Embed Size (px)
Citation preview

Structural and Functional Relationships between the Lectinand Arm Domains of Calreticulin*□S
Received for publication, May 5, 2011, and in revised form, May 31, 2011 Published, JBC Papers in Press, June 7, 2011, DOI 10.1074/jbc.M111.258467
Cosmin L. Pocanschi‡1,2, Guennadi Kozlov§1, Ulf Brockmeier‡3, Achim Brockmeier‡, David B. Williams‡4,5,and Kalle Gehring§4,6
From the ‡Departments of Biochemistry and Immunology, University of Toronto, Toronto, Ontario M5S 1A8, Canada and the§Groupe de Recherche Axe sur la Structure des Proteines, Department of Biochemistry, McGill University,Montreal, Quebec H3G 0B1, Canada
Calreticulin and calnexin are key components in maintainingthe quality control of glycoprotein folding within the endoplas-mic reticulum. Although their lectin function of bindingmono-glucosylated sugar moieties of glycoproteins is well docu-mented, their chaperone activity in suppressing proteinaggregation is lesswell understood.Here, we use a series of dele-tion mutants of calreticulin to demonstrate that its aggregationsuppression function resides primarily within its lectin domain.Using hydrophobic peptides as substrate mimetics, we showthat aggregation suppression is mediated through a single poly-peptide binding site that exhibits a Kd for peptides of 0.5–1 �M.This site is distinct from the oligosaccharide binding site anddiffers from previously identified sites of binding to thrombo-spondin and GABARAP (4-aminobutyrate type A receptor-as-sociated protein). Although the arm domain of calreticulin wasincapable of suppressing aggregation or binding hydrophobicpeptides on its own, it did contribute to aggregation suppressionin the context of the whole molecule. The high resolution x-raycrystal structure of calreticulin with a partially truncated armdomain reveals a marked difference in the relative orientationsof the arm and lectin domains when compared with calnexin.Furthermore, a hydrophobic patch was detected on the armdomain that mediates crystal packing and may contribute tocalreticulin chaperone function.
Soluble calreticulin (Crt)7 and membrane-bound calnexin(Cnx) are glycoprotein-specific chaperones of the endoplasmicreticulum (ER). They are components of the quality controlmachinery that prevents the premature release of folding inter-mediates from the ER, and they promote proper folding by pre-venting aggregation and by providing a suitable environment inwhich the associated ERp57 enzyme catalyzes thiol oxidationand isomerization. If proper folding cannot be achieved, theglycoproteins are marked for ER-associated degradation (1–4).Crt and the ER luminal portion of Cnx share a similar struc-
ture (5–7). They consist of a globular lectin domain and anelongated hairpin-like arm domain, the tip of which comprisesthe binding site for ERp57 (8, 9). The arm domain is also knownas the P domain because it consists of two proline-richsequencemotifs that are repeated in tandem three or four timesin Crt and Cnx, respectively. Whereas the structure of the ERluminal portion of Cnx has been solved (7), only the individuallectin and armdomain structures have been determined for Crtby x-ray (6) and NMR (5) methods, respectively. These studiesrevealed the presence of a bound Ca2� ion within the lectindomain that is crucial for chaperone stability (10, 11) as well asthe location of the oligosaccharide binding site. The lectindomain of both chaperones binds monoglucosylated N-linkedoligosaccharides on newly synthesized glycoproteins (12–14),and the removal and re-addition of the terminal glucose residueon these oligosaccharides regulates lectin-based cycles of chap-erone interaction (15). Cnx and Crt also interact with manyfolding glycoproteins through polypeptide-based interactions(for review, see Ref. (4). This has been demonstrated in in vitroexperiments where both Cnx and Crt can suppress the aggre-gation of non-glycosylated substrates (16, 17) as well as in cellswhere lectin-deficient mutants of the chaperones retain func-tion in the biogenesis of class I histocompatibility molecules(18, 19).The lectin sites of Cnx and Crt have been well characterized
through structural and mutagenesis studies, including therecent determination of the structure of the Crt lectin domainin complex with Glc1Man3 tetrasaccharide (6, 7, 9, 20, 21). Incontrast, the polypeptide binding sites are less well character-ized. Early experiments in this regard used in vitro aggregation
* This work was supported by Canadian Institutes of Health Research GrantsMOP-53310 and MOP-81277 (to D. B. W. and K. G.) and the Canadian Can-cer Society (to D. B. W.).
□S The on-line version of this article (available at http://www.jbc.org) containssupplemental Figs. S1–S4.
The atomic coordinates and structure factors (code 3RG0) have been deposited inthe Protein Data Bank, Research Collaboratory for Structural Bioinformatics,Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
1 Both authors contributed equally to this work.2 Recipient of a fellowship from the Canadian Institutes of Health Research
Strategic Training Program in Protein Folding and Interaction Dynamics:Principles and Diseases. Present addresses: Centre for Research in Neuro-degenerative Diseases, University of Toronto, Toronto, Ontario M5S 3H2,Canada and Program in Molecular Structure and Function, Hospital for SickChildren, Toronto, M5G 1X8, Canada.
3 Present address: Institut fur Physiologie, Universitat Duisburg-Essen, Hufe-landstrasse 55, 45122 Essen, Germany.
4 Both are joint corresponding authors.5 To whom correspondence may be addressed: Dept. of Biochemistry, Medi-
cal Sciences Bldg., Rm. 5316, University of Toronto, Toronto, Ontario,M5S 1A8 Canada. Tel.: 416-978-2546; Fax: 416-978-8548; E-mail: [email protected].
6 To whom correspondence may be addressed: Dept. of Biochemistry, McGillUniversity, Rm. 469, Bellini Life Science Complex, 3649 Promenade Sir Wil-liam Osler, Montreal, Quebec H3G 0B1 Canada. Tel.: 514-398-7287; Fax:514-398-2983; E-mail: [email protected].
7 The abbreviations used are: Crt, calreticulin; Cnx, calnexin; ER, endoplasmicreticulum; FL, firefly luciferase; GABARAP, 4-aminobutyrate type A (GABAA)receptor-associated protein; RCMLA, reduced and carboxymethylated�-lactalbumin; G1M3 oligosaccharide (Glc�1-3Man�1-2Man�1-2Man-OH;Tev, tobacco etch virus.
THE JOURNAL OF BIOLOGICAL CHEMISTRY VOL. 286, NO. 31, pp. 27266 –27277, August 5, 2011© 2011 by The American Society for Biochemistry and Molecular Biology, Inc. Printed in the U.S.A.
27266 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 286 • NUMBER 31 • AUGUST 5, 2011
by guest on June 4, 2018http://w
ww
.jbc.org/D
ownloaded from

suppression assays that were performed either in the absence ofCa2� or at temperatures ranging from 42 to 50 °C (9, 20,22–25). The fact thatCa2�plays amajor role in stabilizing thesechaperones as well as their relatively low melting temperatures(46–49 °C) suggest that the observed polypeptide-based inter-actions may have been influenced by partial protein unfolding.However, more recent studies have focused on characterizingpolypeptide-based interactions under physiological conditionsof the ER lumen. In the case of Cnx, it potently suppresses the invitro aggregation of non-glycosylated firefly luciferase at 37 °Cand 0.4 mM Ca2� (10). Furthermore, deletion mutagenesismapped a single polypeptide binding site to its lectin domain ata location distinct from the oligosaccharide binding site.Detailed substrate specificity studies were not undertaken, buttwo hydrophobic peptides were shown to bind to this site withmicromolar affinity (10). Crt also suppresses the in vitro aggre-gation of non-glycosylated proteins under physiological condi-tions (17, 26). The specificity of its polypeptide binding site hasbeen extensively characterized in vitro using a competitiveenzyme-linked immunosorbent assay. Crt binds to denaturedbut not native proteins, and analysis of a broad range of peptidesubstrates revealed that binding required a length of at least fiveresidues that was hydrophobic in character (27, 28). Severalbinding sites for peptides or proteins have been mapped ontoCrt (29–31), but the relationship between these sites and thesite that mediates suppression of protein aggregation remainsunknown. Furthermore, the relative roles of the arm and lectindomains in either aggregation suppression or direct peptidebinding are unclear.In this study we investigate structural and functional rela-
tionships of the arm and lectin domains of Crt. We use variousarm truncation mutants to show that the arm domain contrib-utes to the aggregation suppression function of Crt, but theprimary polypeptide binding site is located within the lectindomain. This site binds hydrophobic peptides withmicromolardissociation constants and is distinct from Crt sites that inter-act with partners such as GABARAP and thrombospondin.Wealso provide the first crystal structure of Crt with a partial armdomain and identify a hydrophobic patch on the arm that maycontribute to Crt chaperone function.
EXPERIMENTAL PROCEDURES
Materials—The hep1 peptide (ELTGAARKGSGRRLVK-GPD), 6KSGG peptide (KKKKKKSGGSGGSGGSC) with an
amidated C terminus and fCLV peptide (FITC-Ahx-CLVLF-VAMWSD, where Ahx is NH2-(CH2)5-COOH) were synthe-sized by GenScript Corp. (Piscataway, NJ). The 6KAAF peptide(KKKKKKAAFAAFAAFAA) and the KHP peptide (KHPYAY-LAAAIAAEVAGTTALKLSK) were provided by Dr. CharlesDeber, SickKids, Toronto. Human GABARAP (TrEMBLMM46, accession number O95166) was obtained from Dr.DieterWillbold, Forschungszentrum Julich, Germany. All pep-tides were purified by reverse-phase HPLC, dissolved in 20 mM
Hepes, pH 7.4, 150mMNaCl, and 0.4 mMCaCl2, separated intoaliquots, and stored at �70 °C. The G1M3 oligosaccharide(Glc�1–3Man�1–2Man�1–2Man-OH) was purchased fromthe Alberta Research Council (Edmonton, Alberta, Canada).Mutagenesis of Calreticulin cDNA, Protein Expression, and
Purification—Amino acid numbering refers to the mouse Crtsequence, with residue 1 corresponding to the first residue ofthe signal sequence. All primers for PCR-based amplification ormutagenesis are listed in Table 1. Full-length Crt (residues18–417) and its arm domain (arm, residues 207–300) wereamplified in standard PCR reactions using as template mouseCrt cDNA in pcDNA3.1 (19). Both PCR-generated fragmentswere digested with NdeI and BamHI and ligated into theexpression vector pET15b-Tev, a modified pET15b vector(Novagen) that encodes an N-terminal His6 tag followed by atobacco etch virus (Tev) protease cleavage site. The unpairedCys at residue 163 inCrt was subsequentlymutated to Ser usingQuikChange mutagenesis with pET15b-Tev-Crt as template.The resulting pET15b-Tev-CrtCS was then used as the tem-plate to generate two variants of Crt with a truncated armdomain, �arm1 (�aa239–272) and �arm2 (�aa223–286). Themutagenic primers for these constructs introduced a GGSGand a GSG linker, respectively, at the site of truncation. Thelectin domain (residues 18–206 and 301–417) was obtained byPCR amplification using pET15b-Tev-CrtCS as template, andthe primers incorporated aGSGSG linker between residues 206and 301. Each construct was verified by DNA sequencing. Crtand its various deletion mutants were expressed in Escherichiacoli BL21-CodonPlus cells (Stratagene) and purified as previ-ously described for calnexin (10). Typical yields from 2 liters ofbacterial culture were 23 mg (Crt), 20 mg (�arm1), 16 mg(�arm2), 7 mg (lectin), and 32 mg (arm).For crystallization, a variant of the Crt �arm1 construct was
created. This variant, termed Crt�arm1�C368, featured the
TABLE 1Oligonucleotides used in this studyRestriction sites are underlined, stop codons are shown in italics, and introduced codons are depicted in bold.
Primer Sequence 5�3 3� Modifications
wt-Crt forward atatatatatcatatggaccctgccatctatttcaaagag NdeIwt-Crt reverse atatatggatccttagagctcatccttggcttggccag BamHI, StopArm forward atatatatatcatatgaagataaaggaccctgatgctg NdeIArm reverse atatatggatccttaggagtattcagggttgtcaatttc BamHI, Stop�arm1 forward ccaagcctgaggactgggacaagggcggtagtggtggcgagtggaaaccacgtcaaattgacaacccag GGSG linker�arm1 reverse gacgtggtttccactcgccaccactaccgcccttgtcccagtcctcaggcttggaatctgtggggtcatcg GGSG linker�arm2 forward gactgggatgaacgaggcagtggcggtacctggatacacccagaaattgacaaccctgaatactcccccgatgc GSG linker�arm2 reverse cagggttgtcaatttctgggtgtatccaggtaccgccactgcctcgttcatcccagtcttccggcttggcagcatc GSG linkerLectin forward gactttctgccacccaagggcagcgggagcggccccgatgcaaatatctatgcctatgatagttttgc GSGSG linkerLectin reverse ggcatagatatttgcatcggggccgctcccgctgcccttgggtggcagaaagtcccaatcatcctcc GSGSG linkerC163S forward gtgctgatcaacaaggatatccggtctaaggatgatg C163SC163S reverse gtgtgtgaattcatcatccttagaccggatatccttg C163S
Structure and Chaperone Functions of Calreticulin Domains
AUGUST 5, 2011 • VOLUME 286 • NUMBER 31 JOURNAL OF BIOLOGICAL CHEMISTRY 27267
by guest on June 4, 2018http://w
ww
.jbc.org/D
ownloaded from

same arm domain truncation as �arm1 as well as the C163Smutation (see above) but also included a shorter C terminusending at residue 368. This variant was inserted into thepET29a vector (Amersham Biosciences) and expressed inE. coli BL21(DE3) in LB medium as an N-terminal and C-ter-minalHis-tag fusion. Cells were harvested and broken in 50mM
Tris, 300mMNaCl, 3mMCaCl2, pH 8.0. The fusion protein waspurified by affinity chromatography on Ni2�-charged Sephar-ose resin, and theN-terminal tag was removed by cleavage withthrombin, leaving a Gly-Ser-Met N-terminal extension and aLeu-Glu-His-His-His-His-His-His C-terminal extension. Thecleaved protein was additionally purified using size-exclusionchromatography with 20 mM Tris, 100 mM NaCl, 3 mM CaCl2,pH 7.5 buffer.Crystallization—Initial crystallization conditions were iden-
tified utilizing hanging drop vapor diffusion using the PACTscreen (Qiagen). The best crystals were obtained at 22 °C bymixing a 0.6-�l drop of Crt�arm1�C368 (10 mg/ml in 20 mM
Tris, 100mMNaCl, 3mMCaCl2, pH 7.5) with 0.6�l of reservoirsolution containing 25% (w/v) polyethylene glycol 1500 and 25mM sodium malonate, 37.5 mM imidazole, 37.5 mM boric acid,pH 9.0, and suspending over 0.6 ml of reservoir solution. Noadditional cryoprotectant was necessary. For data collection,crystals were picked up in a nylon loop and flash-cooled in a N2cold stream (Oxford Cryosystem). The crystals contain onemolecule in the asymmetric unit (Z � 4) corresponding toVm � 2.2 Å3 Da�1 and a solvent content of 44%.Structure Solution and Refinement—The native dataset from
a crystal of Crt�arm1�C368 was collected using a single wave-length (0.9779 Å) regime on an ADSC Quantum-210 CCDdetector (Area Detector Systems Corp.) at beamline A1 at theCornell High Energy Synchrotron Source (Table 2). Data proc-essing and scaling were performed with HKL2000 (32). Thestarting phases were obtained using molecular replacementwith the Crt lectin domain structure (PDB code 3o0v) using
PHASER (33). The resulting model was extended manuallywith the help of the program Coot (34) and was improved byseveral cycles of refinement using the program REFMAC (35)followed by the translation-libration-screw (TLS) refinement(36). Of 332 residues in the construct, the final model does notinclude the N-terminal GSM residues remaining after throm-bin cleavage, residuesAsp-226—Lys-238, theGGSG linker thatreplaces the outermost repeat motifs of the arm domain, Gly-273–Pro-283, and the C-terminal residues 368KLEHHHHHH.In addition, 1 calcium ion and 26 water molecules wereincluded in the model. The final model has good stereo-chemistry with no outliers in the Ramachandran plot com-puted using PROCHECK (37).Aggregation Assay—Firefly luciferase (FL, Promega) was dis-
solved at a concentration of 300�M in 20mMHepes, pH7.4, 150mM NaCl, 1 mM CaCl2, 0.5% glycerol, and stored in aliquots at�70 °C. They were thawed only once before use. Crt constructswere equilibrated in 20 mM Hepes, pH 7.4, 150 mM NaCl, and0.4mMCaCl2 in the presence or absence of peptides (20�M) for10 min at 37 °C. FL was subsequently added to 3 �M final con-centration in a total volume of 150 �l. Aggregation was moni-tored continuously over 1 h at 37 °C bymeasuring light scatter-ing at 360 nm using a Shimadzu 1601 spectrophotometerequipped with a temperature-controlled cuvette holder. Datawere recorded every 6 s.Fluorescence Measurements—Crt constructs (1 �M) were
equilibrated for a period of 1 h at 25 °C in 20mMHepes, pH 7.4,150 mM NaCl, and 0.4 mM CaCl2 in the presence or absence ofvarious peptides (20 �M). Tryptophan fluorescence emissionspectra were then recorded at 25 °C from 290 to 390 nm usingan excitationwavelength of 280 nm.AFluorolog 3Horiba JobinYvon fluorescence spectrofluorometer was used with excita-tion and emission slit widths set to 2 and 5 nm, respectively.When titrating with various peptides, samples were equili-brated for 10 min between the titration steps. Fluorescencemeasurements were corrected by subtracting the peptide con-tribution to fluorescence intensity.Circular Dichroism Experiments—Solutions of Crt or dele-
tion mutants (3.5 �M) were prepared in 8 mM Hepes, pH 7.4,150 mMNaCl, 1 mM EDTA, and 1.4 mMCaCl2 and equilibratedfor 10 min at 37 °C (room temperature for lectin and arm con-structs) before CD spectra were recorded between 200 and 260nm. In thermal denaturation experiments, Crt or its deletionmutants (3.5�M)were equilibrated in 20mMHepes, pH7.4, 150mM NaCl, 1 mM EDTA containing either 1.0 or 1.4 mM CaCl2for 10 min at room temperature. Melting curves were recordedin a 1-mm path length cuvette from 20 to 70 °C at 228 nm witha scan rate of 2 °C/min. SigmaPlot 2004 Version 9.0 was used tofit the thermal denaturation data to a standard equation bynonlinear least-squares regression assuming a two-state transi-tion process. Tm values represent the midpoint temperature ofthe thermal unfolding transition. A Jasco J-810 spectropolarim-eter equipped with a PTC-423S temperature controlling unitwas used for all CD measurements. The results are shown asmean residue molar ellipticity [�] with units converted todegrees cm2 dmol�1.
TABLE 2Data collection and refinement statistics
Crt�arm1C368
Data collectionSpace group P212121Cell dimensions, a, b, c (Å) 43.06, 71.70, 108.39Resolution (Å) 50-2.55 (2.59-2.55)aRsym 0.053 (0.408)I/�I 23.6 (2.2)Completeness (%) 96.9 (77.9)Redundancy 3.7 (2.3)
RefinementResolution (Å) 59.8-2.57No. reflections 10,321Rwork/Rfree 0.234/0.285No. atomsProtein 2332Calcium ions 1Water 26
B-FactorsProtein 44.9Calcium ions 54.0Water 54.3
Root mean square deviationsBond lengths (Å) 0.006Bond angles (°) 1.06
Ramachandran statistics (%)Most favored regions 87.8Additional allowed regions 12.2
a The highest resolution shell is shown in parentheses.
Structure and Chaperone Functions of Calreticulin Domains
27268 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 286 • NUMBER 31 • AUGUST 5, 2011
by guest on June 4, 2018http://w
ww
.jbc.org/D
ownloaded from

FITC Labeling of Crt Substrates and Anisotropy Mea-surements—Various peptide and protein substrates of Crt weredissolved in 1ml of 50mMHEPES, pH 7.5, at a concentration of200 �M. Tris(2-carboxyethyl)phosphine (1mM) was added, andthe reaction mixtures were incubated for 15 min at room tem-perature. Fluorescein-5-maleimide dissolved in DMF wasadded at 5:1 molar excess to the Crt substrate, and the sampleswere incubated for 2 h in the dark. The reactions werequenched by adding a 20-fold molar excess of �-mercaptoeth-anol over labeling reagent and incubated for 30min in the dark.The samples were then centrifuged at 16,000 � g for 10 min.The supernatants were loaded on a preparative C-18 reversephase column, and the peptides/proteins were eluted using agradient of acetonitrile and water, both with 0.1% trifluoroace-tic acid. Initially, a small amount of the reactionmixture (50�l)was run to identify the elution time of the labeled peptides/proteins (at 215 nm). Subsequently, the entire reactionmixturewas run in the dark without using the UV detector, and theeluate corresponding to the labeled peptides/proteins was col-lected. Peptide and protein concentrations were determinedusing the bicinchoninic acid assay.Fluorescence Anisotropy Measurements—Various FITC-la-
beled Crt substrates were mixed with individual Crt constructsin 150 �l of 20 mM Hepes, pH 7.4, 150 mM NaCl, and 0.4 mM
CaCl2 at the indicated concentrations and incubation times asdescribed in the figure legends corresponding to each specificexperiment. Using a 10-mm quartz cuvette, fluorescence ani-sotropy was measured at 25 °C on a Fluorolog 3 Horiba JobinYvon fluorescence spectrofluorometer equipped with polariz-ers with excitation and emission slit widths set to 3.75 nm. Theexcitation and emission wavelengths were 491 and 518 nm,respectively. The increment was set to 0.5 nm, and the integra-tion timewas set to 2000ms. A total of five scans were recordedand averaged.Reduction and Carboxymethylation of �-Lactalbumin—Bo-
vine �-lactalbumin (500 �M, Sigma) was reduced and car-boxymethylated (RCMLA) as previously described (38).RCMLA was dialyzed, separated into aliquots, and stored at�70 °C in 20 mM Hepes, pH 7.4, 150 mM NaCl, 1 mM CaCl2.
RESULTS
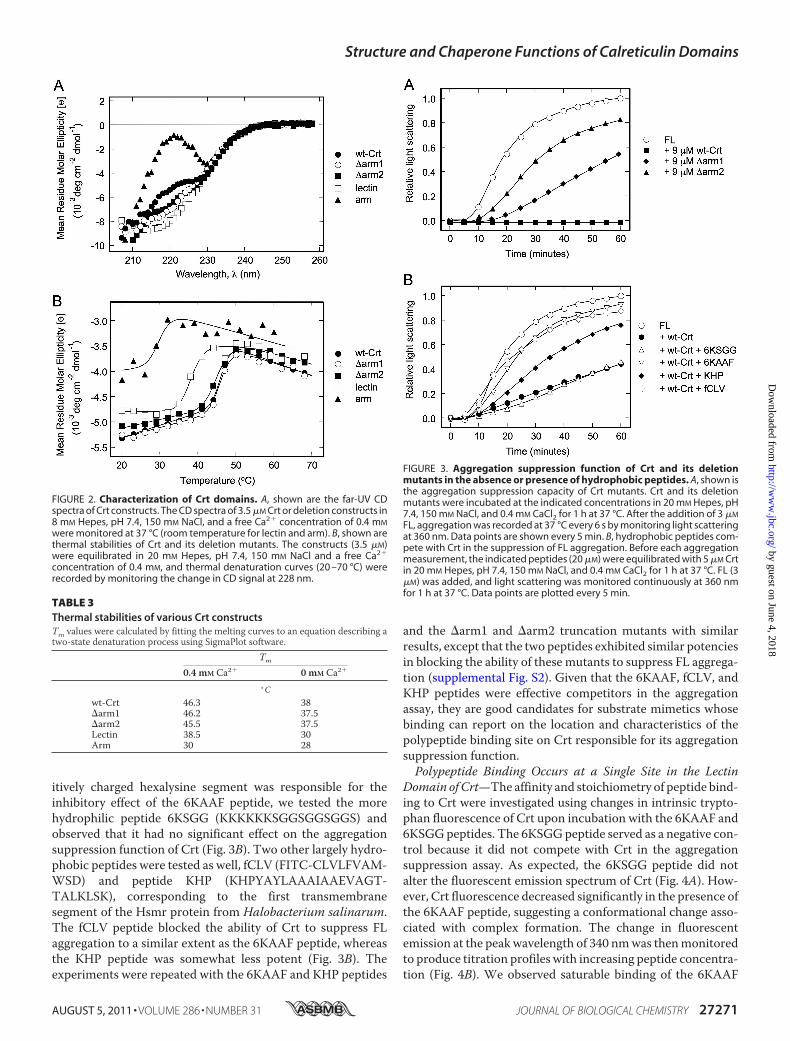
Conformational Assessment of Calreticulin DeletionMutants—To localize the polypeptide binding site thatmediates the chap-erone function of Crt, several deletion mutants were con-structed that included the individual arm and lectin domains aswell as progressive truncations of the arm domain (Fig. 1A).The structural integrity of these constructs was assessed usingfar-UV CD spectroscopy. The CD spectrum of wild type Crtshown in Fig. 2A had a negative band at �226 nm and closelyresembled that described previously for Crt in the presence ofCa2� (11). Likewise, the CD spectra of the isolated lectindomain and the arm domain, the latter with its strong negativemaximum at 230 nm, were similar to published spectra (39).The �arm1 and �arm2 constructs exhibited spectra interme-diate between that of wild type Crt and its lectin domain, con-sistent with stepwise truncations of the arm (Fig. 2A).To assess the stabilities of the Crt constructs, their thermal
unfolding curves were measured from 20 to 70 °C. In the pres-
ence of Ca2�, a midpoint temperature of unfolding (Tm) of46.3 °C was calculated for wild type Crt, in agreement with pre-vious findings (11, 26) (Fig. 2B and Table 3). Partial truncationof the arm domain by roughly 33 and 66% in the �arm1 and�arm2 constructs, respectively, did not lead to significantdestabilization of Crt, whereas complete removal of the armdomain reduced the Tm to 38.5 °C. The isolated arm domainalso exhibited a markedly lower Tm of 30 °C. The reduced sta-bilities of the isolated lectin and arm domain have been notedpreviously (39) and contrast with Cnx where the individualdomains exhibit only modestly reduced stabilities (10). Despitethe reduced stabilities of the lectin and arm domains of Crt, wedemonstrated previously that they retain the ability to bindmonoglucosylated oligosaccharide and ERp57, their respectiveligands (6, 9). In the absence of Ca2�, the thermal stabilities ofthe Crt constructs were significantly decreased except for thearm domain (Table 3). This indicates that all constructs con-taining the lectin domain bind Ca2�, consistent with the boundCa2� ion detected in the crystal structure of the Crt lectindomain (6). Collectively, these findings indicate that the trun-cation mutants used in this study retain native structure.Participation of Crt Domains in Suppressing Firefly Lucifer-
ase Aggregation—To localize the domain responsible for theaggregation suppression (chaperone) function of Crt, the vari-ous truncationmutants were tested for their abilities to preventthe aggregation of FL. FL is a non-glycosylated protein that hasbeen used in the past as a substrate in aggregation suppressionassays to characterize polypeptide-based interactions with Cnxunder physiological conditions of the ER, i.e. 37 °C and 0.4 mM
Ca2� (10, 16). Initially, we compared the abilities of Crt and itstruncationmutants to suppress the aggregation of FL at a 3-foldmolar excess of chaperone over substrate. As shown in Fig. 3A,wild type Crt completely suppressed FL aggregation underthese conditions, whereas the �arm1 construct only partiallysuppressed aggregation; the �arm2 construct was even lesspotent than �arm1. To express their relative potencies in amore quantitative manner, we compared the concentrations ofCrt and its truncation mutants required to suppress the aggre-gation of 3�MFL to 40–45%of the level observedwith FL alone(supplemental Fig. S1), which is a more sensitive assay than thecomplete suppression of FL aggregation. For wild type Crt, a 5�M concentration suppressed FL aggregation to 40–45%. Theconcentrations of �arm1 and �arm2 required to suppress FLaggregation to the same extent were 11 and 14�M, respectively.In other words, �arm1 and �arm2 were 2.2- and 2.8-fold lesspotent than wild type Crt, respectively, indicative of a role forthe armdomain in aggregation suppression.Unfortunately, dueto their reduced thermal stabilities, neither the individual armnor lectin domains could be tested in this 37 °C assay. Conse-quently, we explored whether small peptides could be used assubstrate mimetics to further characterize the polypeptidebinding site of Crt at temperatures where all of the deletionconstructs remained stable.Hydrophobic Peptides Block the Ability of Crt to Suppress FL
Aggregation—In an effort to identify substrate mimetics, wetested whether several hydrophobic peptides could competewith Crt in suppressing FL aggregation. A Crt:FL ratio wasagain chosen to provide only �40% aggregation suppression as
Structure and Chaperone Functions of Calreticulin Domains
AUGUST 5, 2011 • VOLUME 286 • NUMBER 31 JOURNAL OF BIOLOGICAL CHEMISTRY 27269
by guest on June 4, 2018http://w
ww
.jbc.org/D
ownloaded from
a sensitive readout of the effects of different peptides. None ofthe four peptides tested affected the aggregation of FL on theirown (data not shown). In contrast, when Crt was preincubated
for 1 h with peptide 6KAAF (KKKKKKAAFAAFAAFAA), itsability to subsequently suppress FL aggregation was almostfully blocked (Fig. 3B). To rule out the possibility that the pos-
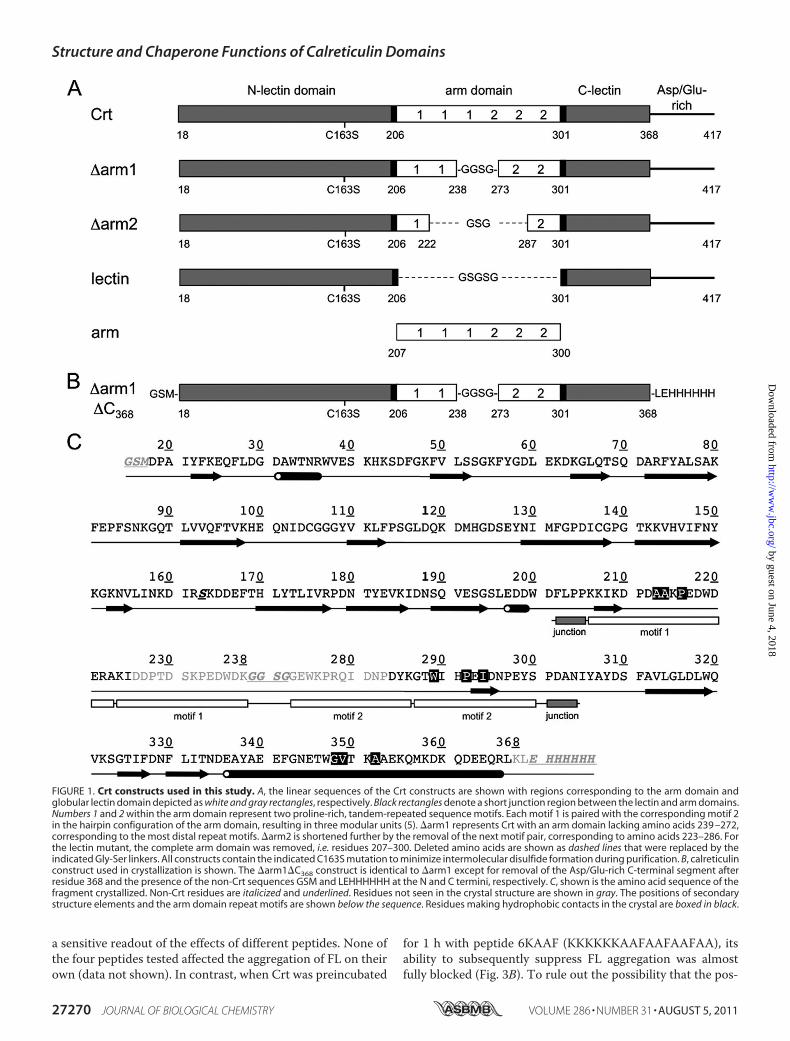
FIGURE 1. Crt constructs used in this study. A, the linear sequences of the Crt constructs are shown with regions corresponding to the arm domain andglobular lectin domain depicted as white and gray rectangles, respectively. Black rectangles denote a short junction region between the lectin and arm domains.Numbers 1 and 2 within the arm domain represent two proline-rich, tandem-repeated sequence motifs. Each motif 1 is paired with the corresponding motif 2in the hairpin configuration of the arm domain, resulting in three modular units (5). �arm1 represents Crt with an arm domain lacking amino acids 239 –272,corresponding to the most distal repeat motifs. �arm2 is shortened further by the removal of the next motif pair, corresponding to amino acids 223–286. Forthe lectin mutant, the complete arm domain was removed, i.e. residues 207–300. Deleted amino acids are shown as dashed lines that were replaced by theindicated Gly-Ser linkers. All constructs contain the indicated C163S mutation to minimize intermolecular disulfide formation during purification. B, calreticulinconstruct used in crystallization is shown. The �arm1�C368 construct is identical to �arm1 except for removal of the Asp/Glu-rich C-terminal segment afterresidue 368 and the presence of the non-Crt sequences GSM and LEHHHHHH at the N and C termini, respectively. C, shown is the amino acid sequence of thefragment crystallized. Non-Crt residues are italicized and underlined. Residues not seen in the crystal structure are shown in gray. The positions of secondarystructure elements and the arm domain repeat motifs are shown below the sequence. Residues making hydrophobic contacts in the crystal are boxed in black.
Structure and Chaperone Functions of Calreticulin Domains
27270 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 286 • NUMBER 31 • AUGUST 5, 2011
by guest on June 4, 2018http://w
ww
.jbc.org/D
ownloaded from
itively charged hexalysine segment was responsible for theinhibitory effect of the 6KAAF peptide, we tested the morehydrophilic peptide 6KSGG (KKKKKKSGGSGGSGGS) andobserved that it had no significant effect on the aggregationsuppression function of Crt (Fig. 3B). Two other largely hydro-phobic peptides were tested as well, fCLV (FITC-CLVLFVAM-WSD) and peptide KHP (KHPYAYLAAAIAAEVAGT-TALKLSK), corresponding to the first transmembranesegment of the Hsmr protein from Halobacterium salinarum.The fCLV peptide blocked the ability of Crt to suppress FLaggregation to a similar extent as the 6KAAF peptide, whereasthe KHP peptide was somewhat less potent (Fig. 3B). Theexperiments were repeated with the 6KAAF and KHP peptides
and the �arm1 and �arm2 truncation mutants with similarresults, except that the two peptides exhibited similar potenciesin blocking the ability of these mutants to suppress FL aggrega-tion (supplemental Fig. S2). Given that the 6KAAF, fCLV, andKHP peptides were effective competitors in the aggregationassay, they are good candidates for substrate mimetics whosebinding can report on the location and characteristics of thepolypeptide binding site on Crt responsible for its aggregationsuppression function.Polypeptide Binding Occurs at a Single Site in the Lectin
Domain of Crt—The affinity and stoichiometry of peptide bind-ing to Crt were investigated using changes in intrinsic trypto-phan fluorescence of Crt upon incubation with the 6KAAF and6KSGGpeptides. The 6KSGGpeptide served as a negative con-trol because it did not compete with Crt in the aggregationsuppression assay. As expected, the 6KSGG peptide did notalter the fluorescent emission spectrum of Crt (Fig. 4A). How-ever, Crt fluorescence decreased significantly in the presence ofthe 6KAAF peptide, suggesting a conformational change asso-ciated with complex formation. The change in fluorescentemission at the peakwavelength of 340 nmwas thenmonitoredto produce titration profiles with increasing peptide concentra-tion (Fig. 4B). We observed saturable binding of the 6KAAF
FIGURE 2. Characterization of Crt domains. A, shown are the far-UV CDspectra of Crt constructs. The CD spectra of 3.5 �M Crt or deletion constructs in8 mM Hepes, pH 7.4, 150 mM NaCl, and a free Ca2� concentration of 0.4 mM
were monitored at 37 °C (room temperature for lectin and arm). B, shown arethermal stabilities of Crt and its deletion mutants. The constructs (3.5 �M)were equilibrated in 20 mM Hepes, pH 7.4, 150 mM NaCl and a free Ca2�
concentration of 0.4 mM, and thermal denaturation curves (20 –70 °C) wererecorded by monitoring the change in CD signal at 228 nm.
TABLE 3Thermal stabilities of various Crt constructsTm values were calculated by fitting the melting curves to an equation describing atwo-state denaturation process using SigmaPlot software.
Tm
0.4 mM Ca2� 0 mM Ca2�
° Cwt-Crt 46.3 38�arm1 46.2 37.5�arm2 45.5 37.5Lectin 38.5 30Arm 30 28
FIGURE 3. Aggregation suppression function of Crt and its deletionmutants in the absence or presence of hydrophobic peptides. A, shown isthe aggregation suppression capacity of Crt mutants. Crt and its deletionmutants were incubated at the indicated concentrations in 20 mM Hepes, pH7.4, 150 mM NaCl, and 0.4 mM CaCl2 for 1 h at 37 °C. After the addition of 3 �M
FL, aggregation was recorded at 37 °C every 6 s by monitoring light scatteringat 360 nm. Data points are shown every 5 min. B, hydrophobic peptides com-pete with Crt in the suppression of FL aggregation. Before each aggregationmeasurement, the indicated peptides (20 �M) were equilibrated with 5 �M Crtin 20 mM Hepes, pH 7.4, 150 mM NaCl, and 0.4 mM CaCl2 for 1 h at 37 °C. FL (3�M) was added, and light scattering was monitored continuously at 360 nmfor 1 h at 37 °C. Data points are plotted every 5 min.
Structure and Chaperone Functions of Calreticulin Domains
AUGUST 5, 2011 • VOLUME 286 • NUMBER 31 JOURNAL OF BIOLOGICAL CHEMISTRY 27271
by guest on June 4, 2018http://w
ww
.jbc.org/D
ownloaded from
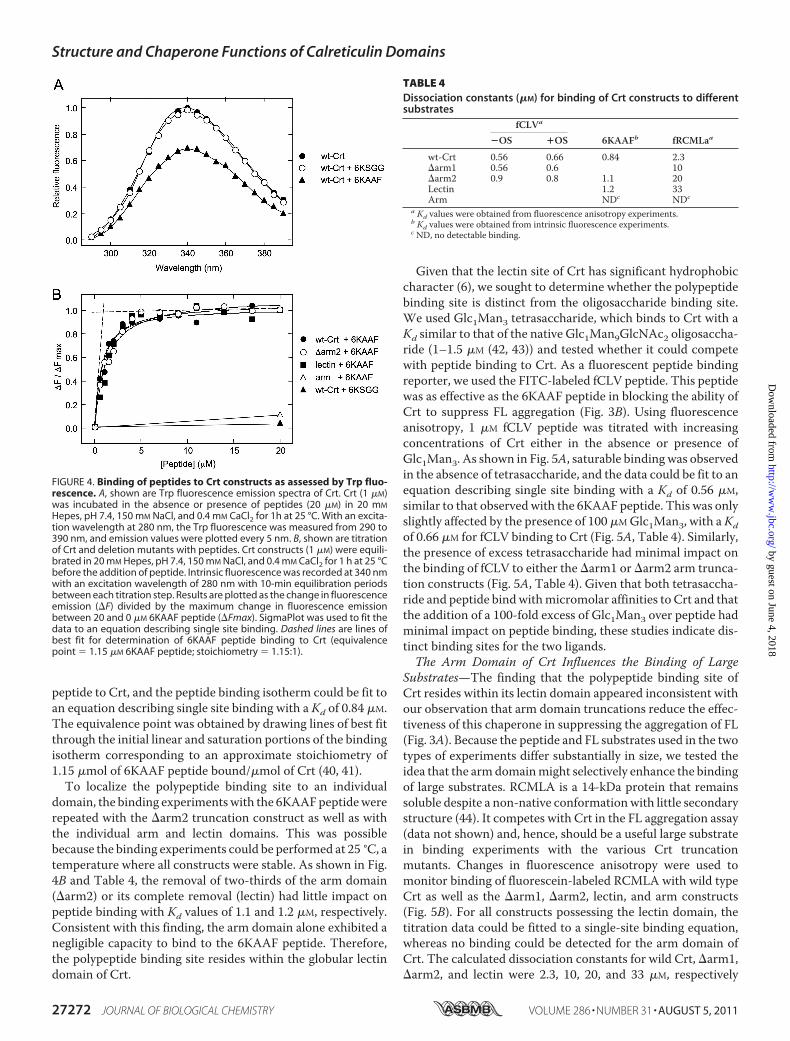
peptide to Crt, and the peptide binding isotherm could be fit toan equation describing single site binding with a Kd of 0.84 �M.The equivalence point was obtained by drawing lines of best fitthrough the initial linear and saturation portions of the bindingisotherm corresponding to an approximate stoichiometry of1.15 �mol of 6KAAF peptide bound/�mol of Crt (40, 41).To localize the polypeptide binding site to an individual
domain, the binding experimentswith the 6KAAFpeptidewererepeated with the �arm2 truncation construct as well as withthe individual arm and lectin domains. This was possiblebecause the binding experiments could be performed at 25 °C, atemperature where all constructs were stable. As shown in Fig.4B and Table 4, the removal of two-thirds of the arm domain(�arm2) or its complete removal (lectin) had little impact onpeptide binding with Kd values of 1.1 and 1.2 �M, respectively.Consistent with this finding, the arm domain alone exhibited anegligible capacity to bind to the 6KAAF peptide. Therefore,the polypeptide binding site resides within the globular lectindomain of Crt.
Given that the lectin site of Crt has significant hydrophobiccharacter (6), we sought to determine whether the polypeptidebinding site is distinct from the oligosaccharide binding site.We used Glc1Man3 tetrasaccharide, which binds to Crt with aKd similar to that of the native Glc1Man9GlcNAc2 oligosaccha-ride (1–1.5 �M (42, 43)) and tested whether it could competewith peptide binding to Crt. As a fluorescent peptide bindingreporter, we used the FITC-labeled fCLV peptide. This peptidewas as effective as the 6KAAF peptide in blocking the ability ofCrt to suppress FL aggregation (Fig. 3B). Using fluorescenceanisotropy, 1 �M fCLV peptide was titrated with increasingconcentrations of Crt either in the absence or presence ofGlc1Man3. As shown in Fig. 5A, saturable binding was observedin the absence of tetrasaccharide, and the data could be fit to anequation describing single site binding with a Kd of 0.56 �M,similar to that observed with the 6KAAF peptide. This was onlyslightly affected by the presence of 100 �MGlc1Man3, with aKdof 0.66 �M for fCLV binding to Crt (Fig. 5A, Table 4). Similarly,the presence of excess tetrasaccharide had minimal impact onthe binding of fCLV to either the �arm1 or �arm2 arm trunca-tion constructs (Fig. 5A, Table 4). Given that both tetrasaccha-ride and peptide bindwithmicromolar affinities to Crt and thatthe addition of a 100-fold excess of Glc1Man3 over peptide hadminimal impact on peptide binding, these studies indicate dis-tinct binding sites for the two ligands.The Arm Domain of Crt Influences the Binding of Large
Substrates—The finding that the polypeptide binding site ofCrt resides within its lectin domain appeared inconsistent withour observation that arm domain truncations reduce the effec-tiveness of this chaperone in suppressing the aggregation of FL(Fig. 3A). Because the peptide and FL substrates used in the twotypes of experiments differ substantially in size, we tested theidea that the armdomainmight selectively enhance the bindingof large substrates. RCMLA is a 14-kDa protein that remainssoluble despite a non-native conformationwith little secondarystructure (44). It competes with Crt in the FL aggregation assay(data not shown) and, hence, should be a useful large substratein binding experiments with the various Crt truncationmutants. Changes in fluorescence anisotropy were used tomonitor binding of fluorescein-labeled RCMLA with wild typeCrt as well as the �arm1, �arm2, lectin, and arm constructs(Fig. 5B). For all constructs possessing the lectin domain, thetitration data could be fitted to a single-site binding equation,whereas no binding could be detected for the arm domain ofCrt. The calculated dissociation constants for wild Crt, �arm1,�arm2, and lectin were 2.3, 10, 20, and 33 �M, respectively
FIGURE 4. Binding of peptides to Crt constructs as assessed by Trp fluo-rescence. A, shown are Trp fluorescence emission spectra of Crt. Crt (1 �M)was incubated in the absence or presence of peptides (20 �M) in 20 mM
Hepes, pH 7.4, 150 mM NaCl, and 0.4 mM CaCl2 for 1h at 25 °C. With an excita-tion wavelength at 280 nm, the Trp fluorescence was measured from 290 to390 nm, and emission values were plotted every 5 nm. B, shown are titrationof Crt and deletion mutants with peptides. Crt constructs (1 �M) were equili-brated in 20 mM Hepes, pH 7.4, 150 mM NaCl, and 0.4 mM CaCl2 for 1 h at 25 °Cbefore the addition of peptide. Intrinsic fluorescence was recorded at 340 nmwith an excitation wavelength of 280 nm with 10-min equilibration periodsbetween each titration step. Results are plotted as the change in fluorescenceemission (�F) divided by the maximum change in fluorescence emissionbetween 20 and 0 �M 6KAAF peptide (�Fmax). SigmaPlot was used to fit thedata to an equation describing single site binding. Dashed lines are lines ofbest fit for determination of 6KAAF peptide binding to Crt (equivalencepoint � 1.15 �M 6KAAF peptide; stoichiometry � 1.15:1).
TABLE 4Dissociation constants (�M) for binding of Crt constructs to differentsubstrates
fCLVa
6KAAFb fRCMLaa�OS �OS
wt-Crt 0.56 0.66 0.84 2.3�arm1 0.56 0.6 10�arm2 0.9 0.8 1.1 20Lectin 1.2 33Arm NDc NDc
a Kd values were obtained from fluorescence anisotropy experiments.b Kd values were obtained from intrinsic fluorescence experiments.c ND, no detectable binding.
Structure and Chaperone Functions of Calreticulin Domains
27272 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 286 • NUMBER 31 • AUGUST 5, 2011
by guest on June 4, 2018http://w
ww
.jbc.org/D
ownloaded from
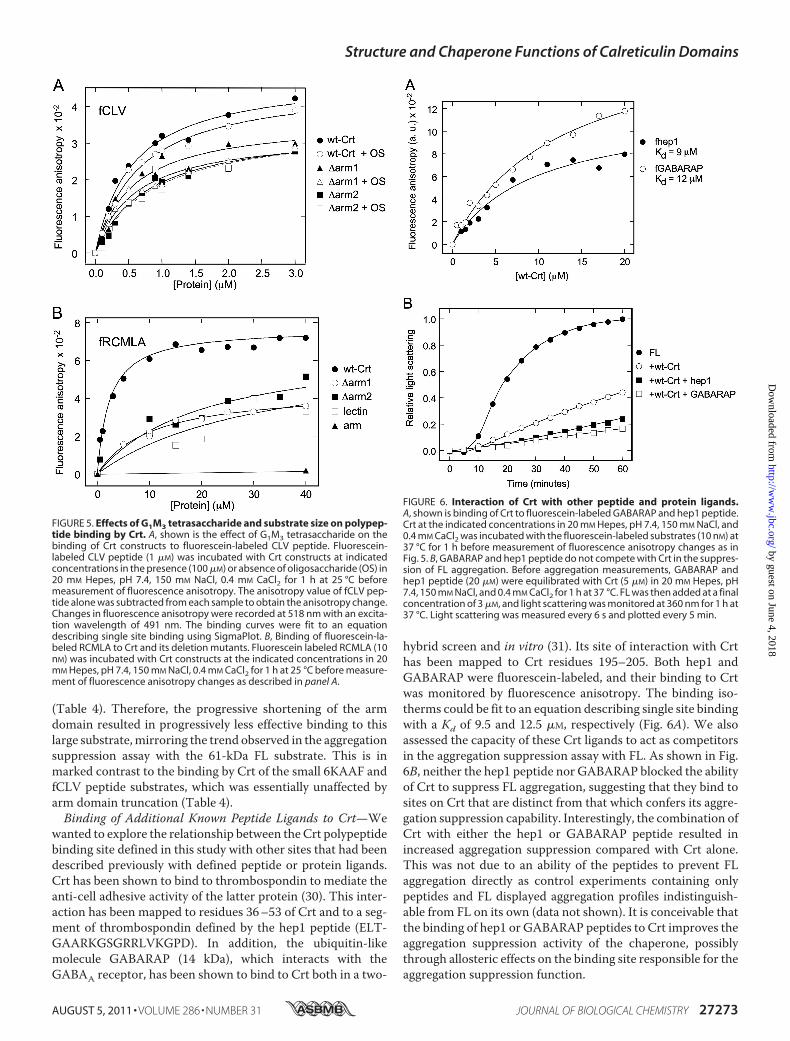
(Table 4). Therefore, the progressive shortening of the armdomain resulted in progressively less effective binding to thislarge substrate,mirroring the trend observed in the aggregationsuppression assay with the 61-kDa FL substrate. This is inmarked contrast to the binding by Crt of the small 6KAAF andfCLV peptide substrates, which was essentially unaffected byarm domain truncation (Table 4).Binding of Additional Known Peptide Ligands to Crt—We
wanted to explore the relationship between the Crt polypeptidebinding site defined in this study with other sites that had beendescribed previously with defined peptide or protein ligands.Crt has been shown to bind to thrombospondin to mediate theanti-cell adhesive activity of the latter protein (30). This inter-action has been mapped to residues 36–53 of Crt and to a seg-ment of thrombospondin defined by the hep1 peptide (ELT-GAARKGSGRRLVKGPD). In addition, the ubiquitin-likemolecule GABARAP (14 kDa), which interacts with theGABAA receptor, has been shown to bind to Crt both in a two-
hybrid screen and in vitro (31). Its site of interaction with Crthas been mapped to Crt residues 195–205. Both hep1 andGABARAP were fluorescein-labeled, and their binding to Crtwas monitored by fluorescence anisotropy. The binding iso-therms could be fit to an equation describing single site bindingwith a Kd of 9.5 and 12.5 �M, respectively (Fig. 6A). We alsoassessed the capacity of these Crt ligands to act as competitorsin the aggregation suppression assay with FL. As shown in Fig.6B, neither the hep1 peptide nor GABARAP blocked the abilityof Crt to suppress FL aggregation, suggesting that they bind tosites on Crt that are distinct from that which confers its aggre-gation suppression capability. Interestingly, the combination ofCrt with either the hep1 or GABARAP peptide resulted inincreased aggregation suppression compared with Crt alone.This was not due to an ability of the peptides to prevent FLaggregation directly as control experiments containing onlypeptides and FL displayed aggregation profiles indistinguish-able from FL on its own (data not shown). It is conceivable thatthe binding of hep1 or GABARAP peptides to Crt improves theaggregation suppression activity of the chaperone, possiblythrough allosteric effects on the binding site responsible for theaggregation suppression function.
FIGURE 5. Effects of G1M3 tetrasaccharide and substrate size on polypep-tide binding by Crt. A, shown is the effect of G1M3 tetrasaccharide on thebinding of Crt constructs to fluorescein-labeled CLV peptide. Fluorescein-labeled CLV peptide (1 �M) was incubated with Crt constructs at indicatedconcentrations in the presence (100 �M) or absence of oligosaccharide (OS) in20 mM Hepes, pH 7.4, 150 mM NaCl, 0.4 mM CaCl2 for 1 h at 25 °C beforemeasurement of fluorescence anisotropy. The anisotropy value of fCLV pep-tide alone was subtracted from each sample to obtain the anisotropy change.Changes in fluorescence anisotropy were recorded at 518 nm with an excita-tion wavelength of 491 nm. The binding curves were fit to an equationdescribing single site binding using SigmaPlot. B, Binding of fluorescein-la-beled RCMLA to Crt and its deletion mutants. Fluorescein labeled RCMLA (10nM) was incubated with Crt constructs at the indicated concentrations in 20mM Hepes, pH 7.4, 150 mM NaCl, 0.4 mM CaCl2 for 1 h at 25 °C before measure-ment of fluorescence anisotropy changes as described in panel A.
FIGURE 6. Interaction of Crt with other peptide and protein ligands.A, shown is binding of Crt to fluorescein-labeled GABARAP and hep1 peptide.Crt at the indicated concentrations in 20 mM Hepes, pH 7.4, 150 mM NaCl, and0.4 mM CaCl2 was incubated with the fluorescein-labeled substrates (10 nM) at37 °C for 1 h before measurement of fluorescence anisotropy changes as inFig. 5. B, GABARAP and hep1 peptide do not compete with Crt in the suppres-sion of FL aggregation. Before aggregation measurements, GABARAP andhep1 peptide (20 �M) were equilibrated with Crt (5 �M) in 20 mM Hepes, pH7.4, 150 mM NaCl, and 0.4 mM CaCl2 for 1 h at 37 °C. FL was then added at a finalconcentration of 3 �M, and light scattering was monitored at 360 nm for 1 h at37 °C. Light scattering was measured every 6 s and plotted every 5 min.
Structure and Chaperone Functions of Calreticulin Domains
AUGUST 5, 2011 • VOLUME 286 • NUMBER 31 JOURNAL OF BIOLOGICAL CHEMISTRY 27273
by guest on June 4, 2018http://w
ww
.jbc.org/D
ownloaded from

Crystallization of Crt�arm1�C368 and Structure Deter-mination—In an effort to gain further insight into how the armand lectin domains of Crt participate in its chaperone func-tions, we crystallized a variant of the �arm1 mutant. This var-iant, termed Crt�arm1�C368 (Fig. 1B and C), lacks the acidic Cterminus that we previously showed interferes with crystal for-mation (6). It also contains the non-Crt sequences Gly-Ser-Metand Leu-Glu-His6 at itsN andC termini, respectively. The crys-tals diffracted to 2.6 Å using synchrotron radiation with onemolecule of Crt�arm1�C368 in the asymmetric unit.The structure was determined by molecular replacement
using the structure of the Crt lectin domain (6). The initialelectron density map showed clear electron density for the firstarm domain module proximal to the lectin domain. This wasfollowed by weak patches of electron density for the secondmodule of the arm domain. No electron density was observedfor Asp-226—Lys-238, Gly-273—Pro-283 or the GGSG linker(Table 2).Structure of Crt with a Partially Truncated Arm Domain—
The structure of the lectin domain of Crt�arm1�C368 is nearlyidentical (root mean square deviation of 0.22 Å over 230 C�atoms) to that previously determined for Crt with the armdomain deleted (6). The lectin domain adopts a fold formed bytwo large �-sheets, a seven-stranded concave �-sheet and asix-stranded convex �-sheet, and three �-helices including along C-terminal �-helix (Fig. 7A). A small �-sheet is positionedon the side of the �-sandwich. The carbohydrate binding site islocated on the concave �-sheet, whereas the arm domain is
inserted into the lectin domain and separates its N- and C-ter-minal parts. There is a single calcium ion bound on the oppositeside of the lectin domain.The Crt�arm1�C368 structure reveals a small structured
region, which we term the junction region, that links the armdomain to the Crt lectin domain. In the Cnx structure, thisregionwas disordered and incompletelymodeled (7). The junc-tion region starts at Leu-203 and ends at Lys-206 on one strandand residues Pro-301 to Asn-304 on the other. The region isstructured by the aromatic ring of Tyr-299 from the armdomain, which is surrounded by the side chains of Leu-203,Lys-206, and Pro-301, forming a small hydrophobic core (Fig.7B). The reason for the difference between the Crt and Cnxstructures is unclear; it could result from crystal packing, or thelinker region could be intrinsically more flexible in Cnx.Sequence alignment betweenCrt andCnx reveals low sequenceconservation that may be responsible for the differences instructure (supplemental Fig. S3).The first arm domain module starts from a short two-
stranded �-sheet formed by Lys-207–Lys-209 and Glu-293–Glu-295. Its conformation is similar to that observed in theNMRstructure of theCrt armdomain (5). The small hydropho-bic core is formed by the two tryptophan residues (Trp-219 andTrp-289) that stack against each other and the side chain of aconserved lysine residue (Lys-215) (Fig. 7C). The second mod-ule of the arm domain could not be modeled due to disorder inthe crystals.
FIGURE 7. Structure of mouse Crt with a partially truncated arm domain. A, shown is a schematic representation of the lectin domain (cyan), arm domain(red/green), and junction (black). For orientation, the position of Glc1Man3 tetrasaccharide previously observed in the structure of the lectin domain is included(6). B, shown is an enlarged view of the junction region where the arm domain leaves the lectin domain. The junction is positioned by lectin domain residuesTyr-150, Phe-202, Ile-305 and the hydrogen bond (dotted line) between Tyr-150 and Asp-302. Residue Tyr-299 of the arm domain is sandwiched betweenPro-301 and Lys-206 to contribute to the rigidity of the junction region. C, shown is an enlarged view of the first module of the arm domain. The arm is stabilizedby a small �-sheet with hydrogen bonds between residues in motif 1 (red) and motif 2 (green) and a small hydrophobic cluster formed by the aromatic rings ofTrp-219 and Trp-289 and the aliphatic part of the side chain of Lys-215. D, shown is an overlay of the lectin domains of the Crt (cyan) and Cnx (PDB entry 1jhn;purple) structures. The C� atoms of corresponding residues near the end of the first modules are separated by �18 Å. E, shown is an overlay of the first modulesof the Crt and Cnx arm domains. Changes in the packing of the tryptophan and lysine residues that form the characteristic small hydrophobic core lead to aroughly 45° angular change in the shape of the arm repeat.
Structure and Chaperone Functions of Calreticulin Domains
27274 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 286 • NUMBER 31 • AUGUST 5, 2011
by guest on June 4, 2018http://w
ww
.jbc.org/D
ownloaded from

The Arm Domains of Crt and Cnx Adopt DifferentOrientations—Comparison of the Crt and Cnx structuresshows that the relative orientations of the arm domains aremarkedly different (Fig. 7D). In Crt, the arm domain is lesscurved, and the angle between the arm domain and the lectindomain is larger by 30°. By the end of the first module, thedistance between analogous residues in Crt and Cnx is �18 Å.Sequence comparison shows that the type 1 repeats of theCrt armdomain are two residues shorter than those in Cnx, which likelyaffects overall curvature of the arm domains (7). Interestingly, theoverlay of the first modules of the Crt and Cnx arm domains alsoreveals differences in packing of aromatic residues, which maycontribute to the differences in curvature (Fig. 7E). Furthermore,this reveals a significant and previously unappreciated conforma-tional plasticity within an individual module.In contrast to the static x-ray structures, NMR studies of the
Crt arm domain suggested the existence of hinge-like motionsbetween the individualmodules (5). In our crystal, thismobilityis likely to be responsible for the lack of electron density for thesecondmodule. The B-factors for theCrt armdomain (�60Å2)are markedly higher than for the globular lectin domain (�40Å2) (supplemental Fig. S4). Given the limited number of struc-tures, the extent to which crystal contacts affect the overallorientation of the arm domain is difficult to assess. In the Cnxstructure, the arm domain wraps around the globular domainof another Cnx molecule. In our structure, the orientation ofthe Crt arm domain may also be affected by intermolecularcontacts (see below and Fig. 8A).The ArmDomain Contains a Putative Substrate Binding Site—
Analysis of the crystal contacts shows that the first module ofthe arm domain interacts with the long C-terminal helix ofanother Crt molecule (Fig. 8A). The contacts are largely hydro-phobic, as the arm domain curls around the helix, forming ashallow groove by the side chains of Ala-213, Ala-214, Pro-216,Trp-289, Pro-292, and Ile-294 (Fig. 8B). The interacting surfaceof the �-helix is centered on Gly-348, Val-349, and Ala-352.This observation may be relevant to our finding that the armdomain, when combinedwith the lectin domain, contributes tothe suppression of FL aggregation and to the binding of largesubstrates (Figs. 3A and 5) but on its own is unable to bind
hydrophobic substrates (Fig. 4 andTable 4). The identified con-tacts result in a relatively small buried surface area of 795 Å2,suggesting a low binding affinity. Thus, in the context of full-length Crt, the hydrophobic site on the arm domain could playa complementary role in binding large non-native proteins butis unlikely to bind hydrophobic substrates on its own.
DISCUSSION
We took a deletion mutagenesis approach to examine thecontribution of the lectin and arm domains to the aggregationsuppression (chaperone) activity of Crt and to localize the poly-peptide binding site responsible for this function. Biophysicalcharacterization of themutants revealed that the individual lec-tin and arm domains were substantially less stable than theintact protein with melting temperatures 8 and 16 °C, respec-tively, below that of wild type Crt. This is in good agreementwith previous studies on the isolated domains by Bouvier andco-workers (39) but contrasts with the findings of Del Cid et al.(26), who noted no loss of stability with the isolated lectindomain. The reason for this discrepancy is unclear but mayreflect differences in the assays used (CD versus fluorescent dyebinding) or slight differences in truncation boundaries and thesize of the bridging linker used to construct the lectin domain.Despite the stabilizing influence of the domains on one another,little difference was noted between the structure of the isolatedlectin domain (6) and the lectin domain solved in this study in thecontext of a partial arm domain. This indicates that the armdomaindoesnot substantially affect the conformationof the lectindomain and is consistent with the fact that the isolated arm andlectin domains retain the ability to bind ERp57 andmonoglucosy-lated oligosaccharide, respectively (9).We also confirmed by ther-mal denaturation studies a previous report showing that the stabi-lizing influence of Ca2� on Crt occurs within the lectin domainandnot the armdomain (39), a finding supported by the detectionin the crystal structure of a Ca2� ion within the lectin domain.Using the various truncation mutants, we demonstrate that
Crt suppresses the aggregation of non-native proteins mainlythrough its lectin domain. To characterize the site(s) responsi-ble, several hydrophobic peptides were used as substratemimetics in direct binding assays, which revealed that the chap-erone activity is conferred through a single polypeptide bindingsite that isdistinct fromthe lectin site.These findingsare strikinglysimilar to our previous studies on the soluble ER luminal segmentofCnx, S-Cnx (10).The lectindomainsof bothproteins bind iden-tical hydrophobic peptides with dissociation constants in themicromolar range, and the binding is unaffected either by theaddition of a 100-foldmolar excess ofmonoglucosylated oligosac-charide or, in the case of Cnx, by ablating lectin function throughmutagenesis (Table 4 and Ref. 10).Numerous studies over the years have implicated Crt in a
bewildering array of processes that occur not onlywithin the ERbut in locations as diverse as the cytosol, nucleus, cell surface,and extracellular environment. These include autoimmune dis-ease, complement activation, tumor immunogenicity, cytolyticT cell activity, apoptosis, angiogenesis, wound healing, cardio-genesis, and adipogenesis among others (45). In many of theseprocesses, interactions between Crt and diverse proteins havebeen documented that are unlikely to involve lectin-oligosac-
FIGURE 8. Crystal contacts identify a putative hydrophobic binding siteon the arm domain. A, the arm domain makes contacts with the C-terminalhelix of the neighboring Crt molecule in the crystal. B, shown is an enlarged viewof the interaction site. Residues involved in the contacts are shown as sticks andlabeled. The surface is colored according to charge distribution: positive charge isin blue, negative charge is in red, and non-polar regions are in gray.
Structure and Chaperone Functions of Calreticulin Domains
AUGUST 5, 2011 • VOLUME 286 • NUMBER 31 JOURNAL OF BIOLOGICAL CHEMISTRY 27275
by guest on June 4, 2018http://w
ww
.jbc.org/D
ownloaded from

charide interactions either due to cellular location or to the factthat the protein or interacting segment is not glycosylated orbears complex glycans. Such interactions include Crt bindingwithin the cytosol to the tail of�-integrins, to the ubiquitin-likeprotein GABARAP (31), and to glucocorticoid and androgenreceptors as well as at the cell surface to complement compo-nent C1q, thrombospondin, CD91, and laminin (for review, seeRef. 45). In two cases, thrombospondin and GABARAP, thebinding site on Crt has been identified as residues 35–53 and195–205, respectively (30, 31). Consequently, it was of interestto explore whether these interaction sites were similar or dis-tinct from the site thatwe identified as being responsible forCrtaggregation suppression function.We were able to reproduce the binding between Crt and
either GABARAP or the thrombospondin-derived peptidehep1. However, unlike peptides such as K6AAF or fCLV thatbind to the site on Crt responsible for its aggregation suppres-sion activity, neitherGABARAPnor hep1 competedwithCrt inthe FL aggregation suppression assay. Thus, we conclude thatneither the GABARAP nor thrombospondin interaction sites isresponsible for the chaperone function of Crt. A recent x-raystructural study of the lectin domain of human Crt suggestedthe presence of an additional peptide binding site that over-lapped with the lectin site (29). This was based on the observa-tion thatwithin the crystal lattice, anN-terminal peptide exten-sion arising from the construction of the recombinant proteinwas found to bind to a portion of the lectin site in a neighboringmolecule defined by residues Phe-46, Phe-74, Cys-105, Met-131, Asp-135, Cys-137, Asp-317, and Trp-319. It was proposedthat this peptide binding sitemight be responsible for the chap-erone function of Crt. However, we think this is unlikely for tworeasons. First, the peptide extension mediating this interaction(KGSIEGR) is muchmore polar than the hydrophobic peptidesshown in the present study to bind to the chaperone site of Crt.Furthermore, many of the residues in the proposed peptidebinding site, including Asp-135, Met-131, Asp-317, and Trp-319,are known to mediate interactions with monoglucosylated oligo-saccharide and thus would be unavailable when the lectin site isoccupied. The present study clearly shows that occupancy of thelectin site with Glc1Man3 tetrasaccharide has no impact on thebinding of hydrophobic peptides to the chaperone site of Crt.Because none of the known peptide binding sitesmediate the
aggregation suppression function of Crt, the precise delinea-tion of the chaperone site remains a high priority. Efforts toaccomplish this through mutagenesis have been made in thepast. For example, mutation of His-153 in Crt (numbered His-170 in the current study) impaired its ability to suppress theaggregation of malate dehydrogenase at 45 °C (46). However,this mutation was found to destabilize Crt, a situation thatwould be further exacerbated by the elevated temperature.Thus, it is unclear whether the impairment of chaperone func-tion is due to the mutated residue or to some indirect confor-mational perturbation. In another studymutagenesis of Crtwasundertaken that targeted hydrophobic residues predicted to besurface-exposed based on a homology model of the Crt struc-ture (17). However, examination of the subsequent crystalstructure of the lectin domain of Crt revealed that the sidechains of about half of the residues selected were directed into
the core of the protein or were buried by oligosaccharide. Neitherthesenor the othermutants impaired the aggregation suppressionfunction of Crt under physiological conditions of the ER (17). Ourefforts to delineate the chaperone site by co-crystallizing hydro-phobic peptides with Crt have been unsuccessful to date. Conse-quently, mutagenesis experiments are ongoing to test candidatesurface-exposed hydrophobic sites for this function.Our current experiments also demonstrate a role for the arm
domain of Crt in suppressing the aggregation of non-nativeprotein substrates such as FL. A progressive reduction in chap-erone potency was noted as the arm domain was increasinglytruncated. Such truncations also progressively reduced thebinding affinity of Crt for the large, non-native protein sub-strate RCMLA but had no effect on its binding to small hydro-phobic peptides. These findings are remarkably similar to ourprevious studies on S-Cnx wherein we showed that the lengthof the arm domain correlates with binding affinity for largesubstrates such as FL and RCMLA but not with small peptides(10). Given that the isolated armdomains of either S-Cnx orCrtexhibit no detectable binding to these large substrates, we pre-viously suggested that the arm domains contribute to bindingaffinity by sterically constraining large protein substrates thatenter the cavity between the arm and lectin domains (10). Thus,protein aggregation would be suppressed by a combination ofbinding to exposed hydrophobic patches on the folding sub-strate (through the polypeptide binding site on Cnx and Crt) aswell as by physically sequestering the substrate from the vicinityof other folding proteins. An additional possibility is suggestedby crystal contacts observed in the structure we solved of Crtwith a partially truncated arm domain. Hydrophobic contactswere observed between the innermost arm domainmodule andthe C-terminal �-helix of another Crt molecule. Such contactsmay also occur between this module and large substrates,thereby contributing to overall binding affinity. However, giventhe small surface area of the contacts, they would be unlikely tobind hydrophobic substrates on their own. Because smallhydrophobic patches are also present in the remaining armdomain modules of Cnx (7) and possibly Crt as well, it is con-ceivable that they may also contribute to the binding of largesubstrates. Extensive mutagenesis studies will be required toevaluate the relative contributions of the “steric constraint” and“multi-point contact” models for the role of the arm domain insuppressing substrate aggregation. Regardless of mechanism, itis likely that the arm domain plays a significant role in substrateinteraction in cells as removal of two-thirds of this domain (Crt�arm2 mutant) results in a loss of Crt interaction with class Ihistocompatibility molecules (47).
Acknowledgments—For providing various Crt substrates, we grate-fully acknowledge Drs. Arianna Rath andCharles Deber (6KAAF andKHP) andDr.DieterWillbold (GABARAP).Wealso thankDr. RonnieLum for the FITC-labeling protocol, initial RCMLA probes, and help-ful discussions as well as Myrna Cohen-Doyle for excellent technicalassistance. Data acquisition at the Macromolecular Diffraction(MacCHESS) facility at the Cornell High Energy Synchrotron Sourcewas supported by the National Science Foundation Award DMR0225180 and the National Institutes of Health Award RR-01646.
Structure and Chaperone Functions of Calreticulin Domains
27276 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 286 • NUMBER 31 • AUGUST 5, 2011
by guest on June 4, 2018http://w
ww
.jbc.org/D
ownloaded from

REFERENCES1. Hebert, D. N., and Molinari, M. (2007) Physiol. Rev. 87, 1377–14082. Rutkevich, L. A., and Williams, D. B. (2011) Curr. Opin Cell Biol. 23,
157–1663. Vembar, S. S., and Brodsky, J. L. (2008)Nat. Rev.Mol. Cell Biol. 9, 944–9574. Williams, D. B. (2006) J. Cell Sci. 119, 615–6235. Ellgaard, L., Riek, R., Herrmann, T., Guntert, P., Braun, D., Helenius, A.,
and Wuthrich, K. (2001) Proc. Natl. Acad. Sci. U.S.A. 98, 3133–31386. Kozlov, G., Pocanschi, C. L., Rosenauer, A., Bastos-Aristizabal, S., Gorelik,
A., Williams, D. B., and Gehring, K. (2010) J. Biol. Chem. 285,38612–38620
7. Schrag, J. D., Bergeron, J. J., Li, Y., Borisova, S., Hahn, M., Thomas, D. Y.,and Cygler, M. (2001)Mol. Cell 8, 633–644
8. Frickel, E. M., Riek, R., Jelesarov, I., Helenius, A., Wuthrich, K., and Ell-gaard, L. (2002) Proc. Natl. Acad. Sci. U.S.A. 99, 1954–1959
9. Leach, M. R., Cohen-Doyle, M. F., Thomas, D. Y., and Williams, D. B.(2002) J. Biol. Chem. 277, 29686–29697
10. Brockmeier, A., Brockmeier, U., and Williams, D. B. (2009) J. Biol. Chem.284, 3433–3444
11. Li, Z., Stafford, W. F., and Bouvier, M. (2001) Biochemistry 40,11193–11201
12. Ware, F. E., Vassilakos, A., Peterson, P. A., Jackson,M. R., Lehrman,M. A.,and Williams, D. B. (1995) J. Biol. Chem. 270, 4697–4704
13. Spiro, R. G., Zhu, Q., Bhoyroo, V., and Soling, H. D. (1996) J. Biol. Chem.271, 11588–11594
14. Hammond, C., Braakman, I., andHelenius, A. (1994) Proc. Natl. Acad. Sci.U.S.A. 91, 913–917
15. Hebert, D. N., Foellmer, B., and Helenius, A. (1995) Cell 81, 425–43316. Brockmeier, A., andWilliams, D. B. (2006)Biochemistry 45, 12906–1291617. Jeffery, E., Peters, L. R., and Raghavan, M. (2011) J. Biol. Chem. 286,
2402–241518. Leach, M. R., and Williams, D. B. (2004) J. Biol. Chem. 279, 9072–907919. Ireland, B. S., Brockmeier, U., Howe, C. M., Elliott, T., andWilliams, D. B.
(2008)Mol. Biol. Cell 19, 2413–242320. Culina, S., Lauvau, G., Gubler, B., and van Endert, P. M. (2004) J. Biol.
Chem. 279, 54210–5421521. Thomson, S. P., and Williams, D. B. (2005) Cell Stress Chaperones 10,
242–25122. Ihara, Y., Cohen-Doyle, M. F., Saito, Y., and Williams, D. B. (1999) Mol.
Cell 4, 331–34123. Martin, V., Groenendyk, J., Steiner, S. S., Guo, L., Dabrowska, M., Parker,
J. M., Muller-Esterl, W., Opas, M., and Michalak, M. (2006) J. Biol. Chem.281, 2338–2346
24. Rizvi, S. M., Mancino, L., Thammavongsa, V., Cantley, R. L., and Ragha-van, M. (2004)Mol. Cell 15, 913–923
25. Thammavongsa, V., Mancino, L., and Raghavan, M. (2005) J. Biol. Chem.280, 33497–33505
26. Del Cid, N., Jeffery, E., Rizvi, S.M., Stamper, E., Peters, L. R., Brown,W. C.,Provoda, C., and Raghavan, M. (2010) J. Biol. Chem. 285, 4520–4535
27. Duus, K., Sandhu, N., Jørgensen, C. S., Hansen, P. R., Steinø, A., Thaysen-Andersen, M., Højrup, P., and Houen, G. (2009) Protein Pept. Lett. 16,1414–1423
28. Sandhu, N., Duus, K., Jørgensen, C. S., Hansen, P. R., Bruun, S. W., Peder-sen, L. Ø., Højrup, P., and Houen, G. (2007) Biochim. Biophys. Acta 1774,701–713
29. Chouquet, A., Païdassi, H., Ling, W. L., Frachet, P., Houen, G., Arlaud,G. J., and Gaboriaud, C. (2011) PLoS One 6, e17886
30. Goicoechea, S., Pallero, M. A., Eggleton, P., Michalak, M., and Murphy-Ullrich, J. E. (2002) J. Biol. Chem. 277, 37219–37228
31. Thielmann, Y.,Weiergraber, O. H., Mohrluder, J., andWillbold, D. (2009)FEBS J. 276, 1140–1152
32. Otwinowski, Z., and Minor, W. (1997)Methods Enzymol. 276, 307–32633. McCoy, A. J., Grosse-Kunstleve, R. W., Adams, P. D., Winn, M. D., Sto-
roni, L. C., and Read, R. J. (2007) J. Appl. Crystallogr. 40, 658–67434. Emsley, P., andCowtan, K. (2004)ActaCrystallogr. D. Biol. Crystallogr. 60,
2126–213235. Murshudov, G. N., Vagin, A. A., Lebedev, A., Wilson, K. S., and Dodson,
E. J. (1999) Acta Crystallogr. D. Biol. Crystallogr. 55, 247–25536. Winn, M. D., Murshudov, G. N., and Papiz, M. Z. (2003) Methods Enzy-
mol. 374, 300–32137. Laskowski, R. A., MacArthur, M. W., Moss, D. S., and Thornton, J. M.
(1993) J. Appl. Crystallogr. 26, 283–29138. Bosl, B., Grimminger, V., and Walter, S. (2005) J. Biol. Chem. 280,
38170–3817639. Tan, Y., Chen, M., Li, Z., Mabuchi, K., and Bouvier, M. (2006) Biochim.
Biophys. Acta 1760, 745–75340. Arnaud, N., and Georges, J. (1997) Analyst 122, 143–14641. Urbanova, N., Kadar, M., Toth, K., Bogati, B., Andruch, V., and Bitter, I.
(2008) Anal Sci. 24, 727–73342. Patil, A. R., Thomas, C. J., and Surolia, A. (2000) J. Biol. Chem. 275,
24348–2435643. Kapoor, M., Srinivas, H., Kandiah, E., Gemma, E., Ellgaard, L., Oscarson,
S., Helenius, A., and Surolia, A. (2003) J. Biol. Chem. 278, 6194–620044. Okazaki, A., Ikura, T., Nikaido, K., and Kuwajima, K. (1994) Nat. Struct.
Biol. 1, 439–44645. Michalak, M., Groenendyk, J., Szabo, E., Gold, L. I., and Opas, M. (2009)
Biochem. J. 417, 651–66646. Guo, L., Groenendyk, J., Papp, S., Dabrowska, M., Knoblach, B., Kay, C.,
Parker, J. M., Opas, M., and Michalak, M. (2003) J. Biol. Chem. 278,50645–50653
47. Zhang, Y., Kozlov, G., Pocanschi, C. L., Brockmeier, U., Ireland, B. S.,Maattanen, P., Howe, C., Elliott, T., Gehring, K., andWilliams,D. B. (2009)J. Biol. Chem. 284, 10160–10173
Structure and Chaperone Functions of Calreticulin Domains
AUGUST 5, 2011 • VOLUME 286 • NUMBER 31 JOURNAL OF BIOLOGICAL CHEMISTRY 27277
by guest on June 4, 2018http://w
ww
.jbc.org/D
ownloaded from

Williams and Kalle GehringCosmin L. Pocanschi, Guennadi Kozlov, Ulf Brockmeier, Achim Brockmeier, David B.
CalreticulinStructural and Functional Relationships between the Lectin and Arm Domains of
doi: 10.1074/jbc.M111.258467 originally published online June 7, 20112011, 286:27266-27277.J. Biol. Chem.
10.1074/jbc.M111.258467Access the most updated version of this article at doi:
Alerts:
When a correction for this article is posted•
When this article is cited•
to choose from all of JBC's e-mail alertsClick here
Supplemental material:
http://www.jbc.org/content/suppl/2011/06/07/M111.258467.DC1
http://www.jbc.org/content/286/31/27266.full.html#ref-list-1
This article cites 47 references, 23 of which can be accessed free at
by guest on June 4, 2018http://w
ww
.jbc.org/D
ownloaded from





























