Embed Size (px)
Citation preview

Structural Mechanisms of DNA Replication, Repair, and Recombination
Melissa E. Stauffer and Walter J. Chazin
Departments of Biochemistry and Physics and Center for Structural Biology
Vanderbilt University, Nashville, TN 37232-8725 USA
DNA processing involves multiple biochemical steps requiring far more functions than
can be easily incorporated into a single protein. Consequently, replication, repair, and
recombination are performed by multi-protein assemblies. Remarkably, there is growing
evidence that these processes share many common features. The enzymes active in replicating
DNA during S phase are needed to synthesize new stretches of DNA during various types of
recombination and repair. Repair of all types must occur in tandem with replication and
transcription, since the exposure of DNA during these processes renders it open to discovery of
common lesions. Recombination is used to catalyze genetic crossover during meiosis, and is also
a major DNA repair mechanism during replicative and later phases of the cell cycle. Thus, the
processes of DNA replication, repair, and recombination are being increasingly viewed as
integrated events in cellular life.
Considerable progress has been made in developing techniques to investigate at the
atomic level how the DNA processing machinery functions. We are now in a position to ask very
basic questions: what are the structural mechanisms that regulate progression from initiation
through termination of a DNA processing pathway, and how does the DNA processing
machinery facilitate cross-talk between replication, recombination, and repair? We highlight here
some of the trends and concepts that are emerging as the structural mechanisms that regulate
DNA processing begin to be elucidated.
1
JBC Papers in Press. Published on April 16, 2004 as Manuscript R400015200
Copyright 2004 by The American Society for Biochemistry and Molecular Biology, Inc.
by guest on June 11, 2018http://w
ww
.jbc.org/D
ownloaded from

Multiple dynamic protein interactions coordinate the ordered progression of DNA
processing events. Working from a basic description of the hand-off of DNA from one protein to
another, we introduce the concepts of modularity of DNA processing proteins and the versatility
of the common folds found in these proteins. This is followed by recently reported examples of
direct competition and allosteric mechanisms promoting specific steps of DNA processing.
These concepts are integrated into a simple working model that provides a general framework for
thinking about the forward progression of DNA replication, repair, and recombination
assemblies.
The Concept of Handing Off
Evidence is mounting in favor of dynamic assembly and disassembly of the DNA
processing machinery at the time it is needed, and against the existence of preformed
holoenzyme complexes that meander around the cell until they are needed (1-3). Multiple
dynamic protein interactions are fundamental to the nature of DNA processing and to
progression through each processing pathway. The weak and/or transient nature of many of the
protein interactions is thought to be the basis for the ability of the DNA processing machinery to
assemble dynamically and to hand off the DNA from one subcomplex to the next. From the
perspective of the DNA, the proteins are sometimes described as ‘trading places’ (4).
A well-known example from replication is the competitive polymerase switching
observed during lagging strand synthesis (Figure 1A) (5). In eukaryotes, DNA polymerase α-
primase complex (pol-prim) synthesizes an RNA-DNA hybrid primer. For processive DNA
synthesis to follow, the sliding clamp, proliferating cell nuclear antigen (PCNA), must be loaded
by the clamp loader protein, replication factor C (RFC). Loading of PCNA triggers the
displacement of pol-prim from the 3’ end of the primer. Processive synthesis by DNA
polymerase δ (Pol δ) requires its direct association with PCNA. RFC is subsequently displaced
from PCNA, but remains stably associated with the PCNA/pol-prim complex during synthesis.
2
by guest on June 11, 2018http://w
ww
.jbc.org/D
ownloaded from

Each polymerase switch is facilitated by protein interactions with replication protein A (RPA), a
highly abundant ssDNA binding protein in eukaryotes (6).
Hand-off is also well documented in eukaryotic nucleotide excision repair (NER), during
which a wide variety of DNA lesions are removed and replaced (Figure 1B) (7). In the global
genome repair pathway, damage is detected by the XPC/hHR23B/centrin 2 heterotrimer. Local
unwinding is effected by recruited TFIIH, a heterodimer composed of XPB and XPD subunits.
Next, XPA and RPA associate with the complex and orient the incoming excision proteins,
ERCC1/XPF and XPG. Binding of XPG signals the release of XPC/hHR23B, while
ERCC1/XPF triggers the excision of damaged DNA and the release of XPA and TFIIH. Of these
proteins, only RPA remains stably bound after excision to facilitate association of the synthetic
factors RFC, PCNA, Pol δ, and DNA ligase.
Even in recombination, for which detailed knowledge lags somewhat behind that of
replication and repair, there are reports of proteins trading places on DNA (Figure 1C) (8,9).
Homologous recombination (HR) is one pathway used to repair double strand DNA breaks. In
eukaryotes, the 5’ ends around the break are resected by the Mre11/Rad50/NBS1 nuclease,
leaving 3’ ssDNA overhangs. These are coated by RPA, which binds with high affinity to
ssDNA. The displacement of RPA from ssDNA is effected by Rad51, the recombinase, with the
help of Rad52, a recombination mediator protein. Nucleation of Rad51 on ssDNA promotes
formation of the nucleoprotein filament that catalyzes the homology search and subsequent
strand exchange.
In each pathway, the sequential nature of the recruitment and dissociation of DNA
processing proteins is well established. The advantages of dynamic assembly and disassembly
are many, not the least of which is that fewer copies of ubiquitous proteins such as RPA and
PCNA are needed, since they can be recycled for use in other reactions. In addition, the ordered
hand-off of DNA from protein to protein (or complex to complex) allows for several levels of
temporal and spatial regulation of the processing pathways, while parallel steps between two (or
more) pathways allow for cross-talk.
3
by guest on June 11, 2018http://w
ww
.jbc.org/D
ownloaded from

Modular Proteins with Multiple Binding Sites
A common feature of proteins involved in DNA processing is modularity, the physical
connection of structural domains that perform different biochemical functions. This
characteristic facilitates the coordination of biochemical activities without constraining
functional modules to fit tightly together. Flexible tethering of domains in modular proteins
allows multiple steps to be carried out independently, yet remain coupled.
A corollary of modularity is the use of multiple contact points between one or more pairs
of interacting proteins. This facilitates the hand-off of processing proteins in situations where
rearrangement is required rather than full dissociation. It also provides a means to achieve a high
overall affinity from a set of weak individual interactions (vide infra). The use of multiple
binding sites also allows for more than one type of regulation at a single step of DNA processing.
During replication, the clamp loader protein RFC binds to RPA using three of its five
subunits (6). Evidence from replication studies suggests that there are two distinct binding sites
for RFC on RPA: one that overlaps with the Pol δ binding site and one that does not. Multiple
points of interaction between RFC and RPA facilitate hand-off of RFC without causing it to
completely dissociate. Pol δ competes with RFC for binding to RPA during the second
polymerase switch of lagging strand synthesis, but after Pol δ displaces RFC, RFC remains
bound to the polymerase holoenzyme complex. The maintenance of RFC in the holoenzyme is
mediated by a second, species-specific interaction with RPA (E. coli SSB cannot substitute) and
is essential for processive DNA synthesis.
During NER, XPA binds to both the 32-kDa and the 70-kDa subunit of RPA (RPA32 and
RPA70, respectively) (10,11). The Kd for the overall interaction measured by surface plasmon
resonance was 1.9 × 10-8 M (12). ELISA results suggested that the binding affinity is partitioned,
with 80% of the affinity assigned to RPA32 and 20% to RPA70 (11). The expected dissociation
constant for the XPA-RPA70 interaction was therefore on the order of 10-7 M. However, NMR
experiments used to characterize the interaction of the isolated domains revealed that their
4
by guest on June 11, 2018http://w
ww
.jbc.org/D
ownloaded from

binding affinity is several orders of magnitude weaker than the intact proteins’ (13). What factors
make the total affinity significantly stronger than the sum of the isolated domain interactions?
The key here is the linkage effect (14), which provides a means to obtain a high affinity
interaction between binding partners from the tethering together of two or more components that
by themselves interact very weakly. For example, the isolated A and B domains of RPA70 each
bind ssDNA with relatively modest affinity (Kd is micromolar). However, when tethered together
by a flexible ~10 residue linker, their overall affinity for ssDNA is nearly three orders of
magnitude higher (Kd is nanomolar) (15).
In recombination events, Rad52 has been shown to bind to both the A domain of RPA70
(RPA70A) and the C-terminal domain of RPA32 (RPA32C) (16). In this case, the different
points of contact between Rad52 and RPA are associated with different regulatory mechanisms.
NMR chemical shift perturbation assays demonstrated that residues 257-274 of Rad52 bind to
RPA32C at the same location as two other repair proteins: XPA and uracil DNA glycosylase
(17). Overlapping binding sites between proteins from three different repair pathways suggested
a competitive mechanism for switching between pathways. The interaction between Rad52 and
RPA32C is also responsible for an allosteric enhancement (5-fold) of the ssDNA binding affinity
of RPA, an effect mediated through the DNA binding domain in RPA32 (16). The same segment
of Rad52 (residues 218-303) binds to RPA70A, and sequence comparison between RPA32C and
RPA70A has predicted that the Rad52 binding site overlaps with the ssDNA binding site on
RPA70A. This interaction is expected to be competitive with both DNA and other proteins
known to bind at this site.
Versatile Protein Domains
Another striking observation about DNA processing proteins is the repeated utilization of
specific interaction modules (folded domains). Remarkably, these domains may interact
exclusively with proteins in one context, but will interact with both proteins and DNA in another,
and their sites of interaction may be similar, overlapping, or unique. Modules that interact with
5
by guest on June 11, 2018http://w
ww
.jbc.org/D
ownloaded from

both proteins and DNA are critical to the recruitment and positioning of peripheral processing
proteins as they assemble on and dissociate from the DNA.
The oligonucleotide/oligosaccharide-binding (OB) fold, the canonical ssDNA binding
domain (18,19), is one example of such a commonly used interaction module. OB-fold domains
are found in DNA processing proteins ranging from nucleases and pyrophosphatases to ssDNA-
and RNA-binding proteins (20). RPA has six OB folds (and one winged helix domain, see
below), four of which are involved in binding to ssDNA. In the crystal structure of the tandem
RPA70AB domains (residues 181-422 of RPA70), an oligonucleotide is bound in the continuous
cleft created by alignment of the tandem OB-fold domains (Figure 2A) (21). While OB-fold
domains are characterized and, in fact, named for binding to nucleotides, they are also involved
in protein interactions. The N-terminal domain of RPA70, for instance, is dedicated to
interactions with pol-prim and p53 (23), whereas the tandem RPA70AB domains are known to
interact with both ssDNA and proteins, including the SV40 large T antigen, XPA, and Rad51
(22).
A recent crystal structure of BRCA2, one of two well-known human breast cancer
associated proteins, reveals a fundamentally different mode of protein binding to an OB-fold
domain (23). BRCA2 possesses three OB-fold domains in its C-terminal region, two of which
are bound to a 9-mer nucleotide in the same manner as observed for RPA70AB (Figure 2B). The
third domain is bound to a fragment of a binding partner, DSS1, which is essential for
recombinational DNA repair (24). The DSS1 peptide binds away from the usual nucleotide
binding site into a positively charged groove that traverses the domain from front to back. Hence,
this OB-fold domain appears to have been adapted for a purpose other than nucleotide binding.
A second example of a versatile protein fold is the winged helix (WH) domain, a member
of the common helix-turn-helix superfamily (25). WH domains were first characterized in 1993,
and are best known for their role in dsDNA binding in transcription factors (26). Numerous
examples of DNA-bound structures are available for a wide functional variety of proteins. A
recently determined structure of the RecQ helicase from E. coli revealed that it contains a WH
6
by guest on June 11, 2018http://w
ww
.jbc.org/D
ownloaded from

domain that possesses the characteristic structural features of a DNA-binding WH domain (27).
Interestingly, a homologous portion of human Werner’s syndrome protein (WRN) is known to
binds proteins, such as Bloom’s syndrome protein and FEN-1, a eukaryotic endo/exonuclease
(28,29). The interaction of WRN with FEN-1 is functionally relevant, as it increases FEN-1’s
DNA cleavage activity 6-8 fold (28). Thus, the WH domain of WRN is either designed
specifically for protein interaction or it is bifunctional, with both protein and DNA binding
activity.
Insight into the structural basis distinguishing DNA and protein binding activities of
different WH domains can be obtained from comparative structural analysis. For example, the
RPA32C WH domain has no DNA binding activity and is strictly used for protein interaction
(17). The electrostatic surface of RPA32C contrasts greatly with that of the RecQ WH (Figure
2C). While the RecQ WH, which purportedly binds DNA, possesses a long tract of positive
charge along a shallow groove, RPA32C is mostly negative over the same surface, with no
observable groove for DNA binding.
A recent example of a dual-function WH domain is found in eukaryotic Cdc6, a
component of the pre-replication complex (30). Cdc6 has been shown to interact with the
minichromosome maintenance helicase in a way that inhibits helicase activity (31). The region of
Cdc6 responsible for the interaction is the conserved C-terminal WH domain. Interestingly, this
domain has also been demonstrated to bind dsDNA (32). The electrostatic surface (data not
shown) for this domain corroborates its dual functionality, since a combination of positive and
negative patches are found, and the positive surface corresponds to the canonical dsDNA binding
site of WH domains.
Competition for Binding Sites
As noted above, proteins in DNA processing assemblies usually have more than one
protein or DNA binding site. Often, the binding sites are clustered together or overlapping on a
single surface of a molecule, setting up a competitive mechanism for regulating progression
7
by guest on June 11, 2018http://w
ww
.jbc.org/D
ownloaded from

through a particular pathway. Direct competition between two or more molecules for a single
binding site facilitates hand-off of DNA and proteins during DNA processing. It allows for cell
cycle-coupled regulation via timed transcription of essential protein factors. It minimizes the
number of possible protein interactions, promoting interplay between alternate regulatory
mechanisms.
In DNA replication, competition is evident in the binding of bacterial polymerases I-V to
the β-clamp processivity factor (33). All the polymerases interact with the same hydrophobic
pocket into which the δ-subunit of the γ-complex clamp-loader binds. Competition for the
hydrophobic pocket of the β-clamp is therefore thought to provide the mechanism for switching
from the loading phase to the synthesis phase of replication. Also, the affinity of the β-clamp for
its partner proteins is modulated by its binding to DNA, and structural features of damaged DNA
may play a role in both successful bypass of lesions and coordination of different types of repair.
In eukaryotic replication, competition for RPA precludes simultaneous binding of pol-
prim, RFC, and Pol δ, promoting progression of replication from priming to synthesis. Each of
these proteins has a binding site on RPA, though the precise locations of binding are unknown
(6). The ability of proteins acting at later steps in the pathway to displace those present at earlier
steps suggests overlapping binding sites, which is a well-established feature of RPA. Mer et al.
showed that the C-terminal domain of the 32-kDa subunit of RPA binds to peptides from three
different repair proteins, all at the same site (17). This early finding suggested that competition
might facilitate handing off the DNA from one repair protein to another.
Two recent studies identify overlap between protein and ssDNA binding sites on RPA.
NMR chemical shift perturbation experiments have indicated that both XPA and Rad51 bind to
the canonical oligonucleotide binding site on RPA70A (13,34). NMR competition experiments
demonstrated that ssDNA could disrupt the preformed complex between RPA70A and the N-
terminal domain of Rad51 (Rad51N), indicating that the binding of Rad51N and ssDNA is
mutually exclusive (34). The competition between Rad51 and ssDNA for RPA is the basis for
8
by guest on June 11, 2018http://w
ww
.jbc.org/D
ownloaded from

the displacement of RPA during initiation of homologous recombination, while XPA affects the
preference of RPA for undamaged ssDNA during NER.
Homologous recombination is also regulated by competition in the case of the interaction
between BRCA2 and Rad51 (35). BRCA2 possesses eight BRC repeats in its central region, six
of which are capable of binding to Rad51 with varying affinities. The interaction is essential for
the transport of Rad51 into the nucleus and is inhibitory to Rad51 filament formation. The basis
for the inhibition is simply that the BRC sequence binds to the site on Rad51 at which it (Rad51)
self-associates. Thus, BRCA2 and Rad51 compete for an overlapping binding site on Rad51.
Still, Rad51 forms oligomeric filaments in vivo. How? One possible explanation is that the low-
affinity BRC repeats release Rad51 first, followed by release of the remaining tightly bound
molecules by allosteric or cooperative means.
Allostery
Allostery (action at a remote site) is a second common mechanism that promotes
progression through a DNA processing pathway. Allostery takes on many forms. Some protein
interactions serve to orient the binding partners for optimal activity at a distant site. In other
cases, ATP binding and/or hydrolysis are coupled to conformational changes that affect the ATP-
binding protein’s oligomerization state or its interactions with other proteins. Allostery can be
used in conjunction with other types of regulation to catalyze a single step of DNA processing.
Allostery is evident in the interactions between the eukaryotic sliding clamp protein,
PCNA, and its binding partners. PCNA is a recruitment factor for repair proteins from several
different pathways, including XPG (NER), uracil DNA glycosylase (base excision repair),
several MSH proteins (mismatch repair), and the Werner helicase (recombination) (36). The
homotrimeric structure of PCNA allows for several regulatory mechanisms, including the use of
multiple binding sites, competition for overlapping sites, and allosteric control of binding
partners. The type of DNA damage to which PCNA is bound plays a role in attracting binding
partners, several of which specifically recognize kinked or bent DNA (37,38). PCNA stimulates
9
by guest on June 11, 2018http://w
ww
.jbc.org/D
ownloaded from

the nuclease activity of flap endonuclease-1 (FEN-1), one of its binding partners for lagging
strand synthesis, by up to 50-fold (39). The interaction between these two proteins and its effect
on FEN-1’s DNA binding ability was recently characterized (40). The PCNA-FEN-1 interaction
induces the formation of a β-zipper, with strands contributed by the C-terminal segments of each
protein. On FEN-1, the β-zipper links the PCNA binding site to a DNA binding site. Formation
of the β-zipper likely enhances the nuclease activity of FEN-1 through allostery by properly
positioning it for cleavage of flapped DNA substrates.
Allosteric regulation is also observed for the Mre11/Rad50 complex, which generates the
3’ ssDNA ends used as templates for homologous recombination (41). Mre11 contains nuclease
activity specific for ssDNA, dsDNA ends, or hairpin DNA structures, while Rad50 plays a
structural role in bringing sister chromatids together. Recent crystal structures have elucidated
the allosteric mechanism whereby the ATPase activity of Rad50 is coupled to the nuclease
activity of Mre11 (42). Mre11 interacts with the ATPase domain of Rad50, which is comprised
of globular and coiled coil regions from two separate chains of the Rad50 homodimer. Catalytic
Rad50 heads are organized into a DNA-binding lobe (I) and a structural coiled coil lobe that
binds Mre11 (II). ATP binding to lobe I causes a 30° rotation of this lobe with respect to lobe II,
repositioning the bound DNA with respect to the Mre11 bound to lobe II (Figure 3). ATP
hydrolysis has the effect of increasing the kcat of nucleolysis by Mre11, which can be interpreted
primarily as an acceleration of product release in preparation for the next cleavage cycle.
During recombination, the binding and hydrolysis of ATP are coupled to strand exchange
through allosteric effects. ATP binding has recently been shown to affect the pitch and
orientation of subunits in the bacterial RecA filament (43). In eukaryotic Rad51, ATP hydrolysis
is coupled to strand exchange by the DNA-binding loop L1 adjacent to the ATP binding Walker
A motif (44). A mutation in this loop retained DNA-dependent ATPase activity, indicating that
primary DNA binding was not affected. However, subsequent measurements of strand exchange
showed no formation of joint molecules by the mutant protein, suggesting that ATP hydrolysis is
allosterically coupled to strand exchange via residues in loop L1.
10
by guest on June 11, 2018http://w
ww
.jbc.org/D
ownloaded from

Concluding Remarks
The accumulated evidence to date confirms that the DNA processing machinery is
dynamically assembled and disassembled in conjunction with the cell cycle and in response to
DNA damage. Assembly and rearrangement of protein assemblies is facilitated by the hand-off
of proteins and DNA from one step to the next. We have discussed five key concepts relevant to
understanding the progression of DNA processing assemblies:
1. Modular organization—DNA processing proteins contain multiple structural
domains with distinct biochemical functions.
2. Multiple contact points—DNA processing proteins interact with their partner
proteins through multiple sites.
3. Modest affinity—The majority of protein interactions characterized to date
have dissociation constants in the micromolar range.
4. Versatile structural modules—DNA processing proteins utilize a common set
of structural frameworks that can interact with DNA and/or protein.
5. Multiple structural mechanisms promote hand-off—Direct competition for
binding sites and allosteric structural rearrangements have been characterized.
The emerging view is that DNA processing assemblies are composed of modular proteins with
multiple, structurally independent functional domains. Individual contact points are characterized
by relatively low (micromolar) binding affinities, which promotes hand-off of DNA and proteins.
The structural modules commonly used by DNA processing proteins are versatile in their
binding preferences and modes. These characteristics are integrated into the structural
mechanisms that mediate the forward progression of DNA processing.
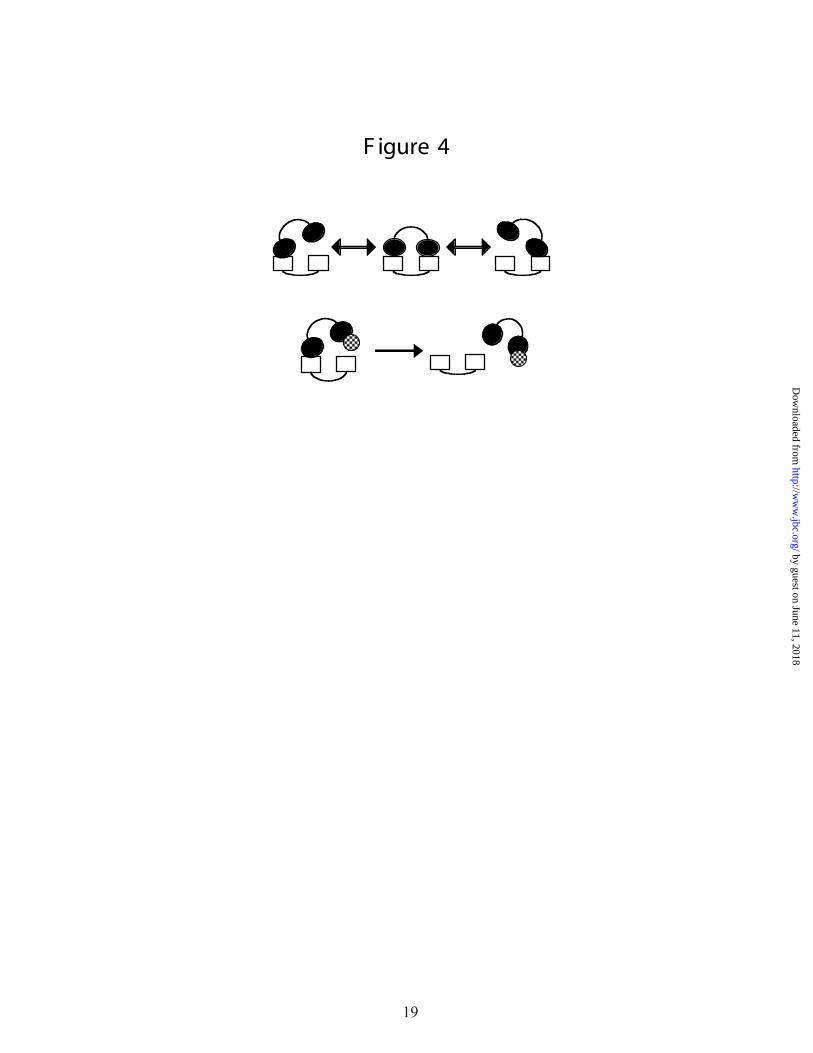
In this model, linked weak interactions provide high overall affinity while maintaining a
significant off-rate for each contact point (Figure 4).1 This in turn provides a ready means for
rapid disassembly: by simply blocking one of the weak interactions, the overall affinity between
two interacting molecules can be reduced by several orders of magnitude almost instantaneously.
11
by guest on June 11, 2018http://w
ww
.jbc.org/D
ownloaded from

The rapid transition between high and low affinity states promotes hand-off and forward
progression of DNA processing. This simple model provides a framework for developing a more
sophisticated and detailed understanding of the structural mechanisms used by the multi-protein
assemblies involved in DNA replication, repair, and recombination.
Figure Legends
Figure 1. Schematic diagrams of sequential hand-off of proteins during (A) replication, (B)
nucleotide excision repair, and (C) recombination. Proteins are labeled at the point at which
they become involved in each pathway.
Figure 2. Versatile proteins folds. OB-fold domains from (A) RPA70AB and (B) BRCA2. The
domains with ssDNA binding activity are colored yellow, with bound ssDNA in blue. The N-
terminal OB-fold from BRCA2 is colored orange, with the bound DSS1 peptide in red. The
black arrow indicates the location of the canonical ssDNA binding site. Panel C compares the
electrostatic surfaces of WH domains from the RecQ helicase (left) and RPA32C (right) in
equivalent orientations. Red and blue indicate negative and positive surface potential,
respectively. The arrows indicate the position of the canonical WH DNA binding helix 3.
Figure 3. Allosteric effect of ATP binding on the subdomain orientation of Rad50. Lobe I of
Rad50 (blue oval) contacts both DNA (black helix) and lobe II (purple oval). When ATP
binds (red cross), lobe I rotates with respect to lobe II, bringing the DNA into position for
nucleolytic cleavage by Mre11 (yellow diamond) bound to lobe II.
Figure 4. Key concepts in DNA processing. The upper panel symbolizes the interaction between
two multidomain proteins (A, two open rectangles; B, two filled ovals) that have two contact
points. The overall affinity between the two proteins results from two modest affinity
interactions, each of which has an appreciable off-rate. This corresponds to one
representation of the linkage effect. The lower panel represents the facilitation of hand-off in
this system. A third protein (checked circle) needs to bind to only one of the two domains of
12
by guest on June 11, 2018http://w
ww
.jbc.org/D
ownloaded from

protein B in order to drastically reduce the overall binding affinity and promote release of B
from A.
References
1. Kowalczykowski, S. C. (2000) Nat Struct Biol 7, 1087-1089
2. Rademakers, S., Volker, M., Hoogstraten, D., Nigg, A. L., Mone, M. J., Van Zeeland, A.
A., Hoeijmakers, J. H., Houtsmuller, A. B., and Vermeulen, W. (2003) Mol Cell Biol 23,
5755-5767
3. Essers, J., Houtsmuller, A. B., van Veelen, L., Paulusma, C., Nigg, A. L., Pastink, A.,
Vermeulen, W., Hoeijmakers, J. H., and Kanaar, R. (2002) Embo J 21, 2030-2037
4. Yuzhakov, A., Kelman, Z., and O'Donnell, M. (1999) Cell 96, 153-163
5. Davey, M. J., and O'Donnell, M. (2000) Curr Opin Chem Biol 4, 581-586
6. Yuzhakov, A., Kelman, Z., Hurwitz, J., and O'Donnell, M. (1999) Embo J 18, 6189-6199
7. Riedl, T., Hanaoka, F., and Egly, J. M. (2003) Embo J 22, 5293-5303
8. Sung, P., Krejci, L., Van Komen, S., and Sehorn, M. G. (2003) J Biol Chem 278, 42729-
42732
9. Valerie, K., and Povirk, L. F. (2003) Oncogene 22, 5792-5812
10. Stigger, E., Drissi, R., and Lee, S. H. (1998) J Biol Chem 273, 9337-9343
11. Walther, A. P., Gomes, X. V., Lao, Y., Lee, C. G., and Wold, M. S. (1999) Biochemistry
38, 3963-3973
12. Saijo, M., Kuraoka, I., Masutani, C., Hanaoka, F., and Tanaka, K. (1996) Nucleic Acids
Res 24, 4719-4724
13. Daughdrill, G. W., Buchko, G. W., Botuyan, M. V., Arrowsmith, C., Wold, M. S.,
Kennedy, M. A., and Lowry, D. F. (2003) Nucleic Acids Res 31, 4176-4183
14. Zhou, H. X. (2001) Biochemistry 40, 15069-15073
15. Arunkumar, A. I., Stauffer, M. E., Bochkareva, E., Bochkarev, A., and Chazin, W. J.
(2003) J Biol Chem 278, 41077-41082
13
by guest on June 11, 2018http://w
ww
.jbc.org/D
ownloaded from

16. Jackson, D., Dhar, K., Wahl, J. K., Wold, M. S., and Borgstahl, G. E. (2002) J Mol Biol
321, 133-148
17. Mer, G., Bochkarev, A., Gupta, R., Bochkareva, E., Frappier, L., Ingles, C. J., Edwards,
A. M., and Chazin, W. J. (2000) Cell 103, 449-456
18. Arcus, V. (2002) Curr Opin Struct Biol 12, 794-801
19. Bochkarev, A., and Bochkareva, E. (2004) Curr Opin Struct Biol 14, 36-42
20. Theobald, D. L., Mitton-Fry, R. M., and Wuttke, D. S. (2003) Annu Rev Biophys Biomol
Struct 32, 115-133
21. Bochkarev, A., Pfuetzner, R. A., Edwards, A. M., and Frappier, L. (1997) Nature 385,
176-181
22. Iftode, C., Daniely, Y., and Borowiec, J. A. (1999) Crit Rev Biochem Mol Biol 34, 141-
180
23. Yang, H., Jeffrey, P. D., Miller, J., Kinnucan, E., Sun, Y., Thoma, N. H., Zheng, N.,
Chen, P. L., Lee, W. H., and Pavletich, N. P. (2002) Science 297, 1837-1848
24. Kojic, M., Yang, H., Kostrub, C. F., Pavletich, N. P., and Holloman, W. K. (2003) Mol
Cell 12, 1043-1049
25. Gajiwala, K. S., and Burley, S. K. (2000) Curr Opin Struct Biol 10, 110-116
26. Giraldo, R. (2003) FEMS Microbiol Rev 26, 533-554
27. Bernstein, D. A., Zittel, M. C., and Keck, J. L. (2003) Embo J 22, 4910-4921
28. Brosh, R. M., Jr., von Kobbe, C., Sommers, J. A., Karmakar, P., Opresko, P. L.,
Piotrowski, J., Dianova, I., Dianov, G. L., and Bohr, V. A. (2001) Embo J 20, 5791-5801
29. von Kobbe, C., Karmakar, P., Dawut, L., Opresko, P., Zeng, X., Brosh, R. M., Jr.,
Hickson, I. D., and Bohr, V. A. (2002) J Biol Chem 277, 22035-22044
30. Liu, J., Smith, C. L., DeRyckere, D., DeAngelis, K., Martin, G. S., and Berger, J. M.
(2000) Mol Cell 6, 637-648
31. Shin, J. H., Grabowski, B., Kasiviswanathan, R., Bell, S. D., and Kelman, Z. (2003) J
Biol Chem 278, 38059-38067
14
by guest on June 11, 2018http://w
ww
.jbc.org/D
ownloaded from

32. Grabowski, B., and Kelman, Z. (2001) J Bacteriol 183, 5459-5464
33. Lopez de Saro, F. J., Georgescu, R. E., Goodman, M. F., and O'Donnell, M. (2003) Embo
J 22, 6408-6418
34. Stauffer, M. E., and Chazin, W. J. (2004) Journal of Biological Chemistry, in press
35. Pellegrini, L., Yu, D. S., Lo, T., Anand, S., Lee, M., Blundell, T. L., and Venkitaraman,
A. R. (2002) Nature 420, 287-293
36. Maga, G., and Hubscher, U. (2003) J Cell Sci 116, 3051-3060
37. Sawaya, M. R., Prasad, R., Wilson, S. H., Kraut, J., and Pelletier, H. (1997) Biochemistry
36, 11205-11215
38. Lee, J. Y., Chang, C., Song, H. K., Moon, J., Yang, J. K., Kim, H. K., Kwon, S. T., and
Suh, S. W. (2000) Embo J 19, 1119-1129
39. Tom, S., Henricksen, L. A., and Bambara, R. A. (2000) J Biol Chem 275, 10498-10505
40. Chapados, B. R., Hosfield, D. J., Han, S., Qiu, J., Yelent, B., Shen, B., and Tainer, J. A.
(2004) Cell 116, 39-50
41. Hopfner, K. P., and Tainer, J. A. (2003) Curr Opin Struct Biol 13, 249-255
42. Hopfner, K. P., Karcher, A., Craig, L., Woo, T. T., Carney, J. P., and Tainer, J. A. (2001)
Cell 105, 473-485
43. VanLoock, M. S., Yu, X., Yang, S., Lai, A. L., Low, C., Campbell, M. J., and Egelman,
E. H. (2003) Structure (Camb) 11, 187-196
44. Shin, D. S., Pellegrini, L., Daniels, D. S., Yelent, B., Craig, L., Bates, D., Yu, D. S.,
Shivji, M. K., Hitomi, C., Arvai, A. S., Volkmann, N., Tsuruta, H., Blundell, T. L.,
Venkitaraman, A. R., and Tainer, J. A. (2003) Embo J 22, 4566-4576
Footnotes 1 We note here there is mixing of the kinetic and thermodynamic views, but the assumption that
weak binding is associated with high on/off rates is logical in this context.
15
by guest on June 11, 2018http://w
ww
.jbc.org/D
ownloaded from

R P A
P C NA R FC αA
B
XP C
T F IIH R P A
XP A
XP F XP G
CR P A
R ad52R ad51
δ
δ
α
F igure 1
16
by guest on June 11, 2018http://w
ww
.jbc.org/D
ownloaded from

A
B
C
F igure 2
A
B
C
17
by guest on June 11, 2018http://w
ww
.jbc.org/D
ownloaded from

50-I50-II
50-I
ATP
11
F igure 3
50-II11
30o
18
by guest on June 11, 2018http://w
ww
.jbc.org/D
ownloaded from

Melissa E. Stauffer and Walter J. ChazinStructural mechanisms of DNA replication, repair, and recombination
published online April 16, 2004J. Biol. Chem.
10.1074/jbc.R400015200Access the most updated version of this article at doi:
Alerts:
When a correction for this article is posted•
When this article is cited•
to choose from all of JBC's e-mail alertsClick here
by guest on June 11, 2018http://w
ww
.jbc.org/D
ownloaded from