Embed Size (px)
Citation preview

STUDIES TOWARD THE TOTAL SYNTHESIS OF MULBERRY DIELS-ALDER ADDUCTS
MORUSALBANOL A AND SOROCEIN B
TEE JIA TI
FACULTY OF SCIENCE UNIVERSITY OF MALAYA
KUALA LUMPUR
2016

STUDIES TOWARD THE TOTAL SYNTHESIS OF
MULBERRY DIELS-ALDER ADDUCTS
MORUSALBANOL A AND SOROCEIN B
TEE JIA TI
THESIS SUBMITTED IN FULFILMENT
OF THE REQUIREMENTS FOR
THE DEGREE OF DOCTOR OF PHILOSOPHY
DEPARTMENT OF CHEMISTRY
FACULTY OF SCIENCE
UNIVERSITY OF MALAYA
KUALA LUMPUR
2016

ii
UNIVERSITY OF MALAYA
ORIGINAL LITERARY WORK DECLARATION
Name of Candidate: TEE JIA TI (I.C/Passport No: 851230-01-5116)
Registration/Matric No: SHC 120015
Name of Degree: DOCTOR OF PHILOSOPHY
Title of Project Paper/Research Report/Dissertation/Thesis (“this Work”):
STUDIES TOWARD THE TOTAL SYNTHESIS OF MULBERRY DIELS-
ALDER ADDUCTS MORUSALBANOL A AND SOROCEIN B
Field of Study: TOTAL SYNTHESIS, ORGANIC CHEMISTRY
I do solemnly and sincerely declare that:
(1) I am the sole author/writer of this Work;
(2) This Work is original;
(3) Any use of any work in which copyright exists was done by way of fair dealing
and for permitted purposes and any excerpt or extract from, or reference to or
reproduction of any copyright work has been disclosed expressly and sufficiently
and the title of the Work and its authorship have been acknowledged in this
Work;
(4) I do not have any actual knowledge nor do I ought reasonably to know that the
making of this work constitutes an infringement of any copyright work;
(5) I hereby assign all and every rights in the copyright to this Work to the
University of Malaya (“UM”), who henceforth shall be owner of the copyright in
this Work and that any reproduction or use in any form or by any means
whatsoever is prohibited without the written consent of UM having been first had
and obtained;
(6) I am fully aware that if in the course of making this Work I have infringed any
copyright whether intentionally or otherwise, I may be subject to legal action or
any other action as may be determined by UM.
Candidate’s Signature Date:
Subscribed and solemnly declared before,
Witness’s Signature Date:.
Name:
Designation:

iii
ABSTRACT
Morusalbanol A (42) and sorocein B (62) are biologically active natural products
isolated from moraceous plants. They are postulated to be biosynthetically derived from an
intramolecular cyclization/ketalization of a cis-trans mulberry Diels-Alder adduct. Thus far,
there has been no report on the synthesis of these compounds since their first isolation in
1991. A model studies on construction of the oxabicyclic [3.3.1] core of morusalbanol A
was conducted prior to embarking on the synthesis of morusalbanol A and sorocein B. The
results showed that the required cis-trans Diels-Alder precursors of morusalbanol A was
obtained via the thermal cycloaddition reaction which was proven to be dependent on the
presence of a hydrogen-bonded ortho OH substituent on the chalcones dienophile. Acid
catalyzed intramolecular cyclization of a cis-trans Diels-Alder adduct (endo-181) afforded
the desired oxabicyclic [3.3.1] core of morusalbanol A in a stereocontrolled manner.
Results from the model study have been applied to the synthesis of morusalbanol A (42).
The requisite cis-trans Diels-Alder precursor (endo-204) of morusalbanol A was
successfully prepared via the thermal cycloaddition reaction between the methyl ether
protected chalcones dienophile 88 and the dehydroprenyl diene 191. Selective removal of
the ortho OMe group of the endo-204 by using MgI2 gave (±) morusalbanol A methyl
ethers 206 and 207. A number of key proton and carbon signals in the NMR spectra of 206
and 207 were absent as a result of atroisomerism due to the rotational hindrance of the
Diels-Alder-rings about the C5´´-15´´ and C4´´-C8´´-C9´´ bond. Global demethylation on
the (±) morusalbanol A methyl ethers 206 and 207 by using MgI2, BCl3, TMSI-quinoline
was unsuccessful. An efficient method for preparing 2´2-dimethyl-2H-chromones via
Pd(II)-catalyzed Heck coupling of o-halophenols with 2-methyl-3-buten-2-ol has been
developed during the synthesis of sorocein B. The method is very general and can be useful
to the synthesis of some natural 2´,2-dimethyl-2H-chromones. Similar strategy was used for
construction of the cis-trans Diels-Alder precursor of sorocein B. The thermal
cycloaddition reaction between chalcones dienophile 89 and dehydroprenyl diene 106
afforded the requisite cis-trans Diels-Alder precursor (endo-241) in 35% yield along with
the trans-trans Diels-Alder diastereomer. Subsequently PdCl2 catalyzed cyclization of the
ortho-prenyl group of endo-241 afforded the required 2,2-dimethylchromenyl ring in 230.
However, attempts to remove the methyl ether group of 230 to form sorocein B with BCl3,
MgI2, and TMSI-quinoline were unsuccessful.

iv
ABSTRAK
Morusalbanol A (42) dan sorocein B (62) adalah produk semula jadi biologi aktif
yang diasingkan daripada tumbuhan moraceous. Biosynthetically, mereka berasal dari
intramolecular cyclization / ketalization daripada cis-trans mulberi Diels-Alder adduct.
Setakat ini, tiada laporan mengenai sintesis sebatian ini sejak pengasingan pertama mereka
pada tahun 1991. Sebelum memulakan sintesis morusalbanol A dan sorocein B, kajian
tentang pembinaan model oxabicyclic [3.3.1] teras bagi morusalbanol A telah dijalankan.
Hasil kajian menunjukkan bahawa kehadiran orto OH hidrogen terikat pada dienophile
chalcone itu diperlukan dalam tindak balas cycloaddition haba. Intramolecular cyclization
daripada cis-trans Diels-Alder adduct (endo-181) yang dimangkin oleh asid berjaya
memberi model oxabicyclic [3.3.1] teras bagi morusalbanol A dengan cara yang
stereocontrolled. Hasil daripada kajian model telah digunakan untuk sintesis morusalbanol
A (42). Cis-trans Diels-Alder (endo-204) dari morusalbanol A telah berjaya disediakan
melalui tindak balas cycloaddition haba antara metil eter chalcone dienophile 88 dan
dehydroprenyl diene 191. Penyingkiran dariapada kumpulan orto OMe (endo-204) dengan
menggunakan MgI2 berjaya memberi (±) morusalbanol A metil eter 205 dan 206. Keadaan
atroisomerism menyebabkan ketidakhadiran beberapa proton dan karbon dalam spektrum
NMR bagi 206 dan 207. Keadaan ini berlaku disebabkan terdapat halangan putaran pada
Diels-Alder ring di bond C5´´-15´´ dan C4´´-C8´´-C9´´ itu. Global demethylation ke atas (±)
morusalbanol metil eter A 206 dan 207 dengan menggunakan MgI2, BCl3, TMSI-quinoline
tidak berjaya. Semasa sintesis sorocein B, satu kaedah yang berkesan untuk menyediakan
2'2-dimetil-2H-chromones melalui Pd (II) gandingan Heck -catalyzed o-halophenols
dengan 2-metil-3-buten-2-ol telah dibangunkan. Kaedah ini sangat umum dan boleh
digunakan untuk sintesis beberapa 2,2-dimetil-2H-chromones yang semulajadi. Strategi
yang sama telah digunakan dalam pembinaan cis-trans Diels-Alder sorocein B. Reaksi
cycloaddition haba antara chalcone dienophile 89 dan dehydroprenyl diene 106 memberi
cis-trans Diels-Alder adduct (endo-241) dalam hasil 35% bersama-sama dengan trans-trans
Diels-Alder diastereomer. Selepas itu, cyclization kumpulan orto-prenyl group daripada
endo-241 yang dimangkin oleh PdCl2 berjaya memberikan 2,2-dimethylchromenyl ring
230. Walau bagaimanapun, usaha untuk menghapuskan kumpulan metil eter 230 bagi
membentuk sorocein B dengan BCl3, MgI2 dan TMSI-quinoline tidak berjaya.

v
ACKNOWLEDGEMENTS
This study would not have been possible without the help and assistance of many.
First and foremost, I would like to express my sincere gratitude to my supervisor, Prof Dr
Noorsaadah Abd. Rahman for her invaluable guidance and enthusiasm throughout the research
project and the completion of this thesis. I gratefully acknowledge her for her constructive advices
and suggestions on my research work. It had been a privilege to join the Drug design and
development research group (DDDRG) and work under her supervision.
I appreciate Dr. Chee Chin Fei for his patients, whose encouragement, guidance and
support from the initial to the final level enabled me to complete this project. I am also grateful to
him for reading through the draft of the thesis and advice in the compilation of the thesis.
I am indebted to Dr. Marzieh Yaeghoobi who gave me an extreme support in showing me
the way to think logically in research. To the DDDRG group members with whom I have worked
directly, I would like to collectively thank the group for providing informational and critical
discussions. I am especially proud to be one of them. In addition, I would like to acknowledge to
my friends and lab-mates, especially Assoc. Prof Dr. Michael James Christopher Buck, Dr. Lee
Yean Kee, Dr. Hamid Khaledi for providing valuable comments, stimulating suggestions and
motivation work atmosphere.
Aside from the DDDRG group, I would like to thank all the staff for their generous help.
This work was made possible with SLAI scholarship and research grants from the University of
Malaya (PG020/2014A).
Finally, I would like to thank my family for all their love and encouragement. This thesis
would not have been possible without their continuous support and caring of them.

vi
TABLE OF CONTENTS
Abstract ........................................................................................................................... iii
Abstrak ............................................................................................................................ iv
Acknowledgements .......................................................................................................... v
Table of Contents ........................................................................................................... vi
List of Figures ................................................................................................................. ix
List of Schemes ............................................................................................................... xi
List of Tables ................................................................................................................ xiii
List of Symbols and Abbreviations ............................................................................. xiv
CHAPTER 1: INTRODUCTION AND LITERATURE REVIEW ........................... 1
1.1 Introduction ............................................................................................................. 1
1.2 Mulberry Diels-Alder adducts ................................................................................. 2
1.3 Absolute configurations of mulberry Diels-Alder type adducts .............................. 9
1.4 Biosynthesis of mulberry Diels-Alder adducts ...................................................... 15
1.5 Biological activity of mulberry Diels-Alder type adducts ..................................... 18
1.5.1 Antioxidation ................................................................................................ 19
1.5.2 Anti-inflammation ........................................................................................ 22
1.5.3 Cytotoxicity .................................................................................................. 23
1.5.4 Antimicrobial and antifungus ....................................................................... 25
1.6 Scope and objectives of this thesis ........................................................................ 26
CHAPTER 2: RELATED SYNTHESIS ..................................................................... 28
2.1 Semisythesis of mulberry Diels-Alder adducts ..................................................... 28

vii
2.2 Total synthesis of mulberry Diels-Alder adducts .................................................. 31
2.2.1 Thermal conditions for Diels-Alder reation ................................................. 31
2.2.2 Catalytic Diels-Alder reaction ...................................................................... 41
2.2.3 Chiral-Boron-complex promoted enantioselective synthesis of MDA ........ 51
CHAPTER 3: APPROACHES TOWARD THE SYNTHESIS OF
MORUSALBANOL A ................................................................ 58
3.1 Model study towards biomimetic Diels-Alder reaction Morusalbanol A ............. 59
3.2 DFT calculation for model study ........................................................................... 68
3.3 Approaches towards the synthesis of morusalbanol A .......................................... 73
3.3.1 Retrosynthetic analysis for morusalbanol A ................................................. 74
3.3.2 Synthesis of diene 189 .................................................................................. 75
3.3.3 Synthesis of dienophile 191 .......................................................................... 76
3.3.4 Diels-Alder reaction between 189 and 191 .................................................. 77
3.3.5 Synthesis of diene 190 .................................................................................. 78
3.3.6 Synthesis of dienophile 87 ............................................................................ 78
3.3.7 Diels-Alder reaction between 190 and 87 .................................................... 79
CHAPTER 4: APPROACHES TOWARD THE SYNTHESIS OF
SOROCEIN B .............................................................................. 88
4.1 Retrosynthetic analysis .......................................................................................... 88
4.1.1 Synthesis of chalcone core as diene 105....................................................... 89
4.1.2 Synthesis of dienophile 208 .......................................................................... 90
4.1.3 Method development for installation of 2,2-Dimethyl-2H-chromenes (pyran
moiety) .......................................................................................................... 91

viii
4.1.4 Cycloaddition reaction between 105 and 208 ............................................... 97
4.2 Cycloaddition reaction of diene 232 and dienophile 233 ...................................... 98
4.2.1 Synthesis of diene 232 .................................................................................. 98
4.2.2 Synthesis of dienophile 233 .......................................................................... 99
4.3 Cycloaddition reaction of diene 105 and dienophile 210 .................................... 101
4.4 Cycloaddition reaction of diene 105 and dienophile 88 ...................................... 102
CHAPTER 5: CONCLUSION AND FUTURE WORK ......................................... 107
5.1 Conclusion ........................................................................................................... 107
5.2 Suggestion for Future work ................................................................................. 108
CHAPTER 6: EXPERIMENTALS ........................................................................... 111
References .................................................................................................................... 138
APPENDIX : SPECTRA OF COMPOUNDS .......................................................... 150
LIST OF PUBLICATIONS ........................................................................................ 193

ix
LIST OF FIGURES
Figure 1.1: Flavonoids from mulberry tree ....................................................................... 2
Figure 1.2: Biogenetic synthesis of MDA and cylised MDA adducts .............................. 3
Figure 1.3: Examples of dehydroprenylchalcone adduct .................................................. 6
Figure 1.4: Examples dehydroprenylflavonoid adduct ..................................................... 7
Figure 1.5: Examples of dehydroprenylstilbene adducts .................................................. 8
Figure 1.6: Examples of prenylarylbenzofuran type adducts ........................................... 9
Figure 1.7:The CD spectra of mulberrofuran C (24) and J (25) and their absolute
configurations……………………………………………………………..10
Figure 1.8: The reduction of mulberrofuran C (24) and mulberrofuran J (25) and their
CD spectra of their reduction products........................................................11
Figure 1.9: The X-ray analysis of mulberrofuran G pentamethyl ether (30) .................. 12
Figure 1.20: Feeding experiments with methyl chalcones 33 and prenyl chalcones 34 to the
Morus alba cell cultures .................................................................................................. 16
Figure 1.21: Biosynthesis of sorocenol B (37) and related natural products .................. 17
Figure 1.22: Biosynthesis of morusalbanol A (42) ......................................................... 17
Figure 1.23: Examples of mulberry Diels-Alder adducts with hypotensive property .... 19
Figure 1.24: Examples of mulberry Diels-Alder adducts with promising inhibition against
malondialdehyde (mda) ................................................................................................... 22
Figure 1.25: The 50% cytotoxicity concentration of sanggenol M (59) and sanggenon C
(45) against human oral squamous cell carcinoma (HSC-2) and human salivary gland
tumuor (HSG) ................................................................................................................. 24
Figure 1.26: Structure of (+)_morusalbanol A (42) and sorocein B (62)… ................... 26
Figure 3.1: Morusalbanol A (42) and related mulberry Diels-Alder adducts ................. 58
Figure 3.2: Chemical structure for CAS 441772-64-4 (185) .......................................... 63
Figure 3.3: Key NOESY correlations leading to relative stereochemistry assignment of 183
......................................................................................................................................... 64

x
Figure 3.4: Semisynthesis of wittiorumin F (8) and mulberrofuran F (9) from
chalcomoracin C (6) ........................................................................................................ 65
Figure 3.5: Energy profile of transition state structure of (a) 179 (b) 180 (c) 188 (d) 189
where black dashed lines denoted the bond formation that led to cyclisation and blue
dashed lines denoted the intermolecular hydrogen bonding ........................................... 69
Figure 3.6: Calculated reaction path for a) 187 and b) 186 ............................................ 71
Figure 3.7: Potential energy scans on dihedral angle together the respective energy barrier
and distance for (a) endo-181 and (b) exo-182. The distances between the carbon and
oxygen atom in red circle were calculated in the plot ..................................................... 72
Figure 3.8: Morusalbanol A (42) ................................................................................... 74
Figure 3.9: Comparison of the 1H NMR spectra for endo-204 at a) day 1 b) day 14 ..... 80
Figure 3.10: Comparison of the 13
C NMR spectra for endo-204 at a) day 1 b) day 14 .. 81
Figure 3.11: NOESY correlation of moruslabanol A pentamethyl ether 206 ................. 84
Figure 4.1: Sorocein B (62) ............................................................................................ 88

xi
LIST OF SCHEMES
Scheme 1.1: The general Diels-Alder reaction of mulberry Diels-Alder adducts ............ 2
Scheme 1.2: Classification of mulberry Diels-Alder adducts ........................................... 5
Scheme 1.3: Establishment of absolute configuration of mulberrofuran C (24) via
mulberrofuran G (29) and aromatized compound 32 ...................................................... 12
Scheme 2.1: Semisynthesis of kuwanons G (45) and H (13) ......................................... 29
Scheme 2.2: Pyrolysis experiment of chalcomoracin (47).............................................. 30
Scheme 2.3: Semisynthesis of mulberrofuran C (24) and chalcomoracin (6) ................ 31
Scheme 2.4: Retrosynthesis of kuwanon V pentamethyl ether (73) and dorsterone
pentamethyl ether (74) via a Diels-Alder reaction .......................................................... 32
Scheme 2.5: Synthesis of diene 75 .................................................................................. 33
Scheme 2.6: Synthesis of dienophile 75 ......................................................................... 34
Scheme 2.7: Synthesis of kuwanon V (73) and dorsterone (74) methyl ether ................ 35
Scheme 2.8: The coupling constants of the cyclohexene ring of 72 and 73 ................... 35
Scheme 2.9: Retrosynthetic analysis of methyl ether mulberrofuran C (85) and methyl
ether chalcomoracin (69)............................................................................ 36
Scheme 2.10: Synthesis of diene 87 ................................................................................ 37
Scheme 2.11: Synthesis of dienophile 89 ....................................................................... 38
Scheme 2.12: Synthesis of mulberrofuran C hexamethyl ether (101), mulberrofuran C
heptamethylether (86), mulberrofuran J hexamethyl ether (102) ............ 39
Scheme 2.13: Synthesis of dien Synthesis of chalcomoracin hexamethyl ether (103),
chalcomoracin heptamethyl ether (70), mongolicin F hexamethyl ether
(104)………………………………………………………….................40
Scheme 2.14: Retrosynthetic scheme of kuwanon J heptamethyl ether (105) ................ 41
Scheme 2.15: Retrosynthetic analysis for sorocenol B (37) ........................................... 42

xii
Scheme 2.16: Synthesis of dienophile 110……………………………………………..43
Scheme 2.17: Synthesis of diene 111 .............................................................................. 43
Scheme 2.18: AgNp’s catalyzed Diels-Alder cycloaddition of 110 and 111 ................. 44
Scheme 2.19: Pd(II)-catalyzed oxidation cyclization 107 and 121 ................................. 45
Scheme 2.20: Key NOE’s leading to relative stereochemistry assignments 107 and 121...
......................................................................................................................................... 46
Scheme 2.21: Pd(II)-catalyzed Oxidation cyclization 109 ............................................. 46
Scheme 2.22: Synthesis of sorocenol B (106) ................................................................ 47
Scheme 2.23: Biomimetic synthetic design for Brosimone A (121) and B (122) .......... 48
Scheme 2.24: Synthesis of Brosimone B (122) .............................................................. 50

xiii
LIST OF TABLES
Table 1.1: The optical rotations [α]D of mulberry Diels-Alder adducts isolated from nature
......................................................................................................................................... 14
Table 1.2: The cytotoxicity of mulberrofuran F1 (62), mulberrofuran F (9), chalcomoracin
(6) .......................................................................................... …………………………25
Table 2.1: Development of the initial methodology employing a model prenylchalcone 49
Table 3.1: Development of the initial methodology employing a model and dienophile 62
Table 3.2: Crystal data and structure refinement for 184 and 186 .................................. 67
Table 3.3: Crystal data and structure refinement for 206 and 207 .................................. 85
Table 3.4: 1H and
13C NMR spectra of morusalbanol A pentamethyl ether (206) ......... 86
Table 3.5: 1H and
13C NMR spectra of compound 207................................................... 87
Table 4.1: Pd-catalysed coupling-condensation of o-iodophenol 212 and 2-methyl-3-buten-
2-ol 82 ............................................................................................................................. 94
Table 4.2: Pd-catalysed condensation of o-halophenols 212 and 2-methyl-3-buten-2-ol 82
......................................................................................................................................... 96
Table 4.3: Attempt to remove the OMe groups of endo-230 ........................................ 105

xiv
LIST OF SYMBOLS AND ABBREVIATIONS
(o-toly)3-P Tri(o-tolyl)phosphine
∆G Gibbs Free Energy of Reaction
∆G* Activation Energy
A2780 Human Ovaries Carcinoma Cell Line
A549 Human Lung Carcinoma Cell Line
AcCl Acetyl chloride
AgBF4 Silver tetrafluoroborate
AgNP Silver nanoparticle
AgOTf Silver trifluromethnesulfonate
AlBr3 Aluminium bromide
BCl3 Boron trichloride
Bel-7402 Human Liver Carcinoma Cell Line
BF3.Et2O Boron trifluoride Diethyl Etherate
BGC-823 Human Stomach Carcinoma Cell Line
BuNBH4 Tetrabutylammonium
CD Circular Dichroism
CH2Cl2 Dichloromethane

xv
COX Cyclooxygenase
DDQ 2,3-dichloro-5,6-dicyanobenzoquinone
DHDA Dehydrogenative Diels-Alder
DMF N, N-Dimethylformamide
DMSO Dimethyl sulfoxide
EOM Ethoxymethyl
EOM-Cl Ethoxymethyl chloride
Et3N Triethylamine
EtOAc Ethyl acetate
EtOH Ethanol
FeCl3 Iron (III) chloride
Ga(OTf)3 Gallium (III) triflate
H2O Water
HCl Hydrochloric acid
HCT-8 Human Colon Carcinoma Cell Line
HIF-1 Hypoxia-Inducible Factor-1
HMBC Heteronuclear Multiple Bond Correlation
HPLC High Performance Liquid Chromatography
HRMS High Resolution Mass Spectroscopy

xvi
HSC-2 Human Oral Squamous Cell Carcinoma
HSG Human Salivary Gland Tumour
I2 Iodine
ICI Iodine monochloride
K2CO3 Potassium carbonate
KIO3 Potassium iodate
KOAc Potassium acetate
LDA Local Density Approximation
LPS Lipopolysaccharide
LST Linear Synchronous Transit
mda Malondialdehyde
MDA Mulberry Diels-Alder
MeOH Methanol
MgI2 Magnesium iodide
MOM Methoxymethyl
MOMCl Methoxymethyl chloride
MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
Na2CO3 Sodium carbonate
NaH Sodium hydride

xvii
NBS N-bromosuccinimide
n-Bu4NCl n-Tetrabutyammonium chloride
n-BuLi n-Butyllithium
NMR Nuclear Magnetic Resonance
NOESY Nuclear Overhauser Effect
Pd(dba)2) Tris[dibenzylideneacetone]dipalladium(0)
Pd(OAc)2 Palladium (II) acetate
Pd(PPh3)4 Tetrakis(triphenylphospine)palladium(0)
Pd(TFA)2 Palladium (II) trifluoroacetate
PdCl2 Palladium (II) chloride
Pt/C Platinum on activated carbon
QST Quadratic Synchronous Transit
SiO2 Silicon dioxide
TCM Traditional Chinese Medicine
THF Tetrahydrofuran
TiCl4 Titanium tetrachloride
TLC Thin Layer Chromatography
TMEDA Tetramethylethylene diamine
TMSI-quinoline 1-Trimethylsilylquinolinium iodide

xviii
TPP Tetraphenylporphyrin
UV Ultraviolet
ZnI2 Zinc iodide

1
CHAPTER 1
INTRODUCTION AND LITERATURE REVIEW
1.1 Introduction
Moraceous plants (family MORACEAE) are a rich source of mulberry Diels-Alder
(MDA) adducts. Moraceae comprises of a large family of sixty genera and nearly 1400
species. Some important genera such as Artocarpus, Ficus and Morus, are found in
temperate and subtropical regions of the world (Venkataraman, 1972). The mulberry tree
has been cultivated for thousands of year and play a key role in agriculture in China,
particularly in providing leaves as indispensable food for silk worms.
The different parts of the mulberry tree, such as root bark, twigs, leaves, and fruits,
have also been commonly used in Traditional Chinese Medicine. Besides Morus alba, the
other species of Morus, such as M. australis and M. cathayana were also used in folk
medicine in China. In the early 1970s, investigation into chemical constituents of the
mulberry tree’s (Morus alba) root bark, which is called “Sang-Bai-Pi” in Traditional
Chinese Medical, was initiated. Many compounds with unique structures were isolated,
such as Diels-Alder type adducts (so-called Mulberry Diels-Alder adducts), benzofurans,
stilbenes and flavonoids (Yang et al. 2014).
Many of mulberry Diels-Alder adducts are biologically active and exhibit a wide
range of pharmacological activities including antibacterial, antioxidant, anti-inflammatory,
anticancer, cyclic AMP inhibition, tyrosinase inhibition, and hypoxia-inducible factor-1
(HIF-1) inhibition (Gan et al. 2010; Nomura, 1988a; Yen, Wu, & Duh, 1996). These
discoveries have stimulated significant research interest into their biogenesis and chemical
synthesis.

2
1.2 Mulberry Diels-Alder adducts
Nomura and co-workers were pioneers in the studies of Mulberry-Diels-Alder
adducts from mulberry tree (Nomura, 1988a). They isolated prenylated flavoids which
displayed an interesting pattern for the isoprenyl group such as, kuwanon A (1), kuwanon B
(2) and kuwanon C (3) (Figure 1.1). Generally, Mulberry Diels-Alder adducts are
trisubstituted methylcyclohexenes derived from intermolecular [4+2] cycloaddition (Diels-
Alder) reaction between a dehydroprenylphenol as the diene and the α,β-unsaturated bond
of a chalcones, as the dienophile (Scheme 1.1).
OHO
OH O
Me
Me
OHO
Me
Me
kuwanon A (1)
OHO
OH O
Me
Me
OHO
kuwanon B (2)
Me
Me
OHO
OH O
Me
Me
OHHO
kuwanon C (3)
MeMe
Figure 1.1. Flavonoids isolated from mulberry tree
OOH
+
Me
Me
O
HO
cis,trans trans-trans
OH
HO
chalcone (dienophile)
dehydroprenylphenol(diene)
+Me
Me
HO
prenylphenol
Me
O
HO
OH
Scheme 1.1. The general Diels-Alder reaction of mulberry Diels-Alder adducts

3
OHOOH
OH Me
OOH
HO
Me
Me OH
OH
+
morachalcone A (4) dehydroprenylmoracin C (5)
Me
OOH
OH
O
HOOH
MeMe
HO
OH
HO
3'' 4''
5''SR
S
[4+2]
intramolecular cyclisation/ketalisation
Me
O
OH
O
O
OHHO
Me Me
OH
OH
HO wittiorumin F (8)
3''4''
5''SR
S+
O O
Me
OH
OH
OH
OH
H
Me
Me
O
HO
SR S
R
3''
4''5''
mulberrofuran F (9)
chalcomoracin (6)(cis-trans, endo)
+
mongolicin F (7) (trans-trans, exo)
Me
OOH
OH
O
HOOH
MeMe
HO
OH
HO
Figure 1.2. Biogenetic synthesis of MDA and cylised MDA adducts
Adducts bearing cis-trans stereochemistry are derived from an endo addition
whereas the trans-trans isomer arises from the exo addition. For example, the reaction of
morachalcone A (4) (as dienophile) and dehydroprenyl moracin C (5) (as diene) in nature
produces a pair of cis-trans and trans-trans adducts, chalcomoracin (6) and mongolicin F
(7), respectively (Figure 1.2). Interestingly, the cis-trans MDA adducts chalcomoracin (6)
underwent acid catalysed regio- and stereo-selective intramolecular cyclization/ketalization

4
in nature to give two different metabolites containing an intriguing oxacyclic core structure
(e.g. wittiorumin F (5) and mulberrofuran F (6) (Figure 1.2)).
According to a report by Nomura and co-workers, the mulberry Diels-Alder adducts
may be classified into four groups on the basis of the phenol nuclei as follows: (a)
dehydroprenylchalcone adducts; (b) dehydroprenylflavonoid adducts; (c)
dehydroprenylstilbene adducts; (d) dehydroprenyl-2-arylbenzofuran adducts (Scheme 1.2)
(Nomura & Hano, 1994).
Amongst the four types of mulberry Diels-Alder adducts listed in Scheme 1.2, the
dehydroprenylchalcone type Diels-Alder type adducts are rare. Kuwanon J (10), kuwanon I
(11) and guangsangon C (12) are some. Examples of these dehydroprenylchalcone type
Diels-Alder adducts as kuwanon J (10), kuwanon I (11) guangsangon C (12) (Figure 1.3).
Kuwanon J (10) and kuwanon I (11) were isolated from the root bark of Morus macroura
and Morus alba (Dai et al. 2004; Nomura & Fukai, 1981), respectively. Guangsangon C
(12), was isolated from the stem bark of Morus macrour (Dai et al. 2004)

5
Scheme 1.2. Classification of mulberry Diesl-Alder adducts

6
OH
OH
Me
Me
O
Me
OH
HO
O OH
OH
HO
HO
3"
4"5"
OH
OH
Me
Me
O
Me
OH
HO
O OH
OH
HO
HO
3"
4"5"
kuwanon J (10) kuwanon I (11)
OH
OH
O
MeHO
OH
OH
HO
HO
3"
4"5"
OH
O
5
3
1
2'
4'
guangsangon C (12)
Figure 1.3. Examples of dehydroprenylchalcone adduct
Dehydroprenylflavonoid Diels-Alder adducts (Scheme 1.2) are abundantly found in
nature. Kuwanon H (13), wittiorumin A (14), wittiorumin B (15), wittiorumin C (16) (Tan
et al., 2009), guangsangon D (17), guangsangon K (18) (Dai et al. 2004) and sanggenon G
(19) (Rollinger et al. 2006) are some of the examples of this type of Diels-Alder adducts
(Figure 1.4).

7
O
OOH
Me
Me
HO OH
Me
O
HO
OH
Me
Me
OH
HO
OHO
OH O
OH
R1 OH
Me
O OH
OH
R2
HO
OH
3''
kuwanon H (13) wittiorumin A (14) R1=R2, 3"Rwittiorumin B (15) R1=OH, R2= prenyl, 3"Rwittiorumin C (16) R1=H, R2= prenyl, 3"S
OHO
O
HOOH
Me
O OH
OH
HO
OH
OHO
O
Me
O OH
OH
HO
OH
OHOH OH
guangsangon D (17) guangsangon K (18)
OH
OH
OMe
Me
HO
OH
OH
O
O
HO
HO OH
sanggenon G (19)
Figure 1.4. Examples of dehydroprenylflavonoid adduct
There are not as many examples of these compounds in the third class of Diels-
Alder adduct, dehydroprenylstilbene. Kuwanon X (20) was isolated from the root bark of
Morus Ihou in Japan and Morus macroura in China. Kuwanon Y (21) was isolated from

8
Morus alba and Morus macroura (Rama Rao et al. 1983). Kuwanon P (22) and
guangsangon B (23) were isolated from Morus Ihou and Morus macroura, respectively
(Dai et al. 2004, (Hano, Tsubura & Nomura, 1986) (Figure 1.5).
O
HO OH
OH
HO
OH
OH
Me
OH
3"
4"
5"
R
OH5
33'
5'
O
HO OH
OH
OH
Me
OH
3"
4"
5"5
3R
HO
OH
kuwanon X (20): 3”α kuwanon P (22): R= OH
kuwanon Y (21): 3”β guangsangon B (23): R= H
Figure 1.5. Examples of dehydroprenylstilbene adducts (Hirakura et al. 1985, Dai et al. 2004)
The fourth class of mulberry Diels-Alder adduct isolated from the genus Morus is
dehydroprenyl-2-arylbenzofuran type. Mulberrofuran C (24), calcomoracin (6),
mulberrofuran J (25), mongolicin F (7) and mulberrofuran U (26) are examples of this type
of Diels-Alder adducts (Basnet et a.l 1993; Nomura et al. 1982; Tan et al. 2009) (Figure
1.6).

9
O
HO OH
OH
O
HO
HO
OH
Me
OH
R
O
HO OH
OH
O
HO
HO
OH
Me
OH
R
O
HO OH
OH
OHO
OH
OH
Me
OH
MeMe
mulberrofuran C (24): R = H mulberrofuran J (25): chalcomoracin (6): R = prenyl mongolicin F (7): R = prenyl
mulberrofuran U (26) Figure 1.6. Examples of prenylarylbenzofuran type adduct (Basnet et al. 1993; Nomura et al. 1982;
Tan et al. 2009)
1.3 Absolute configurations of mulberry Diels-Alder adducts
The absolute configurations of C3”, C4” and C5” in the methylcyclohexene ring of
mulberry Diels-Alder adducts were systematically investigated by the Nomura group. In
general, the stereochemistry and absolute configuration of mulberry Diels-Alder adducts
have been confirmed by two methods (a) circular dichroism (CD) spectroscopic studies and
(b) X-ray analysis (Nomura, 1988b).
The magnitude of ∆ε values in the circular dichroism (CD) spectra of
mulberrofurans C (24) and J (25) is known to be larger than any other mulberry Diels-Alder
adducts (Figure 1.7) (Hano et al. 1988). Mulberrofurans C and J exhibited strong split
Cotton effect in the region of 280-350 nm in the UV for both of the compounds. It has
been suggested that this effect may originated from exciton coupling between 2,4-
dihydroxybenzoyl and 2-arylbenzofuran chromophores (Hano et al. 1988). In order to test
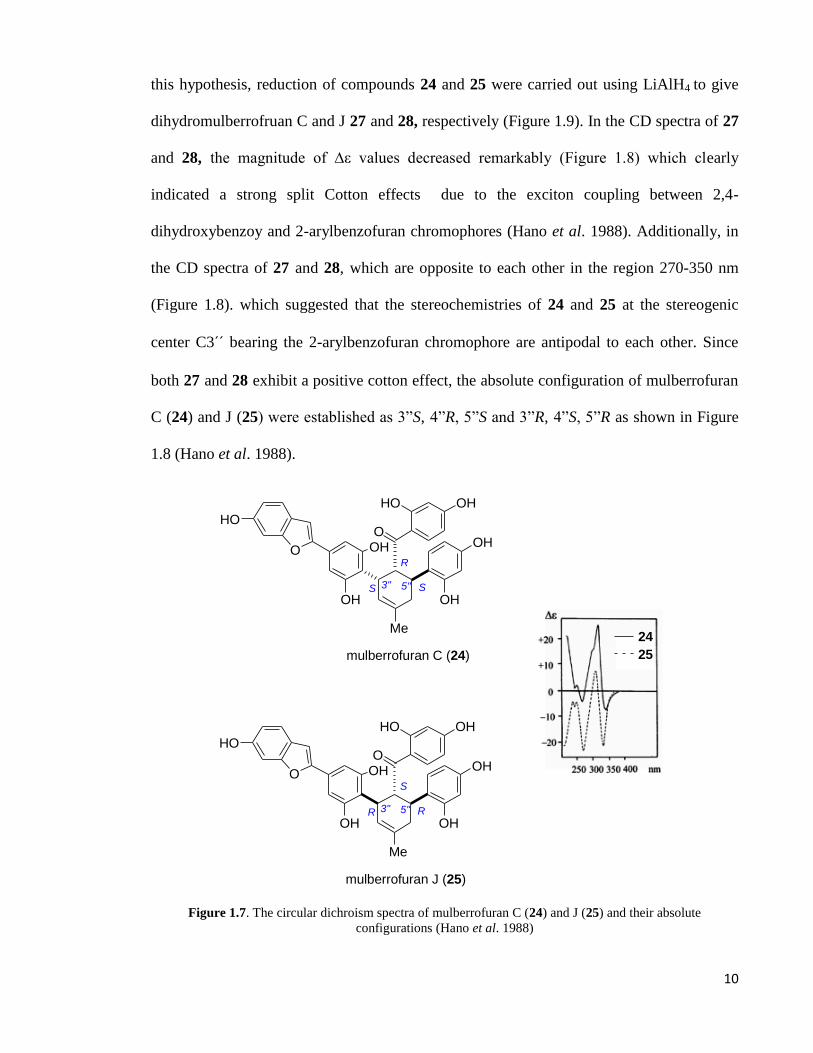
10
this hypothesis, reduction of compounds 24 and 25 were carried out using LiAlH4 to give
dihydromulberrofruan C and J 27 and 28, respectively (Figure 1.9). In the CD spectra of 27
and 28, the magnitude of ∆ε values decreased remarkably (Figure 1.8) which clearly
indicated a strong split Cotton effects due to the exciton coupling between 2,4-
dihydroxybenzoy and 2-arylbenzofuran chromophores (Hano et al. 1988). Additionally, in
the CD spectra of 27 and 28, which are opposite to each other in the region 270-350 nm
(Figure 1.8). which suggested that the stereochemistries of 24 and 25 at the stereogenic
center C3´´ bearing the 2-arylbenzofuran chromophore are antipodal to each other. Since
both 27 and 28 exhibit a positive cotton effect, the absolute configuration of mulberrofuran
C (24) and J (25) were established as 3”S, 4”R, 5”S and 3”R, 4”S, 5”R as shown in Figure
1.8 (Hano et al. 1988).
Me
O
HO OH
OH
OH
OH
OHO
HO
S
R
S3" 5"
mulberrofuran C (24)
Me
O
HO OH
OH
OH
OH
OHO
HO
R
S
R3" 5"
mulberrofuran J (25)
24
25
Figure 1.7. The circular dichroism spectra of mulberrofuran C (24) and J (25) and their absolute configurations (Hano et al. 1988)

11
Me
O
HO OH
OH
OH
OH
OHO
HO
3"
27
28
LiAlH4
Me
HO
HO OH
OH
OH
OH
O
HO
3"
OH H
mulberrofuran C (24): 3”α dihydromulberrofuran C (27): 3”α
mulberrofuran J (25): 3”β dihydromulberrofuran J (28): 3”β
Figure 1.8. Reduction of mulberrofuran C (24) and mulberrofuran J (25) and the CD spectra of
dihydromulberrofuran C and J (Hano et al. 1988)
Another method to determine the absolute configuration of mulberry Diels-Alder
adducts is through X-ray crystallographic analysis. The absolute configuration of
mulberrofuran G pentamethyl ether (30) has been determined by X-ray crystallographic
analysis (Rama Rao et al. 1983) where the relative configuration of the stereogenic center
at C-8” was determined as shown in Figure 1.9. (Rama Rao et al. 1983). Absolute
configuration of mulberrofuran C (24) was confirmed from the following procedure.
Conversion of 24 to mulberrofuran G (29) was achieved under acidic conditions as
described in Scheme 1.3 followed by methylation of 29 to give the mulberrofuran G
pentamethyl ether (30). Crystal structure of the ether 30 was confirmed by X-ray
crystallographic analysis. Treatment of 30 with N-bromosuccinimide (NBS) gave
bromomulberrofuran G pentamentyl ether (31) which was then converted to an aromatic

12
compound 32 through dehydrogenation by 2,3-dichloro-5,6-dicyanobenzoquinone DDQ
(Scheme 1.3). The X-ray crystallographic analysis of 32 showed the absolute configuration
at C-8” to be R. As the correlation between 32 and 24 through 30 was confirmed, the
absolute configuration of mulberrofuran C (24) was determined to be 3”S, 4”R, 5”S (Hano
et al. 1988).
O
HO OH
OH
O
HO
HO
OH
Me
1.5% H2SO4
EtOH
55%
O O
OR1
O
R1O
OR1
Me
OR1
H
H H
OH
3" 5"
4"
3" 20"
17"
13" 11"
OR1R2
8"
6'
mulberrofuran C (24) CH3Imulberrofuran G (29)
30 R1= Me, R2= HNBS
31 R1= Me, R2= Br
O O
OMe
O
MeO
OMe
Me
OMe
3"5" 20"
17"
13" 11"
OMe
8"
6' BrDDQ
32
5"
Scheme 1.3. Establishment of absolute configuration of mulberrofuran C (24) via mulberrofuran G (29)
and aromatized compound 32 (Hano et al. 1988)

13
Figure 1.9. The X-ray analysis of mulberrofuran G pentamethyl ether (30) (Rama Rao et al. 1983)
Optical rotations of some mulberry Diels-Alder adducts isolated from Morus
species are summarized in Table 1.1. The relationships of the absolute configuration,
optical rotation and CD spectrum, provided a more convenient way to classify mulberry
Diels-Alder-type adducts. In case of a negative optical rotation or negative Cotton effects, a
maximum UV absorption will be observed in the CD spectrum, and the relative
configuration of a mulberry Diels-Alder adduct will be trans-trans. Otherwise, if both the
optical rotation and Cotton effect shown in the CD spectrum were positive, then the relative
configuration of a mulberry Diels-Alder adduct will be cis-trans (Hano et al. 1998).
Configuration at the C-3´´ stereogenic center will influence the sign of optical rotation.
Therefore, absolute configurations of the three stereogenic centers (3´´, 4´´, 5´´) in the
methylcyclohexene ring of a cis-trans mulberry Diels-Alder adduct may be specified as 3”S,
4”R, 5”S, and those of the trans-trans adducts as 3´´R, 4´´S, 5´´R.

14
TABLE 1.1. The optical rotations [α]D of some mulberry Diels-Alder adducts isolated from nature
Name
Relative
configuration
Optical
rotation [α]D Name
Relative
configuration
Optical
rotation [α]D australisin C cis-trans +340 (MeOH) mongolicn F trans-trans -283 (MeOH)
australisin C cis-trans +340 (MeOH) mongolicn F trans-trans -283 (MeOH)
cathayanon A cis-trans -194 (MeOH) moracenin D trans-trans -388 (MeOH)
chacomoracin cis-trans +194 (acetone) mulberrofuran J trans-trans -341 (MeOH)
guangsangon E cis-trans +140 (MeOH) mulberrofuran J trans-trans -341 (MeOH)
mulberrofuran C cis-trans +153 (MeOH) sanggenon D trans-trans -145 (MeOH)
mulberrofuran E cis-trans +302 (MeOH) sanggenon E trans-trans -86 (MeOH)
mulberrofuran O cis-trans +196 (MeOH) sanggenon M trans-trans -126 (MeCN)
mulberrofuran T cis-trans +139 (MeOH) sanggenon T trans-trans -194 (EtOH)
sanggenon C cis-trans +304 (MeOH) wittionrumin A trans-trans -415 (MeOH)
sanggenon J cis-trans +98 (MeOH) wittionrumin B trans-trans -443 (MeOH)
sanggenon O cis-trans -64 (MeOH) wittiorumin D trans-trans -575 (MeOH)
wittionrumin C cis-trans +420 (MeOH) wittiorumin E trans-trans -296 (MeOH)
wittiorumin G cis-trans +87 (MeOH) yunanensin B trans-trans -183 (MeOH)
albafuran C trans-trans -302 (MeOH) yunanensin B trans-trans -183 (MeOH)
cathayanon B trans-trans -734 (MeOH) yunanensin C trans-trans +439 (MeOH)
guangsangon A trans-trans -409 (MeOH) albanol B a +118 (CHCl3)
guangsangon D trans-trans -108 (MeOH) australisine A a +523 (MeOH)
guangsangon F trans-trans -112 (MeOH) australisine B a +191 (MeOH)
guangsangon G trans-trans -469 (MeOH) cathayanon C a +11 (MeOH)
guangsangon H trans-trans -128 (MeOH) mongolicn A a +650 (MeOH)
guangsangon I trans-trans -471 (MeOH) mongolicn C a +160 (MeOH)
guangsangon J trans-trans -420 (MeOH) mulberrofuran F a +412 (MeOH)
guangsangon K trans-trans -179 (MeOH) mulberrofuran G a -546 (MeOH)
guangsangon M trans-trans -277 (MeOH) mulberrofuran K a +425 (MeOH)
guangsangon N trans-trans -335 (MeOH) wittiorumin F a +299 (MeOH)
kuwanon G trans-trans -534 (MeOH) yunanensin A a +12 (MeOH)
kuwanon H trans-trans -536 (MeOH) yunanensin D a +161 (MeOH)
kuwanon L trans-trans -227 (MeOH) yunanensin E a +675 (MeOH)
kuwanon O trans-trans -243 (MeOH) mulberrofuran Q a +182 (EtOH)
mongolicn D trans-trans -227 (MeOH) cathayanon C a +11 (MeOH)
a non cis-trans or trans-trans

15
1.4 Biosynthesis of mulberry Diels-Alder adducts
Morus alba callues tissues exhibit a high productivity of mulberry Diels-Alder
adducts. The production of kuwanon J (10) and chalcomoracin (6) as the major secondary
metabolites in Morus alba cell cultures was about 100-1000 times more than that of the
intact plant. Therefore, biosynthesis of mulberry Diels-Alder adducts was studied with the
aid of Morus alba cell cultures (Ueda et al. 1982).
The biosynthesis of the mulberry Diels-Alder adducts was conducted by feeding
experiments of methyl chalcones 33 in Morus alba cell cultures. Chalcone 33 was
administered into cell cultures and this resulted in the isolation of prenyl chalcone 34,
kuwanon J methyl ether 36 and chalcomoracin methyl ether (35) (Figure 1.20). The
formation of 34 from 33 in the cell cultures indicated prenylation occurred first before the
formation of 35 and 36.

16
OOH
HO
OH
OMe
methyl chalcone 33
OOH
HO
OH
OMe
Me
Me
prenyl chalcone 34
M. albacell cultures
M. albacell cultures
OOH
HO
OH
OMe
Me
Me
prenyl chalcone 34
OOH
HO
OH
OMeMe
HO
MeO
O
OH
OH Me
Me
OH
HOMe
HO
MeO
O
OH
OH Me
Me
methyl chalcomoracin 35
methyl kuwanon J 36
O
OH
Figure 1.20. Feeding experiments with chalcone 33 and prenyl chalcone 34 to the Morus alba cell cultures
Biosynthesis pathway of cyclized MDA such as morusalbanol A (42), sorocenol B
(37) (Hano, et al. 1995), mulberrofuran I (39) (Hano et al. 1984), australisin B (40) (Zhang
et al. 2007) and mongolicin C (7) (Kang, Chen, & Yu, 2006) consists of intramolecular
cyclization or oxidative cyclization of their corresponding mulbery Diels-Alder adduct
precursors (Figure 1.21, Figure 1.22).

17
O
OH
OH
OMe
MeMe
OHHO
OOH
OMe
MeMe
OHHO
O
HO
OH
Me
OH
HO
OHO
R
HO
OH
HO
O
OH
[O]
MeO
HO
R
HO
OH
HO
O
OH
-H2O
O
OH
MeO
HO
HO
OH
O
OH
O
OH
sorocenol B (37)
mulberrofuran I (39)R= prenyl, australisin B (40)R= H, mongolicin C (7)
R= prenyl, chalcomoracin (6)R= H, mulberrofuran C (24)
38
[o]
Figure 1.21. Biosynthesis of sorocenol B (37) and related natural products
Me
OHHO
OOH
HO
OH
HO
OHO
OCH3
Me
OH
HO
O
HO
HO
O
HO
OH
O
OCH3
41 42
Figure 1.22. Biosynthesis of morusalbanol A (42)

18
1.5 Biological activities of mulberry Diels-Alder type adducts
Mulberry tree is a highly valuable plant and has been widely used as important
sources in Traditional Chinese Medicine (TCM) (Nomura, 1988b). Studies of phenolic
constituents of Morus root bark were originally undertaken to characterize the components
of the root bark responsible for hypotensive activity. Kuwanon G (43), kuwanon H (13),
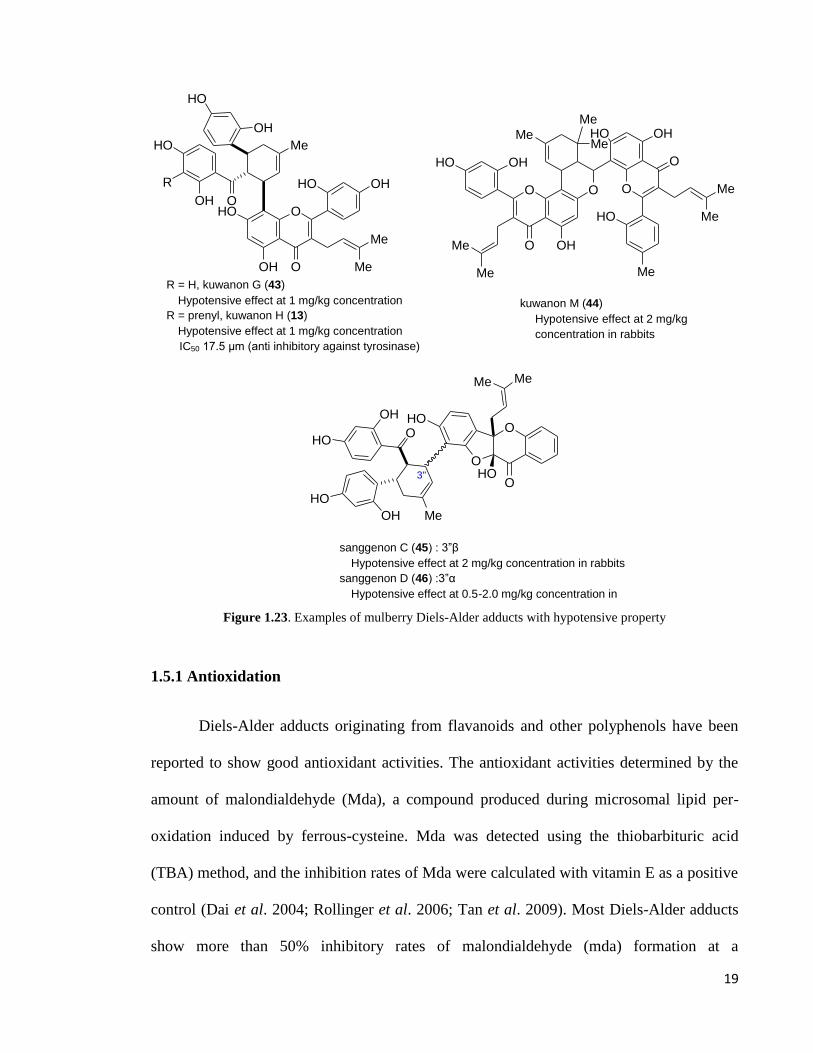
kuwanon M (44), mulberrofuran C (24), mulberrofuran F (9), and mulberrofuran G (29)
were shown to have hypotensive properties (Figure 1.23). Compounds 9, 13, 24, 29 and 43
also showed an almost equal transient decrease in arterial blood pressure in doses of 0.1-1
mg/kg in rabbits (Nomura & Fukai, 1980) while compound 44 showed hypotensive action
in hypertensive rats (2 mg/kg). Sanggenon C (45) and sanggenon D (46) have been
characterised as the hypotensive compounds of the crude medicine “Sang-Bai-Pi”.
Sanggenon C (45) showed at 2 mg/kg to have hypotensive effect in rabbits while
sanggenon D (46) required about 0.5-2.0 mg/kg (Figure 1.23) for the same activity in rats.
The promising anti-hypotensive properties of these mulberry Diels-Alder adducts warrant
further investigation for their biological activities.
19
Me
OH
HO
OOH
HO
HO
OH
O
O
HO OH
Me
Me
RO
Me
HO
O
Me
Me
OHHO
O
Me
OH
O
OMe
Me
OHHO
Me
Me
O
O
HO
Me Me
Me
O
OH
HO
OH
HO
3" HOO
R = H, kuwanon G (43)
Hypotensive effect at 1 mg/kg concentration
R = prenyl, kuwanon H (13)
Hypotensive effect at 1 mg/kg concentration
IC50 17.5 μm (anti inhibitory against tyrosinase)
kuwanon M (44)
Hypotensive effect at 2 mg/kg
concentration in rabbits
sanggenon C (45) : 3”β
Hypotensive effect at 2 mg/kg concentration in rabbits
sanggenon D (46) :3”α
Hypotensive effect at 0.5-2.0 mg/kg concentration in
rats Figure 1.23. Examples of mulberry Diels-Alder adducts with hypotensive property
1.5.1 Antioxidation
Diels-Alder adducts originating from flavanoids and other polyphenols have been
reported to show good antioxidant activities. The antioxidant activities determined by the
amount of malondialdehyde (Mda), a compound produced during microsomal lipid per-
oxidation induced by ferrous-cysteine. Mda was detected using the thiobarbituric acid
(TBA) method, and the inhibition rates of Mda were calculated with vitamin E as a positive
control (Dai et al. 2004; Rollinger et al. 2006; Tan et al. 2009). Most Diels-Alder adducts
show more than 50% inhibitory rates of malondialdehyde (mda) formation at a

20
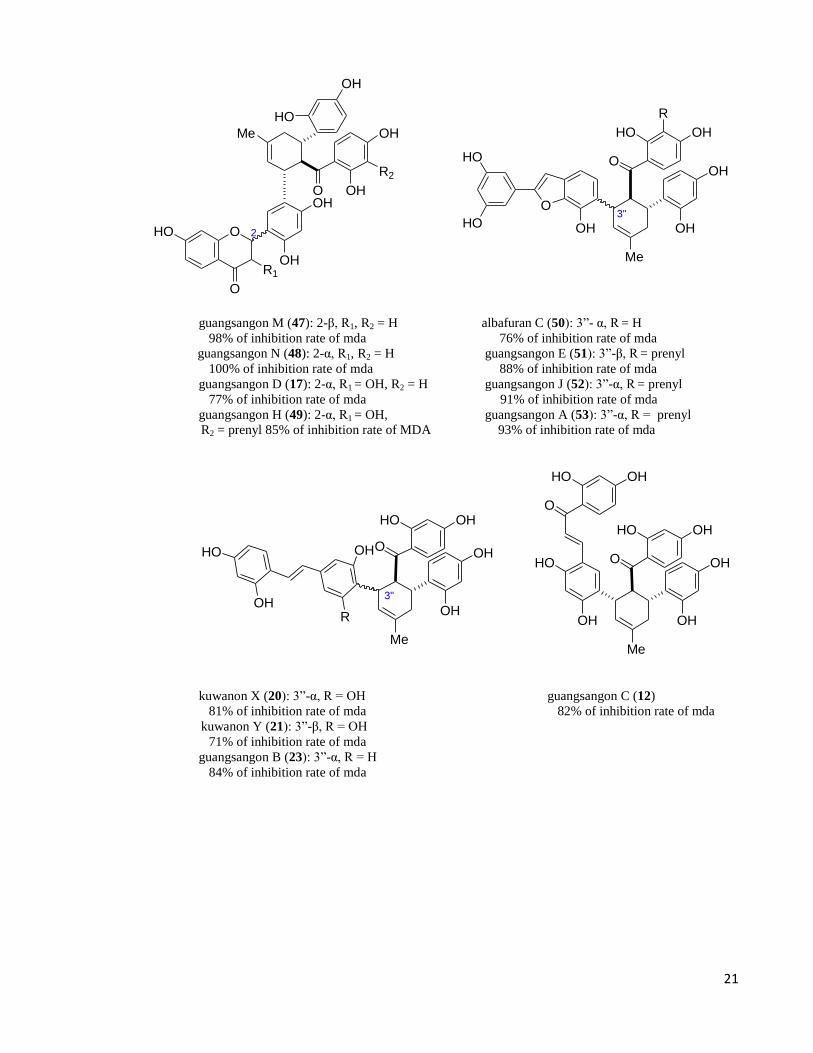
concentration of 10µM. Dehydroprenylflavonoid adducts such as guangsangon M (47),
guangsangon N (48), guangsangon D (17), guangsangon H (49), and guangsangon K (18)
exhibited good antioxidant activity with inhibition of mda ranging from 77-100% at the
concentration of 10 µM. Dehydroprenylarylbenzofuran adducts such is albafuran C (50),
guangsangon E (51), guangsangon J (52) and guangsangon A (53) showed inhibitor activity
ranging 76 - 91% at 10 µM (Dai et al. 2004). Dehydroprenylstilbene addcuts such as
kuwanon X (20), kuwanon Y (21), guangsangon B (23) and cathayanon D (54) displayed
inhibition mda ranging from 71-100% at 10 µM (Dai et al. 2004), (Zhang et al. 2009).
Dehydroprenylchalcone adduct such as guangsangon C (12) displayed inhibition rate of
mda 82% at 10 µM (Figure 1.22) (Dai et al. 2004).
Wittionrumin A (14), wittionrumin B (15), wittionrumin C (16), wittionrumin D
(57) also displayed potent antioxidant activities with inhibitory rates of 73%, 82%, 82%
and 62.5%, respectively, at a concentration of 10 μM. The relatively high inhibitory rates
indicated compounds 14, 15, and 16 possess better antioxidant activities than vitamin E
(Tan et al. 2009).
21
O
OH
Me OH
OHOH
OH
HO
O
O
HO 2
R1
Me
OH
OHO
HO OH
R
OH
O
HO
HO
R2
3"
guangsangon M (47): 2-β, R1, R2 = H albafuran C (50): 3”- α, R = H
98% of inhibition rate of mda 76% of inhibition rate of mda
guangsangon N (48): 2-α, R1, R2 = H guangsangon E (51): 3”-β, R = prenyl
100% of inhibition rate of mda 88% of inhibition rate of mda
guangsangon D (17): 2-α, R1 = OH, R2 = H guangsangon J (52): 3”-α, R = prenyl
77% of inhibition rate of mda 91% of inhibition rate of mda
guangsangon H (49): 2-α, R1 = OH, guangsangon A (53): 3”-α, R = prenyl
R2 = prenyl 85% of inhibition rate of MDA 93% of inhibition rate of mda
Me
OH
OHO
HO OH
ROH
HO OH
3"
Me
OH
OHO
HO OH
OH
HO
O
HO OH
kuwanon X (20): 3”-α, R = OH guangsangon C (12)
81% of inhibition rate of mda 82% of inhibition rate of mda
kuwanon Y (21): 3”-β, R = OH
71% of inhibition rate of mda
guangsangon B (23): 3”-α, R = H
84% of inhibition rate of mda

22
O O
OH
OHOH
OH
HO
Me
HO
HO
O
O OH
R1
OH
Me
O
OH
R2
OH
HO
OH
3"
cathayanon D (54) wittiorumin A (14): 3”-β, R1 = R2 = H
100% of inhibition rate of mda 73% of inhibition rate of mda
wittiorumin B (15): 3”-β, R1 = OH, R2 = prenyl
82% of inhibition rate of mda
wittiorumin C (16): 3”-α, R1 = H, R2 = prenyl
82% of inhibition rate of mda
HO
O
O
HO
OH
Me
O
OH
R
OH
HO
OH
wittiorumin D (55): R = H
62.5% of inhibition rate of mda
Figure 1.24. Examples of mulberry Diels-Alder adducts with promising inhibition against malondialdehyde
(mda)
1.5.2 Anti-inflammation
Cyclooxygenase (COX) is an enzyme, responsible for the formation of prostanoids
which are important biological mediators. The relief from symptoms of pain and
inflammation can be provided from pharmacological inhibition of COX. There are
three known cyclooxygenase isoenzymes, namely COX-1, COX-2, and COX-3.

23
Sanggenon C (45), sanggenon E (56), and sanggenon O (57) have been reported to
show inhibitions against COX-1 and COX-2 with IC50 values ranging from 10-14 and 40-
50 µM, respectively. It is noteworthy that compounds with prenyl group improved the
hydrophobicity, stability and penetrativity of the cell membranes (Rollinger et al. 2005).
Sanggenon B (58) and sanggenon D (46) showed the inhibition of lipopolysaccharide
(LPS)-induced NO production in RAW 264.7 cells, with IC50 values of 18.3 and 59.3 µM,
respectively (Cheon et al. 2000).
1.5.3 Cytotoxicity
Optically active mulberry Diels-Alder adducts have also been examined for
cytotoxicity properties. Shi and co workers reported that sanggenol M (59) and sanggenol C
(45) displayed potent cytotoxicity effect against human oral squamous cell carcinoma
(HSC-2) (CC50 13.0 μM and 18.0 μM) and human salivary gland tumour (HSG) (CC50 13.0
μM and 23.0 μM) (Shi et al. 2001).
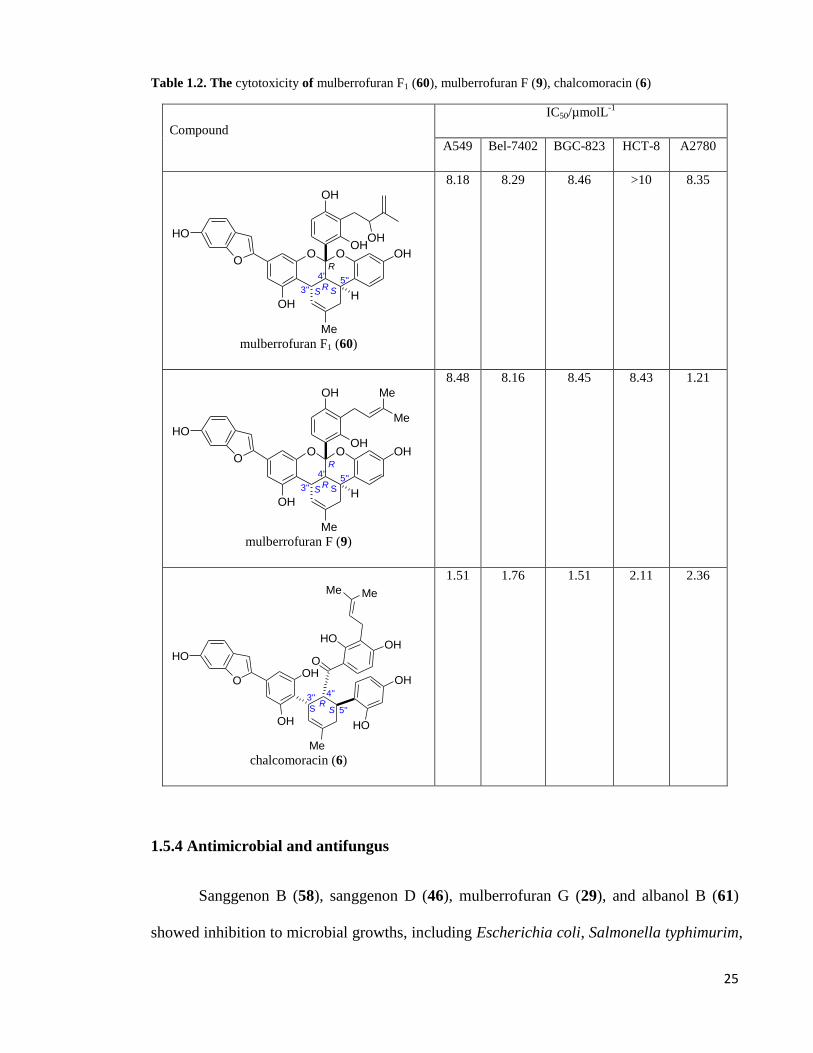
Mulberrofuran F1 (60), mulberrofuran F (9), chalcomoracin (6) and kuwanon J (11)
were evaluated for their cytotoxic activities against five human cancer cell lines [A549
(human lung carcinoma cell line), Bel-7402 (human liver carcinoma cell line), BGC-823
(human stomach carcinoma cell line), HCT-8 (human colon carcinoma cell line), and
A2780 (human ovaries carcinoma cell line)] by means of the (3-(4,5-dimethylthiazol-2-yl)-
2,5-diphenyltetrazolium bromide (MTT) cell viability assay (Zhang et al. 2007). It was
found that most of the Diels-Alder type adducts showed moderate cytotoxicities with IC50
values ranging from 1-10 µM. Ethanol extract of leaves of Morus alba yielded four Diels-
Alder type adducts, mulberrofuran F1 (60), mulberrofuran F (9), chalcomoracin (6) and

24
kuwanon J (11) but only compounds (6, 9 and 60) exhibited moderate cytotoxicity effects
as shown in Table 1.2 (Yang, Wang, & Chen 2010).
O
HO OH
HO
OH OMe
Me
OH
OH Me
Me
OHO
HO
O
O
OOH
HO
OH
MeMe
MeOH
HO
OH
HOO
sanggenon M (59) sanggenon C (45) CC50 = 13.0 μM against HSC-2 cell CC50 = 13.0 μM against HSC-2 cell
CC50 = 13.0 μM against HSG cell CC50 = 23.0 μM against HSG cell
Figure 1.25. The 50% cytotoxicity concentration of sanggenol M (59) and sanggenon C (45) against human
oral squamous cell carcinoma (HSC-2) and human salivary gland tumuor (HSG).
25
Table 1.2. The cytotoxicity of mulberrofuran F1 (60), mulberrofuran F (9), chalcomoracin (6)
Compound
IC50/µmolL-1
A549 Bel-7402 BGC-823 HCT-8 A2780
O O
Me
OH
OH
OH
OH
H
O
HO
SR S
R
3''
4''5''
OH
mulberrofuran F1 (60)
8.18 8.29 8.46 >10 8.35
O O
Me
OH
OH
OH
OH
H
Me
Me
O
HO
SR S
R
3''
4''5''
mulberrofuran F (9)
8.48 8.16 8.45 8.43 1.21
Me
OOH
OH
O
HOOH
MeMe
HO
OH
HO
3'' 4''
5''SR
S
chalcomoracin (6)
1.51 1.76 1.51 2.11 2.36
1.5.4 Antimicrobial and antifungus
Sanggenon B (58), sanggenon D (46), mulberrofuran G (29), and albanol B (61)
showed inhibition to microbial growths, including Escherichia coli, Salmonella typhimurim,

26
Staphylococcus epidermidis, and Staphylococcus aureus, with MIC values in the range of
5-50 µg/ml (Sohn et al. 2004). Chalcomoracin (6) completely inhibited the germination of
spore of Fusarium roseum and Bipolaris leersiae at a concentration of 10-100 µM (Yang et
al. 2010).
1.6 Scope and objectives of this thesis
Traditionally, natural products have played an important role in drug discovery and
have been the basis of most early medicines. However, the bioactive ingredients, often the
secondary metabolites isolated from the nature products are usually obtained in minute
quantity. Total synthesis of bioactive complex natural products is a significant challenge in
synthetic organic chemistry. Thus, our obejectives for this study are:
a) To synthesis two mulberry Diels-Alder adduct, morusalbanol A (42) and
sorocein B (62) (Figure 1.26) via a biomimetic Diels-Alder reaction to enable
access to these compounds for future bioactivity testing.
b) To investigate the intramolecular cyclization of their cis-trans and trans-trans
precursors and understand the reason for failure of the trans-trans mulberry
Diels-Alder to cylized in nature.
Figure 1.26. Structure of (+)-morusalbanol A (42) and (+)-sorocein B (62).
HO
OH
O
O
OH
OH
OHHO
(+)-morusalbanol A (42)
OMeO
O O
OH
OH
OOH
HO
OH
O
(+)-sorocein B (62)
HH
H

27
In this thesis, we described the work carried out to achieve the objectives that we set
out for the research. In the first chapter describes the introduction and literature review.
Chapter 2 introduces the other syntheses related to preparation of mulberry adduct. Chapter
3 discussed the approaches towards morusabanol A (42) synthesis while Chapter 4
describes the approaches towards sorocein B (62) synthesis. Chapter 5 is the conclusion of
the work as well as some suggestions for future work and the experimental work and data is
described in Chapter 6.

28
CHAPTER 2
RELATED SYNTHESIS
This chapter presents the semi-synthesis of mulberry Diels-Alder adducts such as
kuwanon G (43), H (13), mulberrofuran C (24), chalcomoracin (7). A brief review of total
syntheses of mulberry Diels-Alder adducts was also described.
2.1 Semisynthesis of mulberry Diels-Alder adducts
Several studies have been reported on the semi-syntheses or partial synthesis of
Mulberry Diels-Alder adduct. Kuwanon G (43) and kuwanon H (13) were the first two
compounds isolated from Morus plant. Kuwanon G (43) and kuwanon H (13) are regarded
as dehydroprenyl flavonoid type Diels-Alder adducts (Nomura et al. 1981). Treatment of
kuwanon G (43) using dimethylsulphate and potassium carbonate in reluxing acetone gave
ether 63 (Scheme 2.1). Pyrolysis of a solution of 63 in toluene in a pressure tube at 280 oC
gave the trans-chalcone tetramethyl ether 65 and the dehydrokuwanon C tetramethyl ether
67. Dehydrokuwanon C tetramethyl ether 67 is a flavonoid which contains a conjugated
diene and a prenyl group. The presence of a diene was supported by signals in the 1H NMR
spectrum at δ 1.91 (br s, Me-5´´), δ 4.88, 4.96 (br s, CH2-4´´), δ 6.80 (d, J = 7.0 Hz, CH2-1´´)
and δ 7.26 (d, J = 16.0 Hz, CH-2´´). Meanwhile the presence of a prenyl group was
supported by signals at δ 1.44 and 1.61 (br s, Me-4´ and 5´), δ 3.09 (br d, J = 7.0 Hz, CH2-
4´) and δ 5.24 (br t, J = 7.0 Hz, CH-2´). Diels-Alder cycloaddition reaction between 65 and
67 at 160 oC in a sealed tube gaved a racemate mixture of cycloaddition products, 63 and
68 in 60% yield. However, detail 1H NMR data was has not been reported for these
products.

29
Similarly, pyrolysis of kuwanon H methyl ethers (62) at 280 oC gave tetramethyl
morachalcone A (66) and dehydrokuwanon C tetramethyl ether (67) (Nomura et al., 1981).
Diels-Alder cycloaddition reaction between 66 and 67 at 160 oC in a sealed tube gave a
racemate mixture of cycloadducts 64 and 69 in 50% yield (Nomura et al. 1981).
O
O
Me
Me
OMeMeO
Me
O
OMeMeO
OMe
R
MeO
63: R = H 64: R = prenyl
toluene
280 oC 65: R = H, 54%66: R = prenyl
O
OMe
OMe
MeO
OMe
R
+
O
O
Me
Me
OMeMeO
Me
1"
3"
8
3 1'
2'
dehydrokuwanon C tetramethyl ether (67)37% from 63
toluene
160 oC
60% (63 and 68)50% (64 and 69)1:1 O
O
Me
Me
OMeMeO
Me
O
OMeMeO
OMe
R
MeO
63: R = H 64: R =prenyl
O
O
Me
Me
OMeMeO
Me
O
OMeMeO
OMe
R
MeO
+
68: R = H 69: R = prenyl
Scheme 2.1. Semisynthesis of kuwanons G (45) and H (13) (Nomura et al. 1981)
The pyrolysis of chalcomoracin (46) has been reported by Takasugi et. al. as shown
in Scheme 2.2. Methylation of chalcomoracin (6) using dimethylsulphate gave the
chalcomoracin methyl ether (69). Subsequently pyrolysis of 70 provided dehydromoracin C
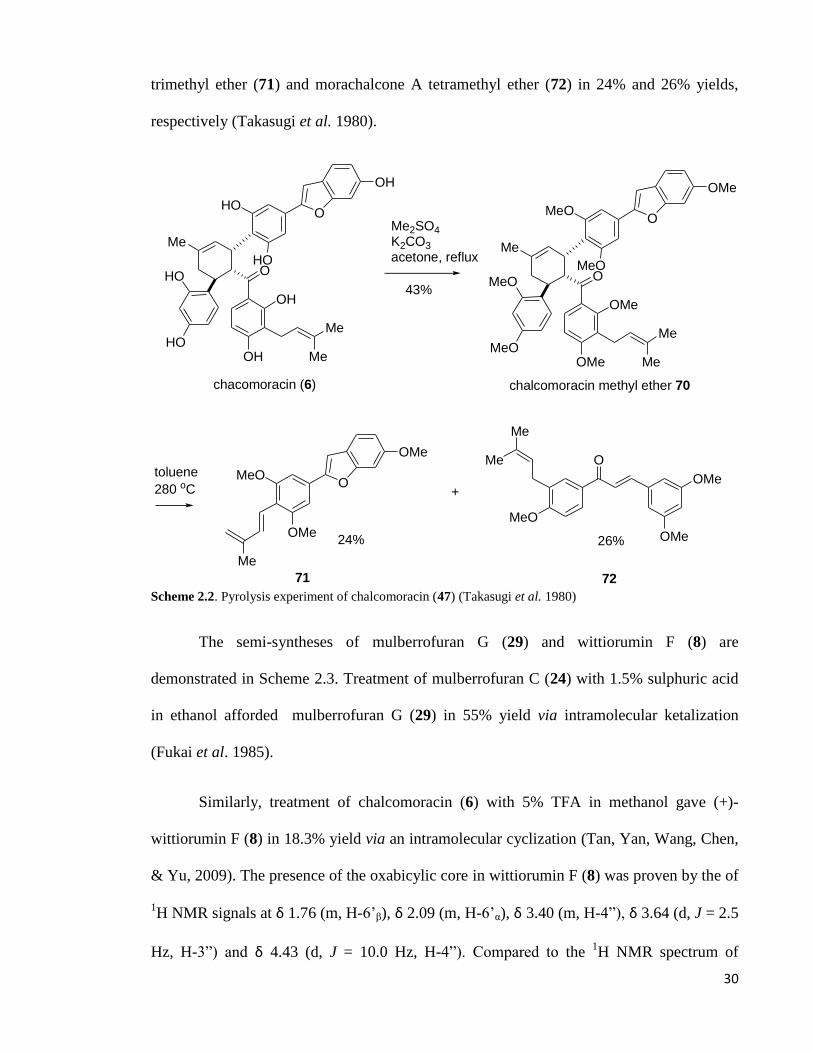
30
trimethyl ether (71) and morachalcone A tetramethyl ether (72) in 24% and 26% yields,
respectively (Takasugi et al. 1980).
Me
HO
HO
O
OH
OH
Me
Me
HO
HO
O
OH
Me2SO4
K2CO3
acetone, reflux
43%
Me
MeO
MeO
O
OMe
OMe
Me
Me
MeO
MeO
O
OMe
toluene
280 oCMeO
Me
OMe
O
OMe
+
O
OMe
OMe
MeO
Me
Me
24% 26%
chacomoracin (6) chalcomoracin methyl ether 70
71 72
Scheme 2.2. Pyrolysis experiment of chalcomoracin (47) (Takasugi et al. 1980)
The semi-syntheses of mulberrofuran G (29) and wittiorumin F (8) are
demonstrated in Scheme 2.3. Treatment of mulberrofuran C (24) with 1.5% sulphuric acid
in ethanol afforded mulberrofuran G (29) in 55% yield via intramolecular ketalization
(Fukai et al. 1985).
Similarly, treatment of chalcomoracin (6) with 5% TFA in methanol gave (+)-
wittiorumin F (8) in 18.3% yield via an intramolecular cyclization (Tan, Yan, Wang, Chen,
& Yu, 2009). The presence of the oxabicylic core in wittiorumin F (8) was proven by the of
1H NMR signals at δ 1.76 (m, H-6’β), δ 2.09 (m, H-6’α), δ 3.40 (m, H-4”), δ 3.64 (d, J = 2.5
Hz, H-3”) and δ 4.43 (d, J = 10.0 Hz, H-4”). Compared to the 1H NMR spectrum of

31
chalcomoracin (6), there are some significant changes for the chemical shifts of the
methylcyclohexene ring. The olefinics proton which signal at δ 5.30 -5.80 ppm as a broad
signlet was disappeared while the chemical shift of the methyl proton was shifted upfield
from δ 1.70 ppm to δ 1.36 ppm as a singlet (Tan et al. 2009).
Me
O
OHHO
OH
OH OH
OH
O
HO
mulberrofuran C (24) mulberrofuran G (29)
1.5% H2SO4
EtOH, 50 oC
24 h
55%
Me
OH
OHO
HO
O O
OH
OH
H
H H
Me
O
OHHO
Me Me
OH
OH OH
OH
O
HO5% TFAMeOH, 50 oC
24 h
HO OH
O OH
OH
Me
Me
Me
O
OHO
HO
18.3%
chalcomoracin (6) wittiorumin F (8)
1"
4"6"5'
Scheme 2.3. Semisynthesis of mulberrofuran C (24) (Fukai et al. 1985) and chalcomoracin (6) (Tan et al. 2009)
2.2 Total syntheses of mulberry Diels-Alder adducts
2.2.1 Thermal Conditions For Diels-Alder Reaction
Thermally promoted Diels-Alder cycloaddition reaction is the most commonly used
strategy for construction of the cyclohexene skeleton. Since Nomura’s pioneering work on
the synthesis of (±)-kuwanon G octamenthyl ether via thermal Diels-Alder reaction was
reported, a number of methods on the total syntheses of mulberry Diels-Alder adducts have
been reported.

32
In 2011, total synthesis of kuwanon V (73) and dorsterone pentamethyl ethers (74)
were reported by Chee and co-workers as outlined in Scheme 2.4 (Chee et al. 2011).
Kuwanon V pentamethyl ether (73) and dorsterone pentamethyl ether (74) were sythesized
via a [4+2] cycloaddition reaction between a chalcone-type diene 75 and a dienophile 76.
The chalcone precursors were prepared from commercially available acetophenone and
aldehyde via Claisen-Schmidt condesation (Scheme 2.4).
MeO
OOMe
Me
OMe
MeO
O
OMe
OH
Me
Me
MeO
OOMe
Me
OMe
MeO
O
OMe
OH
Me
Me
+
kuwanon V pentamethyl ether (73) dorsterone pentamethyl ether (74)
MeO
OOMe
Me
OMe
MeO
O
OMe
OH
Me
Me
75
76
Scheme 2.4. Retrosynthesis of kuwanon V pentamethyl ether (73) and dorsterone pentamethyl ether (74)
via a Diels-Alder reaction (Chee et al. 2011)
The synthesis of diene 75 was accomplished in five steps (Scheme 2.5). First,
acetophenone 77 was iodinated with ICl/CH2Cl2 to give the C-3 iodinated acetophenone 78
in 40% yield. Methylation of aryl iodide 78 followed by Claisen-Schmidt condensation
with 4-methoxybenzaldehyde led to the formation of chalcone 81 in 85% yield. Subjecting
chalcone 81 to Heck coupling [Pd(OAc)2, (o-toly)3-P, Et3N, DMF] (Arkoudis et al., 2009)

33
with 2-methylbut-3-en-2-ol (82) followed by dehydration with acetyl chloride/pyridine
gave the diene 75 (Harrington, Hegedus, & McDaniel, 1987).
O Me
OH
OH
O Me
OH
OH
CH2Cl2, r.t40% I
OMe
O H
KOH, EtOH
acetone, rt
Me2SO4, K2CO3
O Me
OMe
OMe
I
rt, 85%
MeO
I
OMe O
OMe MeO
OMe O
OMe
1) (82)
Pd(OAc)2,(o-tolyl)3P,
Et3N, DMF, 90 oC, 18 h
2) AcCl, pyridine
C6H6, 60 oC
95 %
Me
ICl
77 78 79
81 75
Me
Me
OH
80
Scheme 2.5. Synthesis of diene 75
Dienophile 76 was synthesized from chalcone 83 as shown in Scheme 2.6. Selective
demethylation of the 2-methoxychalcone 83 gave chalcone 84 which was subjected to
prenylation leading to the formation of prenyl ether 85. Then, the prenyl ether 85 then was
treated with Montmorillonite K10 to give dienophile 76 in 45% yield via [1,3]-sigmatropic
rearrangement (Sugamoto et al. 2008).

34
MeO
OMe O
OMe MeO
OH O
OMeBCl3, CH2Cl20 oC, 24 h
70%
MeO
OH O
OMe
Me
Me
Montmorillonite K10,
CH2Cl2, 0 oC, 2 hMeO
O O
OMe
prenyl bromide, K2CO3,acetone, reflux, 8 h
Me
Me
85%
45%
83 84
85 76
Scheme 2.6. Synthesis of dienophile 75
Diels-Alder reaction between diene 75 and dienophile 76 in toluene in a sealed tube
at 160oC afforded a mixture of 73 and 74 in 3:2 ratio in 55% yield (Scheme 2.7). The
coupling constant J3’’,4’’ is 6.8 Hz in 73 (cis) and 10 Hz in the 74 (trans) (Scheme 2.8).
Meanwhile, the coupling constant J4’’,5’’ is 10 Hz (trans) in both 73 and 74. Dorsterone
pentamethyl ether 74 shows evidence of atroisomerism as majority of the signals in the 1H-
NMR spectrum were doubled at room temperature. However, the NMR signals were
resolved by measurement in DMSO-d6 at 80 oC (Chee et al. 2011).

35
MeO
OOMe
Me
OMe
MeO
O
OMe
OH
Me
Me
+
kuwanon V pentamethyl ether (73)
dorsterone pentamethyl ether (74)
75
76
toluene
160 oC, 18 h
55%, 73/74 (3:2)
Me
O
HO OMe
MeMe
OMe
O
MeO
OMe
OMe
+
Me
O
HO OMe
MeMe
OMe
O
MeO
OMe
OMe
Scheme 2.7. Synthesis of kuwanon V (73) and dorsterone (74) methyl ether (Chee et al. 2011)
MeH3"
H4"
O
"H5
(6.8Hz)
(9.6Hz)
MeH3"
H4"
O
"H5
(10Hz)
(10Hz)
R
SR
S
SR
NOESY
Me
O
HO OMe
MeMe
OMe
O
MeO
OMe
OMe
dorsterone pentamethyl ether (74)
Me
O
HO OMe
MeMe
OMe
O
MeO
OMe
OMe
kuwanon V pentamethyl ether (73)
Scheme 2.8 The coupling constants of the cyclohexene ring of 72 and 73 (Chee et al. 2011)

36
In 2010, Rizzacasa and co-workers reported the synthesis of methyl ether
derivatives of mulberrofuran C (86) and chalcomoracin (70) via thermal Diels-alder
reaction. Their retrosynthetic analysis involved a biogenesis-inspired intermolecular [4+2]-
cycloaddition reaction between a dehydroprenylbenzofuran 87 and chalcones dienophile 88
and 89, respectively as shown in Scheme 2.9 (Gunawan & Rizzacasa, 2010).
Me
O
OMe
OMe
OMe
R
MeO
OMe
OMeO
MeO
methyl ether mulberrofuran C (86) : R = H methyl ether chalcomoracin (70) : R = prenyl
Diels-Alder
OMe
OMeO
MeO
Me
OMe
OMe
OMeO
I + BO
O
HO
OH
OH
COOH
OHHO
Me
OH
R
MeO
O
MeO OMe
OH O
Me
HO
O H
OH
OH
+
+
HO
R1
R2O
O
R2O
OR2
Ar
Me
88 R = H89 R = prenyl
87
90 91
77 92
92 93
Scheme 2.9. Retrosynthetic analysis of methyl ether mulberrofuran C (85) and methyl ether chalcomoracin
(69) (Gunawan & Rizzacasa, 2010)

37
Synthesis of the dehydroprenylbenzofuran diene 87 is outlined in Scheme 2.10. A
selective (Fürstner, Heilmann, & Davies, 2007) Sonogashira coupling (Sonogashira, Tohda,
& Hagihara, 1975) between the alkyne 94 and iodide 95 using Cs2CO3 (Rathwell, et al.
2009) afforded the alkyne product 96 in 65% yield. Methanolysis of the acetate 96 provided
phenol 97 which was then cyclized by using TBAF (Hiroya et al. 2000) to produce the
arylbenzofuran halide 90 in good yield. Finally, Suzuki-Miyaura (Miyaura & Suzuki, 1995)
(Coleman, Lu, & Modolo, 2007) coupling was performed on iodide 90 with
pinacolboronate 91 to give diene 87 in 82% yield.
MeO OMe
I
I
OAcMeO
65%
OMe
I
OMe
ORMeO
K2CO3
MeOH
96 R = Ac
97 R = H; 78%
TBAF, THFreflux
82%
MeO OI
OMe
OMe
MeO O
OMe
OMe
Me
BO
O
Me
Pd2(dba)3, AsPh3
K3PO4, DMF, 50 oC
82%
95
94
Pd(PPh3)4, CuICs2CO3, DMF, r.t
90 87
91
Scheme 2.10. Synthesis of diene 87 (Gunawan & Rizzacasa, 2010)
As shown in Scheme 2.11, the synthesis of required dienophile 89 began with an
aldol condensation reaction between 98 and 99 (Ahmed, Wagner, & Razaq, 1978) to afford
the chalcone 88 in 90% yield. Chalcone 88 was utilized for the synthesis of chalcomoracin

38
(6) through O-prenylation, followed by [1,3]-rearrangement using Florisil®
to give 89
(Talamás et al. 1997).
O
OH
OMe
+
O H
OMe
OMe
OH
MeO
O OMe
OMe
KOH, EtOH, r.t.
90%
O
MeO
O OMe
OMe
prenylchalcone,K2CO3, acetone,
84%
Me
Me
OH
MeO
O OMe
OMe
florisil, toulene,
100 oC, [1,3]
Me
Me
98 99
88
100 89
65MeIK2CO3
Scheme 2.11. Synthesis of dienophile 89 (Gunawan & Rizzacasa, 2010)
The Diels-Alder reaction between dehydroprenylbenzofuran 87 and chalcone 88 in
toluene (Scheme 2.12) in a sealed tube at 180 oC gave the endo-adduct 101 and exo-adduct
102, respectively in a 1:1 ratio after separation by preparative HPLC. The endo-adduct 101
corresponded to the hexamethyl ether derivative of mulberrofuran C (24) while the exo-
adduct 102 is the hexamethyl ether derivative of mulberrofuran J (25). Methylation of 101
gave mulberrofuran C heptamethyl ether (86) (Gunawan & Rizzacasa, 2010). The isomers
101 and 102 could be distinguished by 1H NMR spectroscopy where the coupling constant
between H3´´ and H4´´ is 5Hz (cis) in the endo-isomer (101) and 10 Hz in the exo-isomer
(102) (Gunawan & Rizzacasa, 2010). Several attempts toward the deprotection of either
mulberrofuran C heptamethyl ether (86) or mulberrofuran C hexamethyl ether (101) to give
mulberrofuran C (24) only led to incomplete demethylation or decomposition of the
compounds (Gunawan & Rizzacasa, 2010).

39
Me
O OMe
OMe
OMeRO
OMeO
MeO
OMe
+
87
toluene,
180 oC
40%, 1:1
MeIK2CO3
R = H (101)mulberrofuran C hexamethylether
86 R = Me; 98%
endo
exo
+
88
102
O
MeO
OH OMe
OMe
O
Me
OMe
OMeMeO
Me
O OMe
OMe
OMeRO
OMeO
MeO
OMe
Scheme 2.12. Synthesis of mulberrofuran C hexamethyl ether (101), mulberrofuran C heptamethyl ether
(86), mulberrofuran J hexamethyl ether (102) (Gunawan & Rizzacasa, 2010)
The synthesis of methyl ether derivatives of chalcomroacin (6) and mongolicin F (7)
is detailed in Scheme 2.13. Cycloaddition reaction between chalcone 89 and diene 87
proceeded at 180 oC to give endo-adduct 103 and exo-adduct 104, respectively in a 1:1 ratio
after separation by preparative HPLC. The endo-adduct 103 corresponded to hexamethyl
ether derivative of chalcomoracin (6) while the exo-adduct 104 is the hexamethyl ether
derivative of mongolicin F (7).

40
Me
O OMe
OMe
OMeHO
OMeO
MeO
OMe
+
toluene, 180 oC
55%, 1:1
MeIK2CO3
103, R = Hchalcomoracin hexamethyl ether
70, R = Me; 56%
exo
MeMe
mongolicin F hexamethyl ether (104)
O
MeO
OH
Me
Me OMe
OMe
+
OMeOMe
OMe
OMe
Me
O OMe
OMe
OMeRO
OMeO
MeO
OMeendo
MeMe
87
89
Scheme 2.13. Synthesis of chalcomoracin hexamethyl ether (103), chalcomoracin heptamethyl ether (70),
mongolicin F hexamethyl ether (104) (Gunawan & Rizzacasa, 2010)
Rizzacasa and co-workers also investigated on the effect of different protecting
groups on the Diels-Alder reaction (Boonsri et al. 2012). They found that fully methylated
chalcone 65 failed to undergo cycloaddition reaction. They reasoned that the presence of a
H-bonded ortho phenol in the chalcone dienophile is essential to the Diels-Alder reaction.
Their density functional theory calculation suggested that their LUMO-lowering effect of
the OH...O=C hydrogen bond and better co-planarity between the diene and its aryl
substituent in the transition state would lower the energy barrier for the Diels-Alder

41
reaction of the 2-hydroxychalcone compared to that of 2’-methoxychalcone (Boonsri et al.
2012).
Dienophile bearing hydroxy group at the ortho position appeared to be very reactive
while in the absent of an ortho hydroxyl group in dienophile, the diene may undergo
intermolecular dimerisation (Boonsri et al. 2012). They furthered demonstrated that Diels-
Alder reaction of diene 106 and dienophile 89 have kuwnaon J heptamethyl ether 105. The
presence of an ortho hydroxyl group in dienophile 89 was essential for this reaction
(Boonsri et al. 2012).
Me
OOMe
MeOO
OMe
OMe
HO OMe
Me Me
OMe
OMe
[4+2]
Me
Me
OO
H
MeO
OMe
OMe
Me
OOMe
MeO
OMe
OMe
+
kuwanon J heptamethyl ether (105)
89
106
Scheme 2.14. Retrosynthetic scheme of kuwanon J heptamethyl ether (105) (Boonsri et al. 2012)
2.2.2 Catalytic Diels-Alder reaction
In 2012, Porco and co-worker reported the first synthesis of (±)-sorocenol B (37)
(Hano, Yamanaka, Nomura, & Momose, 1995) by employing silver nanoparticle (AgNp)-
catalyzed Diels-Alder reaction (H. Cong & Porco, 2012). The AgNP was prepared from a
3:1 ratio of AgBF4/Bu4NBH4 in CH2Cl2 and then coated on silica gel. The solid product

42
was filtered and then calcinated at 220 oC to give AgNP (Cong et al. 2010). Retrosynthetic
analysis for sorocenol B (37) is shown in Scheme 2.15.
The synthesis of the acetylated chalcone 110 started with Claisen-Schmidt
condensation between ketone 112 and aldehyde 114 with NaH in THF (Nishida &
Kawabata, 2006) to afford chalcone 115 in 96 % yield. Hydrolysis of the MOM protecting
group with 3 M aqueous HCl in refluxing methanol, followed by acetylation provided
chalcone 110 in 79 % yield (two steps) (Scheme 2.16) (Cong et al. 2008).
O
OH OOHHO
MeO
OH
O
OH OOMOMMOMO
MeO
OH
O
OH OOMOMMOMO
Me
OH
HO
O
OH O OAc
OAc+
OMOM
OMOM
Me
HO OH
O
OH O
Me
sorocenol B (37) 107
110111
112 113
3''
Scheme 2.15. Retrosynthetic analysis for sorocenol B (37)
108: 3”-Hβ, endo
109: 3”-Hα, exo
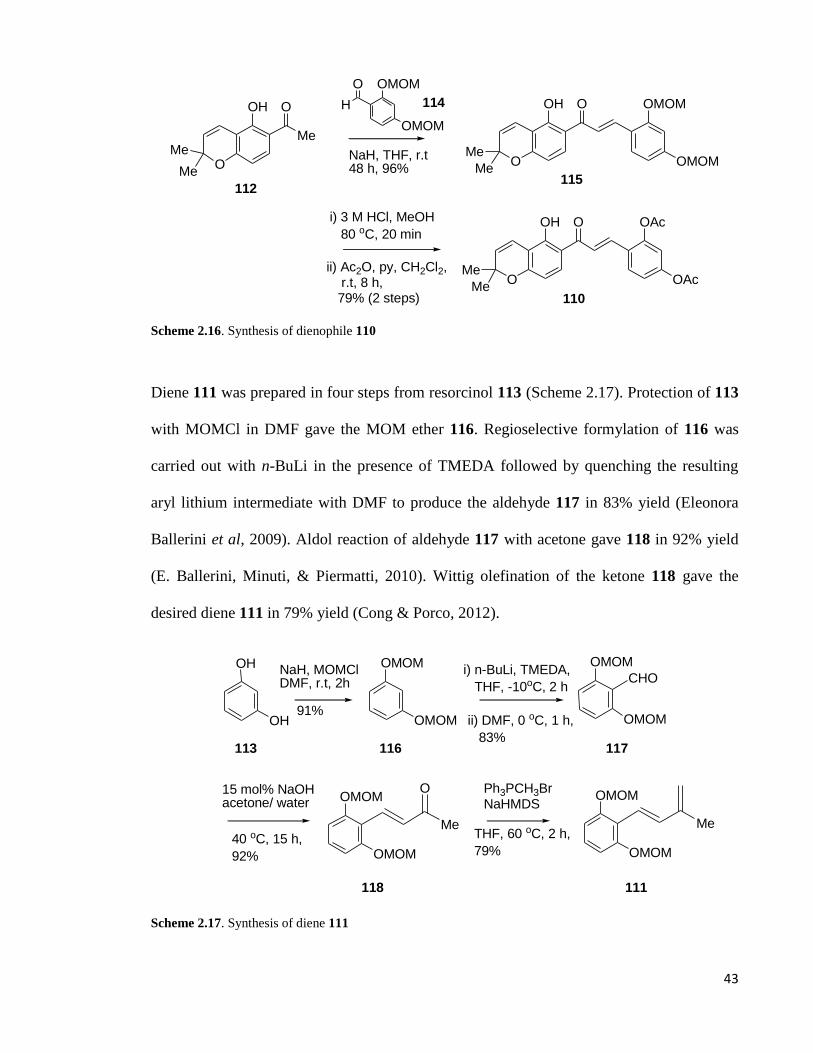
43
OMe
OH O
MeMe
OMe
Me
OH O OMOM
OMOM
OMOM
OMOM
O
H
NaH, THF, r.t48 h, 96%
OMe
Me
OH O OAc
OAc
i) 3 M HCl, MeOH
80 oC, 20 min
ii) Ac2O, py, CH2Cl2, r.t, 8 h, 79% (2 steps)
112115
110
114
Scheme 2.16. Synthesis of dienophile 110
Diene 111 was prepared in four steps from resorcinol 113 (Scheme 2.17). Protection of 113
with MOMCl in DMF gave the MOM ether 116. Regioselective formylation of 116 was
carried out with n-BuLi in the presence of TMEDA followed by quenching the resulting
aryl lithium intermediate with DMF to produce the aldehyde 117 in 83% yield (Eleonora
Ballerini et al, 2009). Aldol reaction of aldehyde 117 with acetone gave 118 in 92% yield
(E. Ballerini, Minuti, & Piermatti, 2010). Wittig olefination of the ketone 118 gave the
desired diene 111 in 79% yield (Cong & Porco, 2012).
NaH, MOMClDMF, r.t, 2h
91%
OH
OH
OMOM
OMOM
OMOM
OMOM
CHO
OMOM
OMOM
Me
OOMOM
OMOM
Me
Ph3PCH3BrNaHMDS
THF, 60 oC, 2 h,
79%
i) n-BuLi, TMEDA,
THF, -10oC, 2 h
ii) DMF, 0 oC, 1 h,
83%
15 mol% NaOHacetone/ water
40 oC, 15 h,
92%
113 116 117
118 111
Scheme 2.17. Synthesis of diene 111

44
The silver nanoparticle-catalyzed Diels-Alder cycloaddition of dienophile 110 and
diene 111 is shown in Shceme 2.18. The reaction using 0.1 mol% catalyst loading of silica-
supported silver nanoparticles (AgNp’s) reacted cleanly in air (50 oC) to afford 90%
combined yield of separable endo/exo diastereomers in a 2:1 ratio (Scheme 2.18).
Meanwhile, the same reaction conducted in thermal condition, without AgNp did not
proceed to completion. As a result, only 53 % combined yield of the endo/exo
diastereomers, 119 and 120 in 2:1 ratio were obtained.
Scheme 2.18. AgNp’s catalyzed Diels-Alder cycloaddition of 110 and 111
Next, deacylation of 119 gave 118 (Scheme 2.19). Oxidative cyclization of 118 by
using Pd(OAc)2, pyridine gave the desired bicylcle [3.3.1] product 107 and its epimer 121
in 50% combined yield. (Trend et al. 2003; Trend, Ramtohul, & Stoltz, 2005) (Scheme
2.19). The relative stereochemistry of 107 and 121 were determined by key NOE signals
(Scheme 2.20) (Cong & Porco, 2012).
119: 3”-Hβ, endo
120: 3”-Hα, exo
119:120 = 2:1

45
O
OH OOMOMMOMO
MeO
OH
MeMe
O
OH OOMOMMOMO
MeO
OH
MeMe6
2622
7
72121
26
6 1+
107 121
50% (two steps)107/121 (2:1 ratio)
Me
OMOMMOMO
AcO OAc
OOH
OMe
Me
3"
Me
OMOMMOMO
HO OH
OOH
OMe
Me
3"
Na2CO3, MeOH/H2O
rt, 12 h
119 108
20 % Pd(OAc)2
40% pyridine
1 atm O2
toluene, 80 oC, 24 h
Scheme 2.19. Pd(II)-catalyzed oxidation cyclization 107 and 121

46
O
OH OOMOMMOMO
MeO
OH
Me
MeO
OH OOMOMMOMO
MeO
OH
Me
Me
4
61
2622
7 721 21
26
61
O
OH
OMe
O
HO MeMe
HH
HH
Ar'
H
HH
3%
2%3%
1%
Me
O
HO
H
HH
Ar'
O
O
Me
Me
HO
HH
2%
10%
3%
NOESY
107 121
Scheme 2.20. Key NOE’s leading to relative stereochemistry assignments 107 and 121 (Cong & Porco, 2012)
However, when the same Stoltz’s conditions for oxidative Wacker cyclization were
applied to 109, it failed to give any cyclize product (Scheme 2.21), possibly due to
stereoelectronic restrictions in the process of oxidative cyclization.
Me
OMOMMOMO
AcO OAc
OOH
OMe
Me
3"
Me
OMOMMOMO
HO OH
OOH
OMe
Me
3"
Na2CO3, MeOH/H2O
rt, 12 h
120 109
20 % Pd(OAc)2
40 % pyridine 1 atm O2
toluene, 80 oC,
24 h
X
Scheme 2.21. Pd(II)-catalyzed oxidative cyclization of 109

47
Finally, treatment of 107 with 3M hydrochloric acid in methanol gave sorocenol B
(106) in 74% without epimerization at C-4 position (Scheme 2.22).
O
OH OOMOMMOMO
MeO
OH
MeMe
3 M HCl, MeOH
80 oC, 10 min
74%
O
OH OOHHO
MeO
OH
MeMe
107 106
Scheme 2.22. Synthesis of sorocenol B (106) (H. Cong & Porco, 2012)
Porco and co-workers also developed another biomimetic and dehydrogenative
Diels-Alder (DHDA) cycloaddition for a total synthesis of the MDA brosimone A (121)
(Messana, Ferrari, & De Araujo, 1988) and brosimone B (122) (Qi et al. 2013). Brosimone
A (121) and B (122) were both isolated from the Brazilian plant, Brosimopsis oblongifolia.
These dimeric structures were derived from protected prenyl chalcones 124 (Scheme 2. 23).
Diene 123 was prepared in situ by dehydrogenation of the prenyl chalcones 124.

48
HO
OH
Me
HO OH
O
Me
OH
HO
O
HO
OH
Me
O OH
OH
Me MeOH
HO
OH
HO
O OH
OH
brosimone A (121) brosimone B (122)
OOH
PgO
Me
OPg
OPg
123
OOH
PgO
Me
OPg
OPg
124Me
Pg = Protecting group
Scheme 2.23. Biomimetic synthetic design for Brosimone A (121) and B (122)(Qi et al. 2013)
Model studies were carried out on a prenyl chalcone 125 with number of catalysts
and oxidants. The results are summarized in Table 2.1. After numerous studies, it was
found that the desired dehydrogenative cycloadducts 126 and 127 can be obtained when
prenyl chalcone 125 was exposed to 10 mol% of platinum on activated carbon (Pt/C)
(Kogan & Herskowitz, 2002; Sebastián et al. 2008) and 0.2 mol% silica-supported AgNPs
in an ambient air atmosphere (Table 1, entry 1). Adding hydrogen scavengers such as
cyclopentene (Lee, Kwon, & Yi, 2012) (entry 3) and norbornene (Wang et al. 1996) (entry
6) in the transfer dehydrogenation reaction gave the cycloadducts 126 and 127 in 61% and
44% yields, respectively. Control experiments showed that reactions with cyclopentene did

49
not proceed without Pt/C (entry 4) and were significantly less efficient in the absence of
AgNPs (entry 5).
Table 2.1. Development of the initial methodology employing a model prenylchalcone (Qi et al. 2013)
Entry Condition details[c]
conv.[a]
endo-126/ exo-127[a]
1 air (1 atm), 10 mol% Pt/C, 88% (52%)[b]
52:48
0.2 mol% AgNP, 90 oC, 24 h
2 O2 (1 atm), 5 mol% Pt/C, 79% (60%)[b]
57:43
0.1 mol% AgNP, 90 oC, 36 h
3 9.0 equiv. cyclopentene, 10 mol% Pt/C, 66% (61%)[b]
42:58
0.1 mol% AgNP, 90 oC, 36 h
4 9.0 equiv. cyclopentene, 0.2 mol% AgNP <10% -
Ar, 90 oC, 36 h
5 9 equiv. cyclopentene, 10 mol% Pt/C, 38% (8%)[b]
72:28
Ar, 90 oC, 36 h
6 10 equiv. norbornene, 10 mol% Pt/C, 76% (44%)[b]
50:50
0.2 mol% AgNP, Ar, 90 oC, 36 h
7 10 mol% Pt/C, 0.2 mol% AgNP, 40%(18%)[b]
55:45
Ar, 90 oC, 36 h
[a] Based on 1H NMR integration.
[b] Yield of isolated product show in parentheses.
With the model reaction established, DHDA cycloaddition of 128 using the
optimized Pt/C–AgNP conditions with cyclopentene as H2 scavenger were carried out to
give the cycloadducts exo-130 and endo-129 (1.2:1) in 64% yield (Qi et al. 2013). Transfer
hydrogenolysis of exo-130 was accomplished by employing 1,4-cyclohexadiene as
hydrogen donor to produce brosimone B. Ammonium formate (HCO2NH4) (Iikubo et al.
2002) was used as additive to further accelerate the hydrogenolysis. Methylation of 122
126: 3”-Hβ, endo
127: 3”-Hα, exo

50
afforded derivative 131. The structure of 131 was determined by X-ray crystal-structure
analysis (Scheme 2.24) (Qi et al., 2013).
OH O
BnO
Me Me
Me
BnO
OH O
O OH
OBn
MeMe
OBn
OBn
OBn
OBn
BnO
BnO
Me
HO
OH O
O OH
OH
MeMe
OH
OH
HO
OH
Brosimone B (122)
i
iii
128
ii
An ORTEP drawing for 131, hydrogen atoms were omitted for clarity
3"
Me
MeO
OMe O
O OMe
OMe
MeMe
OMe
OMe
MeO
MeO
131
Scheme 2.24. Synthesis of Brosimone B (122) (Qi et al., 2013). Reagents and conditions: i)Pt/C (10 mol%),
AgNP (0.2 mol%), cyclopentene, 1,2-dichloroethane (DCE), 110 oC, 48 h, 64%, endo-129:exo-130= 1:1.2 ii)
Pd/C (40 mol%), HCO2NH4 (2 equiv), 1,4-cyclohexadiene, acetone, 40 oC, 24 h, 85% iii) K2CO3, Me2SO4,
acetone, 40 oC, 4 h, 15%.
Brosimine A (121), which biosynthetically originated from brosimone B (102), was
prepared via a second intramolecular dehydrogenative Diels-Alder reaction (Scheme 2.25).
Dehydrogenative Diels-Alder (DHDA) cycloaddition of exo-130 in the presence of DDQ-
AgNps in chlorobenzene at 130 oC gave the exo-exo cycloadduct 132 in 62% yield. Finally,
hydrogenolysis of 132 afforded brosimone A (121) in 91% yields. Methylation of 121 gave
133 as single crystal, thus allow stereochemical assignment for the synthetic brosimone A
(Qi et al. 2013).
129: 3”Hβ endo 130: 3”Hα exo

51
Me
BnO
OH O
O OH
OBn
MeMe
OBn
OBn
BnO
BnO
BnO
BnO
OHO
OBn
Me
BnO
OH
OMe
OBn
BnO
MeO
MeO
OMeO
OMe
Me
MeO
OMe
OMe
OMe
MeO
brosimone A (121) exo-exo 132
i ii
iii
exo-130
HO
HO
OHO
OH
Me
HO
OH
OMe
OH
HO
133
An ORTEP drawing for 133, hydrogen atoms were omitted for clarity
Scheme 2.25 Synthesis of brosimone A (121) (Qi et al., 2013). Reagents and conditions: i) DDQ (1.5
equiv), AgNp (0.3 mol%), PhCl, 130 oC, 72 h, 62%, d.r > 20: 1 ii) Pd/C (60 mol%), HCO2NH4 (2 equiv),
HCO2H, 1,4-cyclohexadiene, acetone, 40 oC, 24 h, 91% iii) K2CO3, Me2I, acetone, 40
oC, 12 h, 13%, Bn
= benzyl.
2.2.3 Chiral-Boron-Complex Promoted enantioselective synthesis
In 2014, Lei and Wulff and their co-workers reported the first enatioselective
total syntheses of (–)-kuwanon I (10), (+)-kuwanon J (9), (–)-brosimone A (121), and (–
)-brosimone B (122) (Han et al., 2014). This successful Diels-Alder cycloaddition
reaction was catalyzed by a chiral boron complex, prepared by coordination of an
axially chiral ligand, such as VANOL or VAPOL (Bao, Wulff, & Rheingold, 1993)
(Heller et al. 2006) to boron. The development of model dienophile 135 and diene 134
using this chiral ligand system was shown in the Scheme 2.26. The enantio- and
diastereoselectivities of the cycloaddition were strongly influenced by the ligand
structure (Han et al., 2014). The reaction in the presence of (R)-VAPOL as ligand gave
a mixture of cycloadducts 136 and 137 in 98% combined yield in a 1:1.4 ratio but

52
moderate ee value (48:72 % ee) (Scheme 2.28, entry 3). When ligand (S)-VANOL was
used, the endo-137 was generated in 80% yield and excellent ee value (97:11 % ee)
(Scheme 2.28, entry 4) (Han et al. 2014).
Me
+
O
OAc
OAc
AcO
OH
134
135
Me
AcO
OAc
O
OH
OAc
entry ligand time (h) yield (%) endo/exo ee (%) endo/exo
1 138 20 92 3.2:1 1:0
2 139 20 97 4.0:1 48:21
3 140 23 98 1.4:1 48:72
4 141 20 99 4.2:1 97:11
O
O
Me
Me OH
OH
(R, R)-TADDOL (138) (S)-BINOL (139) (R)-VAPOL (140) (S)-VANOL (141)
OH
OH Ph
Ph
OH
OH Ph
Ph OH
OH
Scheme 2.26. Chiral-boron-complex-promoted asymmetric Diels-Alder cycloaddition
(Han et al. 2014)
Scheme 2.27 shows the mechanism proposed by Lei and co-workers (Han, Jones,
& Lei, 2015) for the enantioselective Diels-Alder cycloaddition. The reaction was
proposed to proceed through the formation of a chiral boron complex 143, followed by
formation of a tetracoordinate boron complex 144 which then underwent a
enantioselective cycloaddition with diene 134. Lei and co-workers suggested that the
136: 3”Hα: exo
137: 3”Hβ: endo
ii) 135, 5 Å MS, r.t. 1.5 h
iii) 134, r.t.
i) Ligand, BH3.THF
AcOH, THF,
r.t., 25 min

53
enantioselective Diels-Alder cycloaddition may be induced by the following factors
(Han et al. 2015).
a) The coordination bond between boron and dienophile 135 which may lower
energy of the LUMO.
b) Mobility of dienophile 135 being reduced upon complexation.
c) The π-π stacking between chiral ligand and dienophile 135 shielding one face of
chalcones to attack by the diene 134.
Scheme 2.27. Proposed mechanism for the enatioselective Diels-Alder cycloaddition
(Han et al. 2014)
This catalyst was then applied to the synthesis of (–)-kuwanon I (10), (+)-
kuwanon J (9), (–)-brosimone A (121), and (–)-brosimone B (122). For the synthesis of
(–)-kuwanon I (10), (+)-kuwanon J (9), prenylation of 145 followed by sigmatropic
rearrangement gave ortho-prenylated chalcone 148 (37%), along with para-prenylated
chalcone 147 (27%). Chalcone 148 was later converted to chalcone 149 in 35% yield
over two steps. Regisoselctive Schenck ene reaction (Helesbeux et al. 2003) with
[Ru(bpy)3Cl2.6H2O] and MeOH of 149 to form secondary allylic alcohol 150 and
π-π stacking
interaction
136: 3”Hα: exo
137: 3”Hβ: endo

54
tertiary allylic alcohol 151 in 62% combined yield. Dehydration of alcohol 151 in the
presence of SOCl2/DBU proceeded smoothly to provide the desired diene 152 in 75%
yield (Scheme 2.28).
MOMO
MOMO
O OH
OMOM
Br
acetone, K2CO3, reflux, 24 h, 95%
MOMO
MOMO
O O
OMOM
montmorillonite K-10
CH2Cl2, 0 oC, 8 hMOMO
MOMO
O OH
OMOM
MOMO
MOMO
O OH
OMOM+
OAc
AcO
O OH
OAc
i) con. HCl, MeOH, r.t., 20 hii) 4 M NaOH, r.t., 2 hiii) Ac2O, pyridine, CH2Cl2, r.t., 35%
OAc
AcO
O OH
OAcOH
+
OAc
AcO
O OH
OAc
OH
OAc
AcO
O OH
OAcSOCl2, DBU, CH2Cl2
-78 oC to r.t., 12 h, 75%
i) Ru(bpy)3Cl2.6H2O, hv O2, MeOH, r.t., 26 h
ii) Ph3P, CH2Cl2, r.t. 16 h, 62%
145 146
147 27%148 37%
149150
151
152
150/151 = 8:1
Scheme 2.28. Synthesis of dienophile 149 and 152 (Han et al. 2014)
Diene 152 and dienophile 149 were then subjected to asymmetric Diels-Alder
reaction as summarized in Scheme 2.29. Based on the reported result, the chiral ligand
strongly influenced the enantioselectivity of the cycloaddition reaction. The optimal
condition for the formation of kuwanon J precursor, 154 (97% ee, 1.1:1 endo/exo) is
when diene 152 and dienophile 149 were subjected in the present of 2.5 equiv. of (R)-
VANOL, whereas for the formation of kuwanon I precursor exo-153 as major isomer
(84% ee, 1.2:1 exo/endo) when diene 152 and dienophile 149 were subjected in the

55
present of 2.5 equiv. of (S)-8,8’-dimethyl-VANOL. A final global deprotection of endo-
154 and exo-153, under mild basic conditions, efficiently furnished the desired natural
products (-)-kuwanon J (9), (+)-kuwanon I (10) respectively.
Scheme 2.29. Synthesis of (–)-kuwanon I and (+)-kuwanon J (Han et al. 2015)
Similar synthetic route was applied in the synthesis of (–)-brosimone A (121)
and (–)-brosimone B (122). For (–)-brosimone B (122), compound 147 was first
converted to 157 in 33% yield (two steps) (Scheme 2.30). Next, Schenck ene reduction
of 157 in the presence of TPP in MeOH gave 158 in 48% yield. Dehydration of 158
produced the diene 159 in 68% yield. Cycloaddition between para-prenylated
dienophile 157 and diene 159 using (S)-VANOL gave a mixture of 160 and 161 in 71%
3”Hα: kuwanon I (10)
3”Hβ: kuwanon J (9)
153: 3”Hα: exo
154: 3”Hβ: endo

56
yield in 1.2:1 ratio. Remarkably, excellent ee values for both compounds were obtained
(98% ee for endo-161, 93% ee for exo-160). Deprotection of the acetyl groups of exo-
160 gave (-)-brosimone B in 70% yield (Han et al. 2014).
146i
AcO
OH O
OAc
OAc
Me Me
AcO
OH O
OAc
OAc
Me Me
ii
OH
iiiAcO
OH O
OAc
OAc
Me
157 158 159
Me
O
OAc
OAc
OH
AcOO
AcO
OH
Me
OAc
AcOMe
157
+
159
iv v Me
O
OH
OH
OH
HOO
HO
OH
Me
OH
HOMe
Brosimone B (122)
Scheme 2.30. Synthesis of (–)-brosimone B. Reagents and conditions: i) a) 3 M HCl, MeOH, resorcinol,
80 oC, 23 min; b) 4 M NaOH, r.t., 2 h c) Ac2O, pyridine, CH2Cl2 ii) a) hv, O2, TPP, MeOH, r.t., 10 h
b)PPh3, CH2Cl2, r.t. 16 h, 48%; iii) SOCl2, DBU, THF, -78 oC to r.t., 16 h, 48% iv) (S)-VANOL,
BH3.THF, AcOH, 5 Å M. S., THF, r.t., 72 h, 71% (160:161 = 1.2:1), 93% ee for exo-140, 98% ee for
endo-141, recovered (S)-VANOL (90%); v) K2CO3, MeOH/H2O (10:1), r.t., 1 h, 70%
The same diene core 159 was used in the synthesis of brosimone A (121) in a
one-pot inter-/intramolecular Diels-Alder cycloaddition cascade strategy (Roush &
Sciotti, 1998) (Yuan et al. 2013). The (S)-VANOL-boron complex efficiently mediated
the cyloaddition reaction to give endo-endo-162, exo-endo-163 and exo-exo-164
(Scheme 2.30). Global deprotection of the exo-exo-164 gave (-)-brosimone A in 70%
yield (Han et al. 2014).
160: 3”Hα: exo
161: 3”Hβ: endo

57
159
OH
AcO
OMe
OAc
OAc
OAc
OHO
Me
AcO
OAc
OH
AcO
O
Me
OAc
OAc
OAc
OHO
Me
AcO
OAc
+
endo-endo-162 exo-endo-163
OH
AcO
O
Me
OAc
OAc
OAc
OHO
Me
AcO
OAc
+
28%, 98% ee 20%
K2CO3, MeOH-H2O
r.t, 1 h, 70%
OH
HO
O
Me
OH
OH
OH
OHO
Me
HO
OH
Brosimone A (121)exo-exo-16413%, 95% ee
Scheme 2.31. Synthesis of (–)-brosimone A
i) (S)-VANOL, BH3.THF, AcOH, THF, r.t., 25 min
ii) 158, THF, 5 Å MS, r.t., 96 h, 61%

58
CHAPTER 3
APPROACHES TOWARD THE SYNTHESIS OF MORUSALBANOL A
Morusalbanol A (42) is a mulberry Diels-Alder adduct isolated from the bark of
Morus alba which exhibits interesting neuroprotective activity (Chen et al. 2012). It is
characterized to contain an oxabicyclic [3.3.1] skeleton that derived from an
intramolecular cyclization of a cis-trans type mulberry Diels-Alder adduct.
Morusalbanol A shows evidence of atropisomerism due to the rotational hindrance of
the D/E-rings about the C5´´-C15´´and C4´´-C8´´-C9´´ bonds (Figure 3.1) (Chen et al.,
2012). Other examples of natural products in this class are cathayanon E (165) (Q. J.
Zhang, Ni, Wang, Chen, & Yu, 2009), wittiorumin F (8) (Tan et al. 2009), and CAS
441772-64-1 (166) (Figure 3.1) (Abegaz et al. 2002).
O
OH
O
O
OHHO
OH
OH
HO wittiorumin F (8)
3"4"
5"SR
S
S
morusalbanol A (42)
OH
OH
O
O
OH
HO
HO OH
MeO O
cathayanon E (165)
OH
OH
O
O
OH
O
HO OH
O
O
HO
OH
RS
OH
O OH
O
O
HO O
OH
CAS 441772-64-1 (166)
D
E
8"
9"
4"
5"
15"
Figure 3.1 Morusalbanol A (42) and related mulberry Diels-Alder adducts

59
3.1 Model study towards biomimetic Diels-Alder reaction Morusalbanol A
An important issue that needed to be addressed during the planning of the
synthesis of morusalbanol A and related mulberry Diels-Alder adducts is the formation
of a cis-trans (endo) Diels-Alder adduct precursor. Our approach hinged on the
hydrogen bond-assisted regioselective biomimetic Diels-Alder reaction between
chalcone type dienophile 169 and dehydroprenyl diene 170 (Scheme 3.1). This would
result in the formation of a cis-trans and a trans-trans type Diels-Alder adducts in one
step. The feasibility of such a [4+2] cycloaddition reaction has recently been
demonstrated by Porco and co-workers (H. Cong & Porco, 2012; Han et al. 2014),
Rizzacasa and co-workers (Boonsri et al. 2012; Gunawan & Rizzacasa, 2010) and Chee
and co-workers (Chee et al. 2011). It was envisaged that the ortho and para OH groups
on the aryl ring of diene 170 could be selectively protected due to their relative
positions related to the carbonyl group. Selective deprotection of the para OH group in
the cis-trans adducts 168 and subsequent intramolecular cyclization would then produce
morusalbanol A (Scheme 3.1)
Scheme 3.1. Retrosynthetic analysis for morusalbanol A and related mulberry Diels-Alder adducts
To test the feasibility of the biomimetic Diels-Alder reaction, we carried out a
model study on simple diene and dienophile. We examined the [4+2] cycloaddition
reactions with the model diene 172 and dienophile 175 (Table 3.1, entry 1). We

60
anticipated that cis-trans and trans-trans type Diels-Alder adducts 179 and 180 would
undergo intramolecular cyclization to produce the oxabicyclic [3.3.1] skeleton found in
the morusalbanol A. In contrast, we presumed that due to the para methyl ether
protection, the cis-trans and trans-trans type Diels-Alder adducts 181 and 182 would
not undergo intramolecular cyclization. This compound could also be used to further
understand the intramolecular cyclization reaction.
The preparation of diene 172 and 174 is outlined in Scheme 3.2. Regioselective
iodination of commercially available 2,4-dihydroxyacetophenone 76 with I2/KIO3 in
EtOH/H2O utilizing the method of Yu (H. Wang et al., 2014) gave iodobenzene 77 in
high yield. Selective protection of the para OH group in 77 with ethoxymethoxy (EOM)
group, followed by methylation of the remaining ortho OH group with dimethylsulfate
gave the iodide 171. Heck coupling of 171 and subsequent dehydration using
AcCl/pyridine (Harrington et al., 1987) provided the desired diene 172 in 81% yield in
two steps.
For diene 174, iodination of the commercially available 2,4-dihydroxyacetophenone
77 with I2/KIO3 in EtOH/H2O (H. Wang et al., 2014) gave iodobenzene 778 Selective
protection of the para OH group in 78 with dimethylsulfate gave 173. Heck coupling of
173 and subsequent dehydration with AcCl/pyridine (Harrington et al., 1987) provided
the desired diene 174 in 87% combined yield (two steps).

61
HO
OH
Me
O
HO
OH
Me
O
I
OMOE
OMe
Me
O
I
I2, KIO3
EtOH, H2O,
acetone,-10oC,
PdCl2, NaHCO3,
81% combined yield (two steps)
2) AcCl, pyridine, C6H6, 80oC
1) K2CO3
EOMO
OMe
Me
O
Me
2) Me2SO4, K2CO3,
acetone, rt, 12h, 85%77 78
171
172
Bu4NCl, DMF,100oC, 12h
15h, 72%
rt, 24h,92%
MeI, K2CO3,
acetone, rt, 8h, 85%
MeO
OH
Me
O
I
173
PdCl2, NaHCO3,
87% combined yield (two steps)
2) AcCl, pyridine, C6H6, 80oC
MeO
OH
Me
O
Me
174
Bu4NCl, DMF,100oC, 12h
Me
Me
OH1)
Me
Me
OH1)
(82)
(82)
O Cl
Scheme 3.2. Syntheses of model dienes 172 and 174
The thermal Diels-Alder reaction of diene 172 with chalcone-type dienophile
175 in toluene at 110°C for 24h afforded an inseparable mixture of cis-trans (endo) and
trans-trans (exo) adducts 179 and 180 in a 3:2 ratio and 55% yield (Table 3.1, entry 1).
The same reaction between diene 174 and dienophile 176 also gave the desired
cycloadducts in 49% combined yield as a 1:1 mixture of inseparable endo/exo
diastereomers (181 and 182), Table 3.1, entry 2. Following Rizzacasa and co-workers’
report of the substantial rate enhancement for the Diels-Alder reaction for an hydrogen-
bonded ortho OH substituent on a chalcone-type dienophile (Boonsri et al. 2012), we
proceeded to examine the thermal Diels-Alder reaction of diene 172 with dienophiles
177 and 178. Both 177 and 178 lacked an ortho hydroxyl substituent on carbonyl group
of deienophile. As expected, all attempts of thermal Diels-Alder reaction between diene
172 and dienophiles 177 or 178 failed to yield any of the desired products (Table 3.1,
entries 3-4). In each case, the dienophile was recovered while the diene decomposed.

62
This clearly indicated that the presence of an ortho OH substituent on the dienophile is
essential for the Diels-Alder reactivity.
Table 3.1. Development of initial methodology employing a model diene and dienophile
Entry Diene Dienophile Conditionsa Yield
b endo/exo
c
R1 R
2 R
3 R
4
1 172 Me EOM- 175 OH H Thermal 55% 3/2
2 174 H Me 176 OH EOMO- Thermal 49% 1/1
3 172 Me EOM- 177 H H Thermal - -
4 172 Me EOM- 178 OMe H Thermal - -
5 172 Me EOM- 175 OH H AgOTf 38% 0/1
6 172 Me EOM- 175 OH H AgBF4 20% 0/1
7 172 Me EOM- 175 OH H AlBr3 - -
8 172 Me EOM-- 175 OH H FeCl3 - -
9 172 Me EOM- 175 OH H Ga(OTf)3 - -
10 172 Me EOM- 175 OH H TiCl4 - -
11 172 Me EOM- 175 OH H BF3.Et2O - - a Thermal reaction conditions: reaction conducted in toluene (2mL/mmol) at 110°C for 24h in a pressure tube. For
catalytic conditions: reaction conducted in CH2Cl2 (4mL/mmol) at rt for 24h. Molar ratio of diene/dienophile/catalyst
= 1.2/1/0.3 bIsolated yield. cendo/exo ratios are based on 1H NMR integration.
Interestingly, the use of silver catalysts (AgOTf and AgBF4) for the Diels-Alder
reaction between diene 172 and dienophile 175 promoted the selective formation of the
trans-trans adduct, exo-180, albeit in low yield (Table 3.1, entries 5-6). The use of other
Lewis acids such as AlBr3, FeCl3, Ga(OTf)3, TiCl4, and BF3.Et2O did not give any of

63
the desired product (Table 3.1, entries 7-11). In these cases, both the diene and
dienophile decomposed.
Cyclization to form the desired oxabicylic ring was achieved by heating a
solution of endo-179 and exo-180 in 3M aqueous HCl in MeOH for 20 min.
Deprotection of the EOM group and subsequent intramolecular cyclization occurred to
give the desired oxabicyclic [3.3.1] compounds in 90% combined yield as a 3:2 mixture
of separable diastereomers (183 and 184, Scheme 3.3). Diastereomers 183 represents
the core skeleton of the natural product 441772-64-14 (185) (Figure 3.2).
Scheme 3.3. Acid catalyzed intramolecular cyclization of endo-adduct 179 and exo-adduct 180
OH
O OH
O
O
HO O
OH
CAS 441772-64-1
Figure 3.2. Chemical structure for CAS 441772-64-4(185).

64
The stereochemistry of each of the bicyclic diastereomers was confirmed by 1H-
NMR and NOESY experiments. The diastereomer 183, which was derived from the
endo-179 showed a large coupling constant (J = 11.4 Hz) from the splitting between
H4´´ and H5´´ (Figure 3.2). This clearly indicated the trans-diaxial arrangement of H4´´
and H5´´ in diastereomer 183. The small coupling constant (J = 2.8 Hz) between H3´´
and H4´´ implied a cis-orientation with H3´´ being in an equatorial position. The
NOESY correlation between the aromatic protons H14´´ and H4´´ on the cyclohexane
ring and between H3´´ and H4´´ also indicated a cis arrangement (Figure 3.3).
Figure 3.3. Key NOESY correlations leading to relative stereochemistry assignment of 183
The structure and stereochemistry of diastereomer 184 were confirmed by single
crystal X-ray crystallography (Table 3.2). The formation of oxabicyclo[3.3.1] 184 from
its trans-trans precursor exo-180 was rather surprising. Natural products containing
similar oxabicyclic core structures including morusalbanol A (44), wittiorumin F (8),
and cathayanon E (165) were all exclusively derived from their cis-trans Diels-Alder
precursors (Chen et al. 2012; Tan et al. 2009; Q. J. Zhang et al. 2009). Yu and co-
workers hypothesized that the trans-trans Diels-Alder adducts could not meet the
spatial requirement to form an oxabicyclic compound (Tan et al. 2009; Zhang et al.
2009). They demonstrated that the treatment of a cis-trans Diels-Alder adduct,
chalcomoracin (6) in 5% triflic acid/MeOH afforded a mixture of oxabicyclic and
ketalized products (wittiorumin F (8) and mulberrofuran F (9), respectively Figure 3.4).

65
However, when they subjected the recovered trans-trans Diels-Alder adduct,
mulberrofuran J (25) to similar reaction conditions, they did not obtain any product
(Tan et al. 2009).
Figure 3.4. Semisynthesis of wittiorumin F (8) and mulberrofuran F (9) from chalcomoracin C (6)
(Tan et al., 2009)
We examined the intramolecular cyclization of endo-181 and exo-182. When a
solution of endo-181 and exo-182 in 3M aqueous HCl in MeOH was heated for 20 min,
a new oxabicylic compound 186 was obtained in 92% yield (based on endo-181) along
with the recovery of 182. According the observation by Yu and co-workers in the
semisynthesis of wittiorumin F and mulberrofuran F (Figure 3.4), we expected that
endo-181 and exo-182 would undergo intramolecular cyclization to produce a ketalized
compound rather than an oxabicyclic compound due to the para methyl ether protecting
group. Although the 1H-NMR spectrum of compound 186 was observed to be quite
similar to that of 183, its 13
C-NMR spectrum however, did not appear to correspond to
either 183 or ketalized compound 187. Instead, the
13C-NMR spectrum showed signals
for an oxygenated sp3 quaternary carbon (c 76.1 ppm) and two carbonyls (c 199.9 and

66
206.0 ppm). The relative configuration of 186 was determined by HMBC correlations
and NOESY correlations. Single-crystal X-ray structure determination confirmed the
assignment as compound 186 (Scheme 3.4). Interestingly, subjecting the recovered
exo-182 (without the EOM protecting group) to similar reaction conditions did not yield
any product.
The formation of oxabicyclo [3.3.1] 186 from endo-181 is quite surprising.
Natural products containing similar cyclohexenyl core structures including kuwanon I
(11), mulberrofuran J (25) and dorsterone show restricted rotation about the C3”-C3’
bond (as shown in Scheme 3.4) due to the unsymmetrical nature of the aryl ring. Most
of the signals in the 1H-NMR spectrum of these natural products were doubled at room
temperature as a result of atropisomerism due to the high rotation barrier. However, in
the case of 181, the barrier for rotation about the C3”-C3’ bond was negligible, allowing
a 180° rotation followed by intramolecular cyclization to form the respective
oxabicyclic [3.3.1] compound 186.
Scheme 3.4. Formation of diastereomer 186.

67
Table 3.2 Crystal data and structure refinement for 184 and 186
184 186
Empirical formula C29H28O5 C30H29Cl3O6
Formula weight 456.51 591.88
Crystal system, Space group Monoclinic, C2/c Monoclinic, P21/c
a (Å) 39.193(4) 15.3140(9)
b (Å) 7.3592(8) 13.9413(8)
c (Å) 16.0729(15) 13.3869(10)
β () 95.317(7) 106.278(7)
Volume (Å3) 4615.9(8) 2743.5(3)
Z 8 4
Calculated density (Mg/m3) 1.314 1.433
Absorption coefficient 0.089 mm-1
0.378 mm-1
F(000) 1936 1232
Crystal size (mm3) 0.690 x 0.510 x 0.050 0.510 x 0.310 x 0.270
Crystal color and habit colorless plate colorless needle
θ range for data collection 2.087 to 29.615° 2.793 to 30.211°
Reflections collected 15853 37196
Independent reflections 6342 (Rint = 0.0419) 11114 (Rint = 0.1283)
Observed reflections [I > 2σ(I)] 3926 7561
Completeness to θ = 25.242° 99.6 % 99.9 %
Data / restraints / parameters 6342 / 0 / 313 11114 / 13 / 399
Goodness-of-fit on F2 1.039 1.021
Final R indices [I > 2σ(I)] R1 = 0.0530,
wR2 = 0.1099
R1 = 0.0650,
wR2 = 0.1737
R indices (all data) R1 = 0.0963,
wR2 = 0.1285
R1 = 0.0935,
wR2 = 0.1867
Largest diff. peak and hole (e Å-3
) 0.570 and -0.256 0.444 and -0.594
CCDC deposit number for 184 and 186 are 1026385 and 1043889 respectively.

68
3.2 DFT calculation for model study
To understand why the Diels–Alder reactivity is so highly dependent on the
presence of the ortho OH group of the chalcones 175 and 178, we carried out density
functional theory (DFT) calculations on the Diels–Alder reactions of 175 and 178 with
the model diene 172 as shown in Scheme 3.5.
Me
O OMe
Me
OMOE
172
OR O
175: R = H 177: R= Me
Me
O
RO
OMOE
OMe
O Me
Me
O
RO
OMOE
OMe
O Me
+
179 R= H 188 R= Me
180 R= H 189 R= Me
Scheme 3.5. Model Diels-Alder reaction between diene 172 with chalcones 175 and 177 for DFT
calculation
Transition state calculation was performed to gain insights on how the ortho OH
group on the dienophile 175 contributed to the formation of Diels–Alder adducts 179
and 180. The endo and exo transition structures and their activation energies and the
overall reaction energies are shown in Figure 3.4. The transition states were concerted
and synchronous and the cycloaddition reactions were observed to be exothermic.
Hydrogen bonding was observed between the ortho OH group of the dienophile 175
with the adjacent carbonyl oxygen (intramolecular) and the carbonyl oxygen of the
diene 172 (intermolecular). This hydrogen bonding is essential as it leads to the shorter
distance between the carbonyl oxygen of the diene 172 and the ortho OH group of the
dienophile 175 and thus promoting the cyclization.

69
Figure 3.5 showed the energy profile of the transition state structure of 179, 180,
188 and 189. The activation energy (∆G*) for the transition state of endo-179 and exo-
180 were 2.95 kcal/mol and 2.57 kcal/mol, respectively. The Gibbs free energy of
reaction (∆G) for endo-179 and exo-180 were 4.64 kcal/mol and 3.73 kcal/mol,
respectively. The low values of activation energy and the Gibbs free energy explain why
endo-179 and exo-180 can undergo intramolecular cyclization to form oxabicylic [3.3.1]
compound 183 and 184.
TS1 endo-179 TS2 endo-180
∆H* = -0.20 ∆H* = -0.12
∆G* = 2.95 ∆G* = 2.57
∆H = -1.34 ∆H = -1.07
∆G = 4.64 ∆G = 3.73
TS3 endo-188 TS4 endo-189
∆H* = 0.61 ∆H* = -0.27
∆G* = 2.49 ∆G* = 4.85
∆H = -1.11 ∆H = -2.60
∆G = 5.05 ∆G = 8.40
Figure 3.5. Energy profile of transition state structure of (a) 179 (b) 180 (c) 188 (d) 189 where black
dashed lines denoted the bond formation that led to cyclisation and blue dashed lines denoted the
intermolecular hydrogen bonding

70
The calculation for reaction pathway of 186 and 187 is shown in Figure 3.6. The
transition state calculations were carried out using linear synchronous transit (LST) and
quadratic synchronous transit (QST) methods to further understand the reaction
mechanism. The lower energy barrier between the transition state and reactant were
found in the formation of the 186 rather than 187.
In order to have a better insight into the how C3”-C3’ bond rotate endo-181 to
facilitate the formation of the endo-186, an energy scan of the optimized cycloadduct
geometry using local density approximation (LDA) or local functional PWC using
Dmol3 module along the coordinates of the dihedral angle was performed. The torsion
for rotation of 181 and 182 were plotted in Figure 3.6. The torsion angle was scanned
from left to right with the initial optimized structure of each form. Eighteen
conformations with a difference dihedral angle of 20° were obtained. The shortest
bridging distance was 3.61 Å and 2.72 Å for endo-181 and exo-182, respectively. As
highlighted in the potential energy scan graph in Figure 3.7, exo-182 has a barrier to
rotate of approximately 9 times higher than that of endo-181. This observation could
explain why no product was obtained from the acid treatment of exo-182.

71
Figure 3.6. Calculated reaction path for (a) 187 and (b) 186
a) b)

72
(a)
(b)
Figure 3.7. Potential energy scans on dihedral angle together the respective energy barrier and distance for (a)
endo-181 and (b) exo-182. The distances between the carbon and oxygen atom in red circle were calculated
in the plot
0
1
2
3
4
5
6
-1 0 1 2 3 4 5 6 7 8 9
74
.49
94
.49
11
4.4
9
13
4.4
9
15
4.4
9
17
4.4
9
-16
5.5
1
-14
5.5
1
-12
5.5
1
-10
5.5
1
-85
.51
-65
.51
-45
.51
-25
.51
-5.5
1
14
.49
34
.49
54
.49
74
.49
Energy
(kcal/mol)
Distance(Å)
0
1
2
3
4
5
6
0
2
4
6
8
10
-54
.76
-30
.31
-1.8
4
28
.69
51
.99
72
.52
90
.36
10
8.7
9
12
6.0
5
14
3.4
7
16
0.8
0
17
8.8
8
-16
3.2
0
-14
5.1
2
-12
7.6
3
-11
1.2
1
-95
.77
-77
.98
-57
.64
Torsion
Angle(o)

73
In conclusion, the model studies provided a useful approach toward the synthesis of
the oxabicyclic [3.3.1] core system found in morusalbanol A and related mulberry Diels-
Alder adducts. In particular, the required cis-trans (endo) Diels-Alder precursors of
morusalbanol A and 441772-64-1 were obtained via the thermal cycloaddition reaction
which was proven to be dependent on the presence of a hydrogen-bonded ortho-OH
subtituent on the chalcones dienophile. Acid-catalyzed intramolecular cyclization of a cis-
trans (endo) Diels-Alder adduct (endo-179) afforded the desired oxabicyclic [3.3.1] core of
441772-64-1 in a stereoselective manner. Additionally, rotation about the C3”-C3’ bond
for cis-trans (endo) Diels-Alder (endo-181) was observed during acid catalyzed
intramolecular cyclization to form an oxabicyclic [3.3.1] compound. Together with the
results from these studies, it provide important insights into to syntheses of morusalbanol A
and related mulberry Diels-Alder adducts.
3.3 Approaches toward the synthesis of morusalbanol A
Having established the method for construction of the oxabicyclo[3.3.1] core of
morusalbanol A (Section 3.2), we then attempted the synthesis of morusalbanol A (44). The
biosynthesis pathway of morusalbanol A was proposed by Chen and co-workers as
discussed in Section 1.4 (Scheme 1.22) (Chen et al. 2012). In this section, we elaborate
several approaches toward the synthesis of morusalbanol A.

74
OH
HO
O
MeO
O
O
Me
OH
OH
HO
HO
morusalbanol A (42)
Figure 3.8. Morusalbanol A (42) (Chen et al., 2012)
3.3.1 Retrosynthetic analysis for morusalbanol A (42)
A retrosynthetic analysis for morusalbanol A synthesis is detailed in Scheme 3.6.
We envisaged that morusalbanol A (42) could be derived from an intramolecular
cyclization of a cis-trans type mulberry Diels-Alder adduct 43. The required dienes 190 and
191 could be made from a C3-iodinated benzoic ester 193 followed by the Heck coupling
with 2-methyl-3-buten-2-ol (82). The chalcone type dienophiles 192 and 88 could be
readily prepared from commercially available acetophenone 77.

75
HO
HO OH
OMe
O
Me
OH
O
HO
HOHO
HO
OH
OH
MeO
O
O OH
OH
OH
HO
OH
HO
O
MeO
O
O
OH
OH
HO
HO Me
morusalbanol A (42)
43
endo-addition
Me
RO OR
OR O
OMe
190: R = EOM191: R = Me
O OR
RO
OH
OR
192: R = EOM 88: R = Me
HO OH
OH
OH
O
193
HO
OH
Me
O
77
Scheme 3.6. Retrosynthetic strategy of morusalbanol A (42) via Diels-Alder reaction
3.3.2 Synthesis of diene 190
We began our synthesis of the diene 190 from commercially available 2,4,6-
trihydroxybenzoic acid 193 (Scheme 3.7). Esterification followed by mono iodination on
193 with I2/KIO3 in EtOH/H2O (Han et al., 2014) gave iodobenzene 195 in 80% yields.

76
Ethoxymethoxy (EOM) protection of 195 gave 196 in 55% yields. Heck coupling of 196
with 2-methyl-3-buten-2-ol (82) and subsequent dehydration with AcCl/pyridine provided
the diene 190 in two steps.
HO OH
OH
OOH
HO OH
OMe
OOH
i
iii EOMO OMOE
OMe
OEOMO
I
HO OH
OMe
OOH
I
ii
iv, vEOMO OMOE
OMe
OEOMO
Me
193 194 195
196 190
Scheme 3.7. Synthesis of diene 190. Reagents and conditions: (i) CH3I, K2CO3, acetone, rt, 15 h, 60% (ii) I2,
KIO3, ethanol, H2O, rt, 14 h, 80% (iii) EOM-Cl, K2CO3, acetone, 0 oC, 48 h, 55% (iv) 2-methylbut-3-en-2-ol
(82), Pd(OAc)2, K2CO3, DMF, 100oC, 7 h (v) AcCl, pyridine, benzene, 60
oC, 4 h, 80% (2-steps yield)
3.3.3 Synthesis of dienophile 192
The synthesis of dienophile 192 is detailed in Scheme 3.8. Selective protection of
the para OH with EOM group on 77 afforded acetophenone 197. Subsequently Claisen-
Schmidt condensation (Fine & Pulaski, 1973) of 197 with benzaldehyde 198 gave
dienophile 192 in 55% yield.

77
HO
OH
Me
O
i
EOMO
OH
Me
O
ii
EOMO
EOMO
H
O EOMO
OH O
OMOE
OMOE
77 197 192
198
Scheme 3.8. Synthesis of dienophile 192. Reagents and conditions: (i) EOM-Cl, K2CO3, acetone, 0 oC, 16 h,
85% (ii) 50% aqueous KOH, EtOH, 42h, 55%
3.3.4 Diels-Alder reaction between 190 and 192
With the diene 190 in hand, we attempted the [4+2] cycloaddition with chalcone
192. Unfortunately, the Diels-Alder reaction between diene 190 and dienophile 192
resulted in decomposition at 130 oC under refluxing toluene (Scheme 3.9). Presumably,
both diene 190 and dienophile 192 needed to be protected with a more thermally stable
protecting group.
EOMO
EOMO
OMOE
Me OMe
O
+
O
EOMO
OH OMOE
OMOE
190
192
toluene
130 oCX
HO
OMOE
OMOE
MeO
O
O OMOE
OMOE
OMOE
EOMO
Me
exo-200
endo-199
HO
OMOE
OMOE
MeO
O
O OMOE
OMOE
OMOE
EOMO
Me
Scheme 3.9. Cycloaddition reaction of diene 190 and dienophile 192

78
3.3.5 Synthesis of diene 191
We then attempted to synthesise the methyl ether diene 191. The preparation of
diene 191 is outlined in Scheme 3.10. Esterification and methyl ether protection of benzoic
acid 193 gave 201 in 60% yield. Regiospecific iodination of 201 with I2/ KIO3 in
EtOH/H2O gave ester 201 in 80% yields (H. Wang et al., 2014). Methylation of the
remaining ortho OH group with iodomethane gave 203 in 95% yield. Subsequent
installation of the diene moiety by a Heck coupling reaction (Arkoudis et al. 2009) onto
203 gave the required diene 191 in 80% yield.
HO OH
OH
OOH
MeO OMe
OMe
OOH
i iiMeO OMe
OMe
OOH
I
iiiMeO OMe
OMe
OOMe
I
iv,vMeO OMe
OMe
OOMe
Me
193 201 202
203 191
Scheme 3.10. Synthesis of Diene 191: Reagents and conditions: (i) CH3I, K2CO3, acetone, rt, 15 h, 60% (ii) I2,
KIO3, ethanol, H2O, rt, 14 h, 80% (iii) CH3I, K2CO3, acetone, rt, 15 h, 95% (iv) 2-methylbut-3-en-2-ol,
Pd(OAc)2, K2CO3, DMF, 100oC, 6 h (v) AcCl, pyridine, benzene, 60
oC, 4 h, 80%.
3.3.6 Synthesis of dienophile 88
Similarly the synthesis of dienophile 88 methyl ether was attempted from
acetophenone 77. Methylation of the para OH group of 77, followed by Claisen-Schmidt
condensation with 2’,4’-dimethoxybenzaldehyde 99 produced the dienophile 88 in 80%
yield (Scheme 3.11) (Fine & Pulaski, 1973).
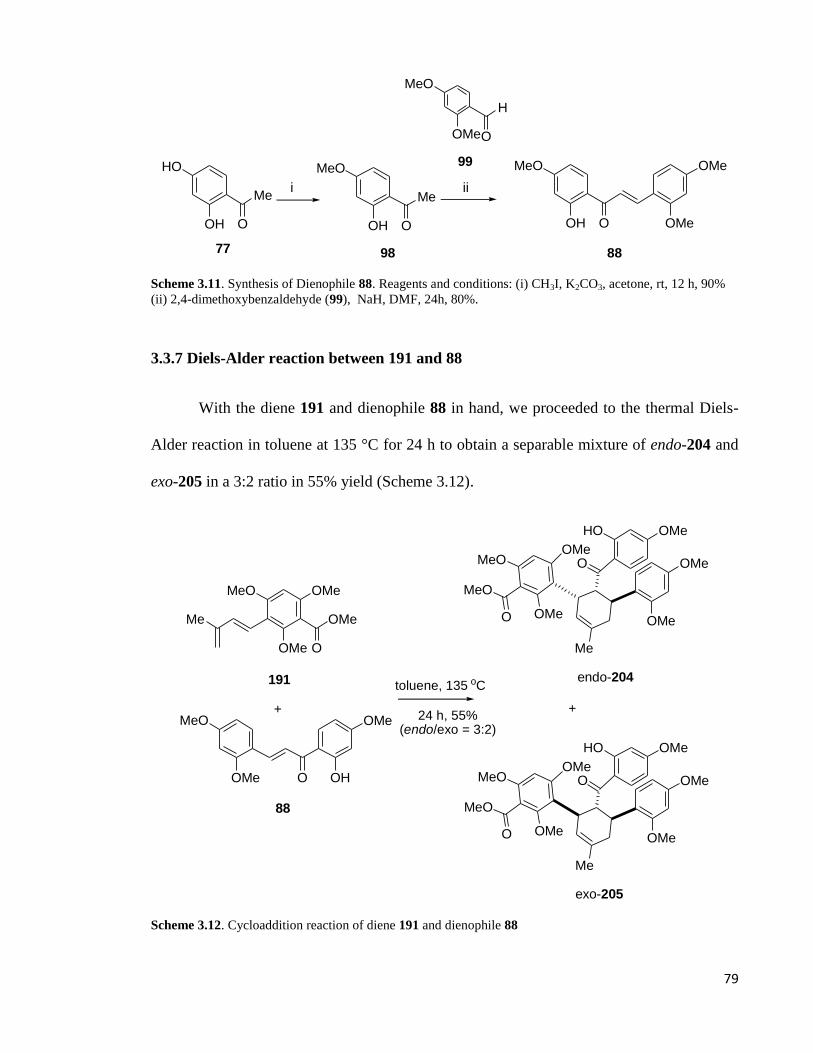
79
MeO
OH
Me
O
MeO
OH O OMe
OMe
98 88
99
MeO
OMe
H
O
HO
OH
Me
O
77
i ii
Scheme 3.11. Synthesis of Dienophile 88. Reagents and conditions: (i) CH3I, K2CO3, acetone, rt, 12 h, 90%
(ii) 2,4-dimethoxybenzaldehyde (99), NaH, DMF, 24h, 80%.
3.3.7 Diels-Alder reaction between 191 and 88
With the diene 191 and dienophile 88 in hand, we proceeded to the thermal Diels-
Alder reaction in toluene at 135 °C for 24 h to obtain a separable mixture of endo-204 and
exo-205 in a 3:2 ratio in 55% yield (Scheme 3.12).
OMe
MeO OMe
OMe
O
Me
OMe
O
MeO
OHOMe
+
191
88
HO
OMe
OMe
MeO
O
O OMe
OMe
OMe
MeO
Me
endo-204
exo-205
toluene, 135 oC
24 h, 55%(endo/exo = 3:2)
HO
OMe
OMe
MeO
O
O OMe
OMe
OMe
MeO
Me
+
Scheme 3.12. Cycloaddition reaction of diene 191 and dienophile 88

80
The proton and carbon-13 NMR spectra for endo-204 showed significant changes at
room temperature after 14 days (Figure 3.9 and 3.10) presumably due to atropisomerism. In
the 1H NMR spectrum, chemical shifts at δ 7.25 (d, J = 2.3 Hz), 6.91 (d, J = 9.2 Hz), 6.49
(d, J = 2.3 Hz), 6.46 (d, J = 2.3 Hz) and 6.39 (d, J = 2.3 Hz) were appeared after 14 days.
Additionally, a doublet at δ 6.38 ppm, two singlets at δ 5.78 ppm and δ 5.42 ppm and a
multiplet at the range of δ 3.88-3.87 ppm were also observed. Likewise, new chemical
shifts at the region of δ 170 -100 ppm were observed in 13
C NMR spectrum.
Figure 3.9. Comparison of the 1H NMR spectra for endo-204 at a) day 1 b) day 14
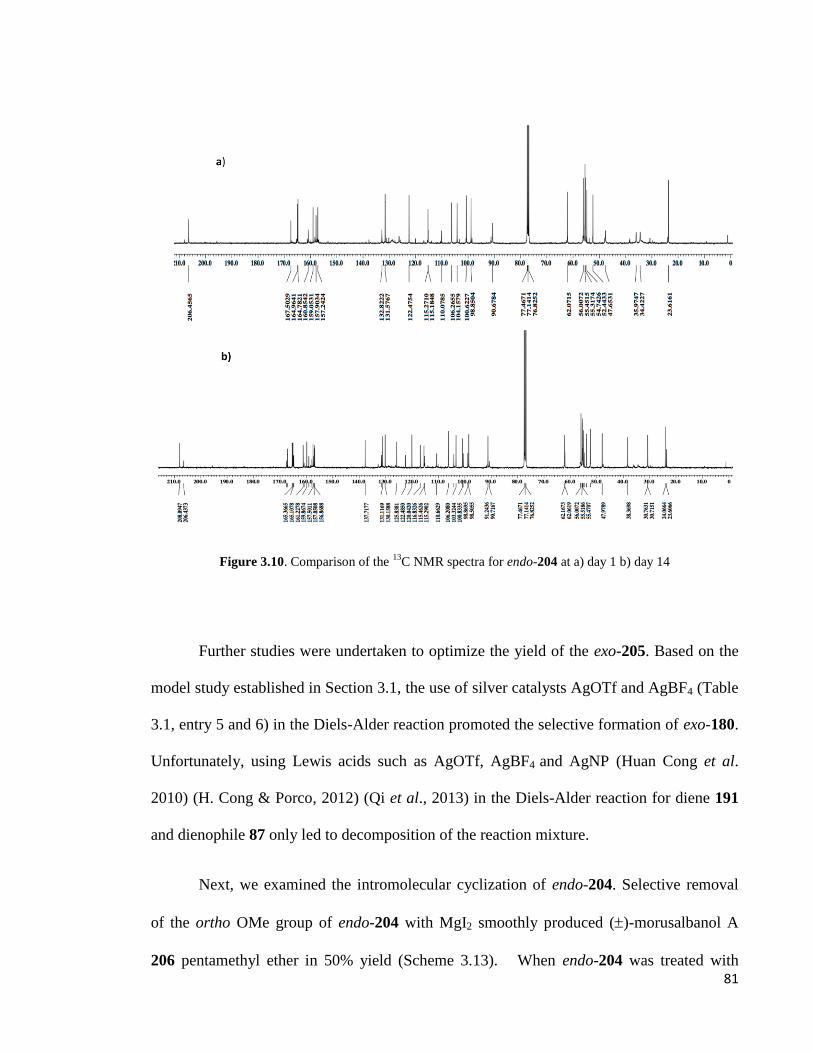
81
Figure 3.10. Comparison of the 13
C NMR spectra for endo-204 at a) day 1 b) day 14
Further studies were undertaken to optimize the yield of the exo-205. Based on the
model study established in Section 3.1, the use of silver catalysts AgOTf and AgBF4 (Table
3.1, entry 5 and 6) in the Diels-Alder reaction promoted the selective formation of exo-180.
Unfortunately, using Lewis acids such as AgOTf, AgBF4 and AgNP (Huan Cong et al.
2010) (H. Cong & Porco, 2012) (Qi et al., 2013) in the Diels-Alder reaction for diene 191
and dienophile 87 only led to decomposition of the reaction mixture.
Next, we examined the intromolecular cyclization of endo-204. Selective removal
of the ortho OMe group of endo-204 with MgI2 smoothly produced ()-morusalbanol A
206 pentamethyl ether in 50% yield (Scheme 3.13). When endo-204 was treated with

82
MgI2 for 6h, a new compound ()-207, was obtained in 36% yield (Scheme 3.14).
Attempts to globally demethylate 206 or 207 with MgI2, BCl3, and 1-
trimethylsilylquinolinium iodide (TMSI-quinoline) to give murasalbanol A were
unsuccessful.
HO
OMe
OMe
MeO
O
O OMe
OMe
OMe
MeO
Me
endo-204
OH
MeO
O
MeO
O
O
Me
OMe
OMe
MeO
MeO
MgI2 (10 equiv)
Et2O: THF (1:1)6 h, 50 %
morusalbanol A pentamethyl ether (206)
Scheme 3.13. Synthesis of morusalbanol A pentamethyl ether (206)
(X-ray )

83
HO
OMe
OMeMeO
O
O OMe
OMe
OMe
MeO
Me
endo-204
OH
MeO
O
O
O
Me
OMe
OMe
MeO
HO
MgI2 (10 equiv)
Et2O: THF (1:1)6 h, 36 %
207
OH
Scheme 3.14. Synthesis of 207.
The relative stereochemistry of 206 was determined by 1H-NMR (Table 3.3) and
NOESY experiments (Figure 3.10). The large coupling constant (11.4 Hz) between H-4”
and H-5” of 206 indicated their trans arrangement. In the NOESY spectrum (Figure 3.11),
correlation between the aromatic protons H15” and H4” on the cyclohexane ring indicated
a cis relationship while correlation between H3” and H4” suggested these were also in a cis
arrangement. The structures of 206 as well as 207 were confirmed by X-ray
crystallography (Table 3.3).
(X-ray )

84
OH
MeO
O
MeO
O
O
Me
OMe
OMe
MeO
MeOO
H
OMe
O
OOMe
O
MeO
OMe
MeO
OMe
H
H
H
H
H
HH
H
HH
H
H
H
H
H
H
H
(NOESY)
1"
3"
4"5"
6"2"
2'
6'
NOEmorusalbanol A pentamethyl ether (206)
9"15"
17"
Figure 3.11. NOESY correlations of morusalbanol A pentamethyl ether (206)
It was noteworthy that a number of proton and carbon signals in the NMR spectra
were absent in compounds 206 and 207. For instance, the absence of proton (H5”) and
carbon (C5” and C20”) resonances in chloroform-d1 at ambient temperature could have
resulted from atropisomerism due to the rotational hindrance of the D/E-rings about the
C5”-C15” and C4”-C8”-C9” bonds. However, when the 1H NMR spectra of 206 was
acquired at 289 K and 353 K in DMSO-d6, resonances of H5 and C20 were observed,
although the resonance for C5 was still not observed. The overall spectral data for ()-206
and ()-207 were in agreement with those reported by Yue et al. for the natural product
(Chen et al. 2012).

85
Table 3.3. Crystal data and structure refinement for 206 and 207
206 207
Empirical formula C69.50H76O20 C31H32O10
Formula weight 1231.30 564.56
Crystal system, Space group Monoclinic, P21/c Triclinic, P-1
a (Å) 13.7493(8) 10.1052(9)
b (Å) 10.2452(5) 11.8237(10)
c (Å) 43.919(2) 12.7062(12)
α () 90 77.712(7)
β () 90.148(5)° 79.804(8)
() 90 70.061(8)
Volume (Å3) 2743.5(3) 1385.4(2)
Z 4 2
Calculated density (Mg/m3) 1.433 1.353
Absorption coefficient (mm-1
) 0.097 0.101
F(000) 2612 596
Crystal size (mm3) 0.320 x 0.120 x 0.090 0.280 x 0.050 x 0.020
Crystal color and habit colorless needle colorless plate
θ range for data collection 2.841 to 27.000° 2.863 to 25.992°
Reflections collected 70030 10730
Independent reflections 13461 (Rint= 0.1344) 5444 (Rint = 0.0566)
Observed reflections [I > 2σ(I)] 7921 2621
Completeness to θ = 25.242° 99.8 % 99.8 %
Data / restraints / parameters 13461 / 0 / 845 5444 / 3 / 378
Goodness-of-fit on F2 1.043 1.023
Final R indices [I > 2σ(I)] R1 = 0.0756, wR2 = 0.1555 R1 = 0.0847, wR2 = 0.1844
R indices (all data) R1 = 0.1362, wR2 = 0.1890 R1 = 0.1783, wR2 = 0.2374
Largest diff. peak and hole (e Å-3
) 0.341 and -0.310 1.460 and -0.319
CCDC deposit number 1402220 (for 206) and 1402153 (for 207).

86
Table 3.4. 1H and
13C NMR spectra of morusalbanol A pentamethyl ether (206)
a signals invisible, bmeasurement at 353 K, ccoupling constant cannot be measured due to broad reasonance.
No. 206 (CDCl3) 206 (toluene) 206 (toluene)b
13C δ(ppm)
1H δ (multiplicity, J in Hz)
13C δ(ppm)
1H δ (multiplicity, J in Hz)
13C δ(ppm)
1H δ (multiplicity, J in Hz)
1” 75.6 - 75.1 - 75.5 -
2” 36.3 1.89 (m)
2.11 (dd, 2.8, 13.3)
36.0 1.60 (bd, 12.8)
1.71 (bd, 11.9)
36.8 1.68 (m)
1.80 (dd,3.2, 13.2)
3” 31.2 3.65 (m) 31.3 3.72 (m) 31.8 3.72 (m)
4” 50.6 4.37 (bs) 51.0 4.21 (m) 51.3 4.26 (d, 11.4)
5” a a a
3.86 (m) a
3.79 (ddd, 4.6, 12.8, 16.9)
6” 45.5 2.19 (m) 45.8 2.30 (bd)c 46.5 2.26 (ddd, 1.8, 4.1, 13.8)
7” 28.6 1.36 (s) 28.4 1.26 (s) 28.7 1.26 (s)
8” 204.1 - 203.9 - 204.2 -
9” 114.3 - 114.5 - 115.3 -
10” 165.4 - 165.4 - 166.0 -
11” 101.1 6.39 (d, 2.3) 100.9 6.35 (d, 2.3) 101.8 6.33 (d, 2.7)
12” 165.3 - 166.2 - 166.4 -
13” 107.1 6.50 (dd, 2.8, 9.1) 107.2 6.45 (d, 8.7) 107.4 6.46 (dd, 2.3, 8.7)
14” 130.5 7.87 (d, 9.1) 130.3 7.79 (d, 9.2) 130.7 7.79 (d, 8.7)
15” 125.6 - 127.6 - 127.6 -
16” 158.5 - 158.3 - 158.4 -
17” 99.1 6.31 (d, 2.3) 99.1 6.16 (d, 1.8) 100.2 6.18 (d, 2.3)
18” 159.1 - 159.3 - 160.0 -
19” 104.1 6.25 (dd, 2.3, 8.2) 104.0 6.06 (dd, 1.8, 8.2) 105.3 6.10 (dd, 2.3, 8.2)
20” a
6.90 (d, 8.2) 125.5 6.83 (d, 8.2) 125.8 6.84 (d, 8.2)
3’ 103.0 - 103.2 - 104.1 -
2’ 154.9 - 155.0 - 155.6 -
1’ 107.1 - 105.5 - 106.6 -
6’ 157.5 - 157.6 - 158.9 -
5’ 86.1 5.88 (s) 86.2 5.60 (s) 87.5 5.68 (s)
4’ 158.3 - 158.4 - 159.1 -
7’ 167.5 - 166.3 - 166.6 -
MeO 54.7 3.38 (s) 54.1 3.18 (s) 54.6 3.20 (s)
MeO 55.3 3.68 (s) 54.2 3.20 (s) 54.9, 3.26 (s)
MeO 55.6 3.74 (s) 54.6 3.21 (s) 55.0 3.26 (s)
MeO 55.5 3.80 (s) 54.6 3.23 (s) 55.3 3.30 (s)
MeO 55.9 3.82 (s) 54.8 3.24 (s) 55.8 3.34 (s)
MeO 52.2 3.88 (s) 51.2 3.72 (s) 51.8 3.72 (s)
OH 12.75 13.36 13.09

87
Table 3.5. 1H and
13C NMR spectra of compound 207
asignals invisible, bmeasurement at 353 K, 13C-NMR was not recorded at 353 K, ccoupling constant cannot be
measured due to broad reasonance. dsignal overlapped.
No. 207 (CDCl3) 207 (DMSO) 207 (DMSO)b
13C δ(ppm)
1H δ (multiplicity, J in Hz)
13C δ(ppm)
1H δ (multiplicity, J in Hz)
1H δ (multiplicity, J in Hz)
1” 80.3 - 77.6 - -
2” 35.9 1.97 (dt, 12.8)
2.22 (dd, 2.7, 13.1)
35.1 1.69 (d, 11.9)
2.38 (d, 11.4)
1.70 (m)
2.39 (d, 11.4)
3” 30.3 3.68 (m)d 30.9 3.45 (m)
d 3.35 (m)
d
4” 49.8 4.37 (bd) c 50.3 4.50 (bd, 11.9) 4.47 (d, 11.2)
5” a a a a a
6” 44.9 2.29 (bd, 12.8) 46.2 2.06 (bd)c 2.09 (bd)
c
7” 28.6 1.56 (s) 28.6 1.35 (s) 1.40 (s)
8” 203.4 - 205.0 - -
9” 114.1 - 114.3 - -
10” 165.5 - 164.9 - -
11” 101.1 6.38 (d, 2.3) 101.4 6.40 (d, 2.3) 6.37 (d, 2.5)
12” 165.0 - 165.8 - -
13” 107.5 6.50 (dd, 1.8, 8.7) 107.5 6.56 (dd, 2.3, 9.2) 6.55 (d, 7.6)
14” 130.3 7.82 (d, 9.1) 132.2 8.19 (d, 8.7) 8.12 (d, 8.7)
15” 122.5 - 124.6 - -
16” 158.2 - 158.1 - -
17” 99.2 6.33 (d, 2.3) 99.0 6.36 (d, 2.3) 6.37 (d, 2.5)
18” 159.3 - 159.1 - -
19” 104.2 6.26 (dd, 2.3, 8.2) 105.0 6.27 (dd, 2.3, 8.7) 6.27 (d, 8.5)
20” a
6.88 (d, 8.7) a
6.98 (d, 8.7) 6.94 (d, 8.7)
3’ 102.0 - 102.6 - -
2’ 155.7 - 157.5 - -
1’ 94.1 - 96.0 - -
6’ 165.5 - 163.5 - -
5’ 92.8 6.02 (s) 91.3 5.91 (s) 5.94 (s)
4’ 162.3 - 161.7 - -
7’ 171.2 - 172.4 - -
MeO 55.2 3.38 (s) 55.4 3.23 (s) 3.28 (s)
MeO 55.3 3.69 (s) 55.4 3.61 (s) 3.62 (s)
MeO 55.5 3.75 (s) 56.0 3.64 (s) 3.67 (s)
MeO 55.6 3.88 (s) 56.2 3.78 (s) 3.80 (s)
OH 12.37 12.13 12.43
OH 12.62 12.54 12.52
COOH 11.38 a a

88
CHAPTER 4
APPROACHES TOWARD THE SYNTHESIS OF SOROCEIN B
Sorocein B (62) was isolated from the root bark of Sorocea bonplandii with an
overall yield of 52 mg per kilogram of dried bark. Thus far, there have been no report on
the synthesis of this bioactive natural product. In this chapter we described several
approaches toward the synthesis of sorocein B.
O O
Me
OH
OH
OOH
HO
OH
OMe
Me
H
H
3"
4"
6"
SR
S
R
Figure 4.1. Sorocein B (62)
4.1 Retrosynthetic analysis
A retrosynthetic analysis towards the synthesis of sorocein B (62) involves a
biogenesis-inspired intermolecular [4+2]-cycloaddition reaction between the chalcone
dienophile 209 and diene 106 as shown in Scheme 4.1. It was envisaged that the
chalcone-type diene 106 could be obtained from a Heck coupling on the chalcone 210 with
2-methyl-3-buten-2-ol (82) (Arkoudis et al., 2009) followed by dehydration (Harrington et
al., 1987). The required dienophile 209 could be readily prepared from commercially
available 2,4-dihydroxyacetophenone 77 and 2´,4´-dihydroxybenzaldehyde 92 via Claisen-
Schmidt condensation (Fine & Pulaski, 1973).
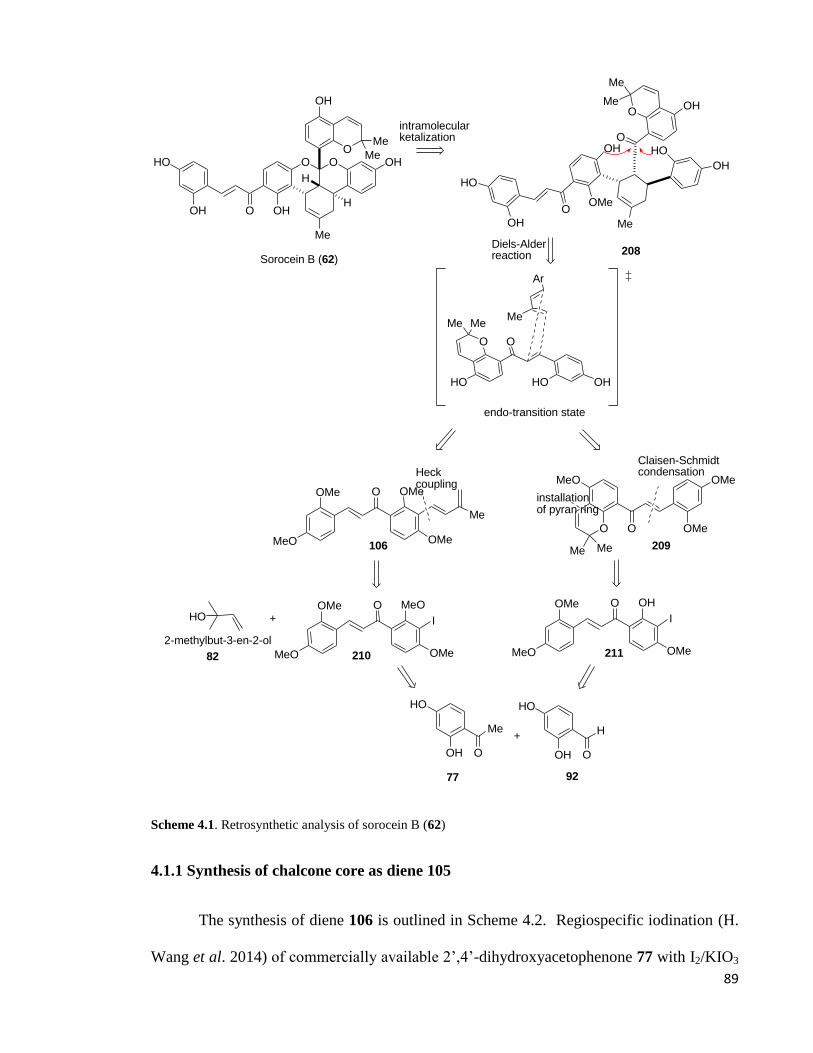
89
HO
OH
Me
O
OH
O
Me
O
O
OMe
HO
OH
OH
OH
Me
HO
OMe
OMeO
MeO
OMe
Me
MeO
O O
OMe
OMe
Me
Diels-Alderreaction
O O
OH
OH
OOH
HO
OH
OMe
Me
H
H
Me
OO
HO HO OH
Me Me
Ar
Me
endo-transition state
Heck coupling
Claisen-Schmidtcondensation
HO
OH
H
O
OMe
MeOO
I
MeO
OMe+HO
OMe
OHO
I
MeO
OMe
Sorocein B (62)208
106 209
2-methylbut-3-en-2-ol
210 211
9277
82
intramolecular ketalization
+
installation of pyran ring
Me
Me
Scheme 4.1. Retrosynthetic analysis of sorocein B (62)
4.1.1 Synthesis of chalcone core as diene 105
The synthesis of diene 106 is outlined in Scheme 4.2. Regiospecific iodination (H.
Wang et al. 2014) of commercially available 2’,4’-dihydroxyacetophenone 77 with I2/KIO3

90
in EtOH/H2O followed by methylation and iodomethane. Subsequent Heck coupling of
210 and dehydration with AcCl/pyridine provided the desired diene 106 in 65% combined
yield in two steps. However, diene 106 was unstable and used immediately after silica gel
purification.
O Me
OH
OH
O Me
OH
OH
I2, KIO3
EtOH, H2Ort, 24 h, 92% I
OMe
OMe
O H
KOH, EtOHacetone, rt15 h, 80%
CH3I, K2CO3
O Me
OMe
OMe
I rt, 70%
MeO
I
OMe O OMe
OMe MeO
OMe O OMe
OMe
1) 2-methylbut-3-en-2-ol (82) PdCl2, K2CO3, DMF, 140oC, 24h
2) AcCl, pyridine
C6H6, 80oC
65 % combined yield
Me
77 78
99
79
210 106
Scheme 4.2. Synthesis of diene 106
4.1.2. Synthesis of dienophile 209
The synthesis of dienophile 209 began with selective iodination of 2´,4´-
dihydroxyacetophenone 77 with I2/KIO3 in EtOH/H2O, followed by methylation to give
acetophenone 173 (Scheme 4.3). Claisen-Schmidt condensation between acetophenone
173 and benzaldehyde 99 furnished the chalcone 211 in 70% yield. Installation of 2,2-
dimethyl chromene ring (pyran moiety) gave chalcone 209 in 65% yield (Scheme 4.3).

91
O Me
OH
OH
O Me
OH
OH
I2, KIO3
EtOH, H2Ort, 24 h, 92% I
OMe
OMe
O H
KOH, EtOHacetone, rt15 h, 80%
CH3I, K2CO3
O Me
OH
OMe
I rt, 70%
MeO
I
OH O OMe
OMeMeO
O O OMe
OMe
Me Me1) 2-methylbut-3-en-2-ol (82)
PdCl2, K2CO3,
DMF, 140oC, 24h
2) SiO2
heat, 130oC
78
99
77 173
211 209
Scheme 4.3. Synthesis of dienophile 209
4.1.3 Method development for installation of 2,2-Dimethyl-2H-chromenes (pyran
moiety)
2,2-Dimethyl-2H-chromenes (referring to the pyran moiety of 209) are commonly
found as parent structure in natural products (Ellis et al. 1977). It is also an important
intermediate for the syntheses of numerous pharmaceutical and biologically active
compounds (Mannhold et al., 1999; Sa e Sant'Anna et al., 2005; Szczepanik et al., 2005).
Therefore, a number of methods have been reported for the construction of 2,2-dimethyl-
2H-chromenes. In particular, condensation of phenols with α,β-unsaturated carbonyls
(aldehydes or acetals) under different reaction conditions, is the most commonly used
method (Adler & Baldwin, 2009; Akanksha & Maiti, 2012; Bröhmer, Volz, & Bräse, 2009;
Chauder et al. 1998; Lamcharfi, Menguy, & Zamarlik, 1993; Lee, Choi, & Yoon, 2005;
North et al. 1995; Prado, Janin, & Bost, 2006; Sartori et al. 1979). Other methods include
condensation of phenols with 2-methyl-3-butyn-2-ol (82) catalyzed by ReCl(CO)5 (Zeng,

92
Ju, & Hua, 2011), BF3.Et2O (Madabhushi et al. 2012) or other Lewis acids (Bigi et al.
1997; Dong, Wang, & Wang, 2008; Gabbutt et al. 2003; Zhao & Carreira, 2003);
condensation of phenols with 3-chloro-3-methylbut-1-yne catalysed by Ph3PAuNTf2
(Lykakis et al. 2011); dehydration of chromanols (Bergmann & Gericke, 1990; Kureshy et
al. 2009); Pd-catalyzed cyclization of o-prenyl phenols (Iyer & Trivedi, 1990; Larock, Wei,
& Hightower, 1998), and photocyclization of 3-aryl-1,1-dimethylprop-2-en-1-ol (Pandey &
Krishna, 1988).
We first attempted to make the chromane (215) via a condensation reaction of a
simple o-iodophenol 212 and 2-methyl-3-buten-2-ol 82 using a variety of Pd catalysts
(PdCl2, Pd(OAc)2, Pd(PPh3)4, Pd(TFA)2, Pd(dba)2), bases (Na2CO3, K2CO3, KOAc),
additive (n-Bu4NCl) and solvents (DMF, DMSO, toluene) and the results are summarised
in Table 4.1. Mixture of 212 (1.0 mmol), 82 (1.5 mmol) and PdCl2 (1 to 10 mol %) in the
present of K2CO3 (3 equiv.) in toluene (1 mL) was heated in a sealed tube under nitrogen at
100C. TLC analysis of the reaction mixture showed 212 to be completely consumed after
18 h. As expected, the Heck coupling product 214 was the major product obtained (35-45%
yield, entries 1-3). The desired product, 2,2-dimethyl-2H-chromene (215), was isolated
only in low yield (5-10%, entries 1-3). Increasing the reaction temperature to 140 C
resulted in a slightly increased yield of 215 (20%, entry 4 vs entry 3). Presumably, a higher
reaction temperature improved the formation of 215, most likely due to increase in the rate
of condensation of 214. Indeed, when the reaction was repeated at 150 C in DMF (entry 5)
and DMSO (entry 6), 215 was obtained in 23% and 25% yields, respectively. Attempts to
increase the yield of 215 through microwave irradiation, however, were unsuccessful
(entries, 7-8). It is noteworthy that identical results are observed under both air and
nitrogen atmosphere (Table 4.1, entries 4 and 5).

93
We next examined the catalytic activity of other Pd catalysts. Notably, the yields of
214 and 215 were slightly increased with catalytic Pd(OAc)2 (Table 4.1, entries 9-10).
Although three other Pd catalysts, Pd(PPh3)4, Pd(TFA)2 and Pd(dba)2, showed activity for
the reaction, they were less effective than Pd(OAc)2 (Table 4.1, entries 11-13). Adding n-
Bu4NCl had no significant effect on formation of 214 and 215 (Table 4.1, entry 10 vs entry
14). The effect of base on the yield of reaction was also evaluated (Table 4.1, entries 15-
17). The yields of 214 and 215 were reduced when Et3N was used (Table 4.1, entry 17). We
also found that both Na2CO3 and KOAc were less efficient for the Heck coupling reactions
(Table 4.1, entries 15-16). Amongst the bases studied, K2CO3 gave the best result (Table
4.1, entries 10 & 14).

94
Table 4.1. Pd-catalysed coupling-condensation of o-iodophenol 212 and 2-methyl-3-buten-2-ol 82
212
+
82: R = H213: R = Ac 214 215
conditions
OH
IMe
Me
OROH
Me
MeOH
+
OMe
Me
aReaction conditions: 212 (1.0 equiv.), 82 (10 equiv.), solvent 1 ml/mmol;
bUnder air condition;
cMicrowave
irradiation; dIsolated yield after chromatography;
eIn present of molecular sieve 4Å (10 equiv.);
f Using 213
(10 equiv.); gSilica gel (SiO2, MW=60, size 230-400 mesh) was added to the reaction mixture after heating for
16 h.
OH
OHMe
Me
SiO2 (20 equiv.)
DMF (1 ml/ mmol)
140 oC, 8 h
96%
OMe
Me
214 215
Scheme 4.4. Controlled experiment
Entry Catalysta
(mol %) Base Additive Solvent Temp
(°C)
Time
(h)
Yield (%)d
(equiv.) (equiv.) (214:215)
1 PdCl2 (1) 81 K2CO3 (1.5) n-Bu4NCl (1.5) toluene 100 24 35:5
2 PdCl2 (5) 81 K2CO3 (2) n-Bu4NCl (1.5) toluene 100 24 40:10
3 PdCl2 (10) 81 K2CO3 (3) n-Bu4NCl (1.5) toluene 100 24 45:10
4 PdCl2 (10) 81 K2CO3 (3) n-Bu4NCl (1.5) DMF 140 24 40:20
5b PdCl2 (10) 81 K2CO3 (3) n-Bu4NCl (1.5) DMF 150 24 43:23
6 PdCl2 (10) 81 K2CO3 (3) n-Bu4NCl (1.5) DMSO 150 24 45:25
7c PdCl2 (10) 81 K2CO3 (3) n-Bu4NCl (1.5) DMF 140 0.5 65:15
8c PdCl2 (10) 81 K2CO3 (3) MS 4Å (10)
e DMF 140 0.5 70:10
9 Pd(OAc)2 (5) 81 K2CO3 (3) n-Bu4NCl (1.5) DMF 140 24 52:15
10 Pd(OAc)2 (10) 81 K2CO3 (3) n-Bu4NCl (1.5) DMF 140 24 55:20
11 Pd(TFA)2 (10) 81 K2CO3 (3) - DMF 140 24 40:12
12 Pd(PPh3)4 (10) 81 K2CO3 (3) - DMF 140 24 35:5
13 Pd(dba)2 (10) 81 K2CO3 (3) - DMF 140 24 38:5
14 Pd(OAc)2 (10) 81 K2CO3 (3) - DMF 140 24 55:15
15 Pd(OAc)2 (10) 81 KOAc (3) - DMF 140 24 40:8
16 Pd(OAc)2 (10) 81 Na2CO3 (3) - DMF 140 24 35:5
17 Pd(OAc)2 (10) 81 Et3N (3) - DMF 140 24 25:3
18f Pd(OAc)2 (10) 212 K2CO3 (3) - DMF 140 24 10: -
19 Pd(OAc)2 (10) 81 K2CO3 (3) SiO2 (10)g DMF 140 24 -:76
20 Pd(OAc)2 (10) 81 K2CO3 (3) SiO2 (20)g DMF 140 24 -:80

95
Subsequently we investigated the condensation reaction of o-iodophenol 212 with
1,1-dimethyl allylic acetate (2-methylbut-3-en-2-yl acetate) 213. Li and co-workers
described a selective Heck reaction of 4-iodoanisole with 213 to give (E)-4-(4-
methoxyphenyl)-2-methylbut-3-en-2-ol in quantitative yield (Liu et al. 2011). Thus we
anticipated that the condensation of 212 with 213 could enhance the formation of 214 and
215. However, condensation of 212 with 213 under the present optimal reaction condition
(Pd(OAc)2 (10 mol%), K2CO3 (3 equiv.), DMF (2M), 140 C) significantly decreased the
yield of 214 and 215 (Table 4.1, entry 18), accompanied with the formation of undesired
side-product.
Since present condensation reaction of 214 and 215 did not proceed well at elevated
temperature or under microwave irradiation, we proceeded to investigated the condensation
under acidic condition, i.e. AcOH (David Cotterill, Iqbal, & Livingstone, 1998), H3PO4
(Goujon, Zammattio, & Kirschleger, 2000; Yus, Foubelo, & Ferrández, 2001).
Considering sensitivity of some functional groups (i.e. ethoxy methoxy group, EOM) in
acidic condition, we decided to use a milder reagent such as molecular sieve or silica gel
(SiO2). However, the condensation reaction of 214 and 215 did not proceed well in present
of molecular sieve (Table 4.1, entry 8). To our delight, the condensation reaction
proceeded extremely well in presence of SiO2 to afford 215 in good yield (76 – 80%, Table
4.1, entries 19-20). A controlled experiment was carried out to examine the role of SiO2 as
a dehydrating agent (Scheme 4.4). Treatment of allylic alcohol 214 in the present of SiO2
afforded chromene 215 in nearly quantitative yield under the standard conditions.

96
Table 4.2. Pd-catalysed condensation of o-halophenols 212 and 2-methyl-3-buten-2-ol 82a
aReaction conditions: Halide (1.0 mmol), 82 (10 mmol), Pd(OAc)2(10 mol %), K2CO3 (3.0 equiv), and DMF (1 mL) at 140 C under air for 8 ~ 14 h. The reaction was monitored by TLC until halide was completely consumed. Silica gel (20 equiv.) then was added to the
reaction mixture, followed by heating at 140°C for another 4 ~ 16h to yield desired product. bIsolated yield.
Entry Halide 1 t
(h)
Product Yield
(%)
1
216
15
215
75
2
217
15
218
80
3
219
12
220
60
4
221
20
222
25
5
223
20
224
70
6
225
20
226
15
7
202
18
227
85
8
228
30
229
17
9
211
24
209
65

97
Consequently, the scope of condensation reaction of o-iodophenols was explored in
order to determine its optimised conditions (Table 4.2). In the presence of Pd(OAc)2 and
K2CO3, a variety of o-iodophenols were reacted with 2-methyl-3-buten-2-ol 82 (Table 4.2,
entries 1-9). The results indicated both electron-rich and electron-deficient aryl iodides to
be suitable for the reaction. Aryl bromide 216 has similar activity as iodide 212. However,
aryl bromides bearing aldehyde and naphthol moieties gave lower product yield (Table 4.2,
entries 4 & 8).
With regards to the synthesis of natural products, isoencecalin (Manners & Jurd,
1976) 220 and 2-demethoxymillepachine 209, it is worthy to note that the present protocol
allows for the installation of 2,2-dimethylchromene ring at the late-stage of the synthesis.
In contrast, the conventional approach, such as in the synthesis of millepachine, the
preparation of 2,2-dimethylchromene ring was performed at an earlier stage of the synthesis
(G. Wang et al., 2012). The late-stage installation of 2,2-dimethylchromene ring was
successfully applied in the synthesis of 209 from chalcones 211 (Scheme 4.3).
4.1.4 Cycloaddition reaction of 106 and 209
With the dienophile 209 in hand, we began to investigate the [4+2] cyloaddition
with diene 106. However, chalcone 209 failed to undergo cycloaddition with diene 106 at
150 oC in refluxing toluene even after several days of reaction (Scheme 4.5). We reasoned
that the ortho OH in chalcone substituent 209 was critical for the success of the [4+2]
cycloaddition as previously described in Chapter 3.

98
O O
OMe
Me Me
OMe
MeO
O OMe
OMe
OMe
MeO
Me
+
toluene
150 oC
endo-230
Me
O
O
Me
Me
OMe
OMe
OMe
OMe
OMe
O
OMe
MeO
x
106
209
exo-231
Me
O
O
Me
Me
OMe
OMe
OMe
OMe
OMe
O
OMe
MeO
+
Scheme 4.5. Cycloaddition reaction of diene 106 with dienophile 209
4.2 Cycloaddition reaction of diene 233 and dienophile 234
The presence of an ortho-OH group in dienophile 209 was found to be essential for
the success of the [4+2] cycloaddition reaction. Thus we directed our attention to a
synthesis of dienophile with an ortho-OH group. Two different protecting groups (EOM
and OMe) were used to protect the other OH groups in dienophile.
4.2.1 Synthesis of diene 233
Synthesis of diene 233 is outlined in Scheme 4.6. Regioselective iodination of
commercially available 2’,4’-dihydroxyacetophenone 77 with I2/KIO3 in EtOH/H2O gave
78. Ethoxymethoxy protection followed by methylation of the phenolic groups of 78 gave
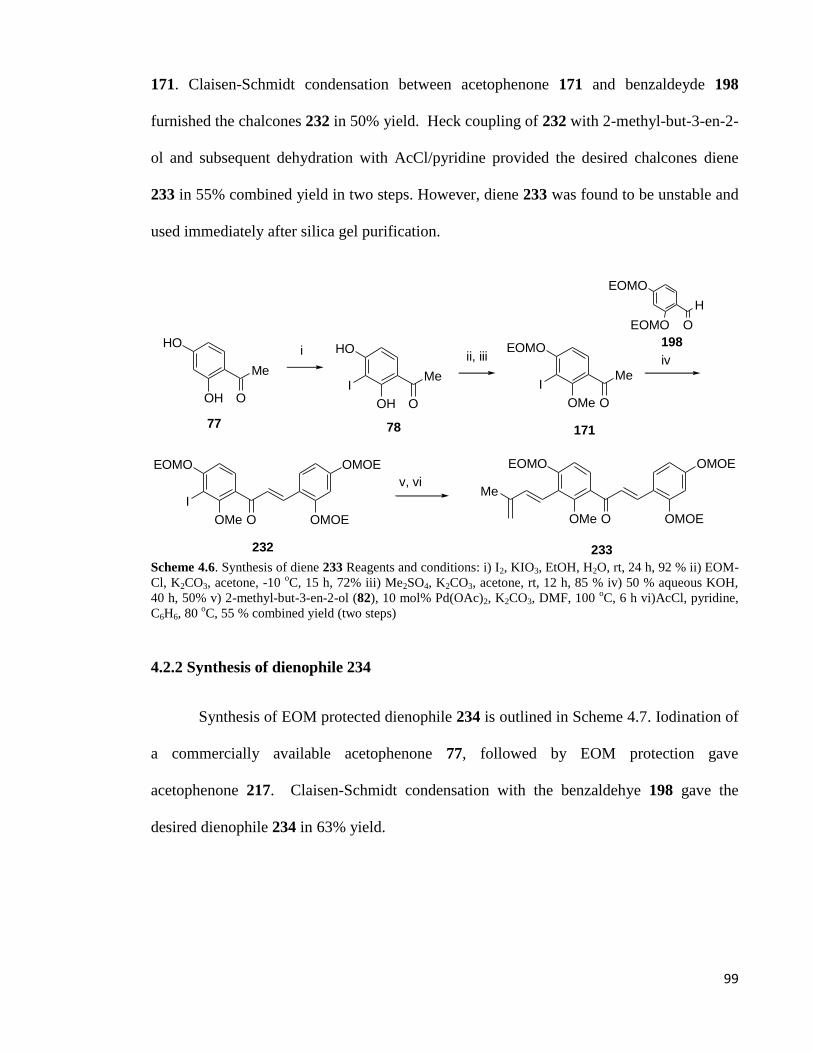
99
171. Claisen-Schmidt condensation between acetophenone 171 and benzaldeyde 198
furnished the chalcones 232 in 50% yield. Heck coupling of 232 with 2-methyl-but-3-en-2-
ol and subsequent dehydration with AcCl/pyridine provided the desired chalcones diene
233 in 55% combined yield in two steps. However, diene 233 was found to be unstable and
used immediately after silica gel purification.
HO
OH
Me
O
HO
OH
Me
O
I
EOMO
OMe
Me
O
I
i ii, iii
EOMO
OMe O
I
EOMO
EOMO
H
O
iv
OMOE
OMOE
v, vi
EOMO
OMe O OMOE
OMOE
Me
77 78 171
198
232 233
Scheme 4.6. Synthesis of diene 233 Reagents and conditions: i) I2, KIO3, EtOH, H2O, rt, 24 h, 92 % ii) EOM-
Cl, K2CO3, acetone, -10 oC, 15 h, 72% iii) Me2SO4, K2CO3, acetone, rt, 12 h, 85 % iv) 50 % aqueous KOH,
40 h, 50% v) 2-methyl-but-3-en-2-ol (82), 10 mol% Pd(OAc)2, K2CO3, DMF, 100 oC, 6 h vi)AcCl, pyridine,
C6H6, 80 oC, 55 % combined yield (two steps)
4.2.2 Synthesis of dienophile 234
Synthesis of EOM protected dienophile 234 is outlined in Scheme 4.7. Iodination of
a commercially available acetophenone 77, followed by EOM protection gave
acetophenone 217. Claisen-Schmidt condensation with the benzaldehye 198 gave the
desired dienophile 234 in 63% yield.

100
O Me
OH
OH
O Me
OH
OH
I2, KIO3
EtOH, H2Ort, 24 h, 92% I
OMOE
OMOE
O H
KOH, EtOH
acetone, -10oC
15 h, 85%
EOM-Cl, K2CO3
O Me
OH
OMOE
I
rt, 50%
EOMO
I
OH O OMOE
OMOE
76 77 216
233
197
Scheme 4.7. Synthesis of dienophile 234
However, subjecting the dienophile 234 to thermal Diels-Alder reaction with
diene 233 resulted in decomposition (Scheme 4.8). Similarly, the Diels-Alder reaction
between diene 233 and dienophile 234 performed in the presence of catalytic amount of
using ZnI2/BuNBH4 and AgBF4/BuNBH4 catalysts also resulted in the decomposition.
O OH
OMOE
OMOE
EOMO
O OMe
OMOE
OMOE
EOMO
Me
+
toluene
150 oC
Me
O
HO
I
OMOE
OMOE
OMOE
OMe
OMOE
OOMOE
EOMO
x
I
Me
O
HO
I
OMOE
OMe
OMOE
OOMOE
EOMO
+
233
234
endo-235
exo-236
OMOE
OMOE
Scheme 4.8. Cycloaddition reaction of diene 233 with dienophile 234

101
4.3 Cycloaddition reaction of diene 106 and dienophile 211
Since the use of EOM protecting group was unstable for the Diels-Alder reaction
between diene 233 and dienophile 234, we decided to change it to a more thermally stable
protecting group with a methyl ether in both diene and dienophile. When the Diels-Alder
reaction between diene 106 and dienophile 211 was performed in toluene at 150 oC in a
pressure tube, a mixture of endo-237 and exo-238 in ratio 3:2 was obtained, respectively
(Scheme 4.9). These adducts were separated by flash chromatography with 30%
EtOAc/hexane as an eluent to give pure endo-237 and exo-238.
O OH
OMe
OMe
MeO
O OMe
OMe
OMe
MeO
Me
+
toluene
150oC
Me
O
HO
I
OMe
OMe
OMe
OMe
OMe
OOMe
MeO
I
Me
O
HO
I
OMe
OMe
OMe
OMe
OMe
OOMe
MeO
+
OOH
MeO
OMe
OMe
O OMe
OMe
OMe
MeOMe
I
endo-addition
OOH
MeO
OMe
OMe
O OMe
OMe
OMe
MeO
Me
I
exo-addition
55%
3:2
106
211
endo-237exo-238
pressure tube
Scheme 4.9. Diels-Alder reaction between diene 106 and dienophile 211

102
Next, we investigated the removal of methyl ether protecting groups in endo-237.
Attempts to use Lewis acid BCl3 at different conditions (-78oC to room temperature) in
CH2Cl2 were unsuccessful. In all cases, endo-237 decomposed (Scheme 4.10).
Me
O
HO
I
OMe
OMe
OMe
OMe
OMe
OOMe
MeOBCl3, CH2Cl2-78 oC - r.t
Me
O
HO
I
OH
OH
OH
OH
OH
OOH
HO
X
237 239
Scheme 4.10. Attempts to remove the OMe groups of endo-237
4.4 Cycloaddition reaction of diene 106 and dienophile 89
Next, we attempted the cycloaddition reaction of diene 106 and dienophile 89. The
dienophile 89 was synthesized from chalcones 88 as shown in Scheme 4.11. Selective
demethylation of the 2’-methoxy group of chalcone 65 afforded chalcone 88. Installation of
prenyl group was applied in chalcones 88 following the method of Romano et al. O-
prenylation of 88 with prenyl chloride in refluxing acetone gave the prenyl ether 100.
Subsequently, the prenyl ether 100 then was subjected to a Montmorillonite K10 promoted
[1,3]-sigmatropic rearangement to give the dienophile 89 in 45% yield.

103
O Me
OH
OH
OMe
OMe
O H
KOH, EtOHacetone, rt15 h, 80%
CH3I, K2CO3
O Me
OMe
OMe
rt, 70%MeO
OMe O OMe
OMe
BCl3, CH2Cl2
0 oC- r.t, 45%MeO
OH O OMe
OMe
MeO
O O OMe
OMe
Me
Me
Cl Me
Me
K2CO3, acetonereflux, 84%
MeO
OH O OMe
OMe
Me
Memontmorillonite K10
CH2Cl2, 0oC,
2 h, 45%
77 240 65
88
100 89
99
Scheme 4.11. Synthesis of dienophile 89
With the dienophile 89 in hand, attention was turned to the Diels-Alder reaction.
Thermal Diels-Alder reaction between 106 and dienophile 89 in toluene at 150 oC in a
pressure tube gave a mixture of the cycloaddition products, endo-241 and exo-242 in 35%
yield in a 1:1 ratio, respectively (Scheme 4.12). The cycloaddition adducts were separated
by flash chromatography with 30% EtOAc/hexane as an eluent to give pure endo-241 and
exo-242.

104
O OH
OMe
OMe
MeO
O OMe
OMe
OMe
MeO
Me
+
toluene
150oC
Me
O
HO OMe
OMe
OMe
OMe
OMe
OOMe
MeOO
HO OMe
OMe
OMe
OMe
OMe
OOMe
MeO
+
O OMe
OMe
OMe
MeOMe
endo-addition
OOH
MeO
OMe
OMe
O OMe
OMe
OMe
MeO
Me
exo-addition
35%
1:1
Me
Me
Me
Me
MeMe MeMe
OHO
MeO
MeO
OMe
Me
Me
106
89
endo-241 exo-242
pressure tube
Scheme 4.12. Diels-Alder reaction between diene 106 and dienophile 89
Installation of the 2,2-dimethylchromene ring (pyran moiety) was performed on
adducts 241 and 242 with 20 mol% PdCl2 in dry ethanol afforded the desired intermediates
230 and 231 in 50% and 45% yield, respectively (Scheme 4.13) after a long period of
reaction.

105
Me
O
HO OMe
OMe
OMe
OMe
OMe
OOMe
MeO
MeMe
20 mol% PdCl2
dry ethanol, r.t 120 h, 50%
dry ethanol, r.t 216 h, 45%
20 mol% PdCl2
Me
O
O OMe
OMe
OMe
OMe
OMe
O
OMe
MeO
Me
Me
242
231
Me
O
O OMe
MeO
OMe
OMe
OMe
O
OMe
MeO
Me
Me
230
Me
O
HO OMe
OMe
OMe
OMe
OMe
OOMe
MeO
MeMe
241
Scheme 4.13. Installation of 2,2-dimethyl chromene in endo-241 and exo-242
Attempt to remove the methyl ether groups of endo-230 and exo-231 with several
reagents such as BCl3, MgI2, TMSI quinoline only resulted in decomposition (Table 4.3).
Table 4.3 Attempt to remove the OMe groups of endo-230
Me
O
O OMe
OMe
OMe
OMe
OMe
OOMe
MeOsee table
Me
O
O OH
OH
OH
OH
OH
OOH
HO
endo-230 243
Entry Reagents Solvent Conditions Result
1 BCl3 CH2Cl2 -78 oC to rt Decomposition
2 MgI2 Et2O:THF 60 oC Decomposition
3 TMSI quinoline net 130 oC Decomposition

106
In summary, a simple and efficient method for preparing 2,2-dimethyl-2H-
chromenes via Pd(II)-catalysed Heck coupling has been developed. Importantly, this
protocol provides a facile synthesis of naturally occurring 2,2-dimethyl-2H-chromenes in
the late stage of synthesis of sorocein B.
Deprotection of endo-237, endo-241 and exo-242 with several reagents such as BCl3,
MgI2, TMSI quinoline were not successful, which only resulted in decomposition. At this
stage, global deprotection remains to be achieved. Effort towards synthesis of sorocien B
was underway.

107
CHAPTER 5
CONCLUSION AND FUTURE WORK
5.1 Conclusion
In conclusion, we have successfully developed a viable method for the synthesis of
morusalbanol A. A model study on construction of the oxabicylic [3.3.1] core system of
morusalbanol A have been developed. The required cis-trans Diels-Alder adduct precursor
of morusalbanol A was obtained via thermal cycloaddition reaction which was proven to be
dependent on the presence of a hydrogen-bonded ortho OH substituent on the chalcone
dienophile. Acid catalyzed intramolecular cyclization of a cis-trans Diels-Alder adduct
afforded the desired oxabicyclic [3.3.1] compound in a stereo-controlled manner.
Additionally, rotation about the C3´´-C3´ bond of a para methyl ether protected, cis-trans
Diels-Alder adduct (endo-186) was observed during acid catalyzed intramolecular
cyclization to form the respective oxabicyclic [3.3.1] core of morusalbanol A.
The strategies that developed from the model study were successfully applied to the
synthesis of ()-morusalbanol A methyl ethers. The key step involved a biomimetic
cycloaddition reaction between a chalcones dienophile and a dehydroprenyl diene.
Intramolecular cyclization of the cis-trans Diels-Alder adduct afforded (±)-morusalbanol A
methyl ethers (endo-204) in a stereo-selective manner. A number of key proton and carbon
signals in the NMR spectra of ()-morusalbanol A methyl ethers were found absent
presumably due to atroisomerism. Global demethylation on (±)-morusalbanol A methyl
ethers (endo-206 or endo-207) with MgI2, BCl3, and 1-trimethylsilylquinolinium iodide
(TMSI-quinoline) were unsuccessful.

108
During the course of synthesis of sorocein B, a method for preparing 2,2-dimethyl-
2H-chromenes via Pd(II)-catalyzed Heck coupling has been developed. The procedure is
simple and efficient. Importantly, the protocol provides a facile synthesis of naturally
occurring 2,2-dimethyl-2H-chromenes, including the precursor of sorocein B.
Similar strategy was used for construction of the cis-trans Diels-Alder precursor of
sorocein B. The thermal cycloaddition reaction between chalcones dienophile 89 and
dehydroprenyl diene 106 afforded the requisite cis-trans Diels-Alder precursor (endo-241)
in 35% yield along with the trans-trans Diels-Alder diastereomer. Subsequently PdCl2
catalyzed cyclization of the ortho-prenyl group of endo-241 afforded the required 2,2-
dimethylchromenyl ring in 230. However, to date attempts to remove the methyl ether
group of 230 to form sorocein B with BCl3, MgI2, and TMSI-quinoline were unsuccessful.
5.2 Suggestion for Future Work
Enantioselective total syntheses of other mulberry Diels-Alder adducts such as
Kuwanon I and J as well as Brosimones A and B utilized a chiral ligand/ boron Lewis acid
was recently reported by Lei’s group (Han, Jones & Lei, 2015). These chiral-boron
complexes promote asymmetric Diels-Alder cycloadition by significantly lower the energy
of the LUMO. Thus, we anticipates that these chiral-boron complexes chiral-boron
complexes would also be useful for the synthesis of morusalbanol A, sorocein B and other
related mulberry Diels-Alder adducts.
For the synthesis of morusalbanol A (42), diene 244 and dienophile 245 were
subjected to asymmetric Diels-Alder reaction as summarized in the Scheme 5.1. The chiral
ligand will strongly influenced the enantioslectivity of the cycloaddition reaction.

109
HO
OH
OH
MeO
O
O OH
OH
OH
HO
OH
HO
O
MeO
O
O
OH
OH
HO
HO
morusalbanol A (42)
Me
PgO OPg
OPg O
OMe
O OPg
PgO
OH
OPg
244
245
+
chiral-boron complex
O OPg
PgO
O
OPg
Me
PgOOPg
PgO O
OMesteric effect
Diels-Alder
B
OO
41
PhPh
Pg = protecting group
Scheme 5.1 Future work for enantioselective total syntheses of morusalbanol A (42)

110
Biosynthesis-inspired asymmetric Diels-Alder cycloaddition promoted by chiral
VANOL@ VAPOL/boron Lewis acid have been recently reported by Lei’s group (Gao,
Han & Lei, 2016). As depicted in Scheme 5.2, the endo-246 could be generated through
asymmetric Diels-Alder cycloadditions from diene 244 and dienophile 245 using (R)-
VANOL as a chiral ligand. With 246 in hand, sulphuric acid was employed to catalyze the
biomimetic intramolecular ketalisation. Following the Lei’s method for ketalisation and
applying our method for a late-stage pyranyl installation of 2,2-dimethylchromene ring on
248 will presumably help to furnish the enantioselective total synthesis of Sorocein B (62).
MOMO
MOMOO
OMOM
OMOM
AcO
OH O
I
OAc
OAc
endo-246
exo-247 (not shown for clarity)
O
HO
I
OAc
AcO
OAc
OMOM
OMOM
OOMOM
MOMO
+
i) (R)-Vanol, AcOH BH3.THF, rt, 25 minii) dienophile, rt, 1.5 h
iii) diene, rt
diene 244
dienophile 245
endo-24610% H2SO4
EtOH, rtO O
OH
OH
OOH
HO
OH
I
OH
HH
H
O O
OH
OH
OOH
HO
OH
O
HH
H
late stage pyran installation
our method
sorocein B (62)
248
Scheme 5.2. Future work for enantioselective total syntheses of sorocein B (62)

111
CHAPTER 6
EXPERIMENTALS
Chemical reagents were purchased from Aldrich, Acros and Merck and were used
as received. Anhydrous solvents were purchased from Merck. HPLC grade tetrahydrofuran,
methylene chloride, diethyl ether, toluene, and benzene were purified and dried by passing
through a PURESOLV® solvent purification system (Innovative Technology, Inc.).
Acetone (HPLC grade) was dried by distillation from activated molecular sieves 4Å. NMR
spectra were obtained using a Jeol ECA 400 (400 MHz) NMR spectrometer with TMS as
the internal standard. All measurements were accomplished in solution in DMSO-d6 or
CDCl3. Chemical shifts are reported in parts per million relative to CDCl3 or TMS. Data
for 1H NMR are reported as follows: chemical shift, integration, multiplicity (br s = broad
singlet, s = singlet, d = doublet, dd = doublet of doublets, ddd= doublet of doublet of
doublets, t = triplet, q = quartet, m = multiplet) and coupling constants. All 13
C NMR
spectra were recorded with complete proton decoupling. Analytical thin layer
chromatography (TLC) was carried out on Merck precoated aluminium silica gel sheets
(Kieselgel 60 F254). Visualization was accomplished under UV light. All products were
purified by silica gel column chromatography using a hexane/EtOAc/chloroform as eluent.
All target compounds were characterized by 1H,
13C, 2D NMR and HRMS (ESI) analyses.

112
Crystallography
Diffraction data for the crystal of 184 was collected on a Bruker SMART Apex II
CCD area-detector diffractometer (graphite-monochromatized Mo-Kα radiation, λ =
0.71073 Å) at 150(2) K. The orientation matrix, unit cell refinement and data reduction
were all handled by the Apex2 software (SAINT integration, SADABS multi-scan
absorption correction). Diffraction data for the crystal of 186, 206, 207 were collected on
an Agilent SuperNova Dual diffractometer with an Atlas detector (graphite-
monochromatized Mo-Kα radiation, λ = 0.71073 Å) at 100(2) K. The data were processed
using CrysAlisPro, Agilent Technologies, Version 1.171.37.34 (release 22-05-2014
CrysAlis171 .NET) and empirical absorption correction using spherical harmonics,
implemented in SCALE3 ABSPACK scaling algorithm (184, 186 and 207) or SADABS
(for 206). The structures were solved using the program SHELXT (Sheldrick, 2012), and
refined by the full matrix least-squares method on F2 with SHELXL-2014/7 (Sheldrick,
2008). All the non-hydrogen atoms were refined anisotropically. All the C-bound hydrogen
atoms were placed at calculated positions and were treated as riding on their parent atoms.
The O-bound hydrogen atoms were found in difference Fourier maps. For all hydrogen
atoms Uiso(H) were set to 1.2-1.5 times Ueq(carrier atom).
The structure of 186 was determined from a two-component twinned crystal with
the twin parameter refined to 0.477(1). The chloroform solvate molecule is disordered over
two positions in a 0.64/0.36 ratio. For the crystal structure of 207, the difference Fourier
map shows one relatively large peak, 1.46 e Å-3
, located at 1.02 Å from H14A and 1.72 Å
from C14. The peak cannot be assigned to any chemical reasonable species in the structure
and could be due to an adventitious incorporation of an impurity during the crystallization
process.

113
Drawing of the molecule was produced with XSEED (Barbour, 2001) (184 and 186),
Mercury (Macrae et al. 2008) (206 and 207). Crystal data and structural refinement
parameters for 184 and 186 are given in Table 3.2, 206 and 207 are given in Table 3.3.
Computational Methodology
Molecular structure for the mechanism study was obtained from trans-chalcone
(CID=637760) with modification. Geometry of the modified structure was then optimized
by using gaussian 09 programme (Frisch et al. 2009). Density Functional Theory (DFT)
method at the B3LYP/6-31G(d,p) level (Becke, 1993; Lee, Yang, and Parr 1988). The
transition state (TS) investigation of the reactants and each of the possible products were
then performed on Material Studio programme 4.3. Geometry optimisation and TS
calculation were carried out using synchronous transit method that embedded in Dmol3
module (Delley, 2000; Delley, 1990) DFT with local density approximation (LDA) of local
functional PWC19
were employed in all the calculations considering the effective core
potential treatment along with the DN or DND basis set. Linear synchronous transit (LST)
accounted for the reaction paths. Meanwhile, optimisation calculation performs a single
interpolation to a maximum energy prior to quadratic synchronous transit (QST) method for
obtaining an energy maximum with constrained minimizations in order to refine the
transition state to a high degree (Perdew & Wang, 1992) Also, each point was treated with
another conjugate gradient minimization. The cycle keeps going until a stationary point is
located or the number of the allowed QST steps has been fully exhausted. The highest
energy points were then optimised to the closest transition state (TS) after the convergence
of the initial paths. Minimum energy path (MEP) between the critical points were
calculated with nudged elastic band (NEB) under TS Optimisation function to ensure the
continuity of the path and projection of the force in order to converge the system to the

114
MEP. On the other hand, potential energy scan was performed on the dihedral angle where
rotations occur under gaussian 09 programme using semi-empirical calculation at PM6
level.
1-(4-(ethoxymethoxy)-3-iodo-2-methoxyphenyl)ethanone (171)
To a solution of 1-(2,4-dihydroxy-3-iodophenyl)ethanone 78 (1.2 g, 4.3 mmol) and K2CO3
(1.5 g, 2.5 equiv) in dry acetone (150 mL) at -10oC was added EOM-Cl (0.44 mL, 4.7
mmol). The reaction mixture was stirred for 15 h. The mixture was diluted with EtOAc,
washed with a sat. NH4Cl aq. Solution, water, and dried over Na2SO4. The crude residue
was purified by flash chromatography eluting with 20% EtOAc/hexane to give 1-(4-
(ethoxymethoxy)-2-hydroxy-3-iodophenyl)ethanone (1.04 g, 72%) as colourless oil. 1H
NMR (400 MHz, CDCl3): δ 13.46 (s, OH, 1H), 7.61 (d, J = 8 Hz, H6, 1H), 6.59 (d, J = 8
Hz, H5, 1H), 5.30 (s, OCH2O, 2H), 3.77 (m, OCH2CH3, 2H), 2.55 (s, Ac, 3H), 1.15 (t, J =
6.9 Hz, OCH2CH3, 3H); 13
C NMR (100 MHz, CDCl3): δ 202.7, 163.4, 162.6, 132.5, 114.9,
105.6, 93.5, 78.1, 65.3, 26.3, 15.1; HRMS calcd for C11H14IO4 [M+H]+
336.9931. Found:
336.9926.
To a solution of 1-(4-(ethoxymethoxy)-2-hydroxy-3-iodophenyl)ethanone (426 mg, 1.27
mmol) and K2CO3 (0.44g, 2.5 equiv) in dry acetone (10 mL) was added dimethyl sulfate
(0.13 ml, 1.1 equiv). The mixture was stirred at r.t. for 12 h. Then, K2CO3 was filtered and

115
the mixture was diluted with EtOAc. The organic phase was extracted with water. The
organic layer was collected, washed with brine, dried on Na2SO4 and evaporated. The
residue was subjected to column chromatography (hexane: EtOAc = 8:2) to afford the
desired compound 171 (378 mg, 85%) as colourless viscous liquid. 1H NMR (400 MHz,
CDCl3): δ 7.61 (d, J = 8.7 Hz, H6, 1H), 6.85 (d, J = 8.7 Hz, H5, 1H), 5.28 (s, OCH2O, 2H),
3.78 (s, OCH3, 3H), 3.71 (m, OCH2CH3, 2H), 2.56 (s, Ac, 3H), 1.15 (t, J = 7.3 Hz,
OCH2CH3, 3H); 13
C NMR (100 MHz, CDCl3): δ 198.1, 160.9, 160.8, 131.9, 127.3, 110.2,
93.6, 86.4, 65.1, 62.6, 24.9, 15.1; HRMS calcd for C12H16IO4 [M+H]+
351.0088. Found:
351.0094.
(E)-1-(4-(ethoxymethoxy)-2-methoxy-3-(3-methylbuta-1, 3-dienyl)phenyl)ethanone
(172)
To a mixture of 2-methyl-3-buten-2-ol (82) (0.52 ml, 5 mmol), Pd(Cl)2 (10 mg, 0.05
mmol) , nBu4NCl (300 mg, 1.08 mmol), NaHCO3 (227 mg, 2.70 mmol), and 171 (378 mg,
1.08 mmol) of in 3 mL of DMF in a pressure tube was heated to 100°C for 12 h. The
mixture was purified by column chromatography eluting with 40% EtOAc/ hexane to yield
a pale yellow viscous liquid (300 mg, 90%). To this liquid (300 mg, 0.97 mmol) in 10 mL
of benzene was added acetyl chloride (87 µL, 1.22 mmol, 1.25 equiv) and pyridine (102 µL,
1.26 mmol, 1.3 equiv). The mixture was then heated at 60°C for 6h. The white precipitate
was filtered, and the organic layers were collected and dried. The residue was purified by
column chromatography (hexane: EtOAc =7:3) to afford 254 mg of diene 1a (90% yield) as
pale yellow viscous liquid. 1H NMR (400 MHz, CDCl3): δ 7.54 (d, J = 9.2 Hz, H6, 1H),
7.25 (d, J = 16.9 Hz, H7, 1H), 6.94 (d, J = 9.2 Hz, H5, 1H), 6.67 (d, J = 16.5 Hz, H8, 1H),

116
5.29 (s, OCH2O, 2H), 5.09 (bs, H11, 2H), 3.73 (m, OCH2CH3, 2H), 3.72 (s, OCH3, 3H),
2.61 (s, Ac, 3H), 1.99 (s, CH3, 3H), 1.21 (t, J = 7.3 Hz, OCH2CH3, 3H); 13
C NMR (100
MHz, CDCl3): δ 199.2, 159.5, 143.1, 137.3, 129.7, 127.1, 120.9, 119.2, 117.7, 110.3, 93.4,
64.9, 61.9, 30.6, 18.3, 15.1; HRMS calcd for C17H23O4 [M+H]+
291.1591. Found: 291.1589.
Endo-179 and exo-180
Diene 172 (160 mg, 0.55 mmol) and dienophile 175 (112 mg, 0.50 mmol) was dissolved in
freshly distilled toluene (1 mL) in a pressure tube. The mixture was heated at 150 oC for 24
h, which afforded a mixture of inseparable endo-179 and exo-180 (ratio 179:180 = 3:2, 140
mg, 54.5% yield). The mixture of endo-179 and exo-180 in 3M aqueous HCl in methanol
was heated for 20 min before addition of water. The residue was extracted with CH2Cl2 (2x).
The organic layers were combined, washed with brine, dried over MgSO4, filtered and
concentrated. The crude residue was purified by flash chromatography eluting with 30%
EtOAc/ hexane to give diastereomers 183 (62 mg) and 184 (50 mg)(90% combined yield)
as viscous liquid.
Diastereomer 183 was obtained as a pale yellow viscous liquid. 1H NMR (400 MHz,
CDCl3): δ 11.94 (s, OH, 1H), 7.89 (d, J = 8.3 Hz, H14”, 1H), 7.48 (d, J = 8.8 Hz, H6’, 1H),
7.40 (t, J = 7.5 Hz, H11”, 1H), 7.13-7.02 (m, H16”-H20”, 5H), 6.91-6.88 (m, H12”, H13”,
2H), 6.62 (d, J = 8.9 Hz, H5’, 1H), 4.11 (dd, J = 2.7, 11.6 Hz, H4”, 1H), 3.64 (d, J = 3.0 Hz,
H3”, 1H), 3.18 (ddd, J= 4.6, 12.5, 16.5 Hz, H5”, 1H), 3.13 (s, OCH3, 3H), 2.44 (s, Ac, 3H),

117
2.27-2.20 (m, H2”, H6”, 2H), 1.98-1.93 (m, H2”, H6”, 2H), 1.41 (s, CH3, 3H); 13
C NMR
(100 MHz, CDCl3): δ 204.4, 199.7, 162.8, 160.6, 159.0, 143.1, 135.8, 130.7, 128.7, 128.5,
128.4, 127.3, 126.6, 124.3, 120.0, 118.7, 118.6, 115.0, 112.0, 75.3, 62.0, 53.1, 48.3, 37.1,
36.1, 32.0, 29.7, 28.5; HRMS calcd for C29H29O5 [M+H]+
457.2010. Found: 457.2018.
Diastereomer 9a was obtained as a pale yellow viscous liquid. Recrystallisation in
methanol gave a pale yellow crystal in very small quantity (not enough for melting point
determination). 1H NMR (400 MHz, CDCl3): δ 12.45 (s, OH), 7.69 (d, J = 8 Hz, H14”, 1H),
7.45 (d, J = 8.8 Hz, H6’, 1H), 7.33 (t, J = 7.6 Hz, H11”, 1H), 7.02 (m, H17”, H19”, 2H),
6.93 (m, H16”, H18”, H20”, 3H), 6.87 (d, J = 8.4 Hz, H12”, 1H), 6.67 (t, J = 7.6 Hz, H13”,
1H), 6.54 (d, J = 8.8 Hz, H5’, 1H), 3.91 (d, J = 8.8 Hz, H4”, 1H), 3.46 (s, OCH3, 3H), 3.30
(br s, H3”, 1H), 3.24 (m, H5”, 1H), 2.50 (s, Ac, 3H), 2.28 (m, H2”, 2H), 2.17 (m, H6”, 2H),
1.55 (s, CH3, 3H); 13
C NMR (100 MHz, CDCl3): δ 209.0, 197.4, 162.0, 157.6, 157.3, 141.5,
135.6, 130.1, 129.4, 127.2, 125.9, 125.4, 122.9, 119.4, 117.7, 117.5, 117.0, 112.3, 74.4,
61.1, 52.4, 39.3, 37.3, 29.8, 28.9, 28.3, 28.2; HRMS calcd for C29H29O5 [M+H]+ 457.2010.
Found: 457.2022.
The procedure using AgOTf and AgBF4 for exo-adduct 180

118
To a solution of diene 172 (60 mg, 0.2 mmol) and dienophile 175 (46 mg, 0.2 mmol) in dry
CH2Cl2 (1mL) was added AgOTf (15 mg, 60 μmol). The reaction mixture was stirred at rt
for 1h. The crude residue was purified by flash chromatography with 30% EtOAc/ hexane
to give exo-180 (39 mg, 38% yield) as a pale yellow viscous liquid. 1H NMR (400 MHz,
CDCl3): δ 11.99 (s, OH, 1H), 7.75 (br d, J = 6.9 Hz, H14”, 1H), 7.37 (d, J = 9.1 Hz, H6’,
1H), 7.31 (t, J = 7.6 Hz, H11”, 1H), 7.02-7.19 (m, H16”, H17”, H19”, H20”, 4H), 6.76-6.73
(m, H12”, H13”, H5’, 3H), 5.49 (br s, H2”, 1H), 4.88 (br s, OCH2O, 2H), 4.55 (br s, H3”,
1H), 4.32 (t, J = 9.6 Hz, H4”, 1H), 3.92 (ddd, J = 6.9, 9.6, 16.0 Hz, H5”, 1H), 3.56 (m,
OCH2CH3, 2H), 3.44 (s, OCH3, 3H), 2.58 (dd, J = 9.6, 17.8, H6”, 1H), 2.44 (s, Ac, 3H),
2.31 (dd, J = 9.1, 17.8 Hz, H6”, 1H), 1.83 (s, CH3, 3H), 1.15 (t, J = 6.9 Hz, OCH2CH3, 3H);
13C NMR (100 MHz, CDCl3): δ 207.3, 200.5, 162.2, 160.1, 145.8, 135.6, 133.3, 130.4,
130.0, 128.5, 127.6, 126.3, 123.1, 122.0, 118.4, 118.0, 109.5, 92.9, 64.4, 63.2, 50.0, 38.9,
34.8, 29.8, 29.7, 23.6, 15.1; HRMS calcd for C32H35O6 [M+H]+ 515.2434, Found: 515.2440.
(E)-1-(2-hydroxy-4-methoxy-3-(3-methylbuta-1,3-dienyl)phenyl)ethanone (174)
To a mixture of 2-methyl-3-buten-2-ol (81) (0.72 ml, 6.85 mmol), Pd(Cl)2 (12 mg, 0.07
mmol), nBu4NCl (190 mg, 0.69 mmol), NaHCO3 (145 mg, 1.73 mmol), and 1-(2-hydroxy-
3-iodo-4-methoxyphenyl)ethanone (200 mg, 0.68 mmol) in 5 mL of DMF was heated at
100°C in a pressure tube for 4 h. The mixture was subjected to column chromatography

119
eluting with 40% EtOAc/ hexane to give a pale yellow liquid (144 mg, 85%). To this
yellow liquid (144 mg, 0.52 mmol) in 10 mL benzene was added acetyl chloride (48 µL,
0.68 mmol, 1.2 equiv) and pyridine (53 µL, 0.68 mmol, 1.2 equiv). The mixture was
heated at 80°C for 2h. Formation of white precipitate was filtered and the organic layer was
collected. The residue was purified by column chromatography (hexane: EtOAc =7:3) to
afford diene 174 (122 mg, 91% yield) as pale yellow viscous liquid. 1H NMR (400 MHz,
CDCl3): δ 13.32 (s, OH, 1H), 7.58 (d, J = 9.2 Hz, H6, 1H), 7.41 (d, J = 16.9 Hz, H7, 1H),
6.80 (d, J = 16.5 Hz, H8, 1H), 6.50 (d, J = 9.2 Hz, H5, 1H), 5.05 (s, H11, 2H), 3.92 (s,
OMe, 3H), 2.55 (s, Ac, 3H), 1.98 (s, Me, H10, 3H); 13
C NMR (100 MHz, CDCl3): δ 203.3,
163.3, 162.5, 143.5, 136.3, 131.0, 118.6, 117.1, 114.3. 113.9, 102.2, 56.0, 26.4, 18.4;
HRMS calcd for C14H17O3 [M+H]+
233.1172, Found: 233.1166.
Endo-181 and exo-182
Diene 174 (128 mg, 0.55 mmol) and dienophile 176 (149 mg, 0.50 mmol) was dissolved in
fresh distilled toluene (1 mL) in a pressure tube. The mixture was heated at 150 oC for 25 h,
which afforded a mixture of inseparable endo-181 and exo-182 (ratio 181:182 = 1:1, 130
mg, 49% yield). The mixture of endo-181 and exo-182 in 5 mL, 3M aqueous HCl in
methanol was heated for 20 min before addition of water. The residue was extracted with
CH2Cl2 (2x). The organic layers were combined, washed with brine, dried over MgSO4,
filtered and concentrated. The crude residue was purified by flash chromatography eluting
with 30% EtOAc/ hexane to give diastereomer 186 (53 mg, 92% based on conversion of
endo-181, which was 65 mg) and the recovery of 182 (40 mg).

120
Diastereomer 186 was obtained as a pale yellow viscous liquid. Recrystallisation in
methanol gave a pale yellow crystal in very small quantity (not enough for melting point
determination). 1H NMR (400 MHz, CDCl3 + MeOD-d4): δ 12.03 (s, OH, 1H), 7.88 (d, J =
7.8 Hz, H14”, 1H), 7.58 (d, J = 8.9 Hz, H6’, 1H), 7.30 (t, J = 7.3 Hz, H11”, 1H), 6.84-6.74
(m, H16”, H17”, H18”, H19”, 4H), 6.51-6.49 (m, H12”, H13”, 2H), 6.20 (d, J = 8.7 Hz,
H5’, 1H), 4.50 (br s, H4”,1H), 3.64 (s, H3”, 1H), 3.30 (br s, H5”, 1H), 3.19 (s, OCH3, 3H),
2.49 (s, Ac, 3H), 2.17-2.14 (m, H2”, H6”, 2H), 1.96 (m, H2”, 1H), 1.78 (d, J =12.8, H6”,
1H), 1.36 (s, CH3, 3H); 13
C NMR (100 MHz, CDCl3): δ 206.0, 199.9, 161.9, 160.8, 157.6,
154.6, 135.7, 130.9, 129.1, 128.8, 127.2, 119.8, 119.7, 119.4, 119.0, 117.9, 115.9, 110.3,
101.4, 76.1, 54.6, 50.8, 44.8, 35.3, 31.9, 31.1, 28.4 (C5 was not observed); HRMS calcd for
C29H29O6 [M+H]+
473.1959. Found: 473.1967.
exo-189 was obtained as a pale yellow viscous liquid. 1H NMR (400MHz, CDCl3): δ 13.0
(s, OH, 1H), 11.95 (s, OH, 1H), 7.33-7.28 (m, H6’, H14”, 2H), 7.17 (d, J = 9.7, H16”, 1H),
7.05 (t, J = 7.8 Hz, H11”, 1H), 6.95-6.88 (m, H19”, 1H), 6.71 (m, H12”, H5’, 2H), 6.56 (d,
J = 8.2 Hz, H17”, 1H), 6.28 (t, J = 7.4 Hz, H13”, 1H), 5.97 (d, J = 8.8 Hz, H18”, 1H), 5.26

121
(s, H2”, 1H), 4.56 (br s, H4”, 1H), 4.27 (br d, H3”, 1H), 3.89 (m, H5”, 1H), 3.53 (s, OCH3,
1H), 2.43 (s, Ac, 3H), 2.17 (m, H6”, 2H), 1.71 (s, CH3, 3H); 13
C NMR (100 MHz, CDCl3):
δ 212.1, 203.2, 163.0, 162.9, 161.6, 153.3, 136.2, 132.3, 131.4, 130.4, 127.6, 123.2, 121.4,
120.8, 118.0, 117.5, 117.2, 117.1, 114.3, 101.8, 55.8, 55.6, 48.5, 38.6, 36.8, 29.8, 26.3, 23.3;
HRMS calcd for C29H29O6 [M+H]+
473.1959. Found: 473.1959.
(E)-methyl-2,4,6-tri(ethoxymethoxy)-3-(3-methylbuta-1,3-dienyl)benzoate (190)
To a mixture of 2-methyl-3-buten-2-ol (82) (0.65 ml, 6.25 mmol), Pd(OAc)2 (29 mg, 0.13
mmol), K2CO3 (345 mg, 2.50 mmol), and 196 (530 mg, 1.25 mmol) of in 1 mL of DMF in
a pressure tube was heated to 100°C for 7 h. The mixture was purified by column
chromatography eluting with 40% EtOAc/ hexane to yield a pale yellow viscous liquid
(387 mg, 70%). To this liquid (387 mg, 0.88 mmol) in 10 mL of benzene was added acetyl
chloride (63 µL, 0.88 mmol, 1.00 equiv) and pyridine (71 µL, 0.88 mmol, 1.00 equiv). The
mixture was then heated at 60°C for 6h. The white precipitate was filtered, and the organic
layers were collected and dried. The residue was purified by column chromatography
(hexane: EtOAc =7:3) to afford 280 mg of diene 190 (75% yield) as pale yellow viscous
liquid. 1H NMR (400 MHz, CDCl3): δ 7.13 (d, J = 16.4 Hz, H7, 1H), 6.83 (s, H5, 1H), 6.59
(d, J = 16.4 Hz, H8, 1H), 5.23 (s, OCH2O, 2H), 5.18 (s, OCH2O, 2H), 5.01 (br s, H11, 2H),
5.00 (s, OCH2O, 2H), 3.87 (s, OCH3, 3H), 3.70 (m, OCH2CH3, 6H), 1.93 (s, CH3, 3H), 1.19
(m, OCH2CH3, 9H); 13
C NMR (100 MHz, CDCl3): δ 166.6, 157.7, 154.3, 153.8, 143.1,

122
135.8, 119.4, 116.8, 115.1, 114.1, 99.1, 98.8, 93.7, 93.6, 65.8, 64.5, 64.4, 52.3, 18.2, 15.1,
15.0, 14.9; HRMS calcd for C22H33O8 [M+H]+
425.2167. Found: 425.2169.
(E)-3-(2,3-bis(ethoxymethoxy)phenyl)-1-1(4-(ethoxymethoxy)-2-hydroxyphenyl)prop-
2-en-1-one (192)
To a solution of acetonphenone 197 (1.1 g, 5 mmol) and benzaldehyde 198 (1.3 g, 5 mmol)
in EtOH (25 mL) was added 50% aqueous KOH (84 mg, 15 mmol). The reaction mixture
was refluxed for 42 h before the addition of water and CH2Cl2. The aqueous phase was
extracted with CH2Cl2 and the combined organic layer were washed with brine, dried over
MgSO4, filtered and concentrated. The crude residue was purified by flash chromatography
eluting with 20% EtOAc/hezane to afford 192 (1.23 g, 55%) as yellow viscous liquid. 1H
NMR (400 MHz, CDCl3): δ 13.48 (s, OH, 1H), 8.17 (d, J = 15.6 Hz, H8, 1H), 7.82 (d, J =
9.2 Hz, H3, 1H), 7.58 (d, J = 16.0 Hz, H9, 1H), 7.53 (d, J = 7.3 Hz, H12, 1H), 6.89 (d, J =
2.8 Hz, H6, 1H), 6.74 (dd, J = 2.8, 9.2 Hz, H5, 1H), 6.63 (d, J = 2.3 Hz, H14, 1H), 6.57 (dd,
J = 2.7, 9.2 Hz, H15, 1H), 5.32 (s, OCH2O, 2H), 5.26 (s, OCH2O, 2H), 5.25 (s, OCH2O,
2H), 3.74 (m, OCH2CH3, 6H), 1.23 (m, OCH2CH3, 9H); 13
C NMR (100 MHz, CDCl3): δ
192.6, 166.2, 163.6, 160.9, 158.2, 140.4, 131.3, 130.1, 118.6, 118.4, 115.1, 109.5, 108.1,
104.0, 103.5, 93.5, 93.1, 92.9, 64.9, 64.8, 64.6, 15.2; HRMS calcd for C24H31O8 [M+H]+
447.2011. Found: 447.2025.

123
Methyl-3-iodo-2,4,6-trimethoxybenzoate (203)
To a solution of 202 (800 mg, 2.37 mmol) and K2CO3 (490 mg, 1.5 equiv) in dry acetone
(50 mL) was added iodomethane (0.16 mL, 1.1 equiv). The mixture was stirred at r.t. for 15
h. Then, K2CO3 was filtred and the organic phase was extracted with EtOAc and water. The
organic layers were collected, washed with brine, dried on MgSO4 and evaporated. The
residue was subjected to column chromatography (hexane: EtOAc= 8:2) to afford 792 mg
compound 203 (95% yield) as colorless viscous liquid. 1H NMR (400 MHz, CDCl3): δ 6.21
(s, H5, 1H), 3.84-3.79 (2s, 4xOMe, 12H); 13
C NMR (100 MHz, CDCl3): δ 165.9, 160.7,
158.9, 158.5, 111.6, 91.5, 72.7, 62.1, 56.5, 56.1, 52.4; HRMS calcd for C11H14IO5 [M+H]+
352.9878. Found: 352.9882.
(E)-methyl-2,3,6-trimethoxy-3-(3-methylbuta-1,3-dienyl)benzoate (191)
To a mixture of 2-methyl-3-buten-2-ol (1.5 ml, 14.2 mmol), Pd(OAc)2 (32 mg, 0.14 mmol),
K2CO3 (400 mg, 2.84 mmol), and 203 (500 mg, 1.42 mmol) of in 1 mL of DMF in a
pressure tube was heated to 100°C for 6 h. The mixture was purified by column
chromatography eluting with 50% EtOAc/hexane to yield pale yellow viscous liquid (242
mg, 55%). To this liquid (242mg, 0.78 mmol) in 10 mL of benzene was added acetyl

124
chloride (69 µL, 0.87 mmol, 1.1 equiv), and pyridine (61 µL, 0.87 mmol, 1.1 equiv). The
mixture was then heated to 60°C for 4h. The white precipitate was filtered, and the organic
layers were collected, dried. The residue was purified by column chromatography (hexane:
EtOAc =7:3) to afford 200 mg of diene 191 (90% yield) as pale yellow viscous liquid. 1H
NMR (400 MHz, CDCl3): δ 7.19 (d, J = 16.5 Hz, H7, 1H), 6.61 (d, J = 16.9 Hz, H8, 1H),
6.26 (s, H5, 1H), 5.03 (d, J = 7.8 Hz, H11, 2H), 3.9-3.7 (4s, 4xOMe, 12H), 1.96 (s, CH3,
3H); 13
C NMR (100 MHz, CDCl3): δ 167.0, 160.3, 157.4, 156.7, 143.4, 135.0, 119.0, 116.7,
112.7, 111.5, 91.6, 62.0, 56.1, 55.9, 52.6, 18.4; HRMS calcd for C16H20O5 [M+H]+
293.1381. Found: 293.1388.
Endo-204 and exo-205
Diene 191 (100 mg, 0.34 mmol) and dienophile 88 (157 mg, 0.34 mmol) were dissolved in
freshly distilled toluene (2 mL) in a pressure tube. The mixture was heated at 135oC for
24h to give a mixture of diastereomers 204 and 205. The mixture of diastereomers 204 and
205 was separated by column chromatography with 80% hexane/EtOAc as eluent to give a
mixture of diastereoisomers 204 (74 mg) and 205 (50 mg) (55% combined yield).
Endo-204 was obtained as a pale yellow viscous liquid. 1H NMR (400 MHz, CDCl3): δ
12.87 (s, OH, 1H), 7.51 (bd, H14”, 1H)*, 6.99 (d, J = 8.8 Hz, H20”, 1H), 6.33 (bd, H11”,
H13”, 2H)*, 6.24 (bd, H17”, H19”, 2H)*, 5.88 (s, H5’, 1H), 5.41 (s, H2”, 1H), 4.23 (bs,
H4”, 1H), 3.84-3.68 (5s, 5xOMe, 15H), 3.40 (s, OMe, 3H), 3.31(s, OMe, 3H), 2.59 (m,

125
H6”, 1H), 2.22(m, H6”, 1H), 1.74 (s, CH3, 3H); 13
C NMR (100 MHz, CDCl3): δ 206.5,
167.5, 165.0, 164.8, 160.9, 159.1, 157.9, 157.2, 132.8, 131.6, 126.8, 122.5, 115.3, 115.2,
110.1, 106.3, 104.2, 100.6, 98.9, 90.7, 62.0, 56.0, 55.5, 55.3, 54.7, 52.4, 47.7, 38.4, 36.0,
23.6; HRMS calcd for C34H38O10Na [M+Na]+ 629.2363. Found: 629.4568.
*coupling constant cannot be measured due to broad resonance
Exo-205 was obtained as a pale yellow viscous liquid. 1H NMR (400 MHz, CDCl3): δ
13.00 (1H, s), 7.39 (bd, H14”, 1H)*, 6.96 (d, J = 8.5 Hz, H20”, 1H), 6.25 (bd, H13”, H11”,
2H)*, 6.21 (bd, H17”, H19”, 2H)*, 6.12 (bd, H2”, 1H)*, 5.82 (s, H5’, 1H), 5.24 (s, H5”,
1H), 4.29 (bd, J = 9.8 Hz, H4”, 1H), 3.92 (s, OMe, 3H), 3.74-3.64 (6s, 6xOMe, 18H), 2.17
(m, H6”, 1H), 1.98 (m, H6”,1H), 1.63 (s, CH3, 3H); 13
C NMR (100 MHz, CDCl3): δ 207.7,
166.2, 164.2, 163.3, 158.5, 17.9, 157.1, 156.1, 131.4, 130.8, 130.5, 123.7, 123.2, 116.6,
114.4, 113.0, 105.8, 105.3, 103.2, 98.9, 97.8, 89.8, 61.0, 54.9, 54.3, 54.1, 51.4, 46.6, 37.3,
30.9, 28.7, 22.2; HRMS calcd for C34H38O10Na [M+Na]+ 629.2363. Found: 629.2402.
Morusalbanol A pentamethyl ether (206) and 207
To a solution of freshly prepared MgI2 (0.23 g, 0.82 mmol) in dry Et2O (10 mL) was added
endo-204 (0.05 g, 0.08 mmol) in THF (10 mL). The mixture was refluxed at 50 oC for 4 h.
Then, the precipitate was filtered, and the organic layers were collected and dried. The
residue was purified by column chromatography (hexane: EtOAc =8:2) to afford 25 mg of

126
Morusalbanol A pentamethyl ether (206) (50% yield) as colorless crystal in very small
quantity (not enough for melting point determination).
Morusalbanol A pentamethyl ether (206)
1H NMR (400 MHz, CDCl3): δ 12.75 (s, OH, 1H), 7.87 (d, J = 9.1 Hz, H14”, 1H), 6.90 (d,
J = 8.2 Hz, H20”, 1H), 6.50 (dd, J = 2.8, 9.1 Hz, H13”, 1H), 6.39 (d, J = 2.3 Hz, H11”, 1H),
6.31 (d, J = 2.3 Hz, H17”, 1H), 6.25 (dd, J = 2.3, 8.2 Hz, H19”, 1H), 5.88 (s, H5’, 1H), 4.37
(bs, H4”, 1H), 3.88-3.68 (5s, 5xOMe, 15H), 3.65 (m, H3”, 1H), 3.38 (s, OMe, 3H), 2.19
(m, H6”, 1H), 2.11(dd, J = 2.8, 13.3 Hz, H2”, 1H), 1.89 (m, H2”, 1H), 1.36 (s, H7”, 3H);
13C NMR (100 MHz, CDCl3): δ 204.1, 167.5, 165.4, 165.3, 159.1, 158.5, 158.3, 157.5,
130.5, 125.6, 114.3, 107.1, 104.1, 103.0, 101.1, 99.1, 86.1, 75.6, 55.9, 55.6, 55.5, 55.3, 54.7,
52.2, 50.6, 45.5, 36.3, 31.2, 28.6; HRMS calcd for C33H37O10 [M+H]+
593.2378. Found:
593.2360.
*coupling constant cannot be measured due to broad resonance
1H NMR (400 MHz, toluene-d8): δ 13.36 (s, OH, 1H), 7.79 (d, J = 9.2 Hz, H14”, 1H), 6.83
(d, J = 8.2 Hz, H20”, 1H), 6.45 (d, J = 8.7Hz, H13”, 1H), 6.35 (d, J = 2.3 Hz, H11”, 1H),
6.16 (d, J = 2.3 Hz, H17”, 1H), 6.06 (dd, J = 1.8, 8.2 Hz, H19”, 1H), 5.60 (s, H5’, 1H), 4.21
(m, H4”, 1H), 3.72-3.21 (5s, 5xOMe, 15H), 3.72 (m, H3”, 1H), 3.18 (s, OMe, 3H), 2.30
(bd, H6”, 1H)*, 1.71 (bd, J = 11.9 Hz, H2”, 1H), 1.60 (bd, J = 12.8 Hz, H2”, 1H), 1.26 (s,
H7”, 3H); 13
C NMR (100 MHz, toluene-d8): δ 203.9, 166.3, 166.2, 165.4, 159.3, 158.4,

127
158.3, 157.6, 130.3, 127.6, 125.5, 114.5, 107.2, 105.5, 104.0, 103.2, 100.9, 99.1, 86.2, 75.1,
54.8, 54.6, 54.2, 54.1, 51.2, 51.0, 45.8, 36.0, 31.3, 28.4.
1H NMR (400 MHz, toluene-d8, 353K): δ 13.09 (s, OH, 1H), 7.79 (d, J = 8.7 Hz, H14”,
1H), 6.84 (d, J = 8.2 Hz, H20”, 1H), 6.46 (dd, J = 2.3, 8.7 Hz, H13”, 1H), 6.33 (d, J = 2.7
Hz, H11”, 1H), 6.18 (d, J = 2.3 Hz, H17”, 1H), 6.10 (dd, J = 2.3, 8.2 Hz, H19”, 1H), 5.68 (s,
H5’, 1H), 4.26 (d, J = 11.4 Hz, H4”, 1H), 3.72-3.26 (5s, 5xOMe, 15H), 3.72 (m, H3”, 1H),
3.20 (s, OMe, 3H), 2.26 (ddd, J = 1.8, 4.1, 13.8 Hz, H6”, 1H), 1.80 (dd, J = 3.2, 13.2 Hz,
H2”, 1H), 1.68 (m, H2”, 1H), 1.26 (s, H7”, 3H); 13
C NMR (100MHz, toluene-d8, 353K): δ
204.2, 166.6, 166.4, 166.0, 165.4, 159.1, 158.9, 158.4, 130.3, 127.6, 125.8, 115.3, 107.4,
106.6, 105.3, 104.1, 101.8, 100.2, 87.5, 75.5, 55.8, 55.3, 55.0, 54.9, 54.6, 51.8, 51.3, 46.5,
36.8, 31.8, 28.7.
Compound 207
To a solution of freshly prepared MgI2 (0.23 g, 0.82 mmol) in dry Et2O (10 mL) was added
to a solution of endo-204 (0.05 g, 0.08 mmol) in THF (10 mL). The mixture was refluxed at
50oC for 6 h. Then, the precipitate was filtered, and the organic layers were collected and
dried. The residue was purified by column chromatography (hexane: EtOAc =8:2) to afford
20 mg of 206 (40% yield) and 18 mg of 207 (36% yield) as colorless crystals in very small
quantity (not enough for melting point determination).

128
1H NMR (400 MHz, CDCl3): δ 12.62 (s, OH, 1H), 12.37 (s, OH, 1H), 11.38 (bs, COOH,
1H), 7.82 (d, J = 9.1 Hz, H14”, 1H), 6.88 (d, J = 8.7 Hz, , H20”, 1H), 6.50 (dd, J = 1.8, 8.7
Hz, H13”, 1H), 6.38 (d, J = 2.3 Hz, H11”, 1H), 6.33 (d, J = 2.3 Hz, H17”, 1H), 6.26 (dd, J
= 2.3, 8.2 Hz, H19”, 1H), 6.02 (s, H5’, 1H), 4.37 (bd, H4”, 1H)*, 3.88-3.69 (3s, 3xOMe,
9H), 3.68 (m, H3”, 1H), 3.38 (s, OMe, 3H), 2.29 (bd, J = 12.8 Hz, H6”, 1H), 2.22 (dd, J =
2.7, 13.1 Hz, H2”, 1H), 1.97 (dt, J = 12.8 Hz, H2”, 1H), 1.56 (s, H7”, 3H); 13
C NMR (100
MHz, CDCl3): δ 203.3, 171.3, 165.7, 165.5, 165.0, 162.3, 159.3, 158.2, 155.7, 130.3, 122.5,
114.1, 107.5, 104.2, 102.0, 101.1, 99.2, 94.1, 92.8, 80.3, 55.6, 55.5, 55.3, 55.2, 49.8, 44.9,
35.9, 30.3, 28.6; HRMS calcd for C31H32O10Na [M+Na]+
587.1893. Found: 587.1890.
1H NMR (400 MHz, DMSO-d6): δ 12.58 (s, OH, 1H), 12.13 (s, OH, 1H), 8.19 (d, J = 8.7
Hz, H14”, 1H), 6.98 (d, J = 8.7 Hz, H20”, 1H), 6.56 (dd, J = 2.3, 9.2 Hz, H13”, 1H), 6.40
(d, J = 2.3 Hz, H11”, 1H), 6.36 (d, J = 2.3 Hz, H17”, 1H), 6.27 (dd, J = 2.3, 8.7 Hz, H19”,
1H), 5.91 (s, H5’, 1H), 4.50 (bd, J = 11.9 Hz, H4”, 1H), 3.78-3.61 (3s, 3xOMe, 9H), 3.45
(m, H3”, 1H), 3.23 (s, OMe, 3H), 2.38 (bd, J = 11.4 Hz, H2”, 1H), 2.06 (bd, H6”, 1H),
1.69 (d, J = 11.9 Hz, H2”, 1H), 1.35 (s, H7”, 3H); 13
C NMR (100 MHz, DMSO-d6): δ
205.0, 172.4, 165.8, 164.9, 163.5, 161.7, 161.7, 159.1, 157.5, 158.1, 124.6, 132.2, 114.3,
107.5, 105.0, 102.6, 101.4, 99.0, 96.0, 77.6, 56.2, 56.0, 55.4, 50.3, 46.2, 35.1, 30.9, 28.6.

129
1H NMR (400MHz, DMSO-d6, 353K): δ 12.52 (s, OH, 1H), 12.43 (s, OH, 1H), 8.12 (d, J =
8.7 Hz, H14”, 1H), 6.94 (d, J = 8.7 Hz, H20”, 1H), 6.55 (d, J = 7.6 Hz, H13”, 1H), 6.37 (d,
J = 2.5 Hz, H11”, 1H), 6.37 (d, J = 2.5 Hz, H17”, 1H), 6.27 (d, J = 8.5 Hz, H19”, 1H), 5.94
(s, H5’, 1H), 4.47 (bd, J = 11.2 Hz, H4”, 1H), 3.80-3.62 (3s, 3xOMe, 9H), 3.35 (m, H3”,
1H), 3.28 (s, OMe, 3H), 2.39 (bd, J = 11.4 Hz, H2”, 1H), 2.09 (bd, H6”, 1H)*, 1.70 (m,
H2”, 1H), 1.40 (s, H7”, 3H). 13
C NMR (100 MHz, DMSO-d6, 353K) not measured.
*coupling constant cannot be measured due to broad resonance
Experiment for compound 224, 226, 227 and 209
A mixture of 2-halophenols (1 eqv.), Pd(OAc)2 (10 mol%), K2CO3 (2 eqv.), and 2-methyl-
3-buten-2-ol (10 eqv.), in 1 mL of DMF in a pressure tube was heated to 100°C for 6 h. The
reaction was monitored by TLC under the starting material completely consumed. 1g of
silica gel then was added to the same mixture, followed by heating at 130°C for another
12h to yield desired product.
8-chloro-2,2-dimethyl-2H-chromene-6-carbaldehyde (224)
1H NMR (400MHz, CDCl3): δ 9.79 (1H, s, CHO), 7.81 (1H, s, phenyl), 7.62 (1H, s,
phenyl), 6.57 (1H, d, J = 9.8 Hz, CH), 5.95 (1H, d, J = 10.1 Hz, CH), 1.46 (6H, s, 2Me);
13C NMR (100MHz, CDCl3): δ 190.5, 153.1, 132.5, 131.3, 130.1, 125.8, 122.5, 120.7,
120.6, 79.5, 28.1; HRMS calcd for C12H12ClO2[M+H]+
223.0518. Found: 223.0512.

130
Methyl 2,2-dimethyl-2H-chromene-8-carboxylate (226)
1H NMR (400MHz, CDCl3): δ 7.47 (dd, J = 1.8, 7.9 Hz, phenyl, 1H), 7.25 (dd, J = 1.8, 7.3
Hz, phenyl, 1H), 6.44 (d, J = 9.8 Hz, CH, 1H), 5.83 (d, J = 10.1 Hz, CH, 1H), 3.77 (s, OMe,
3H), 1.38 (s, 2Me, 6H); 13
C NMR (100MHz, CDCl3): δ 165.7, 152.0, 131.7, 130.0, 122.2,
121.5, 120.1, 119.2, 76.8, 51.7, 27.5; HRMS calcd for C13H15O3[M+H]+
219.1013. Found:
219.1021.
Methyl-5,7-dimethyoxy-2,2-dimethyl-2H-chromene-8-carboxylate (227)
1H NMR (400MHz, CDCl3): δ 6.48 (d, J = 9.8 Hz, CH, 1H), 6.27 (s, phenyl, 1H), 5.86 (d, J
= 10.4 Hz, CH, 1H), 3.84-3.72 (3s, 3OMe, 9H), 1.36 (s, 2Me, 6H); 13
C NMR (100MHz,
CDCl3): δ 165.5, 157.3, 156.2, 150.9, 127.1, 115.8, 105.4, 103.2, 88.6, 76.4, 55.8, 51.7,
27.1; HRMS calcd for C15H19O5[M+H]+
279.1224. Found: 279.1223.
(E)-3-(2,4-dimethyoxyphenyl)-1-(5-methoxy-2,2-dimethyl-2H-chromen-8-yl)prop-2-
en-1-one (209)

131
1H NMR (400MHz, CDCl3): δ 7.72 (d, J = 16.0 Hz, Hβ, 1H), 7.56 (d, J = 9.16 Hz, H5’,1H),
7.52-7.47 (overlap, Hα, H6’,2H), 6.65 (d, J = 9.16 Hz, H6, 1H), 6.60-6.56 (overlap, CH, H3,
H5, 3H), 5.73 (d, J= 10.1 Hz, CH, 1H), 3.83-3.80 (3s, 3OMe, 9H), 1.40 (s, 2Me, 6H); 13
C
NMR (100MHz, CDCl3): δ 189.7, 163.2, 160.3, 158.2, 153.0, 137.1, 131.6, 130.4, 129.9,
125.2, 122.0, 116.6, 106.8, 104.6, 99.1, 77.2, 56.5, 56.3, 56.0, 27.9. HRMS calcd for
C23H25O5[M+H]+
381.1694. Found: 381.1706.
(E)-3-(2,4-bis(ethoxymethoxy)phenyl)-1-(4-(ethoxymethoxy)-3-iodo-2-
methoxyphenyl)prop-2-en-1-one (232)
To a solution of acetophenone 171 (1.51 g, 4.5 mmol) in ethanol (2.5mL/mmol), 50% KOH
(3 eq) was added. After 10 min appropriated benzaldehyde 198 (1.08 g, 4.5 mmol) was
added and the solutions were stirred at room temperature for 40 h. After cooling the
reaction mixtures with ice, the mixtures were neutralized carefully with 1N hydrochloric
acid. The crude mixture was extracted with ethyl acetate, washed with water and brine
afforded chalcones 232 (1.32g, 50% yield). 1H NMR (400 MHz, CDCl3): δ 8.04 (d, J =
16.2 Hz, Hβ, 1H), 7.64 (d, J = 8.6 Hz, H6’, 1H), 7.57 (d, J = 8.8 Hz, H6, 1H), 7.44 (d, J =
15.9 Hz, Hα, 1H), 6.92 (d, J = 8.8 Hz, H5’, 1H), 6.86 (d, J = 2.4 Hz, H3, 1H), 6.70 (dd, J =
2.4 Hz, 8.8 Hz, H5, 1H), 5.34 (s, OCH2O, 2H), 5.27 (s, OCH2O, 2H), 5.22 (s, OCH2O, 2H),
3.76 (s, OMe, 3H), 3.74(m, OCH2CH3, 6H), 1.22 (m, OCH2CH3, 9H); 13
C NMR (100 MHz,
CDCl3): δ 191.3, 160.9, 160.6, 160.4, 158.1, 139.5, 132.3, 129.7, 128.3, 124.1, 118.6, 110.4,
109.6, 103.6, 93.9, 93.6, 93.2, 86.5, 65.2, 64.9, 64.7, 63.0, 15.3; HRMS calcd for C25H32IO8
[M-H]+
585.0994. Found: 585.2892.

132
(E)-3-(2,4-bis(ethoxymethoxy)phenyl)-1-(4-(ethoxymethoxy)-2-methoxy-3-((E)-3-
methylbutan-1,3-dienyl)phenyl)prop-2-en-1-one (233)
To a mixture of 2-methyl-3-buten-2-ol (1.8 ml, 17.1 mmol), Pd(OAc)2 (38 mg, 0.17 mmol),
K2CO3 (472 mg, 3.42 mmol), and 232 (1.0 g, 1.71 mmol) of in 1 mL of DMF in a pressure
tube was heated to 100°C for 6 h. The mixture was purified by column chromatography
eluting with 50% EtOAc/hexane to yield pale yellow viscous liquid (698 mg, 75%). To this
liquid (698 mg, 1.28 mmol) in 10 mL of benzene was added acetyl chloride (0.11 mL, 1.41
mmol, 1.1 equiv), and pyridine (0.10 mL, 1.41 mmol, 1.1 equiv). The mixture was then
heated to 60°C for 4h. The white precipitate was filtered, and the organic layers were
collected, dried. The residue was purified by column chromatography (hexane: EtOAc =7:3)
to afford 495 mg of diene 233 (55% yield, over two steps) as pale yellow viscous liquid. 1H
NMR (400 MHz, CDCl3): 1
H NMR (400 MHz, CDCl3): δ 7.98 (d, J = 15.6 Hz, Hβ, 1H),
7.56 (d, J = 8.3 Hz, H6’, 1H), 7.48 (d, J = 8.8 Hz, H6, 1H), 7.44 (d, J = 16.1 Hz, Hα, 1H),
7.31 (d, J = 16.6 Hz, H7’, 1H), 6.96 (d, J = 9.8 Hz, H5’, 1H), 6.85 (d, J = 2.4 Hz, H3, 1H),
6.74 (d, J = 16.6 Hz, H8’, 1H), 6.70 (dd, J = 2.4 Hz, 8.8 Hz, H5, 1H), 5.30 (s, OCH2O, 2H),
5.26 (s, OCH2O, 2H), 5.21 (s, OCH2O, 2H), 3.72(m, OCH2CH3, 6H), 3.69 (s, OMe, 3H),
1.99 (s, H10’, 3H), 1.20 (m, OCH2CH3, 9H); 13
C NMR (100 MHz, CDCl3): δ 192.5, 160.5,
158.8, 158.7, 157.9, 143.2, 138.8, 129.9, 129.5, 128.1, 124.9, 120.3, 118.6, 116.2, 110.3,
109.5, 103.5, 93.4, 93.3, 93.1, 71.6, 64.8, 64.7, 64.6, 62.2, 29.9, 29.8, 15.2; HRMS calcd
for C30H37O8 [M-H]+
525.2497. Found: 525.2888.

133
(E)-3-(2,4-bis(ethoxymethoxy)phenyl)-1-(4-(ethoxymethoxy)-2-hydroxy-3-
iodophenyl)prop-2-en-1-one (234)
To a solution of acetophenone 217 (0.94 g, 2.8 mmol) in ethanol (2.5mL/mmol), 50% KOH
(3 eq) was added. After 10 min appropriated benzaldehyde 198 (0.71 g, 2.8 mmol) were
added and the solutions were stirred at room temperature for 40 h. After cooling the
reaction mixtures with ice, the mixtures were neutralized carefully with 1N hydrochloric
acid. The crude mixture was extracted with ethyl acetate, washed with water and brine
afforded chalcones 234 in 50% yield. 1H NMR (400 MHz, CDCl3): δ 14.47 (s, OH, 1H),
8.17 (d, J = 15.4 Hz, Hβ, 1H), 7.84 (d, J = 9.0 Hz, H6’, 1H), 7.56 (d, J = 15.4 Hz, Hα, 1H),
7.55 (d, J = 8.8 Hz, H6 , 1H), 6.86 (d, J = 2.2 Hz, H3, 1H), 6.72-6.66 (overlap, H5’, H5,
2H), 5.35 (s, OCH2O, 2H), 5.29 (s, OCH2O, 2H), 5.21 (s, OCH2O, 2H), 3.70(m, OCH2CH3,
6H), 1.21 (m, OCH2CH3, 9H); 13
C NMR (100 MHz, CDCl3): δ 192.3, 164.8, 164.4, 161.2,
158.5, 141.1, 131.3, 130.5, 118.2, 117.9, 115.6, 109.6, 103.5, 93.6, 93.5, 93.1, 78.5, 65.3,
64.9, 64.7, 15.2; HRMS calcd for C24H30IO8 [M+H]+
573.0980. Found: 573.0965.
Endo-237 and exo-238
Diene 105 (79 mg, 0.20 mmol) and dienophile 211 (88 mg, 0.20 mmol) were dissolved in
freshly distilled toluene (2 mL) in a pressure tube. The mixture was heated at 150oC for
24h to give a mixture of diastereomers 237 and 238. The mixture of diastereomers 237 and
238 was separated by column chromatography with 80% hexane/EtOAc as eluent to give a
mixture of diastereoisomers 237 (55 mg) and 238 (37 mg) (55% combined yield, 3:2 ratio).

134
Endo-237 was obtained as a pale yellow viscous liquid. 1H NMR (400 MHz, CDCl3): δ
13.57 (s, OH, 1H), 7.83 (d, J = 15.9 Hz, Hβ, 1H), 7.46 (d, J = 8.6 Hz, H6, 1H), 7.31-7.22
(overlap, Hα, H20”,H5’, 3H), 6.87 (d, J = 8.3 Hz, H14”, 1H), 6.44 (dd, J = 2.1, 8.6 Hz,
H17”, 1H), 6.43-6.40 (overlap, H4’, 1H), 6.37 (d, J = 1.8 Hz, H3, 1H), 6.32 (d, J = 8.6 Hz,
H19”, 1H), 6.24 (bd, *, H5, 1H), 5.41 (s, H2”, 1H), 4.38 (s, H4”, 1H), 3.98 (bd, J = 6.7 Hz,
H3”, 1H), 3.75-3.58 (5s, 5OMe, 15H), 3.52 (bs, H5” , 1H), 3.40 (s, OMe, 3H), 3.18
(overlap, H6”, 1H), 3.17 (s, OMe, 3H), 1.76 (bs, H6”, 1H), 1.73 (s, CH3, 3H); 13
C NMR
(100 MHz, CDCl3): δ 207.6, 192.4, 163.6, 163.5, 163.1, 162.0, 160.3, 159.9, 158.7, 157.5,
157.5, 138.9, 137.7, 131.3, 130.4, 130.3, 130.1, 126.9, 125.9, 124.3, 124.1, 120.1, 117.2,
116.3, 106.5, 105.5, 103.4, 101.4, 98.5, 98.4, 76.4, 62.5, 56.7, 55.6, 55.5, 55.3, 55.2, 54.2,
48.5, 38.2, 31.2, 30.3, 24.2; HRMS calcd for C42H44IO10 [M+H]+
835.1971. Found:
835.1969.

135
Exo-238 was obtained as a pale yellow viscous liquid. 1H NMR (400 MHz, CDCl3): δ 7.89
(d, J = 16.5 Hz, Hβ, 1H), 7.68 (d, J = 8.6 Hz, H20”, 1H), 7.54 (d, J = 8.2 Hz, H6, 1H), 7.41
(d, J = 16.0 Hz, Hα, 1H), 7.26 (overlap, H3, 1H), 6.99 (d, J = 8.2 Hz, H5’. 1H), 6.63-6.41
(overlap, H4’, H14”, H17”, 3H), 6.32 (bd, *, H13”, 1H), 6.23 (bd, *, H19”, 1H), 6.07 (bd, *,
H5, 1H), 5.24 (s, H2”, 1H), 4.81 (m, H4”, 1H), 4.45 (m, H3”, 1H), 3.95-3.65 (5s, 5OMe,
15H), 3.74 (overlap, H5” , 1H), 3.57 (s, OMe, 3H), 3.47 (s, OMe, 3H), 2.24 (m, H6”, 1H),
1.73 (s, CH3, 3H); 13
C NMR (100 MHz, CDCl3): δ 208.7, 192.8, 163.7, 163.2, 162.8, 161.9,
160.3, 160.1, 159.1, 158.2, 139.5, 138.6, 132.6, 132.0, 130.5, 130.3, 127.1, 126.7, 125.0,
124.5, 123.9, 117.1, 116.2, 107.5, 105.7, 104.4, 101.6, 98.9, 98.5, 75.7, 63.8, 63.2, 56.6,
56.4, 56.1, 55.7, 55.6, 55.3, 47.7, 38.8, 29.7, 23.4; HRMS calcd for C42H44IO10 [M+H]+
835.1971. Found: 835.1970.
*coupling constant cannot be measured due to broad resonance
Endo-230
Mixture of endo-241 (39 mg, 0.50 mmol) and PdCl2 (18 mg, 0.01 mmol) in dry
ethanol (20 mL) were stirred in room temperature for 120 h, which smoothly afforded the
desired intermediates 230 in 19 mg, 50% yield.

136
Endo-230 was obtained as a pale yellow viscous liquid. 1H NMR (400 MHz, CDCl3): δ
7.91 (d, J = 16.0 Hz, Hβ, 1H), 7.49-7.42 (overlap, Hα, H6, H20”, 3H), 6.99 (d, J = 7.8 Hz,
H14”, 1H), 6.65 (d, J = 10.0 Hz, CH, 1H), 6.49 (overlap, H4’, 1H), 6.45 (dd, J = 2.3, 8.2
Hz, H17”, 1H), 6.40 (d, J = 2.3 Hz, H3, 1H), 6.30 (bs, H19”, 1H), 6.24 (bd, J = 8.2 Hz, H5,
1H), 5.61 (d, J = 10.0, CH), 5.39 (bs, H2”, 1H), 4.87 (m, H5”, 1H), 4.75 (s, H4”, 1H), 4.26
(bs, H3”, 1H), 3.80 (s, 2OMe, 6H), 3.68-3.63 (4s, 4OMe, 12H), 3.44 (s, OMe, 3H), 2.26
(dd, J = 5.5, 16.0 Hz, H6”, 1H), 2.08 (m, H6”, 1H), 1.72 (s, H7”, 3H), 1.61 (s, CH3, 3H),
1.55 (s, CH3, 3H); 13
C NMR (100 MHz, CDCl3): δ200.6, 193.1, 162.8, 160.4, 158.6, 158.5,
157.7, 153.2, 138.6, 133.6, 131.7, 131.0, 130.5, 128.4, 125.1, 123.8, 122.8, 122.2, 117.7,
117.1, 109.8, 106.6, 105.5, 104.4, 103.0, 99.2, 98.4, 76.9, 63.1, 63.0, 55.8, 55.7, 55.4, 53.6,
46.3, 38.8, 33.8, 29.9, 29.5, 28.4, 28.3, 23.9; HRMS calcd for C47H51O10 [M+H]+
775.3474.
Found: 775.3499.
Exo-231
Mixture of exo-242 (31 mg, 0.40 mmol) and PdCl2 (14 mg, 0.008 mmol) in dry
ethanol (20 mL) were stirred in room temperature for 216 h, which smoothly afforded the
desired intermediates 231 in 14 mg, 45% yield.

137
Exo-231 was obtained as a pale yellow viscous liquid 1
H NMR (400 MHz, CDCl3): δ 7.96
(d, J = 15.6 Hz, Hβ, 1H), 7.73 (d, J = 9.2, H20”, 1H), 7.55-7.46 (overlap, Hα, H6, 2H), 7.40
(d, J = 8.2 Hz, H3, 1H), 7.13 (d, J = 8.7 Hz,H5’, 1H), 6.53-6.30 (overlap, H4’,H13”, H14”,
H17”, H19”, CH, 6H), 6.02 (bd, J = 8.7 Hz, H5, 1H), 5.47 (d, J = 9.6, CH), 5.16 (bs, H2”,
1H), 4.87 (t, J = 10.5 Hz, H4”, 1H), 4.25 (bs, H3”, 1H), 3.95 (bs, H5”, 1H), 3.84 (3s, 3OMe,
9H), 3.70 (2s, 2OMe, 6H), 3.56 (s, OMe, 3H), 3.53 (s, OMe, 3H), 2.27 (bs, H6”, 2H), 1.70
(s, H7”, 3H), 1.54 (s, CH3, 3H), 1.47 (s, CH3, 3H); 13
C NMR (100 MHz, CDCl3): δ 205.4,
191.4, 162.9, 162.6, 160.3, 158.9, 158.7, 158.3, 157.1, 152.4, 137.8, 132.9, 132.8, 131.8,
131.3, 130.6, 130.4, 126.5, 125.6, 124.6, 124.1, 124.0, 117.5, 117.0, 109.9, 107.4, 105.4,
104.3, 102.4, 98.6, 76.3, 63.6, 63.2, 56.9, 55.9, 55.8, 55.6, 55.3, 51.4, 39.2, 38.7, 32.0, 29.8,
28.1, 26.7, 26.1, 23.1; HRMS calcd for C47H51O10 [M+H]+
775.3474. Found: 775.3495.

138
REFERENCES
Abegaz, B. M., Ngadjui, B. T., Dongo, E., Ngameni, B., Nindi, M. N., & Bezabih, M.
(2002). Chalcones and other constituents of Dorstenia prorepens and Dorstenia
zenkeri. Phytochemistry, 59(8), 877-883.
Adler, M. J., & Baldwin, S. W. (2009). Direct, regioselective synthesis of 2,2-dimethyl-2H-
chromenes. Total syntheses of octandrenolone and precocenes I and II. Tetrahedron
Letters, 50(36), 5075-5079.
Ahmed, S., Wagner, H., & Razaq, S. (1978). Synthesis of polyhydroxy-flavone
methylethers with potential cytotoxic activity—III: Synthesis of 5,7-dihydroxy
6,2′.4′,5′-tetramethoxyflavone (tabularin) from chukrasia tabularis a. juss and
5,6,7,2′,4′,5′-hexamethoxyflavone (tabularin dimethylether). Tetrahedron, 34(10),
1593-1594.
Akanksha, & Maiti, D. (2012). Microwave-assisted palladium mediated decarbonylation
reaction: synthesis of eulatachromene. Green Chemistry, 14(8), 2314-2320.
Arkoudis, E., Lykakis, I. N., Gryparis, C., & Stratakis, M. (2009). Biomimetic Synthesis of
Dimeric Metabolite Acremine G via a Highly Regioselective and Stereoselective
Diels−Alder Reaction. Organic Letters, 11(14), 2988-2991.
Ballerini, E., Minuti, L., & Piermatti, O. (2010). High-pressure Diels-Alder cycloadditions
between benzylideneacetones and 1,3-butadienes: application to the synthesis of
(R,R)-(-)- and (S,S)-(+)-Delta8-tetrahydrocannabinol. J Org Chem, 75(12), 4251-
4260. doi: 10.1021/jo100785w
Ballerini, E., Minuti, L., Piermatti, O., & Pizzo, F. (2009). High Pressure Diels−Alder
Approach to Hydroxy-Substituted 6a-Cyano-tetrahydro-6H-benzo[c]chromen-6-
ones: A Route to Δ6-Cis-Cannabidiol. The Journal of Organic Chemistry, 74(11),
4311-4317.
Bao, J., Wulff, W. D., & Rheingold, A. L. (1993). Vaulted biaryls as chiral ligands for
asymmetric catalytic Diels-Alder reactions. Journal of the American Chemical
Society, 115(9), 3814-3815.
Barbour, L. J. (2001). X-Seed -A Software Tool for Supramolecular Crystallography.
Journal of Supramolecular Chemistry, 1, 189-191.

139
Basnet, P. K. S., Terashima, S, Shimizu, M., Namba, T. (1993). Two new 2-arylbenzofuran
derivatives from hypoglycemic activity-bearing fractions of Morus insignis.
Chemical Phamaceutical Bulletin, 41, 1238-1243.
Becke, A. D. (1993a).A new mixing of Hartree–Fock and local density‐functional theories.
Journal Chemical Physics, 98, 1372-1377.
Becke, A. D. (1993b). Density‐functional thermochemistry. III. The role of exact exchange.
Journal Chemical Physics, 98, 5648-5652.
Bergmann, R., & Gericke, R. (1990). Synthesis and antihypertensive activity of 4-(1,2-
dihydro-2-oxo-1-pyridyl)-2H-1-benzopyrans and related compounds, new
potassium channel activators. J Med Chem, 33(2), 492-504.
Bigi, F., Carloni, S., Maggi, R., Muchetti, C., & Sartori, G. (1997). Zeolite-Induced
Heterodomino Reaction. Regioselective Synthesis of 2H-1-Benzopyrans from
Phenols and α-Alkynols. The Journal of Organic Chemistry, 62(20), 7024-7027.
Boonsri, S., Gunawan, C., Krenske, E. H., & Rizzacasa, M. A. (2012). Synthetic studies
towards the mulberry Diels-Alder adducts: H-bond accelerated cycloadditions of
chalcones. Org Biomol Chem, 10(30), 6010-6021.
Bröhmer, M. C., Volz, N., & Bräse, S. (2009). Thieme Chemistry Journal Awardees -
Where Are They Now? Microwave-Assisted Rhodium-Catalyzed Decarbonylation
of Functionalized 3-Formyl-2H-chromenes: A Sequence for Functionalized
Chromenes like Deoxycordiachromene. Synlett, 2009(09), 1383-1386.
Chauder, B. A., Lopes, C. C., Lopes, R. S. C., da Silva, Alcides J. M., & Snieckus, V.
(1998). Phenylboronic Acid-Mediated Synthesis of 2H-Chromenes. Synthesis,
1998(03), 279-282.
Chee, C. F., Lee, Y. K., Buckle, M. J. C., & Abdul Rahman, N. (2011). Synthesis of (±)-
kuwanon V and (±)-dorsterone methyl ethers via Diels–Alder reaction. Tetrahedron
Letters, 52(15), 1797-1799.
Chen, H-D, Ding, Y-Q., Yang, S-P, Li, X-C., Wang, X-J., Zhang, H-Y., Ferreira, D., Yue,
J-M.. (2012). Morusalbanol A, a neuro-protective Diels–Alder adduct with an
unprecedented architecture from Morus alba. Tetrahedron, 68(30), 6054-6058.

140
Coleman, R. S., Lu, X-L., & Modolo, I. (2007). Total Synthesis of 2‘-O-
Methylmyxalamide D and (6E)-2‘-O-Methylmyxalamide D. Journal of the
American Chemical Society, 129(13), 3826-3827.
Cong, H., & Porco, J. A. Jr. (2012). Total synthesis of (+/-)-sorocenol B employing
nanoparticle catalysis. Org Lett, 14(10), 2516-2519.
Cong, H., Becker, C. F., Elliott, S. J., Grinstaff, M. W., & Porco, J. A. Jr. (2010). Silver
Nanoparticle-Catalyzed Diels−Alder Cycloadditions of 2′-Hydroxychalcones.
Journal of the American Chemical Society, 132(21), 7514-7518.
Cong, H., Ledbetter, D., Rowe, G. T., Caradonna, J. P., & Porco, J. A. Jr. (2008). Electron
Transfer-Initiated Diels−Alder Cycloadditions of 2′-Hydroxychalcones. Journal of
the American Chemical Society, 130(29), 9214-9215.
Dai, S. J., Ma, Z. B., Wu, Y., Chen, R. Y., & Yu, D. Q. (2004). Guangsangons F-J, anti-
oxidant and anti-inflammatory Diels-Alder type adducts, from Morus macroura Miq.
Phytochemistry, 65(23), 3135-3141.
Dai, S. J., Mi, Z. M., Ma, Z. B., Li, S., Chen, R. Y., & Yu, D. Q. (2004). Bioactive diels-
alder type adducts from the stem bark of Morus macroura. Planta Med, 70(8), 758-
763.
David, C. W., Iqbal, M, & Livingstone, R. (1998). Preparation of Some 6-Substituted 2,2-
Dimethyl- and 2,2- and 2,4-Diphenyl-naphtho[1,2-b]pyrans. Journal of Chemical
Research, Synopses(1), 2-3.
Delley B. Delley (1990). An All-Electron Numerical Method for Solving the Local Density
Functional for Polyatomic Molecules. Journal of Chemical Physics, 92, 508-517.
Delley B. (2000). From molecules to solids with the DMol3 approach. Journal of Chemical
Physics, 113(18), 7756-7764.
Dong, Y-W, Wang, G-W., & Wang, L. (2008). Solvent-free synthesis of naphthopyrans
under ball-milling conditions. Tetrahedron, 64(44), 10148-10154.
Fine, S. A., & Pulaski, P. D. (1973). Reexamination of the Claisen-Schmidt condensation
of phenylacetone with aromatic aldehydes. The Journal of Organic Chemistry,
38(9), 1747-1749.
Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J.
R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G. A.; Nakatsuji, H.; Caricato,

141
M.; Li, X.; Hratchian, H. P.; Izmaylov, A. F.; Bloino, J.; Zheng, G.; Sonnenberg, J.
L.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima,
T.; Honda, Y.; Kitao, O.; Nakai, H.; Vreven, T.; Montgomery, J. A., Jr.; Peralta, J. E.;
Ogliaro, F.; Bearpark, M.; Heyd, J. J.; Brothers, E.; Kudin, K. N.; Staroverov, V. N.;
Kobayashi, R.; Normand, J.; Raghavachari, K.; Rendell, A.; Burant, J. C.; Iyengar, S.
S.; Tomasi, J.; Cossi, M.; Rega, N.; Millam, J. M.; Klene, M.; Knox, J. E.; Cross, J.
B.; Bakken, V.; Adamo, C.; Jaramillo, J.; Gomperts, R.; Stratmann, R. E.; Yazyev,
O.; Austin, A. J.; Cammi, R.; Pomelli, C.; Ochterski, J. W.; Martin, R. L.; Morokuma,
K.; Zakrzewski, V. G.; Voth, G. A.; Salvador, P.; Dannenberg, J. J.; Dapprich, S.;
Daniels, A. D.; Farkas, Ö.; Foresman, J. B.; Ortiz, J. V.; Cioslowski, J.; Fox, D. J.
(2009) Gaussian 09, Inc. Wallingford CT.
Fukai, T., Hano, Y., Hirakura, K., Nomura, T, Uzawa, J., & Fukushima, K. (1985).
Structures of Two Natural Hypotensive Diels-Alder Type Adducts, Mulberrofurans
F and G, from the Cultivated Mulberry Tree (Morus lhou KOIDZ.). Chemical &
Pharmaceutical Bulletin, 33(8), 3195-3204.
Fürstner, A., Heilmann, E. K., & Davies, P. W. (2007). Total Synthesis of the Antibiotic
Erypoegin H and Cognates by a PtCl2-Catalyzed Cycloisomerization Reaction.
Angewandte Chemie International Edition, 46(25), 4760-4763.
Gabbutt, C. D, Heron, B. Mark, I., Alicia C., Thomas, D. A., Partington, S. M., Hursthouse,
M. B., & Gelbrich, T. (2003). Observations on the Synthesis of Photochromic
Naphthopyrans. European Journal of Organic Chemistry, 2003(7), 1220-1230.
Gan, R. Y., Kuang, L., Xu, X. R., Zhang, Y., Xia, E. Q., Song, F. L., & Li, H. B. (2010).
Screening of natural antioxidants from traditional Chinese medicinal plants
associated with treatment of rheumatic disease. Molecules, 15(9), 5988-5997.
Gao, L., Han, J & Lei, X. (2016). Enantioselective Total Syntheses of Kuwanon X,
Kuwanon Y, and Kuwanol A. Organic Letter, 18 (3), 360–363.
Goujon, J. Y., Zammattio, F., & Kirschleger, B. (2000). Synthesis of various 2H-
benzopyran compounds and their kinetic resolution by asymmetric hydrolysis of
their racemic acetates mediated by lipases. Tetrahedron: Asymmetry, 11(11), 2409-
2420.
Gunawan, C., & Rizzacasa, M. A. (2010). Mulberry Diels−Alder Adducts: Synthesis of
Chalcomoracin and Mulberrofuran C Methyl Ethers. Organic Letters, 12(7), 1388-
1391.

142
Halgren, T. A. and Lipscomb, W. N.(1977). The Synchronous Transit Method for
Determining Reaction Pathways and Locating Transition States. Chemical Physics
Letters, 49, 225-232.
Han, J., Jones, A. X., & Lei, X. (2015). Recent Advances in the Total Synthesis of
Prenylflavonoid and Related Diels–Alder Natural Products. Synthesis, 47(11), 1519-
1533.
Han, J., Li, X., Guan, Y., Zhao, W-J., Wulff, W. D., & Lei, X. (2014). Enantioselective
Biomimetic Total Syntheses of Kuwanons I and J and Brosimones A and B.
Angewandte Chemie, 126(35), 9411-9415.
Hano, Y., Fukai, T., Nomura, T., Uzawa, J., & Fukushima, K. (1984). Structure of
mulberrofuran I, a novel 2-arylbenzofuran derivative from the cultivated mulberry
tree (morus bombycis koidz). Chemical & Pharmaceutical Bulletin , 32(3), 1260-
1263.
Hano, Y., Tsubura, H., Nomura, T. (1986). Constituents of the cultivated mulberry tree. 38.
Structures of kuwanons Y and Z, two new stilbene derivatives from the cultivated
mulberry tree (Morus alba L.) Heterocycles, 24, 2603-2610.
Hano, Y., Suzuki, S., Nomura, T., & Iitaka, Y. (1988). Absolute configuration of natural
Diels-Alder type adducts from the morus root bark. Heterocycles, 27(10), 2315-
2325.
Hano, Y., Yamanaka, J., Nomura, T., & Momose, Y. (1995). Sorocenols A and B, two new
isoprenylated phenols from the root bark of Sorocea bonplandii Baillon.
heterocycles 41(5), 8.
Harrington, P. J., Hegedus, L. S., & McDaniel, K. F. (1987). Palladium-catalyzed reactions
in the synthesis of 3- and 4-substituted indoles. 2. Total synthesis of the N-acetyl
methyl ester of (±)-clavicipitic acids. Journal of the American Chemical Society,
109(14), 4335-4338.
Helesbeux, J-J., Duval, O., Guilet, D., Séraphin, D., Rondeau, D., & Richomme, P. (2003).
Regioselectivity in the ene reaction of singlet oxygen with ortho-prenylphenol
derivatives. Tetrahedron, 59(27), 5091-5104.
Heller, D. P., Goldberg, D. R., Wu, H., & Wulff, W. D. (2006). An examination of
VANOL, VAPOL, and VAPOL derivatives as ligands for asymmetric catalytic
Diels–Alder reactions. Canadian Journal of Chemistry, 84(10),1487-1503.
Hirakura, K., Hano, Y., Fukai, T., Nomura, T., Uzawa, J., Fukushima, K. (1985).
Constituents of the cultivated mulberry tree 21. Structures of three new natural

143
Diels-Alder type adducts, kuwanons P and X, and mulberrofuran J from cultivated
mulberry tree (Morus lhou Koidz.) Chemical Phamaceutical Bulletin, 33, 1088-
1096
Hirakura, K., Hano, Y., Fukai, T., Nomura, T., Uzawa, J., Fukushima, K. (1985). CHiroya,
K., Suzuki, N., Yasuhara, A., Egawa, Y., Kasano, A., & Sakamoto, T. (2000). Total
syntheses of three natural products, vignafuran, 2-(4-hydroxy-2-methoxyphenyl)-6-
methoxybenzofuran-3-carboxylic acid methyl ester, and coumestrol from a common
starting material. Journal of the Chemical Society, Perkin Transactions 1(24), 4339-
4346.
Iikubo, K., Ishikawa, Y., Ando, N., Umezawa, K., & Nishiyama, S. (2002). The first direct
synthesis of α-mangostin, a potent inhibitor of the acidic sphingomyelinase.
Tetrahedron Letters, 43(2), 291-293.
Iyer, M., & Trivedi, G. K. (1990). Synthesis of 2, 2-Dimethyl-2H-chromenes via a
Palladium (II) Catalysed Reaction. Synthetic Communications, 20(9), 1347-1351.
Kang, J., Chen, R. Y., & Yu, D. Q. (2006). Five new diels-alder type adducts from the stem
and root bark of Morus mongolica. Planta Med, 72(1), 52-59.
Kogan, S. B., & Herskowitz, M. (2002). Dehydrogenation of Neohexane to Neohexene on
Platinum Polymetallic Catalysts. Industrial & Engineering Chemistry Research,
41(24), 5949-5951.
Kureshy, R. I., Ahmad, I., Pathak, K., Khan, N. H., Abdi, Sayed H. R., & Jasra, R. V.
(2009). Sulfonic acid functionalized mesoporous SBA-15 as an efficient and
recyclable catalyst for the synthesis of chromenes from chromanols. Catalysis
Communications, 10(5), 572-575.
Lamcharfi, E., Menguy, L., & Zamarlik, H. (1993). One-Pot Synthesis of Analogues of
Precocenes and Isoprenyl-Chromenes. Synthetic Communications, 23(21), 3019-
3028.
Larock, R. C., Wei, L., & Hightower, T. R. (1998). Synthesis of 2H-1-Benzopyrans by Pd-
Catalyzed Cyclization of o-Allylic Phenols. Synlett, 1998(05), 522-524.
Lee, C. T., Yang, W. T., and Parr R. G. (1988). Development of the Colle-Salvetti
correlation-energy formula into a functional of the electron density. Physical
Review B, 37, 785-789.

144
Lee, D-H., Kwon, K-H., & Yi, C. S. (2012). Dehydrative C–H Alkylation and Alkenylation
of Phenols with Alcohols: Expedient Synthesis for Substituted Phenols and
Benzofurans. Journal of the American Chemical Society, 134(17), 7325-7328.
Lee, Y-R., Choi, J-H., & Yoon, S-H. (2005). Efficient and general method for the synthesis
of benzopyrans by ethylenediamine diacetate-catalyzed reactions of resorcinols with
α,β-unsaturated aldehydes. One step synthesis of biologically active (±)-confluentin
and (±)-daurichromenic acid. Tetrahedron Letters, 46(44), 7539-7543.
Liu, Y., Yao, B., Deng, C-L., Tang, R-Y., Zhang, X-G., & Li, J.-H. (2011). Palladium-
Catalyzed Selective Heck-Type Diarylation of Allylic Esters with Aryl Halides
Involving a β-OAc Elimination Process. Organic Letters, 13(5), 1126-1129.
Lykakis, I. N., Efe, C., Gryparis, C., & Stratakis, M. (2011). Ph3PAuNTf2 as a Superior
Catalyst for the Selective Synthesis of 2H-Chromenes: Application to the Concise
Synthesis of Benzopyran Natural Products. European Journal of Organic Chemistry,
2011(12), 2334-2338.
Madabhushi, S., Jillella, R., Godala, Kondal R., Mallu, K. K. R., Beeram, C. R., &
Chinthala, N. (2012). An efficient and simple method for synthesis of 2,2-
disubstituted-2H-chromenes by condensation of a phenol with a 1,1-disubstituted
propargyl alcohol using BF3·Et2O as the catalyst. Tetrahedron Letters, 53(39),
5275-5279.
Manners, G. D., & Jurd, L. (1976). Isomerization of phenolic chromens. Journal of the
Chemical Society, Chemical Communications(12), 448-448.
Mannhold, R., Cruciani, G., Weber, H., Lemoine, H., Derix, A., Weichel, C., & Clementi,
M. (1999). 6-Substituted benzopyrans as potassium channel activators: synthesis,
vasodilator properties, and multivariate analysis. Journal of medicinal chemistry,
42(6), 981-991.
Macrae, C. F.; Bruno, I. J.; Chisholm, J. A.; Edgington, P. R.; McCabe, P.; Pidcock, E.;
Rodriguez-Monge, L.; Taylor, R.; van de Streek, J.; Wood, P. A. (2008). Mercury
CSD 2.0 – new features for the visualization and investigation of crystal structures.
Journal of Applied Crystallography, 41, 466-470
Messana, I., Ferrari, F., & De A., Maria D. C. M. (1988). Constituents of brosimopsis
oblongifolia.31: Structure of a new diels-alder type adduct, brosimone a.
Tetrahedron, 44(21), 6693-6698.

145
Miyaura, N., & Suzuki, A. (1995). Palladium-Catalyzed Cross-Coupling Reactions of
Organoboron Compounds. Chemical Reviews, 95(7), 2457-2483.
Nishida, J., & Kawabata, J. (2006). DPPH radical scavenging reaction of hydroxy- and
methoxychalcones. Biosci Biotechnol Biochem, 70(1), 193-202.
Nomura, t., Fukai, T., Matsumoto, J., Ohmori, T. (1982). Constituents of the cultivated
mulberry tree. VIII. Components of root barks of Morus bombycis. Planta Medica,
46, 28-32.
Nomura, T. (1988a). Phenolic Compounds of the Mulberry Tree and Related Plants. In W.
Herz, H. Grisebach, G. W. Kirby & C. Tamm (Eds.), Fortschritte der Chemie
organischer Naturstoffe / Progress in the Chemistry of Organic Natural Products
(Vol. 53, pp. 87-201): Springer Vienna.
Nomura, T. (1988b). Phenolic compounds of the mulberry tree and related plants. Fortschr
Chem Org Naturst, 53, 87-201.
Nomura, T., & Fukai, T. (1981). Constituents of the cultivated mulberry tree. Planta Med,
42(1), 79-88.
Nomura, T., & Hano, Y. (1994). Isoprenoid-substituted phenolic compounds of moraceous
plants. Natural Product Reports, 11(0), 205-218.
Nomura, T., & Fukai, T. (1980). Kuwanon G, a New Flavone Derivative from the Root
Barks of the Cultivated Mulberry Tree (Morus alba L.). Chemical &
Pharmaceutical Bulletin, 28(8), 2548-2552.
Nomura, T., Fukai, T., Narita, T., Terada, S., Uzawa, J., Iitaka, Y., Takasugi, M., Ishikawa,
S., Nagao, S., Masamune, T. (1981). Confirmation of the structures of kuwanons G
and H (albanins F and G) by partial synthesis. Tetrahedron Letters, 22(23), 2195-
2198.
North, J. T., Kronenthal, D. R., Pullockaran, A. J., Real, S. D., Chen, H. Y. (1995).
Synthesis of 6-Cyano-2,2-dimethyl-2H-1-benzopyran and Other Substituted 2,2-
Dimethyl-2H-1-benzopyrans. The Journal of Organic Chemistry, 60(11), 3397-
3400.
Pandey, G., & Krishna, A. (1988). Synthetic application of photoinduced single electron
transfer reactions: a convenient synthetic approach for the 2,2-dimethyl-2H-
chromene system. The Journal of Organic Chemistry, 53(10), 2364-2365.

146
Perdew J. P. and Wang, Y. (1992). Accurate and simple analytic representation of the
electron-gas correlation energy. Physical Review B, 45(23), 13244-13249.
Prado, S., Janin, Y. L., & Bost, P-E. (2006). A simple two steps ytterbium triflate-catalysed
preparation of 2,2-dimethyl-2h-chromenes from salicylaldehydes and 2-
methylpropene. Journal of Heterocyclic Chemistry, 43(6), 1605-1608. .
Qi, C., Cong, H., Cahill, K. J., Muller, P., Johnson, R. P., & Porco, J. A., Jr. (2013).
Biomimetic dehydrogenative Diels-Alder cycloadditions: total syntheses of
brosimones A and B. Angew Chem Int Ed Engl, 52(32), 8345-8348. .
Rama Rao, A. V., Deshpande, V. H., Shastri, R. K., Tavale, S. S., & Dhaneshwar, N. N.
(1983). Structures of albanols a and b, two novel phenols from Morus alba bark.
Tetrahedron Letters, 24(29), 3013-3016.
Rathwell, D. C., Yang, S. H., Tsang, K. Y., & Brimble, M. A. (2009). An efficient formal
synthesis of the human telomerase inhibitor (+/-)-gamma-rubromycin. Angew Chem
Int Ed Engl, 48(43), 7996-8000.
Rollinger, J. M., Bodensieck, A., Seger, C., Ellmerer, E. P., Bauer, R., Langer, T., &
Stuppner, H. (2005). Discovering COX-inhibiting constituents of Morus root bark:
activity-guided versus computer-aided methods. Planta Med, 71(5), 399-405.
Rollinger, J. M., Spitaler, R., Menz, M., Marschall, K., Zelger, R,. Ellmerer, E. P.,
Schneider, P., Stuppner, H. (2006). Venturia inaequalis-inhibiting Diels-Alder
adducts from Morus root bark. J Agric Food Chem, 54(22), 8432-8436.
Roush, W. R., & Sciotti, R. J. (1998). Enantioselective Total Synthesis of (−)-
Chlorothricolide via the Tandem Inter- and Intramolecular Diels−Alder Reaction of
a Hexaenoate Intermediate. Journal of the American Chemical Society, 120(30),
7411-7419.
Sa e Sant'Anna, S., Evangelista, E. A., Alves, R. B., & Raslan, D. S. (2005). Synthesis of
New (±) Pterocarpans by Heck Oxyarylation. Chemistry of Natural Compounds,
41(4), 385-387.
Sartori, G., Casiraghi, G., Bolzoni, L., & Casnati, G. (1979). General synthesis of 2H-
benzo[b]pyrans (chrom-3-enes) from metal phenoxides and .alpha.,.beta.-
unsaturated carbonyl compounds. The Journal of Organic Chemistry, 44(5), 803-
805.

147
Sebastián, D., Bordejé, E. G., Calvillo, L., Lázaro, M. J., & Moliner, R. (2008). Hydrogen
storage by decalin dehydrogenation/naphthalene hydrogenation pair over platinum
catalysts supported on activated carbon. International Journal of Hydrogen Energy,
33(4), 1329-1334.
Sheldrick, G. M. (2008). A short history of SHELX. Acta Crystallographica Section A.
Foundation and Advances, 64, 112-122.
Sheldrick, G. M. (2012). SHELXT, Universität Göttingen, Germany.
Shi, Y-Q., Fukai, T., Sakagami, H., Chang, W-J., Yang, P-Q., Wang, F-P., & Nomura, T.
(2001). Cytotoxic Flavonoids with Isoprenoid Groups from Morus mongolica1.
Journal of Natural Products, 64(2), 181-188.
Sohn, H. Y., Son, K. H., Kwon, C. S., Kwon, G. S., & Kang, S. S. (2004). Antimicrobial
and cytotoxic activity of 18 prenylated flavonoids isolated from medicinal plants:
Morus alba L., Morus mongolica Schneider, Broussnetia papyrifera (L.) Vent,
Sophora flavescens Ait and Echinosophora koreensis Nakai. Phytomedicine, 11(7-8),
666-672.
Sonogashira, K., Tohda, Y., & Hagihara, N. (1975). A convenient synthesis of acetylenes:
catalytic substitutions of acetylenic hydrogen with bromoalkenes, iodoarenes and
bromopyridines. Tetrahedron Letters, 16(50), 4467-4470.
Sugamoto, K., Kurogi, C., Matsushita, Y., & Matsui, T. (2008). Synthesis of 4-
hydroxyderricin and related derivatives. Tetrahedron Letters, 49(47), 6639-6641.
Szczepanik, M., Obara, R., Szumny, A., Gabrys, B., Halarewicz-Pacan, A., Nawrot, J., &
Wawrzenczyk, C. (2005). Synthesis and insect antifeedant activity of precocene
derivatives with lactone moiety. J Agric Food Chem, 53(15), 5905-5910.
Takasugi, M., Nagao, S., Masamune, T., Shirata, A., & Takahashi, K. (1980).
Chalcomoracin, a natural Diels-Alder adduct from diseased mulberry. Chem Lett, 9,
1573-1576.
Talamás, F. X., Smith, D. B., Cervantes, A., Franco, F., Cutler, S. T., Loughhead, D. G.,
Morgans Jr, D. J., Weikert, R. J. (1997). The Florisil® catalyzed [1,3]-sigmatropic
shift of allyl phenyl ethers — An entryway into novel mycophenolic acid analogues.
Tetrahedron Letters, 38(27), 4725-4728.
Tan, Y. X., Yan, R. Y., Wang, H. Q., Chen, R. Y., & Yu, D. Q. (2009). Wittiorumins A - F,
antioxidant diels-alder-type adducts from Morus wittiorum. Planta Med, 75(3), 249-
255.

148
Trend, R. M., Ramtohul, Y. K., Ferreira, E. M., & Stoltz, B. M. (2003). Palladium-
Catalyzed Oxidative Wacker Cyclizations in Nonpolar Organic Solvents with
Molecular Oxygen: A Stepping Stone to Asymmetric Aerobic Cyclizations.
Angewandte Chemie International Edition, 42(25), 2892-2895.
Trend, R. M., Ramtohul, Y. K., & Stoltz, B. M. (2005). Oxidative Cyclizations in a
Nonpolar Solvent Using Molecular Oxygen and Studies on the Stereochemistry of
Oxypalladation. Journal of the American Chemical Society, 127(50), 17778-17788.
Ueda, S., Nomura, T., Fukai, T., & Matsumoto, J.. (1982). Kuwanon J, a new diesl-alder
adduct and chalcomoracin from callus culture of morus alba L. Chemical &
Pharmaceutical Bulletin, 30(8), 3042-3045. doi: 10.1248/cpb.30.3042
Venkataraman, K. (1972). Wood phenolics in the chemotaxonomy of the moraceae.
Phytochemistry, 11(5), 1571-1586.
Wang, G., Wu, W., Peng, F., Cao, D., Yang, Z., Ma, L., Qiu, N., Ye, H., Han, X., Chen,
J., Qiu, J., Sang, Y., Liang, X., Ran, Y., Peng, A., Wei, Y., Chen, L. (2012). Design,
synthesis, and structure–activity relationship studies of novel millepachine
derivatives as potent antiproliferative agents. European Journal of Medicinal
Chemistry, 54(0), 793-803.
Wang, H., Yan, Z., Lei, Y., Sheng, K., Yao, Q., Lu, K., & Yu, P. (2014). Concise synthesis
of prenylated and geranylated chalcone natural products by regiospecific iodination
and Suzuki coupling reactions. Tetrahedron Letters, 55(4), 897-899.
Wang, K., Goldman, M. E., Emge, T. J., & Goldman, A. S. (1996). Transfer-
dehydrogenation of alkanes catalyzed by rhodium(I) phosphine complexes. Journal
of Organometallic Chemistry, 518(1–2), 55-68.
Yang, Y., Tan, Y. X., Chen, R. Y., & Kang, J. (2014). The latest review on the polyphenols
and their bioactivities of Chinese Morus plants. J Asian Nat Prod Res, 16(6), 690-
702.
Yang, Y., Wang, H. Q., & Chen, R. Y. (2010). [Flavonoids from the leaves of Morus alba
L]. Yao Xue Xue Bao, 45(1), 77-81.

149
Yen, G-C., Wu, S-C., & Duh, P-D. (1996). Extraction and Identification of Antioxidant
Components from the Leaves of Mulberry (Morus alba L.). Journal of Agricultural
and Food Chemistry, 44(7), 1687-1690.
Yuan, C., Du, B., Yang, L., & Liu, B. (2013). Bioinspired Total Synthesis of Bolivianine:
A Diels– Alder/Intramolecular Hetero-Diels–Alder Cascade Approach. Journal of
the American Chemical Society, 135(25), 9291-9294.
Yus, M., Foubelo, F., & Ferrández, José V. (2001). Stereoselective Reductive Opening of
2,3-Benzofuran − A Two-Step Synthesis of 2H-Chromenes Including
Deoxycordiachromene. European Journal of Organic Chemistry, 2001(15), 2809-
2813.
Zeng, H., Ju, J, & Hua, R. (2011). ReCl(CO)5-catalyzed cyclocondensation of phenols with
2-methyl-3-butyn-2-ol to afford 2,2-dimethyl-2H-chromenes. Tetrahedron Letters,
52(30), 3926-3928.
Zhang, Q. J., Ni, G., Wang, Y. H., Chen, R. Y., & Yu, D. Q. (2009). Three new Diels-Alder
type adducts from the stem bark of Morus cathayana. J Asian Nat Prod Res, 11(3),
267-273.
Zhang, Q-J., Tang, Y-B., Chen, R-Y., & Yu, D-Q. (2007). Three New Cytotoxic Diels–
Alder-Type Adducts from Morus australis. Chemistry & Biodiversity, 4(7), 1533-
1540.
Zhao, W., & Carreira, E. M. (2003). Facile one-pot synthesis of photochromic pyrans. Org
Lett, 5(22), 4153-4154.

150
APPENDIX
SPECTRA OF COMPOUNDS
1H NMR and
13C NMR of 1-(4-(ethoxymethoxy)-2-hydroxy-3-iodophenyl)ethanone

151
1H NMR and
13C NMR of compound 171
*impurity
*

152
1H NMR and
13C NMR of 172

153
1H NMR and
13C NMR of 174

154
1H NMR and
13C NMR of exo-adduct 180
*impurity
* *

155
1H NMR and
13C NMR of diastereomer 182 (without the EOM protecting group)
*impurities
**
*
*
*
* *

156
1H NMR and
13C NMR of diastereomer 183
*impurities
* *
*

157
1H NMR and
13C NMR of diastereomer 184

158
1H NMR and
13C NMR of diastereomer 186
*MeOD-d4

159
1H NMR and
13C NMR of compound 190

160
1H NMR and
13C NMR of compound 192

161
1H NMR of compound 202

162
13C NMR of compound 203

163
1H NMR and
13C NMR of compound 191

164
1H NMR and
13C NMR of compound 204
*impurities and solvents peaks
*

165
1H NMR and
13C NMR of compound 205
*impurities/ grease peaks
*
*
* *
166
1H NMR of compound 206
*impurities
*
*

167
13C NMR of compound 206
*
*impurities
*

168
HSQC for morusalbanol A pentamethyl ether 206 (CDCl3)

169
HMBC for morusalbanol A pentamethyl ether 206 (CDCl3)

170
Comparison of 1H NMR of morusalbanol A pentamethyl ether 206 in CDCl3 (top), and toluene-d8 (bottom)

171
Comparison of 13
C NMR of morusalbanol A pentamethyl ether 206 in CDCl3 (top), and toluene-d8 (bottom)

172
DEPT-135 for morusalbanol A pentamethyl ether 206 in toluene-d8

173
Comparison of 1H NMR of morusalbanol A pentamethyl ether 206 in toluene-d8 at 25
oC (top) and 80
oC (bottom)

174
Comparison of 1H NMR of morusalbanol A pentamethyl ether 205 at 25
oC and 80
oC (Expansion in the region 1.5 – 4.5 ppm, toluene-d8)

175
13C NMR for morusalbanol A pentamethyl ether 206 at 80
oC (toluene-d8)

176
1H NMR for 207(400 MHz, CDCl3)
*impurities
*
*
*

177
13C NMR (bottom) and DEPT-135 (top) for 207 (CDCl3)

178
HSQC NMR for 207 (CDCl3)

179
HMBC NMR for 207 (CDCl3)

180
Comparison of 1H NMR of 207 in CDCl3 (top) and DMSO-d6 (bottom)
impurities and solvents peaks
*
*
*
*
*
* *
*

181
Comparison of 13
C NMR of 207 in CDCl3 (top) and DMSO-d6 (bottom)
impurities and solvents peaks
*
*

182
1H NMR and
13C NMR of compound 224
183
1H NMR and
13C NMR of compound 226

184
1H NMR and
13C NMR of compound 227

185
1H NMR and
13C NMR of compound 209

186
1H NMR and
13C NMR of compound 232

187
1H NMR and
13C NMR of compound 233

188
1H NMR and
13C NMR of compound 234

189
1H NMR and
13C NMR of compound 237

190
1H NMR and
13C NMR of compound 238

191
1H NMR and
13C NMR of compound 230

192
1H NMR and
13C NMR of compound 231

193
LIST OF PUBLICATIONS
1) Efficient one-pot synthesis of 2,2-dimethyl-2H-chromenes via Pd(II)catalyzed
coupling and SiO2-promoted condensation of o-halophenols with 2-methy-2-buten-
2-ol. Jia Ti Tee, Marzieh Yaeghoobi, Chin Fei Chee, and Noorsaadah Abd.
Rahman, Accepted in Syn. Commun. 2015
2) Model studies on construction of the oxabicyclic [3.3.1] core of mulberry Diels-
Alder adduct morusalbanol A. Jia Ti Tee, Chin Fei Chee, Michael J. C. Buckle,
Vannajan Sanghiran Lee, Wei Lim Chong, Hamid Khaledi, and Noorsaadah Abd
Rahman, Accepted in Tetrahedron Lett. 2015
3) A short diastereoselective synthesis of ()-morusalbanol A methyl ethers. Jia Ti
Tee, Chin Fei Chee, Hamid Khaledi, and Noorsaadah Abd Rahman, (Accpeted in
Synthesis, 2016)
Presentation at the following conferences:
1) Oral Presentation, 2nd
Junior International Conference on Cutting Edge Organic
Chemistry in Asia, 2nd
ICCEOCA (11-14 December 2012), Nanyang Technology
University (Singapore)
2) Poster Presentation, “Studies towards the synthesis of biological active flavonoid
Diels-Alder”, at 5th
HOPE Meeting (26 February - 2 March, 2013), Grand Prince
Hotel New Takanawa (Tokyo, Japan)
3) Oral Presentation, ‘Approach towards the Total Synthesis of Morusalbanol A and
Sorocein B’ 3rd
Junior International Conference on Cutting Edge Organic Chemistry
in Asia, 3rd
ICCEOCA (22-25 November 2013), Seimei-No-Mori, Nanyang
Technology University (Chiba, Japan)

194
4) Poster Presentation, ‘Total Synthesis of Morusalbanol A and Sorocein B’ 5th
UM-
NUS-Chulalongkorn Trilateral Meeting/Seminar (10-12 February 2014), University
Malaya, Malaysia
5) Poster Presentation, ‘Approach towards the Total Synthesis of Morusalbanol A and
Sorocein B’, 4th
Junior International Conference on Cutting Edge Organic
Chemistry in Asia, JICCEOCA-1 (28-30 November 2014), Chulabhorn Research
Institute (CRI) (Bangkok, Thailand)
![TANTAI{GAN GLOBALISASI TERIIADAP DT]NIA PENDIDIKAN ...€¦ · Saja Saja Held. kbas- Disini ini. ini ini hidup Hal dinilai œtiap ini ini yang ini lain (David s. Hal . húek.t kit](https://img.pdfslide.us/doc/110x75/5fdb7e4b80d1e77e8c548e4f/tantaigan-globalisasi-teriiadap-dtnia-pendidikan-saja-saja-held-kbas-disini.jpg)









![Ministry of Public Health · (tnj) OK. (tnj) (Inj) ob. (Ini) (Inj) od. (IthXj) (Inj) 100. (Ini) 190. (Inj) (Ini) lob. (In]) (Ini) Ism. ('tnj) (Ini) bd. (Inj) bd. (tni) (Inj)](https://img.pdfslide.us/doc/110x75/5f0f92947e708231d444d415/ministry-of-public-tnj-ok-tnj-inj-ob-ini-inj-od-ithxj-inj-100.jpg)


















