Embed Size (px)
Citation preview

Tetrahedron: Asymmetry 24 (2013) 515–525
Contents lists available at SciVerse ScienceDi rect
Tetrahedro n: Asym metry
journal homepage: www.elsevier .com/locate / tetasy
Study of asymmetric aldol and Mannich reactions catalyzed by proline–thiourea host–guest complexes in nonpolar solvents
Ayhan Sıtkı Demir a,z, Sinan Basceken a,b,⇑a Department of Chemistry, Middle East Technical University, 06531 Ankara, Turkey b Department of Chemistry, Hitit University, 19030 Corum, Turkey
a r t i c l e i n f o a b s t r a c t
Article history:Received 26 February 2013 Accepted 13 March 2013
0957-4166/$ - see front matter � 2013 Elsevier Ltd. Ahttp://dx.doi.org/10.1016/j.tetasy.2013.03.014
⇑ Corresponding author. Tel.: +90 (0)364 227 7000;E-mail address: [email protected] (S. Bas
z Deceased on Professor Dr. Ayhan Sıtkı Demir (1950
A proline–thiourea host–guest complex has been described as a good catalyst for asymmet ric reactions such as aldol and Mannich reactions. High stereoselectivities were obtained under optimal conditions.Thiourea was observed to have an important effect on the reactivity and selectivity, even in an unconven- tional nonpolar reaction medium and without the need to utilize low temperatures. This proline–thio-urea host–guest system has the ability to participate in a hydrogen bonding network.
� 2013 Elsevier Ltd. All rights reserved.
1. Introduction
Asymmetric catalysis with proline was first realized in the Hajos–Parrish–Eder–Sauer–Wiechert reaction, an enantiogroup differentiating aldol cyclization.1 The field of enamine catalysis was effectively developed by Barbas, Lerner, and List;1 theyshowed that small organic molecule s, such as proline, could cata- lyze some chemical reactions as well as larger organic molecules (enzymes) by using similar mechanis ms. The works of McMillan et al.2 on iminium catalysis, urea derivatives from Jacobsen 3 andothers have provided interesting contributi ons in this field of organocatal ysis.
Although proline is a relatively good catalyst, it is not without some potential drawbacks, such as (1) low solubility, which thus limits reactivity in typical organic solvents; (2) potential side reac- tions and established parasitic equilibria with substrates;4 and (3)low selectiviti es with planar, aromatic aldehydes in direct organo- catalytic reactions. Therefore, a considerable effort has been direc- ted toward the developmen t of proline analogs in order to improve the reactivity, selectivity , and scope. Consideri ng the practical syn- thetic issues, the carboxyli c acid moiety of the proline has been tar- geted as a site for modifications, in which the reactivity and selectivity of proline were enhanced in custom-made catalysis,even though the identification of a good catalyst in turn required the synthesis of various analogue s of a proposed catalyst design in order to identify the optimal one. Moreover, the improved cata- lyst was usually obtained through modification of proline with
ll rights reserved.
fax: +90 (0)364 227 7005.ceken).–2012).
chiral molecules with additional functionality , which are much more precious than the proline itself.5
The basic catalyst system for developing catalysts is modular in nature, and offers an opportunity for structural diversity. However,our main interest lies in using these catalysts in combination with a library of achiral thiourea additives in order to multiply the num- ber of new catalysts available by means of complementar y hydro- gen bonding interactions . Thioureas are readily available , and the use of amine and isothiocyan ates allows for a range of these addi- tives to be readily synthesized.6 For our proof of concept studies, arelatively small library was generated, and the discussion herein focuses on the catalysts generate d from proline and other amino acids and achiral thiourea derivatives.
2. Results and discussion
Herein we report the synthesis of two types of bipyridine de- rived achiral thioureas and their application as co-catalysts (guest)in the proline (host) catalyzed asymmetric aldol and Mannich reactions .
2.1. Synthesi s of bipyridine derived achiral thioureas
The preparation of bipyridine derived thiourea 5 was per- formed from commerc ially available 4,4 0-dimethyl-2,20-bipyridine1, according to the seven-ste p route illustrate d in Scheme 1.7 Thisprocedure involves the oxidation of the methyl groups to carbox- ylic acids with potassium permanganate . The subsequent esterifi-cation with methanol, reduction of the ester to the alcohol with sodium borohydride , substitution with hydrobromic acid, reaction with phthalimide potassium salt, and then reaction with hydra- zine monohydrate gave 4,4 0-bis(aminomethyl)-2,20-bipyridine 4.7
As a result, 1,1 0-([2,20-bipyridine]- 4,4 0-diylbis(methylene))bis

N
N
1
i, ii, iii
N
N
2
OH
OH
N
N
3
Br
Br
iv
N
N
4
NH2
NH2
v, vi
NCS
CF3F3C
CHCl3, 6 h, rt85%
CF3
NH
F3C NH
S
N
NHN
S
HN CF3
CF3
5
Scheme 1. Synthesis of novel thioureas. Reagents and conditions:7 (i) KMnO 4, H2SO4, D, 24 h, 50%; (ii) MeOH, H2SO4, D, 98%; (iii) EtOH, NaBH 4, D, 97%; (iv) HBr, D, 60%; (v)phthalimide potassium salt, DMF; (vi) H2NNH2.H2O, EtOH, D, 98%.
516 A. S. Demir, S. Basceken / Tetrahedron: Asymmetry 24 (2013) 515–525
(3-(3,5-bis(triflu-oromethyl)phenyl)-thiourea) 5 was obtained by reaction of 3,5-bis(trifluoromethyl)phenyl isothiocyan ate with 4,40-bis(aminomethyl)-2,20-bipyridine 4 in chloroform.
N
N
NH2
H2N
N
N
HN
HN
S
CF3
CF3
NH
NH
S
CF3
F3C
F3C CF3
NCS
acetone, 48 h, rt
6 7
70%
1,10-([2,2 0-Bi pyri dine ]-4,40-di yl)bis(3-(3,5-bi s(trifluorom et- hy l)phenyl)thiourea) 7 was prepared by the reaction of 3,5-bis(trifluo-romethyl)phenyl isothiocyan ate with commerciall y available 4,4 0-diamino-2,20-bipyridine 6 in acetone, according to the step shown in Scheme 1.
2.2. Direct asymmetric aldol reaction
The first amine-catal yzed, direct asymmetric intermolecu lar al- dol reaction was develope d by List et al. in 2000.8 Proline catalyzed direct aldol reactions have been shown experimental ly and com- putationally to proceed through enamine intermediates, in which the transition state (TS)-A is highly stabilized by hydrogen bond donation from a carboxylic acid moiety as can be seen in Figure 1.8
NO
OHO
RH
δ-δ+
A
NO
OHO
RH
SHN
HN
N
F3C
CF3
BHost
Gue
st
δ+δ-
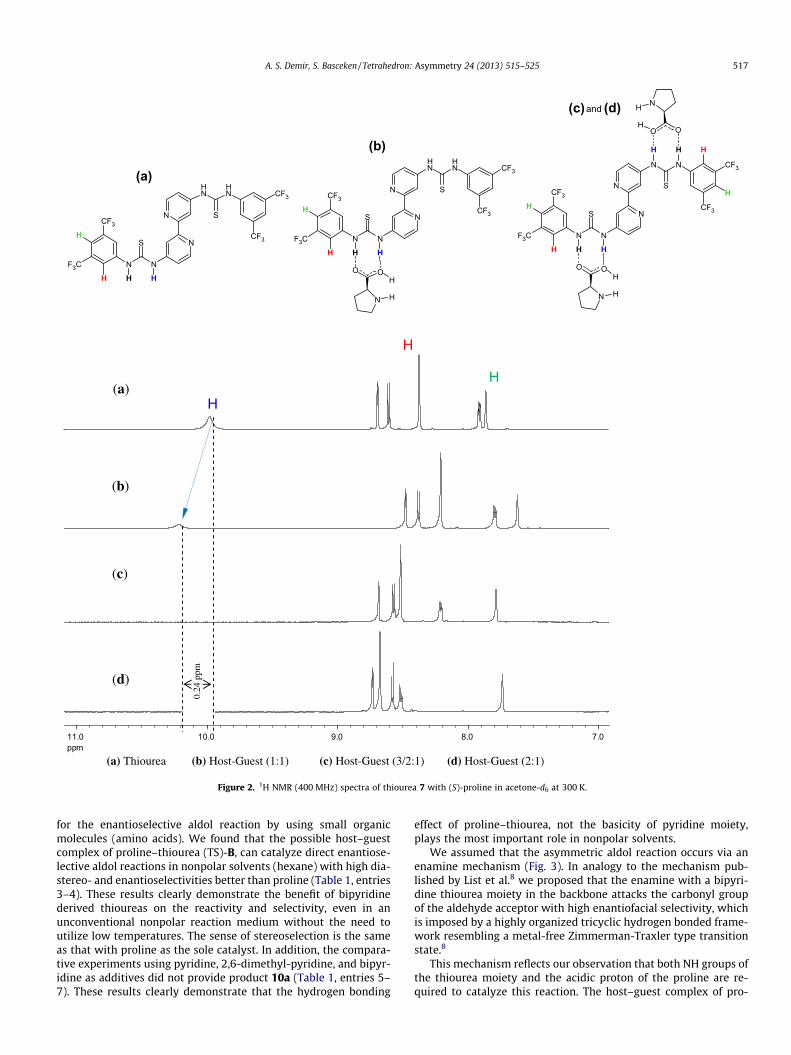
Figure 1. (A) The List–Houk model of (S)-proline catalyzed intermolecular aldol reactions,8 (B) The possible interaction of the bipyridine derived thiourea moiety 7with (S)-proline in the transition state.
In order to elucidate the structural changes upon binding be- tween host and guest, we examined the hydrogen bond interac- tions of bipyridine derived thiourea (guest) 7 with proline (host)
in solution. Some information about the nature of the proline–thio- urea species was obtained from the 1H NMR experiments (Fig. 2),with spectra being recorded in acetone- d6. The results of these experime nts showed that following the addition of proline in the mixture, the N–H proton of the thiourea underwent a downfieldshift (Dd = 0.24 ppm) and decrease d in intensity due to the domi- nant interactions (Fig. 2), that are present in the formatio n of ahydrogen bonding network, as shown in Figure 2.
The data collected thus far indicated that a specific proline–thiourea interaction may operate to afford an enantioselective reaction. Exactly which intermediates and transition states oper- ate, or whether an alternative transition state is involved , is cur- rently under investigation in our laborator y. The possibility of fine-tuning similar proline–thiourea interactions also presents apossible strategy for asymmetric catalyst developmen t.8
In order to optimize the asymmetric catalyst, the role of suitable additives or co-cataly sts can be crucial in enhancing the reactivity and stereoselectivity of the catalytic system. For example, it has been shown that the addition of a small amount of water often accelerates the reaction rate and increases the enantioselectivi ty of proline-catal yzed aldol reactions.8 Recently, Shan et al. have shown that using chiral diols as additives can improve the enanti- oselectiv ity of proline catalyzed aldol reactions, probably through their involvement in the transition state, via the formation of ahydrogen bonding network.8
As a testing ground for the new approach we decided to study the enantioselecti ve aldol reaction of ketones to aldehydes. This was done with the idea of developing a simple catalytic solution
ppm7.08.09.010.011.0
(a) Thiourea (b) Host-Guest (1:1) (c) Host-Guest (3/2:1) (d) Host-Guest (2:1)
H
(a)
(b)
(c)
(d)
H
0.24
ppm
H
N
N
HN
HN
S
CF3
CF3
NN
S
CF3
F3C
H
H
H H
(a)N
N
HN
HN
S
CF3
CF3
NN
S
CF3
F3C
H
H
H H
N
O O
H
H
(b)
N
N
N N
S
CF3
CF3
NN
S
CF3
F3C
H
H
H H
N
O O
H
H
(c)and (d) N
OO
H
H
H H H
H
Figure 2. 1H NMR (400 MHz) spectra of thiourea 7 with (S)-proline in acetone- d6 at 300 K.
A. S. Demir, S. Basceken / Tetrahedron: Asymmetry 24 (2013) 515–525 517
for the enantioselecti ve aldol reaction by using small organic molecules (amino acids). We found that the possible host–guestcomplex of proline–thiourea (TS)-B, can catalyze direct enantiose- lective aldol reactions in nonpolar solvents (hexane) with high dia- stereo- and enantioselecti vities better than proline (Table 1, entries 3–4). These results clearly demonstrat e the benefit of bipyridin ederived thioureas on the reactivity and selectivity, even in an unconventiona l nonpolar reaction medium without the need to utilize low temperat ures. The sense of stereoselect ion is the same as that with proline as the sole catalyst. In addition, the compara- tive experiments using pyridine, 2,6-dimethyl-py ridine, and bipyr- idine as additives did not provide product 10a (Table 1, entries 5–7). These results clearly demonstrat e that the hydrogen bonding
effect of proline–thiourea, not the basicity of pyridine moiety,plays the most important role in nonpolar solvents.
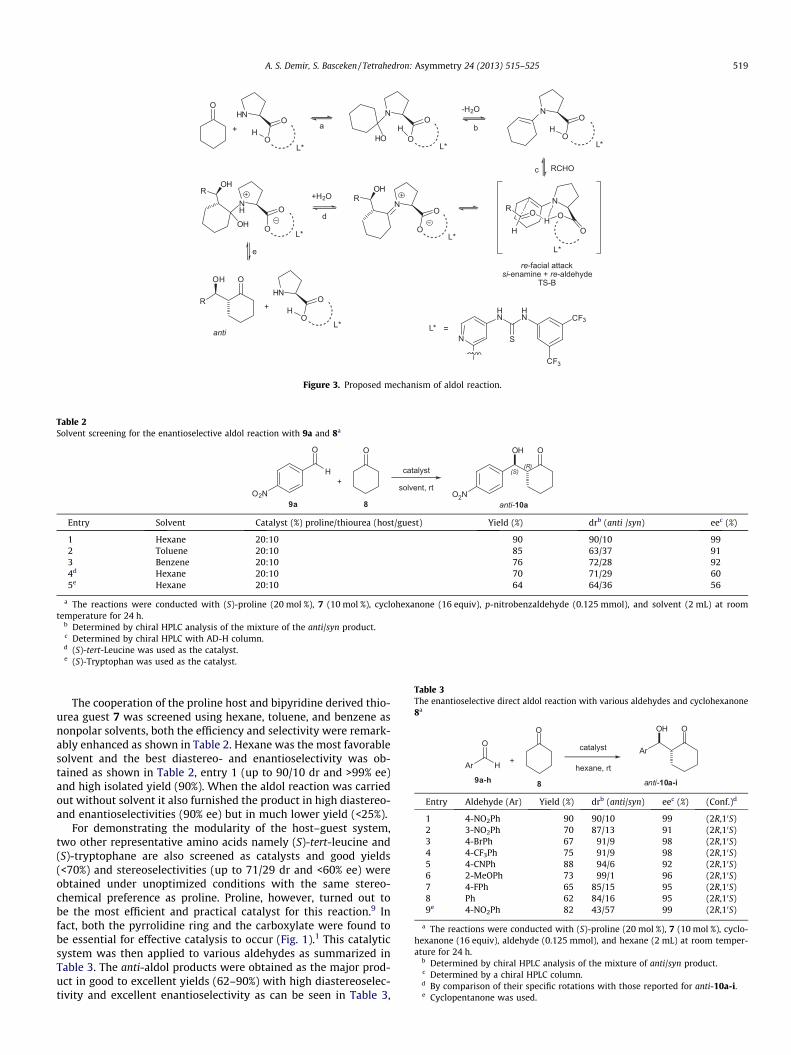
We assumed that the asymmetric aldol reaction occurs via an enamine mechanism (Fig. 3). In analogy to the mechanism pub- lished by List et al.8 we proposed that the enamine with a bipyri- dine thiourea moiety in the backbone attacks the carbonyl group of the aldehyde acceptor with high enantiofacia l selectivity, which is imposed by a highly organized tricyclic hydrogen bonded frame- work resembling a metal-free Zimmerman- Traxler type transition state.8
This mechanism reflects our observation that both NH groups of the thiourea moiety and the acidic proton of the proline are re- quired to catalyze this reaction. The host–guest complex of pro-

Table 1Enantioselective direct aldol reaction with p-nitrobenzaldehyde 9a and cyclohexanone 8 by using various catalysts in hexane a
O2N
H
O O
(S)(R)
OOH
O2N+
anti-10a9a 8
hexane, rt
catalyst
Entry Guest Catalyst loading (mol %) Yield (%) dr b (anti:syn) eec (%)
1N N
HNH
SI
20 40 55:45 46
2
N NH
NH
S
CF3
CF3
II 20 60 75:25 79
3
N
N
NH
HN
NH
HN
S
S
F3C
CF3
F3C
CF3
5 10 75 74:26 92
4
N
N
HN
NH
HN
NH
S
S
CF3
CF3
F3C
F3C
7 10 90 90:10 99
5N
20 0 — —
6N
20 0 — —
7 NN 20 0 — —
a The reactions were conducted with (S)-proline (20 mol %), thiourea or pyridine derivatives (10–20 mol %), cyclohexanone (16 equiv), p-nitrobenzaldehyde (0.125 mmol)and hexane (2 mL) at room temperature for 24 h.
b Determined by chiral HPLC analysis of the mixture of anti/syn product.c Determined by a chiral HPLC column.
518 A. S. Demir, S. Basceken / Tetrahedron: Asymmetry 24 (2013) 515–525
line–thiourea may facilitate each individual step of the mechanism (Fig. 3), including the nucleophi lic attack of the amino group (a),the dehydration of the carbinol amine intermediate (b), the car- bon–carbon bond forming step (c), and both steps of the hydrolysis of the iminium–aldol intermediate (d and e).8 Typically, anti-dia-stereoselect ivity is observed in the proline–thiourea catalyzed al- dol reaction, although other possible mechanism s cannot be ruled out at this stage.
We began the investigations with donor ketone cyclohexanone 8 and acceptor aldehyde p-nitrobenzalde hyde 9a under proline catalysis and pyridine derived thioureas (Table 1, entries 1–2) as the additive in a nonpolar solvent. After the first results of the aldol
reaction (46% ee, Table 1, entry 1), we started to develop multi- functiona l thioureas with bipyridine moieties to examine several functiona lities for further modification and applied them to enan- tioselective aldol reactions . Among them, four functionalized thio- ureas showed high ee and yields as shown in Table 1. The catalyst combinati ons of proline and the synthesized bipyridine bridged bis-thiour eas (Table 1, entries 3–4) showed better catalytic activi- ties and stereoselectiviti es than those of the co-catalysts with asingle thiourea moiety (Table 1, entries 1–2). Although the exact mechanis m is not clear, the possible cooperation of the two thio- urea moieties may account for this advantag e. The selected thio- urea 7 (Table 1, entry 4) was the optimum choice.
OHN
O
O+
NO
OHO
-H2O NO
O
N
O
OHH
OR
RCHO
re-facial attacksi-enamine + re-aldehyde
TS-B
NO
O
ROH
NH O
OOH
ROH
OHN
O
O+R
OH
L*
H
L*
H
L*L*
+H2O
L* =N
HN
HN
S
CF3
CF3
a b
c
d
e
L*
H
L*
L*
H
anti
Figure 3. Proposed mechanism of aldol reaction.
Table 2Solvent screening for the enantioselective aldol reaction with 9a and 8a
O2N
H
O O
(S)(R)
OOH
O2N+
anti-10a9a 8
solvent, rt
catalyst
Entry Solvent Catalyst (%) proline/thiourea (host/guest) Yield (%) dr b (anti /syn) eec (%)
1 Hexane 20:10 90 90/10 99 2 Toluene 20:10 85 63/37 91 3 Benzene 20:10 76 72/28 92 4d Hexane 20:10 70 71/29 60 5e Hexane 20:10 64 64/36 56
a The reactions were conducted with (S)-proline (20 mol %), 7 (10 mol %), cyclohexanone (16 equiv), p-nitrobenzaldehyde (0.125 mmol), and solvent (2 mL) at room temperature for 24 h.
b Determined by chiral HPLC analysis of the mixture of the anti/syn product.c Determined by chiral HPLC with AD-H column.d (S)-tert-Leucine was used as the catalyst.e (S)-Tryptophan was used as the catalyst.
Table 3The enantioselective direc t aldol reaction with various aldehydes and cyclohexanone 8a
Ar H
OO
Ar
OOH
+
anti-10a-i9a-h 8
hexane, rt
catalyst
Entry Aldehyde (Ar) Yield (%) dr b (anti/syn) eec (%) (Conf.)d
1 4-NO 2Ph 90 90/10 99 (2R,10S)2 3-NO 2Ph 70 87/13 91 (2R,10S)3 4-BrPh 67 91/9 98 (2R,10S)4 4-CF 3Ph 75 91/9 98 (2R,10S)5 4-CNPh 88 94/6 92 (2R,10S)6 2-MeOPh 73 99/1 96 (2R,10S)7 4-FPh 65 85/15 95 (2R,10S)8 Ph 62 84/16 95 (2R,10S)9e 4-NO2Ph 82 43/57 99 (2R,10S)
a The reactions were conducted with (S)-proline (20 mol %), 7 (10 mol %), cyclo- hexanone (16 equiv), aldehyde (0.125 mmol), and hexane (2 mL) at room temper- ature for 24 h.
b Determined by chiral HPLC analysis of the mixture of anti/syn product.c Determined by a chiral HPLC column.d By comparison of their specific rotations with those reported for anti-10a-i.e Cyclopentanone was used.
A. S. Demir, S. Basceken / Tetrahedron: Asymmetry 24 (2013) 515–525 519
The cooperation of the proline host and bipyridine derived thio- urea guest 7 was screened using hexane, toluene, and benzene as nonpolar solvents, both the efficiency and selectivity were remark- ably enhanced as shown in Table 2. Hexane was the most favorable solvent and the best diastereo- and enantioselectivi ty was ob- tained as shown in Table 2, entry 1 (up to 90/10 dr and >99% ee)and high isolated yield (90%). When the aldol reaction was carried out without solvent it also furnished the product in high diastereo- and enantioselectivi ties (90% ee) but in much lower yield (<25%).
For demonstrat ing the modularity of the host–guest system,two other representat ive amino acids namely (S)-tert-leucine and (S)-tryptophane are also screened as catalysts and good yields (<70%) and stereoselectiviti es (up to 71/29 dr and <60% ee) were obtained under unoptimized conditions with the same stereo- chemical preferenc e as proline. Proline, however, turned out to be the most efficient and practical catalyst for this reaction.9 Infact, both the pyrrolidine ring and the carboxylate were found to be essential for effective catalysis to occur (Fig. 1).1 This catalytic system was then applied to various aldehydes as summarized in Table 3. The anti-aldol products were obtained as the major prod- uct in good to excellent yields (62–90%) with high diastereoselec- tivity and excellent enantioselecti vity as can be seen in Table 3,

NO
OH
H R
N
R'
NO
OH
H R
NH2PMP
R'
NN
S
N
CF3
CF3
HH
AB
Guest
HostH3CO a
Figure 4. (A) The List model of the TS of the Mannich reaction, (B) The possible interaction of the bipyridine derived thiourea 7 with (S)-proline in the TS.
520 A. S. Demir, S. Basceken / Tetrahedron: Asymmetry 24 (2013) 515–525
entries 1–8. The reactions with aldehydes bearing high electron-wi thdrawing groups proceeded smoothly to afford the al- dol adducts in 24 h (Table 3, entries 1–5 and 7), with a good level of diastereosel ectivity. For the neutral and electron-rich aromatic aldehydes (Table 3, entries 6 and 8), the reactions commonly re- quired a longer time (48 h or 72 h).
Concerning the anti/syn and absolute configuration, the same preference as proline is obtained. The modular catalysts can be ap- plied to aldol type C–C bond forming reactions in high conversion,high diastereo- and enantioselectivi ty.
2.3. Asymmetric Mannich reactions
The Mannich reaction is a highly useful transformation for the construction of nitrogenous molecules.10 In this transformation ,three components, namely two carbonyl compounds and an amine,react to form a b-amino-car bonyl compound. List et al. found the first proline-catal yzed Mannich reaction; that proline catalyzed the direct asymmetr ic three component Mannich reaction of ke- tones with aldehydes and p-anisidine with high ee’s and dr’s,11
with opposite enantiofacia l selectivity compared to the proline- catalyzed aldol reaction. Consequently, the major product had the opposite absolute configuration at the stereogenic center bear- ing the heteroatom . The diastereosel ectivity was also inverted with syn-products in the Mannich and anti-products in the aldol reaction.
In our initial experiment, we found that the possible proline–thiourea host–guest system (TS)-B (Fig. 4), can catalyze the asym- metric Mannich reaction of p-nitrobenzalde hyde 9a, p-anisidine11, and acetone in benzene and toluene to give the expected b-amino-carb onyl compound 12a in 45% yield. The correspond ing al- dol product 13 was formed as well under those conditions but in much lower yield (<21%) as shown in Table 4. Among the nonpolar solvents used, the best results were obtained in toluene as can be seen in Table 4, entry 2. Most importantly, high enantioselecti vity was observed in the Mannich products (91% ee) and the absolute configuration was found to be (S).
Table 4Solvent screening of the asymmetri c three component Man nich reactions a
O
H
O
NO2
OMe
H2N
+ +
119a
Entry Solvent Catalyst (%) proline/thiourea (host/g
1 Hexane 20:20 2 Toluene 20:20 3 Benzene 20:20 4d Toluene 20:20
a The reactions were conducted with (S)-proline (20 mol %), 7 (20 mol %), acetone (50temperature for 48 h.
b Determined by chiral HPLC with an AD column.c By comparison of their specific rotations with those reported.d (S)-tert-Leucine was used as the catalyst.
By using aliphatic aldehydes instead of p-nitro-benzaldehy de 9a under the same reaction condition only the Mannich product was obtained. This reaction then carried out with various aliphatic aldehydes 14a–c and the Mannich products 15a–c was obtained in high ee’s as summarized in Table 5.
We were particularly fascinated by the prospect of replacing the aldehyde with imines. The aldimine from p-nitrobenzal dehyde 9aand p-anisidine 11, with acetone in nonpolar solvents such as ben- zene, and toluene with 20 mol % of the proline–thiourea host–guest catalyst furnished only the Mannich product. Toluene was the most favorable solvent and showed the highest effectivity con- cerning of yield and ee’s (91% ee) in Table 6, entry 1. We found that with 20 mol% of catalyst and hexane as a nonpolar solvent the Mannich product is not formed. Under similar conditions, the Man- nich reaction without thiourea gives, after 6 h, a product which was isolated and HPLC analysis showed 20% ee; then adding bipyr- idine derived thiourea 7 in the reaction mixture increased the ee in 48 h to 91%.
Other ketones can readily be used in proline–thiourea catalyzed Mannich reactions with excellent results as summarized in Table 6.Reacting two different ketones (2-butanone, and hydroxyaceton e,each 20 vol %) with the aldimine in toluene furnished the desired products in good yields and with excellent ee’s (95–99%).
catalyst
solvent, rt
NO2
HN
OMe
O
NO2
OHO
+
12a
13
uest) Yield (%) eeb (%) (Conf.)c
n.d n.d —65 91 (S)62 88 (S)50 n.d —
lL), p-anisidine (0.063 mmol), aldehyde (0.063 mmol), and solvent (2 mL) at room

Table 5Three components Mannich reactions with various aliphatic aldehydes (PMP = p-Methoxyphenyl)a
+
O 10 mol% 710 mol% (S)-proline
toluene, rt
(R) R
O HN+ H R
O
OMe
15a-cOMe
NH2
14a-c
Entry Aldehyde (R) Product Yield (%)
eeb
(%)(Conf.)c
1 (CH3)2CHCH2– O HNPMP
90 81 (R)
2 CH 3(CH2)3– O HN
PMP
88 81 (R)
3 (CH3)2CH–O HN
PMP
85 90 (S)
a The reactions were conducted with (S)-proline (10 mol %), 7 (10 mol %), acetone (20 lL), p-anisidine (0.063 mmol), aldehyde (0.063 mmol), and toluene (2 mL) at room temperature for 24 h.
b Determined by chiral HPLC with an OD-H column.c By comparison of their specific rotations with those reported.
Table 6Two component Mannich reactions a
+
O cata
tolue
16a-c
H
N
NO2
R
MeO
Entry Ketone (R) Product
1 –H
O HNPMP
NO2
(S)
2d –CH3
O HNPMP
NO2
(S)(S)
O HNPMP
NO2
(S)
3 –OH
O HNPMP
NO2OH
(S)(R)
a The reactions were conducted with (S)-proline (20 mol %), 7 (20 mol %), ketone (50–b Determined by chiral HPLC with AD or AD-H column.c By comparison of their specific rotations with those reported in the literature.d Two inseparable regioisomeric products.
A. S. Demir, S. Basceken / Tetrahedron: Asymmetry 24 (2013) 515–525 521
Addition ally, in the case of 2-butano ne 16b and hydroxyaceton e16c excellent diastereos electivities were observed. High regiose- lectivities generally favoring products resulting from the higher substitut ed side of the ketone were found with oxygenated ke- tones, while butanone furnished a 2.5:1 regioisomer ic mixture which could not be isolated. The chemoselectivi ty in these reac- tions was also high, and essentially no aldol product was formed in all three cases. Under similar conditions using (S)-tert-leucinefurnished product in 24% ee.
In order to explain the observed stereoselect ivities, it was pro- posed that the reaction follows an enamine mechanism and in- volves a transition state (TS)-A11 and B similar to the earlier proposed transition state for the correspond ing aldol reaction as can be seen in Figure 1.
The proposed mechanism of the proline–thiourea catalyzed Mannich reaction is depicted in Figure 5. Accordingly , the nucleo- philic attack of the proline (a), the dehydration of the carbinol amine to give an enamine (b), the carbon–carbon bond forming be- tween imine and enamine in the transition state (c), and then reac- tion to give, after hydrolysi s, the enantiomerical ly enriched Mannich product (d and e). This mechanism is similar to the one we proposed for the proline-c atalyzed aldol reaction (Fig. 3) with the only difference being that the aldehyde is first converte d into an imine before reacting with the presumed proline enamine.11
Typically, Mannich products are formed via si-face attack on an imine. Accordingly, in the Mannich transition state we assume (E)-configurations of both the proline enamine and the imine.11 The si-face of the imine is selective ly attacked by the enamine to allow for
lyst
ne, rt
12a-d
O HN
OMe
NO2
R
Yield (%) eeb (%) (Conf.)c
65 91 (S)
70 95 (3S,4S)
99 (S)
62 97 (3S,4R)
100 lL), aldimine (0.0625 mmol), and toluene (2 mL) at room temperature for 48 h.

OHN
O
O+
NO
OHO
-H2O NO
O
N
O
OHR
NH
RCHO
si-facial attacksi-enamine + si-imine
TS-B
NO
O
RNH
NH O
OOH
RNH
OHN
O
O
+R
NH
L*
H
L*
H
L*L*
+H2O
a b
c
d
e
L*
H
L*
L*H
R H
NAr+
ArNH2
-H2O
Ar
ArAr
Ar
R1
R1
R1
R1R1
R1
R1
R1 = -H, -CH3, -OH
syn
Fig. 5. Proposed mechanism of the Mannich reaction.
522 A. S. Demir, S. Basceken / Tetrahedron: Asymmetry 24 (2013) 515–525
protonation of its lone pair and compensati on of negative charge formation. Attack of the imine re-face would result in unfavorable steric interactions between the pyrrolidine and the aromatic ring (Fig. 4, arrow a). Moreove r, if substituted ketone donors are used excellent syn-selectivity was typically observed 11 (Table 6, entries 2 and 3).
3. Conclusion
The scope, optimization, and application of the proline–thiourea complex catalyzed asymmetric two and three component Mannich reactions have been described herein. The developmen t of this reaction constitutes a major advancemen t over prior asymmetric Mannich reaction technologie s. Exceptionally high enantio-, dia- stereo-, regio-, and chemoselecti vities have been obtained. The reactions are operation ally simple and use an inexpensive modular and nontoxic catalyst that is available in both enantiomeri c forms in a nonpolar medium.
In summary, the synthesis of novel achiral bipyridine derived thioureas and their application as co-catalysts in the proline cata- lyzed asymmetric aldol and Mannich reactions were described. The proline–thiourea system demonst rated excellent reactivity, diaste- reoselectivity , and enantio-selecti vity in nonpolar solvents. Thio- ureas may be synthesized easily in good yields, and to the best of our knowledge this is the first report of organocatal ysis by these host–guest complexes. The success of this catalyst design will open up new perspectives in asymmetric organocatal ysis. More detailed studies regarding the mechanism and synthetic applications of this catalyst on enantioselecti ve organoca talytic reactions are currently under investigatio n.
4. Experimental
4.1. General procedure for the asymmetric aldol reaction
The mixture of (S)-proline (0.025 mmol, 2.8 mg, 20 mol %), thio- urea 7 (0.0125 mmol, 9.1 mg, 10 mol %), and 2 mL hexane was placed in a screw capped vial, then cyclohexanone (2 mmol,
200 lL, 16 equiv) was added, in which the resulting mixture was stirred for 15 min at ambient temperature followed by the addition of aldehyde (0.125 mmol) wherein stirring was continued for 24 h(TLC monitoring). The reaction mixture was then treated with sat- urated aqueous ammonium chloride solution and the whole mix- ture was extracted with ethyl acetate. The organic layer was washed with brine, dried, and concentrated to give a crude residue,which was purified with column chromatography over silica gel using hexane–ethyl acetate as an eluent to afford pure product.Diastereos electivity was determined by 1H NMR analysis of the crude aldol product. The enantiomeric excess (ee) of products was determined by chiral-phase HPLC analysis. The absolute con- figuration of the aldol products was determined by comparing the values with those previousl y reported in the literature.12
4.2. General procedur e for the asymme tric Mannich reaction
The mixture of (S)-proline (0.0125 mmol, 1.4 mg, 10 mol%),thiourea 7 (0.0125 mmol, 9.1 mg, 10 mol%), and 2 mL toluene was placed in a screw capped vial, then aldimine (0.0625 mmol,16 mg) was added, in which the resulting mixture was stirred for 30 min at ambient temperat ure followed by addition of 50–100 lL ketone (acetone, 2-butanone, hydroxya cetone) wherein stirring was continued for 48 h (TLC monitoring). The reactions were worked up by adding phosphat e-buffered saline (PBS) solu- tion (pH 7.4), extractin g with ethyl acetate, drying the organic layer with Na 2SO4, purifying the crude product by column chroma- tography over silica gel using hexane-ethyl acetate as an eluent to afford pure product. The enantiomer ic excess of products was determined by chiral-phase HPLC analysis. The absolute configura-tions of products were determined by comparing the values with those previously reported in the literature.11,13
4.3. Thioureas I and II are known compounds 14
1-Phenyl -3-(pyridin-2-yl)thiourea I and 1-(3,5-bis(tri-fluorom-ethyl)phenyl)-3-(pyridin-2-yl)thiourea II were prepared according to literature methods .14

A. S. Demir, S. Basceken / Tetrahedron: Asymmetry 24 (2013) 515–525 523
4.3.1. 1-Phenyl -3-(pyridin-2-yl)thiourea I1H NMR (CDCl3): d 13.7 (s, 1H), 9.6 (s, 1H), 8.1 (s, 1H), 7.6 (m,
3H), 7.3 (m, 2H), 7.1 (m, 1H), 6.9 (m, 2H); 13C NMR (CDCl3): d178.8 (s, C@S), 153.4 (s, pyridine- C-2), 145.6, 139.0 (d, Ar- C),138.7 (s, Ar C-NHR), 128.8, 126.3, 125.1, 118.3, 112.7 (d, Ar- C).
4.3.2. 1-(3,5-Bis(trifluoromethyl )phenyl)-3-(pyridin-2-yl)-thiou-rea II
1H NMR (CDCl3): d 14.2 (s, 1H), 9.0 (s, 1H), 8.2 (m, 3H), 7.7 (m,2H), 7.1 (m, 1H), 6.8 (d, J = 8.3 Hz, 1H); 13C NMR (CDCl3): d 179.2 (s,C@S), 152.8 (s, pyridine- C-2), 145.8 (d, Ar- C), 140.2 (s, Ar C-NHR),139.5 (d, Ar- C), 132.1, 131.8 (s, Ar C-CF3), 124.5 (d, Ar- C), 124.4 (s,C-F), 118.9, 112.5 (d, Ar- C).
4.4. 1,1 0-([2,20-Bipyridine]-4,40-diylbis(methylene))bis(3-(3,5- bis(trifluoromethyl)phenyl)thiourea) 5
4,40-Dicarboxy-2,20-bipyridine (not shown), 4,4 0-dimethoxy-carboyl-2,20-bipyridine (not shown), 4,4 0-bis(hydroxymethyl)-2,20-bipyridine 2, 4,4 0-bis(bromomethyl)-2,20-bipyridine 3, and 4,40-bis(aminomethyl)-2,20-bipyridine 4 were prepared accordin gto literature methods.7
To a solution of 4,4 0-bis(aminomethyl)-2,20-bipyridine 4 (50 mg,0.23 mmol) in dried chloroform (2 mL) was added dropwise 3,5- bis(trifluoromethyl)phenyl isothiocyan ate (150 lL, 0.86 mmol).The reaction mixture was stirred at room temperature and sud- denly a precipita te formed. The suspension was then stirred for 6 h and added on dried hexane (15 mL). The resulting precipitate was filtered off, washed with dry hexane, and dried to give the compound 1,1 0-([2,20-bipyridine]-4,40-diylbis(methylene))bis(3-(3,5-bis(trifluoromethyl)phenyl-)thiourea) 5 (149 mg, 85%) as aslightly yellow solid; [Found: C, 47.43; H, 2.62; N, 11.0; S, 8.46.C30H20F12N6S2 requires C, 47.62; H, 2.66; N, 11.11; S, 8.48%]; mp 225–228 �C; 1H NMR (CD3COCD3): d 9.8 (br s, 2H), 8.6 (app d,J = 5.0 Hz, 2H), 8.5 (app s, 2H), 8.4 (app d, J = 4.8 Hz, 4H), 7.7 (apps, 2H), 7.4 (app dd, J = 1.5, 5.0 Hz, 2H) , 5.07 (app d, J = 4.8 Hz,4H); 13C NMR (CD3COCD3): d 183.2 (s, C@S), 156.9 (s, bipyridine- C-2, bipyridin e- C-20), 150.1 (d, bipyridine- C-6, bipyridine- C-60),149.7 (s, bipyridine- C-4, bipyridine- C-40), 142.8 (s, Ar C-NHR),132.2, 131.9 (s, Ar C-CF3), 128.4 (d, Ar- C), 125.7 (s, C-F), 123.8,123.5, 123.0, 120.2, 117.8 (d, Ar- C), 47.6 (t, Ar- CH2R); IR (ITR) mmax
(cm�1): 3238, 3105, 3034, 2917, 2851, 1598, 1573, 1527, 1382,1273, 1174, 1132; m/z (ESI-TOF) 757 (100, MH +), 758 (40), 759 (18), 487 (30%); HRMS (ESI-TOF): MH +, found 757.1052.C30H20F12N6S2 requires 757.1078.
4.5. 1,1 0-([2,20-Bipyridine]-4,40-diyl)bis(3-(3,5-bis(trifluoro- methyl)phenyl)thiourea) 7
To a suspensi on of the commerc ially available 4,4 0-diamino-2,20-bipyridine 6 (250 mg, 1.34 mmol) in acetone (50 mL) was added dropwise 3,5-bis(trifluoromethyl)phenyl isothiocyan ate (500 lL, 2.7 mmol). The reaction mixture was stirred at room tem- perature for 48 h, until a clear solution was formed. After evapora- tion of the excess acetone, the resulting product was purified by chromatograp hy on silica gel. The crude product 1,1 0-([2,20-bipyri-dine]-4,40-diyl)bis(3-(3,5-bis-(trifluoromethyl)phenyl)thio-urea) 7was obtained as a slightly green solid (689 mg, 70%); [Found: C,46.10; H, 2.19; N, 11.51; S, 8.77. C28H16F12N6S2 requires C, 46.16;H, 2.21; N, 11.53; S, 8.80%]; mp 124–126 �C; 1H NMR (CD3COCD3):d 10.1 (br s, 2H), 8.6 (app d, J = 2.0 Hz, 2H), 8.5 (app d, J = 5.5 Hz,2H), 8.3 (app s, 4H), 7.9 (app dd, J = 2.0, 5.5 Hz, 2H), 7.8 (app s,2H); 13C NMR (CD3COCD3): d 181.1 (s, C@S), 150.7 (s, bipyridine- C-4, bipyridine- C-40), 142.2 (s, Ar C-NHR), 132.7 (d, bipyridin e- C-6,bipyridine-C-60), 132.3, 131.9 (s, Ar C-CF3), 125.7 (s, C-F), 124.9,
122.9, 118.8, 117.3, 114.2 (d, Ar- C); IR (ITR) mmax (cm�1): 3202,3035, 3004, 2917, 2851, 1590, 1575, 1527, 1382, 1275, 1133; m/z(ESI-TOF) 729 (60, MH +), 757 (30), 759 (18), 500 (100), 360 (60%); HRMS (ESI-TOF): MH +, found 729.0762. C28H16F12N6S2 re-quires 729.0764.
4.6. (2R,10S)-2-[Hydroxy-(4-nitrophenyl)methyl]cyclohex- an-1- one 10a:12
Following the general procedure, compound 10a was obtained as a slightly yellow solid, 90%, in a maximum of 99% ee. Rf = 0.4 (EtOAc/hexanes 1:2 v/v); The enantiomeric purity was determined by HPLC on AD-H, hexanes/ i-PrOH = 90:10, flow rate = 0.5 mL/min,UV = 254 nm, anti/syn = 90/10, syn-10a tR = 36.2 min (major) and tR = 42.5 min (minor), anti-10a tR = 62.1 min (major). 1H NMR (CDCl3): d 8.21 (2H, d, J = 8.7 Hz), 7.51 (2H, d, J = 8.7 Hz), 4.90 (1H, d, J = 8.3 Hz), 4.09 (1H, s), 2.63–2.58 (1H, m), 2.53–2.47 (1H,m), 2.41–2.33 (1H, m), 2.15–2.08 (1H, m), 1.85-1.36 (6H, m). 13CNMR (CDCl3): d 214.7 (s, C@O), 148.4, 147.6, 127.9, 123.6, 74.0,57.2, 42.7, 30.8, 27.6, 24.7.
4.7. (2R,10S)-2-[Hydroxy-(3-nitrophenyl)methyl]cyclohex- an-1- one 10b 12
Following the general procedure, compound 10b was obtained as a slightly yellow solid, 70%, in a maximum of 91% ee. Rf = 0.4 (EtOAc/hexanes 1:2 v/v); The enantiomeric purity was determined by HPLC on AD-H, hexanes/ i-PrOH = 95/5, flow rate = 1.0 mL/min,UV = 254 nm, anti/syn = 87/13, syn-10b tR = 26.5 min (major) and tR = 29.7 min (minor), anti-10b tR = 33.5 min (major) and tR = 43.3 min. 1H NMR (CDCl3): d 8.21 (1H, s), 8.15 (1H, d,J = 8.2 Hz), 7.67 (1H, d, J = 7.6 Hz), 7.53 (1H, t, J = 7.8 Hz), 4.90 (1H, d, J = 8.4 Hz), 4.17 (1H, s), 2.67–2.61 (1H, m), 2.52–2.48 (1H,m), 2.42–2.34 (1H, m), 2.13–2.10 (1H, m), 1.83 (1H, d,J = 12.8 Hz), 1.73–1.53 (3H, m), 1.44–1.34 (1H, m). 13C NMR (CDCl3): d 214.8 (s, C@O), 148.2, 143.3, 133.2, 129.2, 122.8, 122,74, 57.1, 42.6, 30.7, 27.6, 24.6.
4.8. (2R,10S)-2-[Hydroxy-(4-bromophenyl)methyl]cyclo-hex an- 1-one 10c 12
Following the general procedure, compound 10c was obtained as a yellow solid, 67%, in a maximum of 98% ee. Rf = 0.4 (EtOAc/hexanes 1:2 v/v); The enantiomeri c purity was determined by HPLC on AD-H, hexanes/ i-PrOH = 95/5, flow rate = 1.0 mL/min,UV = 220 nm, anti/syn = 91/9, syn-10c tR = 17.8 min and tR = 21.1 min, anti-10c tR = 27.9 min (minor) and tR = 32.5 min (ma-jor). 1H NMR (CDCl3): d 7.47 (2H, d, J = 8.4 Hz), 7.19 (2H, d,J = 8.3 Hz), 4.75 (1H, dd, J = 8.7, 2.6 Hz), 4.01 (1H, d, J = 2.7 Hz),2.58-2.31 (3H, m), 2.11–2.06 (1H, m), 1.81–1.51 (3H, m), 1.34–1.23 (1H, m). 13C NMR (CDCl3): d 215.2 (s, C@O), 140.1, 131.5,128.6, 121.7, 74.2, 57.3, 42.7, 30.8, 27.7, 24.7.
4.9. (2R,10S)-2-[Hydroxy-(4-(trifluoromethyl)phenyl)methyl]- cyclohex an-1-one 10d 12
Following the general procedure, compound 10d was obtained as a slightly yellow solid, 75%, in a maximum of 98% ee. Rf = 0.4 (EtOAc/hexanes 1:2 v/v); The enantiomeric purity was determined by HPLC on OD-H, hexanes/ i-PrOH = 95/5, flow rate = 1.0 mL/min,UV = 220 nm, anti/syn = 91/9, syn-10d tR = 8.7 min and tR = 9.5 min,anti-10d tR = 11.2 min (major) and tR = 14.7 min (minor). 1H NMR (CDCl3): d 7.60 (2H, d, J = 8.1 Hz), 7.44 (2H, d, J = 8.1 Hz), 4.85 (1H, dd, J = 8.6, 2.8 Hz), 4.09 (1H, d, J = 3.0 Hz), 2.63–2.57 (1H, m),2.50–2.47 (1H, m), 2.40–2.32 (1H, m), 2.12–2.07 (1H, m), 1.82–1.79 (1H, m), 1.71–1.49 (2H, m), 1.38–1.29 (1H, m). 13C NMR

524 A. S. Demir, S. Basceken / Tetrahedron: Asymmetry 24 (2013) 515–525
(CDCl3): d 215.1 (s, C@O), 145.0, 127.4, 126.1, 125.3, 74.2, 57.3,42.7, 30.7, 27.7, 24.7.
4.10. (2R,10S)-2-[Hydroxy-(4-cyanophenyl)methyl]cyclo-hexan- 1-one 10e 12
Following the general procedure, compound 10e was obtained as a white solid, 88%, in a maximum of 92% ee. Rf = 0.3 (EtOAc/hex-anes 1:2 v/v); The enantiomer ic purity was determined by HPLC on AD-H, hexanes/ i-PrOH = 90/10, flow rate = 0.5 mL/min, UV =220 nm, anti/syn = 94/6, syn-10e tR = 41.3 min (minor) and tR = 34.9 min (major), anti-10e tR = 59.5 min (major) and tR = 47.3 min. 1H NMR (CDCl3): d 7.65 (2H, d, J = 8.3 Hz), 7.45 (2H,d, J = 8.2 Hz), 4.85 (1H, dd, J = 8.4, 2.8 Hz), 4.06 (1H, d, J = 3.1 Hz),2.61–2.46 (2H, m), 2.40–2.32 (1H, m), 2.14–2.08 (1H, m), 1.85–1.80 (1H, m), 1.68–1.54 (2H, m), 1.40-1.33 (1H, m). 13C NMR (CDCl3): d 214.8 (s, C@O), 146.4, 132.2, 127.8, 111.7, 74.2, 57.1,42.7, 30.7, 27.7, 24.7.
4.11. (2R,10S)-2-[Hydroxy-(2-methoxyphenyl)methyl]cyclo- hexan-1-one 10f 12
Following the general procedure, compound 10f was obtained as a slightly yellow solid, 73%, in a maximum of 96% ee. Rf = 0.4 (EtOAc/hexanes 1:2 v/v); The enantiomeri c purity was determined by HPLC on OD-H, hexanes/ i-PrOH = 95/5, flow rate = 0.5 mL/min,UV = 280 nm, anti/syn = 99/1, anti-10f tR = 26.2 min (major) and tR = 36.7 min (minor). 1H NMR (CDCl3): d 7.34 (1H, dd, J = 7.6,1.7 Hz), 7.19–7.15 (1H, dd, J = 7.5, 1.7 Hz), 6.92–6.89 (1H, t,J = 7.5 Hz), 6.79–6.78 (1H, d, J = 8.1 Hz), 5.20–5.17 (1H, dd, J = 8.5,2.5 Hz), 3.78 (1H, d, J = 3.5 Hz), 3.73 (3H, s), 2.69–2.62 (1H, m),2.42–2.36 (2H, m), 2.31–2.22 (1H, m), 2.00–1.94 (1H, m),1.73–1.35 (3H, m). 13C NMR (CDCl3): d 214.5 (s, C@O), 155.7,128.6, 127.6, 126.7, 119.9, 109.5, 67.6, 56.3, 54.4, 41.5, 29.5, 26.9,23.7.
4.12. (2R,10S)-2-[Hydroxy-(4-fluorophenyl)methyl]cyclo-hexan- 1-one 10g 12
Following the general procedure, compound 10g was obtained as a slightly yellow solid, 65%, in a maximum of 95% ee. Rf = 0.6 (EtOAc/hexanes 1:2 v/v); The enantiomeri c purity was determined by HPLC on AD-H, hexanes/ i-PrOH = 95/5, flow rate = 1.0 mL/min,UV = 220 nm, anti/syn = 85/15, syn-10g tR = 12.5 min and tR = 14.8 min, anti-10g tR = 23.1 min (major) and tR = 20.5 min (minor). 1H NMR (CDCl3): d 7.23–7.20 (2H, dd, J = 8.0, 5.7 Hz),6.98-6.93 (2H, t, J = 8.6 Hz), 4.70 (1H, d, J = 8.6 Hz), 3.96 (1H, s),2.53–2.39 (2H, m), 2.32–2.24 (1H, m), 2.04–1.99 (1H, m), 1.73–1.45 (2H, m), 1.25–1.18 (1H, m). 13C NMR (CDCl3): d 215.4 (s,C@O), 163.6, 161.2, 136.8, 128.7, 115.3, 74.1, 57.5, 42.6, 30.8,27.7, 24.7.
4.13. (2R,10S)-2-[Hydroxy-(phenyl)methyl]cyclohexa n-1-one 10h12
Following the general procedure, compound 10h was obtained as a white solid, 62%, in a maximum of 95% ee. Rf = 0.4 (EtOAc/hexanes1:2 v/v); The enantiomeri c purity was determined by HPLC on AS-H,hexanes/i-PrOH = 90/10, flow rate = 0.5 mL/min, UV = 220 nm, anti/syn = 84/16, syn-10h tR = 20.7 min and tR = 22.9 min, anti-10htR = 26.3 min (major) and tR = 24.8 min (minor). 1H NMR (CDCl3): d7.36–7.26 (5H, m), 4.80–4.77 (1H, dd, J = 8.8, 2.7 Hz), 4.00 (1H, d,
J = 2.8 Hz), 2.65–2.58 (1H, m), 2.50–2.44 (1H, m), 2.39–2.31 (1H, m),2.09–2.04 (1H, m), 1.79-1.48 (3H, m), 1.34–1.26 (1H, m). 13C NMR (CDCl3): d 213.1 (s, C@O), 138.6, 126, 125.5, 124.7, 72.3, 55.1, 40.2,28.5, 25.4, 22.3.
4.14. (2R,10S)-2-[Hydroxy-(4-nitrophenyl)methyl]cyclo-pen tan- 1-one 10i 12
Following the general procedure, compound 10i was obtained as a white solid, 82%, in a maximum of 99% ee. Rf = 0.4 (EtOAc/hex-anes 1:2 v/v); The enantiomeric purity was determined by HPLC on AD-H, hexanes/ i-PrOH = 95/5, flow rate = 0.5 mL/min,UV = 254 nm, anti/syn = 43/57, syn-10i tR = 48.4 min (major) and tR = 67.2 min (minor), anti-10i tR = 88.1 min (major). 1H NMR (CDCl3): d 8.19–8.17 (2H, dd, J = 6.2, 3.2 Hz), 7.53 (2H, d,J = 8.4 Hz), 5.42 (1H, s), 4.88 (1H, d, J = 8.8 Hz), 2.49–1.55 (6H, m).13C NMR (CDCl3): d 219.5 (s, C@O), 150.2, 148.6, 147.2, 127.4,126.4, 123.7, 123.6, 74.4, 70.5, 56.1, 55.1, 38.9, 38.6, 26.8, 22.4,20.4, 20.3.
4.15. (S)-4-(4-Nitrophenyl)-4-(4-methoxy-phenyla mino)-butan-2-one 12a 11,13
Following the general procedure, compound 12a was obtained as yellow oil, 65%, in 91% ee. The enantiomer ic purity was determined by HPLC on AD, hexanes/ i-PrOH = 50/50, flow rate = 0.5 mL/min,UV = 280 nm, tR(R) = 15.7 min and tR(S) = 18.6 min. 1H NMR (CDCl3): d 8.17 (2H, d, J = 8.8 Hz), 7.55 (2H, d, J = 8.8 Hz), 6.69 (2H,d, J = 8.9 Hz), 6.46 (2H, d, J = 8.9 Hz), 4.85 (1H, t, J = 6.4 Hz), 4.24 (1H, br), 3.69 (3H, s), 2.94 (2H, d, J = 6.4 Hz), 2.15 (3H, s). 13C NMR (CDCl3): d 206.1 (s, C@O), 152.8, 150.7, 147.2, 140.1, 127.4, 124.1,115.4, 114.8, 55.6, 54.6, 50.7, 30.7.
4.16. (3S,4S)-4-(4-Methoxy-pheny lamino)-3-methyl-4-(4-nitro-phenyl)-butan-2-one 12b and (S)-1-(4-methoxy-pheny lamino)-1-(4-nitro-phenyl)-pentan-3-one 12c 11,13
Following the general procedure, compound 12b and 12c wereobtained as slightly yellow oil, 70%, in 95% ee and 99% ee, respec- tively. The enantiomeri c purity was determined by HPLC on AD,hexanes/i-PrOH = 85/15, flow rate = 1.0 mL/min, UV = 280 nm,tR(3R,4R-12b) = 17.4 min, tR(3S,4S-12b) = 20.1 min and tR(S-12c) = 28.6 min. 1H NMR (CDCl3): d 8.18 (2H, d, J = 8.7 Hz), 7.52 (2H, d, J = 8.7 Hz), 6.67 (2H, d, J = 8.9 Hz), 6.40 (2H, d, J = 8.9 Hz),4.86 (1H, t, J = 6.6 Hz), 4.76 (1H, d, J = 5.1 Hz), 3.68 (3H, s), 3.02 (1H, m), 2.91 (2H, d, J = 6.2 Hz), 2.43-2.37 (2H, m), 2.17 (3H, s),1.09 (3H, d, J = 7.1 Hz), 1.01 (3H, t, J = 7.2 Hz). 13C NMR (CDCl3): d209.7, 208.9 (s, C@O), 152.8, 152.7, 150.8, 149.4, 147.3, 140.2,127.9, 127.4, 124.0, 123.9, 115.4, 115, 114.8, 59.2, 55.7, 54.9,52.5, 49.4, 36.9, 29.4, 10.9, 7.5.
4.17. (3S,4R)-3-Hydroxy-4-(4-methoxy-phenylamin o)-4-(4-nitrophen yl)-butan-2-one 12d 11,13
Following the general procedure, compound 12d was obtained as slightly yellow foam, 62%, in 97% ee. The enantiomeric purity was determined by HPLC on AD-H, hexanes/ i-PrOH = 85/15, flowrate = 1.0 mL/min, UV = 254 nm, anti/syn = 3/97; tR(3S,4R) =38.9 min (major) and tR(3R,4S) = 44.4 min (minor). 1H NMR (CDCl3): d 8.17 (2H, d, J = 8.8 Hz), 7.54 (2H, d, J = 8.7 Hz), 6.68 (2H, d, J = 9.0 Hz), 6.45 (2H, d, J = 9.0 Hz), 5.02 (1H, d, J = 2.2 Hz),4.43 (1H, d, J = 2.3 Hz), 4.25 (1H, s), 3.67 (3H, s), 2.37 (3H, s). 13CNMR (CDCl3): d 206.5 (s, C@O), 152.9, 147.4, 139.2, 128.2, 123.8,115.2, 115, 80.0, 58.8, 55.6, 24.9.

A. S. Demir, S. Basceken / Tetrahedron: Asymmetry 24 (2013) 515–525 525
4.18. (R)-4-(4-Methoxy-pheny lamino)-6-methyl-heptan -2-one 15a11,13
Following the general procedure, compound 15a was obtained as oil, 90%, in 81% ee. The enantiomeric purity was determined by HPLC on OD-H, hexanes/ i-PrOH = 90/10, flow rate = 1.0 mL/ min, UV = 254 nm, tR(S) = 6.2 min (minor) and tR(R) = 10.0 min (major). 1H NMR (CDCl3): d 6.77 (2H, d, J = 8.2 Hz), 6.57 (2H, d,J = 8.7 Hz), 3.83–3.77 (1H, m), 3.74 (3H, s), 3.35 (1H, br), 2.67–2.63 (1H, dd, J = 16.3, 4.9 Hz), 2.58–5.52 (1H, dd, J = 16.3, 6.4 Hz),2.13 (3H, s), 1.78–1.71 (1H, m), 1.51–1.44 (1H, m), 1.34–1.29(1H, m), 0.93 (3H, d, J = 6.8 Hz), 0.91 (3H, d, J = 6.6 Hz). 13C NMR (CDCl3): d 208.4 (s, C=O), 152.2, 141.4, 115.0, 114.9, 55.8, 49.2,48.1, 44.8, 31.0, 25.1, 23.0, 22.3.
4.19. (R)-4-(4-Methoxy-pheny lamino)-octan-2-one 15b 11,13
Following the general procedure, compound 15b was obtained as oil, 88%, in 81% ee. The enantiomeri c purity was determined by HPLC on OD-H, hexanes/ i-PrOH = 90/10, flow rate = 1.0 mL/ min, UV = 254 nm, tR(S) = 6.7 min (minor) and tR(R) = 10.1 min (major). 1H NMR (CDCl3): d 6.77 (2H, d, J = 8.4 Hz), 6.57 (2H, d,J = 8.9 Hz), 3.74 (3H, s), 3.74 (1H, m), 2.65 (1H, dd, J = 16.3,5.4 Hz), 2.57 (1H, dd, J = 16.3, 6.4 Hz), 2.14 (3H, s), 1.53 (2H, dd,J = 13.7, 6.5 Hz), 1.44–1.24 (4H, m), 0.88 (3H, t, J = 6.8 Hz). 13CNMR (CDCl3): d 208.4 (s, C@O), 159.0, 152.3, 141.4, 115.1, 115.0,55.8, 51.0, 48.0, 34.9, 30.9, 28.4, 22.7, 14.0.
4.20. (S)-4-(4-Methoxy-phenylami no)-5-methyl-hexan-2-one 15c11,13
Following the general procedure, compound 15c was obtained as yellow oil, 85%, in 90% ee. The enantiomeric purity was deter- mined by HPLC on OD-H, hexanes/ i-PrOH = 90/10, flowrate = 1.0 mL/min, UV = 254 nm, tR(R) = 6.5 min (minor) and tR(S) = 8.6 min (major). 1H NMR (CDCl3): d 6.75 (2H, d, J = 8.9 Hz),6.58 (2H, d, J = 8.9 Hz), 3.73 (3H, s), 3.68–3.65 (1H, m), 3.38 (1H,br), 2.61–2.49 (1H, m), 2.14 (3H, s), 1.97–1.87 (1H, m), 0.96 (3H,d, J = 6.9 Hz), 0.89 (3H, d, J = 6.8 Hz). 13C NMR (CDCl3): d 208.5 (s,C@O), 152.1, 141.7, 115.0, 56.2, 55.8, 45.2, 31.3, 30.6, 18.8, 18.4.
Acknowled gements
Financial support from the Scientific and Technological Research Council of Turkey (TÜBITAK), the Turkish Academy of Sciences (TÜBA), and the Middle East Technical University (METU)
is gratefully acknowledged . Thanks are also due to Professor Metin Balci for editorial assistance.
References
1. (a) Eder, U.; Sauer, G.; Wiechert, R. Angew. Chem., Int. Ed. Engl. 1971, 10, 496; (b)Hajos, Z. G.; Parrish, D. R. J. Org. Chem. 1974, 39, 1615; (c) List, B.; Lerner, A. R.;Barbas, C. F., III J. Am. Chem. Soc. 2000, 122, 2395; (d) Sakthivel, K.; Notz, W.;Bui, T.; Barbas, C. F., III J. Am. Chem. Soc. 2001, 123, 5260; (e) Mukherjee, S.;Yang, J. W.; Hoffmann, S.; List, B. Chem. Rev. 2007, 107, 5471; (f) Bisai, V.; Bisai,A.; Singh, V. K. Tetrahedron 2012, 68, 4541.
2. Ahrendt, K. A.; Borths, C. J.; MacMillan, D. W. C. J. Am. Chem. Soc. 2000, 122,4243.
3. Sigman, M.; Jacobsen, E. N. J. Am. Chem. Soc. 1998, 120, 4901.4. (a) Isart, C.; Bures, J.; Vilarrasa, J. Tetrahedron Lett. 2008, 49, 5414; (b) List, B.;
Hoang, L.; Martin, H. J. Proc. Natl. Acad. Sci. U.S.A. 2004, 101, 5839; (c) Orsini, F.;Pelizzoni, F.; Forte, M.; Destro, R.; Gariboldi, P. Tetrahedron 1988, 44, 519.
5. (a) Tang, Z.; Jiang, F.; Cui, X.; Gong, L. Z.; Mi, A.-Q.; Jiang, Y.-Z.; Dong, Y. Proc.Natl. Acad. Sci. U.S.A. 2004, 101, 5755; (b) Hartikka, A.; Arvidsson, P. I. Eur. J. Org.Chem. 2005, 11, 4287; (c) Berkessel, A.; Koch, B.; Lex, J. Adv. Synth. Catal. 2004,346, 1141; (d) Vishnumaya, M. R.; Ginotra, S. K.; Singh, V. K. Org. Lett. 2006, 8,4097.
6. (a) Schreiner, P. R.; Wittkopp, A. Org. Lett. 2002, 4, 217; (b) Schreiner, P. R.;Wittkopp, A. Chem. Eur. J. 2003, 9, 407; For a review, see: (c) Schreiner, P. R.Chem. Soc. Rev. 2003, 22, 289.
7. (a) Gauthier, G. _I.; Odobel, F.; Alebbi, M.; Argazzi, R.; Costa, E.; Bignozzi, C. A.;Qu, P.; Meyer, G. J. Inorg. Chem. 2001, 40, 6073; (b) Fukuda, S.; Satake, A.;Kobuke, Y. Thin Solid Films 2006, 499, 263.
8. (a) Hoang, L.; Bahmanyar, S.; Houk, K. N.; List, B. J. Am. Chem. Soc. 2003, 125, 16;(b) Bahmanyar, S.; Houk, K. N.; Martin, H. J.; List, B. J. Am. Chem. Soc. 2003, 125,2475; (c) Clemente, F. R.; Houk, K. N. Angew. Chem., Int. Ed. 2004, 37, 5766; (d)Allemann, C.; Gordillo, R.; Clemente, F. R.; Cheong, Y.; Houk, K. N. Acc. Chem.Res. 2004, 37, 558; (e) Notz, W.; List, B. J. Am. Chem. Soc. 2000, 122, 7386; (f)Lippert, K. M.; Hof, K.; Gerbig, D.; Ley, D.; Hausmann, D.; Guenther, S.;Schreiner, P. R. Eur. J. Org. Chem. 2012, 5919; (g) Martinez-Castaneda, A.;Rodriguez-Solla, H.; Concellon, C.; del Amo, V. J. Org. Chem. 2012, 77, 10375; (h)Reis, Ö.; Eymur, S.; Reis, B.; Demir, A. S. Chem. Commun. 2009, 9, 1088; (i) Zhou,Y.; Shan, Z. J. Org. Chem. 2006, 71, 9510; (j) Nyberg, A. I.; Usano, A.; Pihko, P. M.Synlett 2004, 1891; (k) Torii, H.; Nakadai, M.; Ishihara, K.; Saito, S.; Yamamoto,H. Angew. Chem., Int. Ed. 2004, 43, 1983; (l) Zimmerman, H. E.; Traxler, M. D. J.Am. Chem. Soc. 1957, 79, 1920.
9. Movassaghi, M.; Jacobsen, E. N. Science 1904, 2002, 298.10. (a) Mannich, C.; Krosche, W. Arch. Pharm. 1912, 250, 647; (b) Arend, M.;
Westermann, B.; Risch, N. Angew. Chem., Int. Ed. 1998, 37, 1044; (c) Marques, M.M. B. Angew. Chem., Int. Ed. 2006, 45, 348; (d) Shibasaki, M.; Matsunaga, S. J.Organomet. Chem. 2006, 691, 2089.
11. (a) List, B. J. Am. Chem. Soc. 2000, 122, 9336; (b) List, B.; Pojarliev, P.; Biller, W.T.; Martin, H. J. J. Am. Chem. Soc. 2002, 124, 827.
12. (a) Mase, N.; Nakai, Y.; Ohara, H.; Yoda, H.; Takabe, K.; Tanaka, F.; Barbas, C. F. J.Am. Chem. Soc. 2006, 128, 734; (b) Cobb, A. J. A.; Shaw, D. M.; Longbottom, D. A.;Gold, J. B.; Ley, S. V. Org. Biomol. Chem. 2005, 3, 84; (c) Hayashi, Y.; Sumiya, T.;Takahashi, J.; Gotoh, H.; Urushima, T.; Shoji, M. Angew. Chem., Int. Ed. 2006, 45,958; (d) Wu, Y.; Zhang, Y.; Yu, M.; Zhao, G.; Wang, S. Org. Lett. 2006, 8(20),4417.
13. (a) Zheng, X.; Qian, Y. B.; Wang, Y. Eur. J. Org. Chem. 2010, 515; (b) Sasaoka, A.;Uddin, M. I.; Shimomoto, A.; Ichikawa, Y.; Shiro, M.; Kotsuki, H. Tetrahedron:Asymmetry 2006, 17, 2963.
14. (a) West, D. X.; Hermetet, A. K.; Ackerman, L. J.; Martínez, J. V.; Ortega, S. H.Acta Cryst. 1999, C55, 811–813; (b) Yue, H.; Wang, Y.; Xia, A.; Luo, S.; Xu, D. ActaCryst. 2008, E64, o858.





























