figure 90b and dispersed, Figure 90c) show higher surface
coverage by corrosion products even after short (90 seconds) immersion times.
Additionally, dispersion of the MWNTs in (Figure 90c) seems to lead to more
homogeneous corrosion product formation over the surface. However, in all cases an
incomplete and inhomogeneous coverage of the surface is observed after 90 s
immersion.

135
Figure 90: SEM images showing surfaces covered with Mg(OH)2 species after exposed in 3.5% NaCl after 90 seconds; a) pure Mg, b) non-dispersed MWNT/Mg and c) dispersed MWNT/Mg.
To elucidate the effect of MWNTs in the corrosion process in more detail,
laterally resolved AES measurements were performed. These analyses were carried
out on samples that were soaked in pure water for 12 h, to simplify the chemical
analysis of the surface. Figure 91a-c shows the carbon signals from AES mappings
and corresponding SEM images (Figure 91d-f) for pure Mg and MWNT/Mg
composites. It is worthwhile to emphasize that even though MWNTs are absent in
the pure Mg matrix, weak carbon signals were detected, mostly due to carbon
contamination (Figure 91a). If pure Mg is selected as a background image, one can
conclude that the high intensity of C signals (yellow pixels in Figure 91b) is
attributed to non-dispersed MWNT agglomerations. Additionally, formation of
Mg(OH)2 layers is also favoured on these MWNT bundles; this could be attributed to
formation of C-Mg red-ox couples and therefore to galvanic corrosion process.
Similar clusters of corrosion products are also observed for the dispersed MWNT/Mg
sample (Figure 91c). However, in this case localized clusters of carbon signals
(yellow) are not detected, indicating that the MWNTs were successfully dispersed
during production of composites (hence, the signals from dispersed MWNTs are
below the detection level of the measurement).

136
Figure 91: AES results (a-c) with corresponding SEM images (d-f) showing distribution of MWNTs through the Mg(OH)2 layers after exposed in pure water after 12 hours; a,d) pure Mg, b,e) non-dispersed MWNT/Mg and c,f) dispersed MWNT/Mg.
The corrosion product layers were further analyzed by FIB-cut SEM images.
Figure 92 shows the cross-section of the corrosion product layers formed during 90-
min immersion in 3.5% NaCl. In all cases, an inner compact and an outer porous
layer is observed. The thickness of the corrosion product layer very strongly varies
for pure Mg and for the composite samples: for pure Mg, an ~1100-nm thick layer is
observed (Figure 92a), a layer thickness of ~510 nm is found for non-dispersed
MWNT/Mg (Figure 92b), and ~350 nm for dispersed MWNT/Mg (Figure 92 c).
The higher the corrosion rate, the thinner the corrosion product layer. The origin of
this behaviour may be related to the vigorous H2 gas production on the Mg
composites.

137
Figure 92: FIB-cut SEM images showing composite cross-section surfaces covered with Mg(OH)2
film after being exposed in NaCl for 90 minutes; a) pure Mg, b) non-dispersed MWNT/Mg and c) dispersed MWNT/Mg.
The strong H2 bubbling enhances transport of dissolution products (e.g. Mg+2
ions) away from the surface. Therefore, precipitation of the corrosion products is
decreased. In addition, H2 bubbling may partially detach the growing corrosion
product layer. This could result not only in thinner corrosion product layers, but
moreover in different surface morphologies as shown in Fig. 8a-c. In the case of
dispersed MWNTs in the composite, a smoother morphology of the corrosion
product layer is observed in the top-view image. A schematic illustration of the
influence of clustered and finely-distributed carbon nanotubes on the anodic and
cathodic reactions on the composite surface is included in Figure 93.

138
Figure 93: SEM Images of a) pure Mg, b) non-dispersed MWNT/Mg and c) dispersed MWNT/Mg after exposed in NaCl for 90 minutes. d) Model showing the relationship between a, b and c.
7.4.2.c Long term corrosion measurements
Figure 94 shows the average corrosion rates of dispersed MWNT/Mg, non-
dispersed MWNT/Mg and pure Mg exposed in NaCl during 1 week. Average rates
were calculated and compared by using two methods; 1) weight loss calculations, and
2) hydrogen gas collection measurements. These two methods were used together to
check validity of the measurements, as both methods have their strengths and
challenges (e.g., proper removal of the corrosion product layers is crucial for correct
weight loss measurement). A small difference between the results of the two
methods was indeed observed, probably due to corrosion products that were not
completely removed during immersion in CrO3 + AgNO3 solution. However, the
general agreement between the two methods was good as shown in Figure 94.

139
Figure 94: Change of average corrosion rates of pure Mg, dispersed MWNT/Mg and non-dispersed MWNT/Mg that were each exposed in NaCl during 1 week.
Similar to the initial stages of corrosion as discussed in Section 5.4.2.a,
Figure 94 indicates that the addition of MWNTs into Mg increases the corrosion rate,
and moreover, the daily corrosion rate increases with immersion time (the trends for
the corrosion rate follow the same behaviour both for the weight loss and for the
volume of produced hydrogen gas). The increase of the corrosion rate with time is
most probably due to an increase of the effective surface area by dissolution-induced
roughening of the surface, similarly as was discussed in the case of short-term
immersion effects on the cathodic current densities (Figure 89).
Qualitatively this increase of the corrosion rate with time is the strongest for
the sample containing well-dispersed MWNTs (see dotted lines in Figure 94 which
are drawn as a guide to the eye), which is in agreement with the formation of a high
number of well-distributed galvanic couples on the surface (i.e., finely distributed
localized dissolution leads to the strongest roughening of the surface). As discussed
previously, the corrosion product layers forming on the surface do not provide high
corrosion protection; hence the surface roughening effects in increasing the corrosion
rate overrun the small barrier effect of the corrosion products in decreasing the
dissolution rate. It is noteworthy that the effect of CNTs on the corrosion behaviour

140
prevails over extended times of dissolution, in spite of the low concentration and the
small size of the local cathodes in the matrix.
7.5 Conclusions
In contrast to the previous report by Endo et al. [331], the present chapter
clearly demonstrates that reinforcement of pure Mg and Mg alloy AZ91D by multi
wall carbon nanotube (MWNT) addition leads to a strong increase of the corrosion
rate by formation of galvanic couples between the pure Mg matrix and multi wall
carbon nanotubes (MWNT). Moreover, pre-dispersion of MWNTs increases the
number of galvanic couples and therefore increasing the corrosion rate.
However, addition of MWNTs into the pure Mg matrix plays an important
role in the formation of the corrosion product layers in the early stage of corrosion,
but even more significantly increases the corrosion rate over extended exposure
periods. Therefore, in order to exploit the outstanding mechanical properties of
MWNT reinforced Mg (alloy) composites, additional corrosion protection
measurements are required.
In regards of modifying bulk structure of magnesium material, it would be
aimed to have controlled and homogenous corrosion morphology (not localized) as it
is demonstrated that the corrosion rate of magnesium increases in presence of CNTs.
Thus, different types of CNTs (e.g., single-wall vs. double-wall carbon nanotubes) or
surface-modified CNTs would be of interest to tailor corrosion properties.
Additionally, it would also be of interest to focus on surface modification of CNTs
by electrically insulating or dispersing layers without deteriorating their mechanical
properties as the distribution of the CNTs in the matrix is randomly and the CNTs
have a tendency to cluster in the present case.

141
8 CONCLUSIONS AND OUTLOOK

142
This work covers new types of surface and bulk modifications of magnesium
and its alloys with the aim of tailoring its corrosion rate. In general, it is found that
surface pre-treatments have a significant effect on corrosion behaviour of
magnesium. For instance, in chapters 3, 4 and 5, two different types of surface pre-
treatments are reported; a conventional NH4F pre-treatment and polypyrrole (PPy)
coatings as a new approach. In chapter 3, it is presented that acidic etching and NH4F
steeping durations are very sensitive parameters with respect to the increase of
corrosion resistance. For instance, although short durations of treatment have a
beneficial effect, longer durations of etching and steeping results in dissolution of
magnesium and does not provide a protection, as the chemical etching starts to take
place.
As another pre-treatment strategy, in chapter 4 and 5, a first time formation of
conducting polymer coating (polypyrrole –PPy-) on Mg alloy AZ91D is reported
from aqueous solutions of sodium salicylate by cyclic voltammetry (CV) method. In
contrast to findings from NH4F steeping, these thin films show better protection and
corrosion resistance. It is also worthwhile to stress that these electrically conductive
and organic layers can be further functionalized by additional functional groups. For
instance, in chapter 5, it is demonstrated that further modification of PPy layers with
albumin layers shows even better corrosion performance by release of dopant anions.
In both cases, PPy layers act as anodic barriers and the corrosion protection is
achieved by release of dopant anions. Moreover, these layers remained very adhesive
over the corrosion period and no peeling-off was observed.
In addition to surface pre-treatment approaches, a small example about tuning
of surface morphology of magnesium alloy WE43 is given in chapter 6. It is
presented that anodisation of a biorelevant magnesium alloy (WE43) in HF
containing non-aqueous electrolyte can lead to growth of self-ordered nanostructures
such as nanopores and nanotubes. These structures have considerable potential in
tailoring and functionalising the surface, for instance in biomedical applications of
Mg and its alloys.
In chapter 7, bulk modification of Mg and Mg alloy AZ91D with multiwall
carbon nanotubes (CNTs) is carried out. Results reveal that although CNTs have
been confirmed as good reinforcements to improve the mechanical properties of light
metal matrixes, addition of these materials into a Mg matrix drastically increases the
corrosion rate as the coupling effect between cathodic CNTs and anodic Mg

143
increases. Moreover, better dispersion of CNTs leads to even more corrosion rates
and reduced corrosion resistivity.
In general, surface modification of magnesium is very open to new
approaches and strategies. To use this metal as a biomedical component or in
industry, a proper coating or surface treatment can be achieved by multi steps of
coatings, as every coating step has its individual protection mechanism and property.
For instance it would be of interest to use acidic etching or ammonium fluoride
treatment before a zinc-phosphate coating or a hard anodisation process.
Additionally, further modification of porous/tubular Mg surfaces or PPy layers with
other biocompatible ceramic layers or biopolymers (e.g., bioglass, chitosan) would be
of interest to extend these approaches to a wide scale of applications.

144

145
References:
1. Witte, F., The history of biodegradable magnesium implants: A review. Acta
Biomaterialia, 2010. 6(5): p. 1680-1692. 2. Cisar, L., et al., Development of high strength and ductile magnesium alloys
for automobile applications. Magnesium Alloys 2003, Pts 1 and 2, 2003. 419-4: p. 249-254.
3. Schumann, S. and H. Friedrich, Current and future use of magnesium in the automobile industry. Magnesium Alloys 2003, Pts 1 and 2, 2003. 419-4: p. 51-56.
4. Goken, J., et al., New development in magnesium technology for light weight structures in transportation industries. Thermec'2003, Pts 1-5, 2003. 426-4: p. 153-160.
5. Heuler, P. and K. Sponheim, Light weight structures by material selection? Fatigue strength of cast magnesium and aluminium parts. Materialprufung, 2001. 43(5): p. 195-200.
6. Magusin, P.C.M.M., et al., Hydrogen sites and dynamics in light-weight hydrogen-storage material magnesium-scandium hydride investigated with H-1 and H-2 NMR. Chemical Physics Letters, 2008. 456(1-3): p. 55-58.
7. Dou, S.X., et al., Aluminium-stabilized magnesium diboride - a new light-weight superconductor. Superconductor Science & Technology, 2006. 19(4): p. 333-337.
8. Beals, R.S., et al., USAMP magnesium powertrain cast components: Fundamental research summary. Jom, 2007. 59(8): p. 43-48.
9. Powell, B.R., The USAMP magnesium powertrain cast components project. Jom-Journal of the Minerals Metals & Materials Society, 2003. 55(11): p. A28-A29.
10. Pekguleryuz, M.O. and A.A. Kaya, Creep resistant magnesium alloys for powertrain applications. Advanced Engineering Materials, 2003. 5(12): p. 866-878.
11. Luo, A.A., Recent magnesium alloy development for automotive powertrain applications. Magnesium Alloys 2003, Pts 1 and 2, 2003. 419-4: p. 57-65.
12. Anyanwu, I.A., et al., Heat resistant magnesium alloys for automotive powertrain applications. Magnesium Alloys 2003, Pts 1 and 2, 2003. 419-4: p. 445-450.
13. Aghion, E., et al., Newly developed magnesium alloys for powertrain applications. Jom-Journal of the Minerals Metals & Materials Society, 2003. 55(11): p. A30-A33.
14. Powell, B.R., The USAMP magnesium powertrain cast components project. Jom-Journal of the Minerals Metals & Materials Society, 2002. 54(2): p. 49-50.
15. Feng, L.J., et al., Investigation on corrosion of yttrium-doped magnesium-based sacrificial anode in ground grid protection. Journal of Rare Earths, 2010. 28: p. 389-392.
16. Little, R.D., et al., The influence of electrogenerated Sm(II), electrogenerated Yb(II), and magnesium ions produced at a sacrificial magnesium anode, upon the diastereoselectivity of electroreductive cyclization reactions. Journal of Electroanalytical Chemistry, 2006. 593(1-2): p. 69-73.

146
17. Bonafoux, D., et al., Stereoselective Electrochemical Synthesis of Silyl Enol Ethers Using a Sacrificial Magnesium Anode. Journal of Organometallic Chemistry, 1995. 493(1-2): p. 27-32.
18. Mcharek, S., et al., Advantage of the Use of a Sacrificial Magnesium Anode in the Electro-Carboxylation of Aliphatic, Aromatic and Vinylic Carbonyl-Compounds. Bulletin De La Societe Chimique De France, 1989(1): p. 95-97.
19. Atrens, A., et al., Calculated phase diagrams and the corrosion of die-cast Mg-Al alloys. Corrosion Science, 2009. 51(3): p. 602-619.
20. Gebert, A., et al., Stability of the bulk glass-forming Mg65Y10Cu25 alloy in aqueous electrolytes. Materials Science and Engineering a-Structural Materials Properties Microstructure and Processing, 2001. 299(1-2): p. 125-135.
21. Morales, E.D., et al., Corrosion behaviour of magnesium alloys with RE additions in sodium chloride solutions. Magnesium Alloys 2003, Pts 1 and 2, 2003. 419-4: p. 867-872.
22. Tong, Z.S., et al., Galvanic corrosion behavior of die cast AZ91D magnesium alloy in chloride solution. Journal of University of Science and Technology Beijing, 2004. 11(2): p. 127-132.
23. Tunold, R., et al., Corrosion of Magnesium in Aqueous-Solution Containing Chloride-Ions. Corrosion Science, 1977. 17(4): p. 353-365.
24. Zhou, W.Q., et al., Corrosion and electrochemical behavior of AZ31D magnesium alloys in sodium chloride. Transactions of Nonferrous Metals Society of China, 2006. 16: p. S1789-S1792.
25. Song, G.L. and A. Atrens, Understanding magnesium corrosion - A framework for improved alloy performance. Advanced Engineering Materials, 2003. 5(12): p. 837-858.
26. Inoue, H., et al., Corrosion rate of magnesium and its alloys in buffered chloride solutions. Corrosion Science, 2002. 44(3): p. 603-610.
27. Atrens, A., Z.M. Shi, and M. Liu, Measurement of the corrosion rate of magnesium alloys using Tafel extrapolation. Corrosion Science, 2010. 52(2): p. 579-588.
28. Song, G.L. and A. Atrens, Corrosion mechanisms of magnesium alloys. Advanced Engineering Materials, 1999. 1(1): p. 11-33.
29. Petrova, L.M. and V.V. Krasnoyarskii, Study of the Differential Effect for Magnesium in Neutral Aqueous-Solutions. Protection of Metals, 1987. 23(3): p. 344-347.
30. Petty, R.L., A.J. Davidson, and J. Kleinberg, Journal of the American Chemical Society, 1954. 76: p. 363.
31. Makar, G.L. and J. Kruger, Corrosion Studies of Rapidly Solidified Magnesium Alloys. Journal of the Electrochemical Society, 1990. 137(2): p. 414-421.
32. Song, G., et al., The anodic dissolution of magnesium in chloride and sulphate solutions. Corrosion Science, 1997. 39(10-11): p. 1981-2004.
33. Turhan, M.C., et al., Effect of acidic etching and fluoride treatment on corrosion performance in Mg alloy AZ91D (MgAlZn). Electrochimica Acta, 2009. 55(1): p. 250-257.
34. Prosek, T., et al., Corrosion mechanism of model zinc-magnesium alloys in atmospheric conditions. Corrosion Science, 2008. 50(8): p. 2216-2231.
35. Gvozdenovic, M., et al., Corrosion behavior of magnesium, aluminum and zinc as anodic materials in chloride based electrolytes for use in primary and

147
secondary electrochemical power sources. Materials & Design, 2009. 30(8): p. 3291-3294.
36. Hausbrand, R., M. Stratmann, and M. Rohwerder, Corrosion of zinc-magnesium coatings: Mechanism of paint delamination. Corrosion Science, 2009. 51(9): p. 2107-2114.
37. Senf, J. and E. Broszeit, Wear and corrosion protection of aluminum and magnesium alloys using chromium and chromium nitride PVD coatings. Advanced Engineering Materials, 1999. 1(2): p. 133-137.
38. Huang, C.A., C.K. Lin, and Y.H. Yeh, Increasing the wear and corrosion resistance of magnesium alloy (AZ91D) with electrodeposition from eco-friendly copper- and trivalent chromium-plating baths. Surface & Coatings Technology, 2010. 205(1): p. 139-145.
39. Huo, H.W., Y. Li, and F.H. Wang, Corrosion of AZ91D magnesium alloy with a chemical conversion coating and electroless nickel layer. Corrosion Science, 2004. 46(6): p. 1467-1477.
40. Song, Y.W., D.Y. Shan, and E.H. Han, High corrosion resistance multilayer nickel coatings on AZ91D magnesium alloys. Surface Engineering, 2007. 23(5): p. 329-333.
41. Gray, J.E. and B. Luan, Protective coatings on magnesium and its alloys - a critical review. Journal of Alloys and Compounds, 2002. 336(1-2): p. 88-113.
42. X.B. Chen, N.B., T.B. Abbott, Review of Corrosion-Resistant Conversion Coatings for Magnesium and Its Alloys. Corrosion 2011. 67(3). 43. Blawert, C., et al., Anodizing treatments for magnesium alloys and their
effecton corrosion resistance in various environments. Advanced Engineering Materials, 2006. 8(6): p. 511-533.
44. Willicut, R.J. and R.L. Mccarley, Electrochemical Polymerization of Pyrrole-Containing Self-Assembled Alkanethiol Monolayers on Au. Journal of the American Chemical Society, 1994. 116(23): p. 10823-10824.
45. Sarac, A.S., et al., Monomer Concentration Effect on Electrochemically Modified Carbon Fiber with Poly[1-(4-Methoxyphenyl)-1H-Pyrrole] as Microcapacitor Electrode. Advances in Polymer Technology, 2009. 28(2): p. 120-130.
46. Sarac, A.S., et al., Effect of Electrolyte on the Electropolymerization of 2,2-Dibutyl-3,4-Propylenedioxythiophene on Carbon Fiber Microelectrodes. Journal of Nanoscience and Nanotechnology, 2009. 9(5): p. 2877-2886.
47. Ates, M., et al., Polycarbazole modified carbon fiber microelectrode: Surface characterization and dopamine sensor. Fibers and Polymers, 2009. 10(1): p. 46-52.
48. Sarac, A.S., et al., Electrochemical impedance spectroscopy and morphological analyses of pyrrole, phenylpyrrole and methoxyphenylpyrrole on carbon fiber microelectrodes. Surface & Coatings Technology, 2008. 202(16): p. 3997-4005.
49. Sarac, A.S., et al., Electrochemical impedance spectroscopy of poly(N-methyl pyrrole) on carbon fiber microelectrodes and morphology. Progress in Organic Coatings, 2008. 62(3): p. 331-335.
50. Barsoukov, E. and J.R. Macdonald, Impedance spectroscopy : theory, experiment, and applications. 2nd ed. 2005, Hoboken, N.J.: Wiley-Interscience. xvii, 595 p.
51. Krane, K.S., Modern physics. 3rd ed. 2012, Hoboken, NJ: Wiley.

148
52. Vernon-Parry, K.D., et al., The use of electron back-scattered diffraction to study the regrowth of amorphised silicon-based heterostructures. Materials Science in Semiconductor Processing, 2001. 4(1-3): p. 121-123.
53. Auger, P., Auger Effect. Surface Science, 1975. 48(1): p. 1-8. 54. Goldstein, J., Scanning electron microscopy and x-ray microanalysis. 3rd ed.
2003, New York: Kluwer Academic/Plenum Publishers. xix, 689 p. 55. Yao, N., Focused ion beam systems : basics and applications. 2007,
Cambridge New York: Cambridge University Press. xi, 395 p. 56. Briggs, D. and J.T. Grant, Surface analysis by Auger and x-ray photoelectron
spectroscopy. 2003, Chichester, West Sussex, U.K.: IM Publications. xi, 899 p.
57. Benninghoven, A., F.G. Rüdenauer, and H.W. Werner, Secondary ion mass spectrometry : basic concepts, instrumental aspects, applications, and trends. Chemical analysis v. 86. 1987, New York: J. Wiley. xxxv, 1227 p.
58. Smith, B.C., Fundamentals of Fourier transform infrared spectroscopy. 1996, Boca Raton: CRC Press. 202 p.
59. Sharma, A.K., Chromate conversion coatings for magnesium-lithium alloys. Metal Finishing, 1989. 87(2): p. 73.
60. Dennis, J.K. and T.E. Such, Nickel and chromium plating. 2nd ed. 1986, London ; Boston: Butterworths. 385 p.
61. Durney, L.J., Electroplating engineering handbook. 4th ed. 1984, New York: Van Nostrand Reinhold. x, 790 p.
62. Lin, C.S., et al., Formation and properties of stannate conversion coatings on AZ61 magnesium alloys. Corrosion Science, 2006. 48(1): p. 93-109.
63. Shashikala, A.R., et al., Chemical Conversion Coatings on Magnesium Alloys - A Comparative Study. International Journal of Electrochemical Science, 2008. 3(9): p. 993-1004.
64. Niu, L.Y., et al., A study and application of zinc phosphate coating on AZ91D magnesium alloy. Surface & Coatings Technology, 2006. 200(9): p. 3021-3026.
65. Zhou, W.Q., et al., Structure and formation mechanism of phosphate conversion coating on die-cast AZ91D magnesium alloy. Corrosion Science, 2008. 50(2): p. 329-337.
66. Duan, H.P., C.W. Yan, and F.H. Wang, Growth process of plasma electrolytic oxidation films formed on magnesium alloy AZ91D in silicate solution. Electrochimica Acta, 2007. 52(15): p. 5002-5009.
67. Chen, X.B.B.N.A.T.B., Review of corrosion-resistant conersion coatings for magnesium and its alloys. Corrosion 2011. 67(3): p. 035005-1.
68. Chen, J., et al., The effect of chemical coating with Ni on the electrode properties of Mg2Ni alloy. Journal of Alloys and Compounds, 1998. 280(1-2): p. 290-293.
69. Elsentriecy, H.H., K. Azumi, and H. Konno, Improvement in stannate chemical conversion coatings on AZ91 D magnesium alloy using the potentiostatic technique. Electrochimica Acta, 2007. 53(2): p. 1006-1012.
70. Liu, X., et al., Effect of fluoride film on bonding of electroless nickel coating with magnesium alloys. Transactions of the Institute of Metal Finishing, 2005. 83(6): p. 286-290.
71. Bartak, D.E., Lemieux B. E., Woolsey E. R., Hard anodic coating for magnesium alloys in USPTO. 1995, Technology Applications Group: United States of America.

149
72. Avedesian, M.M., H. Baker, and ASM International. Handbook Committee., Magnesium and magnesium alloys. ASM specialty handbook. 1999, Materials Park, OH: ASM International. ix, 314 p.
73. Mathieu, S., et al., Corrosion behaviour of high pressure die-cast and semi-solid cast AZ91D alloys. Corrosion Science, 2002. 44(12): p. 2737-2756.
74. Bursik, J. and M. Svoboda, A HREM and analytical STEM study of precipitates in an AZ91 magnesium. Mikrochimica Acta, 2002. 139(1-4): p. 39-42.
75. Celotto, S., TEM study of continuous precipitation in Mg-9 Wt%Al-l Wt%Zn alloy. Acta Materialia, 2000. 48(8): p. 1775-1787.
76. Gonzalez-Martinez, R., et al., Influence of aging on damping of the magnesium-aluminium-zinc series. Journal of Alloys and Compounds, 2007. 437(1-2): p. 127-132.
77. Gharghouri, M.A., G.C. Weatherly, and J.D. Embury, The interaction of twins and precipitates in a Mg-7.7 at.% Al alloy. Philosophical Magazine a-Physics of Condensed Matter Structure Defects and Mechanical Properties, 1998. 78(5): p. 1137-1149.
78. Celotto, S. and T.J. Bastow, Study of precipitation in aged binary Mg-Al and ternary Mg-Al-Zn alloys using Al-27 NMR spectroscopy. Acta Materialia, 2001. 49(1): p. 41-51.
79. Braszczynska-Malik, K.N., Spherical shape of gamma-Mg(17)Al(12) precipitates in AZ91 magnesium alloy processed by equal-channel angular pressing. Journal of Alloys and Compounds, 2009. 487(1-2): p. 263-268.
80. Song, G.L., et al., Corrosion behaviour of AZ21, AZ501 and AZ91 in sodium chloride. Corrosion Science, 1998. 40(10): p. 1769-1791.
81. Baril, G. and N. Pebere, The corrosion of pure magnesium in aerated and deaerated sodium sulphate solutions. Corrosion Science, 2001. 43(3): p. 471-484.
82. Baril, G., C. Blanc, and N. Pebere, AC impedance spectroscopy in characterizing time-dependent corrosion of AZ91 and AM50 magnesium alloys - Characterization with respect to their microstructures. Journal of the Electrochemical Society, 2001. 148(12): p. B489-B496.
83. Epelboin, I. and R. Wiart, Mechanism of Electrocrystallization of Nickel and Cobalt in Acidic Solution. Journal of the Electrochemical Society, 1971. 118(10): p. 1577-&.
84. Baril, G., et al., An impedance investigation of the mechanism of pure magnesium corrosion in sodium sulfate solutions. Journal of the Electrochemical Society, 2007. 154(2): p. C108-C113.
85. Wang, J.Q., et al., AC impedance spectroscopy study of the corrosion behavior of an AZ91 magnesium alloy in 0.1 M sodium sulfate solution. Electrochimica Acta, 2007. 52(9): p. 3299-3309.
86. Anik, M. and G. Celikten, Analysis of the electrochemical reaction behavior of alloy AZ91 by EIS technique in H3PO4/KOH buffered K2SO4 solutions. Corrosion Science, 2007. 49(4): p. 1878-1894.
87. Rossi, A., et al., XPS, AES and ToF-SIMS investigation of surface films and the role of inclusions on pitting corrosion in austenitic stainless steels. Surface and Interface Analysis, 2000. 29(7): p. 460-467.
88. Alexander, M.R., C. Fotea, and J. Callaway, Characterisation of the surface chemistry of magnesium exposed to the ambient atmosphere. Surface and Interface Analysis, 2006. 38(10): p. 1363-1371.

150
89. Verdier, S., et al., The surface reactivity of a magnesium-aluminium alloy in acidic fluoride solutions studied by electrochemical techniques and XPS. Applied Surface Science, 2004. 235(4): p. 513-524.
90. Shirakawa, H., et al., Synthesis of Electrically Conducting Organic Polymers - Halogen Derivatives of Polyacetylene, (Ch)X. Journal of the Chemical Society-Chemical Communications, 1977(16): p. 578-580.
91. Gerard, M., A. Chaubey, and B.D. Malhotra, Application of conducting polymers to biosensors. Biosensors & Bioelectronics, 2002. 17(5): p. 345-359.
92. Smela, E., Conjugated polymer actuators for biomedical applications. Advanced Materials, 2003. 15(6): p. 481-494.
93. Brunel, L., et al., Oxygen transport through laccase biocathodes for a membrane-less glucose/O-2 biofuel cell. Electrochemistry Communications, 2007. 9(2): p. 331-336.
94. Feng, C.H., P.C.H. Chan, and I.M. Hsing, Catalyzed microelectrode mediated by polypyrrole/Nafion (R) composite film for microfabricated fuel cell applications. Electrochemistry Communications, 2007. 9(1): p. 89-93.
95. Perepichka, I.F., et al., Light-emitting polythiophenes. Advanced Materials, 2005. 17(19): p. 2281-2305.
96. Cebeci, F.C., E. Sezer, and A.S. Sarac, A novel EDOT-nonylbithiazole-EDOT based comonomer as an active electrode material for supercapacitor applications. Electrochimica Acta, 2009. 54(26): p. 6354-6360.
97. Sharma, R.K., A.C. Rastogi, and S.B. Desu, Pulse polymerized polypyrrole electrodes for high energy density electrochemical supercapacitor. Electrochemistry Communications, 2008. 10(2): p. 268-272.
98. Fan, L.Z. and J. Maier, High-performance polypyrrole electrode materials for redox supercapacitors. Electrochemistry Communications, 2006. 8(6): p. 937-940.
99. Shirakawa, H., Nobel Lecture: The discovery of polyacetylene film - the dawning of an era of conducting polymers. Reviews of Modern Physics, 2001. 73(3): p. 713-718.
100. Shirakawa, H., The discovery of polyacetylene film: The dawning of an era of conducting polymers (Nobel lecture). Angewandte Chemie-International Edition, 2001. 40(14): p. 2575-2580.
101. Genies, E.M., G. Bidan, and A.F. Diaz, Spectroelectrochemical Study of Polypyrrole Films. Journal of Electroanalytical Chemistry, 1983. 149(1-2): p. 101-113.
102. Waltman, R.J. and J. Bargon, Electrically Conducting Polymers - a Review of the Electropolymerization Reaction, of the Effects of Chemical-Structure on Polymer Film Properties, and of Applications Towards Technology. Canadian Journal of Chemistry-Revue Canadienne De Chimie, 1986. 64(1): p. 76-95.
103. Waltman, R.J. and J. Bargon, Reactivity Structure Correlations for the Electropolymerization of Pyrrole - an Indo Cndo Study of the Reactive Sites of Oligomeric Radical Cations. Tetrahedron, 1984. 40(20): p. 3963-3970.
104. Sadki, S., et al., The mechanisms of pyrrole electropolymerization. Chemical Society Reviews, 2000. 29(5): p. 283-293.
105. Skotheim, T.A., Handbook of conducting polymers. 1986, New York: M. Dekker. 2 v. (1417 p.).
106. Geetha, S., et al., Biosensing and drug delivery by polypyrrole. Analytica Chimica Acta, 2006. 568(1-2): p. 119-125.

151
107. Xiao, Y.H., et al., Preparation of nano-tentacle polypyrrole with pseudo-molecular template for ATP incorporation. Journal of Biomedical Materials Research Part A, 2007. 80A(4): p. 925-931.
108. Svirskis, D., et al., Electrochemically controlled drug delivery based on intrinsically conducting polymers. Journal of Controlled Release, 2010. 146(1): p. 6-15.
109. Pournaghi-Azar, M.H. and H. Nahalparvari, Zinc hexacyanoferrate film as an effective protecting layer in two-step and one-step electropolymerization of pyrrole on zinc substrate. Electrochimica Acta, 2005. 50(10): p. 2107-2115.
110. Arenas, M.A., et al., Synthesis and electrochemical evaluation of polypyrrole coatings electrodeposited onto AA-2024 alloy. Progress in Organic Coatings, 2008. 62(1): p. 79-86.
111. Le, H.N.T., et al., Corrosion protection and conducting polymers: polypyrrole films on iron. Electrochimica Acta, 2001. 46(26-27): p. 4259-4272.
112. Duran, B., et al., Electropolymerization, characterization and corrosion performance of poly(N-ethylaniline) on copper. Electrochimica Acta, 2009. 55(1): p. 104-112.
113. Guo, X.W., et al., Preparation of even polyaniline film on magnesium alloy by pulse potentiostatic method. Synthetic Metals, 2003. 135(1-3): p. 169-170.
114. Jiang, Y.F., et al., Corrosion protection of polypyrrole electrodeposited on AZ91 magnesium alloys in alkaline solutions. Synthetic Metals, 2003. 139(2): p. 335-339.
115. Luo, X.L. and X.Y.T. Cui, Electrochemical deposition of conducting polymer coatings on magnesium surfaces in ionic liquid. Acta Biomaterialia, 2011. 7(1): p. 441-446.
116. Petitjean, J., et al., Ultra-fast electropolymerization of pyrrole in aqueous media on oxidizable metals in a one-step process. Journal of Electroanalytical Chemistry, 1999. 478(1-2): p. 92-100.
117. Hermelin, E., et al., Ultrafast electrosynthesis of high hydrophobic polypyrrole coatings on a zinc electrode: Applications to the protection against corrosion. Chemistry of Materials, 2008. 20(13): p. 4447-4456.
118. Cascalheira, A.C., et al., Electropolymerization of pyrrole on oxidizable metals: Role of salicylate ions in the anodic behavior of copper. Russian Journal of Electrochemistry, 2004. 40(3): p. 294-298.
119. Klampfer, L., et al., Sodium salicylate activates caspases and induces apoptosis of myeloid leukemia cell lines. Blood, 1999. 93(7): p. 2386-2394.
120. Rae, C., et al., Elevated NF-kappa B responses and FLIP levels in leukemic but not normal lymphocytes: reduction by salicylate allows TNF-induced apoptosis. Proceedings of the National Academy of Sciences of the United States of America, 2007. 104(31): p. 12790-12795.
121. Redondo, M.I. and C.B. Breslin, Polypyrrole electrodepo sited on copper from an aqueous phosphate solution: Corrosion protection properties. Corrosion Science, 2007. 49(4): p. 1765-1776.
122. Alvarez-Ros, M.C., S. Sanchez-Cortes, and J.V. Garcia-Ramos, Vibrational study of the salicylate interaction with metallic ions and surfaces. Spectrochimica Acta Part a-Molecular and Biomolecular Spectroscopy, 2000. 56(12): p. 2471-2477.

152
123. dos Santos, L.M.M., et al., Electrochemical synthesis of polypyrrole films on copper electrodes in acidic and neutral aqueous media. Journal of Electroanalytical Chemistry, 2006. 587(1): p. 67-78.
124. Schoenbe.Ln, Structure of Complexed Copper Species in Electroless Copper Plating Solutions. Journal of the Electrochemical Society, 1971. 118(10): p. 1571-&.
125. Chasteen, N.D. and R.L. Belford, Triplet State of a Binuclear Copper Dl-Tartrate and Electron Paramagnetic Resonance Spectra of Copper Tartrates. Inorganic Chemistry, 1970. 9(1): p. 169-&.
126. Davidson, R.G. and T.G. Turner, An Ir Spectroscopic Study of the Electrochemical Reduction of Polypyrrole Doped with Dodecyl-Sulfate Anion. Synthetic Metals, 1995. 72(2): p. 121-128.
127. Singh, R., et al., Growth kinetics of polypyrrole, poly(N-methyl pyrrole) and their copolymer, poly(N-methyl pyrrole-pyrrole): Effect of annealing on conductivity and surface structure. Synthetic Metals, 1996. 79(1): p. 1-6.
128. Kaynak, A., Effect of synthesis parameters on the surface morphology of conducting polypyrrole films. Materials Research Bulletin, 1997. 32(3): p. 271-285.
129. Kaplin, D.A. and S. Qutubuddin, Electrochemically Synthesized Polypyrrole Films - Effects of Polymerization Potential and Electrolyte Type. Polymer, 1995. 36(6): p. 1275-1286.
130. Otero, T.F. and C. Santamaria, Dependence of Polypyrrole Production on Potential. Synthetic Metals, 1992. 51(1-3): p. 313-319.
131. Wadhwa, R., C.F. Lagenaur, and X.T. Cui, Electrochemically controlled release of dexamethasone from conducting polymer polypyrrole coated electrode. Journal of Controlled Release, 2006. 110(3): p. 531-541.
132. Kontturi, K., P. Pentti, and G. Sundholm, Polypyrrole as a model membrane for drug delivery. Journal of Electroanalytical Chemistry, 1998. 453(1-2): p. 231-238.
133. Wallace, G.G., et al., Conductive electroactive polymers. 2009, Florida: Taylor& Francis Group. Ch. 3. .
134. Thompson, B.C., et al., Optimising the incorporation and release of a neurotrophic factor using conducting polypyrrole. Journal of Controlled Release, 2006. 116(3): p. 285-294.
135. Williams, G. and H.N. McMurray, Factors affecting acid-base stability of the interface between polyaniline emeraldine salt and oxide covered metal. Electrochemical and Solid State Letters, 2005. 8(9): p. B42-B45.
136. Rohwerder, M. and A. Michalik, Conducting polymers for corrosion protection: What makes the difference between failure and success? Electrochimica Acta, 2007. 53(3): p. 1300-1313.
137. Rohwerder, M., Conducting polymers for corrosion protection: a review. International Journal of Materials Research, 2009. 100(10): p. 1331-1342.
138. Stoye, D. and W. Freitag, Paints, coatings, and solvents. 2nd, completely rev. ed. 1998, Weinheim ; New York: Wiley-VCH. xvii, 414 p.
139. Eftekhari, A., Nanostructured conductive polymers. 2010, Chichester, West Sussex, U.K. ; Hoboken, N.J.: Wiley. xxiii, 776 p.
140. Malshe, V.C. and N.S. Sangaj, Effect of introduction of structural defects on protective ability of polyesters. Progress in Organic Coatings, 2006. 57(1): p. 37-43.

153
141. Sangaj, N.S. and V.C. Malshe, Permeability of polymers in protective organic coatings. Progress in Organic Coatings, 2004. 50(1): p. 28-39.
142. Thomas, N.L., The Barrier Properties of Paint Coatings. Progress in Organic Coatings, 1991. 19(2): p. 101-121.
143. Bierwagen, G.R. and D. Wang, Sol-gel coatings on metals for corrosion protection. Progress in Organic Coatings, 2009. 64(4): p. 327-338.
144. Pagliaro, M., R. Ciriminna, and G. Palmisano, Silica-based hybrid coatings. Journal of Materials Chemistry, 2009. 19(20): p. 3116-3126.
145. Deberry, D.W., Modification of the Electrochemical and Corrosion Behavior of Stainless-Steels with an Electroactive Coating. Journal of the Electrochemical Society, 1985. 132(5): p. 1022-1026.
146. Cheung, K.M., D. Bloor, and G.C. Stevens, Characterization of Polypyrrole Electropolymerized on Different Electrodes. Polymer, 1988. 29(9): p. 1709-1717.
147. Trochnagels, G., et al., Electron Conducting Organic Coating of Mild-Steel by Electropolymerization. Journal of Applied Electrochemistry, 1992. 22(8): p. 756-764.
148. Ferreira, C.A., et al., Electropolymerization of Pyrrole on Iron Electrodes - Influence of Solvent and Electrolyte on the Nature of the Deposits. Journal of Electroanalytical Chemistry, 1990. 284(2): p. 351-369.
149. Ferreira, C.A., et al., Electrosynthesis of strongly adherent polypyrrole coatings on iron and mild steel in aqueous media. Electrochimica Acta, 1996. 41(11-12): p. 1801-1809.
150. Bereket, G., et al., Electropolymerization, characterization and corrosion performance of poly(N-ethylaniline) on copper. Electrochimica Acta, 2009. 55(1): p. 104-112.
151. Lacroix, J.C., et al., Aniline electropolymerization on mild steel and zinc in a two-step process. Journal of Electroanalytical Chemistry, 2000. 481(1): p. 76-81.
152. Rajagopalan, R. and J.O. Iroh, Development of polyaniline-polypyrrole composite coatings on steel by aqueous electrochemical process. Electrochimica Acta, 2001. 46(16): p. 2443-2455.
153. Iroh, J.O., et al., Electrochemical synthesis: a novel technique for processing multi-functional coatings. Progress in Organic Coatings, 2003. 47(3-4): p. 365-375.
154. Hasanov, R. and S. Bilgic, Monolayer and bilayer conducting polymer coatings for corrosion protection of steel in 1 M H(2)SO(4) solution. Progress in Organic Coatings, 2009. 64(4): p. 435-445.
155. Michalik, A. and M. Rohwerder, Conducting polymers for corrosion protection: A critical view. Zeitschrift Fur Physikalische Chemie-International Journal of Research in Physical Chemistry & Chemical Physics, 2005. 219(11): p. 1547-1559.
156. Bernard, M.C., et al., Polyaniline layer for iron protection in sulfate medium. Journal of the Electrochemical Society, 1999. 146(3): p. 995-998.
157. Cook, A., A. Gabriel, and N. Laycock, On the mechanism of corrosion protection of mild steel with polyaniline. Journal of the Electrochemical Society, 2004. 151(9): p. B529-B535.
158. Leng, A., H. Streckel, and M. Stratmann, The delamination of polymeric coatings from steel. Part 1. Calibration of the Kelvinprobe and basic delamination mechanism. Corrosion Science, 1999. 41(3): p. 547-578.

154
159. Lu, W.K., R.L. Elsenbaumer, and B. Wessling, Corrosion Protection of Mild-Steel by Coatings Containing Polyaniline. Synthetic Metals, 1995. 71(1-3): p. 2163-2166.
160. Meneguzzi, A., et al., Electroactive poly(aromatic amine) films for iron protection in sulfate medium. Journal of the Electrochemical Society, 2001. 148(4): p. B121-B126.
161. Stratmann, M. and H. Streckel, On the Atmospheric Corrosion of Metals Which Are Covered with Thin Electrolyte Layers .1. Verification of the Experimental-Technique. Corrosion Science, 1990. 30(6-7): p. 681-696.
162. Wessling, B., Scientific and commercial breakthrough for organic metals. Synthetic Metals, 1997. 85(1-3): p. 1313-1318.
163. Cook, A., et al., Corrosion protection of low carbon steel with polyaniline: passivation or inhibition? Current Applied Physics, 2004. 4(2-4): p. 133-136.
164. Kendig, M., M. Hon, and L. Warren, 'Smart' corrosion inhibiting coatings. Progress in Organic Coatings, 2003. 47(3-4): p. 183-189.
165. Barisci, J.N., et al., Conducting polymers as a basis for responsive materials systems. Journal of Intelligent Material Systems and Structures, 1998. 9(9): p. 723-731.
166. de Souza, S., et al., Polyaniline based acrylic blends for iron corrosion protection. Electrochemical and Solid State Letters, 2001. 4(8): p. B27-B30.
167. Chang, A.C. and L.L. Miller, Electrochemically Controlled Binding and Release of Salicylate, Tcnq(.-) and Ferrocyanide from Films of Oligomeric 3-Methoxythiophene. Journal of Electroanalytical Chemistry, 1988. 247(1-2): p. 173-184.
168. Weidlich, C., K.M. Mangold, and K. Juttner, EQCM study of the ion exchange behaviour of polypyrrole with different counterions in different electrolytes. Electrochimica Acta, 2005. 50(7-8): p. 1547-1552.
169. Saji, V.S., A Review on Recent Patents in Corrosion Inhibitors. Recent Patents on Corrosion Science, 2010. 2(6): p. 6.
170. Le, H.N.T., et al., Corrosion protection of iron by polystyrenesulfonate-doped polypyrrole films. Journal of Applied Electrochemistry, 2002. 32(1): p. 105-110.
171. Paliwoda-Porebska, G., et al., Release mechanism of electrodeposited polypyrrole doped with corrosion inhibitor anions. Journal of Solid State Electrochemistry, 2006. 10(9): p. 730-736.
172. Tallman, D.E., et al., Electroactive conducting polymers for corrosion control Part 1. General introduction and a review of non-ferrous metals. Journal of Solid State Electrochemistry, 2002. 6(2): p. 73-84.
173. Rammelt, U., P.T. Nguyen, and W. Plieth, Corrosion protection by ultrathin films of conducting polymers. Electrochimica Acta, 2003. 48(9): p. 1257-1262.
174. Lacaze, P.C., et al., Ultrafast electrosynthesis of high hydrophobic polypyrrole coatings on a zinc electrode: Applications to the protection against corrosion. Chemistry of Materials, 2008. 20(13): p. 4447-4456.
175. Otero, T.F. and J.M. Sansinena, Artificial Muscles Based on Conducting Polymers. Bioelectrochemistry and Bioenergetics, 1995. 38(2): p. 411-414.
176. Abidian, M.R. and D.C. Martin, Multifunctional Nanobiomaterials for Neural Interfaces. Advanced Functional Materials, 2009. 19(4): p. 573-585.

155
177. Sarac, A.S. and M. Ates, Conducting polymer coated carbon surfaces and biosensor applications. Progress in Organic Coatings, 2009. 66(4): p. 337-358.
178. Pei, Q.B. and P. Brochu, Advances in Dielectric Elastomers for Actuators and Artificial Muscles. Macromolecular Rapid Communications, 2010. 31(1): p. 10-36.
179. Collier, J.H., et al., Synthesis and characterization of polypyrrole-hyaluronic acid composite biomaterials for tissue engineering applications. Journal of Biomedical Materials Research, 2000. 50(4): p. 574-584.
180. Abidian, M.R., D.H. Kim, and D.C. Martin, Conducting-polymer nanotubes for controlled drug release. Advanced Materials, 2006. 18(4): p. 405-+.
181. Sundarrajan, S., et al., Applications of conducting polymers and their issues in biomedical engineering. Journal of the Royal Society Interface, 2010. 7: p. S559-S579.
182. Garg, S., et al., Electrochemically controlled drug delivery based on intrinsically conducting polymers. Journal of Controlled Release, 2010. 146(1): p. 6-15.
183. Zinger, B. and L.L. Miller, Timed Release of Chemicals from Polypyrrole Films. Journal of the American Chemical Society, 1984. 106(22): p. 6861-6863.
184. Pernaut, J.M. and J.R. Reynolds, Use of conducting electroactive polymers for drug delivery and sensing of bioactive molecules. A redox chemistry approach. Journal of Physical Chemistry B, 2000. 104(17): p. 4080-4090.
185. Zhou, Q.X., L.L. Miller, and J.R. Valentine, Electrochemically Controlled Binding and Release of Protonated Dimethyldopamine and Other Cations from Poly(N-Methylpyrrole) Polyanion Composite Redox Polymers. Journal of Electroanalytical Chemistry, 1989. 261(1): p. 147-164.
186. Serruys, P.W. and A.H. Gershlick, Handbook of Drug-eluting Stents. 2005: Taylor & Francis Routledge.
187. Vogt, F., et al., A novel biodegradable coronary polymer stent with drug delivery capacities: Paclitaxel loading inhibits neointimal hyperplasia in a porcine model of coronary restenosis. Journal of the American College of Cardiology, 2003. 41(6): p. 58a-58a.
188. Vogt, F., et al., Long-term assessment of a novel biodegradable paclitaxel-eluting coronary polylactide stent. European Heart Journal, 2004. 25(15): p. 1330-1340.
189. Kumar, V. and A.P. Gupta, New emerging trends in synthetic biodegradable polymers - Polylactide: A critique. European Polymer Journal, 2007. 43(10): p. 4053-4074.
190. Li, W.J., et al., Fabrication and characterization of six electrospun poly(alpha-hydroxy ester)-based fibrous scaffolds for tissue engineering applications. Acta Biomaterialia, 2006. 2(4): p. 377-385.
191. Witte, F., et al., Biodegradable magnesium scaffolds: Part I: Appropriate inflammatory response. Journal of Biomedical Materials Research Part A, 2007. 81A(3): p. 748-756.
192. Witte, F., et al., Biodegradable magnesium scaffolds: Part II: Peri-implant bone remodeling. Journal of Biomedical Materials Research Part A, 2007. 81A(3): p. 757-765.
193. Krtschil, A., et al., Electrical characterization of magnesium implanted gallium nitride. Journal of Applied Physics, 2002. 91(1): p. 178-182.

156
194. Feyerabend, F., F. Witte, and R. Willumeit, Magnesium supports chondrocyte proliferation and redifferentiation - A new approach for tissue engineering? Cytotherapy, 2006. 8: p. 60-60.
195. Witte, F., et al., Degradable biomaterials based on magnesium corrosion. Current Opinion in Solid State & Materials Science, 2008. 12(5-6): p. 63-72.
196. Witte, F., et al., In vivo corrosion and corrosion protection of magnesium alloy LAE442. Acta Biomaterialia, 2010. 6(5): p. 1792-1799.
197. Cui, F.Z., J.X. Yang, and I.S. Lee, Surface Modifications of Magnesium Alloys for Biomedical Applications. Annals of Biomedical Engineering, 2011. 39(7): p. 1857-1871.
198. Virtanen, S., et al., Control of magnesium corrosion and biocompatibility with biomimetic coatings. Journal of Biomedical Materials Research Part B-Applied Biomaterials, 2011. 96B(1): p. 84-90.
199. Chen, X.B., N. Birbilis, and T.B. Abbott, A simple route towards a hydroxyapatite-Mg(OH)(2) conversion coating for magnesium. Corrosion Science, 2011. 53(6): p. 2263-2268.
200. Ratner, B.D., Biomaterials science : an introduction to materials in medicine. 1996, San Diego: Academic Press. xi, 484 p.
201. Virtanen, S. and R. Rettig, Time-dependent electrochemical characterization of the corrosion of a magnesium rare-earth alloy in simulated body fluids. Journal of Biomedical Materials Research Part A, 2008. 85A(1): p. 167-175.
202. Yamazoe, H., et al., Reduced platelet adhesion and blood coagulation on cross-linked albumin films. Materials Science & Engineering C-Materials for Biological Applications, 2010. 30(6): p. 812-816.
203. Yamazoe, H. and T. Tanabe, Drug-Carrying Albumin Film for Blood-Contacting Biomaterials. Journal of Biomaterials Science-Polymer Edition, 2010. 21(5): p. 647-657.
204. Virtanen, S., et al., Optimization of electrochemical polymerization parameters of polypyrrole on Mg-Al alloy (AZ91D) electrodes and corrosion performance. Electrochimica Acta, 2011. 56(15): p. 5347-5354.
205. Virtanen, S., et al., Electrochemical polymerization and characterization of polypyrrole on Mg-Al alloy (AZ91D). Synthetic Metals, 2011. 161(3-4): p. 360-364.
206. Muller, L. and F.A. Muller, Preparation of SBF with different HCO3- content and its influence on the composition of biomimetic apatites. Acta Biomaterialia, 2006. 2(2): p. 181-189.
207. Schmuki, P., M.S. Killian, and H.M. Krebs, Protein Denaturation Detected by Time-of-Flight Secondary Ion Mass Spectrometry. Langmuir, 2011. 27(12): p. 7510-7515.
208. Virtanen, S., et al., Functionalization of Metallic Magnesium with Protein Layers via Linker Molecules. Langmuir, 2010. 26(14): p. 12044-12048.
209. Kunze, J., et al., ToF-SIMS and XPS Studies of the Adsorption Characteristics of a Zn-Porphyrin on TiO(2). Langmuir, 2010. 26(5): p. 3531-3538.
210. Henry, M. and P. Bertrand, Surface composition of insulin and albumin adsorbed on polymer substrates as revealed by multivariate analysis of ToF-SIMS data. Surface and Interface Analysis, 2009. 41(2): p. 105-113.
211. Castner, D.G. and R. Michel, Advances in time-of-flight secondary ion mass spectrometry analysis of protein films. Surface and Interface Analysis, 2006. 38(11): p. 1386-1392.

157
212. Ratner, B.D., Biomaterials science : an introduction to materials in medicine. 2nd ed. 2004, Amsterdam ; Boston: Elsevier Academic Press. xii, 851 p.
213. Keenan, M.R. and P.G. Kotula, Accounting for Poisson noise in the multivariate analysis of ToF-SIMS spectrum images. Surface and Interface Analysis, 2004. 36(3): p. 203-212.
214. Henry, M. and P. Bertrand, Influence of polymer surface hydrophilicity on albumin adsorption. Surface and Interface Analysis, 2004. 36(8): p. 729-732.
215. Poleunis, C., et al., ToF-SIMS chemical mapping study of protein adsorption onto stainless steel surfaces immersed in saline aqueous solutions. Applied Surface Science, 2003. 203: p. 693-697.
216. Henry, M., C. Dupont-Gillain, and P. Bertrand, Conformation change of albumin adsorbed on polycarbonate membranes as revealed by ToF-SIMS. Langmuir, 2003. 19(15): p. 6271-6276.
217. Wagner, M.S. and D.G. Castner, Characterization of adsorbed protein films by time-of-flight secondary ion mass spectrometry with principal component analysis. Langmuir, 2001. 17(15): p. 4649-4660.
218. Tidwell, C.D., et al., Static time-of-flight secondary ion mass spectrometry and x-ray photoelectron spectroscopy characterization of adsorbed albumin and fibronectin films. Surface and Interface Analysis, 2001. 31(8): p. 724-733.
219. Lhoest, J.B., et al., Characterization of adsorbed protein films by time of flight secondary ion mass spectrometry. Journal of Biomedical Materials Research, 2001. 57(3): p. 432-440.
220. Lhoest, J.B., et al., Fibronectin adsorption, conformation, and orientation on polystyrene substrates studied by radiolabeling, XPS, and ToF SIMS. Journal of Biomedical Materials Research, 1998. 41(1): p. 95-103.
221. Ratner, B.D., et al., TOF-SIMS studies of adsorbed protein films. Abstracts of Papers of the American Chemical Society, 1996. 211: p. 325-POLY.
222. Mantus, D.S., et al., Static Secondary-Ion Mass-Spectrometry of Adsorbed Proteins. Analytical Chemistry, 1993. 65(10): p. 1431-1438.
223. Horbett, T.A., Principles Underlying the Role of Adsorbed Plasma-Proteins in Blood Interactions with Foreign Materials. Cardiovascular Pathology, 1993. 2(3): p. S137-S148.
224. Virtanen, S., et al., Effect of acidic etching and fluoride treatment on corrosion performance in Mg alloy AZ91D (MgAlZn). Electrochimica Acta, 2009. 55(1): p. 250-257.
225. Stern, M. and G. A., Journal of Electrochemical Society 1957. 104(56). 226. Hoyer, P., Formation of a titanium dioxide nanotube array. Langmuir, 1996.
12(6): p. 1411-1413. 227. Nakato, Y., et al., Photooxidation Reaction of Water on an N-Tio2 Electrode -
Improvement in Efficiency through Formation of Surface Micropores by Photo-Etching in H2so4. Journal of Electroanalytical Chemistry, 1995. 396(1-2): p. 35-39.
228. Schmuki, P., R. Hahn, and J.M. Macak, Rapid anodic growth of TiO(2) and WO(3) nanotubes in fluoride free electrolytes. Electrochemistry Communications, 2007. 9(5): p. 947-952.
229. Shin, H.J., et al., Formation of TiO2 and ZrO2 nanotubes using atomic layer deposition with ultraprecise control of the wall thickness. Advanced Materials, 2004. 16(14): p. 1197-+.

158
230. Daub, M., et al., Ferromagnetic nanotubes by atomic layer deposition in anodic alumina membranes. Journal of Applied Physics, 2007. 101(9).
231. Kasuga, T., et al., Formation of titanium oxide nanotube. Langmuir, 1998. 14(12): p. 3160-3163.
232. Sander, M.S., et al., Template-assisted fabrication of dense, aligned arrays of titania nanotubes with well-controlled dimensions on substrates. Advanced Materials, 2004. 16(22): p. 2052-+.
233. Ghicov, A. and P. Schmuki, Self-ordering electrochemistry: a review on growth and functionality of TiO2 nanotubes and other self-aligned MOx structures. Chemical Communications, 2009(20): p. 2791-2808.
234. Schmuki, P., P.R. Roy, P., and S. Berger, TiO(2) Nanotubes: Synthesis and Applications. Angewandte Chemie-International Edition, 2011. 50(13): p. 2904-2939.
235. Keller, F., M.S. Hunter, and D.L. Robinson, Structural Features of Oxide Coatings on Aluminum. Journal of the Electrochemical Society, 1953. 100(9): p. 411.
236. Linsebigler, A.L., G.Q. Lu, and J.T. Yates, Photocatalysis on Tio2 Surfaces - Principles, Mechanisms, and Selected Results. Chemical Reviews, 1995. 95(3): p. 735-758.
237. Yates, J.T. and T.L. Thompson, Surface science studies of the photoactivation of TiO2-new photochemical processes. Chemical Reviews, 2006. 106(10): p. 4428-4453.
238. Hoffmann, M.R., et al., Environmental Applications of Semiconductor Photocatalysis. Chemical Reviews, 1995. 95(1): p. 69-96.
239. Fujishima, A., X.T. Zhang, and D.A. Tryk, TiO(2) photocatalysis and related surface phenomena. Surface Science Reports, 2008. 63(12): p. 515-582.
240. Xie, Y.B., Photoelectrochemical application of nanotubular titania photoanode. Electrochimica Acta, 2006. 51(17): p. 3399-3406.
241. Lai, Y.K., et al., Effects of the structure of TiO2 nanotube array on Ti substrate on its photocatalytic activity. Journal of the Electrochemical Society, 2006. 153(7): p. D123-D127.
242. Fujishima, A. and K. Honda, Electrochemical Photolysis of Water at a Semiconductor Electrode. Nature, 1972. 238(5358): p. 37-+.
243. Gobrecht, J., H. Gerischer, and H. Tributsch, Electrochemical Solar-Cell Based on the D-Band Semiconductor Tungsten-Diselenide. Berichte Der Bunsen-Gesellschaft-Physical Chemistry Chemical Physics, 1978. 82(12): p. 1331-1335.
244. Oregan, B. and M. Gratzel, A Low-Cost, High-Efficiency Solar-Cell Based on Dye-Sensitized Colloidal Tio2 Films. Nature, 1991. 353(6346): p. 737-740.
245. Schmuki, P., et al., Dye-sensitized solar cells using anodic TiO(2) mesosponge: Improved efficiency by TiCl(4) treatment. Electrochemistry Communications, 2010. 12(4): p. 574-578.
246. Schmuki, P., et al., Bamboo-Type TiO(2) Nanotubes: Improved Conversion Efficiency in Dye-Sensitized Solar Cells. Journal of the American Chemical Society, 2008. 130(49): p. 16454-+.
247. Schmuki, P., et al., Efficient solar energy conversion using TiO2 nanotubes produced by rapid breakdown anodization - a comparison. Physica Status Solidi-Rapid Research Letters, 2007. 1(4): p. 135-137.
248. Schmuki, P., et al., Fast formation of aligned high-aspect ratio TiO(2) nanotube bundles that lead to increased open circuit voltage when used in

159
dye sensitized solar cells. Electrochemistry Communications, 2011. 13(3): p. 302-305.
249. Schmuki, P., et al., TiO(2) nanotubes and their application in dye-sensitized solar cells. Nanoscale, 2010. 2(1): p. 45-59.
250. Huang, S.Y., et al., Rocking Chair Lithium Battery Based on Nanocrystalline Tio2 (Anatase). Journal of the Electrochemical Society, 1995. 142(9): p. L142-L144.
251. Arico, A.S., et al., Nanostructured materials for advanced energy conversion and storage devices. Nature Materials, 2005. 4(5): p. 366-377.
252. Ohzuku, T., K. Sawai, and T. Hirai, Electrochemistry of L-Niobium Pentoxide in a Lithium Nonaqueous Cell. Journal of Power Sources, 1987. 19(4): p. 287-299.
253. Granqvist, C.R., Electrochromic materials: Out of a niche. Nature Materials, 2006. 5(2): p. 89-90.
254. von Wilmowsky, C., et al., In Vivo Evaluation of Anodic TiO(2) Nanotubes: An Experimental Study in the Pig. Journal of Biomedical Materials Research Part B-Applied Biomaterials, 2009. 89B(1): p. 165-171.
255. Bauer, S., et al., Size selective behavior of mesenchymal stem cells on ZrO(2) and TiO(2) nanotube arrays. Integrative Biology, 2009. 1(8-9): p. 525-532.
256. von der Mark, K., et al., Narrow Window in Nanoscale Dependent Activation of Endothelial Cell Growth and Differentiation on TiO(2) Nanotube Surfaces. Nano Letters, 2009. 9(9): p. 3157-3164.
257. Schmuki, P., et al., Improved attachment of mesenchymal stem cells on super-hydrophobic TiO2 nanotubes. Acta Biomaterialia, 2008. 4(5): p. 1576-1582.
258. Schmuki, P., et al., Another look at "Stem cell fate dictated solely by altered nanotube dimension''. Proceedings of the National Academy of Sciences of the United States of America, 2009. 106(24): p. E60-E60.
259. Schmuki, P., et al., TiO(2) Nanotube Surfaces: 15 nm - An Optimal Length Scale of Surface Topography for Cell Adhesion and Differentiation. Small, 2009. 5(6): p. 666-671.
260. Schmuki, P., et al., Magnetically Guided Titania Nanotubes for Site-Selective Photocatalysis and Drug Release. Angewandte Chemie-International Edition, 2009. 48(5): p. 969-972.
261. Schmuki, P., et al., Amphiphilic TiO(2) Nanotube Arrays: An Actively Controllable Drug Delivery System. Journal of the American Chemical Society, 2009. 131(12): p. 4230-+.
262. Schmuki, P. and Y.Y. Song, Modulated TiO(2) nanotube stacks and their use in interference sensors. Electrochemistry Communications, 2010. 12(4): p. 579-582.
263. Varghese, O.K., et al., Hydrogen sensing using titania nanotubes. Sensors and Actuators B-Chemical, 2003. 93(1-3): p. 338-344.
264. Varghese, O.K., et al., Highly ordered nanoporous alumina films: Effect of pore size and uniformity on sensing performance. Journal of Materials Research, 2002. 17(5): p. 1162-1171.
265. Dickey, E.C., et al., Room temperature ammonia and humidity sensing using highly ordered nanoporous alumina films. Sensors, 2002. 2(3): p. 91-110.
266. Masuda, H., et al., Highly ordered nanochannel-array architecture in anodic alumina. Applied Physics Letters, 1997. 71(19): p. 2770-2772.

160
267. Macak, J.M., H. Tsuchiya, and P. Schmuki, High-aspect-ratio TiO2 nanotubes by anodization of titanium. Angewandte Chemie-International Edition, 2005. 44(14): p. 2100-2102.
268. Zwilling, V., et al., Structure and physicochemistry of anodic oxide films on titanium and TA6V alloy. Surface and Interface Analysis, 1999. 27(7): p. 629-637.
269. Macak, J.M., et al., Smooth anodic TiO2 nanotubes. Angewandte Chemie-International Edition, 2005. 44(45): p. 7463-7465.
270. Schmuki, P., S.P. Albu, and A. Ghicov, High aspect ratio, self-ordered iron oxide nanopores formed by anoclization of Fe in ethylene glycol/NH(4)F electrolytes. Physica Status Solidi-Rapid Research Letters, 2009. 3(2-3): p. 64-66.
271. Burleigh, T.D., P. Schmuki, and S. Virtanen, Properties of the Nanoporous Anodic Oxide Electrochemically Grown on Steel in Hot 50% NaOH. Journal of the Electrochemical Society, 2009. 156(1): p. C45-C53.
272. Sieber, I., B. Kannan, and P. Schmuki, Self-assembled porous tantalum oxide prepared in H2SO4/HF electrolytes. Electrochemical and Solid State Letters, 2005. 8(3): p. J10-J12.
273. Tsuchiya, H., et al., Self-organized porous TiO2 and ZrO2 produced by anodization. Corrosion Science, 2005. 47(12): p. 3324-3335.
274. Tsuchiya, H., et al., Self-organized high-aspect-ratio nanoporous zirconium oxides prepared by electrochemical anodization. Small, 2005. 1(7): p. 722-725.
275. Sieber, I., et al., Formation of self-organized niobium porous oxide on niobium. Electrochemistry Communications, 2005. 7(1): p. 97-100.
276. Tsuchiya, H. and P. Schmuki, Self-organized high aspect ratio porous hafnium oxide prepared by electrochemical anodization. Electrochemistry Communications, 2005. 7(1): p. 49-52.
277. Brunner, J.G., et al., Porosity Tailored Growth of Black Anodic Layers on Magnesium in an Organic Electrolyte. Journal of the Electrochemical Society, 2009. 156(2): p. C62-C66.
278. Asoh, H. and S. Ono, Anodizing of magnesium in amine - ethylene glycol electrolyte. Magnesium Alloys 2003, Pts 1 and 2, 2003. 419-4: p. 957-961.
279. Ono, S., et al., Microstructure of anodic films grown on magnesium-lithium-yttrium ultra light alloy. Thermec'2003, Pts 1-5, 2003. 426-4: p. 581-586.
280. Ono, S. and H. Asoh, Mater. Sci. Forum, 2009. 957 (419). 281. Ono, S., H. Kijima, and N. Masuko, Microstructure and voltage-current
characteristics of anodic films formed on magnesium in electrolytes containing fluoride. Materials Transactions, 2003. 44(4): p. 539-545.
282. Ono, S., M. Miyake, and H. Asoh, Effects of formation voltage and electrolyte ions concentration on the structure and passivity of anodic films on magnesium. J. Jap. Ins. Light Met. , 2004. 54(11): p. 544.
283. Park, J., et al., Nanosize and vitality: TiO2 nanotube diameter directs cell fate. Nano Letters, 2007. 7(6): p. 1686-1691.
284. Tsuchiya, H., et al., Hydroxyapatite growth on anodic TiO2 nanotubes. Journal of Biomedical Materials Research Part A, 2006. 77A(3): p. 534-541.
285. Schmuki, P., et al., Time-dependent growth of biomimetic apatite on anodic TiO2 nanotubes. Electrochimica Acta, 2008. 53(23): p. 6995-7003.

161
286. Schmuki, P., et al., Bioactivation of titanium surfaces using coatings of TiO(2) nanotubes rapidly pre-loaded with synthetic hydroxyapatite. Acta Biomaterialia, 2009. 5(6): p. 2322-2330.
287. Virtanen, S., et al., Effect of surface pre-treatments on biocompatibility of magnesium. Acta Biomaterialia, 2009. 5(7): p. 2783-2789.
288. Arrabal, R., et al., Corrosion resistance of WE43 and AZ91D magnesium alloys with phosphate PEO coatings. Corrosion Science, 2008. 50(6): p. 1744-1752.
289. Rzychon, T., et al., Microstructural stability and creep properties of die casting Mg-4Al-4RE magnesium alloy. Materials Characterization, 2009. 60(10): p. 1107-1113.
290. Rzychon, T., A. Kielbus, and M. Serba, The Influence of Pouring Temperature on the Microstructure and Fluidity of Elektron 21 and We54 Magnesium Alloys. Archives of Metallurgy and Materials, 2010. 55(1): p. 7-13.
291. Rettig, R. and S. Virtanen, Composition of corrosion layers on a magnesium rare-earth alloy in simulated body fluids. Journal of Biomedical Materials Research Part A, 2009. 88A(2): p. 359-369.
292. Zheng, Y.F., et al., Corrosion fatigue behaviors of two biomedical Mg alloys-AZ91D and WE43-In simulated body fluid. Acta Biomaterialia, 2010. 6(12): p. 4605-4613.
293. Di Mario, C., P. Barlis, and J. Tanigawa, Coronary bioabsorbable magnesium stent: 15-month intravascular ultrasound and optical coherence tomography findings. European Heart Journal, 2007. 28(19): p. 2319-2319.
294. Yasuda, K., et al., Mechanistic aspects of the self-organization process for oxide nanotube formation on valve metals. Journal of the Electrochemical Society, 2007. 154(9): p. C472-C478.
295. Schmuki, P., S. Bauer, and S. Kleber, TiO2 nanotubes: Tailoring the geometry in H3PO4/HF electrolytes. Electrochemistry Communications, 2006. 8(8): p. 1321-1325.
296. Schmuki, P., W. Wei, and J.M. Macak, High aspect ratio ordered nanoporous Ta(2)O(5) films by anodization of Ta. Electrochemistry Communications, 2008. 10(3): p. 428-432.
297. Dieringa, H., Properties of magnesium alloys reinforced with nanoparticles and carbon nanotubes: a review. Journal of Materials Science, 2011. 46(2): p. 289-306.
298. Ferkel, H. and B.L. Mordike, Magnesium strengthened by SiC nanoparticles. Materials Science and Engineering a-Structural Materials Properties Microstructure and Processing, 2001. 298(1-2): p. 193-199.
299. Ugandhar, S., M. Gupta, and S.K. Sinha, Enhancing strength and ductility of Mg/SiC composites using recrystallization heat treatment. Composite Structures, 2006. 72(2): p. 266-272.
300. Poddar, P., S. Mukherjee, and K.L. Sahoo, The Microstructure and Mechanical Properties of SiC Reinforced Magnesium Based Composites by Rheocasting Process. Journal of Materials Engineering and Performance, 2009. 18(7): p. 849-855.
301. Wong, W.L.E., S. Karthik, and M. Gupta, Development of high performance Mg-Al2O3 composites containing Al2O3 in submicron length scale using microwave assisted rapid sintering. Materials Science and Technology, 2005. 21(9): p. 1063-1070.

162
302. Wong, W.L.E., S. Karthik, and M. Gupta, Development of hybrid Mg/Al2O3 composites with improved properties using microwave assisted rapid sintering route. Journal of Materials Science, 2005. 40(13): p. 3395-3402.
303. Han, B.Q. and D.C. Dunand, Creep of magnesium strengthened with high volume fractions of yttria dispersoids. Materials Science and Engineering a-Structural Materials Properties Microstructure and Processing, 2001. 300(1-2): p. 235-244.
304. Han, B.Q. and D.C. Dunand, Microstructure and mechanical properties of magnesium containing high volume fractions of yttria dispersoids. Materials Science and Engineering a-Structural Materials Properties Microstructure and Processing, 2000. 277(1-2): p. 297-304.
305. Lee, C.J., J.C. Huang, and P.J. Hsieh, Mg based nano-composites fabricated by friction stir processing. Scripta Materialia, 2006. 54(7): p. 1415-1420.
306. Li, Q.Q., C.A. Rottmair, and R.F. Singer, CNT reinforced light metal composites produced by melt stirring and by high pressure die casting. Composites Science and Technology, 2010. 70(16): p. 2242-2247.
307. Hall, I.W., Corrosion of Carbon Magnesium Metal Matrix Composites. Scripta Metallurgica, 1987. 21(12): p. 1717-1721.
308. Bakkar, A. and V. Neubert, Corrosion behaviour of carbon fibres/magnesium metal matrix composite and electrochemical response of its constituents. Electrochimica Acta, 2009. 54(5): p. 1597-1606.
309. Iijima, S., Helical Microtubules of Graphitic Carbon. Nature, 1991. 354(6348): p. 56-58.
310. Hata, K., et al., Water-assisted highly efficient synthesis of impurity-free single-waited carbon nanotubes. Science, 2004. 306(5700): p. 1362-1364.
311. Bethune, D.S., et al., Cobalt-Catalyzed Growth of Carbon Nanotubes with Single-Atomic-Layerwalls. Nature, 1993. 363(6430): p. 605-607.
312. Iijima, S. and T. Ichihashi, Single-Shell Carbon Nanotubes of 1-Nm Diameter. Nature, 1993. 363(6430): p. 603-605.
313. Coleman, J.N., et al., Small but strong: A review of the mechanical properties of carbon nanotube-polymer composites. Carbon, 2006. 44(9): p. 1624-1652.
314. Elliott, J.A., et al., Collapse of single-wall carbon nanotubes is diameter dependent. Physical Review Letters, 2004. 92(9).
315. Wei, B.Q., R. Vajtai, and P.M. Ajayan, Reliability and current carrying capacity of carbon nanotubes. Applied Physics Letters, 2001. 79(8): p. 1172-1174.
316. Durkop, T., B.M. Kim, and M.S. Fuhrer, Properties and applications of high-mobility semiconducting nanotubes. Journal of Physics-Condensed Matter, 2004. 16(18): p. R553-R580.
317. Tang, Z.K., et al., Superconductivity in 4 angstrom single-walled carbon nanotubes. Science, 2001. 292(5526): p. 2462-2465.
318. Brodie, I. and C.A. Spindt, Vacuum Microelectronics. Advances in Electronics and Electron Physics, Vol 83, 1992. 83: p. 1-106.
319. Wang, Q.H., et al., A nanotube-based field-emission flat panel display. Applied Physics Letters, 1998. 72(22): p. 2912-2913.
320. Choi, W.B., et al., Fully sealed, high-brightness carbon-nanotube field-emission display. Applied Physics Letters, 1999. 75(20): p. 3129-3131.
321. Rosen, R., et al., Application of carbon nanotubes as electrodes in gas discharge tubes. Applied Physics Letters, 2000. 76(13): p. 1668-1670.

163
322. Britto, P.J., et al., Improved charge transfer at carbon nanotube electrodes. Advanced Materials, 1999. 11(2): p. 154-157.
323. Che, G.L., et al., Carbon nanotubule membranes for electrochemical energy storage and production. Nature, 1998. 393(6683): p. 346-349.
324. Planeix, J.M., et al., Application of Carbon Nanotubes as Supports in Heterogeneous Catalysis. Journal of the American Chemical Society, 1994. 116(17): p. 7935-7936.
325. Ajayan, P.M. and O.Z. Zhou, Applications of carbon nanotubes. Carbon Nanotubes, 2001. 80: p. 391-425.
326. Li, C.Y. and T.W. Chou, A structural mechanics approach for the analysis of carbon nanotubes. International Journal of Solids and Structures, 2003. 40(10): p. 2487-2499.
327. Yao, Z.H., et al., Mechanical properties of carbon nanotube by molecular dynamics simulation. Computational Materials Science, 2001. 22(3-4): p. 180-184.
328. Treacy, M.M.J., T.W. Ebbesen, and J.M. Gibson, Exceptionally high Young's modulus observed for individual carbon nanotubes. Nature, 1996. 381(6584): p. 678-680.
329. Wong, E.W., P.E. Sheehan, and C.M. Lieber, Nanobeam mechanics: Elasticity, strength, and toughness of nanorods and nanotubes. Science, 1997. 277(5334): p. 1971-1975.
330. Yu, M.F., et al., Strength and breaking mechanism of multiwalled carbon nanotubes under tensile load. Science, 2000. 287(5453): p. 637-640.
331. Endo, M., et al., An anticorrosive magnesium/carbon nanotube composite. Applied Physics Letters, 2008. 92(6).
332. Fukuda, H., et al., The influence of carbon nanotubes on the corrosion behaviour of AZ31B magnesium alloy. Corrosion Science, 2010. 52(12): p. 3917-3923.
333. Zhou, W., et al., Effect of carbon nanotubes on corrosion of Mg-CNT composites. Corrosion Science, 2010. 52(5): p. 1551-1553.
334. Li, Q.Q., M. Zaiser, and V. Koutsos, Carbon nanotube/epoxy resin composites using a block copolymer as a dispersing agent. Physica Status Solidi a-Applied Research, 2004. 201(13): p. R89-R91.
335. Montemor, M.F. and M.G.S. Ferreira, Analytical characterisation and corrosion behaviour of bis-aminosilane coatings modified with carbon nanotubes activated with rare-earth salts applied on AZ31 Magnesium alloy. Surface & Coatings Technology, 2008. 202(19): p. 4766-4774.
336. Li, Q.Q., et al., Improved processing of carbon nanotube/magnesium alloy composites. Composites Science and Technology, 2009. 69(7-8): p. 1193-1199.
337. Ogle, K., J. Swiatowska, and P. Volovitch, The anodic dissolution of Mg in NaCl and Na(2)SO(4) electrolytes by atomic emission spectroelectrochemistry. Corrosion Science, 2010. 52(7): p. 2372-2378.
338. Chen, J., et al., AC impedance spectroscopy study of the corrosion behavior of an AZ91 magnesium alloy in 0.1 M sodium sulfate solution. Electrochimica Acta, 2007. 52(9): p. 3299-3309.
339. Liu, M., et al., Calculated phase diagrams and the corrosion of die-cast Mg-Al alloys. Corrosion Science, 2009. 51(3): p. 602-619.

164
340. Sudholz, A.D., et al., Electrochemical Properties of Intermetallic Phases and Common Impurity Elements in Magnesium Alloys. Electrochemical and Solid State Letters, 2011. 14(2): p. C5-C7.
341. Shi, Z.M., M. Liu, and A. Atrens, Measurement of the corrosion rate of magnesium alloys using Tafel extrapolation. Corrosion Science, 2010. 52(2): p. 579-588.

165
List of Abbreviations:
AES : Auger Electron Spectroscopy
Alb : Albumin
ALD : Atomic Layer Deposition
ATP : Adenosine Triphosphate
BSE : Back-Scattered Electrons
CNT(s) : Carbon Nanotube(s)
CR : Corrosion Rate
CV : Cyclic Voltammetry
DC : Direct Current
DES : Drug Eluting Stent
EDAX : Energy Dispersive X-Ray Spectroscopy
EIS : Electrochemical Impedance Spectroscopy
EPR : Electron Paramagnetic Resonance
FIB : Focused Ion Beam
FT-IR : Fourier Transform Infrared Spectroscopy
HF : High Frequency
ICP(s) : Intrinsically Conductive Polymer(s)
LF : Low Frequency
MF : Middle Frequency
MO : Metal Oxide
MS : Mild steel
MWNT(s) : Multi-wall Carbon Nanotube(s)
NDE : Negative Difference Effect
NSAID : Non-steroidal Anti-inflammatory Drug
OCP : Open Circuit Potential
PAni : Polyaniline
PPy : Polypyrrole
PT : Polythiophene
SBF : Simulated Body Fluid
SE : Secondary Electrons
SEM : Scanning Electron Microscopy

166
SS : Stainless steel
SWNT(s) : Single-wall Carbon Nanotube(s)
ToF-SIMS : Time of Flight Secondary Ion Mass Spectroscopy
USB-GPIB : Universal Serial Bus - General Purpose Interface Bus
XPS : X-Ray Photoelectron Spectroscopy

167
List of Symbols:
ESHE : Standard Electrode Potential
Ia : Anodic Current Density
Ic : Cathodic Current Density
Ucorr : Corrosion Potential
Uapplied : Applied Potential
Ie,a : Expected Anodic Current Density
Ie,c : Expected Cathodic Current Density
Im,c : Measured Cathodic Current Density
Im,a : Measured Anodic Current Density
Et : Potential Applied at t
Eo : Initial Potential
It : Measured Current Density at t
I0 : Initial Current Density at t
Z : Impedance
Φ : Phase Angle
ω : Frequency
t : Time
Rp : Polarisation Resistance
RCT, RT : Charge Transfer Resistance
Eo : Ground State
E1 : First Excited State
h : Planck’s Constant
c : Speed of Light
ΩΩΩΩ : Ohm
βa : Anodic Tafel Constant
βc : Cathodic Tafel Constant
u : Mass unit
Vmax : Maximum Potential