Embed Size (px)
Citation preview

1
Supplementary Information
Reconstructing metabolic pathways of hydrocarbon-degrading
bacteria from the Deepwater Horizon oil spill
Nina Dombrowski1, John A. Donaho1, Tony Gutierrez2, Kiley W. Seitz1, Andreas P.
Teske3, Brett J. Baker1*
1Department of Marine Science, University of Texas Austin, Marine Science Institute,
Port Aransas, TX, USA; 2School of Life Sciences, Heriot-Watt University, Edinburgh,
UK; 3Department of Marine Sciences, University of North Carolina at Chapel Hill,
Chapel Hill, NC, USA
*corresponding author; [email protected]
Reconstructing metabolic pathways of hydrocarbon-degrading bacteria from the Deepwater Horizon oil spill
SUPPLEMENTARY INFORMATIONARTICLE NUMBER: 16057 | DOI: 10.1038/NMICROBIOL.2016.57
NATURE MICROBIOLOGY | www.nature.com/naturemicrobiology 1
2
Supplementary Tables 1-2
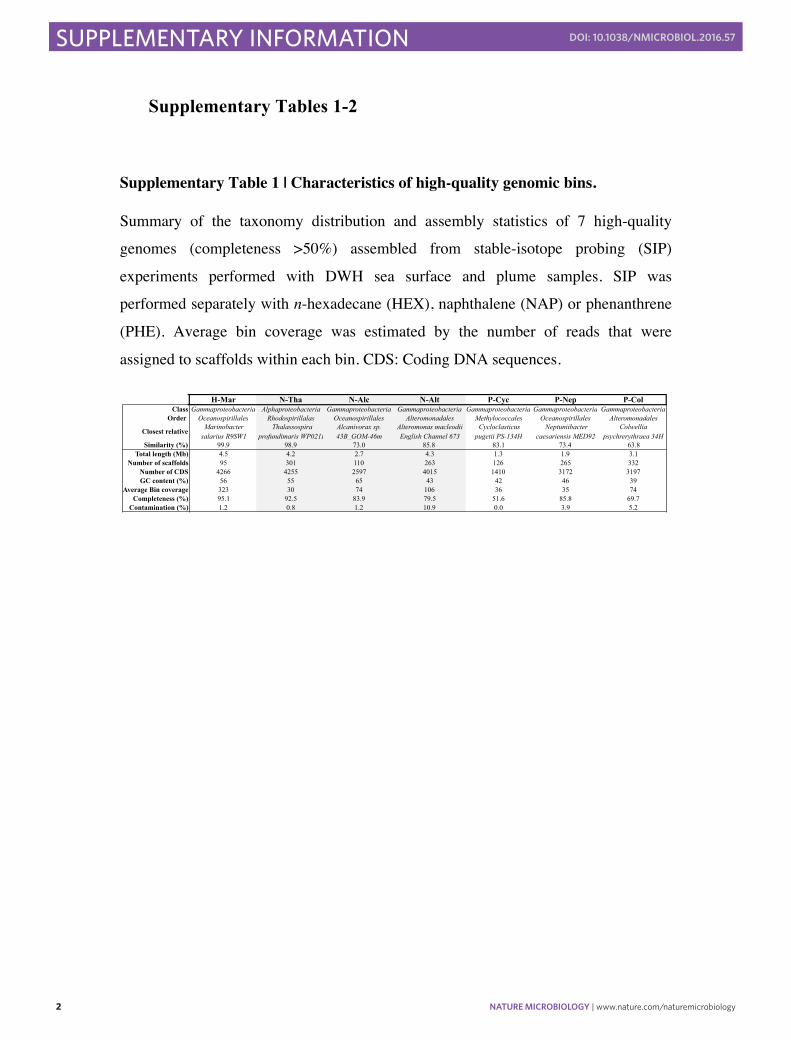
Supplementary Table 1 | Characteristics of high-quality genomic bins.
Summary of the taxonomy distribution and assembly statistics of 7 high-quality
genomes (completeness >50%) assembled from stable-isotope probing (SIP)
experiments performed with DWH sea surface and plume samples. SIP was
performed separately with n-hexadecane (HEX), naphthalene (NAP) or phenanthrene
(PHE). Average bin coverage was estimated by the number of reads that were
assigned to scaffolds within each bin. CDS: Coding DNA sequences.
H-Mar N-Tha N-Alc N-Alt P-Cyc P-Nep P-ColClass Gammaproteobacteria Alphaproteobacteria Gammaproteobacteria Gammaproteobacteria Gammaproteobacteria Gammaproteobacteria Gammaproteobacteria
Order Oceanospirillales Rhodospirillalas Oceanospirillales Alteromonadales Methylococcales Oceanospirillales Alteromonadales
Closest relative Marinobacter salarius R9SW1
Thalassospira profundimaris WP0211
Alcanivorax sp. 43B_GOM-46m
Alteromonas macleodii English Channel 673
Cycloclasticus pugetii PS-134H
Neptuniibacter caesariensis MED92
Colwellia psychrerythraea 34H
Similarity (%) 99.9 98.9 73.0 85.8 83.1 73.4 63.8Total length (Mb) 4.5 4.2 2.7 4.3 1.3 1.9 3.1
Number of scaffolds 95 301 110 263 126 265 332Number of CDS 4266 4255 2597 4015 1410 3172 3197GC content (%) 56 55 65 43 42 46 39
Average Bin coverage 323 30 74 106 36 35 74Completeness (%) 95.1 92.5 83.9 79.5 51.6 85.8 69.7
Contamination (%) 1.2 0.8 1.2 10.9 0.0 3.9 5.2
2 NATURE MICROBIOLOGY | www.nature.com/naturemicrobiology
SUPPLEMENTARY INFORMATION DOI: 10.1038/NMICROBIOL.2016.57

3
Supplementary Table 2 | Read mapping of assembled SIP-enrichments and 7
high-quality bins against published DWH metagenomic and metatranscriptomic datasets.
Percentage of mapped reads of SIP-enrichments or 7 high-quality genomes against
previously published plume-derived DWH metagenomic and metatranscriptomic
datasets1. Read mapping was performed using the n-hexadecane (HEX), naphthalene
(NAP) and phenanthrene (PHE) SIP-enrichments (scaffolds ≥ 2,500 bp) or the
individual metagenomic bins as reference database. Read mapping was performed
using BWA (Burrows-Wheeler Alignment Tool) using standard settings.
Metagenome Metatranscriptome plume uncontaminated proximal plume distal plume HEX 2.7 0.8 6.3 1.3 NAP 3.2 2.6 6.6 8.2 PHE 2.5 2.5 8.2 8.2 H-Mar 0.2 0.03 0.07 0.05 N-Tha 0.1 0.02 0.01 0 N-Alc 0.3 0.04 0.1 0 N-Alt 0.2 0.04 0.1 0 P-Nep 0.3 0.3 0.1 0 P-Cyc 0.1 0.2 0.04 0 P-Col 0.2 0.1 0.08 0
NATURE MICROBIOLOGY | www.nature.com/naturemicrobiology 3
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NMICROBIOL.2016.57

4
Supplementary Figures 1-6
Supplementary Figure 1 | Metagenomic binning using Emergent Self-Organizing Maps (ESOM).
ESOM of genomic sequence fragments based on their tetranucleotide frequency
considering all scaffolds in the SIP-enrichments with a length >4,000 bp. a, Topography (U-Matrix) representing the structure of the underlying tetranucleotide
frequency data. Large distances in tetranucleotide frequencies mark natural divisions
between taxonomic groups. b, ESOM map highlighting hydrocarbon-degrading
bacterial bins as reconstructed from the U-matrix depicted in a. Each square equals a
sequence fragment and color-coding refers to individual metagenomic bins. Bold:
high-quality bins (completeness >50%).
4 NATURE MICROBIOLOGY | www.nature.com/naturemicrobiology
SUPPLEMENTARY INFORMATION DOI: 10.1038/NMICROBIOL.2016.57

5
Supplementary Figure 2 | Relative abundance of bacterial taxa detected in SIP-
enrichments and average bin coverage of reconstructed genomic bins.
a-b, Relative abundance (%) of detected bacterial orders (a) and genera (b) in n-
hexadecane (HEX), naphthalene (NAP) and phenanthrene (PHE) SIP-enrichments.
Only the 20 most abundant taxa are depicted. c, Average bin coverage of assembled
high-quality and fragmentary genomes (colored and grey, respectively). High-quality
genomes are defined by a completeness >50% and colors refer to HEX (purple), NAP
(green) and PHE (blue) degraders. Average bin coverage was estimated by the
number of reads that were assigned to scaffolds within each bin.
HE
X
NA
P
PH
E
HE
X
NA
P
PH
E0
25
50
75
100 MethylococcalesGammaproteobacteriaFlavobacteriiaLactobacillalesClostridialesChromatialesEnterobacterialesRhizobialesBurkholderialesVibrionalesActinomycetalesBacillalesPseudomonadalesFlavobacterialesThiotrichalesRhodospirillalesRhodobacteralesOceanospirillalesAlteromonadalesunassigned
0
25
50
75
100 ThalassotaleaVibrioPaenibacillusGlaciecolaThalassolituusMaricaulisPseudomonasOlleyaPseudophaeobacterPseudoalteromonasColwelliaNeptuniibacterCitreicellaCycloclasticusMarinobacterThalassospiraHalomonasAlcanivoraxAlteromonasunassigned
0
50
100
150
200
250
300
350
0 2 4 6 8 10 12 14 16 18 20
H-Mar
N-Alt
N-Alc
P-Col
P-Nep
P-Cyc
N-ThaAv
erag
e bi
n co
vera
geR
elat
ive
abun
danc
e (%
)
Rel
ativ
e ab
unda
nce
(%)
Metagenomic bins
c
a b
NATURE MICROBIOLOGY | www.nature.com/naturemicrobiology 5
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NMICROBIOL.2016.57

6
Supplementary Figure 3 | Phylogenetic 16S rRNA gene tree.
Maximum-likelihood based phylogenetic tree of 16S rRNA genes retrieved from n-
hexadecane (HEX), naphthalene (NAP) and phenanthrene (PHE)-degrading
communities (red). For comparison, sequences from previously published datasets
were included, comprising previous SIP-clone libraries as well as plume and sea
surface samples collected during the DWH spill (green, dark blue and light blue,
respectively)2–4. Black, grey and white circles: bootstrap support in percent. Black,
grey and white circles: nodes with bootstrap values of 100, 75-100 and <75% (1,000
replicates).
PHE_SIP: AB071990.1.1532
NAP_SIP: EU799707.1.1502Gammaproteobacterium HTCC2207, AAPI0100
JN018705.1| Uncultured bacterium clone V−S−32 16S ribosomal RNA gene, par, 34766354
Thalassomonas sp. M−M1, HM237288Alteromonas litorea, AY428573
NAP_SIP: DQ836765.1.1479
JN018698.1| Uncultured bacterium clone 85−S−69 16S ribosomal RNA gene
JN018730.1| Uncultured bacterium clone V−S−59 16S ribosomal RNA geneHEX_SIP:GU145456.1.1494JN018694.1| Uncultured bacterium clone 73−S−61 16S ribosomal RNA gene
AM279755.1DQ513004.1
HEX_SIP: AM286690.530914.532455
Halomonas sp. TGOS−10, JQ246432Betaproteobacteria
Rhodobacteraceae bacterium enrichment culture clone TBNAP74, JN166983JN018671.1| Uncultured bacterium clone 47−S−27 16S ribosomal RNA gene
Thalassobius sp. J86, EU143368JN018675.1| Uncultured bacterium clone 47−S−72 16S ribosomal RNA gene
Thalassobius mediterraneus, AB607868HEX_SIP: scaffold_905 length_3138, scaffold
Maricaulis sp. MCS28, AJ227810Maricaulis maris, AB008849JN018674.1| Uncultured bacterium clone 47−S−68 16S ribosomal RNA geneThalassospira profundimaris, HQ425693
Thalassospira xianhensis, HM587995Rhodospirillaceae bacterium 03PA−Blue−76, AB371589
SAR116 cluster Alphaproteobacterium HIMB100, AFXB0100JN018691.1| Uncultured bacterium clone 73−S−52 16S ribosomal RNA gene
Rhodospirillales bacterium 2622, EU83398Salibacter luridus, AM712887
PHE_SIP: scaffold_4625 length_2834,Flavobacterium psychrophilum, AB297
Olleya marilimosa, AY58652Olleya_marilimosa_SWPHE27_AY586527
Olleya sp. L−4, FJ88671JN018732.1| Uncultured bacterium clone V−S−61 16S r
FirmicutesSynechococcus sp. MI0608F, FJ763774
Synechococcus sp. ACT 0615, HQ859461JN018688.1| Uncultured bacterium clone 73−S−72 16S ribosomal RNA gene
Thermotogae10
BacteroidetesAlphaproteobacteria
Gammaproteobacteria
ThiotrichalesAlteromonadales
Oceanospirillales
100% probability
75-100% probability
Metagenomic binsSIPPlumeSea surface <75% probability
Cycloclasticus sp. E3, AB080113SIP-Cycloclasticus_SWNAP12_KC242319
Cycloclasticus pugetii, CS187042Cyzanc gi, ARB_4AEF
Colwellia hornerae, JN175346Colwellia piezophila, AB094412SIP_Colwellia_SPHE03_KC242316
Pseudoalteromonas sp., X86462 AM747817.1Oceaniserpentilla haliotis, AM747817Oceaniserpentilla haliotis, AB540014
AM117931.1HM587890.1
AF468253, AF468253.1, AF468253 Oleibacter marinus, AB435650 Oleibacter marinus, AB4356491494_gi240119438Oleibacter marinus, AB435651
AJ302699.1
match_AM286690.530914.532455 Alcanivorax borkumensis, Y12579
SIP_Alcanivorax_DWHEX05_KC242317Alcanivorax sp. I4, AB053125
Marinobacter algicola, AY258116Marinobacter algicola DG893, ABCP0100SIP_Marinobacter_DWHEX95_KC242320
Halomonas campisalis, DQ077909
Thermotogae
SIP_Alteromonas_SWPHE06_KC242318 Alteromonas marina, AF529060Pseudoalteromonas marina, FJ040185
6 NATURE MICROBIOLOGY | www.nature.com/naturemicrobiology
SUPPLEMENTARY INFORMATION DOI: 10.1038/NMICROBIOL.2016.57

7
Supplementary Figure 4 | Read mapping of SIP-assemblies against
metatranscriptomic DWH plume samples.
Total numbers of reads (log2-transformed) of DWH metatranscriptomic data mapped
against SIP-assemblies. Read mapping was performed using the n-hexadecane (HEX),
naphthalene (NAP) and phenanthrene (PHE) SIP-enrichments as reference database
against reads from proximal (grey) or distal (black)-derived metatranscriptomic
samples from Mason et al., 2012. Read mapping was performed using BWA
(Burrows-Wheeler Alignment Tool) using standard parameters.
Genes
HEX
0
5
10
15
Cou
nts
(log 2
)
proximal plume
distal plume
NAP
0
5
10
15
Cou
nts
(log 2
)
PHE
0
5
10
15
Cou
nts
(log 2
)
Genes
Genes
NATURE MICROBIOLOGY | www.nature.com/naturemicrobiology 7
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NMICROBIOL.2016.57

8
Supplementary Figure 5 | Overlapping gene annotations between metagenomic
bins with metatranscriptomic DWH plume samples.
Overlap between gene annotations from 7 high-quality draft genomes and a published
metatranscriptomic dataset consisting of samples collected proximal and distal from
the plume as published by Mason et al., 2012. Depicted are the 50 most abundant
detected gene annotations. Read mapping was performed using RapSearch2 using the
amino acid sequences from the 7 draft genomes as reference database (e-value
threshold of 0.001). Total counts refer to the total number of mapped reads.
H-Mar
N-ThaN-AlcN-AltP-CycP-NepP-Col
8 NATURE MICROBIOLOGY | www.nature.com/naturemicrobiology
SUPPLEMENTARY INFORMATION DOI: 10.1038/NMICROBIOL.2016.57

9
Supplementary Figure 6 | Overlapping and unique gene annotations retrieved
from different hydrocarbon-degrading bacterial communities.
a, Overlap between retrieved gene annotations derived from n-hexadecane (HEX),
phenanthrene (PHE) and naphthalene (NAP) SIP-enrichments. b, Heatmap depicting
the relative abundance and distribution of the 100 most abundant gene annotation
terms across the HEX-, NAP- and PHE-assemblies. Overlap and unique gene
annotations depicted in a, are detailed in Supplementary Data S1.
Transcriptional regulatorUncharacterized protein conserved in bacteriaAraC−type DNA−binding domain−containing proteinsAcyl−CoA dehydrogenasesSignal transduction histidine kinasePredicted hydrolases or acyltransferases (alpha/beta hydrolase superfamily) − COG0596Response regulator containing a CheY−like receiver domain and a GGDEF domainUncharacterized conserved proteinResponse regulator containing CheY−like receiver, AAA−type ATPase, and DNA−binding domainsCation/multidrug efflux pumpTransposase and inactivated derivativesGlutathione S−transferaseMethyl−accepting chemotaxis proteinDehydrogenases with different specificities (related to short−chain alcohol dehydrogenases)FOG: GGDEF domainNAD−dependent aldehyde dehydrogenasesGlycosyltransferaseResponse regulators consisting of a CheY−like receiver domain and a winged−helix DNA−binding domainFOG: WD40 repeatSerine/threonine protein kinaseProtein kinase domainABC−type multidrug transport system, ATPase componentSuperfamily II DNA and RNA helicasesCation transport ATPaseADP−ribose pyrophosphataseDnaJ−class molecular chaperone with C−terminal Zn finger domainIsopropylmalate/homocitrate/citramalate synthasesMembrane protein involved in the export of O−antigen and teichoic acidCo/Zn/Cd efflux system componentMembrane proteins related to metalloendopeptidasesTranscription elongation factorMembrane protease subunits, stomatin/prohibitin homologstRNA_Met_CATRhodanese−related sulfurtransferaseUniversal stress protein UspA and related nucleotide−binding proteinsAspartate/tyrosine/aromatic aminotransferaseATPase components of ABC transporters with duplicated ATPase domainsRestriction endonuclease S subunitsType I restriction−modification system methyltransferase subunitFatty acid desaturaseAsp−tRNAAsn/Glu−tRNAGln amidotransferase A subunit and related amidasesPredicted thioesterase − COG0824Short−chain alcohol dehydrogenase of unknown specificityABC−type branched−chain amino acid transport system, permease componentBranched−chain amino acid ABC−type transport system, permease componentsABC−type branched−chain amino acid transport systems, periplasmic componentTonB−dependent Receptor Plug DomainTRAP−type C4−dicarboxylate transport system, large permease componentOuter membrane receptor for ferrienterochelin and colicinsABC−type dipeptide/oligopeptide/nickel transport systems, permease componentsTRAP−type C4−dicarboxylate transport system, small permease componentTRAP−type C4−dicarboxylate transport system, periplasmic componentAcetylornithine deacetylase/Succinyl−diaminopimelate desuccinylase and related deacylasescAMP−binding proteins − catabolite gene activator and regulatory subunit of cAMP−dependent protein kinasesMultidrug resistance efflux pumpPutative threonine efflux proteinMethyltransferase domainNADPH:quinone reductase and related Zn−dependent oxidoreductasesPhosphoglycerate dehydrogenase and related dehydrogenasesGlycine/D−amino acid oxidases (deaminating)Beta−lactamase class C and other penicillin binding proteinsABC−type uncharacterized transport system, permease componentNADH:flavin oxidoreductases, Old Yellow Enzyme familyABC−type multidrug transport system, ATPase and permease componentsPredicted permeases − COG0730FAD/FMN−containing dehydrogenasesNTP pyrophosphohydrolases including oxidative damage repair enzymesNa+/proline symporterLactoylglutathione lyase and related lyasesShort−chain dehydrogenases of various substrate specificitiesABC−type branched−chain amino acid transport systems, ATPase componentCholine dehydrogenase and related flavoproteins1−acyl−sn−glycerol−3−phosphate acyltransferaseUncharacterized protein, possibly involved in aromatic compounds catabolismAcetyltransferasesOuter membrane receptor proteins, mostly Fe transportFOG: EAL domainMajor Facilitator SuperfamilyHistidine kinase−, DNA gyrase B−, and HSP90−like ATPaseAcyl−CoA synthetases (AMP−forming)/AMP−acid ligases IIOuter membrane proteinSite−specific recombinase XerDNucleoside−diphosphate−sugar epimerasesPyruvate/2−oxoglutarate dehydrogenase complex, dihydrolipoamide dehydrogenase (E3) component, and related enzymesPutative silver efflux pumpAcetyl−CoA acetyltransferaseOuter membrane protein and related peptidoglycan−associated (lipo)proteinsATPases involved in chromosome partitioningEAL domainSmall−conductance mechanosensitive channelPeroxiredoxinZn−dependent hydrolases, including glyoxylasesDNA−directed RNA polymerase specialized sigma subunit, sigma24 homologGlycosyltransferases involved in cell wall biogenesisResponse regulator containing a CheY−like receiver domain and an HTH DNA−binding domainArabinose efflux permeaseABC−type amino acid transport/signal transduction systems, periplasmic component/domainPredicted transcriptional regulators − COG0789Enoyl−CoA hydratase/carnithine racemase
HEX PH
E
NAP
0 2 4 6 8 10
relative abundance (%)12
1 9 0 9
2 2 3 6 4 0
1 1 51 8 8 1 0 5 4
6 2 7
Hex Phe
Nap
a bMembrane-fusion protein
NATURE MICROBIOLOGY | www.nature.com/naturemicrobiology 9
SUPPLEMENTARY INFORMATIONDOI: 10.1038/NMICROBIOL.2016.57

10
References
1. Mason, O. U. et al. Metagenome, metatranscriptome and single-cell sequencing reveal
microbial response to Deepwater Horizon oil spill. ISME J. 6, 1715–1727 (2012).
2. Gutierrez, T. et al. Hydrocarbon-degrading bacteria enriched by the Deepwater Horizon
oil spill identified by cultivation and DNA-SIP. ISME J. 7, 2091–2104 (2013).
3. Hazen, T. C. et al. Deep-sea oil plume enriches indigenous oil-degrading bacteria.
Science 330, 204–208 (2010).
4. Redmond, M. C. & Valentine, D. L. Natural gas and temperature structured a microbial
community response to the Deepwater Horizon oil spill. Proc. Natl. Acad. Sci. 109,
20292–20297 (2012).
10 NATURE MICROBIOLOGY | www.nature.com/naturemicrobiology
SUPPLEMENTARY INFORMATION DOI: 10.1038/NMICROBIOL.2016.57
![REAMBLE ARC2471C MEDICAIDWAIVERSERVICES - … · MEDICAIDWAIVERSERVICES PREAMBLE ... [ARC2471C,IAB3/30/16,effective5/4/16] DIVISIONI—HCBSHEALTHANDDISABILITYWAIVERSERVICES 441—83.1(249A)](https://img.pdfslide.us/doc/110x75/5adb4cd67f8b9a6d318de199/reamble-arc2471c-medicaidwaiverservices-preamble-arc2471ciab33016effective5416.jpg)



![MIC: Mining Interclass Characteristics for Improved Metric ... › content_ICCV_2019 › papers › Roth_MI… · MIC+ProxyNCA 128 60.6 72.2 81.5 64.9 Margin[39] 128 63.6 74.4 83.1](https://img.pdfslide.us/doc/110x75/60c1a11601c66a5ef1527f7e/mic-mining-interclass-characteristics-for-improved-metric-a-contenticcv2019.jpg)
























