Embed Size (px)
Citation preview

States of Water in Poly(vinyl alcohol) Derivative Hydrogels
J. RUIZ, A. MANTECON, V. CADIZ
Universitat Rovira i Virgili, Departament de Quımica Analıtica i Quımica Organica, Facultat de Quımica,Placa Imperial Tarraco 1, 43005 Tarragona, Spain
Received 20 May 2002; revised 25 March 2003; accepted 25 March 2003
ABSTRACT: We studied the behavior of absorbed water in equilibrium-swollen poly(vi-nyl alcohol) derivative hydrogels by differential scanning calorimetry (DSC), 1H nuclearmagnetic resonance, and wide-angle X-ray diffraction. By DSC, three types of waterwere detected, and their relative fractions were estimated. With this technique we alsocalculated the pore size for every sample. From the nonexponential decay of thespin–spin relaxation data, we distinguished two environmental states of the absorbedwater in the samples. The relaxation times were determined. From these data, wecalculated the fractions of each type of water for every hydrogel and related them to thedegree of crosslinking. The X-ray study indicated that the water absorbed in thesehydrogels forms a single crystalline phase on cooling. © 2003 Wiley Periodicals, Inc. J PolymSci Part B: Polym Phys 41: 1462–1467, 2003Keywords: hydrogel; poly(vinyl alcohol); DSC; NMR; X-ray
INTRODUCTION
The water in polymers affects their properties inseveral ways (e.g., its plasticizing effect can con-siderably decrease their strength). An under-standing of the properties of water in gel mem-branes is important if the transport behavior ofsolutes in separation processes is to be inter-preted and the mechanism of membrane perme-ability elucidated.1 Moreover, by studying thestate of water absorbed in hydrophilic materialssuch as hydrogels, we can learn more about theinternal structure of the network.
Several techniques can be used to examine thedifferent organization of solvent molecules in thehydrogel. These techniques provide complemen-tary information. Differential scanning calorime-try (DSC),2–4 solution, solid nuclear magnetic res-onance (NMR),5,6 wide-angle X-ray diffraction,7
and other less common techniques such as ther-
mogravimetric analysis,8 positron annihilationlifetime spectroscopy,9 light scattering,10 andsmall-angle neutron scattering11,12 have beenused to study the various states of water in hy-drogels and relate them with different environ-ments in the internal structure of the material.
In this article we explore the states of water inpreviously synthesized hydrogels obtained bychemical crosslinking of poly(vinyl alcohol) (PVA)with ethylenediaminetetraacetic dianhydride(EDTAD).13 The nature and amount of differenttypes of water determined in polymers dependson the experimental methods used and other fac-tors such as swelling state, temperature, densityof crosslinking, and so forth. Thus, we used dif-ferent techniques to determine the different typesof water and compare the information obtainedfrom them.
The most extensively technique used is DSC,which has been used successfully to analyze theamount and states of water in polymers andmembranes.14 We used this technique to examinehow water is incorporated into the network byevaluating three types of water. It is widely ac-
Correspondence to: V. Cadiz (E-mail: [email protected])Journal of Polymer Science: Part B: Polymer Physics, Vol. 41, 1462–1467 (2003)© 2003 Wiley Periodicals, Inc.
1462

cepted that the states of absorbed water are clas-sified mainly as “bound”, “intermediate”, and“free”. Evidence of the existence of bound water invarious water polymer systems, as revealed inmost DSC experiments, is based on the presenceof nonfreezable water with constrained motionrelative to free water, at temperatures far belowambient. These experiments that imply the freez-ing of the absorbed water have also been used tocalculate some network parameters as pore size.
We also used other techniques such as NMRspectroscopy and X-ray diffraction (XRD). 1HNMR spectroscopy allowed us to characterize wa-ter behavior by determining the spin–lattice (T1)and spin–spin (T2) relaxation times. This tech-nique, which does not imply water freezing, mea-sures a magnetic property and therefore shouldprovide a different view of the water-moleculeorganization. Normally, XRD studies are directedto characterize the matrix, but we also used it toobtain information about the absorbed water inthe swollen hydrogel.
EXPERIMENTAL
Materials
PVA (Fluka) had a degree of hydrolysis of 86–89% and a degree of polymerization of 300. ED-TAD (Aldrich) and dimethyl sulfoxide (DMSO)(Panreac) were used as received.
Preparation of Gel Films: Crosslinking with EDTAD
PVA (18 mmol of the OH group) was dissolved in5 mL of DMSO at room temperature. The dian-hydride compound was dissolved in 2 mL ofDMSO at room temperature and added in an OH/anhydride group ratio of 1/0.250 (PVA-EDTA25),1/0.100 (PVA-EDTA10), 1/0.050 (PVA-EDTA5),1/0.033 (PVA-EDTA3.3), and 1/0.025 (PVA-EDTA2.5). The reaction mixture was then stirredfor 2 min and introduced between two 170 � 120� 3 mm glasses separated by microscope slides toadjust the thickness (ca. 1 mm). Gels were ob-tained in a few minutes, but the reaction wasconsidered complete after 24 h. To remove DMSOof the gels, they were immersed in deionized wa-ter to equilibrium, and this water was changedthree to four times.
Instrumentation
Thermal data were evaluated on a MettlerDSC-30 thermal analyzer with hydrogel samples
of known weight (ca. 5 mg) in sealed aluminiumpans. The temperature was cooled to �40 °C andthen held for 15 min to freeze the water, as indi-cated by an exothermic peak. The samples werethen heated at a rate of 10 °C/min from �40 to�10 °C and then at 0.3 °C/min from �10 to �5 °C.
Powder XRD measurements were performed atseveral temperatures on a Siemens D5000 diffrac-tometer with a �-� configuration fitted with anAnton Parr TTK low-temperature chamber with25 �m capton windows. Cu K� radiation wasused, and graphite was the secondary monochro-mator. The Bragg angle step was 0.05°, and thetime per step was 3 s. The samples were placedfilling the cavity of the sample holder (chromium-plated copper 14 � 10 � 1 mm).
1H NMR spectra were obtained with a Gemini300 spectrometer, operating at 300 MHz, fromswollen samples with D2O in 5-mm tubes. T1 andT2 relaxation times were measured at a temper-ature of 20 °C. Relaxation curves were obtainedwith T1 saturation recovery pulse sequence for T1and Carr–Purcell–Meiboom–Gil pulse sequencefor T2.
The equilibrium absorption of water was mea-sured for all the samples with an electronic mi-crobalance (Mettler AB204) and a microbalancefrom Sartorius GMBH, both of which had an ac-curacy of �10�4 g. We averaged the equilibriumwater uptakes over nine measurements, and foreach type of sample, water absorption was repli-cated three times.
RESULTS AND DISCUSSION
How water and polymers interact with hydro-philic groups has been researched extensively byDSC.3,5,8,15–17
This technique is often used to estimate theamounts of free, freezable-bound, and nonfreez-able water in water-swollen polymers. Free water(type I) is defined as the water with the samephase-transition temperature as bulk water.Freezable-bound water (type II) is water whosephase transition is lower than 0 °C. This depres-sion is usually ascribed to the fact that the waterinteracts weakly with the polymer chain of themembrane and/or to capillary condensation in themembranes. Nonfreezing water (type III) is de-fined as water with no detectable phase transitionfrom 0 to �100 °C. This water is assumed to beinfluenced by a strong interaction with the polarmoieties of the polymer chain.
STATES OF WATER 1463
With DSC analysis we can calculate theamount of free water and freezable-bound waterfrom the enthalpies of melting or crystallizationof water associated with the polymer. The amountof nonfreezable water can be calculated by thedifference with the total weight of water. We cal-culated the enthalpy of melting of different statesof freezable water from the area of each peak ofthe DSC curve and the amount of free water fromthe enthalpy of melting of pure water (332 J/g) byintegration of the peak at 0 °C.
As suggested by Kuhn et al.,18 the meltingtemperature depression, that is, the difference oftemperature between the endotherms of free andfreezable-bound water, can be used to examinepore size. In additional theoretical studies, Brunet al.19 established the relationship between thepore radius and the heat of fusion for the pene-trant liquid with the melting point depression.We therefore used eqs 1 and 2
Rp � A �B
�T (1)
�H�T� � �Hf � C�T � D��T�2 (2)
where �T is the melting point depression, whichis negative; and �Hf is the heat of fusion for thewater under normal conditions. The numericalvalues of the constants A–D depended on whetherthe measurements were made in heating or cool-ing, pore geometry, and penetrant liquid. In ourcase in which we assumed spherical geometry andthe thermogram was obtained by heating, A� 0.57 nm, B � 32.33 nm, C � 11.39 J/gK, and D� 0.155 J/gK2.20,21 The existence of nonfreezingwater was confirmed by the relationship betweenthe freezable water content estimated by DSC
and the total water content. We calculated thequantity of nonfreezing water from the differencebetween the sum of the weights of the free andbound water as well as the weight of the totalwater content, determined in the experiments ofwater absorption.
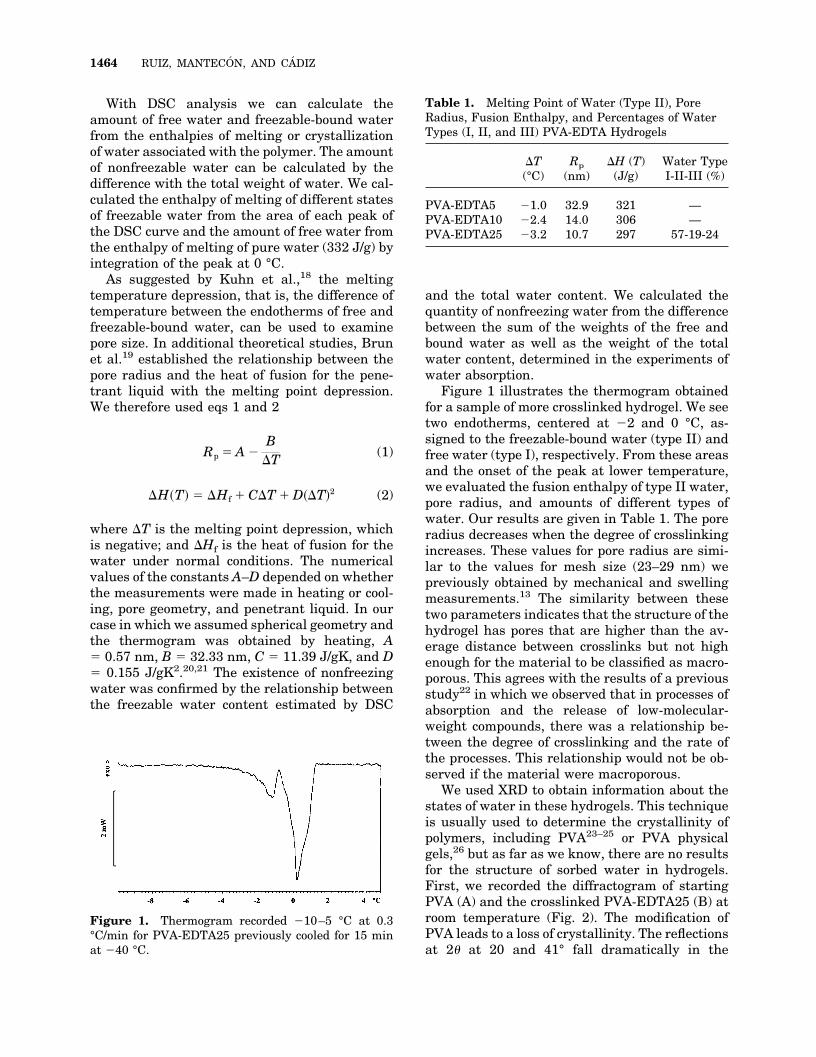
Figure 1 illustrates the thermogram obtainedfor a sample of more crosslinked hydrogel. We seetwo endotherms, centered at �2 and 0 °C, as-signed to the freezable-bound water (type II) andfree water (type I), respectively. From these areasand the onset of the peak at lower temperature,we evaluated the fusion enthalpy of type II water,pore radius, and amounts of different types ofwater. Our results are given in Table 1. The poreradius decreases when the degree of crosslinkingincreases. These values for pore radius are simi-lar to the values for mesh size (23–29 nm) wepreviously obtained by mechanical and swellingmeasurements.13 The similarity between thesetwo parameters indicates that the structure of thehydrogel has pores that are higher than the av-erage distance between crosslinks but not highenough for the material to be classified as macro-porous. This agrees with the results of a previousstudy22 in which we observed that in processes ofabsorption and the release of low-molecular-weight compounds, there was a relationship be-tween the degree of crosslinking and the rate ofthe processes. This relationship would not be ob-served if the material were macroporous.
We used XRD to obtain information about thestates of water in these hydrogels. This techniqueis usually used to determine the crystallinity ofpolymers, including PVA23–25 or PVA physicalgels,26 but as far as we know, there are no resultsfor the structure of sorbed water in hydrogels.First, we recorded the diffractogram of startingPVA (A) and the crosslinked PVA-EDTA25 (B) atroom temperature (Fig. 2). The modification ofPVA leads to a loss of crystallinity. The reflectionsat 2� at 20 and 41° fall dramatically in the
Figure 1. Thermogram recorded �10–5 °C at 0.3°C/min for PVA-EDTA25 previously cooled for 15 minat �40 °C.
Table 1. Melting Point of Water (Type II), PoreRadius, Fusion Enthalpy, and Percentages of WaterTypes (I, II, and III) PVA-EDTA Hydrogels
�T(°C)
Rp
(nm)�H (T)
(J/g)Water TypeI-II-III (%)
PVA-EDTA5 �1.0 32.9 321 —PVA-EDTA10 �2.4 14.0 306 —PVA-EDTA25 �3.2 10.7 297 57-19-24
1464 RUIZ, MANTECON, AND CADIZ
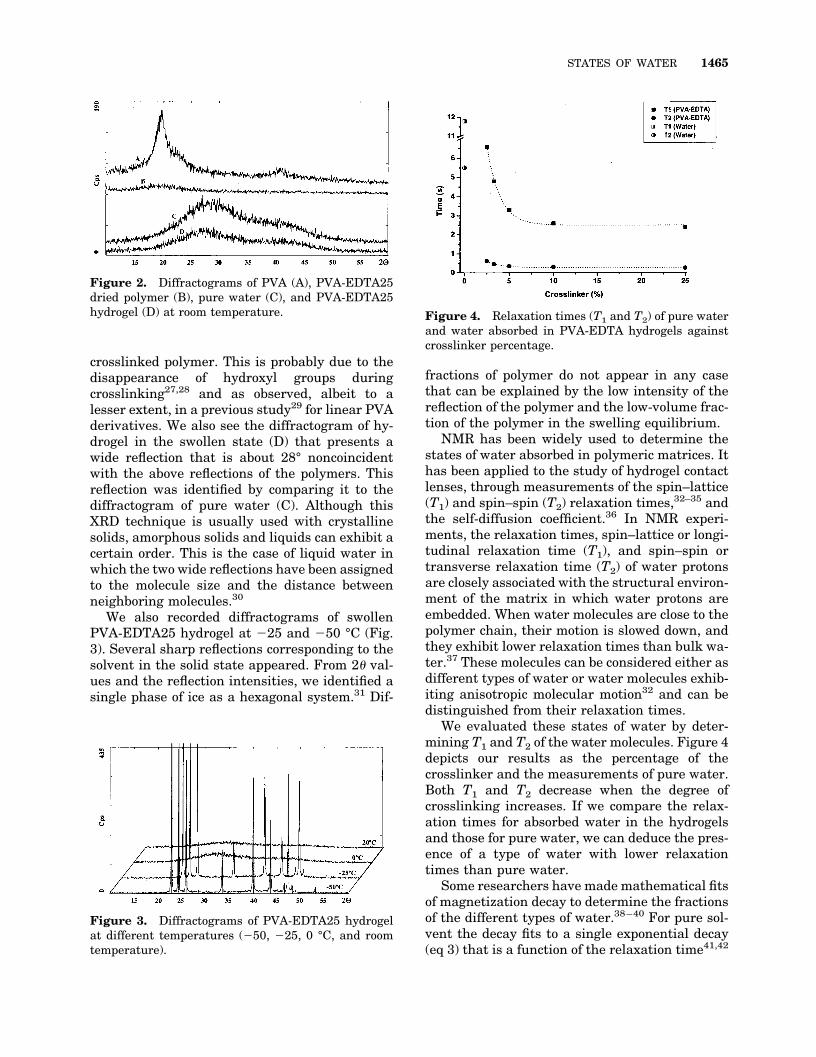
crosslinked polymer. This is probably due to thedisappearance of hydroxyl groups duringcrosslinking27,28 and as observed, albeit to alesser extent, in a previous study29 for linear PVAderivatives. We also see the diffractogram of hy-drogel in the swollen state (D) that presents awide reflection that is about 28° noncoincidentwith the above reflections of the polymers. Thisreflection was identified by comparing it to thediffractogram of pure water (C). Although thisXRD technique is usually used with crystallinesolids, amorphous solids and liquids can exhibit acertain order. This is the case of liquid water inwhich the two wide reflections have been assignedto the molecule size and the distance betweenneighboring molecules.30
We also recorded diffractograms of swollenPVA-EDTA25 hydrogel at �25 and �50 °C (Fig.3). Several sharp reflections corresponding to thesolvent in the solid state appeared. From 2� val-ues and the reflection intensities, we identified asingle phase of ice as a hexagonal system.31 Dif-
fractions of polymer do not appear in any casethat can be explained by the low intensity of thereflection of the polymer and the low-volume frac-tion of the polymer in the swelling equilibrium.
NMR has been widely used to determine thestates of water absorbed in polymeric matrices. Ithas been applied to the study of hydrogel contactlenses, through measurements of the spin–lattice(T1) and spin–spin (T2) relaxation times,32–35 andthe self-diffusion coefficient.36 In NMR experi-ments, the relaxation times, spin–lattice or longi-tudinal relaxation time (T1), and spin–spin ortransverse relaxation time (T2) of water protonsare closely associated with the structural environ-ment of the matrix in which water protons areembedded. When water molecules are close to thepolymer chain, their motion is slowed down, andthey exhibit lower relaxation times than bulk wa-ter.37 These molecules can be considered either asdifferent types of water or water molecules exhib-iting anisotropic molecular motion32 and can bedistinguished from their relaxation times.
We evaluated these states of water by deter-mining T1 and T2 of the water molecules. Figure 4depicts our results as the percentage of thecrosslinker and the measurements of pure water.Both T1 and T2 decrease when the degree ofcrosslinking increases. If we compare the relax-ation times for absorbed water in the hydrogelsand those for pure water, we can deduce the pres-ence of a type of water with lower relaxationtimes than pure water.
Some researchers have made mathematical fitsof magnetization decay to determine the fractionsof the different types of water.38–40 For pure sol-vent the decay fits to a single exponential decay(eq 3) that is a function of the relaxation time41,42
Figure 2. Diffractograms of PVA (A), PVA-EDTA25dried polymer (B), pure water (C), and PVA-EDTA25hydrogel (D) at room temperature.
Figure 3. Diffractograms of PVA-EDTA25 hydrogelat different temperatures (�50, �25, 0 °C, and roomtemperature).
Figure 4. Relaxation times (T1 and T2) of pure waterand water absorbed in PVA-EDTA hydrogels againstcrosslinker percentage.
STATES OF WATER 1465
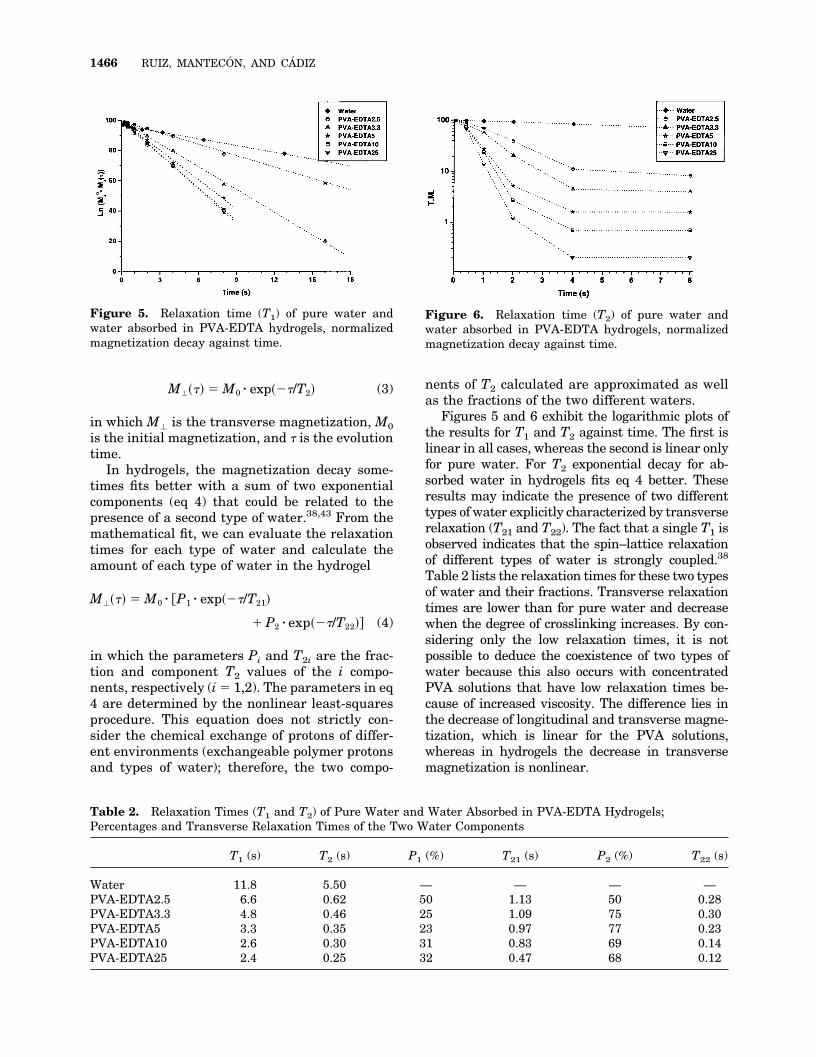
M���� � M0 � exp���/T2� (3)
in which M� is the transverse magnetization, M0is the initial magnetization, and � is the evolutiontime.
In hydrogels, the magnetization decay some-times fits better with a sum of two exponentialcomponents (eq 4) that could be related to thepresence of a second type of water.38,43 From themathematical fit, we can evaluate the relaxationtimes for each type of water and calculate theamount of each type of water in the hydrogel
M���� � M0 � �P1 � exp���/T21�
� P2 � exp���/T22� (4)
in which the parameters Pi and T2i are the frac-tion and component T2 values of the i compo-nents, respectively (i � 1,2). The parameters in eq4 are determined by the nonlinear least-squaresprocedure. This equation does not strictly con-sider the chemical exchange of protons of differ-ent environments (exchangeable polymer protonsand types of water); therefore, the two compo-
nents of T2 calculated are approximated as wellas the fractions of the two different waters.
Figures 5 and 6 exhibit the logarithmic plots ofthe results for T1 and T2 against time. The first islinear in all cases, whereas the second is linear onlyfor pure water. For T2 exponential decay for ab-sorbed water in hydrogels fits eq 4 better. Theseresults may indicate the presence of two differenttypes of water explicitly characterized by transverserelaxation (T21 and T22). The fact that a single T1 isobserved indicates that the spin–lattice relaxationof different types of water is strongly coupled.38
Table 2 lists the relaxation times for these two typesof water and their fractions. Transverse relaxationtimes are lower than for pure water and decreasewhen the degree of crosslinking increases. By con-sidering only the low relaxation times, it is notpossible to deduce the coexistence of two types ofwater because this also occurs with concentratedPVA solutions that have low relaxation times be-cause of increased viscosity. The difference lies inthe decrease of longitudinal and transverse magne-tization, which is linear for the PVA solutions,whereas in hydrogels the decrease in transversemagnetization is nonlinear.
Figure 5. Relaxation time (T1) of pure water andwater absorbed in PVA-EDTA hydrogels, normalizedmagnetization decay against time.
Figure 6. Relaxation time (T2) of pure water andwater absorbed in PVA-EDTA hydrogels, normalizedmagnetization decay against time.
Table 2. Relaxation Times (T1 and T2) of Pure Water and Water Absorbed in PVA-EDTA Hydrogels;Percentages and Transverse Relaxation Times of the Two Water Components
T1 (s) T2 (s) P1 (%) T21 (s) P2 (%) T22 (s)
Water 11.8 5.50 — — — —PVA-EDTA2.5 6.6 0.62 50 1.13 50 0.28PVA-EDTA3.3 4.8 0.46 25 1.09 75 0.30PVA-EDTA5 3.3 0.35 23 0.97 77 0.23PVA-EDTA10 2.6 0.30 31 0.83 69 0.14PVA-EDTA25 2.4 0.25 32 0.47 68 0.12
1466 RUIZ, MANTECON, AND CADIZ

In conclusion, the obtained results with thethree techniques provided different aspects. XRDonly revealed a diffraction pattern at tempera-tures below 0 °C indicative of a single crystallinephase in the solid state. NMR experiments con-firmed the coexistence of more than one type ofwater in the liquid state and allowed us to calcu-late their relative fractions. By DSC we deter-mined three types of water. Two of them, in thesolid state, were observed in the thermogram,whereas the third, in the liquid state, was calcu-lated gravimetrically. Data obtained from solidphases, undistinguishable by XRD, allowed thedetermination of pore size of the network.
The authors thank the Comision de Investigacion Ci-entıfica y Tecnologica (CICYT, MAT 99-1113) and theComissio Interdepartamental de Recerca i InnovacioTecnologica (CIRIT, 2000 SGR 00100) for their finan-cial support.
REFERENCES AND NOTES
1. Zhang, W.-Z.; Satoh, M.; Komiyama, J. J MembrSci 1989, 42, 303.
2. Carles, E.; Scallan, A. M. J Appl Polym Sci 1971, 7,1855.
3. Nakamura, K.; Hatakeyama, T.; Hatakeyama, H.Polymer 1981, 22, 473.
4. Gref, R.; Nguyen, Q. T.; Rault, J.; Neel, J. EurPolym J 1992, 28, 1007.
5. Hatakeyama, T.; Yamahuchi, A.; Hatakeyama, H.Eur Polym J 1984, 20, 61.
6. Horii, F.; Hu, S.; Deguchi, K.; Sugisawa, H.; Ohgi,H.; Sato, T. Macromolecules 1996, 29, 3330.
7. Hodge, R. M.; Edward, J. H.; Simon, G. P. Polymer1996, 37, 1371.
8. Hatakeyama, T.; Yamahuchi, A.; Hatakeyama, H.Eur Polym J 1987, 23, 361.
9. Liu, W. G.; Yao, K. D. Polymer 2001, 42, 3943.10. Takeshita, H.; Kanaya, T.; Nishida, K.; Kaji, K.
Macromolecules 1999, 32, 7815.11. Shibayama, M. Polymeric Materials Encyclopedia;
Salomone, J. C., Ed.; CRC: Boca Roton, FL, 1996;Vol. 9, p 7020.
12. Evmenenko, G.; Alexeev, V.; Budtova, T.; Buyanov,A.; Frenkel, S. Polymer 1999, 40, 2975.
13. Ruiz, J.; Mantecon, A.; Cadiz, V. Polymer 2001, 42,6347.
14. Bershtein, V. A.; Egorov, V. M. Differential Scan-ning Calorimetry of Polymers; Ellis Horwood:Hemel Hempstead, England, 1994, pp 224–226.
15. Ikada, Y.; Suzuki, M.; Iwata, H. Water in Poly-mers. ACS Symposium Series 127; Rowland, S. P.,
Ed.; American Chemical Society: Washington, DC,1980; p 287.
16. Higuchi, A.; Iijima, T. Polymer 1985, 26, 1207.17. Sivashinsky, N.; Tanny, G. B. J Appl Polym Sci
1981, 26, 2625.18. Kuhn, W.; Peterli, E.; Majer, H. J Polym Sci 1955,
16, 539.19. Brun, M.; Lallemand, A.; Quinson, J. F.; Eyraud, C.
Thermochim Acta 1977, 21, 59.20. Reymond, J. P.; Biay, I.; Quinson, J. F. J Sol-Gel
Sci Technol 1997, 8, 189.21. Iza, M.; Woerly, S.; Danumah, C.; Kaliaguine, S.;
Bousmina, M. Polymer 2000, 41, 5885.22. Ruiz, J.; Mantecon, A.; Cadiz, V. J Appl Polym Sci
2002, 85, 1644.23. Hodge, R. M.; Bastow, T. J.; Edward, J. H.; Simon,
G. P.; Hill, A. J. Macromolecules 1996, 29, 8137.24. Bin, Y.; Tanabe, Y.; Nakabayashi, C.; Korosu, H.;
Matsuo, M. Polymer 2001, 42, 1183.25. Lyoo, W. S.; Ha, W. S. Polymer 1996, 37, 3121.26. Hong, P. D.; Chen, J. H.; Wu, H. L. J Appl Polym
Sci 1988, 69, 2477.27. Mooney, R. C. J Am Chem Soc 1941, 63, 2828.28. Polyvinyl Alcohol. Basic Properties and Uses; Prit-
chard, J. G., Ed.; McDonald Technology & Science:London, 1970; p 38.
29. Gimenez, V. Ph.D. Thesis, Roviri i Virgili Univer-sity, Tarragona, Spain, 1997.
30. Fundamentals of Crystallography; Giacovazzo, C.,Ed., Oxford University Press: Oxford, 1995; p 212.
31. Rundle, R. E. J Chem Phys 1953, 21, 1311.32. McConville, P.; Whittaker, M. K.; Pope, J. M. Mac-
romolecules 2002, 35, 6961.33. McConville, P.; Pope, J. M. Polymer 2001, 42, 3559.34. Barbieri, R.; Quaglia, M.; Delfini, M.; Brosio, E.
Polymer 1998, 39, 1059.35. McBrierty, V. J.; Martin, S. J.; Karasz, F. E. J Mol
Liq 1999, 80, 179.36. McConville, P.; Pope, J. M. Polymer 2000, 41, 9081.37. Woesner, D. E. Relaxation Effects of Chemical Ex-
change. In Encyclopedia of Nuclear Magnetic Reso-nance; Harris, R., Ed.; Wiley: New York, 1996; p 4018.
38. Li, B.; Ding, D.; Wang, Y.; Sun, P.; Ma, J.; He, B.J Appl Polym Sci 1999, 72, 1203.
39. Nagura, M.; Takagi, N.; Katakami, H.; Gotoh, Y.;Ohkoshi, Y.; Koyano, T.; Minoura, N. Polym GelsNetworks 1997, 5, 455.
40. Bodurka, J.; Buntkowsky, G.; Olechnowicz, R.;Gutsze, A.; Limbach, H. H. Colloids Surf A 1996,115, 55.
41. NMR Spectroscopy; Gunther, H., Ed.; Wiley:Chichester, England, 1987; p 217.
42. NMR Spectroscopy: Modern Spectral Analysis; We-ber, U.; Thiele, H., Eds.; Wiley-VCH: Weinheim,1998; p 317.
43. Ruan, R. R.; Han, J.; Chen, P. L.; Martinez, B. C.Biotechnol Tech 1997, 11, 257.
STATES OF WATER 1467





![PHYSICAL CHEMISTRY · 2012. 10. 5. · PHYSICAL CHEMISTRY SOME METHODS FOR DOPING POLY(VINYL ALCOHOL) HYDROGELS [PVA-HG]* S. PATACHIA, M. RINJA, L. ISAC “Transilvania” University](https://img.pdfslide.us/doc/110x75/5fd2418d3213667bb909c002/physical-2012-10-5-physical-chemistry-some-methods-for-doping-polyvinyl-alcohol.jpg)























