Embed Size (px)
Citation preview

S
SE
a
ARRA
KPAAABM
1
id[ow
(
sS
0d
Journal of Steroid Biochemistry & Molecular Biology 132 (2012) 93– 104
Contents lists available at SciVerse ScienceDirect
Journal of Steroid Biochemistry and Molecular Biology
jo u r n al hom epage: www.elsev ier .com/ locate / j sbmb
teroid derivatives as pure antagonists of the androgen receptor
ylvain Gauthier, Céline Martel, Fernand Labrie ∗,1
ndoresearch Inc., 2989, de la Promenade, Quebec City, QC, Canada G1W 2J5
r t i c l e i n f o
rticle history:eceived 20 October 2011eceived in revised form 27 February 2012ccepted 28 February 2012
eywords:rostate cancerntiandrogenndrogen receptorndrogen blockadeicalutamideDV3100
a b s t r a c t
Background: While the androgens of testicular origin (representing about 50% of total androgens in menover 50 years) can be completely eliminated by surgical or medical castration with GnRH (gonadotropin-releasing hormone) agonists or antagonists, the antiandrogens currently available as blockers of androgenbinding to the androgen receptor (AR), namely bicalutamide (BICA), flutamide (FLU) and nilutamidehave too weak affinity to completely neutralize the other 50% of androgens made locally from dehy-droepiandrosterone (DHEA) in the prostate cancer tissue by the mechanisms of intracrinology.Materials and methods: Series of steroid derivatives having pure and potent antagonistic activity on thehuman and rodent AR were synthesized. Assays of AR binding and activity in carcinoma mouse Shionogiand human LNCaP cells as well as in vivo bioavailability measurements and in vivo prostate weight assaysin the rat were used.Results: The chosen lead steroidal compound, namely EM-5854, has a 3.7-fold higher affinity than BICA forthe human AR while EM-5855, an important metabolite of EM-5854, has a 94-fold higher affinity for thehuman AR compared to BICA. EM-5854 and EM-5855 are 14 times more potent than BICA in inhibitingandrogen (R1881)-stimulated prostatic specific antigen (PSA) secretion in human prostatic carcinomaLNCaP cells in vitro. MDV3100 has a potency comparable to bicalutamide in these assays. Depending uponthe oral formulation, EM-5854 is 5- to 10-times more potent than BICA to inhibit dihydrotestosterone(DHT)-stimulated ventral prostatic weight in vivo in the rat while MDV3100 has lower activity than BICAin this in vivo model. These data are supported by respective 40-fold and 105-fold higher potencies ofEM-5854 and EM-5855 compared to BICA to inhibit cell proliferation in the androgen-sensitive Shionogi
carcinoma cell model.Conclusions: Although the present preclinical results data need evaluation in clinical trials in men, com-bination of the data obtained in vitro in human LNCaP cells as indicator of potency in the human prostateand the data on metabolism evaluated in vivo on ventral prostate weight in the rat, could suggest thepossibility of a 70- to 140-fold higher potency of EM-5854 compared to bicalutamide (Casodex) for thetreatment of prostate cancer in men.. Introduction
Prostate cancer is the second leading cause of cancer deathsn men with 248 890 new cases to be diagnosed and 33 720eaths estimated to occur in the United States alone in 2011
1]. Although improvements in surgery and radiotherapy haveccurred, National Cancer Institute data from 2.1 million patientsith cancer in the USA between 1975 and 1995 have led toAbbreviation: GnRH, gonadotropin-releasing hormone.∗ Corresponding author. Tel.: +1 418 652 0197; fax: +1 418 651 1856.
E-mail addresses: [email protected], [email protected]. Labrie).
1 Professor Emeritus, Laval University, Québec, Canada (2011) and Visiting Profes-or, College of Medicine, Al Imam Mohammed Ibn Saud Islamic University, Riyadh,audi Arabia (2010).
960-0760/$ – see front matter © 2012 Elsevier Ltd. All rights reserved.oi:10.1016/j.jsbmb.2012.02.006
© 2012 Elsevier Ltd. All rights reserved.
the conclusion that “cancer-fighting drugs improved survivalrates, especially for cancer of the prostate, where drug innova-tions have been the greatest” [2]. In prostate cancer, the mostimportant drugs have clearly been gonadotropin-releasing hor-mone (GnRH) agonists [3] and pure antiandrogens [4] permittingthe co-administration of a pure antiandrogen and medical or sur-gical castration in order to achieve combined androgen blockade(CAB) simultaneously at start of treatment [5]. However, despitesignificant improvements in diagnosis and treatment, prostate can-cer remains the second most common cause of death after lung can-cer in American men [1]. Most importantly, since prostate cancer isthe most sensitive of all cancers to hormone therapy, every effortshould be made to take advantage of this unique characteristic
[6].In 1980, it was discovered that medical castration is easilyachieved in men by chronic administration of GnRH agonists [3].Although GnRH agonists rapidly became the standard treatment

9 mistr
wstm([ca
dtCaiwottbpola
wptosftermb
amfcthoa
bnh
4 S. Gauthier et al. / Journal of Steroid Bioche
orldwide to eliminate testicular androgens, early biochemicaltudies performed with human prostatic tissue have demonstratedhat androgens were also synthesized in the prostate by the
echanism of intracrinology [7,8] from dehydroepiandrosteroneDHEA), a precursor of sex steroids produced by the adrenal glands5,7,9–13]. With this new information, the scientifically basedonsequence is to use CAB to achieve appropriate blockade ofndrogens [5].
While the percentage of objective responses is higher and theuration of response is longer when the androgens of both tes-icular and adrenal origins are blocked at start of treatment byAB, namely surgical or medical castration combined with a purentiandrogen [5,14–17], progression of the disease always occursf the patients have metastatic disease at start of treatment. Some-
hat surprisingly, however, it was found that a large percentagef patients show a positive clinical response upon discontinua-ion of the antiandrogen with a decrease in serum PSA by morehan 90% in 47% of patients [18]. This paradoxical phenomenonecame generally recognized [19–22]. A possible explanation of thearadoxical effect of antiandrogen withdrawal is the developmentf hypersensitivity to low androgens [23–25], and/or intracellu-ar changes making the antiandrogen act as a partial androgengonist.
The traditional therapeutic approach for metastatic diseasehich has become resistant to a specific androgen blockade, usuallyartial androgen blockade secondary to medical or surgical castra-ion or an antiandrogen alone, deserves reevaluation following thebservations of higher AR levels and/or maintenance of respon-iveness to androgens in treatment-resistant cancer [26–32]. Inact, elevated AR expression has been found to lead to resistanceo antiandrogen therapy in mouse xenograft prostate cancer mod-ls [26]. In any case, such data indicate that AR blockade shouldemain an important therapeutic target even at the last stage ofetastatic disease when resistance to various forms of androgen
lockade has developed.Although the available antiandrogens flutamide, bicalutamide
nd nilutamide have pure AR antagonistic activity and have shownajor benefits in prostate cancer therapy [5,14,15,17], their affinity
or AR is low [33–36] and leaves an estimated 5–10% of DHT free toontinue to stimulate AR and prostate cancer growth [37]. There ishus the need to discover and develop novel antiandrogens havingigher affinity for the human AR in order to take optimal advantagef the well demonstrated high responsiveness of prostate cancer tondrogen blockade.
Taking advantage of the structural information on the ligandinding domain of the human AR [38,39], we have synthesizedovel molecules which have higher or much higher affinity for theuman AR than flutamide, bicalutamide or nilutamide, the only
Fig. 1. Chemical structure of the ant
y & Molecular Biology 132 (2012) 93– 104
pure AR antagonists presently available for therapeutic use in mensuffering from prostate cancer. Comparison is also made in thesame assays with MDV3100, an antiandrogen with recently avail-able data [40–45].
2. Materials and methods
2.1. Tested compounds
EM-5854 and EM-5855 were synthesized in our laboratoriesaccording to Labrie F, Gauthier S, Cloutier J, Mailhot J, PotvinS, Dion S, Sancéau JY. Endorecherche Inc., United States PatentApplication Publication US2009/0042844, February 12, 2009. Bica-lutamide, hydroxyflutamide and MDV3100 were also synthesizedin our laboratories. Analytical data (spectral NMR data) of bica-lutamide, hydroxyflutamide and MDV3100 synthesized in-houseshow structures identical to the original drugs. The synthesis ofMDV3100 was recently described in the literature in 6 steps includ-ing 5 linear steps with an overall 27% yield [46,47]. The startingmaterial is 2-fluoro-4-nitrobenzoic acid was transformed to N-methyl-2-fluoro-4-(1,1-dimethylcyanomethyl)aminobenzamidein 4 steps. The benzamide derivative was then condensed with4-isothiocyanato-2-trifluoromethylbenzonitrile (prepared from4-amino-2-trifluoromethylbenzonitrile and thiophosgene) indimethylformamide under microwave irradiation at 100 ◦C fol-lowed by imine hydrolysis to give the thiohydantoin MDV3100at a 51% yield. We have performed the last step by purchasingthe benzamide derivative (AB Chem, Inc., Dorval, Quebec, Canada;AB3604). MDV3100 is now commercially available according toSciFinder. Flutamide was kindly supplied by Dr. Rudolph O. Neri,Schering-Plough Research Institute (Kenilworth, NJ) (see Fig. 1).
2.2. Androgen receptor (AR) assays
2.2.1. AR transfection2.2.1.1. Preparation of human embryonic kidney (HEK-293) cellstransfected with the human androgen receptor (hAR). Cells are cul-tured in 6-well Falcon flasks to approximately 3 × 105 cells/well inDulbecco’s modified Eagle’s medium (DMEM) supplemented with10% calf fetal serum at 37 ◦C under a 95% air, 5% CO2 humidi-fied atmosphere. Five �g of pCMVneo-hAR plasmid are transfectedusing the lipofectin transfection kit (Life Technologies, Ontario,Canada). After 6 h of incubation at 37 ◦C, the transfection mediumis removed and 2 ml of DMEM are added. Cells are further cul-
tured for 48 h and then transferred into 10 cm petri dishes andcultured in DMEM containing 700 �g/ml of G-418 in order to inhibitthe growth of non-transfected cells. Medium containing G-418 ischanged every 2 days until resistant colonies are observed. Positiveiandrogens under evaluation.

mistry
cf
2tssmic
2tgi
2
aedcwa(apb(Hte6iwt
pol
f
R
2r
t[ipcsa
2
u[
ac
S. Gauthier et al. / Journal of Steroid Bioche
lones are selected by PCR. HEK 293 cells transfected with hAR arerozen until being used for the binding assay.
.2.1.2. HEK-293 hAR cell cytosol preparation. On the morning ofhe binding assay, a pellet of HEK-293 hAR cells is thawed anduspended in buffer A (25 mM Tris–HCl, 1.5 mM EDTA disodiumalt, 10 mM �-monothioglycerol, 10% glycerol, and 10 mM sodiumolybdate, pH 7.4; 625 000 cells/0.1 ml). The cell suspension is son-
cated for three periods of 30 s (with intervals for cooling) and thenentrifuged at 105 000 × g for 90 min.
.2.1.3. Rat prostate cytosol preparation. On the morning ofhe binding assay, ventral prostates collected from 24 h-onadectomized rats were homogenized in buffer A (1 g of tissuen 5 ml) and the homogenate was centrifuged as described above.
.2.2. Androgen receptor assaysAndrogen binding is measured using the hydroxylapatite (HAP)
ssay. In brief, the radioactive steroid [3H]R1881 solubilized inthanol is diluted with buffer B (10 mM Tris–HCl, 1.5 mM EDTAisodium salt, 10 mM �-monothioglycerol, pH 7.4). Aliquots of theell or prostate cytosol preparation (0.1 ml) are then incubatedith 5 nM [3H]R1881 (0.1 ml, ∼100 000 cpm) in the presence or
bsence of the indicated concentrations of unlabeled compounds0.1 ml, prepared in buffer B containing 30% ethanol) for 16–18 ht 0–4 ◦C. Triamcinolone acetonide (TAC; 100 nM) is added to maskrogesterone receptors. Unbound steroids are separated by incu-ation for 40 min at 0–4 ◦C with 0.3 ml HAP prepared in buffer P50 mM Tris–HCl, 10 mM KH2PO4, pH 7.4). After incubation withAP and 10 min of centrifugation at 1000 × g, the pellet is washed
hree times with 1 ml of buffer P. Thereafter, the radioactivity isxtracted from the pellet by incubation at room temperature for0 min with 1 ml of ethanol. After centrifugation, the supernatant
s decanted into a scintillation vial and the pellet is extracted againith ethanol. After the addition of scintillation liquid, the radioac-
ivity is measured in a liquid scintillation counter.Dose–response curves as well as IC50 values of the tested com-
ounds (concentration of the compound causing a 50% displacingf [3H]R1881) were calculated using a weighted iterative nonlineareast-square regression.
Relative binding affinity (RBA) was calculated by the followingormula:
BA (%) =[
IC50 (R1881)IC50 (compound)
]× 100.
.3. Estrogen (ER), progesterone (PR) and glucocorticoid (GR)eceptors assays
The binding affinity assays on estrogen and progesterone recep-ors from rat uterus were performed as previously described48,49]. For the glucocorticoid receptor from rat liver, the affin-ty binding assay was done using a slightly modified version of therocedure described by Asselin et al. [50]. Herein, a dextran-coatedharcoal adsorption (as used for ER and PR), instead of a protamineulfate precipitation, was used to achieve the separation of boundnd free steroids.
.4. In vitro assay of androgenic/antiandrogenic activity
The in vitro androgenic/antiandrogenic activity was measuredsing Shionogi mouse mammary carcinoma cells (clone 107)
24,51,52].Minimal essential culture medium (MEM) and non-essentialmino acids were purchased from Gibco BRL (NY, USA) whileharcoal-stripped fetal calf serum (FBS) was purchased from
& Molecular Biology 132 (2012) 93– 104 95
Wisent Inc. (Montreal, Canada). Dihydrotestosterone (DHT) wasobtained from Steraloids (Wilton, NH).
Shionogi cells were routinely grown in MEM supplemented with100 nM DHT, 5% (v/v) charcoal-stripped FBS, 100 IU penicillin/ml,50 �g streptomycin sulfate/ml, and 1% (v/v) non-essential aminoacids, as previously described [24,51,52]. Cells were incubated at37 ◦C in a humidified atmosphere of 5% CO2 and 95% air. Cellswere subcultured at near-confluence by gentle digestion in a solu-tion of 0.1% trypsin (Wisent Inc.) in Hepes buffer containing 3 mMethylenediaminetetraacetic acid (EDTA) (pH 7.2). Cells were thenpelleted by centrifugation, resuspended in culture medium, andreplated.
Cells were plated in 24-well plates at a density of18 000 cells/well and allowed to adhere to the surface of theplates for 24 h. Thereafter, medium was replaced with freshmedium containing 2% (v/v) charcoal-stripped FBS and the indi-cated concentrations of compounds diluted from stock solutions ata x 1000 concentration in 99% redistilled ethanol in the presence orabsence of DHT (0.3 nM). Control cells received only the ethanolicvehicle (0.1% EtOH, v/v). Such a concentration of ethanol doesnot affect cell growth. The indicated increasing concentrations ofagents were added to triplicate dishes, and cells were grown for10 days with changes of medium every 2–3 days. Cell numberwas determined by measurement of DNA content as previouslydescribed [53].
Dose–response curves as well as IC50 values of the testedcompounds are calculated using a weighted iterative nonlinearleast-squares regression. All results are expressed as means ± SEM,except when SEM overlaps with the symbol used in whichinstances only the symbol is illustrated. The apparent inhibitionKi values were calculated according to the following equation:Ki = IC50/(1 + S/K). In this equation, S represents the concentrationof DHT (0.3 nM), K is the apparent KD of DHT action on cell pro-liferation in Shionogi cells (0.1 nM) and IC50 is the concentrationof the compound giving a 50% inhibition of DHT action on cellgrowth.
2.5. Measurement of prostate specific antigen (PSA) in LNCaP cells
LNCaP cells were cultured as previously described [54]. In brief,the LNCaP cells were cultured for 6 days in 1.0 ml RPMI 1640 supple-mented with hormone-depleted 0.25% FCS before each experiment.At the start of the experiment, half of the medium (0.5 ml) wasreplaced with 0.5 ml of identical medium containing the appropri-ate concentrations of tested compounds, in the presence or absenceof 1.0 nM of R1881. After 72 h of incubation of LNCaP cells withcompounds, 0.5 ml of culture medium was removed for PSA deter-mination. PSA levels were measured using the PSA [125I] IRMA KIT(REF: RK-10CT) from Izotop (Institute of Isotopes Ltd., Budapest,Hungary).
2.6. Determination of oral absorption of compounds
Castrated male Sprague–Dawley rats (Crl:CD(SD)Br) weighing275–375 g were used for pharmacokinetic studies. Animals werefasted (access to water only) from around 16h00 the afternoon priorto the dosing day.
Tested compounds were administered orally by gavage(in the morning) at a dose of 0.5 mg/animal (1.0 ml/animal;3 animals/compound). EM-5854 and EM-5855 were dissolved indimethylsulfoxide (DMSO, 10% final concentration) and admin-istered as a solution/suspension in 0.9% NaCl–1% gelatin while
bicalutamide, flutamide and hydroxyflutamide were administeredas a suspension in 0.4% aqueous methylcellulose. Blood samples(∼0.5 ml/timepoint) were collected by jugular venipuncture on ani-mals under isoflurane anesthesia at 1, 2, 3, 4, 7 and 24 h post-dosing.
9 mistr
Bcripot2nEimcd
bmtcm1mwtwwTmeor(uawatu[t
a(72cswtbwPop
2o
oa(el(g
6 S. Gauthier et al. / Journal of Steroid Bioche
lood samples were put into tubes containing EDTA (K3) as anti-oagulant and centrifuged at 4 ◦C for 10 min at 1700–2400 g. Theesulting plasma was frozen on dry ice and kept at −80 ◦C pend-ng analysis. After the blood collection 7 h post-dosing, the ventralrostate was collected from one rat per group for determinationf the intraprostatic concentration of the tested compound whilehe ventral prostate of the two other rats were collected after the4 h post-dosing blood collection. Prostates were frozen in liquiditrogen and kept at −80 ◦C until being used. Oral absorption ofM-5854 in the monkey was determined following oral admin-stration of 1.0 mg/kg (2 ml/kg) of EM-5854 suspended in 0.4%
ethylcellulose to a single intact male animal. Blood samples wereollected at 0, 1, 2, 4, 6 and 24 h post-dosing and were processed asescribed above for the rats.
Plasma concentrations of tested compounds were determinedy a liquid chromatography and mass spectrometry (LC–MS/MS)ethod using TurboIonSpray [55]. On each day of analysis, calibra-
ion standards were prepared using male rat plasma. Briefly, plasmaoncentrations of EM-5854 and its metabolite EM-5855 were deter-ined as follows: a 0.05 ml plasma sample was added to 0.6 ml of a
mM ammonium acetate solution. A methanolic solution (0.05 ml;ethanol:H2O (1:1)) containing an internal standard (50 ng/ml)as then added to each tube and vortexed for 30 s. Samples were
hen transferred to 60 mg Varian Plexa cartridges pre-conditionedith methanol (2 ml) and water (2 ml). Each column was thenashed with 2 ml of a solution of methanol:water (10:90, v:v).
he analytes of interest were then eluted using a solution (5 ml) ofethanol containing 5 mM ammonium acetate. The eluates were
vaporated to dryness at 45 ◦C under nitrogen and then 0.2–0.3 mlf methanol was added, vortexed and re-evaporated. The driedesidue was reconstituted in a solution (0.2 ml) of methanol:water85:15, v:v). The HPLC system used a 75 mm × 4.6 mm ACE C18 col-mn at a flow rate of 0.8 ml/min. Compounds were detected using
Sciex API 3000 triple quadrupole mass spectrometer, equippedith TurboIonSprayTM. A similar procedure was used for BICA, FLU
nd OH-FLU using optimized conditions. The plasma concentra-ion of each compound versus time was used to calculate the areander the plasma concentration curve from 0 to 24 h post-doseAUC(0–24 h)]. AUC(0–24 h) values were calculated using the linearrapezoidal method.
Intraprostatic concentrations of EM-5854 and EM-5855 werelso determined by LC–MS/MS [55]. Briefly, 0.5 ml of PBS buffer137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4 and 2 mM KH2PO4; pH–7.4) and 2 ml of an ethanol:acetone solution (1:1) was added to5–50 mg of crushed prostatic tissue previously weighted with pre-ision. The mixture was homogenized for 20 s with a Polytron at apeed of 20 000 to 24 000 rpm. The stem of the Polytron was washedith 2 ml of an ethanol:acetone solution (1:1) at high speed and
his solution was combined with the previous homogenate beforeeing centrifuged for 10 min at 3000 rpm at 4 ◦C. The supernatantas collected, evaporated to dryness and reconstituted in 0.4 ml
BS buffer. The internal standard was added and then the analytesf interest were extracted and analyzed as described above for thelasma samples.
.7. Systemic antiandrogenic/androgenic activity inrchidectomized and intact immature male rats
Immature male rats (Crl:CD(SD)Br) 22–24-day old werebtained from Charles-River, Inc. (St-Constant, Quebec, Canada)nd housed up to 5 per cage in plastic bins in a temperature23 ± 1 ◦C)- and light (12 h light/day, lights on at 7h15)-controlled
nvironment. The rats were fed rodent chow and tap water adibitum. Compounds were tested in castrated rats supplementedantagonistic activity) or not (agonistic activity) with an andro-en as well as in intact animals. The day following their arrival,y & Molecular Biology 132 (2012) 93– 104
the designed animals were orchidectomized (CX) under isoflu-rane anesthesia (Study Day 1) via the scrotal route and werethen randomly assigned to groups of 3–7 animals. At the time oforchidectomy, one silastic implant of dihydrotestosterone (DHT;1 cm length of pure DHT in silastic tubing having inner and outerdiameter of 0.078 and 0.125 in., respectively) was inserted subcu-taneously in the dorsal area of animals assigned to the evaluationof antiandrogenic activity.
Tested compounds were administered orally once daily for7 days from Study Day 2 to Study Day 8 at the indicated doses. Forthe experiment illustrated in Fig. 6A, compounds were solubilizedin dimethylsulfoxide (DMSO, 10% final concentration) and adminis-tered as a solution/suspension in 0.9% NaCl–1% gelatin. For the twoexperiments presented in Fig. 6B and D, compounds were admin-istered as suspension in aqueous 0.4% methylcellulose (0.4% MeC).Animals of the control groups received the vehicle alone during the7-day period. Some animals were treated with the antiandrogenflutamide, bicalutamide (BICA) and/or MDV3100 as reference. Theanimals under isoflurane anesthesia were killed by cervical disloca-tion on day 9 of the study, approximately 24 h after the last dosing.The ventral prostates were rapidly dissected and weighed.
For antagonistic activity, the percentage of inhibition (% inhib) iscalculated using the following formula:
% inhib=100−[[
W (compound) − W (control CX)W (control DHT) − W (control CX)
]×100
].
For agonistic activity, the percentage of stimulation versus DHT iscalculated by the following formula:
% stimul =[
W (compound) − W (control CX)W (control DHT) − W (control CX)
]× 100.
W is the weight of the prostate.
2.8. Systemic antiandrogenic activity in mature male rats
Mature male rats (Crl:CD(SD)Br) weighing 250–275 g wereobtained from Charles-River, Inc. (St-Constant, Quebec, Canada)and housed up to 3 per cage as described above. The day follow-ing their arrival, the animals were orchidectomized (CX) underisoflurane anesthesia (Study Day 1) via the scrotal route and wererandomly assigned to groups of 5 animals. At the time of orchidec-tomy, two silastic implants of androstenedione (4-dione; length ofpure 4-dione in implant: 1.5 cm, tubing inner and outer diameters:0.062 and 0.125 in., respectively) was inserted subcutaneously inthe dorsal area of animals.
The compound EM-5854 was administered orally once dailyfor 7 days from Study Day 2 to Study Day 8 at doses of 0.2 and1 mg/animal while the reference compounds flutamide and bica-lutamide (Casodex) were administered orally at doses of 1 and5 mg/animal during the same period. Compounds were adminis-tered in 10% DMSO, 1% gelatin, 0.9% NaCl (Fig. 6C). The animalsunder isoflurane anesthesia were killed by cervical dislocation onday 9 of the study, approximately 24 h after the last dosing. Theventral prostates were rapidly dissected and weighed.
3. Results
As can be seen in Fig. 2 and Table 1, EM-5854 shows a RBA (rel-ative binding affinity to the human AR) of 0.66 compared to 0.17for hydroxyflutamide (OH-FLU) and 0.18 for bicalutamide (BICA)for a 3.7-fold higher affinity of EM-5854 compared to BICA. Theaffinity of MDV3100 for the human AR, on the other hand, is mea-
sured at 0.07% that of R1881, a value 2.6 times lower than BICA.The binding of R1881, a well-known synthetic and metabolism-resistant androgen agonist is arbitrarily taken as reference at 100%[56]. It is of interest that the affinity of R1881 for the human AR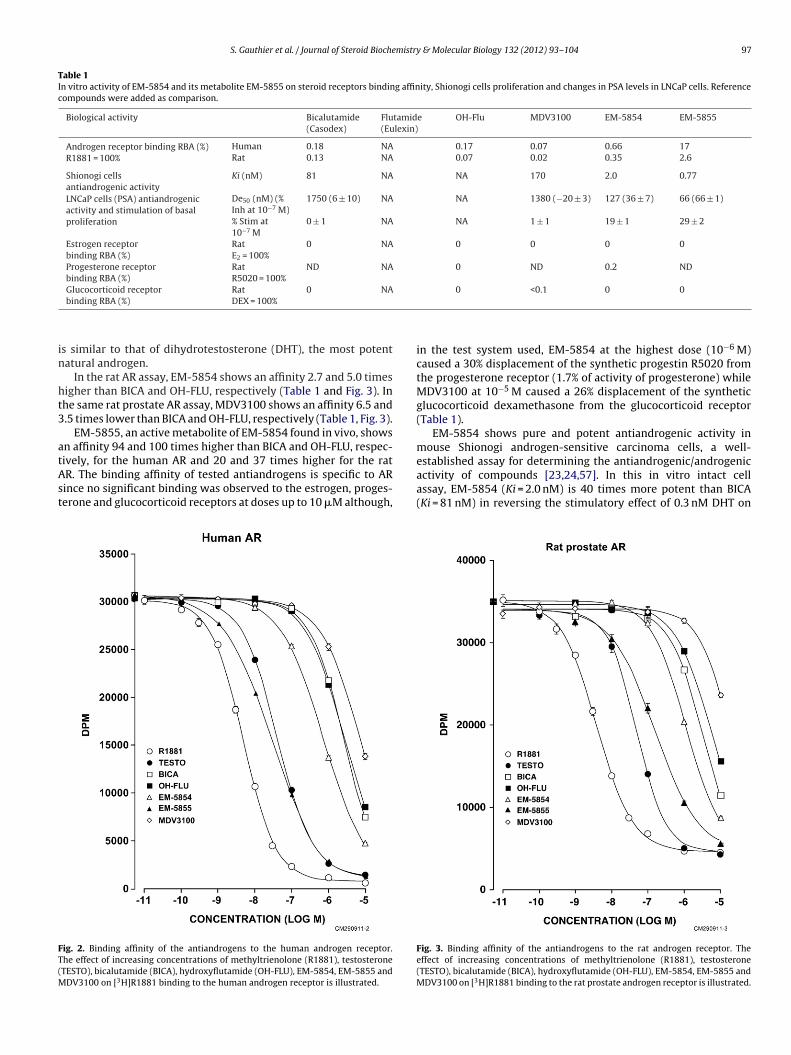
S. Gauthier et al. / Journal of Steroid Biochemistry & Molecular Biology 132 (2012) 93– 104 97
Table 1In vitro activity of EM-5854 and its metabolite EM-5855 on steroid receptors binding affinity, Shionogi cells proliferation and changes in PSA levels in LNCaP cells. Referencecompounds were added as comparison.
Biological activity Bicalutamide(Casodex)
Flutamide(Eulexin)
OH-Flu MDV3100 EM-5854 EM-5855
Androgen receptor binding RBA (%)R1881 = 100%
Human 0.18 NA 0.17 0.07 0.66 17Rat 0.13 NA 0.07 0.02 0.35 2.6
Shionogi cellsantiandrogenic activity
Ki (nM) 81 NA NA 170 2.0 0.77
LNCaP cells (PSA) antiandrogenicactivity and stimulation of basalproliferation
De50 (nM) (%Inh at 10−7 M)
1750 (6 ± 10) NA NA 1380 (−20 ± 3) 127 (36 ± 7) 66 (66 ± 1)
% Stim at10−7 M
0 ± 1 NA NA 1 ± 1 19 ± 1 29 ± 2
Estrogen receptorbinding RBA (%)
RatE2 = 100%
0 NA 0 0 0 0
Progesterone receptorbinding RBA (%)
RatR5020 = 100%
ND NA 0 ND 0.2 ND
Glucocorticoid receptor Rat 0 NA 0 <0.1 0 0
in
ht3
atAst
FT(M
binding RBA (%) DEX = 100%
s similar to that of dihydrotestosterone (DHT), the most potentatural androgen.
In the rat AR assay, EM-5854 shows an affinity 2.7 and 5.0 timesigher than BICA and OH-FLU, respectively (Table 1 and Fig. 3). Inhe same rat prostate AR assay, MDV3100 shows an affinity 6.5 and.5 times lower than BICA and OH-FLU, respectively (Table 1, Fig. 3).
EM-5855, an active metabolite of EM-5854 found in vivo, showsn affinity 94 and 100 times higher than BICA and OH-FLU, respec-
ively, for the human AR and 20 and 37 times higher for the ratR. The binding affinity of tested antiandrogens is specific to ARince no significant binding was observed to the estrogen, proges-erone and glucocorticoid receptors at doses up to 10 �M although,ig. 2. Binding affinity of the antiandrogens to the human androgen receptor.he effect of increasing concentrations of methyltrienolone (R1881), testosteroneTESTO), bicalutamide (BICA), hydroxyflutamide (OH-FLU), EM-5854, EM-5855 and
DV3100 on [3H]R1881 binding to the human androgen receptor is illustrated.
in the test system used, EM-5854 at the highest dose (10−6 M)caused a 30% displacement of the synthetic progestin R5020 fromthe progesterone receptor (1.7% of activity of progesterone) whileMDV3100 at 10−5 M caused a 26% displacement of the syntheticglucocorticoid dexamethasone from the glucocorticoid receptor(Table 1).
EM-5854 shows pure and potent antiandrogenic activity inmouse Shionogi androgen-sensitive carcinoma cells, a well-established assay for determining the antiandrogenic/androgenic
activity of compounds [23,24,57]. In this in vitro intact cellassay, EM-5854 (Ki = 2.0 nM) is 40 times more potent than BICA(Ki = 81 nM) in reversing the stimulatory effect of 0.3 nM DHT onFig. 3. Binding affinity of the antiandrogens to the rat androgen receptor. Theeffect of increasing concentrations of methyltrienolone (R1881), testosterone(TESTO), bicalutamide (BICA), hydroxyflutamide (OH-FLU), EM-5854, EM-5855 andMDV3100 on [3H]R1881 binding to the rat prostate androgen receptor is illustrated.
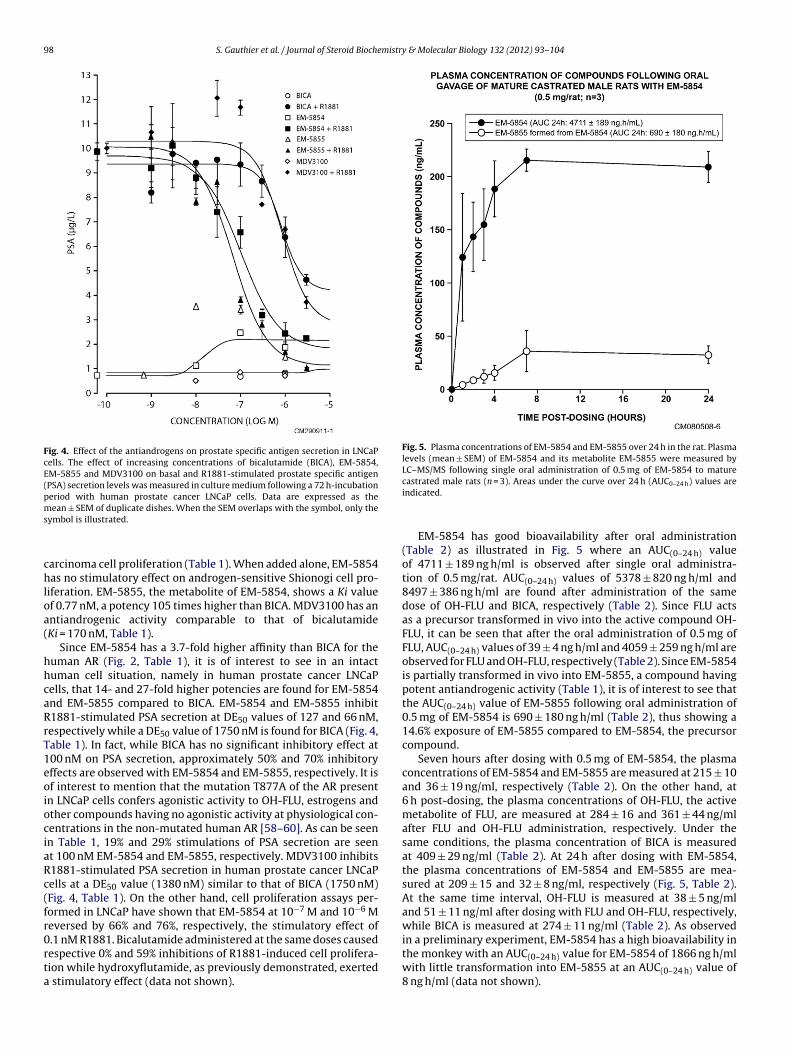
98 S. Gauthier et al. / Journal of Steroid Biochemistry & Molecular Biology 132 (2012) 93– 104
Fig. 4. Effect of the antiandrogens on prostate specific antigen secretion in LNCaPcells. The effect of increasing concentrations of bicalutamide (BICA), EM-5854,EM-5855 and MDV3100 on basal and R1881-stimulated prostate specific antigen(PSA) secretion levels was measured in culture medium following a 72 h-incubationperiod with human prostate cancer LNCaP cells. Data are expressed as thems
chloa(
hhcaRrT1eoiociaRc(fr0rta
Fig. 5. Plasma concentrations of EM-5854 and EM-5855 over 24 h in the rat. Plasmalevels (mean ± SEM) of EM-5854 and its metabolite EM-5855 were measured byLC–MS/MS following single oral administration of 0.5 mg of EM-5854 to mature
ean ± SEM of duplicate dishes. When the SEM overlaps with the symbol, only theymbol is illustrated.
arcinoma cell proliferation (Table 1). When added alone, EM-5854as no stimulatory effect on androgen-sensitive Shionogi cell pro-
iferation. EM-5855, the metabolite of EM-5854, shows a Ki valuef 0.77 nM, a potency 105 times higher than BICA. MDV3100 has anntiandrogenic activity comparable to that of bicalutamideKi = 170 nM, Table 1).
Since EM-5854 has a 3.7-fold higher affinity than BICA for theuman AR (Fig. 2, Table 1), it is of interest to see in an intactuman cell situation, namely in human prostate cancer LNCaPells, that 14- and 27-fold higher potencies are found for EM-5854nd EM-5855 compared to BICA. EM-5854 and EM-5855 inhibit1881-stimulated PSA secretion at DE50 values of 127 and 66 nM,espectively while a DE50 value of 1750 nM is found for BICA (Fig. 4,able 1). In fact, while BICA has no significant inhibitory effect at00 nM on PSA secretion, approximately 50% and 70% inhibitoryffects are observed with EM-5854 and EM-5855, respectively. It isf interest to mention that the mutation T877A of the AR presentn LNCaP cells confers agonistic activity to OH-FLU, estrogens andther compounds having no agonistic activity at physiological con-entrations in the non-mutated human AR [58–60]. As can be seenn Table 1, 19% and 29% stimulations of PSA secretion are seent 100 nM EM-5854 and EM-5855, respectively. MDV3100 inhibits1881-stimulated PSA secretion in human prostate cancer LNCaPells at a DE50 value (1380 nM) similar to that of BICA (1750 nM)Fig. 4, Table 1). On the other hand, cell proliferation assays per-ormed in LNCaP have shown that EM-5854 at 10−7 M and 10−6 Meversed by 66% and 76%, respectively, the stimulatory effect of.1 nM R1881. Bicalutamide administered at the same doses caused
espective 0% and 59% inhibitions of R1881-induced cell prolifera-ion while hydroxyflutamide, as previously demonstrated, exertedstimulatory effect (data not shown).
castrated male rats (n = 3). Areas under the curve over 24 h (AUC0–24 h) values areindicated.
EM-5854 has good bioavailability after oral administration(Table 2) as illustrated in Fig. 5 where an AUC(0–24 h) valueof 4711 ± 189 ng h/ml is observed after single oral administra-tion of 0.5 mg/rat. AUC(0–24 h) values of 5378 ± 820 ng h/ml and8497 ± 386 ng h/ml are found after administration of the samedose of OH-FLU and BICA, respectively (Table 2). Since FLU actsas a precursor transformed in vivo into the active compound OH-FLU, it can be seen that after the oral administration of 0.5 mg ofFLU, AUC(0–24 h) values of 39 ± 4 ng h/ml and 4059 ± 259 ng h/ml areobserved for FLU and OH-FLU, respectively (Table 2). Since EM-5854is partially transformed in vivo into EM-5855, a compound havingpotent antiandrogenic activity (Table 1), it is of interest to see thatthe AUC(0–24 h) value of EM-5855 following oral administration of0.5 mg of EM-5854 is 690 ± 180 ng h/ml (Table 2), thus showing a14.6% exposure of EM-5855 compared to EM-5854, the precursorcompound.
Seven hours after dosing with 0.5 mg of EM-5854, the plasmaconcentrations of EM-5854 and EM-5855 are measured at 215 ± 10and 36 ± 19 ng/ml, respectively (Table 2). On the other hand, at6 h post-dosing, the plasma concentrations of OH-FLU, the activemetabolite of FLU, are measured at 284 ± 16 and 361 ± 44 ng/mlafter FLU and OH-FLU administration, respectively. Under thesame conditions, the plasma concentration of BICA is measuredat 409 ± 29 ng/ml (Table 2). At 24 h after dosing with EM-5854,the plasma concentrations of EM-5854 and EM-5855 are mea-sured at 209 ± 15 and 32 ± 8 ng/ml, respectively (Fig. 5, Table 2).At the same time interval, OH-FLU is measured at 38 ± 5 ng/mland 51 ± 11 ng/ml after dosing with FLU and OH-FLU, respectively,while BICA is measured at 274 ± 11 ng/ml (Table 2). As observedin a preliminary experiment, EM-5854 has a high bioavailability in
the monkey with an AUC(0–24 h) value for EM-5854 of 1866 ng h/mlwith little transformation into EM-5855 at an AUC(0–24 h) value of8 ng h/ml (data not shown).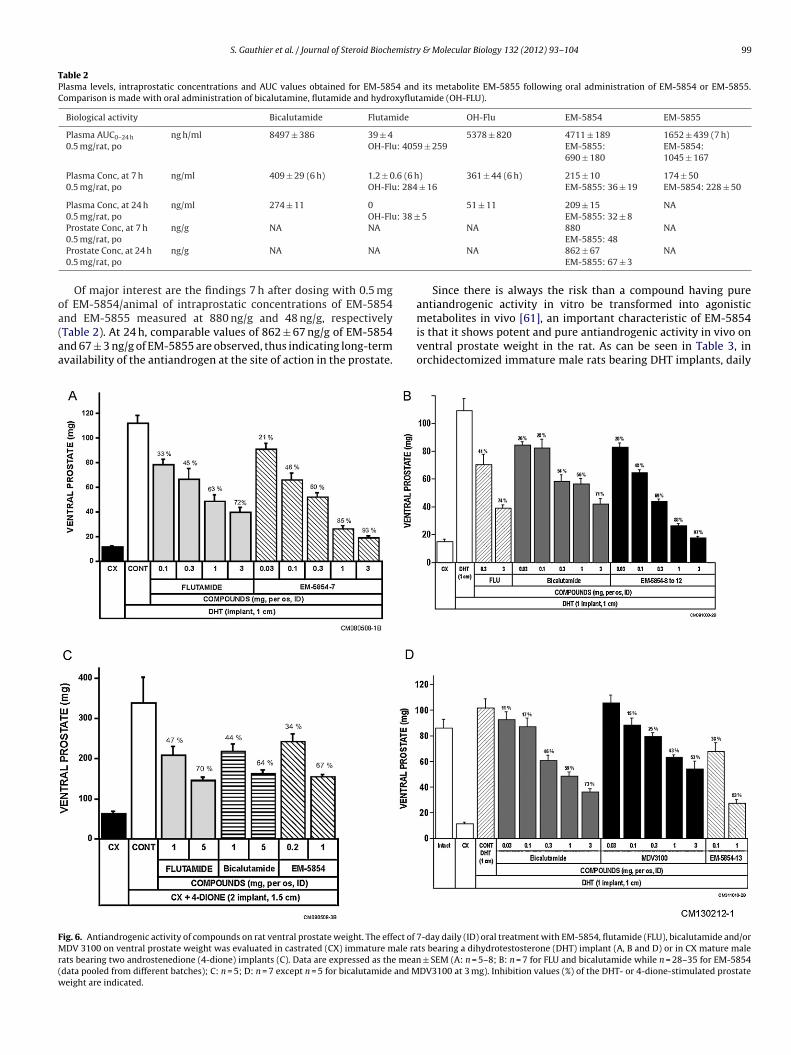
S. Gauthier et al. / Journal of Steroid Biochemistry & Molecular Biology 132 (2012) 93– 104 99
Table 2Plasma levels, intraprostatic concentrations and AUC values obtained for EM-5854 and its metabolite EM-5855 following oral administration of EM-5854 or EM-5855.Comparison is made with oral administration of bicalutamine, flutamide and hydroxyflutamide (OH-FLU).
Biological activity Bicalutamide Flutamide OH-Flu EM-5854 EM-5855
Plasma AUC0–24 h
0.5 mg/rat, pong h/ml 8497 ± 386 39 ± 4
OH-Flu: 4059 ± 2595378 ± 820 4711 ± 189
EM-5855:690 ± 180
1652 ± 439 (7 h)EM-5854:1045 ± 167
Plasma Conc, at 7 h0.5 mg/rat, po
ng/ml 409 ± 29 (6 h) 1.2 ± 0.6 (6 h)OH-Flu: 284 ± 16
361 ± 44 (6 h) 215 ± 10EM-5855: 36 ± 19
174 ± 50EM-5854: 228 ± 50
Plasma Conc, at 24 h0.5 mg/rat, po
ng/ml 274 ± 11 0OH-Flu: 38 ± 5
51 ± 11 209 ± 15EM-5855: 32 ± 8
NA
Prostate Conc, at 7 h ng/g NA NA NA 880 NA
oa(aa
FMr(w
0.5 mg/rat, poProstate Conc, at 24 h0.5 mg/rat, po
ng/g NA NA
Of major interest are the findings 7 h after dosing with 0.5 mgf EM-5854/animal of intraprostatic concentrations of EM-5854nd EM-5855 measured at 880 ng/g and 48 ng/g, respectively
Table 2). At 24 h, comparable values of 862 ± 67 ng/g of EM-5854nd 67 ± 3 ng/g of EM-5855 are observed, thus indicating long-termvailability of the antiandrogen at the site of action in the prostate.ig. 6. Antiandrogenic activity of compounds on rat ventral prostate weight. The effect of
DV 3100 on ventral prostate weight was evaluated in castrated (CX) immature male raats bearing two androstenedione (4-dione) implants (C). Data are expressed as the meandata pooled from different batches); C: n = 5; D: n = 7 except n = 5 for bicalutamide and Meight are indicated.
EM-5855: 48NA 862 ± 67
EM-5855: 67 ± 3NA
Since there is always the risk than a compound having pureantiandrogenic activity in vitro be transformed into agonisticmetabolites in vivo [61], an important characteristic of EM-5854
is that it shows potent and pure antiandrogenic activity in vivo onventral prostate weight in the rat. As can be seen in Table 3, inorchidectomized immature male rats bearing DHT implants, daily7-day daily (ID) oral treatment with EM-5854, flutamide (FLU), bicalutamide and/orts bearing a dihydrotestosterone (DHT) implant (A, B and D) or in CX mature male
± SEM (A: n = 5–8; B: n = 7 for FLU and bicalutamide while n = 28–35 for EM-5854DV3100 at 3 mg). Inhibition values (%) of the DHT- or 4-dione-stimulated prostate

100 S. Gauthier et al. / Journal of Steroid Biochemistry & Molecular Biology 132 (2012) 93– 104
Table 3In vivo activity of EM-5854 and its metabolite EM-5855 on androgen-sensitive parameters in rats. Reference compounds are added as comparison.
Biological activity Bicalutamide Flutamide MDV3100 EM-5854 EM-5855
AntagonisticactivityImmature rat(CX + DHT)0.1 mg/rat/po/ID[0.5 mg/rat/po/ID]
Ventral prostate% of control
−26 ± 6 (n = 4)[−53 ± 4] (n = 13)
−27 ± 2 (n = 7)[−48 ± 1] (n = 131)
−15 ± 6 −45 ± 2 (n = 16)[−68 ± 5] (n = 3)
−43 ± 5 (n = 2)[−63 ± 4] (n = 2)
AntagonisticactivityIntact immature rat0.1 mg/rat/po/ID[0.5 mg/rat/po/ID]
Ventral prostate% change versusintact Cont (IntactCont = 0%; CXCont = −100%)
[−36 ± 0.5] (n = 13) [−24 ± 4] (n = 7) NA −30 ± 6 (n = 5)[−46 ± 6] (n = 2)
NA
Agonistic activityImmature rat (CX)
Ventral prostate %of control
(0; 0.2 mg, sc) (1; 0.2 mg, sc) (1 ± 1; 0.3 mg, po) 0 1 ± 2
I s obta
o(wwaoEvBtd(Ecpssmw
9oFis0tdEDdtoibc
iodlhp
ota(
0.1 mg/rat/po/ID
n this table, “n” indicates the number of separate studies performed (the result wa
ral administration of 0.1 mg/rat of EM-5854 reversed by 45 ± 2%n = 16) the stimulatory effect of DHT on ventral prostate weight,hile an approximately 5-time higher dose of FLU (0.5 mg/rat)as required to achieve comparable inhibition (48 ± 1%, n = 131)
nd a 53 ± 4% (n = 13) inhibition was obtained with daily 0.5 mgral BICA. At the 0.1 mg dose, EM-5855, the metabolite ofM-5854, caused a 43 ± 5% (n = 2) inhibition of DHT-stimulatedentral prostate weight. At the same 0.1 mg daily oral dose, FLU andICA inhibited DHT-stimulated ventral prostatic weight in imma-ure rats by 27 ± 2% (n = 7) and 26 ± 6% (n = 4), respectively. At theose of 0.5 mg/rat, the inhibition achieved by EM-5854 was 68 ± 5%n = 3) on ventral prostate-DHT stimulated weight while that ofM-5855 was 63 ± 4% (n = 2) (Table 3), thus showing similar poten-ies of EM-5854 and its metabolites EM-5855 in vivo in the rat. Inarallel to ventral prostate weights, all tested compounds exertedimilar inhibitory effects on the weight of two other androgen-ensitive tissues, namely seminal vesicles and bulbocavernosususcles and the relative potency of compounds was similar to whatas observed on ventral prostate (data not shown).
As can be seen in Fig. 6A, at the 3 mg daily dose of EM-5854, a3% reversal of the stimulatory effect of DHT was achieved whilenly a 72% inhibitory effect was observed at the same dose ofLU. One hundred percent (100%) inhibition is the value observedn castrated animals or in the complete absence of androgenictimulation. When EM-5854 was administered as suspension in.4% methylcellulose (MeC) (instead of dimethylsulfoxide (DMSO))o orchidectomized immature male rats bearing DHT implants,aily oral administration of 0.3 mg/rat of four different batches ofM-5854 reversed by an average of 69% the stimulatory effect ofHT on ventral prostate weight (Fig. 6B), while a 10-time higherose of BICA (3 mg/rat) was required to achieve comparable inhibi-ion (71%; Fig. 6B). At the same dose, FLU achieved a 74% inhibitionf DHT-stimulated prostate weight. These data show that EM-5854s 10 times more potent than BICA and FLU. Interestingly, a 97% inhi-ition of the stimulatory effect of DHT on ventral prostate weightan be achieved at the highest dose of EM-5854 used (3 mg/animal).
In orchidectomized mature rats supplemented with 4-dionemplants, the efficacy of EM-5854 was five times superior to thatf the two best known antiandrogens, namely FLU and BICA. At theaily oral dose of 1 mg/rat, EM-5854 reversed by 67% the stimu-
atory effect of 4-dione on ventral prostate weight while a 5-foldigher dose (5 mg/rat) was required to obtain similar inhibitions ofrostate weight by BICA (64%) or FLU (70%) (Fig. 6C).
Most importantly, the daily oral administration of EM-5854 to
rchidectomized immature rats has no stimulatory effect on ven-ral prostate weight, thus showing that EM-5854 exerts a purentiandrogenic activity without any intrinsic androgenic activityTable 3).ined from a mean of all studies performed).
On the other hand, when BICA was administered as suspensionin 0.4% methylcellulose to orchidectomized immature male ratsbearing DHT implants, daily oral administration of 0.03, 0.1, 0.3,1 and 3 mg/rat reversed by 11%, 17%, 46%, 59% and 73%, respec-tively, the stimulatory effect of DHT on ventral prostate weight,while MDV3100 administered at the same doses inhibited ventralprostate weight by 0%, 15%, 25%, 43% and 53%, respectively (Fig. 6D).A 3-fold higher dose of MDV3100 was thus required to achievecomparable inhibition compared to BICA (Fig. 6D). In the sameexperiment, the daily oral administration of 0.1 and 1 mg/rat ofthe steroidal antiandrogen EM-5854 reversed the DHT-stimulatedventral prostate weight by 38% and 83%, respectively, compared to17% and 59% at the same doses of BICA (Fig. 6D). The daily oraladministration of 0.3 and 3 mg/rat of MDV3100 and BICA aloneto orchidectomized immature rats has no stimulatory effect onventral prostate weight (Table 3), thus indicating the absence ofintrinsic androgenic activity of the two compounds in vivo in therat. In previous experiments, we have shown that EM-5854 alsoexerts a pure antiandrogenic activity without any intrinsic andro-genic activity (Table 3).
Finally, in intact immature male rat, the administration of0.5 mg/rat of EM-5854 inhibited ventral prostate weight by 46%compared to 36% and 24% following the administration of the samedose of BICA and FLU, respectively (Table 3).
4. Discussion
The present data show that EM-5854 is 14 times more potentthan BICA while its metabolite EM-5855, which represents 15% ofthe 24 h serum concentration of EM5854, is 27 times more potentthan BICA in the in vitro PSA assay in human LNCaP prostatic carci-noma cells, the model which most closely mimics the intraprostaticsituation in men treated for prostate cancer. In addition, depend-ing upon the oral formulation used, EM-5854 is 5–10 times morepotent than the currently available antiandrogens for the treat-ment of prostate cancer, namely BICA and FLU, in the in vivo ratventral prostate weight assay where metabolism plays an impor-tant role. The finding of a slightly higher affinity of EM-5854 forthe human compared to rat AR can suggest a further greater effi-cacy of the antiandrogen at the level of AR in men compared to therat model. Combination of the 14 times higher activity of EM-5854in the human prostatic LNCaP cells and the 5- to 10-fold higheractivity in vivo in the rat where metabolism plays a major role,could suggest the possibility of a 70–140 times higher potency in
men of EM-5854 compared to bicalutamide, the most currentlyused antiandrogen for the treatment of prostate cancer. The highpotency of EM-5854 observed in LNCaP cells is well supported bythe 40-fold higher potency of EM-5854 compared to BICA in the
S. Gauthier et al. / Journal of Steroid Biochemistry
Table 4Potency of EM-5854 relative to bicalutamide (taken as one) in in vitro and in vivoassays.
EM-5854 EM-5855a MDV3100
Androgen receptor (AR): Human
Rat3.7 2.7
94 20 }4.7
0.4 0.15
Shionogi cells 0.5 105 40
PSA 1.3 26 14
iw
ptolrifcdr[
mfhdhtccbnCbipttcmiaptta
trhlbsiatda
In vivo – Rat Prostate 0.3 5 5-10 aMetabolite of EM-5854.
n vitro androgen-sensitive mouse Shionogi carcinoma cell modelhile EM-5855 is 105 times more potent (Table 4).
Upon binding of agonist molecules to their ligand-bindingocket (LBP), the nuclear receptors (NRs) undergo an impor-ant conformational change that notably affects the positionf the carboxy-terminal �-helix (helix 12, H12) located in theigand-binding domain (LBD). When bound by an agonist, nucleareceptors become active and transcriptional factors are able tonteract directly with DNA at specific response elements (REs)ound in the regulatory regions of target genes. These DNA-NRomplexes can then recruit coactivators through their ligand-ependent transactivation function (AF-2) formed upon H12epositioning [62], and hence control transcription of specific genes38,39].
The current drugs bicalutamide and OH-flutamide (activeetabolite of flutamide) unfortunately possess very weak affinity
or the human AR [33–36], thus indicating the need to develop newigh-affinity steroidal antiandrogens. Our compounds are speciallyesigned to impede repositioning of the mobile carboxy-terminalelix 12, which blocks the ligand-dependent transactivation func-ion (AF-2) located in the AR ligand-binding domain (ARLBD). Usingrystal structures of the human ARLBD, we first found that H12ould be directly reached from the ligand-binding pocket (LBP)y a chain positioned on the C18 atom of an androgen steroiducleus [38,39]. A set of DHT-derived molecules bearing various18 chains were thus synthesized and tested for their capacity toind human AR and act as antagonists. Although most of those hav-
ng very high affinity for human AR were agonists, several veryotent antagonists were obtained, confirming the structural impor-ance of the C18 chain. To understand the role of the C18 chain inheir agonistic/antagonistic properties, the structure of the hARLBDomplexed with one of these agonists, namely EM-5744, was deter-ined at a 1.65 A resolution. We could identify new interactions
nvolving Gln738, Met742 and His874 that explain both the high-ffinity of this compound and the inability of its bulky chain torevent the repositioning of H12 [39]. These structural informa-ions were helpful to refine the structure of the chains placed onhe C18 atom in order to obtain efficient H12-directed steroidalntiandrogens.
With the aim of designing such new antiandrogens, we decidedo make use of earlier structural findings on the human estrogeneceptor (hER), a receptor structurally related to the human AR. Theuman ER is unable to interact with coactivator partners when a
igand bearing a well-oriented bulky chain is bound to its ligand-inding site [63]. Indeed, as revealed by comparison of the crystaltructures of the human ER ligand-binding domain (human ERLBD)n complex with a natural estrogen (estradiol – E2) and a potent
ntiestrogen (raloxifene), agonist and antagonist molecules bind athe same site within the ligand binding domain (LBD). However,ifferent compounds exhibit different binding modes, inducingdistinct conformation in the transactivation domain (AF-2)
& Molecular Biology 132 (2012) 93– 104 101
characterized by a different positioning of H12. More precisely, thesize and structure of raloxifene prevent the molecule from beingcompletely confined within the steroid-binding cavity. Conse-quently, its bulky side-chain protrudes from the cavity and impedesH12 from adopting the position found in the E2-human ERLBDcomplex structure, a conformation essential for interaction withtranscriptional coactivators. Concerning human AR, the crystalstructures of its LBD (hARLBD) in complex with the natural andro-gens DHT and testo [38,64] have shown that H12 occupies the sameposition as observed in the E2-human ERLBD structure. Such datasuggest that this helix is also essential for the function of the AF-2of human AR and, like for the human ER, participates in the inter-action with coactivators. This has been confirmed by the structureof the liganded hARLBD in complex with a peptide derived fromphysiological coactivators [65–67].
Using all the available structural information of the humanAR, we then proceeded to molecular modeling studies to findthe best position on an androgen nucleus for introducing a bulkychain able to reach the site normally occupied by H12. Finally,the combined data from molecular modeling and structure/activityrelationship studies served as a basis for the design and improve-ment of the chain structure, with the aim of maximizing theaffinity of these steroidal-based compounds for the human AR.This rational approach yielded several different DHT-based (andother steroidal scaffolds) ligands able to bind human AR with highaffinity (many folds over that of DHT). In our in vitro tests, how-ever, the majority of the synthesized compounds failed to inhibitthe growth of DHT-stimulated cells or were potent agonists. How-ever, a small subgroup proved to be very efficient antagonists ofDHT-stimulation, thus indicating that the particular structure ofthe bulky chain is of paramount importance for its activity [F. Labrieet al., USA Patent No. 20050250749 (November 10, 2005)].
Concerning EM-5854, a potent steroidal antiandrogen pos-sessing a chain at the 17�-position, a different design approachfrom the 18-substituted steroidal antiandrogens was followed. Themain objective was to mimic the natural androgenic steroidalbackbone which contains a ketone at the 3-position by an1,3,5(10)-estratriene backbone substituted by a nitrile group at the3-position. The nitrile function has the capacity to form hydro-gen bonds like ketones. We can mention that bicalutamide alsohas an aromatic nitrile on the left side of the molecule in analogywith EM-5854 where, however, the chain is in the � position. Afterexploring different substitutions on this backbone, we found thata 17�-chain, specially 4-picoline and 4-picoline N-oxide, finallygave positive data. Substitution at the 4-position, such as with afluorine atom, was very important and was preferred for optimiza-tion of the biological activity. The structure–activity relationshipson this new series of steroidal antiandrogens will be reportedelsewhere.
The explanation of the antiandrogenic activity of EM-5854 at themolecular level remains hypothetical. According to our molecularmodeling, the 17�-4-picolyl N-oxide chain could displace helix 11from the ligand-binding domain of the AR, thus probably resultingin helix 12 repositioning, as discussed previously. Moreover, Bohlet al. have reported the crystal structure of the T877A human ARligand-binding domain complexed to cyproterone acetate whichsuggests an additional pocket at the 17�-position of the steroidalarea and a Ser-778 that could be a potential hydrogen bond partner[68]. All the reported data indicate a large range of conformationalflexibility in the ligand-binding pocket.
For the treatment of prostate cancer, it is extremely impor-tant to use compounds completely free of any intrinsic androgenic
activity [25]. For example, some compounds used as blockers ofandrogen action possess intrinsic androgenic activity well demon-strated in preclinical assays [69–71]. Such findings observed in vitroand confirmed in vivo in the ventral rat prostate weight assay can
1 mistr
ttwmg
w[Iiiehicflp
miaiSted
cgralidtib[
caatIctofbtphoaco
tfcgcwfil
[
[
[
[
[
02 S. Gauthier et al. / Journal of Steroid Bioche
ranslate at the clinical level since one of these progestin deriva-ives (cyproterone acetate) has been observed to shorten survivalhen combined with castration compared to castration alone inetastatic prostate cancer patients while the two pure antiandro-
ens flutamide and nilutamide prolong survival [5,14,16,17,72].Resistance to hormone therapy is the standard observation
hen treatment is started at the metastatic stage in prostate cancer25], thus creating a major and unresolved therapeutic challenge.n fact, a large amount of evidence indicates that AR functionings required for the growth of prostate cancer at all stages includ-ng castration-resistant disease [25,73]. Recent data show thatndocrine therapy-resistant prostatic carcinomas generally displayigh AR expression [74,75]. In a recent study, a significant increase
n AR mRNA levels was observed in the cancerous prostatic cellsompared with the benign tissue biopsies [76]. Treatment withutamide has already been shown to decrease AR expression inrostatic carcinoma tissue [77].
The role in prostate cancer growth of the intraprostatic for-ation of androgens from DHEA [5,7,78,79] by the process of
ntracrinology is now well recognized. In fact, following castrationlone, approximately 40% of DHT, the most potent androgen, is leftn the normal human prostate and prostate cancer [5,9,10,80,81].uch data explain why combined androgen blockade with cas-ration and a pure antiandrogen as first treatment shows greaterfficacy than castration alone or than administration of an antian-rogen alone [5,14–17,72,82].
Even at time of progression in patients with metastatic prostateancer treated by castration alone, the benefits of additional andro-en blockade are illustrated by the observation of a positiveesponse in 30–60% of patients in progression by hypophysectomy,drenalectomy or aminoglutethimide [5,83–85]. In a relativelyarge scale study of 209 metastatic prostate cancer patients show-ng disease progression after orchiectomy, treatment with highoses of estrogens or an GnRH agonist alone, the addition of flu-amide permitted to achieve complete, partial and stable responsesn 6.2%, 9.6% and 18.7% of cases, respectively, for a total clinicalenefit of 34.5%. The mean duration of response was 24 months86].
Contrary to the generalized opinion that patients in relapse afterastration have exclusively “androgen-insensitive” tumors, thebove-mentioned data suggest that “androgen-sensitive tumoursre present at all stages of prostate cancer in all patients andhat maximal androgen blockade should always be administered.nstead of being “androgen-insensitive”, most of the tumours whichontinue to grow after castration are androgen-sensitive and ableo grow in the presence of the “low” level of androgens of adrenalrigin left after castration” [86]. “Control of their growth requiresurther androgen blockade”. This affirmation was well supportedy already available clinical data and fundamental [23] observa-ions. As evidence for the androgen sensitivity of prostate cancerrogressing under androgen blockade, Fowler and Whitmore [87]ave observed a rapid and severe exacerbation of the disease in 33ut of 34 patients in relapse within the first 3 days of testosteronedministration, thus clearly showing that at least part of prostateancer cells remain androgen-sensitive even at the advanced stagef progression under androgen blockade.
Based upon the above-summarized data, it becomes impor-ant to develop more potent but always pure blockers of androgenormation or action. In this context of new compounds, positivelinical data have been obtained in studies with the new antiandro-en MDV3100 in patients with CRPC (castration-resistant prostateancer), showing a decrease in serum PSA in CRPC patients treated
ith the drug [40–42,44,45]. No other novel antiandrogen has soar shown efficacy in clinical trials. Studies of abiraterone treatmentn patients who had previously been treated with antiandrogens,ow-dose steroids, estrogens, ketoconazole, docetaxel and other
[
y & Molecular Biology 132 (2012) 93– 104
chemotherapies, showed a ≥50% decrease in PSA in 50–60%, anda ≥90% decrease in PSA in 20–30% of treated patients, respectively.In a study in CRPC patients who had previously been treated withcastration and docetaxel chemotherapy, a significant improvementin overall survival in the group of patients treated with abirateroneplus prednisone compared to prednisone plus placebo has beenobserved [88]. Considering the well demonstrated clinical bene-fits and the important decrease in serum PSA which follow simplecessation of administration of the antiandrogen [18,22], the clini-cal benefits of new compounds used in CRPC patients need carefulevaluation. Moreover, although some compounds have pure antag-onistic activity in vitro in some assays, metabolism can generateagonistic compounds which compromise the usefulness of poten-tially pure antiandrogens [61]. It is clear, however, that a morepotent blocker of AR having pure antagonistic activity is neededand is likely to play a major role in prostate cancer therapy at allstages of the disease.
Acknowledgments
The authors wish to thank Dr Rock Breton for important dis-cussions and data on molecular modeling as well as Ms DianeMichaud and Mr Alain St-Pierre for their assistance with in vitroand in vivo experiments. The authors are grateful to the chemists ofthe medicinal chemistry division of Endoresearch Inc. for synthesisof compounds.
References
[1] R. Siegel, E. Ward, O. Brawley, A. Jemal, Cancer statistics, 2011: the impact ofeliminating socioeconomic and racial disparities on premature cancer deaths,CA Cancer J. Clin. 61 (4) (2011) 212–236.
[2] F. Lichtenberg, Measuring the health impacts of medical innovation and expen-diture, in: Health Services Research Seminal Series 2002–2003, University ofMinnesota, Minneapolis, MN, USA, 2002.
[3] F. Labrie, A. Bélanger, L. Cusan, C. Séguin, G. Pelletier, P.A. Kelly, J.J. Reeves, F.A.Lefebvre, A. Lemay, J.P. Raynaud, Antifertility effects of LHRH agonists in themale, J. Androl. 1 (1980) 209–228.
[4] R. Neri, M.D. Monahan, J.G. Meyer, B.A. Afonso, I.A. Tabachnick, Biological stud-ies of an antiandrogen (SH-714), Eur. J. Pharmacol. 1 (1967) 438–444.
[5] F. Labrie, A. Dupont, A. Bélanger, Complete androgen blockade for the treatmentof prostate cancer, in: V.T. de Vita, S. Hellman, S.A. Rosenberg (Eds.), ImportantAdvances in Oncology, J.B. Lippincott, Philadelphia, 1985, pp. 193–217.
[6] F. Labrie, Editorial: the major role of androgens in prostate cancer and the needfor more efficient blockade, Expert Rev. Endocrinol. Metab. 6 (2011) 313–316.
[7] F. Labrie, Intracrinology, Mol. Cell. Endocrinol. 78 (1991) C113–C118.[8] F. Labrie, A. Bélanger, V. Luu-The, C. Labrie, J. Simard, L. Cusan, J.L. Gomez, B.
Candas, Gonadotropin-releasing hormone agonists in the treatment of prostatecancer, Endocr. Rev. 26 (3) (2005) 361–379.
[9] B. Bélanger, A. Bélanger, F. Labrie, A. Dupont, L. Cusan, G. Monfette, Comparisonof residual C-19 steroids in plasma and prostatic tissue of human, rat and guineapig after castration: unique importance of extratesticular androgens in men, J.Steroid Biochem. 32 (1989) 695–698.
10] F. Labrie, L. Cusan, J.L. Gomez, C. Martel, R. Bérubé, P. Bélanger, A. Bélanger,L. Vandenput, D. Mellström, C. Ohlsson, Comparable amounts of sex steroidsare made outside the gonads in men and women: strong lesson for hormonetherapy of prostate and breast cancer, J. Steroid Biochem. Mol. Biol. 113 (2009)52–56.
11] F. Labrie, Endocrinology and the prostate. Preface, Best Pract. Res. Clin.Endocrinol. Metab. 22 (2) (2008) vii–ix.
12] F. Labrie, Hormonal therapy of prostate cancer, in: L. Martini, G.P. Chrousos, F.Labrie, K. Pacak, D.E. Pfaff (Eds.), Neuroendocrinology, The Normal Neuroen-docrine System, Progress in Brain Research, Elsevier, 2010, pp. 321–341.
13] C. Labrie, J. Simard, H.F. Zhao, A. Bélanger, G. Pelletier, F. Labrie, Stimu-lation of androgen-dependent gene expression by the adrenal precursorsdehydroepiandrosterone and androstenedione in the rat ventral prostate,Endocrinology 124 (6) (1989) 2745–2754.
14] E.D. Crawford, M.A. Eisenberger, D.G. McLeod, J.T. Spaulding, R. Benson, F.A.Dorr, B.A. Blumenstein, M.A. Davis, P.J. Goodman, A controlled trial of leuprolidewith and without flutamide in prostatic carcinoma, N. Engl. J. Med. 321 (7)(1989) 419–424.
15] R.A. Janknegt, C.C. Abbou, R. Bartoletti, L. Bernstein-Hahn, B. Bracken, J.M. Bris-set, F.C. Da Silva, G. Chisholm, E.D. Crawford, F.M.J. Debruyne, G.D. Dijkman, J.Frick, L. Goedhals, H. Knönagel, P.M. Venner, Orchiectomy and nilutamide orplacebo as treatment of metastatic prostatic cancer in a multinational double-blind randomized trial, J. Urol. 149 (1993) 77–83.

mistry
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
S. Gauthier et al. / Journal of Steroid Bioche
16] C.L. Bennett, T.D. Tosteson, B. Schmitt, P.D. Weinberg, M.S. Ernstoff, S.D. Ross,Maximum androgen-blockade with medical or surgical castration in advancedprostate cancer: a meta-analysis of nine published randomized controlled trialsand 4128 patients using flutamide, Prostate Cancer Prostatic Dis. 2 (1999) 4–8.
17] Prostate Cancer Triallists’ Collaborative Group, Maximum androgen blockadein advanced prostate cancer: an overview of the randomised trials, Lancet 355(2000) 1491–1498.
18] A. Dupont, J.L. Gomez, L. Cusan, M. Koutsilieris, F. Labrie, Response to flutamidewithdrawal in advanced prostate cancer in progression under combinationtherapy, J. Urol. 150 (1993) 908–913.
19] M.P. Collinson, F. Daniel, C.J. Tyrrell, C. Teasdale, Response of carcinoma of theprostate to withdrawal of flutamide, Br. J. Urol. 72 (5 Pt 1) (1993) 662–663.
20] J. Herrada, P. Dieringer, C.J. Logothetis, Characterization of patients withandrogen-independent prostatic carcinoma whose serum prostate specificantigen decreased following flutamide withdrawal, J. Urol. 155 (2) (1996)620–623.
21] W.K. Kelly, H.I. Scher, Prostate specific antigen decline after antiandrogen with-drawal: the flutamide withdrawal syndrome, J. Urol. 149 (1993) 607–609.
22] W.K. Kelly, S. Slovin, H.I. Scher, Steroid hormone withdrawal syndromes. Patho-physiology and clinical significance, Urol. Clin. N. Am. 24 (1997) 421–431.
23] F. Labrie, R. Veilleux, A wide range of sensitivities to androgens develops incloned Shionogi mouse mammary tumor cells, Prostate 8 (1986) 293–300.
24] F. Labrie, R. Veilleux, A. Fournier, Low androgen levels induce the develop-ment of androgen-hypersensitive cells clones in Shionogi mouse mammarycarcinoma cells in culture, J. Natl. Cancer Inst. 80 (1988) 1138–1147.
25] F. Labrie, Blockade of testicular and adrenal androgens in prostate cancer treat-ment, Nat. Rev. Urol. 8 (2) (2011) 73–85.
26] C.D. Chen, D.S. Welsbie, C. Tran, S.H. Baek, R. Chen, R. Vessella, M.G. Rosenfeld,C.L. Sawyers, Molecular determinants of resistance to antiandrogen therapy,Nat. Med. 10 (1) (2004) 33–39.
27] H.I. Scher, C.L. Sawyers, Biology of progressive, castration-resistant prostatecancer: directed therapies targeting the androgen-receptor signaling axis, J.Clin. Oncol. 23 (32) (2005) 8253–8261.
28] M.E. Taplin, S.P. Balk, Androgen receptor: a key molecule in the progressionof prostate cancer to hormone independence, J. Cell. Biochem. 91 (3) (2004)483–490.
29] P.F. Mulders, J.A. Schalken, Measuring therapeutic efficacy in the changingparadigm of castrate-resistant prostate cancer, Prostate Cancer Prostatic Dis.12 (3) (2009) 241–246.
30] M.J. McPhaul, Mechanisms of prostate cancer progression to androgen inde-pendence, Best Pract. Res. Clin. Endocrinol. Metab. 22 (2) (2008) 373–388.
31] W.P. Harris, E.A. Mostaghel, P.S. Nelson, B. Montgomery, Androgen deprivationtherapy: progress in understanding mechanisms of resistance and optimizingandrogen depletion, Nat. Clin. Pract. Urol. 6 (2) (2009) 76–85.
32] E.A. Mostaghel, B. Montgomery, P.S. Nelson, Castration-resistant prostate can-cer: targeting androgen metabolic pathways in recurrent disease, Urol. Oncol.27 (3) (2009) 251–257.
33] F. Labrie, J. Simard, S.M. Singh, B. Candas, Estimated potency of Casodex: aproblematic design, Urology 50 (1997) 309–313.
34] F. Labrie, J. Simard, A. Coquet, G. Leblanc, B. Candas, Reply by the authors,Urology 54 (1999) 195–197.
35] J. Simard, S.M. Singh, F. Labrie, Comparison of in vitro effects of the pureantiandrogens OH-flutamide, Casodex and nilutamide on androgen-sensitiveparameters, Urology 49 (1997) 580–589.
36] S. Luo, C. Martel, G. Leblanc, B. Candas, S.M. Singh, C. Labrie, J. Simard, A.Bélanger, F. Labrie, Relative potencies of flutamide and Casodex: preclinicalstudies, Endocr. Relat. Cancer 3 (1996) 229–241.
37] F. Labrie, I. Luthy, R. Veilleux, J. Simard, A. Bélanger, A. Dupont, New conceptson the androgen sensitivity of prostate cancer, in: G.P. Murphy, S. Khoury, R.Kuss, C. Chatelain, L. Denis (Eds.), Progress in Clinical and Biological Research.Prostate Cancer Part A: Research. Endocrine Treatment and Histopathology,1987, pp. 145–172.
38] K. Pereira de Jesus-Tran, P.L. Cote, L. Cantin, J. Blanchet, F. Labrie, R. Breton,Comparison of crystal structures of human androgen receptor ligand-bindingdomain complexed with various agonists reveals molecular determinantsresponsible for binding affinity, Protein Sci. 15 (5) (2006) 987–999.
39] L. Cantin, F. Faucher, J.F. Couture, K.P. de Jesus-Tran, P. Legrand, L.C. Ciobanu, Y.Frechette, R. Labrecque, S.M. Singh, F. Labrie, R. Breton, Structural characteriza-tion of the human androgen receptor ligand-binding domain complexed withEM5744, a rationally designed steroidal ligand bearing a bulky chain directedtoward helix 12, J. Biol. Chem. 282 (42) (2007) 30910–30919.
40] C. Tran, S. Ouk, N.J. Clegg, Y. Chen, P.A. Watson, V. Arora, J. Wongvipat, P.M.Smith-Jones, D. Yoo, A. Kwon, T. Wasielewska, D. Welsbie, C.D. Chen, C.S.Higano, T.M. Beer, D.T. Hung, H.I. Scher, M.E. Jung, C.L. Sawyers, Development ofa second-generation antiandrogen for treatment of advanced prostate cancer,Science 324 (5928) (2009) 787–790.
41] H.I. Scher, T.M. Beer, C.S. Higano, A. Anand, M.E. Taplin, E. Efstathiou, D.Rathkopf, J. Shelkey, E.Y. Yu, J. Alumkal, D. Hung, M. Hirmand, L. Seely, M.J.Morris, D.C. Danila, J. Humm, S. Larson, M. Fleisher, C.L. Sawyers, Antitumouractivity of MDV3100 in castration-resistant prostate cancer: a phase 1–2 study,Lancet 375 (9724) (2010) 1437–1446.
42] D. Mukherji, C.J. Pezaro, J.S. De-Bono, MDV3100 for the treatment of prostatecancer, Expert Opin. Invest. Drugs 21 (2) (2012) 227–233.
43] N.J. Clegg, J. Wongvipat, J.D. Joseph, C. Tran, S. Ouk, A. Dilhas, Y. Chen, K. Grillot,E.D. Bischoff, L. Cai, A. Aparicio, S. Dorow, V. Arora, G. Shao, J. Qian, H. Zhao,G. Yang, C. Cao, J. Sensintaffar, T. Wasielewska, M.R. Herbert, C. Bonnefous, B.
[
& Molecular Biology 132 (2012) 93– 104 103
Darimont, H.I. Scher, P. Smith-Jones, M. Klang, N.D. Smith, E. de Stanchina, N.Wu, O. Ouerfelli, P.J. Rix, R.A. Heyman, M.E. Jung, C.L. Sawyers, J.H. Hager, ARN-509: a novel antiandrogen for prostate cancer treatment, Cancer Res. 72 (6)(2012) 1494–1503.
44] C. Fee, H. Burstin, J.H. Maselli, R.Y. Hsia, Association of emergency departmentlength of stay with safety-net status, JAMA 307 (5) (2012) 476–482.
45] C. Pezaro, G. Attard, Prostate cancer in 2011: redefining the therapeutic land-scape for CRPC, Nat. Rev. Urol. 9 (2) (2012) 63–64.
46] M.E. Jung, S. Ouk, D. Yoo, C.L. Sawyers, C. Chen, C. Tran, J. Wongvipat,Structure–activity relationship for thiohydantoin androgen receptor antago-nists for castration-resistant prostate cancer (CRPC), J. Med. Chem. 53 (7) (2010)2779–2796.
47] C.L. Sawyers, M.E. Jung, C.D. Chen, S. Ouk, D. Welsbie, C. Tran, J. Wongvipat, D.Yoo, Diarylhydantoin Compounds, U.S. Patent Appl. 2007/0004753 (2007).
48] C. Martel, L. Provencher, X. Li, A. St-Pierre, G. Leblanc, S. Gauthier, Y. Mérand,F. Labrie, Binding characteristics of novel nonsteroidal antiestrogens to the ratuterine estrogen receptors, J. Steroid Biochem. Mol. Biol. 64 (1998) 199–205.
49] S. Luo, C. Martel, A. Sourla, S. Gauthier, Y. Mérand, A. Bélanger, C. Labrie, F.Labrie, Comparative effects of 28-day treatment with the new antiestrogenEM-800 and tamoxifen on estrogen-sensitive parameters in the intact mouse,Int. J. Cancer 73 (1997) 381–391.
50] J. Asselin, R. Mélanc on, G. Moachon, A. Bélanger, Characteristics of bindingto estrogen, androgen, progestin and glucocorticoid receptors in 7,12-dimethylbenz(a)anthracene-induced mammary tumors and their hormonalcontrol, Cancer Res. 40 (1980) 1612–1622.
51] F. Labrie, R. Veilleux, Maintenance of androgen responsiveness by glucocorti-coids in Shionogi mammary carcinoma cells in culture, J. Natl. Cancer Inst. 80(1988) 966–970.
52] F. Labrie, R. Veilleux, A. Fournier, Glucocorticoids stimulate the growth ofmouse mammary carcinoma Shionogi cells in culture, Mol. Cell. Endocrinol.58 (1988) 207–211.
53] J. Simard, S. Dauvois, D.E. Haagensen, C. Lévesque, Y. Mérand, F. Labrie, Reg-ulation of progesterone-binding breast cyst protein GCDFP-24 secretion byestrogens and androgens in human breast cancer cells: a new marker of steroidaction in breast cancer, Endocrinology 126 (1990) 3223–3231.
54] H. Qi, Y. Labrie, J. Grenier, A. Fournier, C. Fillion, C. Labrie, Androgens induceexpression of SPAK, a STE20/SPS1-related kinase, in LNCaP human prostatecancer cells, Mol. Cell. Endocrinol. 182 (2001) 181–192.
55] F. Labrie, A. Bélanger, P. Bélanger, R. Bérubé, C. Martel, L. Cusan, J.L. Gomez, B.Candas, I. Castiel, V. Chaussade, C. Deloche, J. Leclaire, Androgen glucuronides,instead of testosterone, as the new markers of androgenic activity in women,J. Steroid Biochem. Mol. Biol. 99 (2006) 182–188.
56] J. Asselin, F. Labrie, Y. Gourdeau, C. Bonne, J.P. Raynaud, Binding of[3H]methyltrienolone (R1881) in rat prostate and human benign prostatichypertrophy (BPH), Steroids 28 (1976) 449–459.
57] I. Luthy, F. Labrie, Development of androgen resistance in mouse mammarytumor cells can be prevented by the antiandrogen flutamide, Prostate 10 (1)(1987) 89–94.
58] Y. de Launoit, R. Veilleux, M. Dufour, J. Simard, F. Labrie, Characteristics ofthe biphasic action of androgens and of the potent antiproliferative effects ofthe new pure antiestrogen EM-139 on cell cycle kinetic parameters in LNCaPhuman prostatic cancer cells, Cancer Res. 51 (19) (1991) 5165–5170.
59] J. Veldscholte, C. Ris-Stalpers, G.G. Kuiper, G. Jenster, C. Berrevoets, E. Claassen,H.C. van Rooij, J. Trapman, A.O. Brinkmann, E. Mulder, A mutation in the ligandbinding domain of the androgen receptor of human LNCaP cells affects steroidbinding characteristics and response to anti-androgens, Biochem. Biophys. Res.Commun. 173 (1990) 534–540.
60] J. Simard, R. Veilleux, Y. de Launoit, D.E. Haagensen, F. Labrie, Stimulation ofapolipoprotein D secretion by steroids coincides with inhibition of cell prolifer-ation in human LNCaP prostate cancer cells, Cancer Res. 51 (1991) 4336–4342.
61] H. Kawata, S. Arai, T. Nakagawa, N. Ishikura, A. Nishimoto, H. Yoshino, T. Shi-raishi, K. Tachibana, R. Nakamura, H. Sato, Biological properties of androgenreceptor pure antagonist for treatment of castration-resistant prostate can-cer: optimization from lead compound to CH5137291, Prostate 71 (12) (2011)1344–1356.
62] D. Moras, H. Gronemeyer, The nuclear receptor ligand-binding domain: struc-ture and function, Curr. Opin. Cell Biol. 10 (3) (1998) 384–391.
63] A.M. Brzozowski, A.C. Pike, Z. Dauter, R.E. Hubbard, T. Bonn, O. Engstrom, L.Ohman, G.L. Greene, J.A. Gustafsson, M. Carlquist, Molecular basis of agonismand antagonism in the oestrogen receptor, Nature 389 (6652) (1997) 753–758.
64] J.S. Sack, K.F. Kish, C. Wang, R.M. Attar, S.E. Kiefer, Y. An, G.Y. Wu, J.E. Schef-fler, M.E. Salvati, S.R. Krystek Jr., R. Weinmann, H.M. Einspahr, Crystallographicstructures of the ligand-binding domains of the androgen receptor and itsT877A mutant complexed with the natural agonist dihydrotestosterone, Proc.Natl. Acad. Sci. U.S.A. 98 (9) (2001) 4904–4909.
65] E. Estebanez-Perpina, J.M. Moore, E. Mar, E. Delgado-Rodrigues, P. Nguyen, J.D.Baxter, B.M. Buehrer, P. Webb, R.J. Fletterick, R.K. Guy, The molecular mech-anisms of coactivator utilization in ligand-dependent transactivation by theandrogen receptor, J. Biol. Chem. 280 (9) (2005) 8060–8068.
66] B. He, R.T. Gampe Jr., A.J. Kole, A.T. Hnat, T.B. Stanley, G. An, E.L. Stewart, R.I.Kalman, J.T. Minges, E.M. Wilson, Structural basis for androgen receptor inter-
domain and coactivator interactions suggests a transition in nuclear receptoractivation function dominance, Mol. Cell 16 (3) (2004) 425–438.67] E. Hur, S.J. Pfaff, E.S. Payne, H. Gron, B.M. Buehrer, R.J. Fletterick, Recognitionand accommodation at the androgen receptor coactivator binding interface,PLoS Biol. 2 (9) (2004) E274.

1 mistr
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
04 S. Gauthier et al. / Journal of Steroid Bioche
68] C.E. Bohl, Z. Wu, D.D. Miller, C.E. Bell, J.T. Dalton, Crystal structure of theT877A human androgen receptor ligand-binding domain complexed to cypro-terone acetate provides insight for ligand-induced conformational changes andstructure-based drug design, J. Biol. Chem. 282 (18) (2007) 13648–13655.
69] M. Plante, S. Lapointe, F. Labrie, Stimulatory effect of synthetic progestinscurrently used for the treatment of prostate cancer on growth of the androgen-sensitive Shionogi tumor in mice, J. Steroid Biochem. 31 (1988) 61–64.
70] C. Labrie, L. Cusan, M. Plante, S. Lapointe, F. Labrie, Analysis of the androgenicactivity of synthetic progestins currently used for the treatment of prostatecancer, J. Steroid Biochem. 28 (1987) 379–384.
71] I.A. Luthy, D.J. Bégin, F. Labrie, Androgenic activity of synthetic progestins andspironolactone in androgen-sensitive mouse mammary carcinoma (Shionogi)cells in culture, J. Steroid Biochem. 31 (1988) 845–852.
72] J.F. Caubet, T.D. Tosteson, E.W. Dong, E.M. Naylon, G.W. Whiting, M.S. Ern-stoff, S.D. Ross, Maximum androgen blockade in advanced prostate cancer:a meta-analysis of published randomized controlled trials using nonsteroidalantiandrogens, Urology 49 (1997) 71–78.
73] M.E. Taplin, Drug insight: role of the androgen receptor in the development andprogression of prostate cancer, Nat. Clin. Pract. Oncol. 4 (4) (2007) 236–244.
74] T.H. van der Kwast, J. Schalken, J.A. Ruizeveld de Winter, Androgen receptorsin endocrine-therapy-resistant human prostate cancer, Int. J. Cancer 48 (1991)189–193.
75] J.A. Ruizeveld de Winter, P.J. Janssen, H.M. Sleddens, M.C. Verleun-Mooijman,J. Trapman, A.O. Brinkmann, A.B. Santerse, F.H. Schroder, T.H. van der Kwast,Androgen receptor status in localized and locally progressive hormone refrac-tory human prostate cancer, Am. J. Pathol. 144 (4) (1994) 735–746.
76] M.H. Lévesque, M. El-Alfy, L. Cusan, F. Labrie, Androgen receptor as a potentialsign of prostate cancer metastasis, Prostate 69 (15) (2009) 1704–1711.
77] T.H. van der Kwast, B. Tetu, Y. Fradet, A. Dupont, J. Gomez, L. Cusan, P. Diamond,F. Labrie, Androgen receptor modulation in benign human prostatic tissue and
prostatic adenocarcinoma during neoadjuvant endocrine combination therapy,Prostate 28 (1996) 227–231.78] C. Labrie, A. Bélanger, F. Labrie, Androgenic activity of dehydroepiandrosteroneand androstenedione in the rat ventral prostate, Endocrinology 123 (1988)1412–1417.
y & Molecular Biology 132 (2012) 93– 104
79] F. Labrie, V. Luu-The, A. Bélanger, S.-X. Lin, J. Simard, C. Labrie, Is DHEA a hor-mone? Starling review, J. Endocrinol. 187 (2005) 169–196.
80] T. Nishiyama, Y. Hashimoto, K. Takahashi, The influence of androgen depriva-tion therapy on dihydrotestosterone levels in the prostatic tissue of patientswith prostate cancer, Clin. Cancer Res. 10 (21) (2004) 7121–7126.
81] E.A. Mostaghel, S.T. Page, D.W. Lin, L. Fazli, I.M. Coleman, L.D. True,B. Knudsen, D.L. Hess, C.C. Nelson, A.M. Matsumoto, W.J. Bremner, M.E.Gleave, P.S. Nelson, Intraprostatic androgens and androgen-regulated geneexpression persist after testosterone suppression: therapeutic implica-tions for castration-resistant prostate cancer, Cancer Res. 67 (10) (2007)5033–5041.
82] L.J. Denis, F. Keuppens, P.H. Smith, P. Whelan, J.L. Carneiro de Moura, D. Newling,A. Bono, R. Sylvester, Maximal androgen blockade: final analysis of EORTC PhaseIII trial 30853, Eur. Urol. 33 (1998) 144–151.
83] J.A. Maddy, W.W. Winternitz, H. Norrell, Cryohypophysectomy in the manage-ment of advanced prostatic cancer, Cancer 28 (2) (1971) 322–328.
84] J.R. Drago, R.J. Santen, A. Lipton, T.J. Worgul, H.A. Harvey, A. Boucher, A. Manni,T.J. Rohner, Clinical effect of aminoglutethimide, medical adrenalectomy, intreatment of 43 patients with advanced prostatic carcinoma, Cancer 53 (7)(1984) 1447–1450.
85] R. Murray, P. Pitt, Treatment of advanced prostatic cancer, resistant to conven-tional therapy, with aminoglutethimide, Eur. J. Cancer Clin. Oncol. 21 (4) (1985)453–458.
86] F. Labrie, A. Dupont, M. Giguère, J.P. Borsanyi, L.Y.G. Monfette, J. Emond, N.Bergeron, Benefits of combination therapy with flutamide in patients relapsingafter castration, Br. J. Urol. 61 (1988) 341–346.
87] J.E. Fowler Jr., W.F. Whitmore Jr., The response of metastatic adenocarci-noma of the prostate to exogenous testosterone, J. Urol. 126 (3) (1981)372–375.
88] G. Attard, A.H. Reid, R. A’Hern, C. Parker, N.B. Oommen, E. Folkerd, C. Mes-
siou, L.R. Molife, G. Maier, E. Thompson, D. Olmos, R. Sinha, G. Lee, M.Dowsett, S.B. Kaye, D. Dearnaley, T. Kheoh, A. Molina, J.S. de Bono, Selectiveinhibition of CYP17 with abiraterone acetate is highly active in the treat-ment of castration-resistant prostate cancer, J. Clin. Oncol. 27 (23) (2009)3742–3748.




























