Embed Size (px)
Citation preview

763
Suppression of Autonomic-Mediated Triggered Firing inPulmonary Vein Preparations, 24 Hours Postcoronary Artery
Ligation in Dogs
EUGENE PATTERSON, PH.D.,∗,† XICHUN YU, M.D.,∗ SHIJUN HUANG, M.D.,MARION GARRETT, B.S.,∗ and DAVID C. KEM, M.D.∗,†
From the ∗University of Oklahoma Health Sciences Center, OU Medical Center, and †Department of Veterans Affairs Medical Center,Oklahoma City, Oklahoma, USA
Autonomic Nerves and Pulmonary Vein Firing. Introduction: Rapid arrhythmia originatingwithin pulmonary veins (PVs) precipitates atrial fibrillation (AF) in man. To determine a possible basisfor an increased incidence of AF observed peri-myocardial infarction in man, we compared AF inductionin vivo and triggered arrhythmia formation within isolated PVs in vitro in normal dogs and dogs studied24 hours postcoronary artery ligation.
Methods and Results: The incidence of AF initiated by atrial premature stimuli was increased in dogspostcoronary artery ligation (6/9) versus normal (2/12) (P=0.03). In isolated PVs from normal hearts, pause-dependent early afterdepolarizations (EADs) were enhanced by catecholamines. Rapid arrhythmia (1,182± 213 beats/min) was triggered during isoproterenol/norepinephrine (32 nM) + acetylcholine (100 nM)(N = 16/23) using the same pacing pauses eliciting EADs. Rapid arrhythmia (802 ± 161 beats/min) wasalso triggered by local autonomic nerve stimulation (ANS; N = 18/23). Despite equivalent pause-dependentafterdepolarization formation in PVs from infarcted hearts, a rightward 45-fold and 28-fold shift in the dose–response curve for afterdepolarization enhancement was observed for isoproterenol and norepinephrine,respectively (P< 0.02). ANS (N= 1/19) and isoproterenol/norepinephrine (32 nM)+ acetylcholine (100 nM)(N = 0/9 and 0/12, respectively) (P = 0.0001) failed to elicit arrhythmia formation. Beta-adrenergic receptordesensitization was associated with a 2.5-fold increase in PV beta-adrenergic receptor kinase (ARK).
Conclusion: The data demonstrate decreased susceptibility of isolated canine PVs for arrhythmia trig-gered by local ANS, or pacing pauses in the presence of a catecholamine + acetylcholine, postmyocardialinfarction, despite a greater susceptibility of the intact heart to AF. The decreased arrhythmia susceptibilitywas observed coincident with an increase in beta-ARK and a decreased responsiveness to beta-adrenergicreceptor agonists. (J Cardiovasc Electrophysiol, Vol. 17, pp. 763-770, July 2006)
atrial fibrillation, pulmonary veins, beta-adrenergic receptor kinase, myocardial infarction, triggered arrhythmia
Introduction
The autonomic nervous system has a well-recognized, butpoorly understood role in the initiation and maintenance ofatrial fibrillation (AF) in man. Increased parasympathetic ner-vous system tone is implicated as a causative factor for AFinitiated during sleep.1,2 Alterations in heart rate variabil-ity suggestive of increased parasympathetic nervous systemactivity,3 increased sympathetic nervous system activity,4 orsimultaneous increases in both parasympathetic and sympa-thetic nervous system activity5 have been observed duringthe immediate time interval preceding paroxysmal AF. AFis also a well-recognized atrial rhythm disorder associated
This study was supported by a grant from the Oklahoma Center for the Ad-vancement of Science and Technology, a grant-in-aid from the AmericanHeart Association (Heartland Affiliate), and research funds from the De-partment of Veterans Affairs.
Address for correspondence: Eugene Patterson, Ph.D., 6E 103 ET CARI,1200 Everett Drive, Oklahoma City, OK 73104. Fax: 405-271-7455; E-mail:[email protected]
Manuscript received 30 November 2005; Revised manuscript received8 February 2006; Accepted for publication 13 February 2006.
doi: 10.1111/j.1540-8167.2006.00507.x
with acute myocardial infarction, a pathology recognized toproduce profound changes in autonomic tone to the heart.6
Local stimulation of intrinsic cardiac ganglionated plexilocated near the antrum of pulmonary veins (PVs) in dogsand local autonomic nerve stimulation (ANS) (stimulatingboth parasympathetic and sympathetic neurons) within my-ocardial sleeve of canine PVs provoke rapid rhythm forma-tion.7,8 Pharmacologic studies suggest that both parasympa-thetic nerve activity (acetylcholine release) and sympatheticnerve activity (norepinephrine release) are essential elementsof the arrhythmia mechanism.8,9 The present studies wereperformed to evaluate both the electrophysiologic alterationsand the response of autonomic interventions of the intact ca-nine heart and the PVs following anterior descending coro-nary artery ligation in dogs. These observations may helpus to better understand changes in the underlying atrial sub-strate sustaining AF and the PV sleeve substrate contributingto an increased triggering of AF, potentiating the spontaneousinitiation of AF following myocardial infarction in man.
Methods
Experiments in the Anesthetized Dog
Male mongrel dogs were anesthetized with intravenoussodium pentobarbital. An endotracheal tube was inserted and

764 Journal of Cardiovascular Electrophysiology Vol. 17, No. 7, July 2006
the animals were respired with room air using a Harvard ven-tilator. Animals were studied in the absence of any previousintervention (normal hearts, N = 12) or 24 hours follow-ing left anterior descending (LAD) coronary artery ligation(N = 9) (described below). Following thoracotomy, bipolarelectrodes (N = 6 recording sites) were placed on the rightand left atria to record activation and/or stimulate atrial my-ocardium. Two surface EKG leads were also recorded. Theleft atrium was paced at 3 Hz from the free wall and prematureatrial stimuli were introduced in decreasing 10-msec intervalsfrom 180 msec until (1) AF was observed, when the intervalwas increased in 1-msec intervals to determine the longestatrial coupling interval producing AF and (2) until atrial re-fractoriness was observed, when the interval was increasedin 1-msec intervals to determine the shortest atrial couplinginterval producing AF. AF was defined as rapid disorganizedatrial activity lasting more than 5 seconds. The window ofvulnerability is defined as the shortest coupling interval foran atrial stimulus producing AF to the longest interval pro-ducing AF. The duration of the window of vulnerability isdefined as the longest coupling interval for an atrial stimulusproducing AF minus the shortest interval producing AF.
The Normal Canine Heart (In Vitro Experiments)
Male dogs were anesthetized with intravenous sodiumpentobarbital. The heart was excised and placed into Tyrode’ssolution containing (mM): NaCl, 130; KCl, 4.0; MgCl2, 1.0;NaHCO3, 20; NaH2PO4, 1.0; glucose, 5.5; and CaCl2, 1.35,bubbled with 95% oxygen:5% CO2 (pH 7.40–7.45). Prepa-rations containing the PV antrum (atrium), the visible PVmyocardial sleeve, and 3–5 mm of the PV distal to the visi-ble sleeve were excised and dissected free of residual adiposeand visceral tissues.
The Canine Heart Post-LAD Coronary Artery Ligation
(Postmyocardial Infarction)
Male dogs were anesthetized with intravenous sodiumpentobarbital. An endotracheal tube was inserted and the an-imals were respired with room air using a Harvard ventilator.Using aseptic technique, a left thoracotomy was performed inthe fifth space and the LAD coronary artery was isolated at thetip of the left atrial appendage. A two-stage ligation was per-formed as described by Harris.10 The incision was closed inlayers. Nalbuphine (0.2–0.4 mg/kg IM) was administered ev-ery 8 hours. The animal was allowed to recover from surgery.At 24 hours, the surviving animals were anesthetized with in-travenous sodium pentobarbital. The heart was removed andPV preparations were prepared as described for the normalgroup.
Electrical Recordings (In Vitro Preparations)
Isolated PV preparations or right atrial free wall tissueswere pinned endocardial side up and superfused with oxy-genated Tyrode’s solution at 37–38◦ (20 mL/min). Bipolarelectrode recordings (0.10 mm diameter Teflon-coated sil-ver wires,7 1 mm apart) were obtained from two to threesites within the tissue preparation, filtered at 10–10,000 Hz,and recorded on a Gould WindowGraf recorder. An intra-cellular recording was obtained using a glass microelec-trode with an intracellular resistance of 10–30 M� (Duo773 electrometer, World Precision Instruments, Sarasota, FL,USA). The preparation was paced at 2–3× diastolic thresh-
old using 4-msec duration stimuli from a Grass model S88stimulator (Quincy, MA, USA) at 1 Hz, unless otherwisestated. All measurements were made from recordings at 50 or100 mm/sec. Intracellular microelectrode recordings wereperformed pre- and post-ANS from the immediate vicinityof the stimulating electrodes (within 2–3 mm).
Autonomic Nerve Stimulation In Vitro Experiments
Selective local ANS was accomplished using high-frequency (100 Hz) trains of 0.05–0.1-msec duration square-wave stimuli introduced at 10–150 V7,8 from a Grass stimula-tor. Stimulus train length was adjusted from 50 to 500 msec,and synchronized to begin with a pacing stimulus introducedat another site within the tissue, 3–5 mm away. Voltage wasmaintained at less than 50% of the threshold voltage requiredto excite local myocardium when introduced as 0.05–0.1-msec duration stimuli during a 2-Hz pacing train. A minimumof three different sites are tested in each preparation.
Determination of G-Receptor Kinase-2/Beta-Adrenergic
Receptor Kinase in Canine PV Sleeves
Beta-adrenergic receptor kinase (beta-ARK) immun-odetection was performed using detergent-solubilized ex-tracts following immunoprecipitation.11 PVs removed fromthe intact canine heart at the conclusion of the ex-periments were homogenized in cold RIPA buffer (50mM Tris, pH 8.0; 5 mM EDTA, 150 mM NaCl, 1%Nonidet P-40, 0.5% sodium deoxycholate, 0.1% SDS,10 mM NaF, 5 mM EGTA, 10 mM sodium pyrophos-phate, and 1 mM phenylmethylsulfonyl fluoride). Beta-ARKwas immunoprecipitated from a 1-mL crude homogenate(250 μg) with 1.2 μg polyclonal anti-GRK-2 antibody(Santa Cruz Biotechnology, Santa Cruz, CA, USA) and20 μL of a 50% slurry of protein A/G agarose conjugateagitated overnight at 4◦C. Immune complexes were washedthree times in ice-cold RIPA, resuspended in 25 μL of load-ing buffer, heated to 95◦C for 5 minutes, and then elec-trophoresed and transferred to nitrocellulose. Beta-ARK wasdetected using monoclonal anti-GRK2 antibody (UpstateBiotechnology, Lake Placid, NY, USA) and enhanced chemi-luminescence. Quantitation was performed by densitometricscanning with ImageQuaNT software (Molecular Dynamics,Sunnyvale, CA, USA) used for analysis.
Statistics
Data are expressed as the mean ± the standard error. Dif-ferences between groups were determined by an unpairedStudent’s t-test. Analysis of variance (repeated measures)was used to determine differences within a group with drugtreatment. Student–Newman–Keuls test was used to deter-mine differences between individual groups. Differences inarrhythmia incidence were determined using Fisher’s exacttest.
Results
Atrial Refractoriness and AF in the Anesthetized Dog
Atrial premature beats were introduced at 2× and 4× di-astolic threshold voltage, producing 3 episodes of AF in 2 of12 normal dogs and 10 episodes of AF in 6 of 9 dogs stud-ied 24 hours post-LAD coronary artery ligation (P = 0.03).The atrial effective refractory period at 2× and 4× diastolic
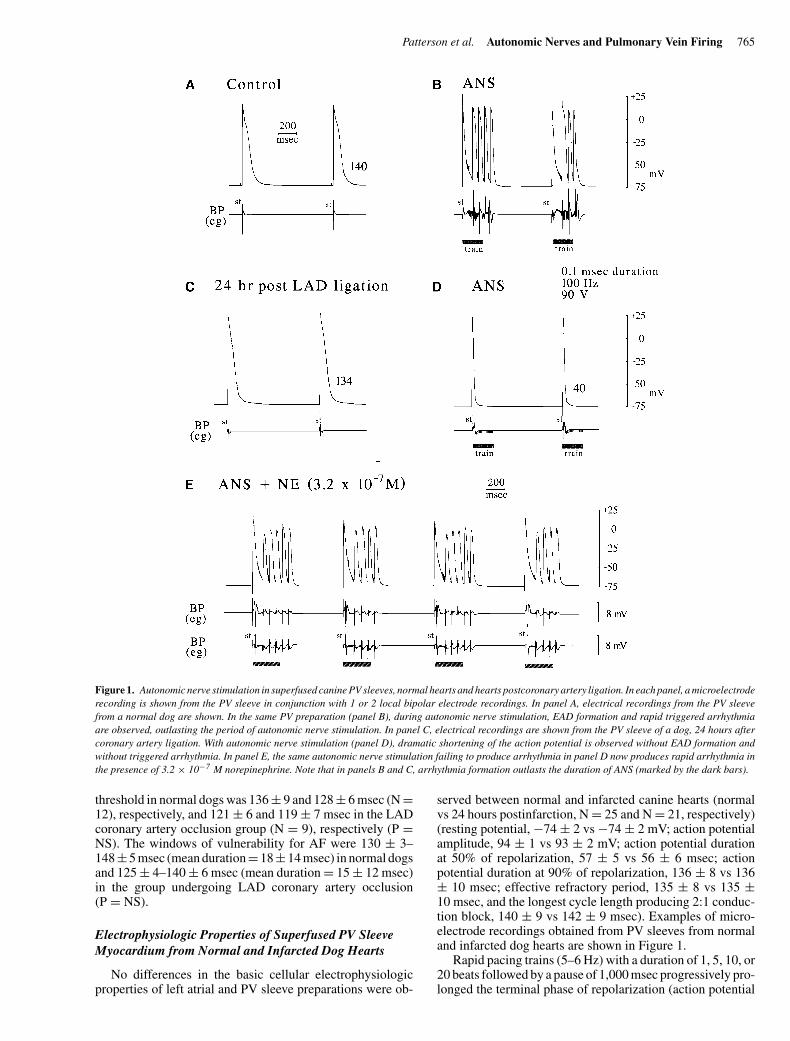
Patterson et al. Autonomic Nerves and Pulmonary Vein Firing 765
Figure 1. Autonomic nerve stimulation in superfused canine PV sleeves, normal hearts and hearts postcoronary artery ligation. In each panel, a microelectroderecording is shown from the PV sleeve in conjunction with 1 or 2 local bipolar electrode recordings. In panel A, electrical recordings from the PV sleevefrom a normal dog are shown. In the same PV preparation (panel B), during autonomic nerve stimulation, EAD formation and rapid triggered arrhythmiaare observed, outlasting the period of autonomic nerve stimulation. In panel C, electrical recordings are shown from the PV sleeve of a dog, 24 hours aftercoronary artery ligation. With autonomic nerve stimulation (panel D), dramatic shortening of the action potential is observed without EAD formation andwithout triggered arrhythmia. In panel E, the same autonomic nerve stimulation failing to produce arrhythmia in panel D now produces rapid arrhythmia inthe presence of 3.2 × 10−7 M norepinephrine. Note that in panels B and C, arrhythmia formation outlasts the duration of ANS (marked by the dark bars).
threshold in normal dogs was 136 ± 9 and 128 ± 6 msec (N =12), respectively, and 121 ± 6 and 119 ± 7 msec in the LADcoronary artery occlusion group (N = 9), respectively (P =NS). The windows of vulnerability for AF were 130 ± 3–148±5 msec (mean duration=18±14 msec) in normal dogsand 125 ± 4–140 ± 6 msec (mean duration = 15 ± 12 msec)in the group undergoing LAD coronary artery occlusion(P = NS).
Electrophysiologic Properties of Superfused PV Sleeve
Myocardium from Normal and Infarcted Dog Hearts
No differences in the basic cellular electrophysiologicproperties of left atrial and PV sleeve preparations were ob-
served between normal and infarcted canine hearts (normalvs 24 hours postinfarction, N = 25 and N = 21, respectively)(resting potential, −74 ± 2 vs −74 ± 2 mV; action potentialamplitude, 94 ± 1 vs 93 ± 2 mV; action potential durationat 50% of repolarization, 57 ± 5 vs 56 ± 6 msec; actionpotential duration at 90% of repolarization, 136 ± 8 vs 136± 10 msec; effective refractory period, 135 ± 8 vs 135 ±10 msec, and the longest cycle length producing 2:1 conduc-tion block, 140 ± 9 vs 142 ± 9 msec). Examples of micro-electrode recordings obtained from PV sleeves from normaland infarcted dog hearts are shown in Figure 1.
Rapid pacing trains (5–6 Hz) with a duration of 1, 5, 10, or20 beats followed by a pause of 1,000 msec progressively pro-longed the terminal phase of repolarization (action potential
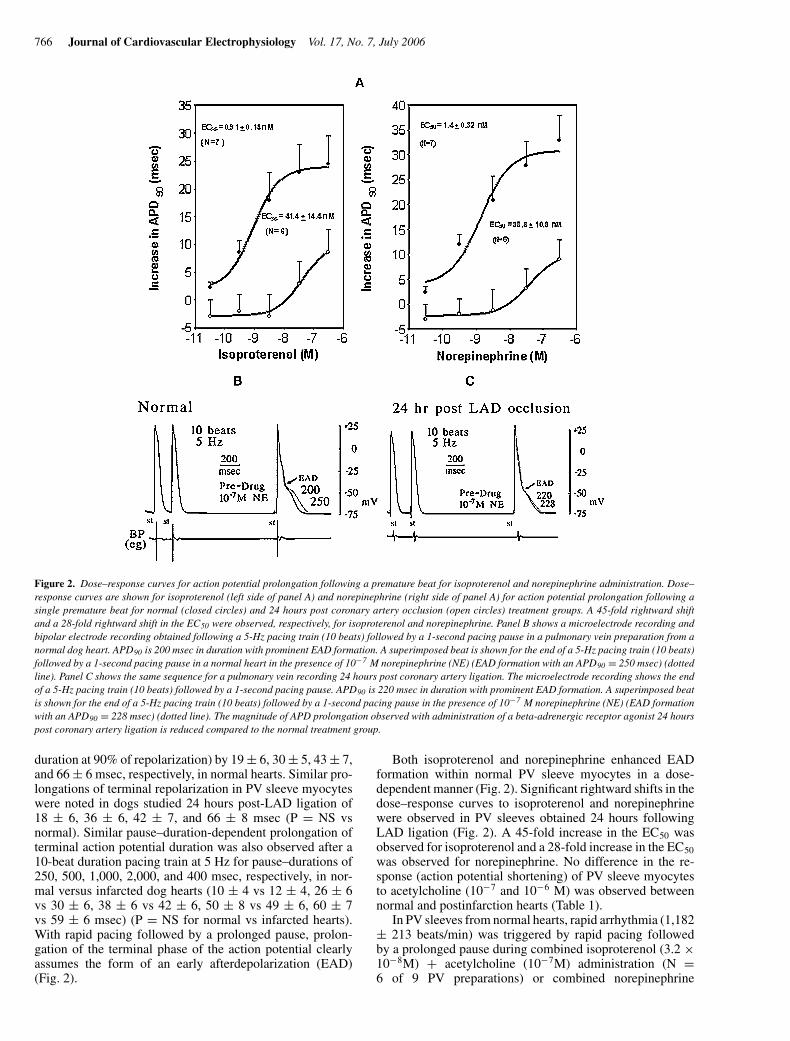
766 Journal of Cardiovascular Electrophysiology Vol. 17, No. 7, July 2006
Figure 2. Dose–response curves for action potential prolongation following a premature beat for isoproterenol and norepinephrine administration. Dose–response curves are shown for isoproterenol (left side of panel A) and norepinephrine (right side of panel A) for action potential prolongation following asingle premature beat for normal (closed circles) and 24 hours post coronary artery occlusion (open circles) treatment groups. A 45-fold rightward shiftand a 28-fold rightward shift in the EC50 were observed, respectively, for isoproterenol and norepinephrine. Panel B shows a microelectrode recording andbipolar electrode recording obtained following a 5-Hz pacing train (10 beats) followed by a 1-second pacing pause in a pulmonary vein preparation from anormal dog heart. APD90 is 200 msec in duration with prominent EAD formation. A superimposed beat is shown for the end of a 5-Hz pacing train (10 beats)followed by a 1-second pacing pause in a normal heart in the presence of 10−7 M norepinephrine (NE) (EAD formation with an APD90 = 250 msec) (dottedline). Panel C shows the same sequence for a pulmonary vein recording 24 hours post coronary artery ligation. The microelectrode recording shows the endof a 5-Hz pacing train (10 beats) followed by a 1-second pacing pause. APD90 is 220 msec in duration with prominent EAD formation. A superimposed beatis shown for the end of a 5-Hz pacing train (10 beats) followed by a 1-second pacing pause in the presence of 10−7 M norepinephrine (NE) (EAD formationwith an APD90 = 228 msec) (dotted line). The magnitude of APD prolongation observed with administration of a beta-adrenergic receptor agonist 24 hourspost coronary artery ligation is reduced compared to the normal treatment group.
duration at 90% of repolarization) by 19 ± 6, 30 ± 5, 43 ± 7,and 66 ± 6 msec, respectively, in normal hearts. Similar pro-longations of terminal repolarization in PV sleeve myocyteswere noted in dogs studied 24 hours post-LAD ligation of18 ± 6, 36 ± 6, 42 ± 7, and 66 ± 8 msec (P = NS vsnormal). Similar pause–duration-dependent prolongation ofterminal action potential duration was also observed after a10-beat duration pacing train at 5 Hz for pause–durations of250, 500, 1,000, 2,000, and 400 msec, respectively, in nor-mal versus infarcted dog hearts (10 ± 4 vs 12 ± 4, 26 ± 6vs 30 ± 6, 38 ± 6 vs 42 ± 6, 50 ± 8 vs 49 ± 6, 60 ± 7vs 59 ± 6 msec) (P = NS for normal vs infarcted hearts).With rapid pacing followed by a prolonged pause, prolon-gation of the terminal phase of the action potential clearlyassumes the form of an early afterdepolarization (EAD)(Fig. 2).
Both isoproterenol and norepinephrine enhanced EADformation within normal PV sleeve myocytes in a dose-dependent manner (Fig. 2). Significant rightward shifts in thedose–response curves to isoproterenol and norepinephrinewere observed in PV sleeves obtained 24 hours followingLAD ligation (Fig. 2). A 45-fold increase in the EC50 wasobserved for isoproterenol and a 28-fold increase in the EC50
was observed for norepinephrine. No difference in the re-sponse (action potential shortening) of PV sleeve myocytesto acetylcholine (10−7 and 10−6 M) was observed betweennormal and postinfarction hearts (Table 1).
In PV sleeves from normal hearts, rapid arrhythmia (1,182± 213 beats/min) was triggered by rapid pacing followedby a prolonged pause during combined isoproterenol (3.2 ×10−8M) + acetylcholine (10−7M) administration (N =6 of 9 PV preparations) or combined norepinephrine

Patterson et al. Autonomic Nerves and Pulmonary Vein Firing 767
TABLE 1
Acetylcholine Administration in Normal and Post-infarction Groups
ACh ACh
Pre-Drug (10−7 M) (10−6 M)
Normal dog hearts†
Resting potential (mV) −74 ± 2 −76 ± 2∗ −78 ± 2∗∗Action potential 94 ± 2 96 ± 2∗ 99 ± 3∗∗
amplitude (mV)Action potential 58 ± 5 45 ± 8∗ 24 ± 9∗∗
duration50 (msec)Action potential 138 ± 8 99 ± 10∗∗ 62 ± 10∗∗
duration90 (msec)Dog hearts, 24 hours
post-LAD ligation‡
Resting potential (mV) −74 ± 2 −76 ± 3∗ −79 ± 3∗∗Action potential 93 ± 2 95 ± 4∗ 98 ± 5∗∗
amplitude (mV)Action potential 56 ± 6 44 + 10∗ 26 ± 10∗∗
duration50 (msec)Action potential 136 ± 10 96 ± 11∗∗ 60 ± 12∗∗
duration90 (msec)
∗P < 0.05 versus pre-drug.∗∗P < 0.01 versus pre-drug.†N = 23.‡N = 21.
(3.2×10−8M)+ acetylcholine (10−7M) administration (N=10 of 14 PV preparations). In superfused PV preparations ob-tained 24 hours after LAD coronary artery ligation, the samepacing sequences uniformly failed to elicit rapid arrhythmiaduring the combined administration of isoproterenol (3.2 ×10−8 M) + acetylcholine (10−7 M) (N = 9) or the combinedadministration of norepinephrine (3.2 × 10−8 M) + acetyl-choline (10−7 M) (N = 12) (P = 0.0009 and P = 0.0002 vsnormal, respectively). An example of arrhythmia triggered bya rapid pacing-prolonged pause pacing sequence in a normalheart (upper panel) and an example of the same pacing se-quence in a PV sleeve preparation postinfarct (lower panel)during combined acetylcholine–norepinephrine administra-tion are shown in Figure 3.
Normal Canine PVs—Autonomic Nerve Stimulation
Each PV preparation studied (N = 23) during superfusionwith oxygenated Tyrode’s solution at 37–38◦C was initiallyquiescent. The initial high-frequency stimulus train testedwas 300 msec in duration, applied immediately after the pac-ing stimulus. The voltage was raised in 10-V steps until amaximal 150 V was employed. Rapid triggered arrhythmia(802 ± 161 beats/min) was observed in 18 of 23 PV prepara-tions at a minimal voltage of 83 ± 23 V (Fig. 1, panel B). Thearrhythmias frequently lasted 3–5 beats following the cessa-tion of the high-frequency pacing train. A stimulus train of0.1-msec duration stimuli applied at a rate of 2 Hz to thePV sleeve at the same voltage producing arrhythmia fails tocapture PV myocardium.
Canine PVs, 24 Hours After LAD Ligation—Autonomic
Nerve Stimulation
Each PV preparation studied (N = 19) during superfusionwith oxygenated Tyrode’s solution at 37–38◦C was initiallyquiescent. High-frequency stimulus train tested was 300 msec
in duration, applied immediately after the pacing stimulus.The voltage was raised in 10-V steps until a maximal 150 Vwas employed. Rapid triggered arrhythmia (765 beats/min)was observed at one site in only 1 of 19 PV sleeve prepara-tions (P < 0.0001 vs normal), with dramatic shortening of theaction potential, but no arrhythmia formation was observedat each of the remaining stimulation sites (Fig. 1, panel D). Inthree of seven preparations tested, the addition of 3.2 × 10−7
M norepinephrine to the superfusion solution allowed high-frequency stimulation trains to produce triggered arrhythmia(763 ± 88 beats/min) (Fig. 1, panel E). High-frequency stim-ulation trains produced similar shortening of action poten-tial duration and hyperpolarization as observed in superfusedPVs from normal dogs (Fig. 4).
Beta-Adrenergic Receptor Kinase Content
A 2.5-fold increase in beta-ARK content by immunoblotwas measured in the canine PV sleeve of dogs, 24 hourspostmyocardial infarction (N = 6), compared to normal PVsleeve myocardium from dogs not undergoing myocardialinfarction (N = 5) (Fig. 5).
Discussion
Basis for Atrial Fibrillation
AF is believed to require both (1) an initiating trigger con-sisting of single or multiple atrial premature beats and (2) anunderlying atrial substrate capable of sustaining fibrillation.In the present studies, we observed an increased frequencyof inducible AF with single premature atrial stimuli intro-duced into canine hearts studied 24 hours post-LAD coronaryartery occlusion as compared to normal hearts, suggesting anenhanced substrate capable of sustaining AF. In the intactdog, however, the initiating event, a premature atrial beatwas introduced early within diastole by the present investi-gators. In man, rapid bursts of electrical activity originatingwithin the PVs have been observed to be the most commonevent initiating AF.12-14 The electrophysiologic mechanismor mechanisms producing the rapid extrasystoles are not well-understood, nor is it well-understood why the PVs are themost common site for rapid rhythms triggering AF. Spon-taneous automaticity (<30 beats/min) can be observed insome superfused rabbit preparations14,15 and less commonlyobserved in superfused canine PV preparations.14-17 Even un-der conditions of beta-adrenergic receptor stimulation thesespontaneous depolarizations are observed only at rates thatare too slow to provide for the observed clinical arrhythmias.Several other electrophysiologic mechanisms have been pro-posed to provide for the rapid rates (>300 beats/min) ob-served clinically. These disparate mechanisms have includedlocalized reentry within the PVs,18 depolarization-inducedautomaticity,19 and triggered activity.7-9,14,17 Triggeredarrhythmias observed within the PV sleeves have beenproposed to originate from both early7,8,14,16 and delayed af-terdepolarizations.17 Each of the proposed arrhythmia mech-anisms would be facilitated by beta-adrenergic receptor stim-ulation. Both reentry- and EAD-facilitated triggered firingmay also be facilitated by action potential shortening inducedby the release of acetylcholine from parasympathetic nerveendings.7-9,15
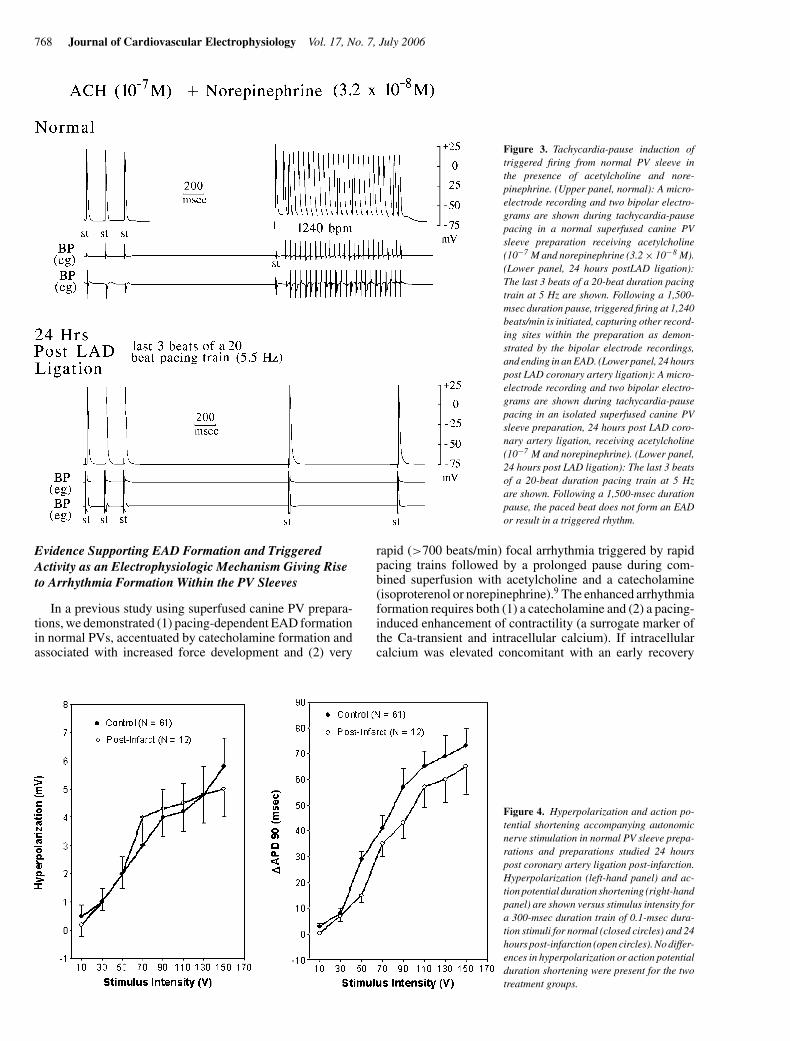
768 Journal of Cardiovascular Electrophysiology Vol. 17, No. 7, July 2006
Figure 3. Tachycardia-pause induction oftriggered firing from normal PV sleeve inthe presence of acetylcholine and nore-pinephrine. (Upper panel, normal): A micro-electrode recording and two bipolar electro-grams are shown during tachycardia-pausepacing in a normal superfused canine PVsleeve preparation receiving acetylcholine(10−7 M and norepinephrine (3.2 × 10−8 M).(Lower panel, 24 hours postLAD ligation):The last 3 beats of a 20-beat duration pacingtrain at 5 Hz are shown. Following a 1,500-msec duration pause, triggered firing at 1,240beats/min is initiated, capturing other record-ing sites within the preparation as demon-strated by the bipolar electrode recordings,and ending in an EAD. (Lower panel, 24 hourspost LAD coronary artery ligation): A micro-electrode recording and two bipolar electro-grams are shown during tachycardia-pausepacing in an isolated superfused canine PVsleeve preparation, 24 hours post LAD coro-nary artery ligation, receiving acetylcholine(10−7 M and norepinephrine). (Lower panel,24 hours post LAD ligation): The last 3 beatsof a 20-beat duration pacing train at 5 Hzare shown. Following a 1,500-msec durationpause, the paced beat does not form an EADor result in a triggered rhythm.
Evidence Supporting EAD Formation and Triggered
Activity as an Electrophysiologic Mechanism Giving Rise
to Arrhythmia Formation Within the PV Sleeves
In a previous study using superfused canine PV prepara-tions, we demonstrated (1) pacing-dependent EAD formationin normal PVs, accentuated by catecholamine formation andassociated with increased force development and (2) very
Figure 4. Hyperpolarization and action po-tential shortening accompanying autonomicnerve stimulation in normal PV sleeve prepa-rations and preparations studied 24 hourspost coronary artery ligation post-infarction.Hyperpolarization (left-hand panel) and ac-tion potential duration shortening (right-handpanel) are shown versus stimulus intensity fora 300-msec duration train of 0.1-msec dura-tion stimuli for normal (closed circles) and 24hours post-infarction (open circles). No differ-ences in hyperpolarization or action potentialduration shortening were present for the twotreatment groups.
rapid (>700 beats/min) focal arrhythmia triggered by rapidpacing trains followed by a prolonged pause during com-bined superfusion with acetylcholine and a catecholamine(isoproterenol or norepinephrine).9 The enhanced arrhythmiaformation requires both (1) a catecholamine and (2) a pacing-induced enhancement of contractility (a surrogate marker ofthe Ca-transient and intracellular calcium). If intracellularcalcium was elevated concomitant with an early recovery
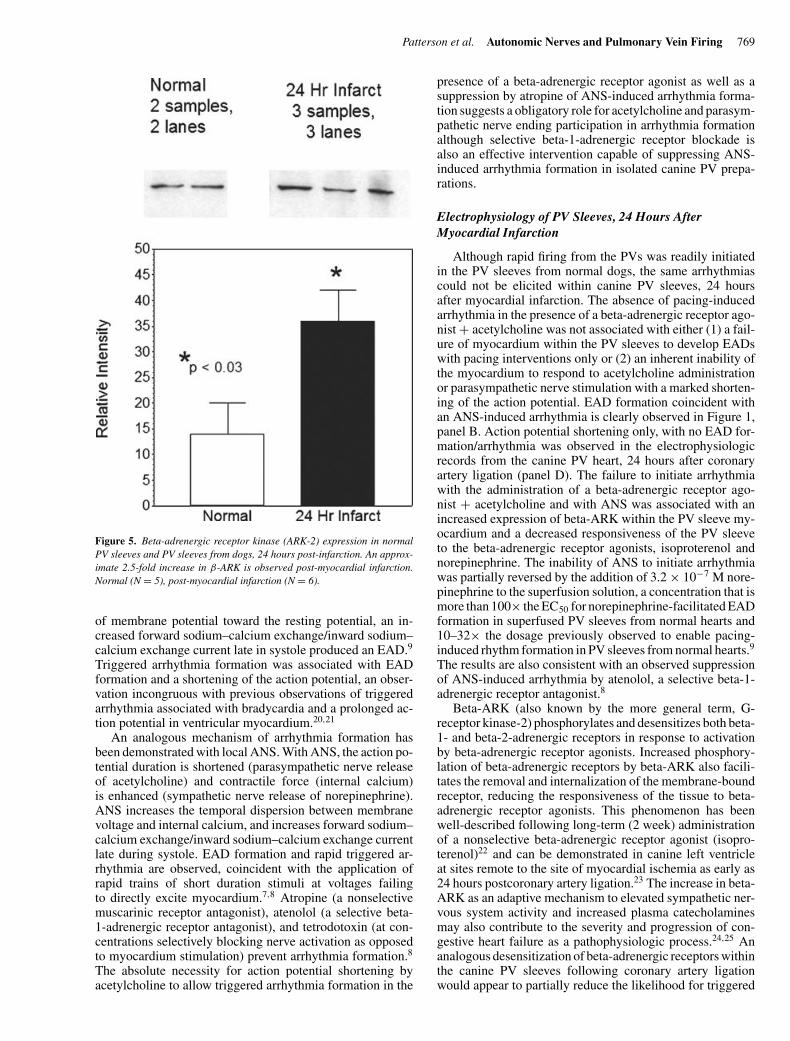
Patterson et al. Autonomic Nerves and Pulmonary Vein Firing 769
Figure 5. Beta-adrenergic receptor kinase (ARK-2) expression in normalPV sleeves and PV sleeves from dogs, 24 hours post-infarction. An approx-imate 2.5-fold increase in β-ARK is observed post-myocardial infarction.Normal (N = 5), post-myocardial infarction (N = 6).
of membrane potential toward the resting potential, an in-creased forward sodium–calcium exchange/inward sodium–calcium exchange current late in systole produced an EAD.9
Triggered arrhythmia formation was associated with EADformation and a shortening of the action potential, an obser-vation incongruous with previous observations of triggeredarrhythmia associated with bradycardia and a prolonged ac-tion potential in ventricular myocardium.20,21
An analogous mechanism of arrhythmia formation hasbeen demonstrated with local ANS. With ANS, the action po-tential duration is shortened (parasympathetic nerve releaseof acetylcholine) and contractile force (internal calcium)is enhanced (sympathetic nerve release of norepinephrine).ANS increases the temporal dispersion between membranevoltage and internal calcium, and increases forward sodium–calcium exchange/inward sodium–calcium exchange currentlate during systole. EAD formation and rapid triggered ar-rhythmia are observed, coincident with the application ofrapid trains of short duration stimuli at voltages failingto directly excite myocardium.7,8 Atropine (a nonselectivemuscarinic receptor antagonist), atenolol (a selective beta-1-adrenergic receptor antagonist), and tetrodotoxin (at con-centrations selectively blocking nerve activation as opposedto myocardium stimulation) prevent arrhythmia formation.8
The absolute necessity for action potential shortening byacetylcholine to allow triggered arrhythmia formation in the
presence of a beta-adrenergic receptor agonist as well as asuppression by atropine of ANS-induced arrhythmia forma-tion suggests a obligatory role for acetylcholine and parasym-pathetic nerve ending participation in arrhythmia formationalthough selective beta-1-adrenergic receptor blockade isalso an effective intervention capable of suppressing ANS-induced arrhythmia formation in isolated canine PV prepa-rations.
Electrophysiology of PV Sleeves, 24 Hours After
Myocardial Infarction
Although rapid firing from the PVs was readily initiatedin the PV sleeves from normal dogs, the same arrhythmiascould not be elicited within canine PV sleeves, 24 hoursafter myocardial infarction. The absence of pacing-inducedarrhythmia in the presence of a beta-adrenergic receptor ago-nist + acetylcholine was not associated with either (1) a fail-ure of myocardium within the PV sleeves to develop EADswith pacing interventions only or (2) an inherent inability ofthe myocardium to respond to acetylcholine administrationor parasympathetic nerve stimulation with a marked shorten-ing of the action potential. EAD formation coincident withan ANS-induced arrhythmia is clearly observed in Figure 1,panel B. Action potential shortening only, with no EAD for-mation/arrhythmia was observed in the electrophysiologicrecords from the canine PV heart, 24 hours after coronaryartery ligation (panel D). The failure to initiate arrhythmiawith the administration of a beta-adrenergic receptor ago-nist + acetylcholine and with ANS was associated with anincreased expression of beta-ARK within the PV sleeve my-ocardium and a decreased responsiveness of the PV sleeveto the beta-adrenergic receptor agonists, isoproterenol andnorepinephrine. The inability of ANS to initiate arrhythmiawas partially reversed by the addition of 3.2 × 10−7 M nore-pinephrine to the superfusion solution, a concentration that ismore than 100× the EC50 for norepinephrine-facilitated EADformation in superfused PV sleeves from normal hearts and10–32× the dosage previously observed to enable pacing-induced rhythm formation in PV sleeves from normal hearts.9
The results are also consistent with an observed suppressionof ANS-induced arrhythmia by atenolol, a selective beta-1-adrenergic receptor antagonist.8
Beta-ARK (also known by the more general term, G-receptor kinase-2) phosphorylates and desensitizes both beta-1- and beta-2-adrenergic receptors in response to activationby beta-adrenergic receptor agonists. Increased phosphory-lation of beta-adrenergic receptors by beta-ARK also facili-tates the removal and internalization of the membrane-boundreceptor, reducing the responsiveness of the tissue to beta-adrenergic receptor agonists. This phenomenon has beenwell-described following long-term (2 week) administrationof a nonselective beta-adrenergic receptor agonist (isopro-terenol)22 and can be demonstrated in canine left ventricleat sites remote to the site of myocardial ischemia as early as24 hours postcoronary artery ligation.23 The increase in beta-ARK as an adaptive mechanism to elevated sympathetic ner-vous system activity and increased plasma catecholaminesmay also contribute to the severity and progression of con-gestive heart failure as a pathophysiologic process.24,25 Ananalogous desensitization of beta-adrenergic receptors withinthe canine PV sleeves following coronary artery ligationwould appear to partially reduce the likelihood for triggered

770 Journal of Cardiovascular Electrophysiology Vol. 17, No. 7, July 2006
arrhythmia and paroxysmal AF. On the surface, this would ap-pear to be a contradiction to the observed increased incidenceof AF with myocardial infarction in man, but the present ob-servation is limited to the initiation of triggered activity withinthe PVs and not to AF initiated by premature beats originatingelsewhere, possibly by different mechanisms.
Conclusions
The present experiments evaluate the ability of early atrialpremature beats to initiate AF in the intact canine heart aswell as the ability of ANS and autonomic nervous systemagonists to initiate and sustain rapid triggering within isolatedsuperfused PVs. Localized reentry may indeed be facilitatedby increased activity of the ganglionated plexi of the intrinsicautonomic nervous system, atrial stretch, atrial infarction,acute inflammation (cytokine release), and other alterationsassociated with coronary artery occlusion. We suggest thatthe profibrillatory effects of myocardial infarction may resultfrom a facilitation of macroreentry within the atria, enhancingthe ability of an early atrial extrasystole to initiate reentrywithin a heterogeneous electrical substrate, as opposed toenhanced triggered activity within the PVs. The answers tothis fundamental question will require further experimentalevaluation.
References
1. Coumel P: Autonomic arrhythmogenic factors in paroxysmal atrial fib-rillation. In: Olsen SB, Alessie MA, Campbell RW, eds. Atrial Fibril-lation: Mechanism and Therapeutic Strategies. Armonk, NY: FuturaPublishing Co., 1994, pp. 171-184.
2. Coumel P: Autonomic influences in atrial tachyarrhythmias. J Cardio-vasc Electrophysiol 1996;7:999-1007.
3. Lombardi F, Tarricone D, Tundo F, Colombo F, Belletti S, Foirentini C:Autonomic nervous system and paroxysmal atrial fibrillation: A studybased on the analysis of RR interval changes before, during, and afterparoxysmal atrial fibrillation. Eur Heart J 2004;25:1242-1248.
4. Amar D, Zhang H, Miodownik S, Kadish AH: Competing autonomicmechanisms precede the onset of postoperative atrial fibrillation. J AmColl Cardiol 2003;42:1262-1268.
5. Chen Y, Chen S, Tai C, Wen Z, Feng A, Ding Y, Chang M: Role ofatrial electrophysiology and autonomic nervous system in patients withsupraventricular tachycardia and paroxysmal atrial fibrillation. J AmColl Cardiol 1998;32:732-738.
6. Webb SW, Adgey AAJ, Pantridge JF: Autonomic disturbances at onsetof acute myocardial infarction. Br Med J 1972;3:89-92.
7. Schauerte PPN, Scherlag BJ, Patterson E, Scherlag MA, MatsudairaK, Nakagawa H, Jackman WM, Lazzara R: Focal atrial fibrilla-tion: Experimental evidence for a pathophysiologic role of the au-tonomic nervous system. J Cardiovasc Electrophysiol 2001;12:592-599.
8. Patterson E, Po SS, Scherlag BJ, Lazzara R: Triggered firing in pul-monary veins initiated by in vitro autonomic nerve stimulation. HeartRhythm 2005;2:624-631.
9. Patterson E, Lazzara R, Szabo B, Liu H, Tang D, Li YH, Scherlag BJ,Po S: Na–Ca exchange initiated by the Ca2+ transient: A trigger forarrhythmia formation in tissues with an abbreviated action potential. JAm Coll Cardiol 2006;47:1196-1206.
10. Harris AS: Delayed development of ventricular ectopic rhythms follow-ing experimental coronary occlusion. Circulation 1950;1:1318-1328.
11. Yu X, Huang S, Patterson E, Garrett M, Kaufman KM, Zhu M, Duhn ST,Kem DC: Proteasome degradation of GRK-2 during ischemia and ven-tricular tachyarrhythmias in a canine model of myocardial infarction.Am J Physiol 2005;299:H1960-1969.
12. Haissaguerre M, Jais P, Shah DC, Takahashi A, Hocini M, Quinou G,Garrigue S, Le Moroux A, Le Metayer P, Clementy J: Spontaneous ini-tiation of atrial fibrillation by ectopic beats originating in the pulmonaryveins. N Engl J Med 1998;339:659-666.
13. Chen S, Hsieh M, Tai C, Tsai C-F, Prakask VS, Yu W-C, Hsu T-L, DingY-A, Chang M-S: Initiation of atrial fibrillation by ectopic beats origi-nating from the pulmonary veins. Circulation 1999;100:1879-1886.
14. Chen YJ, Chen SA: Electrophysiology of pulmonary veins. J CardiovascElectrophysiol 2006;17:220-224.
15. Cheung DW: Electrical activity of the pulmonary vein and its interactionwith the right atrium in the guinea-pig. J Physiol 1980;314:445-456.
16. Po SS, Li Y, Tang D, Liu H, Geng N, Jackman WM, Scherlag BJ,Lazzara R, Patterson E: Rapid and stable reentry within the pulmonaryvein as a mechanism initiating paroxysmal atrial fibrillation. J Am CollCardiol 2005;45:1871-1877.
17. Chen Y, Chen S, Chang M, Lin C: Arrhythmogenic activity of cardiacmuscle in pulmonary veins of the dog: Implication for the genesis ofatrial fibrillation. Cardiovasc Res 2000;48:265-273.
18. Arora R, Verheule S, Luis S, Navarrete A, Katari V, Wilson E, Vaz D,Olgin JE: Arrhythmogenic substrate of the pulmonary veins assessedby high-resolution optical mapping. Circulation 2003;107:1816-1821.
19. Honjo H, Boyett MR, Niwa R, Inada S, Yamamoto M, Mitsui K,Horiuchi T, Shibata N, Kamiya K, Kodama I: Pacing-induced sponta-neous activity in myocardial sleeves of pulmonary veins after treatmentwith ryanodine. Circulation 2003;107:1937-1943.
20. January CT, Riddle JM: Early afterdepolarizations: Mechanism ofinduction and block: A role for L-type Ca2+ current. Circ Res1989;64:977-990.
21. Szabo B, Sweidan R, Rajagopalan CV, Lazzara R: Role of Na+:Ca2+exchange current in CS
+-induced early afterdepolarizations in Purkinjefibers. J Cardiovasc Electrophysiol 1994;5:933-944.
22. Iaccarino G, Tomhave ED, Lefkowitz RJ, Koch WJ: Reciprocal in vivoregulation of myocardial G protein-coupled receptor kinase expres-sion by beta-adrenergic receptor stimulation and blockade. Circulation1998;98:1783-1789.
23. Yu X, Zhang M, Yu X, Kyker K, Benovic JL, Kem DC: Ischemic inacti-vation of G protein-coupled receptor kinase and altered desensitizationof canine cardiac β-adrenergic receptors. Circulation 2000;102:2535-2540.
24. Ungerer M, Kesselbohm K, Kronsbein K, Lohse MJ, Richardt G: Ac-tivation of beta-adrenergic receptor kinase during ischemia. Circ Res1996;79:455-460.
25. Choi DJ, Koch WJ, Hunter JJ, Rockman HA: Mechanism of beta-adrenergic receptor desensitization in cardiac hypertrophy is increasedbeta-adrenergic receptor kinase. J Biol Chem 1997;272:17223-17229.





























