Embed Size (px)
Citation preview

Analytical Biochemistry 383 (2008) 243–254
Contents lists available at ScienceDirect
Analytical Biochemistry
journal homepage: www.elsevier .com/ locate/yabio
Structural analysis of the glycosylation of gene-activated erythropoietin(epoetin delta, Dynepo)
Esther Llop a, Ricardo Gutiérrez-Gallego a,b,*, Jordi Segura a,b, Joaquim Mallorquí a, José A. Pascual a,b
a Bioanalysis Research Group, Neuropsycho-pharmacology Program, IMIM–Hospital del Mar, Barcelona Biomedical Research Park (PRBB), 08003 Barcelona, Spainb Department of Experimental and Health Sciences, Pompeu Fabra University, Barcelona Biomedical Research Park (PRBB), 08003 Barcelona, Spain
a r t i c l e i n f o
Article history:Received 16 June 2008Available online 3 September 2008
Keywords:ErythropoietinEpoetin deltaGlycosylation2-DEMass spectrometryMALDISialic acid
0003-2697/$ - see front matter � 2008 Published bydoi:10.1016/j.ab.2008.08.027
* Corresponding author. Address: Department oSciences, Pompeu Fabra University, Barcelona Biom08003 Barcelona, Spain. Fax: +34 93 316 04 67.
E-mail address: [email protected] (R. Gutiérrez-G1 Abbreviations used: EPO, erythropoietin; rEPO, reco
d, epoetin delta; CHO, Chinese hamster ovary; BHK,CMP-N-acetylneuraminic acid hydroxylase; IEF, isopeptide-N4-(acetyl–glucosaminyl)-asparagine amidaseacid; sinapinic acid, 3,5-dimethoxy-4-hydroxycinnamicAB, 2-aminobenzamide; NaBH3CN, sodium cyanobosulfoxide; DTT, dithiothreitol; SDS, sodium dodecyl sumethylethylenediamine; DMB, 1,2-diamino-4,5-metrifluoroacetic acid; UPLC–ESI–TOF, ultra-performancetrospray ionization–time-of-flight; MS, mass specperformance liquid chromatography–fluorescence deteelectrophoresis; PVDF, polyvinylidene fluoride; mAb,polyacrylamide gel electrophoresis; MALDI, matrix-asstion; WAX, weak anion exchange; NP, normal phaseN-acetyllactosamine; Neu5Gc, N-glycolylneuraminic acinic acid; RAAM, reagent array analysis method; ELISAbent assay.
a b s t r a c t
Recently, a novel recombinant human erythropoietin (epoetin delta, Dynepo) has been marketed in theEuropean Union for the treatment of chronic kidney disease, cancer patients receiving chemotherapy,and so forth. Epoetin delta is engineered in cultures of the human fibrosarcoma cell line HT-1080 byhomologous recombination and ‘‘gene activation.” Unlike recombinant erythropoietins produced in othermammalian cells, epoetin delta is supposed to have a human-type glycosylation profile. However, the iso-electric focusing profile of epoetin delta differs from that of endogenous erythropoietin (both urinary andplasmatic). In this work, structural and quantitative analysis of the O- and N-glycans of epoetin delta wasperformed and compared with glycosylation from recombinant erythropoietin produced in Chinese ham-ster ovary (CHO) cells. From the comparison, significant differences in the sialylation of O-glycans werefound. Furthermore, the N-glycan analysis indicated a lower heterogeneity from epoetin delta when com-pared with its CHO homologue, being predominantly tetraantennary without N-acetyllactosaminerepeats in the former. The sialic acid characterization revealed the absence of N-glycolylneuraminic acid.The overall sugar profiles of both glycoproteins appeared to be significantly different and could be usefulfor maintaining pharmaceutical quality control, detecting the misuse of erythropoietin in sports, andestablishing new avenues to link glycosylation with biological activity of glycoproteins.
� 2008 Published by Elsevier Inc.
Human erythropoietin (EPO)1 was the first hematopoieticgrowth factor to be cloned, and the recombinant molecule becameavailable as a drug in 1988 [1]. Recombinant erythropoietin (rEPO)and analogues are frequently used in the treatment of anemia inchronic kidney disease, AIDS, prior autologous blood transfusions,
Elsevier Inc.
f Experimental and Healthedical Research Park (PRBB),
allego).mbinant erythropoietin; rEPObaby hamster kidney; CMAH,electric focusing; PNGase F,F; DHB, 2,5-dihydroxybenzoic
acid; IAA, iodoacetamide; 2-rohydride; DMSO, dimethyllfate; TEMED, N,N,N,N’-tetra-
thylene dioxybenzene; TFA,liquid chromatography–elec-trometry; HPLC–FLD, high-ction; 2-DE, two-dimensionalmonoclonal antibody; PAGE,isted laser desorption/ioniza-; GU, glucose units; LacNAc,id; Neu5Ac, N-acetylneuram-, enzyme-linked immunosor-
and many other malignancies [2]. In sports, the misuse of these sub-stances has also been demonstrated [3,4]. With the key patents forrEPO expired, the market has opened for biosimilar recombinantproducts from the same cell line with identical or improved pharma-cokinetic properties and less immunogenic response [5]. Also, com-pletely new products are produced, for example, epoetin delta (rEPOd, Dynepo) from Shire Pharmaceuticals. The latter is homologouslyexpressed by gene activation in a human fibrosarcoma cell line(HT-1080) [6]. rEPO d, as well as other epoetins, has the same 165-amino-acid polypeptide chain as the naturally occurring humanforms (urinary and plasmatic). However, from a biochemical view-point, there is an interesting diversity in their posttranslational mod-ification, most likely in glycosylation. Like endogenous EPO, epoetinscontain one O-linked oligosaccharide (Ser126) and three N-linkedoligosaccharides (Asn24, -38, and -83) [7,8], and these account forapproximately 40% of the total molecular weight (� 30 kDa). Thebiosynthesis of glycans is species, tissue, and cell type dependent,but the culture conditions may also contribute to the so-calledmicroheterogeneity, resulting in a large diversity of glycan struc-tures [9–17]. Chinese hamster ovary (CHO) and baby hamster kidney(BHK) cells are the hosts used for the production of rEPO pharmaceu-ticals. The enzymatic endowment of these frequently used host cellsis similar to that of human cells. However, some human tissue-spe-

Fig. 1. IEF profiles obtained for analysis of reference preparations of endogenousurinary EPO (uEPO, international reference preparation from National Institute forBiological Standards and Control, UK), recombinant erythropoietins (rEPO d, epoetindelta, Dynepo, Shire Pharmaceuticals, UK), rEPO a/b (biological reference prepara-tion of the European Pharmacopoeia containing epoetin alpha and epoetin beta inequal amounts), and new erythropoiesis-stimulating protein (NESP, darbepoetinalpha, Aranesp, Amgen, USA). The different areas and identification of bandsestablished by doping control laboratories are indicated.
244 Glycosylation of Dynepo / E. Llop et al. / Anal. Biochem. 383 (2008) 243–254
cific terminal carbohydrate motifs are not synthesized by BHK andCHO cells because they lack the proper sugar-transferring enzymes[18] (e.g., a2-6 sialyltransferase, a1-3/4 fucosyltransferase, bisectingN-acetylglucosamine transferase). On the contrary, these cells con-tain the enzyme responsible for N-glycolylneuraminic acid synthesis(CMP-N-acetylneuraminic acid hydroxylase [CMAH]) that is absentin humans [19]. The isoelectric focusing (IEF) analysis of rEPO dis-plays a profile in which more acidic isoforms can be observed withrespect to CHO-derived marketed rEPOs, albeit not the most acidicas isoforms present in endogenous EPO (urinary or plasmatic) [20–22] (Fig. 1). These different IEF profiles suggest that the glycosylationcould be different. Even though several articles have been publishedon the gene expression of rEPO d and its use in medicine [23,24], de-tails on the glycosylation affecting the biological activity have notyet been reported. In the current article, we describe the glycosyla-tion of rEPO d as compared with CHO cell-derived epoetins.
Materials and methods
Materials
Recombinant human EPO (rEPO produced in CHO cells) was ob-tained from the European Pharmacopoeia Commission (rEPO a/b,Salisbury, UK), and epoetin delta (rEPO produced in human fibro-sarcoma HT-1080 cells) was obtained from Shire Pharmaceuticals(rEPO d, Dynepo, Hampshire, UK). Recombinant peptide-N4-(acet-yl-b-glucosaminyl)-asparagine amidase F (PNGase F, EC 3.1.27.5),recombinant b-1,4-galactosidase (EC 3.2.1.23), recombinant b1-R-N-acetyl-glucosaminidase (EC 3.2.1.97), and recombinant a2-3,6,8,9-sialidase (EC 3.2.1.18) were purchased from Calbiochem(La Jolla, CA, USA). a2-3 Sialidase (EC 3.2.18) was obtained fromTakara Biotechnology (Shiga, Japan). Carbograph graphitized car-bon ultraclean columns (150 mg) were purchased from Alltech(Deerfield, IL, USA). 2,5-Dihydroxybenzoic acid (DHB), 3,5-dime-thoxy-4-hydroxycinnamic acid (sinapinic acid), iodoacetamide
(IAA), bovine serum albumin, bovine fetuin, and trypsin obtainedfrom bovine pancreas (EC 3.4.21.4) were purchased from Sigma(Barcelona, Spain). 2-Aminobenzamide (2-AB), sodium cyanoboro-hydride (NaBH3CN), and dimethyl sulfoxide (DMSO) were obtainedfrom Fluka (Barcelona, Spain). Dithiothreitol (DTT) was obtainedfrom GE Healthcare (Cerdanyola, Spain). Sialic acid reference panelwas obtained from Glyco (Ely, UK). Acrylamide-bis (97:3, w/w), sil-ver nitrate, and sodium dodecyl sulfate (SDS) were obtained fromMerck (Barcelona, Spain). N,N,N,N’-Tetramethylethylenediamine(TEMED) and ammonium persulfate were obtained from Bio-Rad(el Prat de Llobregat, Spain). GELoader tips were purchased fromEppendorf (Madrid, Spain), Quartz microfiber filters QMA were ob-tained from Whatman (Maidstone, UK). All other chemicals were ofthe highest purity commercially available.
Sialic acid analysis
For the study of the sialic acid heterogeneity, sialic acids werereleased from the carbohydrate chains and derivatized with 1,2-diamino-4,5-methylene dioxybenzene (DMB) [25]. Briefly, hydro-lysis of 1 lg of each sample was performed, incubating sampleswith trifluoroacetic acid (TFA) for 1 h at 50 �C. Next, a 7 mMDMB solution in 1.4 M aqueous acetic acid containing 18 mM so-dium hydrosulfite and 1 M b-mercaptoethanol was added. Themixture was kept for 2 h at 50 �C. Fluorescently labeled residueswere analyzed by reversed-phase chromatography on a capillarycolumn (Zorbax SB-C18, 150 � 0.3 mm, 3.5 lm) using MeCN/H2O(20:80) as eluent (mobile phase) and a flow rate of 4 ll/min. Chro-matographic analyses were performed on an Agilent 1100 seriescapillary instrument equipped with a Jasco micro21FP capillaryfluorescence detector (kex = 373 nm, kem = 448 nm).
DMB derivatives of sialic acids were also analyzed by ultra-per-formance liquid chromatography–electrospray ionization–time-of-flight (UPLC–ESI–TOF) mass spectrometry (MS) [26] using anAcquity ultra-performance liquid chromatograph (Waters) coupledto an LCT premier XE ESI–TOF instrument (Micromass). The proce-dure for sialic acid hydrolysis was similar to that described abovebut used 2 M aqueous acetic acid solution for 3 h at 80 �C insteadof TFA [27]. The derivatization protocol was identical, but sampleamounts prepared for LC–ESI–TOF were 10 times higher (10 lg)than in high-performance liquid chromatography–fluorescencedetection (HPLC–FLD) analysis due to the lower sensitivity of theformer technique. LC analyses were performed at a flow rate of0.2 ml/min using an Acquity UPLC BEH C18 column(100 � 2.1 mm, 1.7 lm). Derivatized sialic acids were eluted iso-cratically employing MeOH/MeCN/H2O (7:9:84) for 5 min, andthen with a 1-min gradient the running buffer was changed toMeOH/MeCN/H2O (7:25:68), maintained for 1 min, returned to ini-tial conditions, and stabilized for 1 min before the next injection.Mass spectra were acquired in negative ion mode over a massrange-to-charge (m/z) ratio of 50 to 650. The capillary voltagewas set at 2800 V (negative), the desolvation temperature wasset at 250 to 300 �C, and the desolvation gas flow was set at300 L/h. The TOF tube voltage was kept at 7200 (reflectron at1800 V) with a pusher setting of 900 V (pusher offset at 0.93). Re-corded data were processed using MassLynx software (version 4.1,Waters).
Glycoform characterization
First dimension (IEF)IEF was performed as described previously by Lasne and
coworkers [3]. In brief, rEPO d and rEPO a/b samples (sampleamounts loaded ranged from 0.3 ng for chemiluminescence detec-tion to 5 lg for two-dimensional electrophoresis [2-DE] and silverstaining) were focused in a polyacrylamide IEF gel with a pH gra-
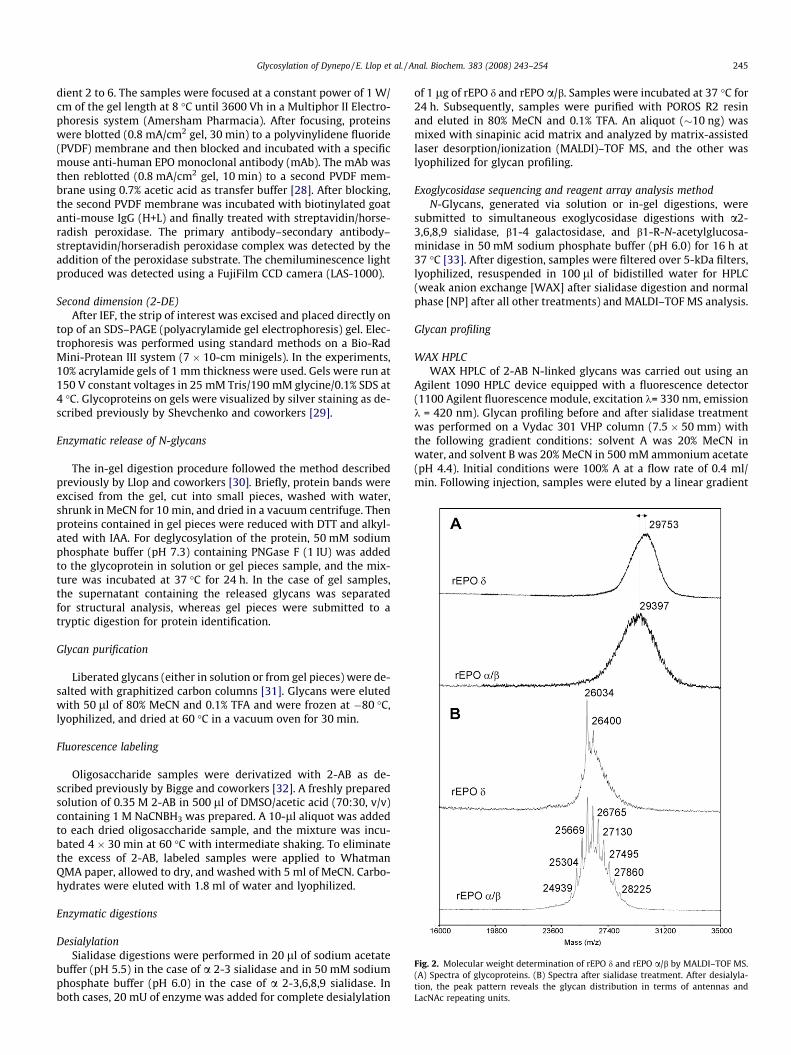
Fig. 2. Molecular weight determination of rEPO d and rEPO a/b by MALDI–TOF MS.(A) Spectra of glycoproteins. (B) Spectra after sialidase treatment. After desialyla-tion, the peak pattern reveals the glycan distribution in terms of antennas andLacNAc repeating units.
Glycosylation of Dynepo / E. Llop et al. / Anal. Biochem. 383 (2008) 243–254 245
dient 2 to 6. The samples were focused at a constant power of 1 W/cm of the gel length at 8 �C until 3600 Vh in a Multiphor II Electro-phoresis system (Amersham Pharmacia). After focusing, proteinswere blotted (0.8 mA/cm2 gel, 30 min) to a polyvinylidene fluoride(PVDF) membrane and then blocked and incubated with a specificmouse anti-human EPO monoclonal antibody (mAb). The mAb wasthen reblotted (0.8 mA/cm2 gel, 10 min) to a second PVDF mem-brane using 0.7% acetic acid as transfer buffer [28]. After blocking,the second PVDF membrane was incubated with biotinylated goatanti-mouse IgG (H+L) and finally treated with streptavidin/horse-radish peroxidase. The primary antibody–secondary antibody–streptavidin/horseradish peroxidase complex was detected by theaddition of the peroxidase substrate. The chemiluminescence lightproduced was detected using a FujiFilm CCD camera (LAS-1000).
Second dimension (2-DE)After IEF, the strip of interest was excised and placed directly on
top of an SDS–PAGE (polyacrylamide gel electrophoresis) gel. Elec-trophoresis was performed using standard methods on a Bio-RadMini-Protean III system (7 � 10-cm minigels). In the experiments,10% acrylamide gels of 1 mm thickness were used. Gels were run at150 V constant voltages in 25 mM Tris/190 mM glycine/0.1% SDS at4 �C. Glycoproteins on gels were visualized by silver staining as de-scribed previously by Shevchenko and coworkers [29].
Enzymatic release of N-glycans
The in-gel digestion procedure followed the method describedpreviously by Llop and coworkers [30]. Briefly, protein bands wereexcised from the gel, cut into small pieces, washed with water,shrunk in MeCN for 10 min, and dried in a vacuum centrifuge. Thenproteins contained in gel pieces were reduced with DTT and alkyl-ated with IAA. For deglycosylation of the protein, 50 mM sodiumphosphate buffer (pH 7.3) containing PNGase F (1 IU) was addedto the glycoprotein in solution or gel pieces sample, and the mix-ture was incubated at 37 �C for 24 h. In the case of gel samples,the supernatant containing the released glycans was separatedfor structural analysis, whereas gel pieces were submitted to atryptic digestion for protein identification.
Glycan purification
Liberated glycans (either in solution or from gel pieces) were de-salted with graphitized carbon columns [31]. Glycans were elutedwith 50 ll of 80% MeCN and 0.1% TFA and were frozen at �80 �C,lyophilized, and dried at 60 �C in a vacuum oven for 30 min.
Fluorescence labeling
Oligosaccharide samples were derivatized with 2-AB as de-scribed previously by Bigge and coworkers [32]. A freshly preparedsolution of 0.35 M 2-AB in 500 ll of DMSO/acetic acid (70:30, v/v)containing 1 M NaCNBH3 was prepared. A 10-ll aliquot was addedto each dried oligosaccharide sample, and the mixture was incu-bated 4 � 30 min at 60 �C with intermediate shaking. To eliminatethe excess of 2-AB, labeled samples were applied to WhatmanQMA paper, allowed to dry, and washed with 5 ml of MeCN. Carbo-hydrates were eluted with 1.8 ml of water and lyophilized.
Enzymatic digestions
DesialylationSialidase digestions were performed in 20 ll of sodium acetate
buffer (pH 5.5) in the case of a 2-3 sialidase and in 50 mM sodiumphosphate buffer (pH 6.0) in the case of a 2-3,6,8,9 sialidase. Inboth cases, 20 mU of enzyme was added for complete desialylation
of 1 lg of rEPO d and rEPO a/b. Samples were incubated at 37 �C for24 h. Subsequently, samples were purified with POROS R2 resinand eluted in 80% MeCN and 0.1% TFA. An aliquot (�10 ng) wasmixed with sinapinic acid matrix and analyzed by matrix-assistedlaser desorption/ionization (MALDI)–TOF MS, and the other waslyophilized for glycan profiling.
Exoglycosidase sequencing and reagent array analysis methodN-Glycans, generated via solution or in-gel digestions, were
submitted to simultaneous exoglycosidase digestions with a2-3,6,8,9 sialidase, b1-4 galactosidase, and b1-R-N-acetylglucosa-minidase in 50 mM sodium phosphate buffer (pH 6.0) for 16 h at37 �C [33]. After digestion, samples were filtered over 5-kDa filters,lyophilized, resuspended in 100 ll of bidistilled water for HPLC(weak anion exchange [WAX] after sialidase digestion and normalphase [NP] after all other treatments) and MALDI–TOF MS analysis.
Glycan profiling
WAX HPLCWAX HPLC of 2-AB N-linked glycans was carried out using an
Agilent 1090 HPLC device equipped with a fluorescence detector(1100 Agilent fluorescence module, excitation k= 330 nm, emissionk = 420 nm). Glycan profiling before and after sialidase treatmentwas performed on a Vydac 301 VHP column (7.5 � 50 mm) withthe following gradient conditions: solvent A was 20% MeCN inwater, and solvent B was 20% MeCN in 500 mM ammonium acetate(pH 4.4). Initial conditions were 100% A at a flow rate of 0.4 ml/min. Following injection, samples were eluted by a linear gradient
246 Glycosylation of Dynepo / E. Llop et al. / Anal. Biochem. 383 (2008) 243–254
of 100 to 0% B over 5 min, followed by a linear gradient of 0 to 100%B over the next 35 min, returning to the start conditions over thenext 15 min. The total running time was 60 min. The column wascalibrated with 2-AB-labeled N-glycans from bovine fetuin [34].
NP HPLCNP HPLC analyses were performed on a capillary system used
for sialic acid analyses. Fluorescence detector parameters werethe ones recommended for 2-AB tag as in WAX analyses. NP profil-ing was carried out on a TSK gel Amide-80 column (0.5 � 150 mm)
Fig. 3. Comparison of O-glycoforms from rEPO d and rEPO a/b by MALDI–TOF MS (
Fig. 4. (A) HPLC–FLD pattern of fluorescent DMB derivatives of sialic acids from rEPO d anlower panel. The identification of the numbered species is given in panel B. The insert isbe appreciated in the rEPO a/b only. (B) Extracted ion chromatograms of LC–MS analysis(right). The total ion chromatogram (TIC) is included at the bottom.
using the following gradient conditions: solvent A was 10% 50 mMammonium formate (pH 4.4) in 90% MeCN, solvent B was 90%50 mM ammonium formate (pH 4.4) in 10% MeCN, and the flowrate was 15 ll/min. Following injection, samples were eluted bya linear gradient of 20 to 55% B over 100 min, followed by a lineargradient of 55 to 100% B over the next 5 min. The column waseluted using 100% B for 2 min and subsequently was reequilibratedin 20% B before injection of the next sample. The system was cali-brated in glucose units (GU) using a 2-AB-labeled dextran hydroly-sate [35]. The total running time was 125 min.
A) and ESI–TOF MS (B). , N-acetylgalactosamine; j, galactose; D, sialic acid.
d rEPO a/b in comparison with the standard sialic acid (SA) mixture included in thea zoom of the chromatograms where the presence of N-glycolylneuraminic acid canof the DMB-derivatized sialic acid residues derived from rEPO d (left) and rEPO a/b

Glycosylation of Dynepo / E. Llop et al. / Anal. Biochem. 383 (2008) 243–254 247
MALDI–TOF MS analyses
Samples (proteins or carbohydrates) were dissolved in water atvarying concentrations. Aliquots were mixed with the correspond-ing matrix solution, and less than 1 ll of these preparations wasapplied to the polished stainless-steel target and allowed to dryat room temperature. A solution of sinapinic acid (10 mg/ml) inMeCN/H2O/TFA (50:50:0.1, v/v/v) was chosen for protein analyses,and a solution of DHB (10 mg/ml) in MeCN/H2O (50:50, v/v) waschosen for N-glycan analyses. Experiments were carried out on aVoyager-DE STR Biospectrometry workstation (Applied Biosys-tems) equipped with an N2 laser (337 nm). Typically, spectra ofsialylated N-glycans were acquired in linear mode with negativepolarity, and spectra of neutral N-glycans were acquired in reflec-tron mode with positive polarity. External calibration of the spec-trometer was performed using a mixture of 2-AB-labeled glucoseoligomers in the positive ion mode and 2-AB-derivatized fetuinN-glycans in the negative mode. Recorded data were processedwith Data Explorer software (Applied Biosystems).
LC–ESI–TOF MS analyses
Positive ion mode LC–ESI–TOF MS analyses of O-glycoproteins,after de-N-glycosylation, were performed using the instrument de-
Fig. 5. WAX HPLC profiles of 2-AB-labeled glycans from rEPO d (upper panel), rEPOa/b (center panel), and Fetuin used as standard (lower panel). The regionscorresponding to neutral (N) glycans and mono-, di-, tri-, and tetra-charged glycans(1, 2, 3, and 4, respectively) are indicated with gray columns. 2-AB indicates theexcess of derivatization reagent. Structures were tentatively assigned based on MSdata (vide infra). h, fucose; d, N-acetylglucosamine; �, mannose; j, galactose; D,sialic acid. The structural assignments for rEPO a/b are based on NMR data. Thestructural assignments for rEPO d have been done by analogy to those of rEPO a/band are indicative only.
scribed earlier in the ‘‘Sialic Acid Analyses” section. An aliquot of1 ll from 0.3 lg/ll sample solution (EPO d and rEPO a/b) was ana-lyzed in flow injection configuration at a flow rate of 0.05 ml/minusing a mobile phase of H2O/MeCN/FA (95:5:0.1, v/v/v). Mass spec-tra were acquired in positive ion mode for 3 min, covering therange of m/z 500 to 1500. The capillary voltage was set at 3000V, the desolvation temperature was set at 350 �C, and the desolva-tion gas flow was set at 400 L/h.
Results
Glycoprotein analysis
Intact rEPO d was analyzed by MALDI–TOF MS to determine themolecular mass and estimate the microheterogeneity of the glyco-protein. The average molecular weight was 29.75 kDa for rEPO d, avalue slightly higher than the reference rEPO a/b (29.39 kDa). Acomparison of the Gaussian distributions of the MS peaks and theirwidths at baseline suggests that rEPO d is less heterogeneous thanrEPO a/b (Fig. 2A). When the molecular weight was measured aftera2-R sialidase digestion (Fig. 2B), the signal became better re-solved, reflecting the number of antennas and/or N-acetyllactos-amine (LacNAc) repeats present in each glycan. The pattern ofpeaks separated by 365 Da consisted of 3 peaks in rEPO d, whereasthis phenomenon was more abundant (11 peaks) in rEPO a/b, indi-cating more structural variety. At this stage, the most intense peakin the mass spectrum of both preparations could be explained bythe peptide backbone plus 3 tetraantennary core-fucosylated N-glycans and 1 core–1 O-glycan. Interestingly, glycoforms contain-ing tri- and diantennary N-glycans are negligible for rEPO d,whereas they constitute an important part of the N-glycans in rEPOa/b. The mass difference between each glycoprotein before andafter sialidase digestion allows estimating the average sialic acidcontent. For rEPO d, the mass shift of approximately 3.5 kDa indi-cates the loss of an average of approximately 12.02 sialic acid res-idues; for rEPO a/b, the mass difference was 3.3 kDa,corresponding to approximately 11.53 sialic acid residues. Thishigher sialic acid content of rEPO d may justify the presence ofmore acidic bands in the IEF profile (Fig. 1). MS analysis after com-plete removal of all N-glycans corroborated that rEPO d containsonly 1 O-glycan and allowed studying the microheterogeneity con-tained within this glycan. The MALDI–TOF analysis (Fig. 3A)showed that 3 main O-glycoforms could be distinguished bearingno sialic acid residue (m/z = 18,616), 1 sialic acid residue (m/z =18,907), or 2 sialic acid residues (m/z = 19,198), as indicated bymass increments of 291. Although heterogeneity was causedmainly by a variable degree of sialylation of the O-glycan struc-tures, non-O-glycosylated molecules could also be detected (m/z =18,249), although the peak intensity was very low. To corroboratethe nature of these differences, the O-glycoprotein was analyzedafter sialidase treatment, showing the disappearance of sialylatedO-glycans and the increment of nonsialylated peak accounting forup to 98% of the total area (data not shown). Although the use ofMALDI–TOF MS reflects the complexity present in the O-glycan,the fact that part of the heterogeneity was caused by fragmentationof sialic acids cannot be excluded. Then a milder ionization tech-nique (LC–ESI–TOF) was used to minimize losses of sialic acids, giv-ing more feasible quantitative results (Fig. 2B). Under theseconditions, mass values and relative abundances obtained by MAL-DI–TOF were confirmed. Also by this technique, rEPO d presentedquantitative differences in sialylation of the O-glycans comparedwith rEPO a/b. The disialylated peak was the only one detected inrEPO d, accounting for the 100% of the signal. For rEPO a/b, both spe-cies (di- and monosialylated O-glycans) could be clearly detected,accounting for 56.36 and 43.64%, respectively. This result suggeststhat even under mild MALDI conditions, a certain degree of

248 Glycosylation of Dynepo / E. Llop et al. / Anal. Biochem. 383 (2008) 243–254
desialylation cannot be avoided [36,37]. Hence, in this article, MAL-DI–TOF MS is proposed to describe the heterogeneity contained inthe glycoprotein and ESI–TOF MS is employed for more accuratequantification.
Sialic acid analysis
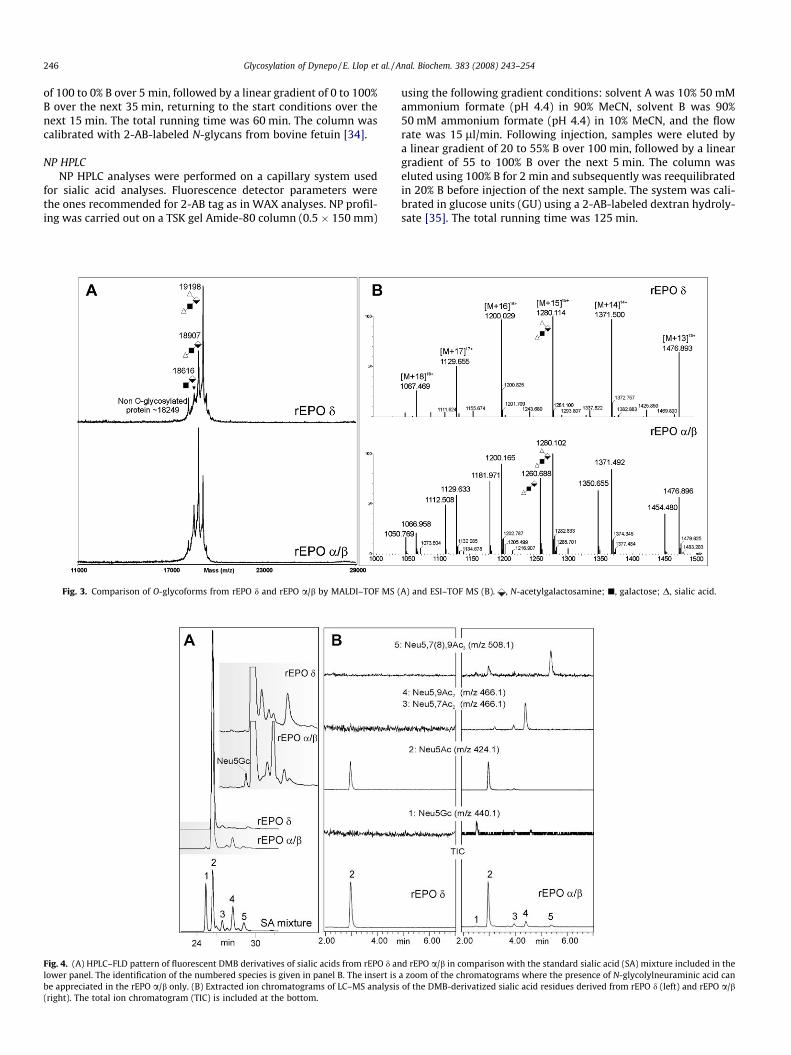
Analysis by reversed-phase HPLC of the fluorescently taggedsialic acids released by mild acid hydrolysis allowed determiningthe percentage of N-glycolylneuraminic acid (Neu5Gc), N-acetyl-neuraminic acid (Neu5Ac), and other variants (Fig. 4A). Identifica-tion was carried out by comparing retention times with referencecompounds [38]. Neu5Ac turned out to be the major constituentin both recombinant preparations, accounting for 98.1 mol% inrEPO d and 87.8 mol% in rEPO a/b. In addition, O-acetyl modifica-tions could also be identified, although they were far less abundantin rEPO d; Neu5,9Ac2 accounted for 1.9 mol% in rEPO-d, whereas itaccounted for up to 7.9 mol% in rEPO a/b. Also, Neu5,7Ac2 (1.2mol%) and Neu5,7(8),9Ac3 (1.7 mol%) could be detected, but inrEPO a/b only. Nevertheless, identification of the exact locationsof the O-acetyl linkage is somewhat cumbersome due to the abilityof these substituents to migrate from one hydroxyl to anotheralong the alkyl structure. Despite the care taken in sample han-dling to avoid migration of O-acetyl groups, the microheterogene-ity observed in replicates could still be affected by the specificbatch of rEPO and/or sample treatment prior to analysis. Finally,an interesting feature in sialic acid speciation is the potential pres-ence of N-glycolylneuraminic acid (Neu5Gc). As expected, rEPO dproduced in human cells (devoid of the corresponding hydroxy-lase) did not contain any Neu5Gc (limit of detection of 6 fmol[i.e., 0.3 mol%] in these experiments), whereas rEPO a/b hadapproximately 1.3 mol% Neu5Gc. To corroborate these results, sia-lic acids were analyzed simultaneously by UPLC–ESI–TOF (Fig. 4B).Confirming what was seen by fluorimetric detection, rEPO dshowed a detectable peak at the trace corresponding to m/z424.141 (protonated pseudomolecular ion of Neu5Ac) but noneat m/z 440.13, characteristic of Neu5Gc. On the contrary, the anal-yses of rEPO a/b showed a clear peak at the trace m/z 440.13, con-firming the presence of Neu5Gc. Furthermore, besides the mostabundant peak of Neu5Ac (at m/z 424.14), other species with acetylgroups at different positions were also found—Neu5,7(9)Ac2 (m/z466.14) and Neu5,7,9Ac3 (m/z 508.15).
Fig. 6. Negative ion mode MALDI mass spectra of 2-AB-labeled glycans from rEPO d (tofucose; d, N-acetylglucosamine; �, mannose; j, galactose; D, sialic acid. The structural asd have been done by analogy to those of rEPO a/b and are indicative only.
N-Glycan analysis
Peptide mapping of rEPO d and rEPO a/b after de-N-glycosyla-tion yielded no structural differences between the two (data notshown). At this point, the study was focused on the N-glycans re-leased from the proteins in an attempt to identify and quantifyall structures present first in the total mixture and then in eachindividual glycoform after 2-DE gel separation. WAX carbohydrateprofiling, standardized against fetuin N-glycans, was chosen toanalyze the charge state of N-glycans (Fig. 5). For rEPO d, 3 majorpeaks were observed, showing a much less heterogeneous profilethan rEPO a/b. The single peak of tetra-charged structures of rEPOd accounted for 44.28%, tri-charged accounted for 37.69%, and di-charged accounted for 16.81%. Although it was supposed to containa low proportion of mono-charged entities, they could not bequantified because of the 2-AB excess. Neutral structures werenearly absent (�1.2%). For rEPO a/b, once the areas of the 2 peaksaccounting for tetrasialylated N-glycans were summed, theyyielded 47.85%. The 5 different peaks of tri-charged species con-tributed 36.85%, and another 4 peaks accounted for 12.63%, of disi-alylated N-glycans. Monosialylated and neutral N-glycans wereminorities, accounting for 0.76 and 1.90%, respectively. Percent-ages of N-glycans with different degrees of sialylation togetherwith percentages of each sialylated O-glycoform allowed calculat-ing the average number of sialic acids. The equation applied is asfollows:
4�%N�4SAþ 3�%N�3SAþ 2�%N�2SAþ 1�%N�1SA4� 100
� �� 12
þ 2�%O�2SAþ 1�%O�1SA2� 100
� �� 2;
where% N�4SA stands for the percentage of N-glycans tetrasialylat-ed,% N�3SA stands for the percentage of N-glycans trisialylated, andso forth. The equivalent is also included for O-glycans. The denom-inator for N-glycans (4 � 100) normalizes the result and then ismultiplied by 12, the maximum number of sialic acids potentiallypresent in all 3 glycans (4 each). For O-glycans the normalizationfactor is then 2 � 100 and the result is multiplied by 2.
To compute the average number of sialic acids in the entire gly-coprotein, the sialic acid content of the O-glycan was calculatedfrom the O-glycoprotein MS analysis and that of the N-glycanswas calculated from the WAX analysis of the derivatized N-glycans.
p) and rEPO a/b (bottom). The depicted structures indicate all possible isomers. h,signments for rEPO a/b are based on NMR data. The structural assignments for rEPO
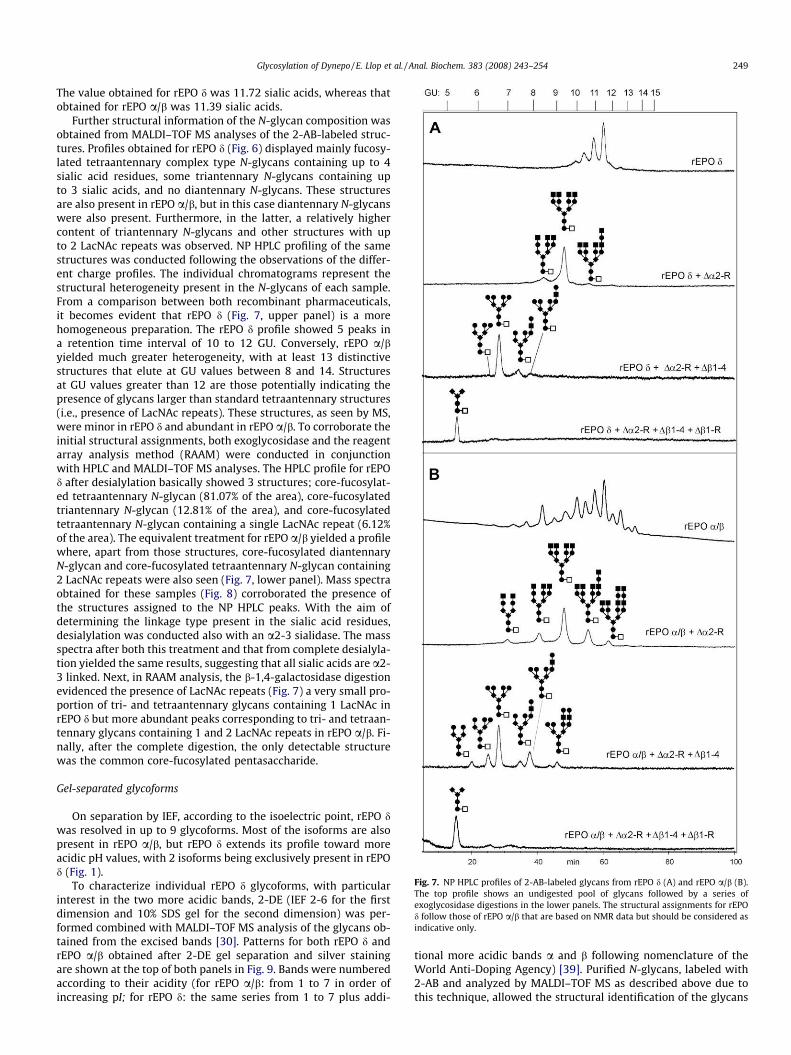
Fig. 7. NP HPLC profiles of 2-AB-labeled glycans from rEPO d (A) and rEPO a/b (B).The top profile shows an undigested pool of glycans followed by a series ofexoglycosidase digestions in the lower panels. The structural assignments for rEPOd follow those of rEPO a/b that are based on NMR data but should be considered asindicative only.
Glycosylation of Dynepo / E. Llop et al. / Anal. Biochem. 383 (2008) 243–254 249
The value obtained for rEPO d was 11.72 sialic acids, whereas thatobtained for rEPO a/b was 11.39 sialic acids.
Further structural information of the N-glycan composition wasobtained from MALDI–TOF MS analyses of the 2-AB-labeled struc-tures. Profiles obtained for rEPO d (Fig. 6) displayed mainly fucosy-lated tetraantennary complex type N-glycans containing up to 4sialic acid residues, some triantennary N-glycans containing upto 3 sialic acids, and no diantennary N-glycans. These structuresare also present in rEPO a/b, but in this case diantennary N-glycanswere also present. Furthermore, in the latter, a relatively highercontent of triantennary N-glycans and other structures with upto 2 LacNAc repeats was observed. NP HPLC profiling of the samestructures was conducted following the observations of the differ-ent charge profiles. The individual chromatograms represent thestructural heterogeneity present in the N-glycans of each sample.From a comparison between both recombinant pharmaceuticals,it becomes evident that rEPO d (Fig. 7, upper panel) is a morehomogeneous preparation. The rEPO d profile showed 5 peaks ina retention time interval of 10 to 12 GU. Conversely, rEPO a/byielded much greater heterogeneity, with at least 13 distinctivestructures that elute at GU values between 8 and 14. Structuresat GU values greater than 12 are those potentially indicating thepresence of glycans larger than standard tetraantennary structures(i.e., presence of LacNAc repeats). These structures, as seen by MS,were minor in rEPO d and abundant in rEPO a/b. To corroborate theinitial structural assignments, both exoglycosidase and the reagentarray analysis method (RAAM) were conducted in conjunctionwith HPLC and MALDI–TOF MS analyses. The HPLC profile for rEPOd after desialylation basically showed 3 structures; core-fucosylat-ed tetraantennary N-glycan (81.07% of the area), core-fucosylatedtriantennary N-glycan (12.81% of the area), and core-fucosylatedtetraantennary N-glycan containing a single LacNAc repeat (6.12%of the area). The equivalent treatment for rEPO a/b yielded a profilewhere, apart from those structures, core-fucosylated diantennaryN-glycan and core-fucosylated tetraantennary N-glycan containing2 LacNAc repeats were also seen (Fig. 7, lower panel). Mass spectraobtained for these samples (Fig. 8) corroborated the presence ofthe structures assigned to the NP HPLC peaks. With the aim ofdetermining the linkage type present in the sialic acid residues,desialylation was conducted also with an a2-3 sialidase. The massspectra after both this treatment and that from complete desialyla-tion yielded the same results, suggesting that all sialic acids are a2-3 linked. Next, in RAAM analysis, the b-1,4-galactosidase digestionevidenced the presence of LacNAc repeats (Fig. 7) a very small pro-portion of tri- and tetraantennary glycans containing 1 LacNAc inrEPO d but more abundant peaks corresponding to tri- and tetraan-tennary glycans containing 1 and 2 LacNAc repeats in rEPO a/b. Fi-nally, after the complete digestion, the only detectable structurewas the common core-fucosylated pentasaccharide.
Gel-separated glycoforms
On separation by IEF, according to the isoelectric point, rEPO dwas resolved in up to 9 glycoforms. Most of the isoforms are alsopresent in rEPO a/b, but rEPO d extends its profile toward moreacidic pH values, with 2 isoforms being exclusively present in rEPOd (Fig. 1).
To characterize individual rEPO d glycoforms, with particularinterest in the two more acidic bands, 2-DE (IEF 2-6 for the firstdimension and 10% SDS gel for the second dimension) was per-formed combined with MALDI–TOF MS analysis of the glycans ob-tained from the excised bands [30]. Patterns for both rEPO d andrEPO a/b obtained after 2-DE gel separation and silver stainingare shown at the top of both panels in Fig. 9. Bands were numberedaccording to their acidity (for rEPO a/b: from 1 to 7 in order ofincreasing pI; for rEPO d: the same series from 1 to 7 plus addi-
tional more acidic bands a and b following nomenclature of theWorld Anti-Doping Agency) [39]. Purified N-glycans, labeled with2-AB and analyzed by MALDI–TOF MS as described above due tothis technique, allowed the structural identification of the glycans

Fig. 8. MALDI mass spectra of rEPO d before exoglycosidase digestions (upper panel, negative ion mode) and after exoglycosidase digestions (lower panels, positive ionmode). The structural assignments for rEPO d follow those of rEPO a/b that are based on NMR data but should be considered as indicative only.
250 Glycosylation of Dynepo / E. Llop et al. / Anal. Biochem. 383 (2008) 243–254
contained in individual bands. Mass spectra of purified N-glycansfrom the least abundant bands (7 and b for rEPO d) could not be ob-tained (Fig. 9). However, the results clearly show the trend towarda higher degree of sialylation following the decreasing pI values ofthe IEF band. Because rEPO d posseses much less heterogeneity,this variation resides mainly on the sialylation degree of tetraan-tennary chains, showing a progressive increase of tetra-chargedstructures and a progressive decrease of mono-charged specieswith increasing acidity of the band (Fig. 9A). Although rEPO a/balso showed this trend (Fig. 9B), the larger structural heterogeneityin this product (the presence of di- and triantennary structures andthe existence of LacNAc repeats) rendered a less pronounced phe-nomenon in individual structures. Overall, these results confirmedthat sialic acid residues are the sole charges present in the glycans.The presence of other charged residues such as sulfates describedfor other human glycoproteins were not observed despite the factthat rEPO d is produced in human cells.
Discussion
Like most glycoproteins, rEPO d and rEPO a/b are also heteroge-neous with respect to the glycosylation, resulting in different iso-forms. Although it is extremely difficult to establish the precisecontribution of individual glycoforms to the overall activity, toxic-ity, and (in some cases) immunogenicity of these biopharmaceuti-cals, analysis of their glycosylation pattern is of utmost importance
in attempting such understanding in addition to guaranteeing drugquality and efficacy [40]. Furthermore, the differential structuralelements could be useful for antidoping purposes through targetedanalyses.
On comparison of the structural features of the recombinantand gene-activated EPOs, the first surprising observation camefrom the molecular weights of both glycoproteins. The slightlyhigher molecular weight found by MALDI–TOF MS for rEPO d whencompared with rEPO a/b contrasted with the molecular weightdetermination by SDS–PAGE [41]. In this technique, rEPO a/b mi-grates slower, indicating higher hydrodynamic volume. MS andHPLC analyses performed in this study suggest LacNAc repeatingunits as being mainly responsible for this effect. This phenomenonwas analyzed in-depth, comparing migration of both rEPOs in a 2-DE gel. Glycoforms from rEPO a/b are observed as a ‘‘train” ofbands that differ not only in pI but also in apparent molecular mass(i.e., diagonality). The latter effect is not observed in urinary EPO[42], and here we have demonstrated that it is also much less pro-nounced in rEPO d. With the obtained knowledge of the structuraldifferences between these two recombinant homologues, the diag-onality in 2-DE and the band broadness in SDS–PAGE can be attrib-uted predominantly to the presence of LacNAc repeats, whereassialic acids play a much less prominent role than is assumed sofar. To validate this assumption, the average mass conferred to aglycoform through the 3 N-glycans in a single band, both withand without sialic acid residues, was calculated. In Fig. 10, a clearly
Glycosylation of Dynepo / E. Llop et al. / Anal. Biochem. 383 (2008) 243–254 251
different slope can be appreciated for the total glycans, and this isfairly consistent with the diagonal trends observed in the 2-DE ofthe intact glycoproteins. Without sialic acid residues, rEPO d showsa nearly horizontal mass trend, as expected given that it containspredominantly tetraantennary N-glycans, whereas rEPO a/b stillshows a diagonal trend albeit with a lower slope.
The difference in microheterogeneity was also observed fromthe molecular weight analysis. The narrower peak width of therEPO d in the MALDI–TOF spectrum suggested lesser heterogeneity
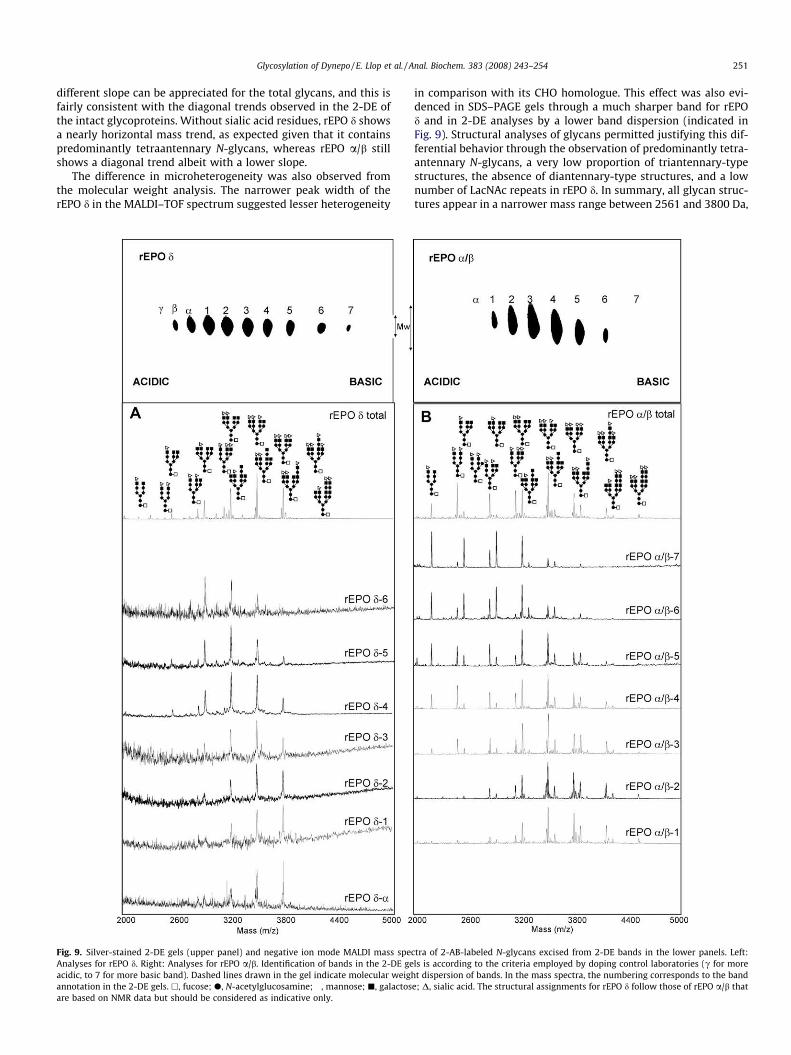
Fig. 9. Silver-stained 2-DE gels (upper panel) and negative ion mode MALDI mass specAnalyses for rEPO d. Right: Analyses for rEPO a/b. Identification of bands in the 2-DE geacidic, to 7 for more basic band). Dashed lines drawn in the gel indicate molecular weighannotation in the 2-DE gels. h, fucose; d, N-acetylglucosamine; �, mannose; j, galactosare based on NMR data but should be considered as indicative only.
in comparison with its CHO homologue. This effect was also evi-denced in SDS–PAGE gels through a much sharper band for rEPOd and in 2-DE analyses by a lower band dispersion (indicated inFig. 9). Structural analyses of glycans permitted justifying this dif-ferential behavior through the observation of predominantly tetra-antennary N-glycans, a very low proportion of triantennary-typestructures, the absence of diantennary-type structures, and a lownumber of LacNAc repeats in rEPO d. In summary, all glycan struc-tures appear in a narrower mass range between 2561 and 3800 Da,
tra of 2-AB-labeled N-glycans excised from 2-DE bands in the lower panels. Left:ls is according to the criteria employed by doping control laboratories (c for moret dispersion of bands. In the mass spectra, the numbering corresponds to the band
e; D, sialic acid. The structural assignments for rEPO d follow those of rEPO a/b that
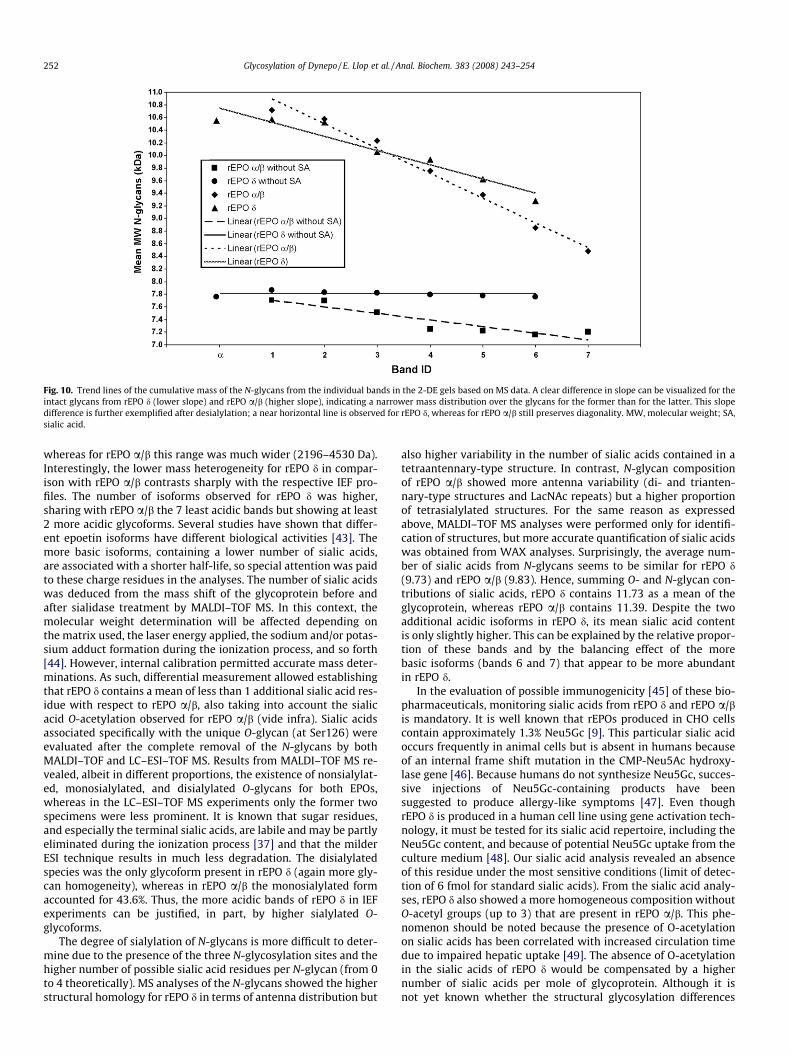
Fig. 10. Trend lines of the cumulative mass of the N-glycans from the individual bands in the 2-DE gels based on MS data. A clear difference in slope can be visualized for theintact glycans from rEPO d (lower slope) and rEPO a/b (higher slope), indicating a narrower mass distribution over the glycans for the former than for the latter. This slopedifference is further exemplified after desialylation; a near horizontal line is observed for rEPO d, whereas for rEPO a/b still preserves diagonality. MW, molecular weight; SA,sialic acid.
252 Glycosylation of Dynepo / E. Llop et al. / Anal. Biochem. 383 (2008) 243–254
whereas for rEPO a/b this range was much wider (2196–4530 Da).Interestingly, the lower mass heterogeneity for rEPO d in compar-ison with rEPO a/b contrasts sharply with the respective IEF pro-files. The number of isoforms observed for rEPO d was higher,sharing with rEPO a/b the 7 least acidic bands but showing at least2 more acidic glycoforms. Several studies have shown that differ-ent epoetin isoforms have different biological activities [43]. Themore basic isoforms, containing a lower number of sialic acids,are associated with a shorter half-life, so special attention was paidto these charge residues in the analyses. The number of sialic acidswas deduced from the mass shift of the glycoprotein before andafter sialidase treatment by MALDI–TOF MS. In this context, themolecular weight determination will be affected depending onthe matrix used, the laser energy applied, the sodium and/or potas-sium adduct formation during the ionization process, and so forth[44]. However, internal calibration permitted accurate mass deter-minations. As such, differential measurement allowed establishingthat rEPO d contains a mean of less than 1 additional sialic acid res-idue with respect to rEPO a/b, also taking into account the sialicacid O-acetylation observed for rEPO a/b (vide infra). Sialic acidsassociated specifically with the unique O-glycan (at Ser126) wereevaluated after the complete removal of the N-glycans by bothMALDI–TOF and LC–ESI–TOF MS. Results from MALDI–TOF MS re-vealed, albeit in different proportions, the existence of nonsialylat-ed, monosialylated, and disialylated O-glycans for both EPOs,whereas in the LC–ESI–TOF MS experiments only the former twospecimens were less prominent. It is known that sugar residues,and especially the terminal sialic acids, are labile and may be partlyeliminated during the ionization process [37] and that the milderESI technique results in much less degradation. The disialylatedspecies was the only glycoform present in rEPO d (again more gly-can homogeneity), whereas in rEPO a/b the monosialylated formaccounted for 43.6%. Thus, the more acidic bands of rEPO d in IEFexperiments can be justified, in part, by higher sialylated O-glycoforms.
The degree of sialylation of N-glycans is more difficult to deter-mine due to the presence of the three N-glycosylation sites and thehigher number of possible sialic acid residues per N-glycan (from 0to 4 theoretically). MS analyses of the N-glycans showed the higherstructural homology for rEPO d in terms of antenna distribution but
also higher variability in the number of sialic acids contained in atetraantennary-type structure. In contrast, N-glycan compositionof rEPO a/b showed more antenna variability (di- and trianten-nary-type structures and LacNAc repeats) but a higher proportionof tetrasialylated structures. For the same reason as expressedabove, MALDI–TOF MS analyses were performed only for identifi-cation of structures, but more accurate quantification of sialic acidswas obtained from WAX analyses. Surprisingly, the average num-ber of sialic acids from N-glycans seems to be similar for rEPO d(9.73) and rEPO a/b (9.83). Hence, summing O- and N-glycan con-tributions of sialic acids, rEPO d contains 11.73 as a mean of theglycoprotein, whereas rEPO a/b contains 11.39. Despite the twoadditional acidic isoforms in rEPO d, its mean sialic acid contentis only slightly higher. This can be explained by the relative propor-tion of these bands and by the balancing effect of the morebasic isoforms (bands 6 and 7) that appear to be more abundantin rEPO d.
In the evaluation of possible immunogenicity [45] of these bio-pharmaceuticals, monitoring sialic acids from rEPO d and rEPO a/bis mandatory. It is well known that rEPOs produced in CHO cellscontain approximately 1.3% Neu5Gc [9]. This particular sialic acidoccurs frequently in animal cells but is absent in humans becauseof an internal frame shift mutation in the CMP-Neu5Ac hydroxy-lase gene [46]. Because humans do not synthesize Neu5Gc, succes-sive injections of Neu5Gc-containing products have beensuggested to produce allergy-like symptoms [47]. Even thoughrEPO d is produced in a human cell line using gene activation tech-nology, it must be tested for its sialic acid repertoire, including theNeu5Gc content, and because of potential Neu5Gc uptake from theculture medium [48]. Our sialic acid analysis revealed an absenceof this residue under the most sensitive conditions (limit of detec-tion of 6 fmol for standard sialic acids). From the sialic acid analy-ses, rEPO d also showed a more homogeneous composition withoutO-acetyl groups (up to 3) that are present in rEPO a/b. This phe-nomenon should be noted because the presence of O-acetylationon sialic acids has been correlated with increased circulation timedue to impaired hepatic uptake [49]. The absence of O-acetylationin the sialic acids of rEPO d would be compensated by a highernumber of sialic acids per mole of glycoprotein. Although it isnot yet known whether the structural glycosylation differences

Glycosylation of Dynepo / E. Llop et al. / Anal. Biochem. 383 (2008) 243–254 253
will lead to any therapeutic advantages of the use of rEPO dover rEPO a/b, articles published recently suggest a similar half-lifeto rEPO b [50] and rEPO a [23] but slightly lower adverse effectsthan rEPO a [51].
Despite the fact that CHO cells glycosylate proteins in a mannerthat is qualitatively similar to that in human cells, some human tis-sue-specific glycan structures are not synthesized by these cells be-cause they lack the proper glycosyl transferases. In this study, glycanmotifs that are present in human cells but absent in CHO cells(bisecting N-acetylglucosamine residues, sialic acids in a2-6, fucoseresidues in a1-3/4, or the presence of charged residues other thansialic acids [e.g., sulfates] in the glycans) were investigated. How-ever, structural analyses following specific exoglycosidase diges-tions depicted in Fig. 8, as well as specific enzyme-linkedimmunosorbent assay (ELISA) and Western analyses (data notshown) for sialyl LewisX and sialyl LewisA, failed to demonstratethe presence of any of these particular structural elements in rEPO d.
With the rigorous structural analysis of rEPO d completed, it canbe concluded that the more acidic bands present in IEF, when com-pared with other recombinant EPOs, are due to a higher degree ofsialylation. The maximum number of sialic acids possible in themolecule of EPO is 14, and the mean number of these residuesfor rEPO d is approximately 11.7. Thus, it can be speculated thatglycans migrating in the band depicted as b contain a maximumof 14 sialic acids. An interesting reflection concerns the fact thatendogenous EPO and rEPOs have the same number of glycosylationsites and that both should present partially overlapping isoformprofiles, with potentially more basic isoforms present in the caseof endogenous EPO because it is not enriched for high sialic acidcontent. However, the IEF analysis (Fig. 1) shows additional non-overlapping isoforms of urinary EPO appearing at more acidic pIvalues. Thus, this pattern cannot be explained in terms of sialicacid residues only and indicates that endogenous EPO containsadditional charges that have not been found in rEPO d despite itshuman origin.
Acknowledgments
The authors thank the World Anti-Doping Agency (WADA) forits financial support. Furthermore, we thank J. Joglar (Departmentof Biological Organic Chemistry, IIQAB–CSIC) for his kind help withthe LC–ESI–TOF MS measurements and C. de Bolós (MunicipalInstitute of Medical Research, IMIM–Hospital del Mar) for the sialylLewisX/A assays. Dynepo was kindly supplied by Christian Reichel(Doping Control Laboratory, ARC Seibersdorf Research GmbH)and Peter Hemmersbach (Hormone Laboratory, Aker UniversityHospital).
References
[1] F.K. Lin, S. Suggs, C.H. Lin, J.K. Browne, R. Smalling, J.C. Egrie, K.K. Chen, G.M.Fox, F. Martín, Z. Stabinsky, Cloning and expression of the humanerythropoietin gene, Proc. Natl. Acad. Sci. USA 82 (1985) 7580–7584.
[2] J.W. Fisher, Erythropoietin: physiology and pharmacology update, Exp. Biol.Med. 228 (2003) 1–14.
[3] F. Lasne, L. Martin, N. Crepin, J. de Ceaurriz, Detection of isoelectric profiles oferythropoietin in urine: differentiation of natural and administeredrecombinant hormones, Anal. Biochem. 311 (2002) 119–126.
[4] D.H. Catlin, A. Breidbach, S. Elliott, J. Glaspy, Comparison of the isoelectricfocusing patterns of darbepoetin alfa, recombinant human erythropoietin, andendogenous erythropoietin from human urine, Clin. Chem. 48 (2002) 2057–2059.
[5] I.C. Macdougall, K.U. Eckardt, Novel strategies for stimulating erythropoiesisand potential new treatments for anaemia, Lancet 368 (2006) 947–953.
[6] R. Deicher, W.H. Horl, Differentiating factors between erythropoiesis-stimulating agents: a guide to selection for anaemia of chronic kidneydisease, Drugs 64 (2004) 499–509.
[7] H. Sasaki, B. Bothner, A. Dell, M. Fukuda, Carbohydrate structure oferythropoietin expressed in Chinese hamster ovary cells by a humanerythropoietin cDNA, J. Biol. Chem. 262 (1987) 12059–12076.
[8] N. Imai, A. Kawamura, M. Higuchi, M. Oh-eda, T. Orita, T. Kawaguchi, N. Ochi,Physicochemical and biological comparison of recombinant humanerythropoietin with human urinary erythropoietin, J. Biochem. (Tokyo) 107(1990) 352–359.
[9] C.H. Hokke, A.A. Bergwerff, G.W. van Dedem, J.P. Kamerling, J.F. Vliegenthart,Structural analysis of the sialylated N- and O-linked carbohydrate chains ofrecombinant human erythropoietin expressed in Chinese hamster ovary cells:sialylation patterns and branch location of dimeric N-acetyllactosamine units,Eur. J. Biochem. 228 (1995) 981–1008.
[10] E. Tsuda, M. Goto, A. Murakami, K. Akai, M. Ueda, G. Kawanishi, N. Takahashi,R. Sasaki, H. Chiba, H. Ishihara, M. Mori, S. Tejima, S. Endo, Y. Arata,Comparative structural study of N-linked oligosaccharides of urinary andrecombinant erythropoietins, Biochemistry 27 (1988) 5646–5654.
[11] M. Takeuchi, S. Takasaki, H. Miyazaki, T. Kato, S. Hoshi, N. Kochibe, A. Kobata,Comparative study of the asparagine-linked sugar chains of humanerythropoietins purified from urine and the culture medium of recombinantChinese hamster ovary cells, J. Biol. Chem. 263 (1988) 3657–3663.
[12] K.B. Linsley, S.Y. Chan, S. Chan, B.B. Reinhold, P.J. Lisi, V.N. Reinhold,Applications of electrospray mass spectrometry to erythropoietin N- and O-linked glycans, Anal. Biochem. 219 (1994) 207–217.
[13] M. Nimtz, W. Martin, V. Wray, K.D. Kloppel, J. Augustin, H.S. Conradt,Structures of sialylated oligosaccharides of human erythropoietin expressedin recombinant BHK-21 cells, Eur. J. Biochem. 213 (1993) 39–56.
[14] C. Neususs, U. Demelbauer, M. Pelzing, Glycoform characterization of intacterythropoietin by capillary electrophoresis–electrospray–time of flight–massspectrometry, Electrophoresis 26 (2005) 1442–1450.
[15] E. Balaguer, U. Demelbauer, M. Pelzing, V. Sanz-Nebot, J. Barbosa, C. Neususs,Glycoform characterization of erythropoietin combining glycan and intactprotein analysis by capillary electrophoresis–electrospray–time-of-flight massspectrometry, Electrophoresis 27 (2006) 2638–2650.
[16] P.E. Groleau, P. Desharnais, L. Cote, C. Ayotte, Low LC–MS/MS detection ofglycopeptides released from pmol levels of recombinant erythropoietin usingnanoflow HPLC-chip electrospray ionization, J, Mass Spectrom. 43 (2008) 924–935.
[17] Y. Takegawa, H. Ito, T. Keira, K. Deguchi, H. Nakagawa, S. Nishimura, Profilingof N- and O-glycopeptides of erythropoietin by capillary zwitterionic type ofhydrophilic interaction chromatography/electrospray ionization massspectrometry, J. Sep. Sci. 31 (2008) 1585–1593.
[18] E. Grabenhorst, P. Schlenke, S. Pohl, M. Nimtz, H.S. Conradt, Geneticengineering of recombinant glycoproteins and the glycosylation pathway inmammalian host cells, Glycoconj. J. 16 (1999) 81–97.
[19] H.H. Chou, T. Hayakawa, S. Diaz, M. Krings, E. Indriati, M. Leakey, S. Paabo, Y.Satta, N. Takahata, A. Varki, Inactivation of CMP-N-acetylneuraminic acidhydroxylase occurred prior to brain expansion during human evolution, Proc.Natl. Acad. Sci. USA 99 (2002) 11736–11741.
[20] V. Skibeli, G. Nissen-Lie, P. Torjesen, Sugar profiling proves that human serumerythropoietin differs from recombinant human erythropoietin, Blood 98(2001) 3626–3634.
[21] J. Mallorqui, J. Segura, C. de Bolos, R. Gutiérrez-Gallego, J.A. Pascual,Recombinant erythropoietin found in seized blood bags from sportsmen,Haematologica 93 (2008) 313–314.
[22] F. Lasne, L. Martin, J.A. Martín, J. de Ceaurriz, Isoelectric profiles of humanerythropoietin are different in serum and urine, Intl. J. Biol. Macromol. 41(2007) 354–357.
[23] K.J. Martin, The first human cell line-derived erythropoietin, epoetin-delta(Dynepo), in the management of anemia in patients with chronic kidneydisease, Clin. Nephrol. 68 (2007) 26–31.
[24] W. Jelkmann, Control of erythropoietin gene expression and its use inmedicine, Methods Enzymol. 435 (2007) 179–197.
[25] S. Hara, Y. Takemori, M. Yamaguchi, M. Nakamura, Y. Ohkura, Fluorometric high-performanceliquidchromatographyofN-acetyl-andN-glycolylneuraminicacidsand its application to their microdetermination in human and animal sera,glycoproteins,andglycolipids,Anal.Biochem.164(1987)138–145.
[26] N. Morimoto, M. Nakano, M. Kinoshita, A. Kawabata, M. Morita, Y. Oda, R.Kuroda, K. Kakehi, Specific distribution of sialic acids in animal tissues asexamined by LC–ESI–MS after derivatization with 1,2-diamino-4,5-methylenedioxybenzene, Anal. Chem. 73 (2001) 5422–5428.
[27] C.H. Hokke, A.A. Bergwerff, G.W. van Dedem, J. van Oostrum, J.P. Kamerling, J.F.Vliegenthart, Sialylated carbohydrate chains of recombinant humanglycoproteins expressed in Chinese hamster ovary cells contain traces of N-glycolylneuraminic acid, FEBS Lett. 275 (1990) 9–14.
[28] F. Lasne, Double-blotting: a solution to the problem of nonspecific binding ofsecondary antibodies in immunoblotting procedures, J. Immunol. Methods 276(2003) 223–226.
[29] A. Shevchenko, M. Wilm, O. Vorm, M. Mann, Mass spectrometric sequencing ofproteins from silver-stained polyacrylamide gels, Anal. Chem. 68 (1996) 850–858.
[30] E. Llop, R. Gutiérrez-Gallego, V. Belalcazar, G.J. Gerwig, J.P. Kamerling, J. Segura,J.A. Pascual, Evaluation of protein N-glycosylation in 2-DE: erythropoietin as astudy case, Proteomics 7 (2007) 4278–4291.
[31] N.H. Packer, M.A. Lawson, D.R. Jardine, J.W. Redmond, A general approach todesalting oligosaccharides released from glycoproteins, Glycoconj. J. 15 (1998)737–747.
[32] J.C. Bigge, T.P. Patel, J.A. Bruce, P.N. Goulding, S.M. Charles, R.B. Parekh,Nonselective and efficient fluorescent labeling of glycans using 2-aminobenzamide and anthranilic acid, Anal. Biochem. 230 (1995) 229–238.

254 Glycosylation of Dynepo / E. Llop et al. / Anal. Biochem. 383 (2008) 243–254
[33] L. Royle, T.S. Mattu, E. Hart, J.I. Langridge, A.H. Merry, N. Murphy, D.J. Harvey,R.A. Dwek, P.M. Rudd, An analytical and structural database provides astrategy for sequencing O-glycans from microgram quantities of glycoproteins,Anal. Biochem. 304 (2002) 70–90.
[34] G.R. Guile, P.M. Rudd, D.R. Wing, S.B. Prime, R.A. Dwek, A rapid high-resolutionhigh-performance liquid chromatographic method for separating glycanmixtures and analyzing oligosaccharide profiles, Anal. Biochem. 240 (1996)210–226.
[35] L. Royle, C.M. Radcliffe, R.A. Dwek, P.M. Rudd, Detailed structural analysis of N-glycans released from glycoproteins in SDS–PAGE gel bands using HPLCcombined with exoglycosidase array digestions, Methods Mol. Biol. 347 (2006)125–143.
[36] E. Mortz, T. Sareneva, I. Julkunen, P. Roepstorff, Does matriz-assisted laserdesorption/ionization mass spectrometry allow analysis of carbohydrateheterogeneity in glycoproteins? A study of natural human interferon-c, J.Mass Spectrom. 31 (1996) 1109–1118.
[37] D.J. Harvey, L. Royle, C.M. Radcliffe, P.M. Rudd, R.A. Dwek, Structural andquantitative analysis of N-linked glycans by matriz-assisted laser desorption/ionization and negative ion nanospray mass spectrometry, Anal. Biochem. 376(2008) 44–60.
[38] K.R. Anumula, Advances in fluorescence derivatization methods for high-performance liquid chromatographic analysis of glycoprotein carbohydrates,Anal. Biochem. 350 (2006) 1–23.
[39] D. Catlin, C. Howe, F. Lasne, G. Nissen-Lie, J. Pascual, M. Saugy, WADA technicaldocument TD2004EPO, World Anti-Doping Agency, 2004.
[40] T. Dingermann, Recombinant therapeutic proteins: production platforms andchallenges, Biotechnol. J. 3 (2008) 90–97.
[41] M. Kohler, C. Ayotte, P. Desharnais, U. Flenker, S. Ludke, M. Thevis, E. Volker-Schanzer, W. Schanzer, Discrimination of recombinant and endogenousurinary erythropoietin by calculating relative mobility values from SDS gels,Intl. J. Sports Med. 29 (2008) 1–6.
[42] A. Khan, J. Grinyer, S.T. Truong, E.J. Breen, N.H. Packer, New urinary EPO drugtesting method using two-dimensional gel electrophoresis, Clin. Chim. Acta358 (2005) 119–130.
[43] P.L. Storring, R.J. Tiplady, R.E. Gaines Das, B.E. Stenning, A. Lamikanra, B.Rafferty, J. Lee, Epoetin alpha and beta differ in their erythropoietin isoformcompositions and biological properties, Br. J. Haematol. 100 (1998) 79–89.
[44] G. Stubiger, M. Marchetti, M. Nagano, C. Reichel, G. Gmeiner, G. Allmaier,Characterisation of intact recombinant human erythropoietins applied indoping by means of planar gel electrophoretic techniques and matriz-assistedlaser desorption/ionisation linear time-of-flight mass spectrometry, RapidCommun. Mass Spectrom. 19 (2005) 728–742.
[45] I.C. Macdougall, Antibody-mediated pure red cell aplasia (PRCA): epidemiology,immunogenicity, and risks, Nephrol. Dial. Transplant 20 (suppl. 4) (2005) iv9–iv15.
[46] E.C. Brinkman-Vander Linden, E.R. Sjoberg, L.R. Juneja, P.R. Crocker, N. Varki, A.Varki, Loss of N-glycolylneuraminic acid in human evolution: implications forsialic acid recognition by siglecs, J. Biol. Chem. 275 (2000) 8633–8640.
[47] B. Byrne, G.G. Donohoe, R. O’Kennedy, Sialic acids: carbohydrate moieties thatinfluence the biological and physical properties of biopharmaceutical proteinsand living cells, Drug Discov. Today 12 (2007) 319–326.
[48] P. Tangvoranuntakul, P. Gagneux, S. Diaz, M. Bardor, N. Varki, A. Varki, E.Muchmore, Human uptake and incorporation of an immunogenic nonhumandietary sialic acid, Proc. Natl. Acad. Sci. USA 100 (2003) 12045–12050.
[49] T. Corfield, Bacterial sialidases: roles in pathogenicity and nutrition,Glycobiology 2 (1992) 509–521.
[50] I.C. Macdougall, Comparison of different dosing regimens (once weekly vs. twiceweekly, and once weekly vs. once every two weeks) with epoetin delta in patientswith chronic kidney disease: a randomized controlled trial, Trials 8 (2007) 35.
[51] W.B. Smith, J.A. Dowell, R.D. Pratt, Pharmacokinetics and pharmacodynamicsof epoetin delta in two studies in healthy volunteers and two studies inpatients with chronic kidney disease, Clin. Ther. 29 (2007) 1368–1380.


![Coordinate Regulation of Metabolite Glycosylation and · Coordinate Regulation of Metabolite Glycosylation and StressHormoneBiosynthesisbyTT8inArabidopsis1[OPEN] Amit Rai2,3, Shivshankar](https://img.pdfslide.us/doc/110x75/60342c778ae2d32d91662064/coordinate-regulation-of-metabolite-glycosylation-coordinate-regulation-of-metabolite.jpg)


























