Embed Size (px)
Citation preview

1
Supplementary Materials for: Ligand-directed tosyl chemistry for protein labeling in vivo Shinya Tsukiji, Masayoshi Miyagawa, Yousuke Takaoka, Tomonori Tamura & Itaru Hamachi* Department of Synthetic Chemistry and Biological Chemistry, Graduate School of Engineering, Kyoto University, Katsura, Nishikyo-ku, Kyoto 615-8510, Japan. Core Research for Evolutional Science and Technology (CREST), Japan Science and Technology Agency, 5 Sanbancho, Chiyoda-ku, Tokyo 102-0075, Japan. *Correspondence should be addressed to I.H. ([email protected])
Nature Chemical Biology: doi:10.1038/nchembio.157

2
Contents:
Supplementary Figures and Table
Supplementary Figure 1 p.3–4 Supplementary Figure 2 p.5 Supplementary Figure 3 p.6
Supplementary Figure 4 p.7 Supplementary Figure 5 p.8 Supplementary Figure 6 p.9–10
Supplementary Figure 7 p.11 Supplementary Table 1 p.12
Supplementary Methods General materials and methods for organic synthesis p.13 Synthesis of LDT reagents and other related compounds p.14–32
General materials and methods for biochemical/biological experiments p.33 Carbonic anhydrase (CA) labeling experiments p.34–37 FK506-binding protein 12 (FKBP12) labeling experiments p.38
Congerin labeling experiments p.39 References p.40
Nature Chemical Biology: doi:10.1038/nchembio.157
3
Nature Chemical Biology: doi:10.1038/nchembio.157

4
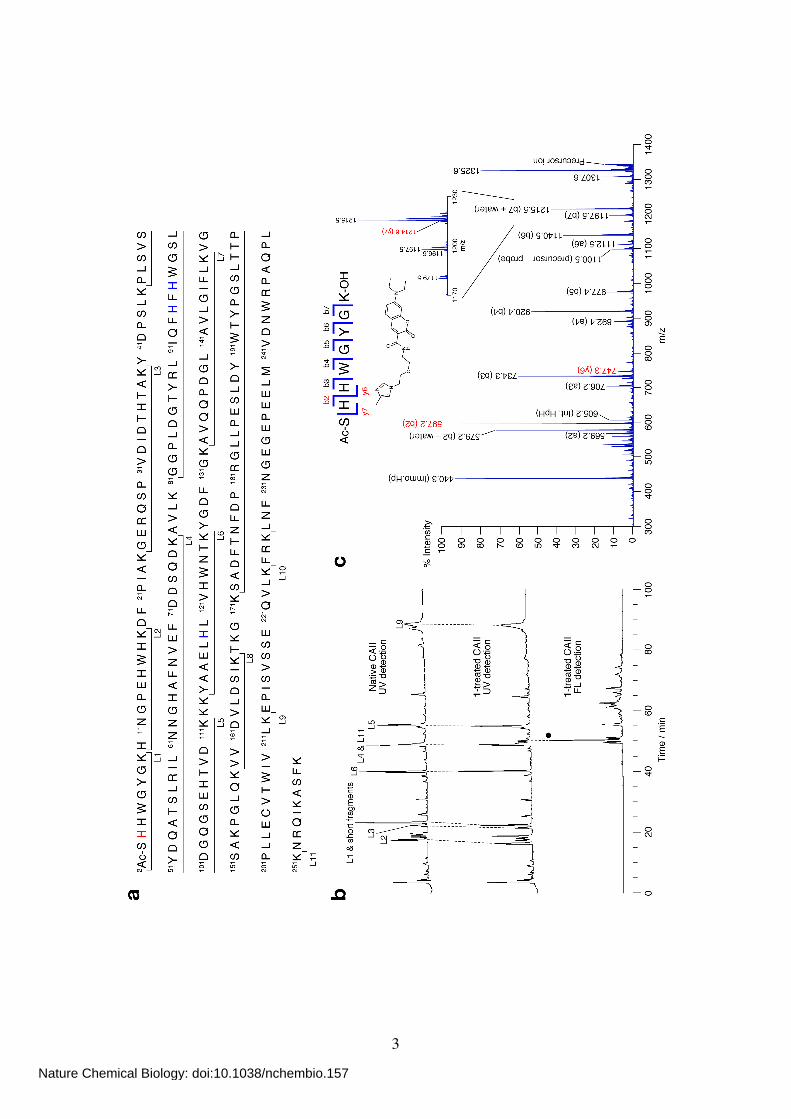
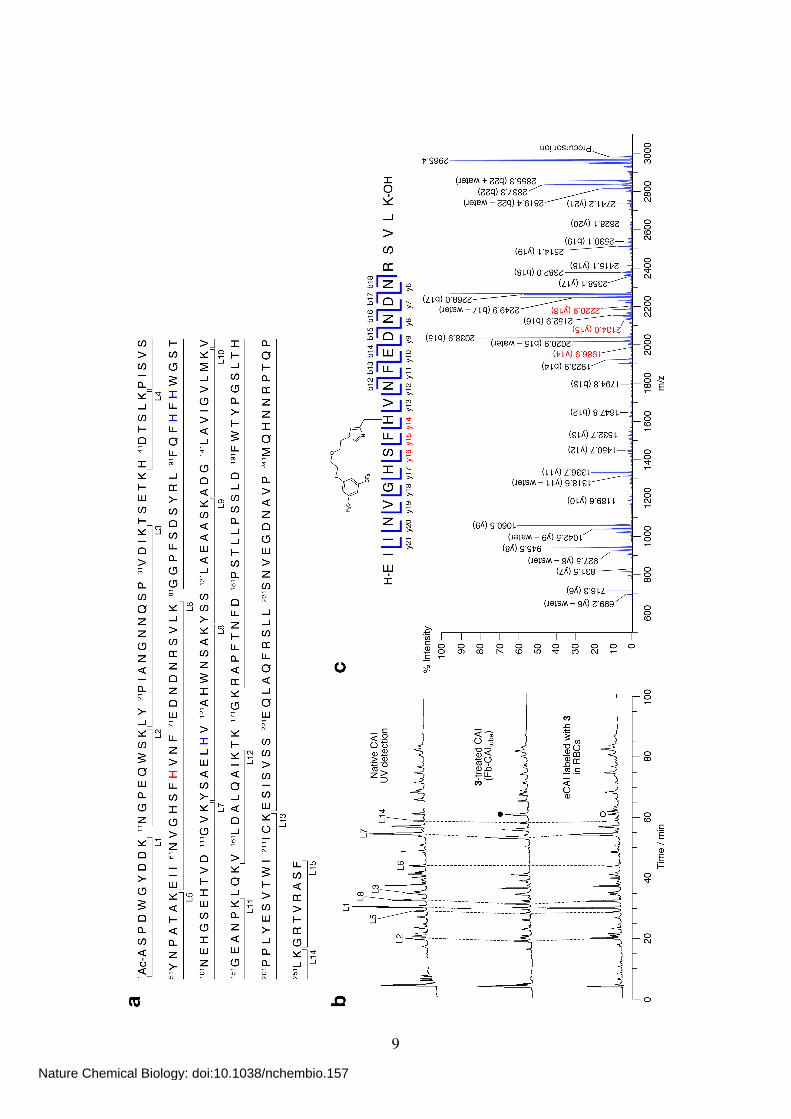
Supplementary Figure 1 (p.3) Identification of the labeling site of CAII treated with SA-Dc (1). (a) The primary sequence of human CAII and the assignment of each fragment generated by lysyl endopeptidase (LEP) digestion. The N-terminal Ser2 is
acetylated. His residues coordinated to a zinc ion (His94, 96 and 119) are shown in blue. His3 shown in red is the labeling site. (b) RP-HPLC traces of LEP-digested native CAII (top) and 1-treated CAII (middle and bottom). A gradient of 5–55% solvent A (see p.33)
over 100 min was used with UV detection at 220 nm (top and middle) and fluorescence detection at 473 nm (excitation at 427 nm, bottom). The peak denoted by , which corresponds to the labeled L1 fragment (the structure is shown in c), was characterized
by MALDI-TOF MS and MALDI-QIT-TOF MS/MS analysis. MALDI-TOF-MS (CHCA): calcd for [M+H]+ = 1343.62, obsd 1343.94. (c) MALDI-QIT-TOF MS/MS analysis of the labeled L1 fragment.
Nature Chemical Biology: doi:10.1038/nchembio.157
5
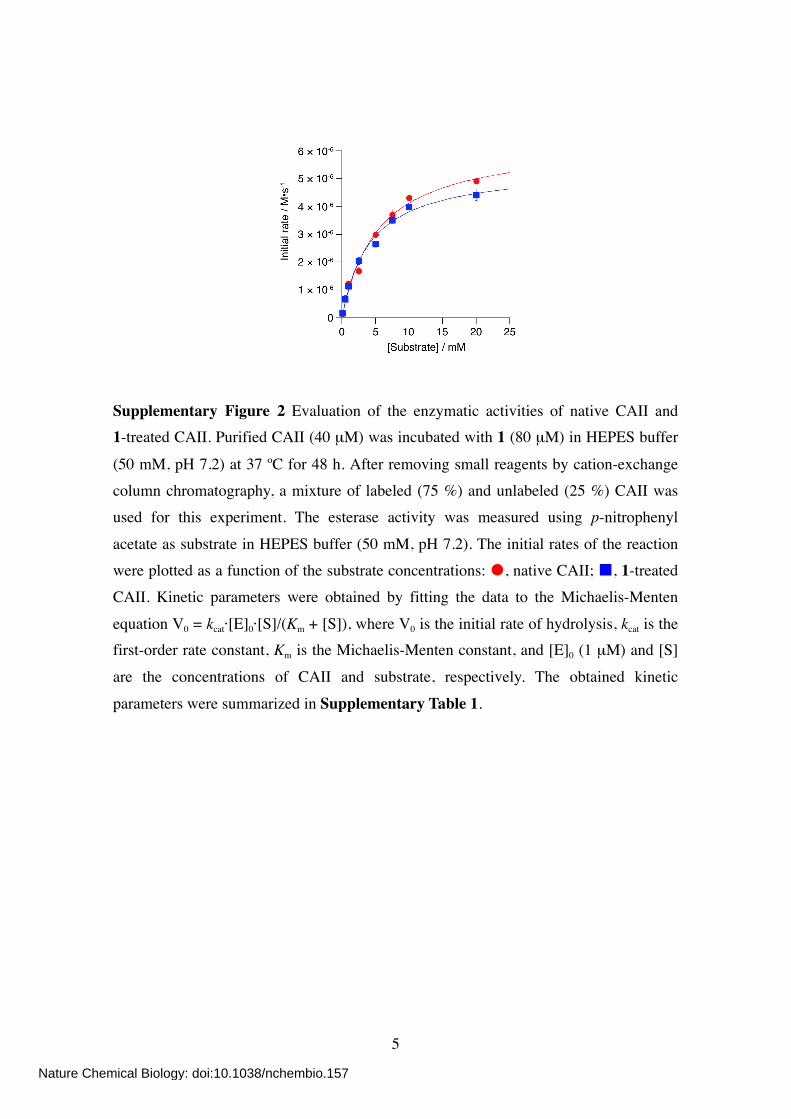
Supplementary Figure 2 Evaluation of the enzymatic activities of native CAII and 1-treated CAII. Purified CAII (40 µM) was incubated with 1 (80 µM) in HEPES buffer
(50 mM, pH 7.2) at 37 ºC for 48 h. After removing small reagents by cation-exchange column chromatography, a mixture of labeled (75 %) and unlabeled (25 %) CAII was used for this experiment. The esterase activity was measured using p-nitrophenyl
acetate as substrate in HEPES buffer (50 mM, pH 7.2). The initial rates of the reaction were plotted as a function of the substrate concentrations: , native CAII; , 1-treated CAII. Kinetic parameters were obtained by fitting the data to the Michaelis-Menten
equation V0 = kcat·[E]0·[S]/(Km + [S]), where V0 is the initial rate of hydrolysis, kcat is the first-order rate constant, Km is the Michaelis-Menten constant, and [E]0 (1 µM) and [S]
are the concentrations of CAII and substrate, respectively. The obtained kinetic
parameters were summarized in Supplementary Table 1.
Nature Chemical Biology: doi:10.1038/nchembio.157

6
Supplementary Figure 3 Western blotting analysis of eCA labeling in mice. After biotinylation detection using SAv-HRP (left), the membrane was washed and reprobed
with anti-mouse CAII antibody (right). The left image is same as that in Fig. 3d.
Nature Chemical Biology: doi:10.1038/nchembio.157
7
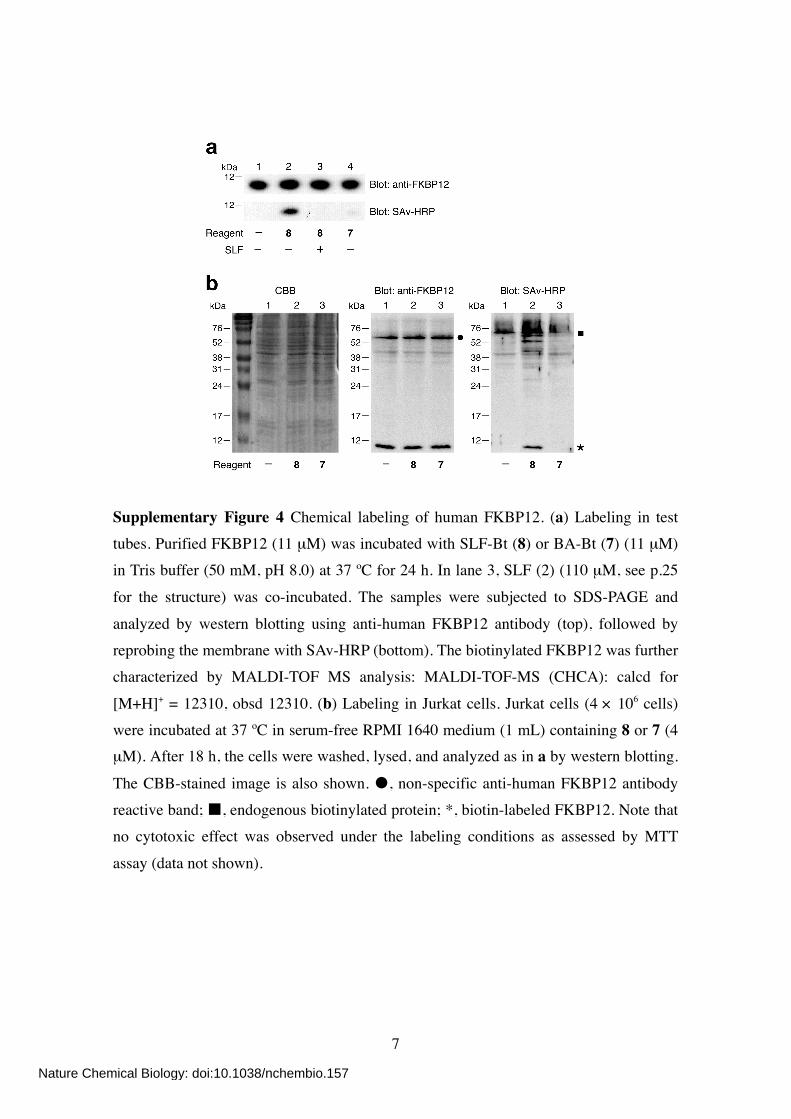
Supplementary Figure 4 Chemical labeling of human FKBP12. (a) Labeling in test tubes. Purified FKBP12 (11 µM) was incubated with SLF-Bt (8) or BA-Bt (7) (11 µM) in Tris buffer (50 mM, pH 8.0) at 37 ºC for 24 h. In lane 3, SLF (2) (110 µM, see p.25
for the structure) was co-incubated. The samples were subjected to SDS-PAGE and
analyzed by western blotting using anti-human FKBP12 antibody (top), followed by reprobing the membrane with SAv-HRP (bottom). The biotinylated FKBP12 was further characterized by MALDI-TOF MS analysis: MALDI-TOF-MS (CHCA): calcd for
[M+H]+ = 12310, obsd 12310. (b) Labeling in Jurkat cells. Jurkat cells (4 × 106 cells) were incubated at 37 ºC in serum-free RPMI 1640 medium (1 mL) containing 8 or 7 (4 µM). After 18 h, the cells were washed, lysed, and analyzed as in a by western blotting.
The CBB-stained image is also shown. , non-specific anti-human FKBP12 antibody reactive band; , endogenous biotinylated protein; *, biotin-labeled FKBP12. Note that no cytotoxic effect was observed under the labeling conditions as assessed by MTT
assay (data not shown).
Nature Chemical Biology: doi:10.1038/nchembio.157
8
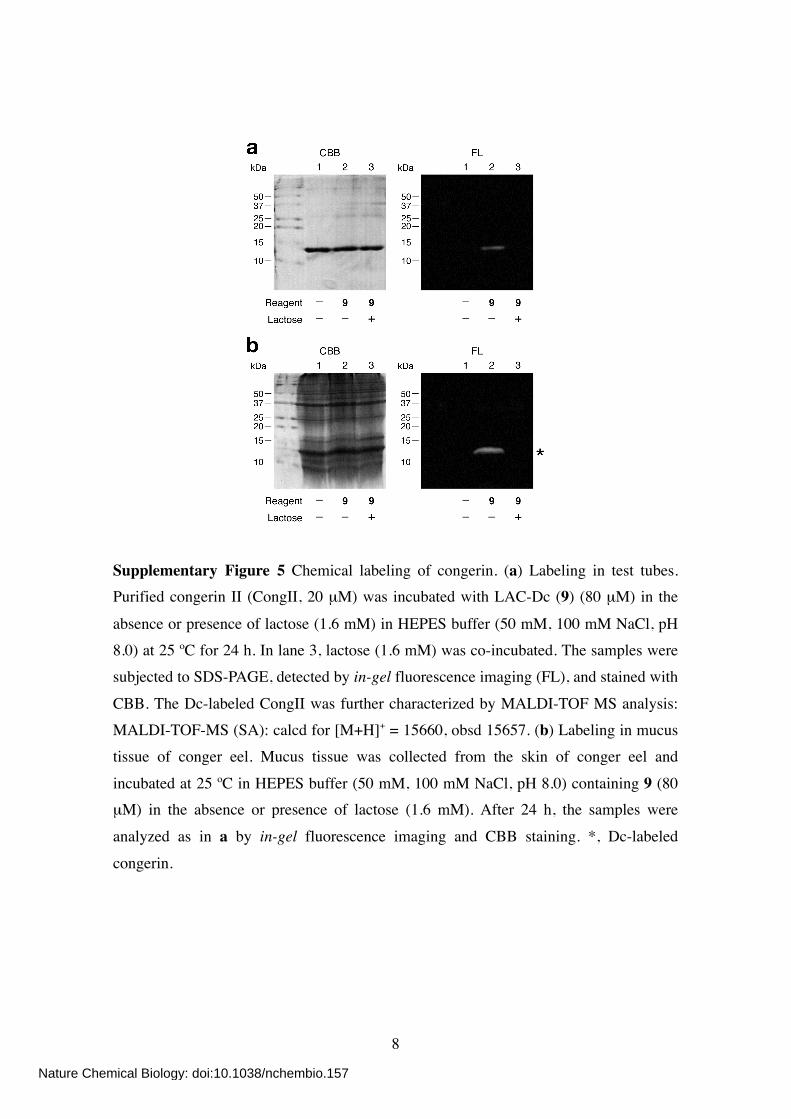
Supplementary Figure 5 Chemical labeling of congerin. (a) Labeling in test tubes. Purified congerin II (CongII, 20 µM) was incubated with LAC-Dc (9) (80 µM) in the
absence or presence of lactose (1.6 mM) in HEPES buffer (50 mM, 100 mM NaCl, pH 8.0) at 25 ºC for 24 h. In lane 3, lactose (1.6 mM) was co-incubated. The samples were subjected to SDS-PAGE, detected by in-gel fluorescence imaging (FL), and stained with
CBB. The Dc-labeled CongII was further characterized by MALDI-TOF MS analysis: MALDI-TOF-MS (SA): calcd for [M+H]+ = 15660, obsd 15657. (b) Labeling in mucus tissue of conger eel. Mucus tissue was collected from the skin of conger eel and
incubated at 25 ºC in HEPES buffer (50 mM, 100 mM NaCl, pH 8.0) containing 9 (80 µM) in the absence or presence of lactose (1.6 mM). After 24 h, the samples were
analyzed as in a by in-gel fluorescence imaging and CBB staining. *, Dc-labeled
congerin.
Nature Chemical Biology: doi:10.1038/nchembio.157
9
Nature Chemical Biology: doi:10.1038/nchembio.157

10
Supplementary Figure 6 (p.9) Identification of the labeling site of the Fb-installed CAI. (a) The primary sequence of human CAI and the assignment of each fragment generated by LEP digestion. The N-terminal Ala1 is acetylated. His residues coordinated to a zinc
ion (His94, 96 and 119) are shown in blue. His67 shown in red is the labeling site. (b) RP-HPLC traces of LEP-digests of native CAI (top), Fb-labeled CAI prepared in a test tube (Fb-CAItube, middle) and in RBCs (bottom). A gradient of 5–55% solvent A (see
p.33) over 100 min was used with UV detection at 220 nm. The peaks denoted by and , both of which correspond to the labeled L6 fragment (the structure is shown in c), were characterized by MALDI-TOF MS and MALDI-QIT-TOF MS/MS analysis.
MALDI-TOF-MS (CHCA) of peak : calcd for [M+H]+ = 2983.38, obsd 2983.35. (c) MALDI-QIT-TOF MS/MS analysis of the labeled L6 fragment . Note that MS analysis of the peak gave identical results as peak .
Nature Chemical Biology: doi:10.1038/nchembio.157

11
Supplementary Figure 7 In vitro NMR detection of EZA using Fb-CAItube. Fb-CAItube (20 µM) was incubated in HEPESDT containing various concentrations of EZA and
analyzed by 19F NMR. Peak , -62.0 p.p.m.; peak , -62.5 p.p.m..
Nature Chemical Biology: doi:10.1038/nchembio.157
12
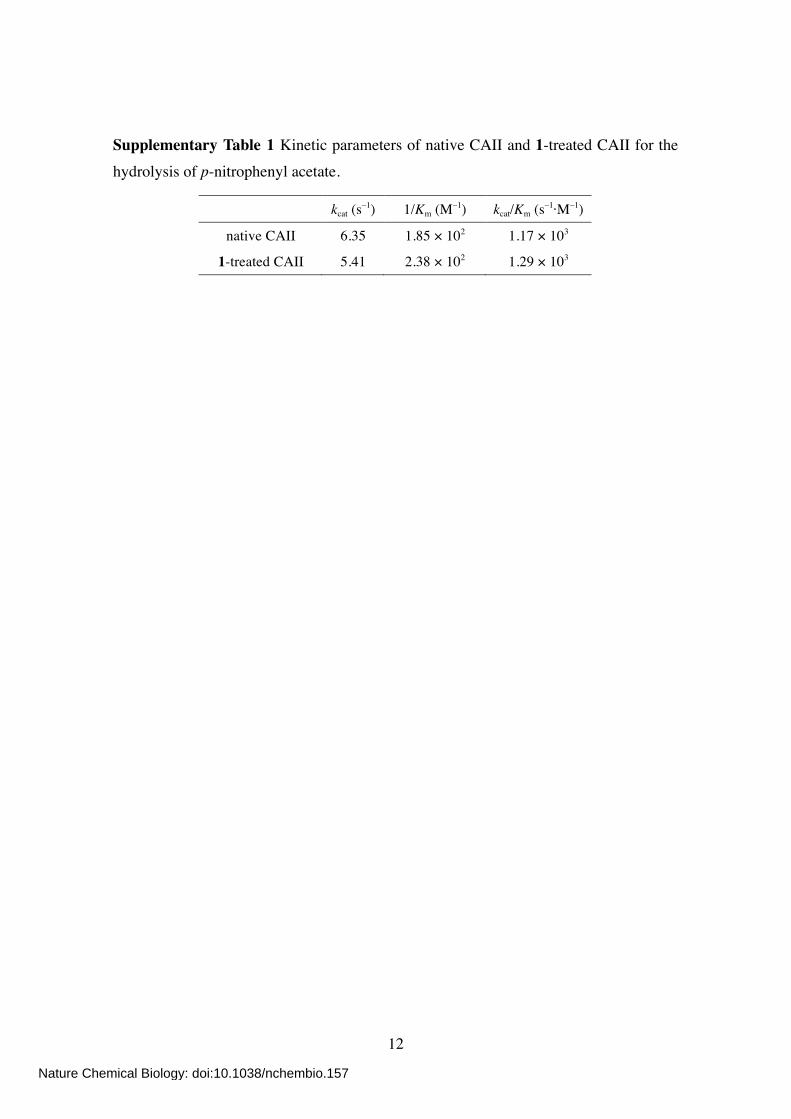
Supplementary Table 1 Kinetic parameters of native CAII and 1-treated CAII for the hydrolysis of p-nitrophenyl acetate.
kcat (s–1) 1/Km (M–1) kcat/Km (s–1·M–1)
native CAII 6.35 1.85 × 102 1.17 × 103
1-treated CAII 5.41 2.38 × 102 1.29 × 103
Nature Chemical Biology: doi:10.1038/nchembio.157

13
Supplementary Methods
General materials and methods for organic synthesis All chemical reagents and solvents were obtained from commercial suppliers (Aldrich, Tokyo Chemical Industry (TCI), Wako Pure Chemical Industries, Acros Organics, Sasaki Chemical, or Watanabe Chemical Industries) and used without further
purification. All reactions were carried out under an atmosphere of argon or nitrogen unless otherwise noted. Thin layer chromatography (TLC) was performed on silica gel 60 F254
precoated aluminum sheets (Merck) and visualized by fluorescence quenching or ninhydrin staining. Chromatographic purification was accomplished using flash column chromatography on silica gel 60 N (neutral, 40–50 µm, Kanto Chemical). 1H NMR
spectra of samples were recorded in deuterated solvents on Varian Mercury 400 (400 MHz) or JEOL JNM-A500 (500 MHz) spectrometers and calibrated to the residual solvent peak or tetramethylsilane (= 0 p.p.m.). Multiplicities are abbreviated as follows:
s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, dd = double doublet, dt = double triplet, br = broad. 13C NMR spectra were recorded in deuterated solvents on JEOL JNM-A400 (100 MHz) or JEOL ECA-600 (150 MHz) spectrometers and
calibrated to the residual solvent peak by Haruo Fujita (Department of Synthetic Chemistry and Biological Chemistry, Graduate School of Engineering, Kyoto University). 19F NMR spectra were recorded on a JEOL EX-400 (376.5 MHz)
spectrometer and calibrated to trifluoroacetic acid (TFA, = –75.6 p.p.m). Matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS) spectra were recorded on an Applied Biosystems Voyager Elite mass spectrometer using α-cyano-4-hydroxycinnamic acid (CHCA) as the matrix. High-resolution fast atomic
bombardment mass spectrometry (HR-FAB MS) spectra and high-resolution electrospray ionization quadrupole fourier transform mass spectrometry (HR-ESI
Qq-LTMS) spectra were acquired on a JEOL JMS-HX110A mass spectrometer with 3-nitrobenzyl alcohol (NBA) as the matrix and on a Bruker apex-ultra (7T) mass spectrometer, respectively, by Dr. Keiko Kuwata (Department of Synthetic Chemistry
and Biological Chemistry, Graduate School of Engineering, Kyoto University). Elementary analysis was carried out using a Yanaco MT-6 CHN corder at the Center for Organic Elemental Microanalysis of Kyoto University.
Nature Chemical Biology: doi:10.1038/nchembio.157
14
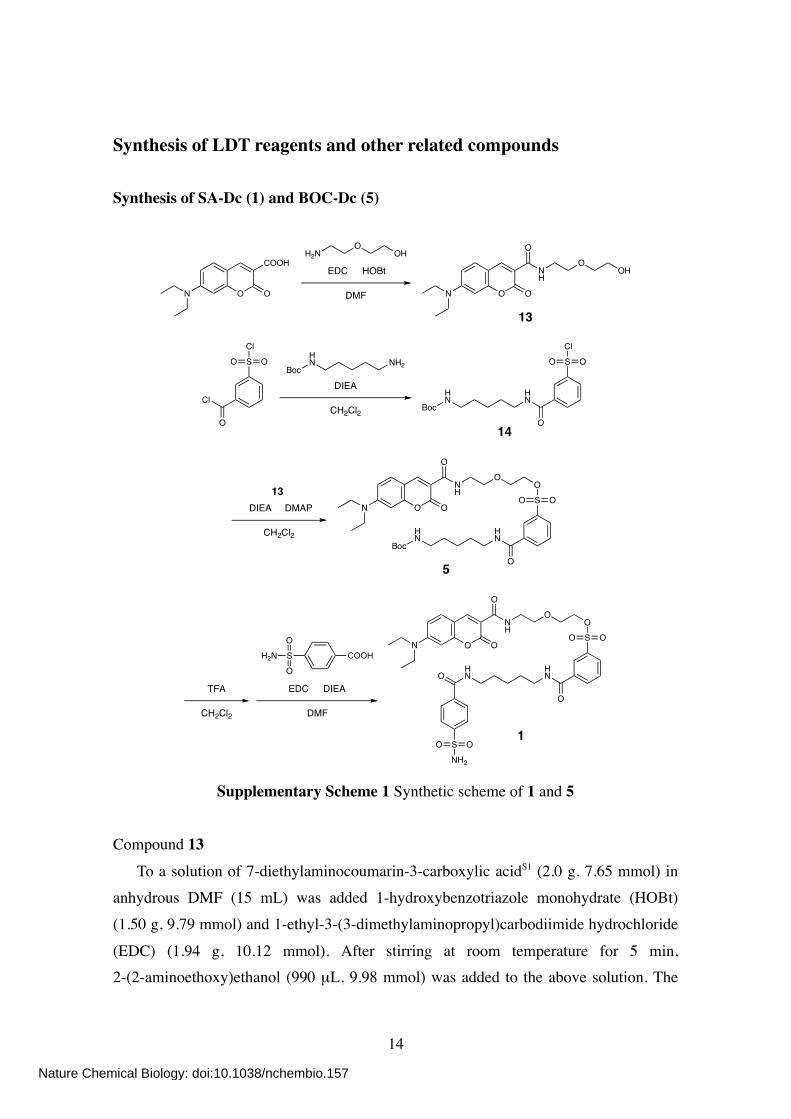
Synthesis of LDT reagents and other related compounds Synthesis of SA-Dc (1) and BOC-Dc (5)
N O O
COOHH2N
OOH
EDC HOBt
DMF
NH
O
O ON
OOH
13
Cl
O
SCl
OOBoc
HN NH2
DIEA
CH2Cl2 BocHN
HN
O
SCl
OO
14
BocHN
HN
O
SO
OO
ONH
O
O ONDIEA DMAP
CH2Cl2
13
5
SNH2
OO
HN
HNO
O
SO
OO
ONH
O
O ONSH2NO
O
EDC DIEA
DMF
TFA
CH2Cl2
COOH
1
Supplementary Scheme 1 Synthetic scheme of 1 and 5 Compound 13
To a solution of 7-diethylaminocoumarin-3-carboxylic acidS1 (2.0 g, 7.65 mmol) in anhydrous DMF (15 mL) was added 1-hydroxybenzotriazole monohydrate (HOBt) (1.50 g, 9.79 mmol) and 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride
(EDC) (1.94 g, 10.12 mmol). After stirring at room temperature for 5 min, 2-(2-aminoethoxy)ethanol (990 µL, 9.98 mmol) was added to the above solution. The
Nature Chemical Biology: doi:10.1038/nchembio.157

15
mixture was stirred at room temperature overnight. After evaporation, the residue was dissolved in CHCl3 and washed three times with saturated aqueous NaHCO3. The organic layer was dried over anhydrous MgSO4, filtered, and concentrated. The
resulting residue was triturated with Et2O to give compound 13 as yellow solid (2.35 g, 88%). 1H NMR (500 MHz; CDCl3): δ 9.11 (br s, 1H), 8.70 (s, 1H), 7.43 (d, 1H, J = 10.0 Hz),
6.65 (dd, 1H, J = 10.0, 5.0 Hz), 6.50 (d, 1H, J = 5.0 Hz), 3.77 (m, 2H), 3.69–3.63 (m, 6H), 3.46 (q, 4H, J = 5.0 Hz), 1.24 (t, 6H, J = 5.0 Hz).
Compound 14 To a solution of 3-(chlorosulfonyl)benzoyl chloride (3.0 mL, 18.82 mmol) in anhydrous CH2Cl2 (10 mL) was added dropwisely a solution of
N-(tert-butoxycarbonyl)-1,5-diaminopentane (1.88 g, 9.29 mmol) and N,N-diisopropylethylamine (DIEA) (3.93 mL, 23.11 mmol) in anhydrous CH2Cl2 (8 mL) at 4 ºC. The mixture was stirred at 4 ºC for 15 min and at room temperature for 1 h.
After evaporation, the crude residue was purified by column chromatography on silica gel (CHCl3) to give compound 14 as pale yellow sticky solid (3.11 g, 83%). 1H NMR (500 MHz; CDCl3): δ 8.43 (s, 1H), 8.22 (d, 1H, J = 7.9 Hz), 8.15 (d, 1H, J =
8.2 Hz), 7.71 (dd, 1H, J = 7.9, 7.9 Hz), 6.83 (br s, 1H), 4.66 (br s, 1H), 3.49 (m, 2H), 3.13 (m, 2H), 1.68 (m, 2H), 1.54 (m, 2H), 1.40 (m + s, 2 + 9H).
Compound 5 To a solution of 14 (3.0 g, 7.41 mmol) in anhydrous CH2Cl2 (10 mL) was added 13 (1.29 g, 3.70 mmol), DIEA (3.13 mL, 18.41 mmol), and 4-dimethylaminopyridine
(DMAP) (45 mg, 0.37 mmol). The mixture was stirred at room temperature for 6 h. After evaporation, the crude residue was purified by column chromatography on silica gel (CHCl3/MeOH, 100:1) to give compound 5 as yellow solid (1.60 g, 60%). 1H NMR (500 MHz; CDCl3): δ 8.91 (br s, 1H), 8.68 (s, 1H), 8.40 (s, 1H), 8.19 (d, 1H, J
= 7.9 Hz), 8.04 (d, 1H, J = 7.6 Hz), 7.62 (dd, 1H, J = 7.9, 7.9 Hz), 7.44 (d, 1H, J = 9.2 Hz), 6.66 (dd, 1H, J = 8.9, 2.5 Hz), 6.48 (d, 1H, J = 2.1 Hz), 4.59 (br s, 1H), 4.23 (m,
2H), 3.68 (m, 2H), 3.55–3.44 (m, 10 H), 3.09 (m, 2H), 1.66 (m, 4H), 1.51 (m, 2H), 1.41 (s, 9H), 1.25 (t, 6H, J = 7.2 Hz). 13C NMR (100 MHz; CDCl3): δ 165.68, 163.44, 162.92, 157.73, 156.11, 152.75, 148.82,
Nature Chemical Biology: doi:10.1038/nchembio.157

16
148.33, 136.40, 136.24, 133.26, 131.30, 130.49, 129.51, 126.11, 110.17, 110.08, 108.44, 96.55, 69.95, 69.72, 68.37, 45.17, 40.39, 40.17, 39.26, 29.80, 29.29, 28.47, 24.18, 12.51.
HR-FAB MS (NBA): calcd for C35H48N4O10S [M]+ = 716.3091; obsd 716.3083. Compound 1
To a solution of 5 (205 mg, 0.29 mmol) in CH2Cl2 (8 mL) was added trifluoroacetic acid (TFA) (1.6 mL). The mixture was stirred at room temperature for 30 min. After co-evaporation twice with toluene (8 mL), the residue was dissolved in anhydrous DMF
(2 mL). To a solution of 4-sulfamoylbenzoic acid (86 mg, 0.43 mmol) in anhydrous DMF (2 mL) was added DIEA (241 µL, 1.42 mmol) and EDC (108 mg, 0.56 mmol). The
mixture was stirred at room temperature for 5 min. To this was added the above solution. The mixture was then stirred at room temperature for 3 h. After evaporation, the residue was dissolved in EtOAc and washed twice with saturated aqueous NaHCO3 and once
with brine. The organic layer was dried over anhydrous MgSO4, filtered, and evaporated. The crude residue was purified by column chromatography on silica gel (CHCl3/MeOH, 25:1). The collected fraction was concentrated and triturated with Et2O to give
compound 1 as yellow solid (60 mg, 26%). 1H NMR (500 MHz; CDCl3): δ 8.87 (br t, 1H, J = 5.3 Hz), 8.59 (s, 1H), 8.34 (s, 1H),
8.05 (d, 1H, J = 7.9 Hz), 7.97 (d, 1H, J = 8.0 Hz), 7.75 (s, 4H), 7.54 (dd, 1H, J = 7.9,
7.9 Hz), 7.39 (d, 1H, J = 9.2 Hz), 6.64 (dd, 1H, J = 9.2, 2.5 Hz), 6.45 (d, 1H, J = 2.5 Hz), 5.99 (s, 2H), 4.19 (m, 2H), 3.63 (m, 2H), 3.49–3.39 (m, 12H), 1.65 (m, 4H), 1.42 (m, 2H), 1.23 (t, 6H, J = 7.2 Hz). 13C NMR (100 MHz; CDCl3): δ 166.67, 166.19, 163.63, 162.93, 157.72, 152.91, 148.39,
144.84, 138.24, 136.30, 136.06, 132.98, 131.41, 130.51, 129.64, 127.88, 126.36, 110.31, 109.59, 108.38, 96.52, 70.07, 69.81, 68.35, 45.21, 39.94, 39.72, 39.30, 29.03, 28.46,
22.95, 12.52. HR-FAB MS (NBA): calcd for C37H45N5O11S2 [M]+ = 799.2557; obsd 799.2559. Elementary Analysis: calcd % for C37H45N5O11S2•H2O; C, 54.33; H, 5.79; N, 8.56. obsd;
C, 54.63; H, 5.71; N, 8.35.
Nature Chemical Biology: doi:10.1038/nchembio.157
17
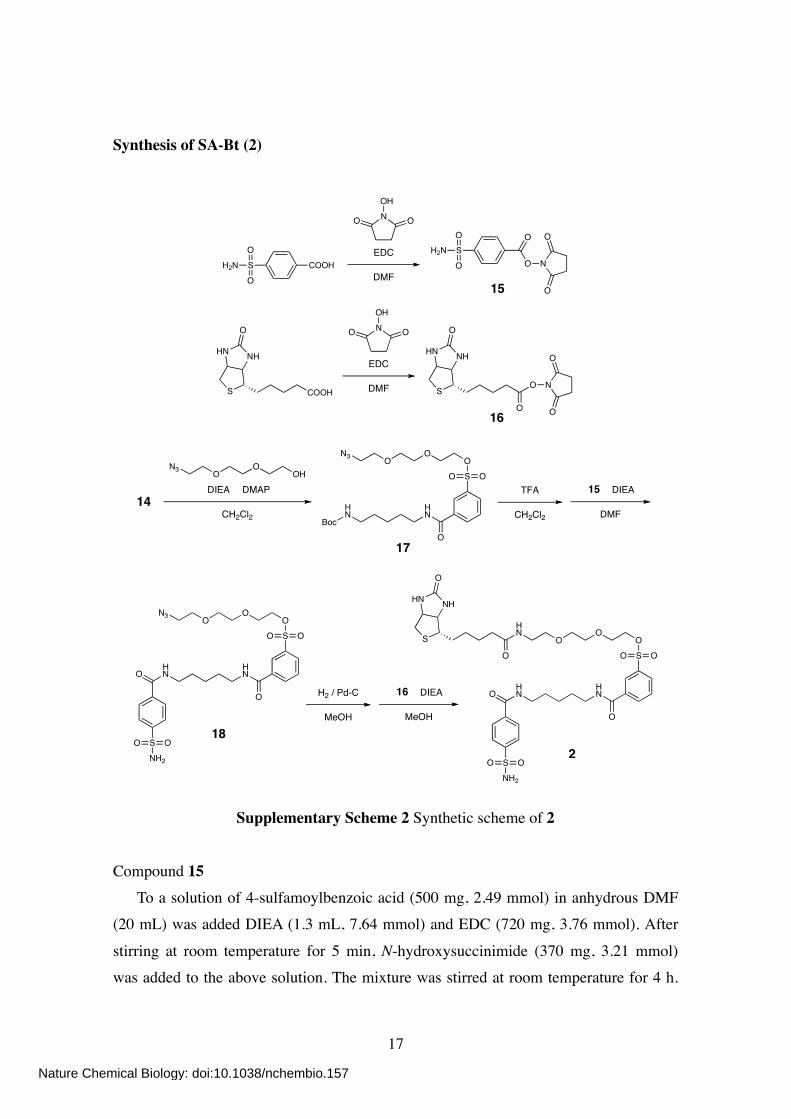
Synthesis of SA-Bt (2)
15
EDC
DMF
NOH
OO
SH2N
O
OCOOH
O
S
NHHN
O
O N
O
O
EDC
DMF
NOH
OO
16
BocHN
HN
O
SO
OO
OO
N3
HN
HN
O
SO
OO
OO
N3
O
SNH2
OO
SNH2
OO
HN
HNO
O
SO
OO
OO
HN
O
S
NHHN
O
14DIEA DMAP
CH2Cl2
OHO
ON3
17
TFA
CH2Cl2
DIEA
DMF
15
18
H2 / Pd-C
MeOH
DIEA
MeOH
16
2
SH2N
O
O O
O N
O
O
COOHS
NHHN
O
Supplementary Scheme 2 Synthetic scheme of 2
Compound 15 To a solution of 4-sulfamoylbenzoic acid (500 mg, 2.49 mmol) in anhydrous DMF (20 mL) was added DIEA (1.3 mL, 7.64 mmol) and EDC (720 mg, 3.76 mmol). After
stirring at room temperature for 5 min, N-hydroxysuccinimide (370 mg, 3.21 mmol) was added to the above solution. The mixture was stirred at room temperature for 4 h.
Nature Chemical Biology: doi:10.1038/nchembio.157

18
After evaporation, the residue was dissolved in EtOAc and washed three times with saturated aqueous NaHCO3 and once with brine. The organic layer was dried over anhydrous MgSO4, filtered, and evaporated to give compound 15 as white solid (650
mg, 88%). 1H NMR (400 MHz; CD3OD): δ 8.28 (d, 2H, J = 8.8 Hz), 8.10 (d, 2H, J = 8.8 Hz), 2.92
(s, 4H).
Compound 16 To a suspension of biotin (1.18 g, 4.83 mmol) in anhydrous DMF (8 mL) was added
N-hydroxysuccinimide (667 mg, 5.80 mmol) and EDC (1.11 g, 5.79 mmol). The mixture was stirred at room temperature for 10 h. After evaporation, the residue was purified by recrystallization from EtOH/AcOH/H2O (95:1:4). White crystals were
filtered and dried under vacuum, affording 16 as white solid (1.30 g, 79%). 1H NMR (400 MHz; CDCl3/CD3OD, 1:1): δ 4.49 (m, 1H), 4.31 (dd, 1H, J = 7.6, 4.4 Hz),
3.17 (m, 1H), 2.91 (dd, 1H, J = 12.8, 5.2 Hz), 2.84 (s, 4H), 2.71 (d, 1H, J = 12.8 Hz),
2.63 (dt, 2H, J = 7.2, 2.0 Hz), 1.83–1.51 (m, 6H). Compound 17
To a solution of 14 (900 mg, 2.22 mmol) in anhydrous CH2Cl2 (5 mL) was added 2-(2-(2-azidoethoxy)ethoxy)ethanolS2 (300 mg, 1.71 mmol), DIEA (872 µL, 5.13 mmol),
and DMAP (63 mg, 0.52 mmol). The mixture was stirred at room temperature overnight.
After evaporation, the crude residue was purified by column chromatography on silica gel (CHCl3/MeOH, 30:1) to give compound 17 as colorless oil (570 mg, 61%). 1H NMR (400 MHz; CDCl3): δ 8.26 (s, 1H), 8.12 (d, 1H, J = 7.6 Hz), 8.04 (d, 1H, J =
8.0 Hz), 7.64 (dd, 1H, J = 8.0, 7.6 Hz), 6.50 (br s, 1H), 4.59 (br s, 1H), 4.23 (m, 2H), 3.71 (m, 4H), 3.63 (t, 2H, J = 5.0 Hz), 3.59 (s, 4H), 3.47 (m, 2H), 3.36 (t, 2H, J = 5.0 Hz), 3.13 (m, 2H), 1.66 (m, 4H), 1.53 (m, 2H), 1.41 (s, 9H).
Compound 18 To a solution of 17 (420 mg, 0.77 mmol) in CH2Cl2 (10 mL) was added TFA (4 mL).
The mixture was stirred at room temperature for 15 min. After co-evaporation twice with toluene (4 mL), the residue was dissolved in anhydrous DMF (2 mL). To this was added 15 (299 mg, 1.00 mmol) and DIEA (390 µL, 2.29 mmol). The mixture was then
Nature Chemical Biology: doi:10.1038/nchembio.157

19
stirred at room temperature for 3 h. After evaporation, the crude residue was purified by column chromatography on silica gel (CHCl3/MeOH, 20:1 to 10:1). The collected fraction was evaporated, dissolved in EtOAc, and washed twice with saturated aqueous
NaHCO3 and once with brine. The organic layer was dried over anhydrous MgSO4, filtered, and evaporated to give compound 18 as white solid (180 mg, 37%). 1H NMR (400 MHz; CD3OD): δ 8.35 (s, 1H), 8.13 (d, 1H, J = 8.0 Hz), 8.07 (d, 1H, J =
8.0 Hz), 7.95 (d, 2H, J = 8.4 Hz), 7.92 (d, 2H, J = 8.8 Hz), 7.71 (dd, 1H, J = 8.0, 8.0 Hz), 4.22 (m, 2H), 3.67 (m, 2H), 3.60 (t, 2H, J = 5.0 Hz), 3.54 (s, 4H), 3.42 (m, 4H), 3.31 (m, 2H), 1.70 (m, 4H), 1.48 (m, 2H).
Compound 2 To a solution of 18 (6.5 mg, 10.4 µmol) in MeOH (2 mL) was added palladium
carbon (Pd-C) (10%, 10 mg). The mixture was stirred at room temperature under H2 atmosphere for 1 h. After changing the atmosphere to N2, 16 (4.5 mg, 13.2 µmol) and DIEA (3.6 µL, 21.2 µmol) were added to the solution. The mixture was then stirred at
room temperature for 1 h. After filtration, the filtrate was evaporated and purified by column chromatography on silica gel (CHCl3/MeOH, 5:1) to give compound 2 as colorless oil (6.4 mg, 75%). 1H NMR (400 MHz; CDCl3/CD3OD, 1:1): δ 8.35 (s, 1H), 8.11 (d, 1H, J = 7.6 Hz), 8.02
(d, 1H, J = 8.0 Hz), 7.92 (d, 2H, J = 8.4 Hz), 7.89 (d, 2H, J = 8.0 Hz), 7.65 (dd, 1H, J = 8.0, 7.6 Hz), 4.47 (m, 1H), 4.28 (dd, 1H, J = 7.8, 4.6 Hz), 4.21 (m, 2H), 3.68 (m, 2H),
3.62–3.48 (m, 8H) 3.42–3.33 (m, 4H), 3.14 (m, 1H), 2.88 (dd, 1H, J = 12.8, 4.4 Hz), 2.69 (d, 1H, J = 12.8 Hz), 2.18 (t, 2H, J = 7.2 Hz), 1.69–1.56 (m, 8H), 1.47–1.38 (m, 4H). 13C NMR (100 MHz; CD3OD): δ 176.41, 168.78, 167.87, 166.09, 147.61, 139.19,
138.11, 137.23, 133.59, 131.63, 131.00, 128.94, 127.81, 127.32, 71.56, 71.52, 71.18, 70.61, 69.74, 63.36, 61.62, 56.99, 41.05, 41.04, 40.96, 40.33, 36.75, 30.07, 29.72, 29.47,
26.84, 25.36. HR-FAB MS (NBA): calcd for C35H50N6O11S3 [M]+ = 826.2700; obsd 826.2672.
Nature Chemical Biology: doi:10.1038/nchembio.157
20
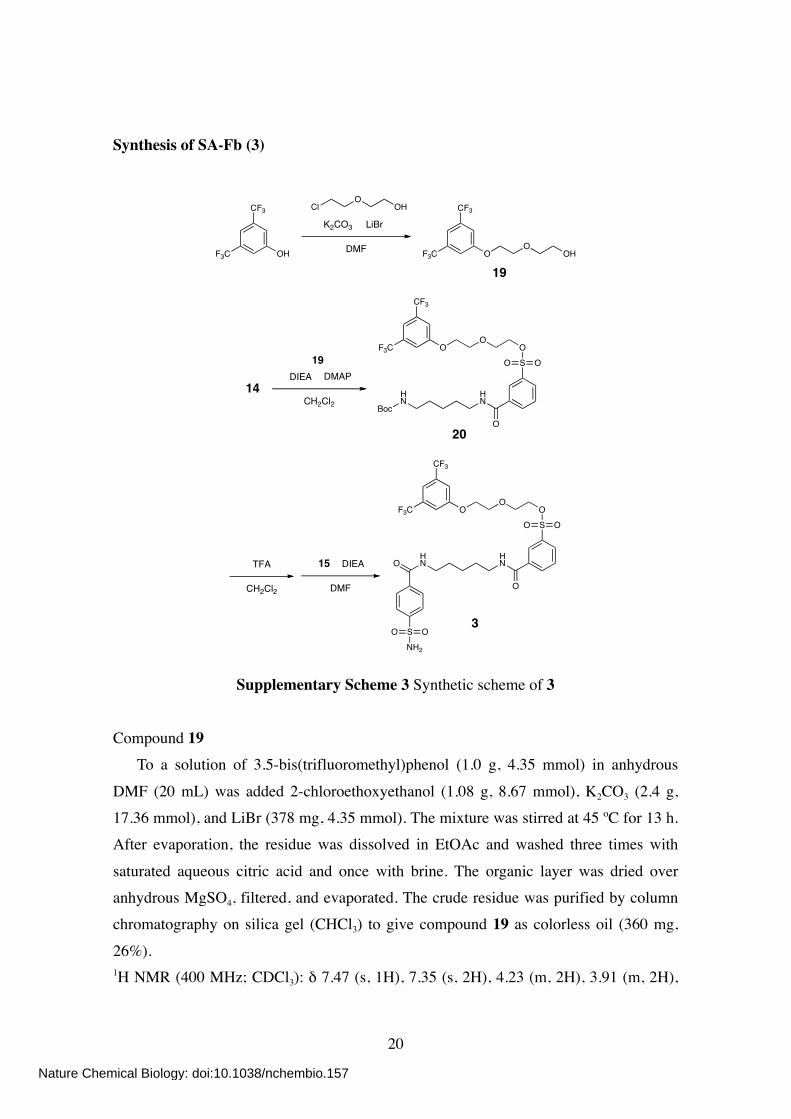
Synthesis of SA-Fb (3)
SNH2
OO
HN
HNO
O
SO
OO
OO
CF3
F3C
BocHN
HN
O
SO
OO
OO
CF3
F3C
OH
CF3
F3C
ClO
OH
K2CO3 LiBr
DMF OHO
O
CF3
F3C
19
14DMAP
20
DIEA
CH2Cl2
19
TFA
CH2Cl2
DIEA
DMF
15
3
Supplementary Scheme 3 Synthetic scheme of 3
Compound 19 To a solution of 3.5-bis(trifluoromethyl)phenol (1.0 g, 4.35 mmol) in anhydrous
DMF (20 mL) was added 2-chloroethoxyethanol (1.08 g, 8.67 mmol), K2CO3 (2.4 g, 17.36 mmol), and LiBr (378 mg, 4.35 mmol). The mixture was stirred at 45 ºC for 13 h. After evaporation, the residue was dissolved in EtOAc and washed three times with
saturated aqueous citric acid and once with brine. The organic layer was dried over anhydrous MgSO4, filtered, and evaporated. The crude residue was purified by column chromatography on silica gel (CHCl3) to give compound 19 as colorless oil (360 mg,
26%). 1H NMR (400 MHz; CDCl3): δ 7.47 (s, 1H), 7.35 (s, 2H), 4.23 (m, 2H), 3.91 (m, 2H),
Nature Chemical Biology: doi:10.1038/nchembio.157

21
3.77 (m, 2H), 3.68 (m, 2H), 2.07 (t, J = 6.0 Hz). Compound 20
To a solution of 14 (115 mg, 0.28 mmol) in anhydrous CH2Cl2 (2 mL) was added 19 (60 mg, 0.19 mmol), DIEA (99 µL, 0.58 mmol), and DMAP (12 mg, 0.10 mmol). The
mixture was then stirred at room temperature for 3 h. After evaporation, the crude
residue was purified by column chromatography on silica gel (CHCl3/MeOH, 30:1) to give compound 20 as colorless oil (98 mg, 76%). 1H NMR (400 MHz; CDCl3): δ 8.28 (s, 1H), 8.07 (d, 1H, J = 8.0 Hz), 8.03 (d, 1H, J =
8.0 Hz), 7.61 (dd, 1H, J = 8.4, 7.6 Hz), 7.47 (s, 1H), 7.31 (s, 2H), 6.42 (br s, 1H), 4.58 (br s, 1H), 4.27 (m, 2H), 4.15 (m, 2H), 3.83 (m, 2H), 3.79 (m, 2H), 3.46 (m, 2H), 3.14 (m, 2H), 1.70–1.48 (m, 4H), 1.41 (m + s, 2 + 9H).
Compound 3 To a solution of 20 (95 mg, 0.14 mmol) in CH2Cl2 (3 mL) was added TFA (1 mL).
The mixture was stirred at room temperature for 1.5 h. After co-evaporation twice with toluene (3 mL), the residue was dissolved in anhydrous DMF (4 mL). To this was added 15 (63 mg, 0.21 mmol) and DIEA (73 µL, 0.43 mmol). The mixture was then stirred at
room temperature for 3 h. After evaporation, the residue was dissolved in EtOAc and washed three times with saturated aqueous NaHCO3 and brine. The organic layer was dried over anhydrous MgSO4, filtered, and evaporated. The crude residue was purified
by column chromatography on silica gel (CHCl3/MeOH, 25:1 to 10:1) to give compound 3 as white solid (73 mg, 69%). 1H NMR (400 MHz; CD3OD): δ 8.35 (s, 1H), 8.09 (d, 1H, J = 7.6 Hz), 8.05 (d, 1H, J =
7.6 Hz), 7.96 (d, 2H, J = 8.8 Hz), 7.92 (d, 2H, J = 8.8 Hz), 7.66 (dd, 1H, J = 7.6, 7.6 Hz), 7.51 (s, 1H), 7.47 (s, 2H), 4.26 (m, 2H), 4.18 (m, 2H), 3.78 (m, 2H), 3.74 (m, 2H), 3.40 (m, 4H), 1.69 (m, 4H), 1.47 (m, 2H). 13C NMR (100 MHz; CD3OD): δ 168.78, 167.85, 161.22, 147.60, 139.18, 138.16,
137.20, 134.42 (q), 133.48, 133.42, 131.55, 130.86, 128.91, 127.81, 127.30, 116.41, 115.04, 71.47, 70.47, 69.92, 69.60, 40.99, 40.93, 30.04, 30.03, 25.34. 19F NMR (376 MHz; CD3OD): δ –62.8.
HR-FAB MS (NBA): calcd for C31H33F6N3O9S2 [M]+ = 769.1562; obsd 769.1549.
Nature Chemical Biology: doi:10.1038/nchembio.157
22
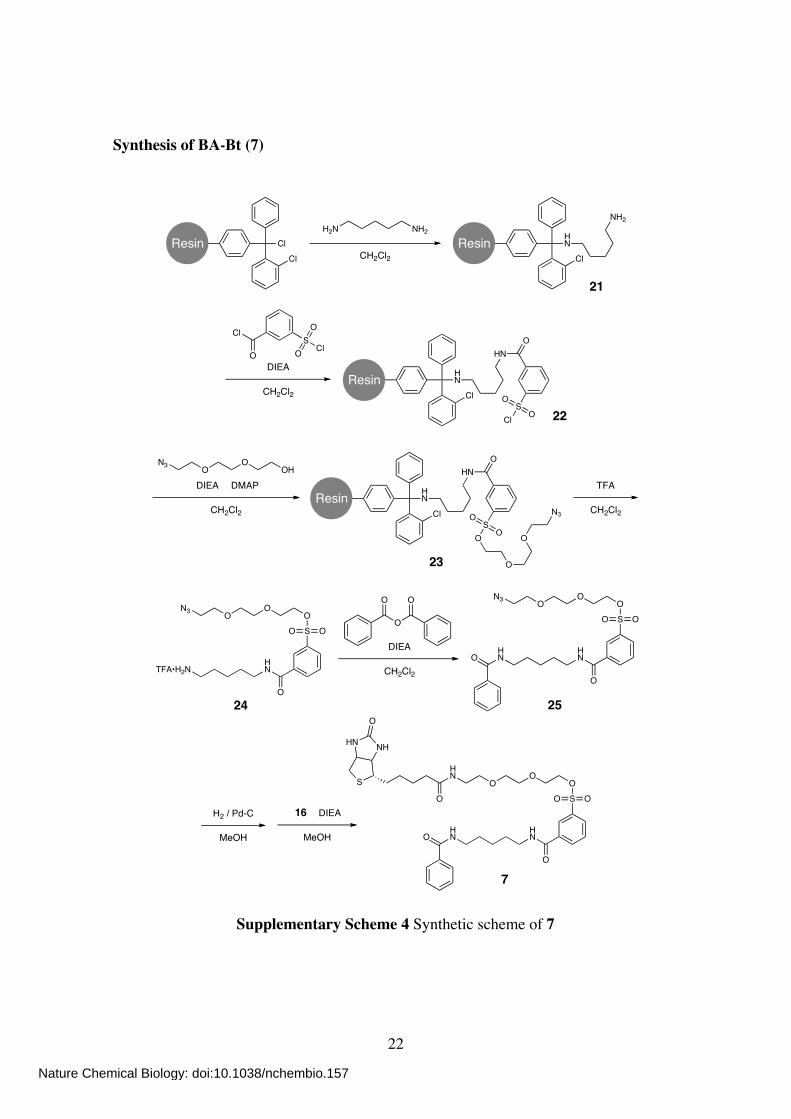
Synthesis of BA-Bt (7)
ClResinCl
NH2H2N
CH2Cl2
HNResin
Cl
NH2
HNResin
Cl
HNO
SCl
O
O
HNResin
Cl
HNO
SO
O
O
O
O
N3
TFA•H2NHN
O
SO
OO
OO
N3
HN
HN
O
SO
OO
OO
N3
O
DIEA
CH2Cl2
SCl
O
O
Cl
O
DIEA DMAP
CH2Cl2
OHO
ON3
TFA
CH2Cl2
DIEA
CH2Cl2
O
O O
21
HN
HNO
O
SO
OO
OO
HN
O
S
NHHN
O
22
H2 / Pd-C
MeOH
DIEA
MeOH
16
23
24 25
7
Supplementary Scheme 4 Synthetic scheme of 7
Nature Chemical Biology: doi:10.1038/nchembio.157

23
Solid-Phase synthesis Step 1: To a suspension of Cl-Trt(2-Cl)-resin (350 mg, 0.52 mmol) in CH2Cl2 (2 mL) was added 1,5-diaminopentane (2.0 mL, 17.03 mmol). The mixture was shaken at room
temperature overnight. The resin was filtered and washed with CH2Cl2 to give resin 21. Step 2: To a suspension of the resin 21 in anhydrous CH2Cl2 (4 mL) was added 3-(chlorosulfonyl)benzoyl chloride (249 µL, 1.56 mmol) and DIEA (438 µL, 2.58
mmol). The mixture was shaken at room temperature for 30 min. The resin was filtered and washed with CH2Cl2 to give resin 22. Step 3: To a suspension of the resin 22 in anhydrous CH2Cl2 (4 mL) was added 2-(2-(2-azidoethoxy)ethoxy)ethanolS2 (274 mg, 1.56 mmol), DIEA (438 µL, 2.58 mmol),
and DMAP (19 mg, 0.16 mmol). The mixture was shaken at room temperature overnight. The resin was filtered and washed with CH2Cl2 to give resin 23.
Step 4: To a suspension of the resin 23 in CH2Cl2 (3 mL) was added TFA (1 mL). The mixture was shaken at room temperature for 15 min. After filtration, the filtrate was co-evaporated with toluene (5 mL) and dried under vacuum, affording crude 24 as
colorless oil (90 mg). Compound 25 To a solution of crude 24 (45 mg, 83.3 µmol) in CH2Cl2 (2 mL) was added benzoic anhydride (23 mg, 101.7 µmol) and DIEA (69 µL, 405.8 µmol). The mixture was stirred
at room temperature for 30 min. After evaporation, the crude residue was purified by
column chromatography on silica gel (CHCl3/MeOH, 30:1) to give compound 25 as colorless oil (14 mg, 31%). 1H NMR (400 MHz; CDCl3): δ 8.30 (s, 1H), 8.10 (d, 1H, J = 8.0 Hz), 8.00 (d, 1H, J =
8.0 Hz), 7.72 (d, 2H, J = 8.0 Hz), 7.57 (dd, 1H, J = 8.0, 7.6 Hz), 7.48 (t, 1H, J = 7.4 Hz), 7.39 (dd, 2H, J = 8.0, 7.4 Hz), 6.85 (br s, 1H), 6.38 (br s, 1H), 4.19 (m, 2H), 3.68 (m, 2H), 3.62 (t, 2H, J = 4.8 Hz), 3.57 (s, 4H), 3.48 (m, 4H), 3.35 (t, 2H, J = 5.0 Hz), 1.70
(m, 4H), 1.47 (m, 2H). Compound 7 To a solution of 25 (14 mg, 25.6 µmol) in MeOH (3 mL) was added Pd-C (10%, 10
mg). The mixture was stirred at room temperature under H2 atmosphere for 1 h. After changing the atmosphere to N2, 16 (13 mg, 38.1 µmol) and DIEA (13.4 µL, 78.8 µmol)
Nature Chemical Biology: doi:10.1038/nchembio.157

24
were added to the solution. The mixture was then stirred at room temperature for 1 h. After filtration, the filtrate was evaporated and purified by column chromatography on silica gel (CHCl3/MeOH, 9:1) to give compound 7 as colorless oil (8.9 mg, 47%). 1H NMR (400 MHz; CDCl3): δ 8.51 (s, 1H), 8.26 (d, 1H, J = 8.0 Hz), 8.16 (br t 1H, J =
5.2 Hz), 8.01 (d, 1H, J = 8.0 Hz), 7.74 (d, 2H, J = 7.6 Hz), 7.61 (dd, 1H, J = 8.0, 7.6 Hz), 7.48 (t, 1H, J = 7.0 Hz), 7.40 (dd, 2H, J = 7.6, 6.8 Hz), 6.54 (br t, 1H, J = 4.8 Hz),
6.50 (br t, 1H, J = 5.6 Hz), 5.90 (s, 1H), 5.48 (s, 1H), 4.49 (m, 1H), 4.30 (m, 1H), 4.19 (m, 2H), 3.68 (m, 4H), 3.59 (s, 4H), 3.55–3.41 (m, 6H), 3.12 (m, 1H), 2.89 (dd, 1H, J = 12.8, 5.2 Hz), 2.75 (d, 1H, J = 12.8 Hz), 2.21 (m, 2H), 1.70 (m, 6H), 1.47 (m, 6H). 13C NMR (100 MHz; CDCl3): δ 173.70, 173.01, 168.05, 165.69, 136.28, 135.95, 134.61,
133.39, 131.44, 130.18, 129.62, 128.55, 127.04, 126.50, 70.59, 70.05, 69.80, 68.52, 61.90, 60.35, 55.64, 40.52, 40.11, 39.74, 39.23, 35.94, 29.13, 28.75, 25.68, 25.52,
24.01. HR-FAB MS (NBA): calcd for C35H49N5O9S2 [M]+ = 747.2972; obsd 747.2972.
Nature Chemical Biology: doi:10.1038/nchembio.157
25
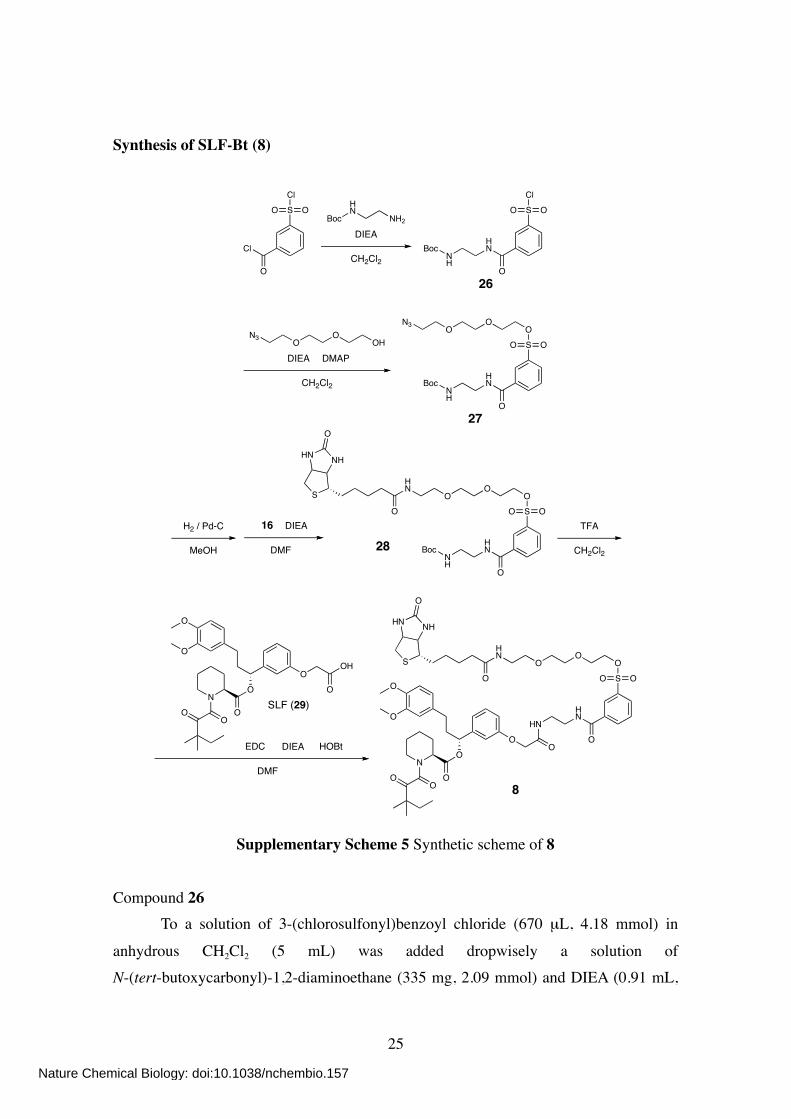
Synthesis of SLF-Bt (8)
Cl
O
SCl
OOBoc
HN
NH2
DIEA
CH2Cl2 NH
HN
O
SCl
OO
27
Boc
BocNH
HN
O
SO
OO
OO
N3
DIEA DMAP
CH2Cl2
OHO
ON3
8
BocNH
HN
O
SO
OO
OO
HN
O
S
NHHN
O
H2 / Pd-C
MeOH
DIEA
DMF
16
O
O
O
ON
OO
OOH
O
TFA
CH2Cl2
DIEA
DMF
HNHN
O
SO
OO
OO
HN
O
S
NHHN
O
O
O
O
ON
OO
OO
EDC HOBt
26
28
SLF (29)
Supplementary Scheme 5 Synthetic scheme of 8
Compound 26
To a solution of 3-(chlorosulfonyl)benzoyl chloride (670 µL, 4.18 mmol) in
anhydrous CH2Cl2 (5 mL) was added dropwisely a solution of N-(tert-butoxycarbonyl)-1,2-diaminoethane (335 mg, 2.09 mmol) and DIEA (0.91 mL,
Nature Chemical Biology: doi:10.1038/nchembio.157

26
5.25 mmol) in anhydrous CH2Cl2 (10 mL) at 4 ºC. The mixture was stirred at 4 ºC for 15 min and at room temperature for 1 h. After evaporation, the crude residue was purified by column chromatography on silica gel (EtOAc/Hexane, 1:1) to give compound 26 as
pale yellow sticky solid (688 mg, 90%). 1H NMR (400 MHz; CDCl3): δ 8.53 (s, 1H), 8.24 (d, 1H, J = 7.6 Hz), 8.14 (d, 1H, J =
8.0 Hz), 7.99 (br s, 1H), 7.71 (dd, 1H, J = 7.6, 8.0 Hz), 5.01 (br s, 1H), 3.59 (m, 2H),
3.44 (m, 2H), 1.44 (s, 9H). Compound 27
To a solution of 26 (688 mg, 1.89 mmol) in anhydrous CH2Cl2 (8 mL) was added 2-(2-(2-azidoethoxy)ethoxy)ethanolS2 (221 mg, 1.26 mmol), DIEA (658 µL, 3.78 mmol),
and DMAP (46 mg, 0.38 mmol). The mixture was stirred at room temperature for 5 h.
After evaporation, the crude residue was purified by column chromatography on silica gel (CHCl3/MeOH, 80:1) to give compound 27 as colorless oil (534 mg, 84%). 1H NMR (400 MHz; CDCl3): δ 8.39 (s, 1H), 8.14 (d, 1H, J = 7.6 Hz), 8.03 (d, 1H, J =
8.0 Hz), 7.68 (br s, 1H), 7.63 (dd, 1H, J = 8.0, 7.6 Hz), 5.06 (br s, 1H), 4.22 (t, 2H, J = 4.4 Hz), 3.75–3.67 (m, 4H), 3.65–3.55 (m, 6H), 3.41 (m, 2H), 3.36 (t, 2H, J = 5.2 Hz), 1.43 (s, 9H).
Compound 28 To a solution of 27 (168 mg, 335 µmol) in MeOH (6 mL) was added Pd-C (10%, 16
mg). The mixture was stirred at room temperature under H2 atmosphere for 1 h. After filtration, the filtrate was evaporated and dissolved in anhydrous DMF (6 mL). To this solution was added 16 (228 mg, 670 µmol) and DIEA (145 µL, 837 µmol). The mixture
was then stirred at room temperature for 2 h. After evaporation, the residue was dissolved in EtOAc and washed once with saturated aqueous NaHCO3 and brine. The organic layer was dried over anhydrous MgSO4, filtered, and evaporated. The crude
residue was purified by column chromatography on silica gel (CHCl3/MeOH, 10:1) to give compound 28 as colorless oil (94 mg, 40%). 1H NMR (400 MHz; CDCl3): δ 8.54 (s, 1H), 8.45 (d, 1H, J = 7.6 Hz), 8.03 (d, 1H, J =
8.0 Hz), 7.66 (dd, 1H, J = 8.0, 7.6 Hz), 6.40 (br s, 1H), 6.01 (br s, 1H), 5.75 (br s, 1H), 5.40 (br s, 1H), 4.52 (m, 1H), 4.35 (m, 1H), 4.20 (m, 2H), 3.74–3.69 (m, 6H), 3.63 (m, 2H), 3.57 (m, 2H), 3.53–3.40 (m, 2H), 3.29 (m, 2H), 3.15 (m, 1H), 2.93 (dd, 1H, J =
Nature Chemical Biology: doi:10.1038/nchembio.157

27
12.8, 5.2 Hz), 2.79 (d, 1H, J = 12.8 Hz), 2.23 (m, 2H), 1.72–1.57 (m, 4H), 1.48–1.28 (m + s, 2 + 9H).
Compound 8 To a solution of 28 (50 mg, 71 µmol) in CH2Cl2 (5 mL) was added TFA (3 mL). The
mixture was stirred at room temperature for 3 h. After co-evaporation twice with
toluene (6 mL), the residue was dissolved in anhydrous DMF (4 mL). To this was added synthetic FKBP ligandS3 (SLF, 29) (42 mg, 71 µmol), HOBt (16 mg, 107 µmol), EDC (20 mg, 107 µmol), and DIEA (37 µL, 214 µmol). The mixture was then stirred at room
temperature for 5 h. After evaporation, the residue was dissolved in EtOAc and washed once with saturated aqueous NaHCO3 and brine. The organic layer was dried over anhydrous MgSO4, filtered, and evaporated. The crude residue was purified by column
chromatography on silica gel (CHCl3/MeOH, 15:1) to give compound 8 as colorless oil (10 mg, 12%). 1H NMR (400 MHz; CDCl3): δ 8.62 (br s, 1H), 8.54 (s, 1H), 8.23 (d, 1H, J = 8.0 Hz),
8.01 (d, 1H, J = 8.0 Hz), 7.63 (dd, 1H, J = 8.0, 8.0 Hz), 7.54 (br s, 1H), 7.26 (dd, 1H, J = 8.0, 7.6 Hz), 6.95–6.91 (m, 2H), 6.86 (d, 1H, J = 7.6 Hz), 6.77 (d, 1H, J = 8.8 Hz), 6.70–6.66 (m, 2H), 6.41 (br s, 1H), 5.75 (m, 1H), 5.68 (br s, 1H), 5.28 (m+br s, 2H),
4.50 (s, 2H), 4.41 (m, 1H), 4.27 (m, 1H), 4.21 (t, 2H, J = 4.4), 3.85 (s, 3H), 3.84 (s, 3H), 3.71 (t, 2H, J = 4.4), 3.67–3.58 (m, 8H), 3.54 (t, 2H, J = 4.8), 3.43 (m, 2H), 3.36 (d, 1H, J = 12.0 Hz), 3.19 (dd, 1H, J = 12.8, 2.8 Hz), 3.12 (m, 1H), 2.87 (dd, 1H, J = 12.8, 5.2
Hz), 2.64 (d, 1H, J = 12.8 Hz), 2.57 (m, 2H), 2.37 (d, 1H, J = 14.4 Hz), 2.27–2.16 (m, 3H), 2.05 (m, 2H), 1.75–1.60 (m, 8H), 1.49–1.32 (m, 4H), 1.20 (s, 6H), 0.88 (t, 3H, J = 8.0 Hz). 13C NMR (150 MHz; CDCl3): δ 207.90, 173.24, 169.65, 169.29, 167.31, 165.70, 163.40,
157.40, 148.86, 147.34, 141.87, 135.85, 135.65, 133.69, 133.29, 130.25, 130.00, 129.62, 126.48, 120.12, 119.97, 114.20, 113.23, 111.68, 111.27, 76.49, 70.63, 70.07, 70.05,
69.76, 68.44, 67.39, 61.61, 60.12, 55.90, 55.83, 55.43, 51.27, 46.69, 44.14, 40.55, 40.05, 39.14, 38.20, 35.89, 32.46, 31.24, 29.68, 28.04, 27.90, 26.40, 25.68, 24.90, 23.39, 23.18, 21.14, 8.75.
HR-FAB MS (NBA): calcd for C57H78N6O16S2 [M+H]+ = 1167.4994; obsd 1167.5007.
Nature Chemical Biology: doi:10.1038/nchembio.157
28
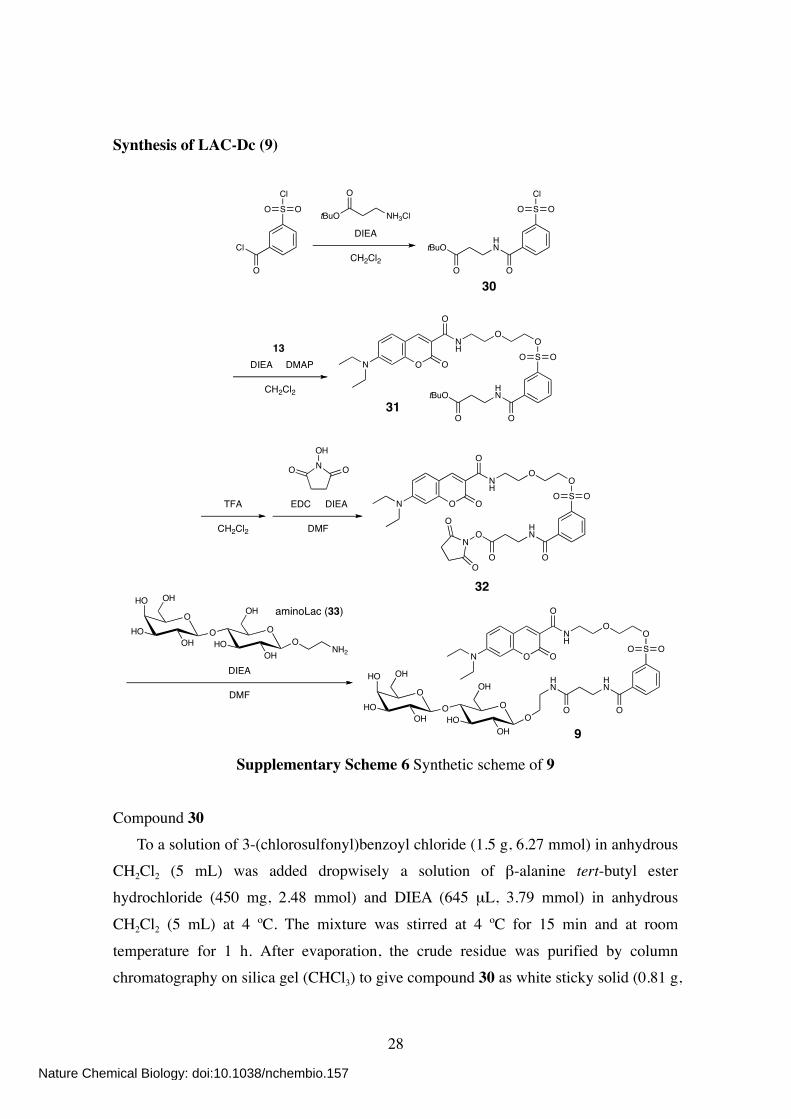
Synthesis of LAC-Dc (9)
Cl
O
SCl
OO
DIEA
CH2Cl2
30
tBuO NH3Cl
O
tBuOHN
O
SCl
OO
O
DIEA DMAP
CH2Cl2
13
tBuOHN
O
SO
OO
ONH
O
O ON
O
EDC DIEA
DMF
TFA
CH2Cl2
NOH
OO
OHN
O
SO
OO
ONH
O
O ON
ON
O
O
OO
O
OH
OHOOH
HO
HO
NH2
OHOH
DIEA
DMFHN
HN
O
SO
OO
ONH
O
O ON
OOO
O
OH
OHOOH
HO
HO
OHOH
31
32
9
aminoLac (33)
Supplementary Scheme 6 Synthetic scheme of 9
Compound 30 To a solution of 3-(chlorosulfonyl)benzoyl chloride (1.5 g, 6.27 mmol) in anhydrous CH2Cl2 (5 mL) was added dropwisely a solution of β-alanine tert-butyl ester hydrochloride (450 mg, 2.48 mmol) and DIEA (645 µL, 3.79 mmol) in anhydrous
CH2Cl2 (5 mL) at 4 ºC. The mixture was stirred at 4 ºC for 15 min and at room
temperature for 1 h. After evaporation, the crude residue was purified by column chromatography on silica gel (CHCl3) to give compound 30 as white sticky solid (0.81 g,
Nature Chemical Biology: doi:10.1038/nchembio.157

29
94%). 1H NMR (400 MHz; CDCl3): δ 8.42 (s, 1H), 8.15 (m, 2H), 7.73 (dd, 1H, J = 7.8, 7.8
Hz), 7.09 (br s, 1H), 3.73 (m, 2H), 2.60 (t, 2H, J = 5.8 Hz), 1.48 (s, 9H).
Compound 31 To a solution of 30 (109 mg, 0.31 mmol) in anhydrous CH2Cl2 (5 mL) was added 13 (80 mg, 0.23 mmol), DIEA (120 µL, 0.71 mmol), and DMAP (18 mg, 0.15 mmol). The
mixture was then stirred at room temperature for 8 h. After evaporation, the crude residue was purified by column chromatography on silica gel (CHCl3/MeOH, 30:1) to
give compound 31 as yellow solid (130 mg, 86%). 1H NMR (400 MHz; CDCl3): δ 8.90 (br s, 1H), 8.69 (s, 1H), 8.36 (s, 1H), 8.12 (d, 1H, J
= 7.6 Hz), 8.06 (d, 1H, J = 7.6 Hz), 7.64 (dd, 1H, J = 7.6, 7.6 Hz), 7.43 (d, 1H, J = 8.8
Hz), 6.65 (dd, 1H, J = 8.8, 2.5 Hz), 6.51 (d, 1H, J = 2.5 Hz), 4.24 (m, 2H), 3.74–3.67 (m, 6H), 3.56 (m, 2H), 3.46 (m, 4H), 2.60 (t, 2H, J = 6.2 Hz), 1.46 (s, 9H), 1.25 (t, 6H, J = 7.0 Hz).
Compound 9 To a solution of 31 (50 mg, 76 µmol) in anhydrous CH2Cl2 (3 mL) was added TFA
(1 mL). The mixture was stirred at room temperature for 2 h. After co-evaporation twice with toluene (2 mL), the residue was dissolved in anhydrous DMF (1 mL). To this was added a solution of N-hydroxysuccinimide (18 mg, 156 µmol), EDC (24 mg, 125 µmol),
and DIEA (50 µL, 0.29 mmol) in anhydrous DMF (1 mL). The mixture was then stirred
at room temperature for 2 h and at 40 ºC for 2h. After evaporation, the crude residue was roughly purified by column chromatography on silica gel (CHCl3/MeOH, 30:1) to
give compound 32 as yellow solid (25 mg) containing some impurities, which was used for the next step without further purification. To a solution of 32 (25 mg) in anhydrous DMF (3 mL) was added 2-aminoethyl lactoseS4 (aminoLac, 33) (13 mg, 34 µmol). The mixture was stirred at room
temperature for 3 h and at 40 ºC for 1h. After evaporation, the crude residue was purified by column chromatography on silica gel (CHCl3/MeOH, 6:1 to 3:1) to give
compound 9 as yellow solid (6 mg, 8% in two steps). 1H NMR (400 MHz; CD3OD): δ 8.63 (s, 1H), 8.37 (s, 1H), 8.10 (m, 2H), 7.71, (dd, 1H,
J = 7.8, 7.8 Hz), 7.57 (d, 1H, J = 9.2 Hz), 6.84 (dd, 1H, J = 9.2, 2.8 Hz), 6.61 (d, 1H, J
Nature Chemical Biology: doi:10.1038/nchembio.157

30
= 2.8 Hz), 4.34 (m, 2H), 4.27 (m, 2H), 3.88–3.52 (m, 20H), 3.48 (m, 6H), 3.14 (m, 2H), 2.54 (t, 2H, J = 6.8 Hz), 1.25 (t, 6H, J = 7.0 Hz). 13C NMR (100 MHz; CD3OD): δ 173.87, 165.42, 159.22, 154.67, 149.39, 138.13,
136.96, 135.89, 132.66, 130.94, 130.01, 129.76, 127.88, 125.87, 111.70, 109.51, 105.13, 97.35, 80.60, 80.43, 77.10, 76.50, 76.33, 74.81, 72.55, 71.52, 70.52, 70.35, 70.31, 69.68, 69.53, 62.52, 61.88, 46.00, 40.33, 37.90, 36.88, 12.72.
HR-ESI Qq-LTMS: calcd for C42H59N4O20S [M+H]+ = 971.3443; obsd 971.3436.
Nature Chemical Biology: doi:10.1038/nchembio.157
31
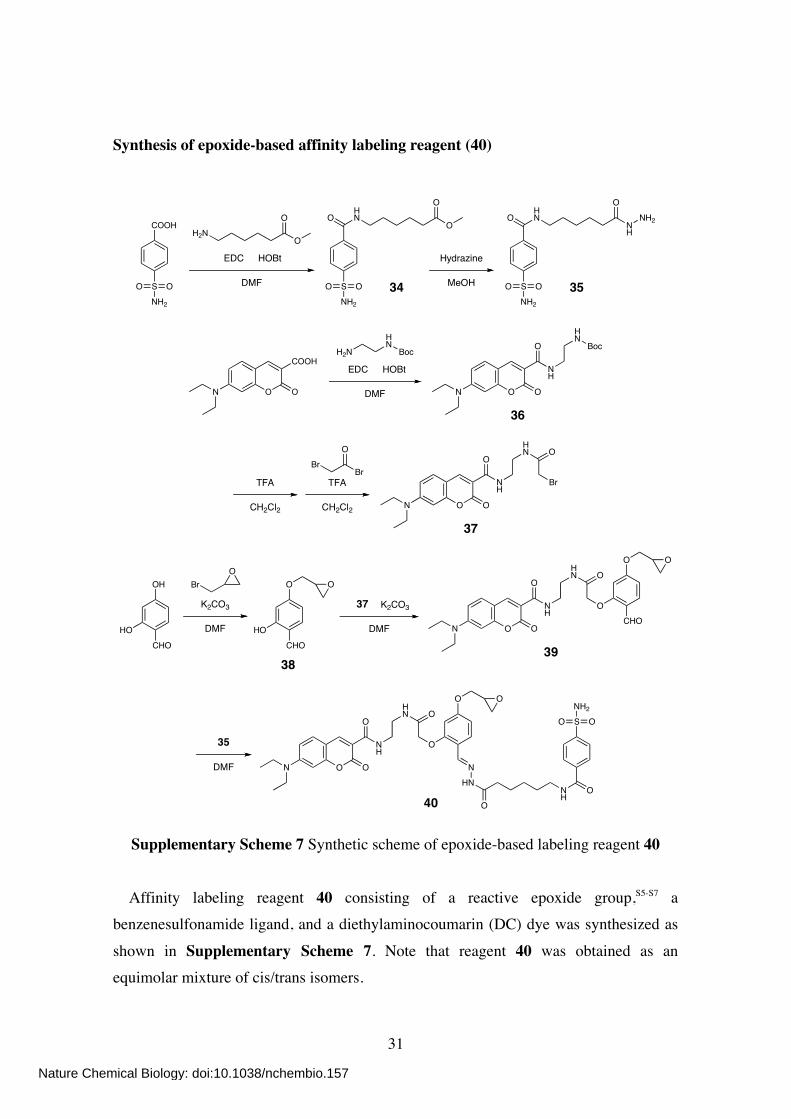
Synthesis of epoxide-based affinity labeling reagent (40)
SNH2
OO
COOH
O
EDC HOBt
DMF SNH2
OO
OHN
O
OO
H2N
SNH2
OO
OHN
NH
NH2
O
Hydrazine
MeOH34 35
NH
O
O ON
HN
BocH2NHN
Boc
N O O
COOHEDC HOBt
DMF
36
NH
O
O ON
HN O
Br
37
TFA
CH2Cl2
TFA
CH2Cl2
BrBr
O
CHO
OH
HO
K2CO3
DMF
BrO
CHO
O
HO
O
NH
O
O ON
HN O
OCHO
O O
K2CO3
DMF
37
3839
NH
O
O ON
HN O
O
O O
NHN
ONH
O
SNH2
OO
DMF
35
40
Supplementary Scheme 7 Synthetic scheme of epoxide-based labeling reagent 40
Affinity labeling reagent 40 consisting of a reactive epoxide group,S5-S7 a benzenesulfonamide ligand, and a diethylaminocoumarin (DC) dye was synthesized as
shown in Supplementary Scheme 7. Note that reagent 40 was obtained as an equimolar mixture of cis/trans isomers.
Nature Chemical Biology: doi:10.1038/nchembio.157

32
1H NMR (400 MHz; (CD3)2SO): δ 11.15 (s, 0.5H), 10.93 (s, 0.5H), 8.74 (br t, 0.5H, J =
5.6 Hz), 8.68 (br t, 0.5H, J = 5.6 Hz), 8.58 (m, 2H), 8.34 (br s, 0.5H), 8.28 (s, 0.5H), 8.27 (s, 0.5H), 8.04 (br t, 0.5H, J = 4.8 Hz), 7.94 (m, 2H), 7.85 (m, 2H), 7.68 (d, 0.5H, J
= 8.4 Hz), 7.62 (m, 1H), 7.54 (d, 0.5H, J = 8.4 Hz), 7.43 (s, 2H), 6.77 (m, 1H), 6.60–6.52 (m, 3H), 4.53 (s, 2H), 4.32 (m, 1H), 3.82 (m, 1H), 3.48–3.22 (m, 11H), 2.81 (m, 1H), 2.67 (m, 1H), 2.14 (t, 2H, J = 7.2 Hz), 1.55 (m, 4H), 1.33 (m, 2H), 1.11 (t, 6H,
J = 7.0 Hz). 13C NMR (150 MHz; (CD3)2SO): δ 168.34, 167.71, 165.02, 161.56, 160.75, 157.20,
157.07, 152.40, 149.71, 146.06, 143.02, 137.55, 131.56, 129.24, 127.76, 125.56, 115.62,
115.56, 110.09, 109.55, 109.33, 107.61, 99.91, 95.80. 69.77, 69.27, 49.52, 44.32, 43.77, 38.70, 38.51, 34.04, 31.84, 28.83, 26.19, 24.81, 12.30. HR-FAB MS (NBA): calcd for C41H49N7O11S [M]+ = 847.3211; obsd 847.3207.
Nature Chemical Biology: doi:10.1038/nchembio.157

33
General materials and methods for biochemical/biological experiments Unless otherwise noted, all proteins/enzymes and reagents were obtained from commercial suppliers (Sigma, Aldrich, Tokyo Chemical Industry (TCI), Wako Pure
Chemical Industries, Pierce Biotechnology, or Calbiochem) and used without further purification. UV-visible spectra were recorded on a Shimadzu UV-2550 spectrophotometer. SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and western
blotting were carried out using a Bio-Rad Mini-Protean III electrophoresis apparatus. Fluorescence gel images were acquired using a Bio-Rad ChemiDoc XRS system with a 480BP70 filter, and analyzed with Quantity One 1-D Analysis Software (Bio-Rad
Laboratories). Analytical reversed-phase HPLC (RP-HPLC) was carried out on a Hitachi LaChrom L-7100 system equipped with LaChrom L-7400 UV and L-7485 fluorescence detectors, and a YMC-Pack ODS-A column (5 µm, 250 × 4.6 mm) at a
flow rate of 1.0 mL/min. UV detection was at 220 nm and fluorescence detection was at 473 nm with excitation at 427 nm. All runs used linear gradients of acetonitrile containing 0.1% TFA (solvent A) and 0.1% aqueous TFA (solvent B). MALDI-TOF MS
spectra were recorded on an Applied Biosystems Voyager Elite mass spectrometer using CHCA or sinapinic acid (SA) as the matrix. Matrix-assisted laser desorption/ionization quadrupole ion trap time-of-flight mass spectrometry/mass spectrometry
(MALDI-QIT-TOF MS/MS) analysis was performed by Dr. Masaki Yamada (Shimadzu Corporation) with a Shimadzu Biotech AXIMA QIT mass spectrometer. Ion-exchange chromatography was performed using an Applied Biosystems BioCad 700E Perfusion
Chromatography Workstation on POROS HS/20 cation-exchange column (100 × 4.6 mm). Protein samples were loaded and washed with HEPES buffer (10 mM, pH 7.0) and eluted by a linear gradient of 0–1.0 M NaCl in HEPES buffer (10 mM, pH 7.0). UV
detection was at 280 and 430 nm.
Nature Chemical Biology: doi:10.1038/nchembio.157

34
Carbonic anhydrase (CA) labeling experiments Purified human carbonic anhydrase I (CAI, C4396) and II (CAII, C6165) were purchased from Sigma. The concentrations of CAI and CAII were determined by
measuring the absorbance at 280 nm using molar absorption coefficients of 49,000 and 54,000 M–1cm–1, respectively.S8 Ethoxzolamide (EZA, 4), 4-sulfamoylbenzoic acid (SBA, 10), benzenesulfonamide (BS, 11) and p-toluenesulfonic acid (TA, 12) were
purchased from Aldrich, TCI, TCI and Wako, respectively. Human blood was taken from one of the authors (healthy male, 26-year-old) by Dr. Eishi Ashihara (Department of Transfusion Medicine and Cell Therapy, Graduate School of Medicine, Kyoto
University Hospital). After heparin treatment, red blood cells (RBCs) were separated from plasma by centrifugation. Specific pathogen-free (SPF) Slc:ICR mice (male, 8-week-old) were purchased from Shimizu Laboratory Suppliers and all animal
experiments were performed at the Mori laboratory (Department of Synthetic Chemistry and Biological Chemistry, Graduate School of Engineering, Kyoto University) in accordance with the Guidelines for Animal Experiments of Kyoto University.
Preparation of a 1:1 conjugate of CAII and Dc dye Purified CAII (20 µM) was incubated with 40 (40 µM) in HEPES buffer (50 mM,
pH 7.2) at 25 ºC. After 35 h, the completion of the labeling was confirmed by MALDI-TOF MS measurement using SA as the matrix (data not shown). The resulting 1:1 conjugate of CAII and Dc dye was purified by size-exclusion chromatography using
a PD-10 column (GE Healthcare) and dialyzed against HEPES buffer (50 mM, pH 7.2) with a Spectra/Por dialysis membrane (MWCO: 10,000) (Spectrum Laboratories).
Labeling of CAII in test tubes Purified CAII (40 µM) was incubated with 1 or 5 (80 µM) in HEPES buffer (50 mM, pH 7.2) at 37 ºC. Control reactions with EZA (400 µM) or GSH (10 mM) were also
included. Aliquots were taken at 3 and 24 h and mixed with an equal volume of 2 × SDS-PAGE loading buffer. The samples were applied to 12.5 % SDS-PAGE and the labeled CAII was detected by in-gel fluorescence imaging. The 1:1 Dc-CAII conjugate
was used as a standard marker to determine the labeling yield based on the relative fluorescence intensities. After fluorescence imaging, the gel was stained with CBB. For labeling in protein mixtures, a mixture of five different proteins (10 µM each; β-Gal,
Nature Chemical Biology: doi:10.1038/nchembio.157

35
OVA, CAII, GST, and Hb) was incubated with 1 (20 µM) in the absence or presence of EZA (100 µM) in HEPES buffer at 37 ºC. After 24 h, the samples were analyzed as
described above.
Peptide mapping of the Dc-labeled CAII Purified CAII (50 µM) was incubated with 1 (50 µM) in HEPES buffer (50 mM, pH
7.2) at 37 ºC. After 72 h, the labeled CAII was purified by size-exclusion chromatography using a PD-10 column and dialyzed against ammonium carbonate buffer (100 mM, pH 8.5) with a Spectra/Por dialysis membrane (MWCO: 10,000). The
resulting solution was concentrated using a Centricon Ultracel YM-10 (Millipore). To this solution was added urea (at a final concentration of 2 M) and lysyl endopeptidase (LEP) (LEP/substrate ratio = 1/50 (w/w)). Native (unlabeled) CAII was also subjected
to LEP digestion. After incubation at 37 ºC for 24 h, the digested samples were applied to RP-HPLC. Collected fractions were analyzed by MALDI-TOF MS using CHCA as the matrix and the labeled fragment was further characterized by MALDI-QIT-TOF
MS/MS. Evaluation of the enzymatic activity of the Dc-labeled CAII Purified CAII (40 µM) was incubated with 1 (80 µM) in HEPES buffer (50 mM, pH
7.2) at 37 ºC. After 48 h, the labeled CAII was purified by cation-exchange column chromatography and obtained as a mixture of 75 % labeled and 25 % unlabeled (native)
CAII. The protein solution was dialyzed against HEPES buffer (50 mM, pH 7.2). The hydrolytic (esterase) activity of the 1-treated CAII was assayed in HEPES buffer (50 mM, pH 7.2) using p-nitrophenyl acetate as substrate.S6 Reaction conditions were as follows: 1 µM CAII, 0.1–20 mM p-nitrophenyl acetate. Initial rates of
p-nitrophenyl acetate hydrolysis were determined by measuring the increase of absorbance at 348 nm (Δε348 = 5,150 M–1cm–1)S9 for initial 30 sec. Kinetic parameters
were obtained by fitting a plot of the initial rates as a function of the substrate concentrations to the Michaelis-Menten Equation (1) using KaleidaGraph (Synergy Software).
V0 = kcat·[E]0·[S] / ([S] + Km) (1) V0 is the initial rate of hydrolysis, kcat and Km are the first-order rate constant from the catalyst-substrate complex and the Michaelis-Menten constant, respectively. [E]0 and
Nature Chemical Biology: doi:10.1038/nchembio.157

36
[S] are the initial concentrations of CAII and substrate, respectively. Chemical labeling of eCA in human RBCs
Sedimented RBCs were resuspended in twice the volume of HEPES buffered saline (HBS, pH 7.4) and incubated with 1 (100 µM) in the absence or presence of EZA (500 µM) at 25 ºC for 48 h. The cells were washed twice with HBS and lysed by sonication.
After centrifugation, the supernatants were mixed with an equal volume of 2 × SDS-PAGE loading buffer, applied to 15 % SDS-PAGE, and analyzed by in-gel fluorescence imaging and CBB-staining.
Chemical biotinylation of eCA in living mice For ex vivo experiments, whole blood (0.4 mL) was taken from a Slc:ICR mouse (SPF grade, male, 8-week-old) and mixed with 2 µL of EDTA solution (0.5 M, pH 8.0).
This blood sample was diluted two-fold with phosphate buffered saline (PBS, pH 7.4). The suspension was incubated with 2 or 7 (5 µM) in the absence or presence of EZA
(500 µM) at 35 ºC. After 20 h, the samples were lysed using NP-40EZA lysis buffer (50
mM Tris•HCl, 150 mM NaCl, 2.5 mM EDTA, 1 % Nonidet P-40, 0.5 mM EZA) and mixed with 2 × SDS-PAGE loading buffer. For in vivo experiments, mice were intravenously injected with 2 or 7 (100 µM in 0.5 mL PBS). Blood (10 µL) was taken
from the tail vain at 4, 8 and 20 h, lysed with NP-40EZA lysis buffer, and mixed with 2 × SDS-PAGE loading buffer. The protein samples were resolved by 15 % SDS-PAGE and
electrotransfered onto an Immun-Blot PVDF membrane (Bio-Rad). The biotinlyated product was detected with SAv-HRP (Invitrogen) using Chemi-Lumi One (Nacalai Tesque). The immunodetection of eCA was accomplished with anti-mouse CAII
antibody and anti-goat IgG-HRP conjugate (both Santa Cruz Biotechnology). Preparation of 19F probe-labeled CAI (Fb-CAItube) Purified CAI (100 µM) was incubated with 3 (200 µM) in HEPES buffer (50 mM,
pH 7.2) at 37 ºC. After 48 h, the completion of the labeling was confirmed by MALDI-TOF MS measurement using SA as the matrix (data not shown). The labeled
CAI was purified by size-exclusion chromatography using a TOYOPEARL HW-40F (Tosoh Corporation).
Nature Chemical Biology: doi:10.1038/nchembio.157

37
Monitoring of 19F-labeling of eCA in RBCs Sedimented RBCs (approximately 0.3 mL packed volume) were resuspended in twice the volume of HBS and incubated with 3 (100 µM) at 25 ºC. At 0, 24 and 48 h,
the cells were washed, resuspended in HBS containing 20% v/v D2O and 167 µM TFA
(internal standard at –75.6 p.p.m.) (HBSDT), and subjected to 19F NMR analysis (approximately 0.6 mL total volume). 19F NMR measurement of purified Fb-CAItube (20 µM) was performed in HEPES buffer (50 mM, pH 7.2) containing 10% v/v D2O and 200 µM TFA (HEPESDT). 19F NMR spectra were recorded on a JEOL EX-400 (376.5
MHz) spectrometer and calibrated to TFA. The number of scans was 1,024 and 2,048
for in vitro and in RBC measurements, respectively. Peptide mapping of the Fb-labeled eCA Sedimented RBCs were resuspended in twice the volume of HBS and incubated with 3 (100 µM) at 25 ºC. After 48 h incubation, the eCA was roughly purified from the
RBCs by a CHCl3/ethanol extraction procedure previously reported.S10 The purified
labeled eCA (predominantly containing CAI), Fb-CAItube, and native (unlabeled) CAI were subjected to LEP digestion. The digested samples were applied to RP-HPLC and analyzed by mass spectrometry as described in the section of “Peptide mapping of the Dc-labeled CAII”.
NMR-based detection of CA inhibitors in RBCs Fb labeling of eCA in RBCs was accomplished as described above. After the cells were washed and resuspended in HBSDT containing various concentrations of inhibitors including EZA, SBA and BS, the samples were subjected to 19F NMR analysis. For in
vitro experiments (Supplementary Fig. 7), samples containing Fb-CAItube (20 µM) and
various concentrations of inhibitors including the above three and TA in HEPESDT were applied to 19F NMR experiments. The number of scans was 1,024 and 2,048 for in vitro
and in RBC measurements, respectively.
Nature Chemical Biology: doi:10.1038/nchembio.157

38
FK506-binding protein 12 (FKBP12) labeling experiments Recombinant human FKBP12 was obtained by bacterial expression as a fusion protein with glutathione S-transferase (GST), followed by cleavage of the GST tag
using Factor Xa (Details of the vector construction and expression will be reported elsewhere). The concentration of FKBP12 was determined by measuring the absorbance at 278 nm using molar absorption coefficient of 9,860 M–1cm–1.S11 Jurkat cells were
cultured in RPMI 1640 medium containing 10% fetal bovine serum at 37 ºC under 5 % CO2. FKBP12 is endogenously expressed in Jurkat cells.S12 Labeling of FKBP12 in test tubes Purified FKBP12 (11 µM) was incubated with 8 (11 µM) in the absence or presence of SLF (29) (110 µM, see p.25 for the structure) in Tris buffer (50 mM, pH 8.0) at 37 ºC.
for 24 h. Aliquots were taken and mixed with an equal volume of 2 × SDS-PAGE loading buffer. The samples were resolved by 15 % SDS-PAGE and electrotransfered onto an Immun-Blot PVDF membrane. The biotinlyated FKBP12 was detected with
SAv-HRP using Chemi-Lumi One. The immunodetection of FKBP12 was accomplished with anti-human FKBP12 antibody (Abcam) and anti-rabbit IgG antibody-HRP conjugate (GE Healthcare). MALDI-TOF MS analysis of the labeled
product was also conducted using CHCA as the matrix. Chemical biotinylation of endogenous FKBP12 in Jurkat cells
Jurkat cells (4 × 106 cells) were incubated at 37 ºC in serum-free RPMI 1640 medium (1 mL) containing 8 or 7 (4 µM). After 18 h, the cells were washed twice with
PBS, lysed using NP-40 lysis buffer (50 mM Tris•HCl, 150 mM NaCl, 2.5 mM EDTA,
1 % Nonidet P-40), and mixed with 2 × SDS-PAGE loading buffer. The samples were analyzed by western blotting using anti-human FKBP12 antibody and SAv-HRP as described in the above section. Cell viability was evaluated by MTT assay using the
Cell Counting Kit-8 (Dojindo).
Nature Chemical Biology: doi:10.1038/nchembio.157

39
Congerin labeling experiments Recombinant congerin II (CongII) was expressed in E. coli strain JM109/pTV-Con II (a kind gift from Prof. Tomohisa Ogawa (Tohoku University)) and purified as
previously described.S13 The concentration of CongII was determined by measuring the absorbance at 280 nm using molar absorption coefficient of 11,500 M–1cm–1.S4 Conger eels were purchased at Nishiki Ichiba (Nishiki Market) in Kyoto, Japan. Congerin (a
mixture of isoform I and II) is naturally expressed in the skin mucus of conger eels.S14 Labeling of CongII in test tubes Purified CongII (20 µM) was incubated with 9 (80 µM) in the absence or presence
of lactose (1.6 mM) in HEPES buffer (50 mM, 100 mM NaCl, pH 8.0) at 25 ºC for 24 h. Aliquots were taken and mixed with an equal volume of 2 × SDS-PAGE loading buffer.
The samples were applied to 15 % SDS-PAGE and the Dc-labeled CongII was detected by in-gel fluorescence imaging. After fluorescence imaging, the gel was stained with CBB. MALDI-TOF MS analysis of the labeled product was also performed using SA as
the matrix. Fluorescent labeling of endogenous congerin in mucus tissue of conger eel skin
Mucus tissue was collected from the skin of conger eel and mixed in HEPES buffer (50 mM, 100 mM NaCl, pH 8.0). The mucus tissue solution was incubated with 9 (80 µM) in the absence or presence of lactose (1.6 mM) at 25 ºC for 24 h. After
homogenization by sonication, aliquots were taken and mixed with 2 × SDS-PAGE loading buffer. The samples were analyzed by in-gel fluorescence imaging and CBB staining as described in the above section.
Nature Chemical Biology: doi:10.1038/nchembio.157

40
References S1. Song, A., Wang, X. & Lam, K.S. Tetrahedron Lett. 44, 1755–1758 (2003) S2. Fernandez-Megia, E., Correa, J., Rodríguez-Meizoso, I. & Riguera, R.
Macromolecules 39, 2113–2120 (2006) S3. Keenan, T., Yaeger, D.R., Courage, N.L., Rollins, C.T., Pavone, M.E., Rivera, V.M.,
Yang, W., Guo, T., Amara, J.F., Clackson, T., Gilman, M. & Holt, D.A. Bioorg. Med.
Chem. 6, 1309–1335 (1998) S4. Koshi, Y., Nakata, E., Miyagawa, M., Tsukiji, S., Ogawa, T. & Hamachi, I. J. Am.
Chem. Soc. 130, 245–251 (2008)
S5. Chen, G., Heim, A., Riether, D., Yee, D., Milgrom, Y., Gawinowicz, M.A. & Sames, D. J. Am. Chem. Soc. 125, 8130–8133 (2003)
S6. Takaoka, Y., Tsutsumi, H., Kasagi, N., Nakata, E. & Hamachi, I. J. Am. Chem. Soc.
128, 3273–3280 (2006) S7. Wakabayashi, H., Miyagawa, M., Koshi, Y., Takaoka, Y., Tsukiji, S. & Hamachi, I.
Chem. Asian J. 3, 1134–1139 (2008)
S8. Supuran, C.T., Briganti, F., Tilli, S., Chegwidden, W.R. & Scozzafava, A. Bioorg.
Med. Chem. 9, 703–714 (2001) S9. Elleby, B., Sjöblom, B. & Lindskog, S. Eur. J. Biochem. 262, 516–521 (1999)
S10. Tashian, R.E., Riggs, S.K. & Yu, Y.-S.L. Arch. Biochem. Biophys. 117, 320–327 (1966)
S11. Standaert, R.F., Galat, A., Verdine, G.L. & Schreiber, S.L. Nature 346, 671–674
(1990) S12. Dumont, F.J., Kastner, C., Iacovone Jr., F. & Fischer, P.A. J. Pharmacol. Exp. Ther.
268, 32–41 (1994).
S13. Ogawa, T., Ishii, C., Suda, Y., Kamiya, H. & Muramoto, K. Biosci. Biotechnol.
Biochem. 66, 476–480 (2002) S14. Muramoto, K., Kagawa, D., Sato, T., Ogawa, T., Nishida, Y. & Kamiya, H. Comp.
Biochem. Physiol. B. 123, 33–45 (1999).
Nature Chemical Biology: doi:10.1038/nchembio.157





























