Embed Size (px)
Citation preview

Standardized reagents and protocols for engineeringzinc finger nucleases by modular assemblyDavid AWright1, Stacey Thibodeau-Beganny2, Jeffry D Sander1,3, Ronnie J Winfrey1, Andrew S Hirsh2,4,Magdalena Eichtinger2,4, Fengli Fu1,3, Matthew H Porteus5, Drena Dobbs1,3, Daniel F Voytas1 & J Keith Joung2,4
1Department of Genetics, Development & Cell Biology, Iowa State University, 1035A Roy J. Carver Co-Lab, Ames, Iowa 50011, USA. 2Molecular Pathology Unit andCenter for Cancer Research, Massachusetts General Hospital, 149 13th Street, Charlestown, Massachusetts 02129, USA. 3Interdepartmental Graduate Program inBioinformatics & Computational Biology, Iowa State University, 2114 Molecular Biology Building, Ames, Iowa 50011, USA. 4Department of Pathology, Harvard MedicalSchool, Boston, Massachusetts 02115, USA. 5Departments of Pediatrics & Biochemistry, University of Texas Southwestern Medical Center, 5323 Harry Hines Boulevard,Dallas, Texas 75390, USA. Correspondence should be addressed to J.K.J. ([email protected]).
Published online 16 November 2006; doi:10.1038/nprot.2006.259
Engineered zinc finger nucleases can stimulate gene targeting at specific genomic loci in insect, plant and human cells. Although
several platforms for constructing artificial zinc finger arrays using ‘‘modular assembly’’ have been described, standardized reagents
and protocols that permit rapid, cross-platform ‘‘mixing-and-matching’’ of the various zinc finger modules are not available. Here we
describe a comprehensive, publicly available archive of plasmids encoding more than 140 well-characterized zinc finger modules
together with complementary web-based software (termed ZiFiT) for identifying potential zinc finger target sites in a gene of
interest. Our reagents have been standardized on a single platform, enabling facile mixing-and-matching of modules and transfer of
assembled arrays to expression vectors without the need for specialized knowledge of zinc finger sequences or complicated
oligonucleotide design. We also describe a bacterial cell-based reporter assay for rapidly screening the DNA-binding activities of
assembled multi-finger arrays. This protocol can be completed in approximately 24–26 d.
INTRODUCTIONZinc finger nucleases (ZFNs) can be used to introducetargeted double-stranded breaks (DSBs) into chromosomalDNA sequences, thereby stimulating homologous recombinationand gene targeting events at specific genomic loci in insect, plantand human cells1–7. ZFNs consist of an engineered Cys2His2 zincfinger DNA-binding domain (typically three8–10 or four6 fingers)fused to a non-specific nuclease domain from the FokI restrictionenzyme (Fig. 1). ZFNs bind as dimers to their target DNA sites,with each monomer using its zinc finger domain to recognizea ‘‘half-site’’ (Fig. 1). Dimerization of ZFNs is mediated bythe FokI nuclease domain, which cleaves within a spacersequence (typically five or six base pairs in length) that separatesthe two inverted half-sites5,10,11 (Fig. 1). Because each zinc fingerrecognizes a three base pair ‘sub-site,’ a three-finger ZFN binds toa nine base pair half-site and a four-finger ZFN binds to atwelve base pair half-site. In principle, ZFN-mediated genetargeting can be used to effect correction, mutation, deletion orinsertion at any endogenous gene, provided that appropriatezinc finger domains can be engineered to target the locus ofinterest. Therefore, although ZFN-based technology has poten-tially important uses in biological research and gene therapy,further application, development and refinement of thisapproach will require a rapid and facile method for engineeringthe zinc finger arrays needed to target specific DNA sequencesof interest.
Several recent reports have demonstrated that ‘‘modular assem-bly’’ methods can yield three-finger arrays that can in turn be usedto generate active ZFNs1–3,7,12. These modular assembly approachesinvolve the joining together of individual, pre-characterized fingermodules to create multi-finger arrays capable of recognizingextended DNA sequences. Three different sets of modules, eachwith well-characterized DNA-binding specificities, have beendescribed in the literature:
1. Carlos Barbas and colleagues have constructed a set ofmodules that target all GNN, most ANN and CNN and a fewTNN triplet subsites13–17. These modules were selected by phagedisplay and in some cases optimized by targeted mutagenesis. TheDNA-binding specificities of these domains were each verifiedusing an in vitro ELISA-based specificity assay. All of these modulesare based on a consensus human zinc finger framework18 and aredesigned to be used at any position (amino terminal, middle orcarboxy terminal) in the context of a multi-finger domain. TheBarbas group has described web-based software for using this setof modules19.
2. Researchers at Sangamo Biosciences used results fromphage display selections together with targeted mutagenesis toconstruct a set of modules that target all GNN and a smallnumber of ANN, CNN and TNN triplet subsites20–22. In vitroSELEX was used to characterize the DNA-binding specificitiesof most of these fingers22. These modules were generated byaltering fingers from the three-finger Sp1 zinc finger domainand have been noted to show position-dependence in their
p
uor
G g
n ih si l
bu
P eru ta
N 600 2©
nat
ure
pro
toco
ls/
moc.er
ut an.
ww
w//:ptt
h
Zinc fingerarray
Foklcleavagedomain
F1
F1
F1F2
F2
F2F3
F3
F3
9 bp9 bp spacer
a b
Figure 1 | Schematic of (a) a zinc finger nuclease and (b) a zinc finger
nuclease dimer bound to its target cleavage site. Individual zinc finger
domains are depicted as colored spheres with F1 representing the
amino-terminal finger, F2 the middle finger and F3 the carboxy-terminal
finger in a three-finger array. The FokI DNA cleavage domain is represented
as a purple colored octagon. Note that the spacer sequence between the
two 9-bp ‘half-sites’ can be five or six base pairs.
NATURE PROTOCOLS | VOL.1 NO.3 | 2006 | 1637
PROTOCOL

behavior (e.g., amino-terminal fingers should only be used inthat position)22.
3. Scientists at ToolGen, Inc. identified a set of naturallyoccurring human zinc fingers that recognize a range ofdifferent triplet subsites23. The DNA-binding specificities of thesemodules were determined using a yeast-based one-hybrid reporterassay. Because these modules are derived from different humantranscription factors, their sequences show little similarity to oneanother outside of the well conserved zinc finger motif residues.A number of studies have shown that many of these modulescan function at different positions within the context of amulti-finger domain23–29.
Although modular assembly has been used successfully to createzinc finger arrays and ZFNs, published reports suggest that theprocess does not always yield functional domains. Barbas andcolleagues made a series of 80 different three-finger domainsusing their modules and found that all of these multi-finger arrayswere able to bind to their intended target DNA site in an in vitroELISA-based DNA-binding assay30. In smaller-scale surveys,Cathomen and colleagues found that two out of four ZFNs theymade using the Barbas modules7 and Porteus found that six out ofsix proteins made using the Sangamo modules12 could stimulategene targeting in human cells. However, these results may reflect apublication bias because unsuccessful attempts may not always bereported. For example, our own collective experience has revealedthat success rates for modular assembly using various publishedmodules may be lower than has been previously described (D.A.W.& D.F.V., unpublished data; M.H.P., unpublished data; A.S.H., S.T.-B., M.E. and J.K.J., unpublished data). We hypothesize that themost likely reason why modular assembly does not always work isthe failure of the method to account for context-dependent effectson DNA-binding that can occur with zinc finger domains31–35.Although the overall success rate of modular assembly across abroad range of potential binding sites remains to be determined,the relative ease of this method compared with other more labor-intensive selection-based approaches makes it a reasonable poten-tial strategy for constructing ZFNs.
Given the imperfect success rate of modular assembly, multi-finger domains constructed using this approach should ideally bescreened for DNA-binding activity before being tested as ZFNs incells. In vitro tests36 or mammalian cell-based reporter assays7 havebeen used for this purpose, but these methods can be timeconsuming and labor intensive. We prefer the use of a bacterialcell-based two-hybrid (B2H) reporter system31,37–39, which tests theability of a zinc finger domain to bind a given target DNA sequence.The B2H reporter assay is rapid, easy to perform and does notrequire protein purification or mammalian cell culture or transfec-tion. In the B2H reporter system, binding of a multi-finger domainto its target site leads to transcriptional activation of a readilyassayable lacZ reporter gene (Fig. 2). Previous work has shownthat multi-finger domains that activate reporter gene expressionefficiently in the B2H also possess excellent DNA-binding affinitiesand specificities for their target sites31. We have also found thatmulti-finger domains that work well in the B2H also yield ZFNsthat mediate gene targeting with high efficiency in human cells(S. Pruett, J.K.J. & M.H.P., unpublished data).
In this protocol, we describe standardized reagents, softwaretools and protocols for constructing ZFNs by modular assemblythat enable the user to leverage the potential strengths of all three
published module sets. We have constructed a set of plasmidsencoding a comprehensive archive of more than 140 zinc fingermodules derived from the Barbas, Sangamo and ToolGen modulesets (Supplementary Table 1 online). We designed these plasmidson a standardized framework, which permits any combination ofmodules to be rapidly assembled into arrays using only standardrestriction digest–based subcloning techniques (Fig. 3). Althoughour assembly method requires two subcloning steps to construct athree-finger array, our strategy and reagents allow researchers toavoid the need for specialized knowledge of zinc finger sequencesand for complicated oligonucleotide design or costly gene synth-esis17,19–21,40. In addition, we describe a complementary web-basedZiFiT (Zinc Finger Targeter) program, which simplifies andautomates the process of identifying target DNA sites that canpotentially be targeted by multi-finger domains made using ourarchive of modules. Following assembly, sequences encoding thesemulti-finger arrays can then be transferred (again using onlyrestriction enzymes) to a B2H expression vector that permitsrapid testing of DNA-binding activities in the B2H reportersystem. Coding sequences for finger arrays that work well in theB2H system can be rapidly transferred by simple restrictiondigest to compatible vectors that permit their expression as ZFNsin plant or human cells. In addition, all of the reagents describedin our manuscript are available as a kit to scientists for non-commercial research through Addgene, a non-profit plasmiddistribution service.
We note that the zinc finger module reagents we describe shouldalso be useful for other applications of engineered zinc fingerproteins32,41–46. For example, many groups have shown thatmulti-finger domains built from these modules can be fusedto transcriptional regulatory domains (e.g., the NF-KB p65 domainor the KRAB repression domain) to create artificial transcriptionfactors capable of regulating specific endogenous genes20,21,40,47–50.In addition, various sets of these modules have been linkedtogether in random combinations to generate libraries ofartificial zinc finger transcription factors24,26,28,29,42,51. Theselibraries have been screened to identify members capable ofinducing specific cellular phenotypes, an approach that maybe useful for functional genomics studies52. The reagents wedescribe here should greatly simplify the construction of both
p
uor
G g
n ih si l
bu
P eru ta
N 600 2©
nat
ure
pro
toco
ls/
moc.er
ut an.
ww
w//:ptt
h
Gal11P
Gal4
F3 F2 F1RNA
polymeraselacZ
Figure 2 | Schematic of the bacterial two-hybrid reporter system.
Coexpression of the zinc finger-Gal11P and alpha-Gal4 hybrid proteins in a
B2H reporter strain leads to activated expression of a lacZ reporter gene if
the multi-finger domain interacts with a target DNA-binding site located
just upstream of the promoter39. This activation is mediated by recruitment
of RNA polymerase complexes (that have incorporated alpha-Gal4) to the
promoter by promoter-bound zinc finger-Gal11P proteins via interaction of
Gal11P and Gal4. A three-finger array is depicted as three colored spheres
(as in Fig. 1) and Gal11P represents a fragment of the yeast Gal11P protein
(aa 263–352)39. The alpha-Gal4 hybrid protein consists of a portion of the
E. coli RNA polymerase alpha-subunit (aa 1–248, encompassing the
amino-terminal domain and inter-domain linker) and a fragment of yeast
Gal4 (aa 58–97)39.
1638 | VOL.1 NO.3 | 2006 | NATURE PROTOCOLS
PROTOCOL

specific individual and combinatorial libraries of zinc finger arraysfor these applications.
Our standardized framework has also been adopted by theinternational group of scientists who participate in the Zinc FingerConsortium (http://www.zincfingers.org). All members of theConsortium have committed to ensuring that future vectorsdeveloped by their labs will be compatible with the plasmidframeworks described in this protocol. By standardizing all ofthe steps, from modular assembly to functional testing, to expres-sion in the ultimate target cells, we believe our reagents andprotocols will define the limitations and strengths of modularassembly and spur the further development of engineered zincfinger and ZFN technologies. We encourage scientists to reportboth positive and negative attempts at designing zinc fingerproteins using modular-assembly (or other techniques) on theZinc Finger Consortium newsgroup (http://www.zincfingers.org/listserv.htm).
MATERIALSREAGENTS.Plasmids and expression vectors (see REAGENT SETUP).Bacterial strain KJBAC1 (F- lacIq DhisB463 D(gpt-proAB-arg-lac)XIII
zaj::Tn10; strain available through Addgene as part of the Zinc FingerConsortium Expression Vector Kit v1.0)
.Bacterial strain Transformax EPI300 (F– mcrA D(mrr-hsdRMS-mcrBC)F80dlacZDM15 DlacX74 recA1 endA1 araD139 D(ara, leu)7697 galU galKl– rpsL nupG trfA dhfr; Epicentre)
.Bacterial strain XL1 Blue (recA1 endA1 gyrA96 thi-1 hsdR17 supE44 relA1 lac[F proAB lacIq lacZDM15 Tn10 (TetR)]; Stratagene)
.Restriction enzymes (all from New England Biolabs): AgeI, BamHI, BsaI,BsgI, EcoRI, HindIII, NotI, XbaI, XhoI, XmaI
.10� restriction enzyme buffers (New England Biolabs and Promega)
.32 mM S-adenosyl methionine (SAM) (New England Biolabs)
.T4 DNA ligase and associated reaction buffer (Promega)
.Calf intestinal phosphatase, CIP (Promega)
.Cloned Pfu polymerase and associated 10� reaction buffer (Stratagene)
.100% ethanol
.QIAquick gel extraction kit (Qiagen)
.QIAprep spin miniprep kit (Qiagen)
.LB medium and LB agar medium (Difco)
.SOB medium (Difco)
.SOC medium (SOB medium with 0.4% glucose)
.Sterile 10% glycerol and sterile 50% glycerol
.Ampicillin (100 mg ml–1 stock solution)
.Chloramphenicol (30 mg ml–1 stock solution in 100% ethanol)
.Kanamycin (30 mg ml–1 stock solution)
.Filter-sterilized 1 M IPTG (isopropyl-beta-D-thiogalactopyranoside; Sigma)
.Sterile 100 mM ZnSO4
.ONPG (o-nitrophenyl-beta-D-galactopyranoside; 4 mg ml–1)
.Z-buffer and Z-buffer with b-mercaptoethanol (see REAGENT SETUP)
.Popculture reagent (Novagen, cat. no. 71092)
.R-lysozyme (30,000 units/ml) and associated dilution buffer (Novagen,cat. no. 71110)
.Lysis master mix (10:1 mixture of Popculture reagent to diluted R-lysozyme,400 units/ml)
.Sterile 1 M MgCl2
.Solution A with 15% glycerol (10 mM MnCl2, 50 mM CaCl2, 10 mM MES,pH 6.3 (with KOH), 15% glycerol; filter sterilize and store at 4 1C)
.Sequencing primer T7-1: 5¢AATACGACTCACTATAG3¢
.Sequencing primer OK163: 5¢CGCCAGGGTTTTCCCAGTCACGAC3¢
EQUIPMENT.Electroporation cuvettes, 2 mm gap (BioRad).Electroporator (Micropulser, BioRad).Sterile 13 ml culture tubes (VWR, cat. no. 60818-667).Standard orbital shaker or roller wheel for growing bacterial cultures.96-well microtiter plates (Corning-Costar, cat. no. 3596).Deep-well 96-well blocks (optional; Greiner, cat. no. 780271).Microtiter plate reader with temperature control option.Sterile 250-ml centrifuge bottlesREAGENT SETUPPlasmids and expression vectors The following kits of plasmids (togetherwith detailed sequence and map information) are available through Addgene(http://www.addgene.org): The ‘‘Zinc Finger Consortium Modular AssemblyKit v1.0’’ which consists of the 141 different zinc finger modules (pc3XB-basedplasmids, AMPs, high copy Co1EI origin of replication). The ‘‘Zinc FingerConsortium Expression Vector Kit v1.0’’ contains the following plasmids:pGP-FB expression plasmid: AMPs, low-copy Co1EI origin of replication.
p
uor
G g
n ih si l
bu
P eru ta
N 600 2©
nat
ure
pro
toco
ls/
moc.er
ut an.
ww
w//:ptt
h
pc3XB-F1pc3XB-F1
pc3XB-F1/F2pc3XB-F1/F2
pc3XB-F1/F2/F3pc3XB-F1/F2/F3
pc3XB-F3pc3XB-F3
pc3XB-F2pc3XB-F2
F1 F2
F1
F1F2
F3
F2 F3
Xba
lX
ma
l Bsg I
Age I
Bam H
I
Xba
lX
ma
l Bsg I
Age I
Bam H
I
Xba
lX
ma
l Bsg I
Age I
Bam H
I
Xba l
Xma
l
Bsg I
Age I
Bam HI
Xba l
Xma l
Bsg I
Age I
Bam HI
Ligate
Ligate
AgeI/BamHI digestIsolate vector backbone
AgeI/BamHI digestIsolate vector backbone
Xmal/BamHI digestIsolate F2-encoding fragment
Xmal/BamHI digestIsolate F3-encoding fragment
Figure 3 | Overview of restriction digest-based modular assembly. Plasmids
pc3XB-F1, -F2 and -F3 encode individual hypothetical finger modules from the
archive cloned into plasmid pc3XB. Each finger coding sequence is flanked on
the 5’ end by unique XbaI and XmaI sites and on the 3’ end by unique AgeI,
BsgI and BamHI sites. The configuration of unique flanking restriction sites in
all pc3XB-based plasmids permits any two fingers (e.g., F1 and F2) to be
joined together by ligating a finger F1-encoding vector backbone (linearized
by digestion with AgeI and BamHI) to a finger F2-encoding fragment
(released from the plasmid by digestion with XmaI and BamHI). The resulting
plasmid encodes a two-finger (F1 followed by F2) array which again is flanked
on the 5’ end by XbaI and XmaI and on the 3’ end by AgeI, BsgI and BamHI.
(Note that ligation of compatible XmaI and AgeI overhangs destroys both
sites.) A third finger (F3) can be added to the array by ligating an
F1/F2-encoding AgeI/BamHI-digested vector backbone to a F3-encoding
XmaI/BamHI-digested fragment. Note that plasmid maps are not to scale.
NATURE PROTOCOLS | VOL.1 NO.3 | 2006 | 1639
PROTOCOL

pGP-FB-origBA expression plasmid: positive control for B2H b-galactosidaseassay; AMPs, ColEI origin of replication. pBAC-lacZ reporter plasmid: CAMs,primary F¢ and secondary oriV origins of replication. pBAC-BA-lacZ reporterplasmid: positive control reporter plasmid with binding site for the zinc fingerarray encoded by positive control plasmid pGP-FB-origBA; CAMs, primary F¢and secondary oriV origins of replication. pAC-KAN-alphaGal4 expressionplasmid: KANs, p15A origin of replication. pDW1775 plant ZFN expression
vector: AMPs, pST1374 ColEI origin of replication. High-copy mammalianZFN expression vector: AMPR, high-copy ColEI origin of replication.Z-buffer To prepare 1 l, use 16.1 g of Na2HPO4-7H20, 5.5 g of NaH2PO4-H20,0.75 g of KCl and 0.246 g of MgSO4-7H20 dissolved in ddH20; filter sterilize andstore at 22–28 1C.Z-buffer with b-mercaptoethanol Prepare fresh by adding 2.7 ml ofb-mercaptoethanol to every 1 ml of Z-buffer.
PROCEDUREIdentification of potential ZFN target sites using ZiFiT software � TIMING o1 d1| Visit the ZiFiT software website at http://bindr.gdcb.iastate.edu/ZiFiT/ (the site can also be accessed from the Zinc FingerConsortium website at http://www.zincfingers.org). From the Introduction page, select ZiFiT from the menu and login. First timeusers will need to create an account.
2| Select the option to design zinc finger nucleases. ZiFiT can also be used to design zinc finger arrays for other applications(e.g., fusion to transcriptional regulatory domains).
3| Check boxes to select the desired module set(s) (Barbas, Sangamo and/or ToolGen). Each module set recognizes differentsets of nucleotide triplets. A more complete description of the module sets is provided in the online instructions, which can beaccessed from the menu (see also Supplementary Table 1).
4| Paste the target nucleotide sequence to be analyzed into the text box labeled ‘sequence’. Sequences may be entered as rawdata or in FASTA format. Spaces and numbers are ignored.
5| Select the number of fingers desired for each array from the Left Array and Right Array drop down boxes. We recommendthe use of three-fingers in each array when using modular assembly. Note that each finger recognizes three base pairs.
6| Select the number of nucleotides between the two arrays from the Spacer drop down box. The double-strand cleavage occursin the spacer. For the ZFNs encoded by the expression vectors described in this protocol6, spacers six base pairs in length workbest, although spacers five base pairs in length also work5,6,10,53,54.
7| If you wish, choose the Advanced button to manually set limits on the number of GNN, TNN, ANN and/or CNN triplets for agiven site. Such limits may be desirable, for example, because most success has been achieved with predominantly GNN fingers.
8| Click Submit. The output provides the nucleotide position and sequence of the target site as well as a list of fingers forarray assembly. Each finger is color-coded to match the nucleotide triplet it recognizes (note that along the primary strand ofDNA [i.e.—the one primarily contacted by the zinc finger array], the amino-terminal finger binds the 3’ most DNA triplet whilethe carboxy-terminal finger binds the 5’ most triplet). If multiple fingers exist for a given triplet, all potential fingers are listed.The identity of each finger is indicated by a standard designation, which can be found in Supplementary Table 1.? TROUBLESHOOTING
Assembly of multi-finger arrays � TIMING 6–7 d9| Linking together F1 and F2. Using the output from ZiFiT as a guide, assemble desired modules into a three-finger domain.(Note that for simplicity in the steps below, we use the naming convention F1, F2, and F3 to specify finger positions within thecontext of a hypothetical three-finger domain where F1 is the amino-terminal finger, F2 is the middle finger and F3 is thecarboxy-terminal finger. This convention for finger position is used throughout this protocol.) Digest 1 mg of plasmid encodingF1 (pc3XB-F1) with AgeI and BamHI (as tabulated below) at 37 1C for 3 h. once digestion is complete, add 0.5 units calfintestinal phosphatase (CIP) and incubate at 37 1C for 5 min.
Also digest 2 mg of plasmid encoding F2 (pc3XB-F2) with XmaI and BamHI (as tabulated below) at 37 1C for 3 h (do not treatthis digest with CIP).
p
uor
G g
n ih si l
bu
P eru ta
N 600 2©
nat
ure
pro
toco
ls/
moc.er
ut an.
ww
w//:ptt
h
Component Amount
F1 Plasmid DNA (pc3XB-F1) 5 ml (1.0 mg)AgeI (5 U/ml) 0.5 mlBamHI (20 U/ml) 0.5 ml10x Buffer (NEB 4) 2.0 mlNuclease-free Water 12.0 mlTotal 20.0 ml
1640 | VOL.1 NO.3 | 2006 | NATURE PROTOCOLS
PROTOCOL

’ PAUSE POINT Digests, once completed and before addition of CIP, can be stored at 4 1C overnight or frozen indefinitelyat –20 1C.
10| Purify the digested vector backbone from pc3XB-F1 and the DNA fragment from pc3XB-F2 by electrophoresis on TAE 1.5%agarose gels. Purify the DNAs using a QIAquick Gel Extraction kit.’ PAUSE POINT Purified DNAs can be stored indefinitely at –20 1C.
11| Ligate purified pc3XB-F1 vector and pc3XB-F2 fragment together using T4 DNA ligase and buffer supplied by themanufacturer, as described in the table below. A 2:1 insert:vector ratio is typically used. As a control, we ligate pc3XB-F1vector alone without any added fragment. Incubate ligations at 15 1C overnight.
’ PAUSE POINT Completed ligations can be stored indefinitely at –20 1C.
12| Transform 1 ml of each of the ligations into 100 ml cells by electroporation. Use all 100 ml of cells in a 2 mm gapelectroporation cuvette at 2.5 kV, 200 Ohms, 25 mF. (See Box 1 for preparation of electrocompetent cells.)
13| Add 1 ml of SOC medium immediately following electroporation and transfer resuspended cells to a sterile 13 ml culturetube. Allow the cells to recover by growing them at 37 1C for 20 min.
14| Transfer the 1 ml of culture to a sterile microfuge tube and centrifuge for 30 s at 16,000g in a microcentrifuge. Remove allbut approximately 100-200 ml of the media and resuspend the cells. Plate the cells on an LB agar plate containing 100 mg ml–1
ampicillin. Incubate overnight (B18 h) at 37 1C.
15| If the transformation of the actual ligation (i.e., the ligation with the fragment) yields at least three-fold more coloniesthan that of the control ligation, inoculate single colonies from the actual ligation/transformation plate into 3 ml of LBcontaining 100 mg ml–1 ampicillin and grow overnight (14–18 h) at 37 1C for plasmid miniprep isolation. We typicallyinoculate at least two colonies from each actual ligation plate.? TROUBLESHOOTING
16| Isolate plasmid DNA from overnight cultures using a QIAprep Spin Miniprep Kit.’ PAUSE POINT Purified DNAs can be stored indefinitely at �20 1C.
p
uor
G g
n ih si l
bu
P eru ta
N 600 2©
nat
ure
pro
toco
ls/
moc.er
ut an.
ww
w//:ptt
h
Component Amount
F2 Plasmid DNA (pc3XB-F2) 5 ml (2.0 mg)XmaI (10 U/ml) 0.5 mlBamHI (20 U/ml) 0.5 ml10x Buffer (NEB 4) 2.0 mlNuclease-free Water 12.0 mlTotal 20.0 ml
Component Volume
Vector DNA (pc3XB-F1) 5.5 mlInsert (F2 fragment) DNA 5.5 ml10X Ligation Enzyme Buffer 2 mlNuclease-free Water 6 mlT4 DNA Ligase (1–3 Weiss units/ml) 1 mlTotal 20 ml
BOX 1 | PREPARING COMPETENT CELLS FOR TRANSFORMATION BY ELECTROPORATION
1. Inoculate a fresh single colony of an appropriate bacterial strain (e.g., XL1 Blue) in 5 ml LB and grow overnight (14–18 h) at 37 1C.2. The next morning, add the saturated overnight culture to 500 ml LB in a sterile 1-l flask. Grow for 2–3 h with agitation at 37 1C untilOD600 ¼ 0.4–0.6.3. Pellet cells in two sterile 250 ml centrifuge tubes at 5,000–6,000g at 4 1C in a tabletop centrifuge.4. Pour off media and resuspend each cell pellet in 50 ml ice cold sterile water.5. Combine the resuspended cells and centrifuge at 5,000–6,000g at 4 1C. Repeat Steps 4 and 5 two more times using 100 ml of ice cold sterile water.6. After the final wash, resuspend the pellet with 25 ml of ice cold, sterile 10% glycerol. Pellet cells by centrifuging at 5,000–6,000g at 4 1C.Pour off supernatant and resuspend pellet in 3 ml ice cold, sterile 10% glycerol.7. Aliquot 100 ml cells into 0.5 ml microcentrifuge tubes. Use immediately for electroporation and freeze the remaining tubes at –80 1C.’ PAUSE POINT Competent cells may be stored for approximately 3 months at –80 1C.
NATURE PROTOCOLS | VOL.1 NO.3 | 2006 | 1641
PROTOCOL

17| Digest candidate plasmid DNAs with XbaI and BamHI at 37 1C for 3 h, as detailed below, and visualize digestion productson a 1.5% agarose gel. The desired plasmid should yield a B200 bp fragment. We will refer to this plasmid as pc3XB-F1/F2in the protocol steps below.
18| Linking together F1/F2 and F3. Digest 1 mg of plasmid encoding the F1-F2 array (pc3XB-F1/F2) with AgeI and BamHI at37 1C for 3 h, as detailed below, and once digestion is complete add 0.5 units CIP to the reaction and incubate at 37 1C for 5 min.
Digest 2 mg of plasmid encoding F3 (pc3XB-F3) with XmaI and BamHI at 37 1C for 3 h, as detailed below.
’ PAUSE POINT Digests, once completed and before addition of CIP, can be stored at 4 1C overnight or frozen indefinitely at –20 1C.
19| Purify the digested vector backbone from pc3XB-F1/F2 and the DNA fragment from pc3XB-F3 by electrophoresis on TAE1.5% agarose gels. Purify the DNAs using a QIAquick Gel Extraction kit.’ PAUSE POINT Purified DNAs can be stored indefinitely at –20 1C.
20| Ligate purified pc3XB-F1/F2 vector and pc3XB-F3 fragment together using T4 DNA ligase and buffer supplied by themanufacturer, as detailed below. A 2:1 insert:vector ratio is typically used. As a control, ligate pc3XB-F1/F2 vector alonewithout any added fragment. Incubate ligations at 15 1C overnight.
’ PAUSE POINT Completed ligations can be stored indefinitely at �20 1C.
21| Transform 1 ml of each of the ligations into 100 ml cells by electroporation and plate on LB plates containing ampicillin asin Steps 12, 13 and 14 above. Incubate overnight (14-18 h) at 37 1C.
22| If the transformation of the actual ligation yields at least three-fold more colonies than that of the control ligation, usesingle colonies from the actual ligation/transformation plate to inoculate two 3-ml cultures of LB containing 100 mg ml–1
ampicillin and grow overnight (14–18 h) at 37 1C for plasmid miniprep isolation.? TROUBLESHOOTING
p
uor
G g
n ih si l
bu
P eru ta
N 600 2©
nat
ure
pro
toco
ls/
moc.er
ut an.
ww
w//:ptt
h
Component Amount
Candidate plasmid DNA 5 ml (0.5-2.0 mg)XbaI (20 U/ml) 0.5 mlBamHI (20 U/ml) 0.5 ml10x Buffer (NEB 2) 2.0 mlNuclease-free water 12.0 mlTotal 20.0 ml
Component Amount
F1/F2 Plasmid DNA (pc3XB-F1/F2) 5 ml (1.0 mg)AgeI (5 U/ml) 0.5 mlBamHI (20 U/ml) 0.5 ml10x Buffer (NEB 4) 2.0 mlNuclease-free water 12.0 mlTotal 20.0 ml
Component Amount
F3 Plasmid DNA (pc3XB-F3) 5 ml (2.0 mg)XmaI (10 U/ml) 0.5 mlBamHI (20 U/ml) 0.5 ml10x Buffer (NEB 4) 2.0 mlNuclease-free water 12.0 mlTotal 20.0 ml
Component Volume
Vector DNA (pc3XB-F1/F2) 5.5 mlInsert (F3 fragment) DNA 5.5 ml10X Ligation Enzyme Buffer 2 mlNuclease-free water 6 mlT4 DNA Ligase (1–3 Weiss units/ml) 1 mlTotal 20 ml
1642 | VOL.1 NO.3 | 2006 | NATURE PROTOCOLS
PROTOCOL

23| Isolate plasmid DNA from overnight cultures using a QIAprep Spin Miniprep Kit.’ PAUSE POINT Purified DNAs can be stored indefinitely at –20 1C.
24| Digest candidate plasmid DNAs with XbaI and BamHI at 37 1C for 3 h and visualize digestion on a 1.5% agarose gel, asdetailed below. The desired plasmid should yield a B300 bp fragment upon digestion with XbaI and BamHI. Although no PCRis used to construct the multi-finger array plasmid, we recommend that it be sequenced to verify the presence of thedesired modules (particularly when constructing multiple arrays). Sequencing can be performed using primer T7-1 which isa sense-strand primer that binds B34 base pairs upstream of the XbaI site. We will refer to the sequence-verified plasmidas pc3XB-F1/F2/F3 in the protocol steps below.
’ PAUSE POINT Purified DNAs can be stored indefinitely at –20 1C.
Testing the DNA-binding activity of a three-finger domain using the B2H reporter assay � TIMING 12–14 d25| Constructing a low-copy expression vector encoding a zinc finger-Gal11P hybrid protein. An overview of the strategy formaking this expression plasmid is shown in Figure 4. Digest the pc3XB-F1/F2/F3 plasmid constructed in Step 24 above withXbaI and BsgI, at 37 1C for 3 h, as detailed below. Purify the B300 bp fragment by electrophoresis through a 1.5% agarose geland isolate the DNA using a QIAquick Gel Extraction kit.
A final concentration of 80 mM S-adenosyl methionine (SAM)is a required buffer component for the restriction endonucleaseBsgI. 32 mM SAM is supplied with BsgI and a sample should bediluted in water to 1 mM prior to use.’ PAUSE POINT Purified DNA can be stored indefinitelyat –20 1C.
26| Digest the B2H expression vector pGP-FB (Fig. 4) withXbaI and BsgI at 37 1C, as detailed below, and purify theB3,400 bp vector backbone by electrophoresis through a1% agarose gel and isolate the DNA using a QIAquick GelExtraction kit (Qiagen).
p
uor
G g
n ih si l
bu
P eru ta
N 600 2©
nat
ure
pro
toco
ls/
moc.er
ut an.
ww
w//:ptt
h
Component Amount
Candidate plasmid DNA 5 ml (0.5–2.0 mg)XbaI (20 U/ml) 0.5 mlBamHI (20 U/ml) 0.5 ml10x Buffer (NEB 2) 2.0 mlNuclease-free water 12.0 mlTotal 20.0 ml
Component Amount
F1/F2/F3 Plasmid DNA (pc3XB-F1/F2/F3) 5 ml (0.5-2.0 mg)XbaI (20 U/ml) 0.5 mlBsgI (3 U/ml) 0.5 ml10x Buffer (NEB 4) 2.0 mlSAM (1 mM) 1.6 mlNuclease-free Water 10.4 mlTotal 20.0 ml pGP-FBpc3XB-F1/F2/F3pc3XB-F1/F2/F3 pGP-FB
pGP-FB-F1/F2/F3pGP-FB-F1/F2/F3
pAC-KAN-alphaGal4pAC-KAN-alphaGal4
Bam HIAge IBsg I
Xma IXbaI
Xba I
Xba
IB
sgI B
anH
I
F1F2
F3
lacU
V5 P
lacU
V5
P
lacU
V5
P
GaI11P
GaI11P
Flag
Flag F1F2
F3
Ligate
Xba l/Bsg l digestIsolate vector backbone
Xbal/Bsg l digestIsolate fragment
Bsg
IB
amH
I
lpp
P
RNAP alpha NTD + linke
rG
al4
B2H reporter strain
Figure 4 | Strategy for constructing B2H expression vectors and
transformation of B2H reporter strains. Zinc finger arrays assembled in the
pc3XB plasmid can be cloned directly into vectors designed to express zinc
finger arrays as a Gal11P hybrid protein (for use in B2H assays) using the
unique XbaI and BsgI restriction sites. To perform B2H assays, the resulting
plasmid (pGP-FB-F1/F2/F3) is co-transformed with the pAC-KAN-alphaGal4
plasmid into a ‘‘B2H reporter strain’’ harboring reporter plasmid
pBAC-ZFBS-lacZ (ZFBS ¼ zinc finger binding site). Note that the
pGP-FB-F1/F2/F3, the pAC-KAN-alphaGal4 and the pBAC-ZFBS-lacZ
plasmids confer resistance to ampicillin, kanamycin and chloramphenicol,
respectively. Note that plasmid maps are not to scale.
NATURE PROTOCOLS | VOL.1 NO.3 | 2006 | 1643
PROTOCOL

A final concentration of 80 mM S-adenosyl methionine (SAM) is a required buffer component for the restriction endonucleaseBsgI. 32 mM SAM is supplied with Bsg I and a sample should be diluted in water to 1 mM prior to use.’ PAUSE POINT Purified DNA can be stored indefinitely at –20 1C.
27| Ligate the DNA fragments purified in Steps 25 and 26 together, as detailed below. As a control, ligate pGP-FB vector alonewithout any added fragment. Incubate ligations at 15 1C overnight.
Transform E. coli strain XL1 Blue with the actual and control ligations and plate on LB plates supplemented with 100 mg ml–1 ampicillin.m CRITICAL STEP Cells used for transformation in this step must express the lacIq allele of lac repressor. This ensures repression ofthe strong lacUV5 promoter, which directs expression of the zinc finger-Gal11P hybrid protein encoded by plasmid pGP-FB (Fig. 4).
28| If the transformation of the actual ligation yields at least three-fold more colonies than that of the control ligation, usesingle colonies from the actual ligation/transformation plate to inoculate two 10-ml cultures of LB containing 100 mg ml–1
ampicillin and grow overnight (14–18 h) at 37 1C for plasmid miniprep isolation.? TROUBLESHOOTING
29| Isolate plasmid DNA from 10 ml of overnight culture using a QIAprep Spin Miniprep Kit.m CRITICAL STEP Plasmid DNA should be isolated from a 10 ml culture because the pGP-FB plasmid is a low copy number plasmid.’ PAUSE POINT Purified DNAs can be stored indefinitely at –20 1C.
30| Digest candidate plasmid DNAs with XbaI and BamHI at 37 1C for 3 h, as detailed below.
Visualize digestion on a 1.5% agarose gel. The desired plasmid should yield a B300 bp fragment. This plasmid expresses a zincfinger-Gal11p fusion protein from a strong lacUV5 promoter that can be repressed by lac repressor. We will refer to this plasmidas pGP-FB-F1/F2/F3 in the protocol steps below.
31| Construction of a single-copy reporter vector bearing a target DNA-binding site for a multi-finger domain positionedupstream of a lacZ reporter gene. Digest single-copy vector pBAC-lacZ with BsaI, at 50 1C for 1 h, as detailed below.
p
uor
G g
n ih si l
bu
P eru ta
N 600 2©
nat
ure
pro
toco
ls/
moc.er
ut an.
ww
w//:ptt
h
Component Volume
Vector DNA (pGP-FB) 5.5 mlInsert (F1/F2/F3 fragment) DNA 5.5 ml10X Ligation Enzyme Buffer 2 mlNuclease-free water 6 mlT4 DNA Ligase (1–3 Weiss units/ml) 1 mlTotal 20 ml
Component Amount
Candidate plasmid DNA 5 ml (0.5–2.0 mg)XbaI (20 U/ml) 0.5 mlBamHI (20 U/ml) 0.5 ml10x Buffer (NEB 2) 2.0 mlNuclease-free water 12.0 mlTotal 20.0 ml
Component Amount
Plasmid DNA (pBAC-lacZ) 5 ml (0.5–2.0 mg)BsaI (10 U/ml) 0.5 ml10x Buffer (NEB 3) 2.0 mlNuclease-free water 12.5 mlTotal 20.0 ml
Component Amount
Plasmid DNA (pGP-FB) 5 ml (0.5–2.0 mg)XbaI (20 U/ml) 0.5 mlBsgI (3 U/ml) 0.5 ml10x Buffer (NEB 4) 2.0 mlSAM (1 mM) 1.6 mlNuclease-free water 10.4 mlTotal 20.0 ml
1644 | VOL.1 NO.3 | 2006 | NATURE PROTOCOLS
PROTOCOL

Purify the B11,100 bp vector backbone by electrophoresisthrough a 0.8% agarose gel and isolate the DNA using aQIAquick Gel Extraction kit.m CRITICAL STEP BsaI digests should be incubatedat 50 1C.’ PAUSE POINT Purified DNA can be stored indefinitelyat –20 1C.
32| Incubate purified, BsaI-digested pBAC-lacZ vectorbackbone with Pfu polymerase in the presence of the dCTPonly (i.e., omit dATP, dGTP and dTTP) at 72 1C for 10 min.The combined 5’ to 3’ polymerase activity and 3’ to 5’exonuclease activity of Pfu will generate the extendedoverhangs illustrated in the bottom part of Figure 5(see also ref. 55)
33| Design and synthesize a pair of oligonucleotides bearingthe target DNA-binding site for a multi-finger domain of interest(see Box 2 and top part of Fig. 5). Anneal 50 fmol of each ofthese oligonucleotides together in 100 ml of annealing buffer(40 mM Tris, pH 8, 20 mM MgCl2, 50 mM NaCl) by heating to 95 1Cfor 2 min and then allowing the mixture to cool slowly (over B1h) to 25 1C. Annealed oligonucleotides will form a short double-stranded DNA fragment with the overhangs shown in Figure 5.
34| Ligate the pBAC-lacZ vector backbone from Step 32 tothe annealed oligonucleotides from Step 33, as detailed below.As a control, perform a ligation with the pBAC-lacZ vectorbackbone only. Incubate ligation reactions at 15 1C overnight.
Transform both ligations into E. coli strain Transformax EPI300 (Epicentre) using standard chemical transformation. Platetransformations on LB plates supplemented with 12.5 mg ml–1 chloramphenicol and incubate overnight (14–18 h) at 37 1C.m CRITICAL STEP The pBAC-lacZ plasmid is a mini-F’ that can be replicated in standard E. coli strains, but because it is maintainedas a single copy episome, it gives low DNA yields. pBAC-lacZ also contains a second, higher copy number origin of replication (oriV)that is only active in the presence of a trans-acting factor encoded by the trfA gene. Transformax EPI300 cells express trfA from aninducible promoter that we have found is inducible with arabinose (S.T.B. and J.K.J., unpublished data). When the trfA gene isinduced in Transformax EPI300 cells, the copy number of pBAC-lacZ plasmid is increased in the cells and reasonable yields of plasmidcan be obtained using a standard miniprep procedure.
35| If the transformation of the actual ligation yields at least three-fold more colonies than that of the control ligation,use single colonies from the actual ligation/transformation plate to inoculate two 3 ml cultures of LB containing 12.5 mg ml–1
chloramphenicol and grow overnight (14–18 h) at 37 1C.? TROUBLESHOOTING
p
uor
G g
n ih si l
bu
P eru ta
N 600 2©
nat
ure
pro
toco
ls/
moc.er
ut an.
ww
w//:ptt
h
Component Volume
Vector DNA (pBAC-lacZ) 5.5 mlInsert DNA (annealed oligos) 5.5 ml10X Ligation Enzyme Buffer 2 mlNuclease-free water 6 mlT4 DNA Ligase (1–3 Weiss units/ml) 1 mlTotal 20 ml
Substitute target site
Synthesize & anneal oligos
5′3′
5′
5′ 3′3′ 5′
3′3′5′
3′5′
LigateReporter plasmidpBAC-ZFBS-lacZ
5′3′
5′3′
5′3′
5′3′
3′5′
3′5′
3′5′
3′5′
lacZ
lacZ
lacZ
Treat withPfu + dCTP
Digest with BsaI
pBAC-lacZ
pBAC-lacZpBAC-lacZ
pBAC-lacZpBAC-lacZ
pBAC-lacZpBAC-lacZ
BsaI BsaI
Figure 5 | Strategy for constructing B2H reporter vectors. The top half of the
figure illustrates the design and annealing of oligonucleotides comprising the
zinc finger binding site as described in Box 2. The bottom half of the figure
illustrates digestion of the plasmid pBAC-lacZ with BsaI and generation of
extended overhangs by treatment with Pfu polymerase in the presence of a
single deoxynucleotide (dCTP). Treatment with Pfu under these conditions
will generate the specific overhangs shown that are complementary to the
overhangs of the annealed oligonucleotide cassette. Ligation of the vector
backbone to the annealed oligonucleotides will generate the desired
pBAC-ZFBS-lacZ reporter vector. Note that plasmid maps are not to scale.
Component Volume
Vector DNA (BsaI-digested vector) 10 ml10 mM dCTP 2 ml10� Pfu reaction buffer 2 mlNuclease-free water 4.8 mlCloned Pfu polymerase 1.2 mlTotal 20 ml
NATURE PROTOCOLS | VOL.1 NO.3 | 2006 | 1645
PROTOCOL

36| Inoculate 1 ml of each overnight culture into 9 ml of LB containing 14 mg ml–1 chloramphenicol and 1.1 mM arabinose.Grow cultures for 6 h at 37 1C and then harvest cells and isolate plasmid DNA using a QIAprep Spin Miniprep Kit.m CRITICAL STEP Subculturing the cells containing the plasmids in arabinose is critical to induce expression of the trfA geneproduct, which in turn increases the copy number of the pBAC-lacZ plasmid.’ PAUSE POINT Plasmids can be stored indefinitely at –20 1C.
37| Digest the candidate reporter plasmids (and the original pBAC-lacZ plasmid as a control) with EcoRI and HindIII at 37 1Cfor 3 h, as detailed below.
Visualize the digestions on a 2% agarose gel. Candidates that have taken up the annealed oligonucleotides should yieldfragments of sizes 159, 3,007 and 7,963 base pairs, whereas the original pBAC-lacZ plasmid should yield fragments ofsizes 182, 3,007 and 7,963 base pairs.
38| Confirm the sequence of the target DNA binding site in the candidate reporter plasmid by sequencing with primer OK163(this primer is an anti-sense primer that anneals B150 bp downstream of the binding site). We will refer to the sequence-confirmed reporter plasmid as the pBAC-ZFBS-lacZ plasmid (ZFBS ¼ Zinc Finger Binding Site) in subsequent steps of the protocol.
39| Transformation of E. coli cells with B2H reporter and expression vectors. Transform strain KJBAC1 (which constitutivelyexpresses high levels of lac repressor) with the pBAC-ZFBS-lacZ reporter plasmid. Also transform strain KJBAC1 with the positivecontrol reporter plasmid pBAC-BA-lacZ (containing a binding site for the zinc finger array encoded by positive control expressionplasmid pGP-FB-origBA). Plate transformations on LB plates containing 12.5 mg ml–1 chloramphenicol and incubate overnight at37 1C. We refer to the resulting KJBAC1 transformants as the ‘‘ZFBS B2H reporter strain’’ (Fig. 4) and the positive control ‘‘BAB2H reporter strain.’’
40| Pick colonies from each of the transformation plates and inoculate each into a 3 ml LB culture with 12.5 mg ml–1
chloramphenicol. Grow cultures overnight (12–16 h) with agitation at 37 1C.
41| Make chemically competent cells from each of the B2H reporter strain overnight cultures (see Box 3 for protocol).Also prepare glycerol stocks of each of the B2H reporter strains for long-term storage at –80 1C.’ PAUSE POINT Competent B2H reporter strain cells can be stored and frozen indefinitely at –80 1C.
42| Doubly transform the ZFBS B2H reporter strain with the pGP-FB-F1/F2/F3 expression vector prepared in Step 30 above andthe compatible pAC-KAN-alphaGal4 expression plasmid encoding the alpha-Gal4 hybrid protein (Fig. 4). As a positive control,doubly transform the BA B2H reporter strain with the pGP-FB-origBA and pAC-KAN-alphaGal4 expression plasmids. As a negativecontrol for assaying basal transcription of the lacZ reporter gene, doubly transform both the ZFBS and BA B2H reporter strains
p
uor
G g
n ih si l
bu
P eru ta
N 600 2©
nat
ure
pro
toco
ls/
moc.er
ut an.
ww
w//:ptt
h
Component Amount
Candidate plasmid DNA 5 ml (0.5–2.0 mg)EcoRI (20 U/ml) 0.5 mlHindIII (20 U/ml) 0.5 ml10x Buffer (NEB 2) 2.0 mlNuclease-free water 12.0 mlTotal 20.0 ml
BOX 2 | DESIGNING OLIGONUCLEOTIDES TO CREATE THE TARGET DNA-BINDING SITEFOR THE B2H REPORTER VECTOR
1. In the double-stranded DNA fragment shown in Figure 5, replace the 9-bp XXXXXXXXX sequence with the 9-bp target site of the three-fingerprotein to be tested. Replace the single bases (x) that flank the 9-bp XXXXXXXXX sequence with the bases that flank the target site in thegenomic sequence.m CRITICAL STEP Note that the XXXXXXXXX site is positioned so that the F3 triplet is on the ‘‘left’’ and the F1 triplet is on the ‘‘right’’ in thedouble-stranded fragment shown in Figure 5. Replacement of the flanking ‘‘x’’ bases with the bases present in the genomic target sequence isimportant because the identities of these adjacent bases can influence binding of the multi-finger domain to the target site. Note also thatsequences should be inspected for the presence of 5¢GATC3¢ sequences which can be methylated by E. coli Dam methylase. Methylation ofadenine in these sequences can inhibit DNA-binding.2. Synthesize oligonucleotides corresponding to the first 29 bases of the top strand of the fragment (starting from the 5’ end) and to the first 26bases of the bottom strand (starting from the 5’ end) (Fig. 5).
1646 | VOL.1 NO.3 | 2006 | NATURE PROTOCOLS
PROTOCOL

with the pGP-FB (expressing the Gal11P fragment with no accompanying zinc finger domain) and pAC-KAN-alphaGal4 plasmids.Plate all transformations on LB plates containing 100 mg ml–1 ampicillin, 30 mg ml–1 kanamycin and 12.5 mg ml–1 chlorampheni-col and incubate overnight (14–18 h) at 37 1C. The pAC-KAN-alphaGal4 expresses the alpha-Gal4 hybrid protein from a strongtandem lpp/lacUV5 promoter (that is repressed by lac repressor) and confers resistance to kanamycin. The p15A origin of replica-tion present in plasmid pAC-KAN-alphaGal4 is compatible with the BAC reporter and zinc finger-Gal11P expression plasmids.m CRITICAL STEP The order in which KJBAC1 is transformed with plasmids is important. Although one can readily obtaintransformants of KJBAC1 that harbor pAC-KAN-alphaGal4, we have sometimes encountered difficulties with introducing thereporter and zinc finger-Gal11P expression plasmids into these cells. However, the procedure works consistently as described(i.e., transforming KJBAC1 first with the reporter plasmid and then double-transforming with the zinc finger-Gal11P expressionplasmid and pAC-KAN-alphaGal4; Fig. 4).’ PAUSE POINT Transformants of the B2H reporter strain can be stored at 4 1C but should be assayed for b-galactosidaseactivity within a few days. We find that transformants often do not grow well, or at all, when left at 4 1C for more than a week.? TROUBLESHOOTING
43| b-galactosidase assay of doubly transformed B2H reporter strain cells. For each transformant to be assayed, inoculatetwo overnight cultures (each from an independent colony) in LB containing 100 mg ml–1 ampicillin, 30 mg ml–1 kanamycin,12.5 mg ml–1 chloramphenicol, 10 mM ZnSO4 and 500 mM IPTG. Grow overnight (14–18 h) with agitation at 37 1C. Culturescan be grown either in standard sterile glass or plastic culture tubes or in 96-well blocks. For growth in deep well blocks,we recommend shaking on a platform with a sufficiently small throw radius to ensure uniform and adequate agitation ofeach well in the block (e.g., the Microtitertron orbital shaker, Appropriate Technical Resources)56.
44| Subculture saturated overnight cultures from Step 43 by diluting them 1:40 into pre-warmed LB-containing 100 mg ml–1
ampicillin, 30 mg ml–1 kanamycin, 12.5 mg ml–1 chloramphenicol, 10 mM ZnSO4 and 500 mM IPTG. Monitor the growth of culturesby measuring OD600 (relative to a media only blank) on a spectrophotometer and harvest them for lysis when they reach logphase (OD600 ¼ 0.3-0.8). Record the OD600 value at which cultures are lysed.? TROUBLESHOOTING
45| Lyse log-phase subcultures from Step 44 in a 96-well microtiter plate by adding 100 ml of culture to 11 ml of Lysis MasterMix56 (already in the plate) and mixing well by pipetting up and down. Allow lysis to proceed for a minimum of 15 min at22–28 1C.’ PAUSE POINT The activity of b-galactosidase is stable in the cell lysates for at least 18 h when stored at room temperature56.Lysates should be covered to prevent evaporation.
46| Set up b-galactosidase assay by adding 15 ml of lysate to a microtiter plate well containing 135 ml of Z buffer withb-mercaptoethanol and 30 ml of 4 mg ml–1 ONPG and mixing well. Perform duplicate assays for each lysate.
47| Place microtiter plate containing b-galactosidase assay reactions in a microtiter plate reader with temperature control(e.g., Biorad Model 680 Microplate Reader). Incubate reactions at 28 1C and take timed serial measurements of absorbanceat 420 nm. Calculate the velocity of ONPG cleavage (v) by plotting A420 vs. time and calculating the slope of the line.m CRITICAL STEP Many microtiter plate readers can be programmed to take absorbance measurements at fixed intervals and tocalculate the velocity of ONPG cleavage. We typically take measurements every 10–30 s. Reactions should not be allowed to proceedfor more than 30 min as substrate can become limiting.
48| Calculate the units of b-galactosidase for each assay using the following formula:
V�1; 000=ðOD600Þ
p
uor
G g
n ih si l
bu
P eru ta
N 600 2©
nat
ure
pro
toco
ls/
moc.er
ut an.
ww
w//:ptt
h
BOX 3 | PREPARING COMPETENT B2H REPORTER STRAINS FOR CHEMICALTRANSFORMATION
1. The next morning, subculture 1 ml of the saturated overnight culture from Step 40 into 50 ml LB containing 12.5 mg ml–1 chloramphenicol and15 mM MgCl2 in a 250 ml flask. Grow for 1 h with agitation at 37 1C.2. Pellet cells in a sterile 50 ml conical tube by centrifuging at 3,000–6,000g in a tabletop centrifuge.3. Pour off media and resuspend cell pellet in 3 ml of ice-cold Solution A with 15% glycerol.4. Leave sitting on ice for 20 min and then aliquot into sterile microcentrifuge tubes.5. Snap-freeze the tubes on dry ice.’ PAUSE POINT Competent cells may be stored indefinitely at –80 1C.
NATURE PROTOCOLS | VOL.1 NO.3 | 2006 | 1647
PROTOCOL

We include the 1,000 multiplier to make all units larger than 1. The final value obtained for each B2H reporter straintransformant is the average of duplicate cultures, which in turn are measured in duplicate.m CRITICAL STEP If a microtiter plate reader is not available, the b-galactosidase assay can be performed in glass tubes as anendpoint assay using a standard spectrophotometer as previously described57.
49| Calculate the fold-activation mediated by each zinc finger protein tested by comparing b-galactosidase units obtained inthe presence of the zinc finger-Gal11P hybrid protein with the units obtained in the presence of the Gal11P control. In ourexperience, zinc finger domains that mediate three-fold activation or higher will have a high probability of generating activeZFNs in human cell-based gene targeting assays. The zinc finger array encoded by positive control plasmid pGP-FB-origBA shouldactivate expression of the lacZ reporter gene on plasmid pBAC-BA-lacZ approximately three-fold.
Construction of ZFN expression vectors � TIMING 3–5 d50| As outlined in Figure 6, DNA sequences encoding zinc finger arrays that show significant activity in the B2H system can betransferred by standard restriction digest-based subcloning techniques to specially designed expression vectors that permit theirexpression as zinc finger nucleases in plant or human cells. Digest the desired pGP-FB-F1/F2/F3 plasmid with XbaI and BamHI(these unique sites flank the zinc finger coding sequence) at 37 1C for 3 h, as detailed below.
Isolate the B300 bp fragment by electrophoresis on a 1.5% agarose gel followed by purification using a QIAquick Gel Extraction kit.
51| Digest the recipient ZFN expression vector with XbaI and BamHI at 37 1C for 3 h, as detailed below.
Once digestion is complete, add 0.5 units CIP and incubate at 37 1C for 5 min. Isolate vector backbone by electrophoresison a 1.5% agarose gel. Purify the vector using a QIAquick Gel Extraction kit. For expression in plants, use vector pDW1775(see Fig. 6). For expression in mammalian cells, use vector pST1374 (see Fig. 6).
52| Ligate the purified expression vector backbone from Step 51 to the purified zinc finger-encoding fragment from Step 50,as detailed below. As a control, also perform a ligation with only the vector backbone. Incubate ligation reactions at 15 1C overnight.
Transform both ligations into E. coli strain XL1 Blue and plate on LB plates supplemented with 100 mg ml–1 ampicillin.
53| If the transformation of the actual ligation yields at least three-fold more colonies than that of the control ligation,use single colonies from the actual ligation/transformation plate to inoculate two 3 ml cultures of LB containing 100 mg ml–1
ampicillin and grow overnight (14–18 h) at 37 1C.? TROUBLESHOOTING
54| Isolate plasmid DNA from overnight cultures using a QIAprep Spin Miniprep Kit.’ PAUSE POINT Plasmids can be stored indefinitely at –20 1C.
p
uor
G g
n ih si l
bu
P eru ta
N 600 2©
nat
ure
pro
toco
ls/
moc.er
ut an.
ww
w//:ptt
h
Component Amount
Expression vector DNA 5 ml (0.5-2.0 mg)XbaI (20 U/ml) 0.5 mlBamHI (20 U/ml) 0.5 ml10x Buffer (NEB 2) 2.0 mlNuclease-free water 12.0 mlTotal 20.0 ml
Component Amount
Plasmid DNA (pGP-FB-F1/F2/F3) 5 ml (0.5–2.0 mg)XbaI (20 U/ml) 0.5 mlBamHI (20 U/ml) 0.5 ml10x Buffer (NEB 2) 2.0 mlNuclease-free water 12.0 mlTotal 20.0 ml
Component Volume
Vector DNA (expression vector) 5.5 mlInsert DNA (FB-F1/F2/F3 fragment) 5.5 ml10X Ligation Enzyme Buffer 2 mlNuclease-free water 6 mlT4 DNA Ligase (1–3 Weiss units/ml) 1 mlTotal 20 ml
1648 | VOL.1 NO.3 | 2006 | NATURE PROTOCOLS
PROTOCOL
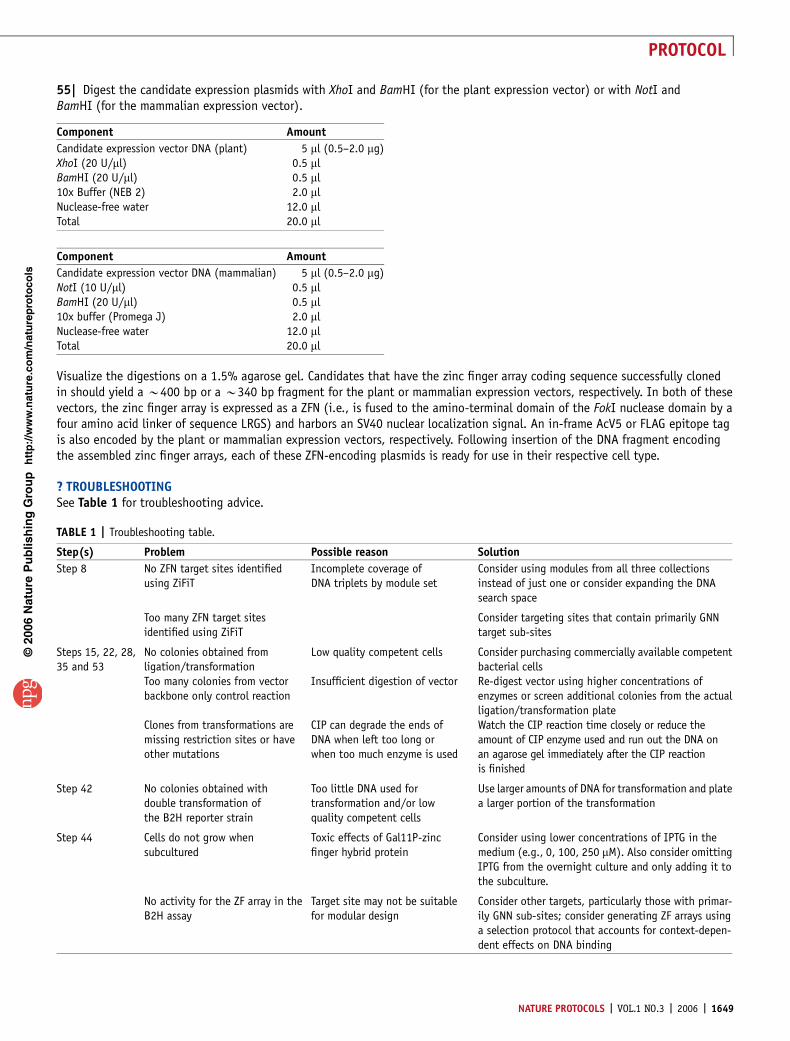
55| Digest the candidate expression plasmids with XhoI and BamHI (for the plant expression vector) or with NotI andBamHI (for the mammalian expression vector).
Visualize the digestions on a 1.5% agarose gel. Candidates that have the zinc finger array coding sequence successfully clonedin should yield a B400 bp or a B340 bp fragment for the plant or mammalian expression vectors, respectively. In both of thesevectors, the zinc finger array is expressed as a ZFN (i.e., is fused to the amino-terminal domain of the FokI nuclease domain by afour amino acid linker of sequence LRGS) and harbors an SV40 nuclear localization signal. An in-frame AcV5 or FLAG epitope tagis also encoded by the plant or mammalian expression vectors, respectively. Following insertion of the DNA fragment encodingthe assembled zinc finger arrays, each of these ZFN-encoding plasmids is ready for use in their respective cell type.
? TROUBLESHOOTINGSee Table 1 for troubleshooting advice.
p
uor
G g
n ih si l
bu
P eru ta
N 600 2©
nat
ure
pro
toco
ls/
moc.er
ut an.
ww
w//:ptt
h
Component Amount
Candidate expression vector DNA (mammalian) 5 ml (0.5–2.0 mg)NotI (10 U/ml) 0.5 mlBamHI (20 U/ml) 0.5 ml10x buffer (Promega J) 2.0 mlNuclease-free water 12.0 mlTotal 20.0 ml
Component Amount
Candidate expression vector DNA (plant) 5 ml (0.5–2.0 mg)XhoI (20 U/ml) 0.5 mlBamHI (20 U/ml) 0.5 ml10x Buffer (NEB 2) 2.0 mlNuclease-free water 12.0 mlTotal 20.0 ml
TABLE 1 | Troubleshooting table.
Step(s) Problem Possible reason Solution
Step 8 No ZFN target sites identifiedusing ZiFiT
Incomplete coverage ofDNA triplets by module set
Consider using modules from all three collectionsinstead of just one or consider expanding the DNAsearch space
Too many ZFN target sitesidentified using ZiFiT
Consider targeting sites that contain primarily GNNtarget sub-sites
Steps 15, 22, 28,35 and 53
No colonies obtained fromligation/transformation
Low quality competent cells Consider purchasing commercially available competentbacterial cells
Too many colonies from vectorbackbone only control reaction
Insufficient digestion of vector Re-digest vector using higher concentrations ofenzymes or screen additional colonies from the actualligation/transformation plate
Clones from transformations aremissing restriction sites or haveother mutations
CIP can degrade the ends ofDNA when left too long orwhen too much enzyme is used
Watch the CIP reaction time closely or reduce theamount of CIP enzyme used and run out the DNA onan agarose gel immediately after the CIP reactionis finished
Step 42 No colonies obtained withdouble transformation ofthe B2H reporter strain
Too little DNA used fortransformation and/or lowquality competent cells
Use larger amounts of DNA for transformation and platea larger portion of the transformation
Step 44 Cells do not grow whensubcultured
Toxic effects of Gal11P-zincfinger hybrid protein
Consider using lower concentrations of IPTG in themedium (e.g., 0, 100, 250 mM). Also consider omittingIPTG from the overnight culture and only adding it tothe subculture.
No activity for the ZF array in theB2H assay
Target site may not be suitablefor modular design
Consider other targets, particularly those with primar-ily GNN sub-sites; consider generating ZF arrays usinga selection protocol that accounts for context-depen-dent effects on DNA binding
NATURE PROTOCOLS | VOL.1 NO.3 | 2006 | 1649
PROTOCOL

� TIMINGIdentification of potential ZFN targetsites using ZiFiT software requires lessthan 1 d. Assembly of multi-fingerarrays requires 6–7 d. Testing the DNA-binding activity of a three-fingerdomain using the B2H reporter assayrequires 12–14 d. Construction of ZFNexpression vectors requires 3–5 d. Notethat because our protocol has simplifiedall of the required steps, the length oftime needed to perform any of thesesteps does not change appreciably evenif one processes more than a singlemulti-finger array.
ANTICIPATED RESULTSAs noted in the Introduction, modular assembly will not always yield a functional zinc finger domain. Screening multi-fingerarrays for function is therefore critical before testing these assembled domains as ZFNs in either plant or human cells. The B2Hsystem provides a rapid and effective method for identifying proteins that possess both good affinities and specificities31.Although E. coli and human genomes differ in their sequence composition and chromatin is present in human cells, ourcollective experience to date suggests that many of the proteins that show at least three-fold transcriptional activation in theB2H system will also show activity in human cells. (Presumably, the B2H system also will serve as a reliable screen for activityin plant cells as well although this remains to be determined.)
We believe that the B2H system provides a rapid and effective alternative to other assays for assessing zinc finger activity.Various in vitro methods have been described for testing binding of zinc finger arrays to target sites (e.g., gel shift assays58,59)or activity of ZFNs (e.g., restriction digest assays with purified target DNA36). Although these assays provide useful functionalreadouts, they only measure the affinity of a zinc finger array for its target site but do not account for its specificity inthe context of genomic DNA sequences. A mammalian cell-based reporter assay for zinc finger binding has also been described7.However, this assay requires transient transfections of human cells and is subject to variability in results depending uponthe absolute and relative concentrations of expression and reporter plasmids introduced into the cells. By contrast, theB2H system only requires transformations of bacterial cells and the reporter episome in this assay is strictly maintainedat single copy. Thus, the B2H system requires a zinc finger array to be able to find its specific, single copy target sequencein the presence of competing chromosomal E. coli DNA, thereby providing a stringent test for both DNA-binding affinityand specificity.
Depending upon the specific application, certain ZFNs generated by modular assembly may not provide sufficiently high ratesof gene targeting and/or may exhibit toxic effects. Both of these issues have been observed with ZFNs created by modularassembly7,12. Because four-finger ZFNs have been reported to work well in human cells6, one possible alternative approach is tosimply add an additional fourth finger to one or both of the ZFNs. However, certain target sites may require zinc finger arrayswith greater affinities and/or specificities than can be achieved with modular assembly. In these cases, one may need toconsider the use of more labor-intensive selection protocols that account for context-dependent effects on DNA-binding31,59,60.In our experience, multi-finger domains identified by such selection methods can exhibit higher efficiencies of gene targetingand reduced toxicities relative to domains created by modular assembly (S. Pruett, J.K.J., & M.H.P., unpublished data).
p
uor
G g
n ih si l
bu
P eru ta
N 600 2©
nat
ure
pro
toco
ls/
moc.er
ut an.
ww
w//:ptt
h
B2H expression vector ZFN cloning vectors
pGP-FB-F1/F2/F3pGP-FB-F1/F2/F3
lacU
V5
P
GaI11P
Flag F1
F2
F3
Xba I
Xba I
Xho I
Not I Xba IBam HI
Xba IBam HI
Bam
HI
Bam H
I
Bsg I
PlantPlant
ZFNExpression
Vector
ZFNExpression
Vector
HumanHuman
NO
Sprom
oter
CM
Vprom
oter
Pro
moter
ATG ATGNLS NLStag
ATGNLS
tagF1 F2
F3
tagFokl nuclease
dom
ain
Fokl nucleas
edo
mai
n
Fokl
nuc.
..edo
mai
nN
os
term
ColEI ori
ColEI oriAMP resistance
BG
HpolyA
OR
Xba l/BamHI digestIsolate fragment
Ligate
Xba l/BamHI digestIsolate vector backbone
AM
Pre
sist
ance
Figure 6 | Strategy for constructing plant or
human ZFN expression vectors. Zinc finger arrays
tested in the B2H system can be directly cloned
from a pGP-FB-F1/F2/F3 expression vector using
the unique XbaI and BamHI sites into a vector
designed to express ZFNs in either plant
(pDW1775) or human (pST1374) cells. The epitope
tag (tag) in the plant ZFN expression vector is
AcV5 whereas it is FLAG in the human ZFN
expression vector. The nuclear localization
signal (NLS) in both vectors is the SV40 NLS.
Restriction sites referred to in the text are
shown. Note that plasmid maps are not to scale.
1650 | VOL.1 NO.3 | 2006 | NATURE PROTOCOLS
PROTOCOL

Another potential reason for the inability of modular assembled ZFNs to mediate high efficiency gene targeting can be poorexpression levels. The codons we used to encode our zinc finger plasmids have not been fully optimized for plant or humanexpression. The steady-state levels of ZFN expression can be monitored using Western blots with an antibody directed againstan epitope tag encoded in all of our expression vectors (AcV5 or FLAG for plants or mammalian cells, respectively). If theexpression level of a ZFN is problematic, one can consider recoding potentially promising zinc finger arrays using fullyoptimized codon sets.
Note: Supplementary information is available via the HTML version of this article.
ACKNOWLEDGMENTS We respectfully acknowledge the previously published workof Carlos Barbas’ group, Sangamo Biosciences and ToolGen, Inc., which describedand characterized the various zinc finger modules we used as the basis for ourcomposite archive. We thank J.-S. Kim for providing the full amino acid sequencesof the ToolGen human zinc finger modules. We thank members of our groups,especially P. Zaback and J. Townsend for helpful suggestions. J.D.S. is supportedby USDA MGET 2001-52100-11506. A.S.H. was supported by NIH T32 CA09216.M.H.P is supported by the NIH (R01 HL0792595 and R21 CA120681). D.F.V. issupported by NSF grant DBI 0501678. J.K.J. is supported by the NIH (R01GM069906 and R01 GM072621) and the MGH Department of Pathology. J.K.J.dedicates this protocol to the memory of Robert L. Burghoff, a patient teacherand friend who always knew how to make molecular biology experiments work.Note added in proof: Caroll, Segal and colleagues have recently describedPCR-boned methods for assembling zinc finger modules into arrays and methodsfor purifying ZFNs and testing their activities in vitro (Caroll, D., Morton,J.J., Beumer, K.J. & Segal, D.J. Design, construction and in vitro testing of zincfinger nucleones. Nat. Protocols 3, 1329–1341).
COMPETING INTERESTS STATEMENT The authors declare that they have nocompeting financial interests.
Published online at http://www.natureprotocols.comReprints and permissions information is available online at http://npg.nature.com/reprintsandpermissions
1. Beumer, K., Bhattacharyya, G., Bibikova, M., Trautman, J.K. & Carroll, D.Efficient gene targeting in Drosophila with zinc-finger nucleases. Genetics 172,2391–2403 (2006).
2. Bibikova, M., Beumer, K., Trautman, J.K. & Carroll, D. Enhancing gene targetingwith designed zinc finger nucleases. Science 300, 764 (2003).
3. Bibikova, M., Golic, M., Golic, K.G. & Carroll, D. Targeted chromosomal cleavageand mutagenesis in Drosophila using zinc-finger nucleases. Genetics 161,1169–1175 (2002).
4. Wright, D.A. et al. High-frequency homologous recombination in plants mediatedby zinc-finger nucleases. Plant J. 44, 693–705 (2005).
5. Porteus, M.H. & Baltimore, D. Chimeric nucleases stimulate gene targeting inhuman cells. Science 300, 763 (2003).
6. Urnov, F.D. et al. Highly efficient endogenous human gene correction usingdesigned zinc-finger nucleases. Nature 435, 646–651 (2005).
7. Alwin, S. et al. Custom zinc-finger nucleases for use in human cells. Mol. Ther. 12,610–617 (2005).
8. Kim, Y.G., Cha, J. & Chandrasegaran, S. Hybrid restriction enzymes: zinc fingerfusions to Fok I cleavage domain. Proc. Natl. Acad. Sci. USA 93, 1156–1160 (1996).
9. Smith, J., Berg, J.M. & Chandrasegaran, S. A detailed study of the substratespecificity of a chimeric restriction enzyme. Nucleic Acids Res. 27, 674–681(1999).
10. Smith, J. et al. Requirements for double-strand cleavage by chimeric restrictionenzymes with zinc finger DNA-recognition domains. Nucleic Acids Res. 28,3361–3369 (2000).
11. Mani, M., Smith, J., Kandavelou, K., Berg, J.M. & Chandrasegaran, S. Binding oftwo zinc finger nuclease monomers to two specific sites is required for effectivedouble-strand DNA cleavage. Biochem. Biophys. Res. Commun. 334, 1191–1197(2005).
12. Porteus, M.H. Mammalian gene targeting with designed zinc finger nucleases.Mol. Ther. 13, 438–446 (2006).
13. Dreier, B., Beerli, R.R., Segal, D.J., Flippin, J.D. & Barbas, C.F. 3rd Development ofzinc finger domains for recognition of the 5’-ANN-3’ family of DNA sequences andtheir use in the construction of artificial transcription factors. J. Biol. Chem. 276,29466–29478 (2001).
14. Dreier, B. et al. Development of zinc finger domains for recognition ofthe 5’-CNN-3’ family DNA sequences and their use in the construction ofartificial transcription factors. J. Biol. Chem. 280, 35588–35597 (2005).
15. Dreier, B., Segal, D.J. & Barbas, C.F., 3rd Insights into the molecular recognitionof the 5’-GNN-3’ family of DNA sequences by zinc finger domains. J. Mol. Biol.303, 489–502 (2000).
16. Segal, D.J., Dreier, B., Beerli, R.R. & Barbas, C.F. 3rd Toward controlling geneexpression at will: selection and design of zinc finger domains recognizing each ofthe 5’-GNN-3’ DNA target sequences. Proc. Natl. Acad. Sci. USA 96, 2758–2763(1999).
17. Segal, D.J. The use of zinc finger peptides to study the role of specific factorbinding sites in the chromatin environment. Methods 26, 76–83 (2002).
18. Desjarlais, J.R. & Berg, J.M. Use of a zinc-finger consensus sequence frameworkand specificity rules to design specific DNA binding proteins. Proc. Natl. Acad. Sci.USA 90, 2256–2260 (1993).
19. Mandell, J.G. & Barbas, C.F. 3rd Zinc Finger Tools: custom DNA-bindingdomains for transcription factors and nucleases. Nucleic Acids Res. 34, W516–523(2006).
20. Liu, P.Q. et al. Regulation of an endogenous locus using a panel of designedzinc finger proteins targeted to accessible chromatin regions. Activation ofvascular endothelial growth factor A. J. Biol. Chem. 276, 11323–11334(2001).
21. Zhang, L. et al. Synthetic zinc finger transcription factor action at an endogenouschromosomal site. Activation of the human erythropoietin gene. J. Biol. Chem.275, 33850–33860 (2000).
22. Liu, Q., Xia, Z., Zhong, X. & Case, C.C. Validated zinc finger proteindesigns for all 16 GNN DNA triplet targets. J. Biol. Chem. 277, 3850–3856(2002).
23. Bae, K.H. et al. Human zinc fingers as building blocks in the construction ofartificial transcription factors. Nat. Biotechnol. 21, 275–280 (2003).
24. Park, K.S. et al. Phenotypic alteration of eukaryotic cells using randomizedlibraries of artificial transcription factors. Nat. Biotechnol. 21, 1208–1214(2003).
25. Kwon, H.S., Shin, H.C. & Kim, J.S. Suppression of vascular endothelial growthfactor expression at the transcriptional and post-transcriptional levels. Nucleic.Acids Res. 33, e74 (2005).
26. Kwon, R.J. et al. Artificial transcription factors increase production ofrecombinant antibodies in Chinese hamster ovary cells. Biotechnol. Lett. 28, 9–15(2006).
27. Lee, D.K. et al. Toward a functional annotation of the human genome usingartificial transcription factors. Genome Res. 13, 2708–2716 (2003).
28. Park, K.S., Jang, Y.S., Lee, H. & Kim, J.S. Phenotypic alteration and target geneidentification using combinatorial libraries of zinc finger proteins in prokaryoticcells. J. Bacteriol. 187, 5496–5499 (2005).
29. Park, K.S. et al. Identification and use of zinc finger transcription factors thatincrease production of recombinant proteins in yeast and mammalian cells.Biotechnol. Prog. 21, 664–670 (2005).
30. Segal, D.J. et al. Evaluation of a modular strategy for the construction of novelpolydactyl zinc finger DNA-binding proteins. Biochemistry 42, 2137–2148(2003).
31. Hurt, J.A., Thibodeau, S.A., Hirsh, A.S., Pabo, C.O. & Joung, J.K. Highly specificzinc finger proteins obtained by directed domain shuffling and cell-basedselection. Proc. Natl. Acad. Sci. USA 100, 12271–12276 (2003).
32. Pabo, C.O., Peisach, E. & Grant, R.A. Design and selection of novel Cys2His2 zincfinger proteins. Annu. Rev. Biochem. 70, 313–340 (2001).
33. Wolfe, S.A., Nekludova, L. & Pabo, C.O. DNA recognition by Cys2His2 zinc fingerproteins. Annu. Rev. Biophys. Biomol. Struct. 29, 183–212 (2000).
34. Isalan, M., Choo, Y. & Klug, A. Synergy between adjacent zinc fingers in sequence-specific DNA recognition. Proc. Natl. Acad. Sci. USA 94, 5617–5621 (1997).
35. Isalan, M., Klug, A. & Choo, Y. Comprehensive DNA recognition through concertedinteractions from adjacent zinc fingers. Biochemistry 37, 12026–12033 (1998).
36. Mani, M., Kandavelou, K., Dy, F.J., Durai, S. & Chandrasegaran, S. Design,engineering, and characterization of zinc finger nucleases. Biochem. Biophys. Res.Commun. 335, 447–457 (2005).
37. Dove, S.L., Joung, J.K. & Hochschild, A. Activation of prokaryotic transcriptionthrough arbitrary protein-protein contacts. Nature 386, 627–630 (1997).
p
uor
G g
n ih si l
bu
P eru ta
N 600 2©
nat
ure
pro
toco
ls/
moc.er
ut an.
ww
w//:ptt
h
NATURE PROTOCOLS | VOL.1 NO.3 | 2006 | 1651
PROTOCOL

38. Joung, J.K. Identifying and modifying protein-DNA and protein-proteininteractions using a bacterial two-hybrid selection system. J. Cell. Biochem.Suppl 37: 53–57 (2001).
39. Joung, J.K., Ramm, E.I. & Pabo, C.O. A bacterial two-hybrid selection system forstudying protein-DNA and protein-protein interactions. Proc. Natl. Acad. Sci. USA97, 7382–7387 (2000).
40. Beerli, R.R., Segal, D.J., Dreier, B. & Barbas, C.F. 3rd Toward controlling geneexpression at will: specific regulation of the erbB-2/HER-2 promoter by usingpolydactyl zinc finger proteins constructed from modular building blocks.Proc. Natl. Acad. Sci. USA 95, 14628–14633 (1998).
41. Beerli, R.R. & Barbas, C.F., 3rd Engineering polydactyl zinc-finger transcriptionfactors. Nat. Biotechnol. 20, 135–141 (2002).
42. Blancafort, P. et al. Genetic reprogramming of tumor cells by zinc fingertranscription factors. Proc. Natl. Acad. Sci. USA 102, 11716–11721 (2005).
43. Jamieson, A.C., Miller, J.C. & Pabo, C.O. Drug discovery with engineeredzinc-finger proteins. Nat. Rev. Drug Discov. 2, 361–368 (2003).
44. Klug, A. Towards therapeutic applications of engineered zinc finger proteins.FEBS Lett 579, 892–894 (2005).
45. Lee, D.K., Seol, W. & Kim, J.S. Custom DNA-binding proteins and artificialtranscription factors. Curr. Topics Med. Chem. 3, 645–657 (2003).
46. Falke, D. & Juliano, R.L. Selective gene regulation with designed transcriptionfactors: implications for therapy. Curr. Opin. Mol. Ther. 5, 161–166 (2003).
47. Beerli, R.R., Dreier, B. & Barbas, C.F., 3rd Positive and negative regulation ofendogenous genes by designed transcription factors. Proc. Natl. Acad. Sci. USA97, 1495–1500 (2000).
48. Ren, D., Collingwood, T.N., Rebar, E.J., Wolffe, A.P. & Camp, H.S. PPARgammaknockdown by engineered transcription factors: exogenous PPARgamma2but not PPARgamma1 reactivates adipogenesis. Genes Dev. 16, 27–32(2002).
49. Liang, Y. et al. Activation of vascular endothelial growth factor A transcription intumorigenic glioblastoma cell lines by an enhancer with cell type-specific DNase Iaccessibility. J. Biol. Chem. 277, 20087–20094 (2002).
50. Falke, D., Fisher, M., Ye, D. & Juliano, R.L. Design of artificial transcription factorsto selectively regulate the pro-apoptotic bax gene. Nucleic Acids Res. 31, e10(2003).
51. Blancafort, P., Magnenat, L. & Barbas, C.F. Scanning the human genomewith combinatorial transcription factor libraries. Nat. Biotechnol. 21, 269–274(2003).
52. Blancafort, P., Segal, D.J. & Barbas, C.F. 3rd Designing transcription factorarchitectures for drug discovery. Mol. Pharmacol. 66, 1361–1371 (2004).
53. Bibikova, M. et al. Stimulation of homologous recombination throughtargeted cleavage by chimeric nucleases. Mol. Cell Biol. 21, 289–297 (2001).
54. Porteus, M.H. & Carroll, D. Gene targeting using zinc finger nucleases.Nat. Biotechnol. 23, 967–973 (2005).
55. Aslanidis, C. & de Jong, P.J. Ligation-independent cloning of PCR products(LIC-PCR). Nucleic Acids Res 18, 6069–6074 (1990).
56. Thibodeau, S.A., Fang, R. & Joung, J.K. High-throughput beta-galactosidaseassay for bacterial cell-based reporter systems. Biotechniques 36, 410–415(2004).
57. Miller, J.H. A Short Course in Bacterial Genetics. (Cold Spring Harbor LaboratoryPress, Cold Spring Harbor, New York, 1992).
58. Rebar, E.J. & Pabo, C.O. Zinc finger phage: affinity selection of fingers with newDNA-binding specificities. Science 263, 671–673 (1994).
59. Greisman, H.A. & Pabo, C.O. A general strategy for selecting high-affinity zincfinger proteins for diverse DNA target sites. Science 275, 657–661 (1997).
60. Isalan, M., Klug, A. & Choo, Y. A rapid, generally applicable method to engineerzinc fingers illustrated by targeting the HIV–1 promoter. Nat. Biotechnol. 19,656–660 (2001).
p
uor
G g
n ih si l
bu
P eru ta
N 600 2©
nat
ure
pro
toco
ls/
moc.er
ut an.
ww
w//:ptt
h
1652 | VOL.1 NO.3 | 2006 | NATURE PROTOCOLS
PROTOCOL





![Journal of Falkenhagen et al, J Antivir Antiretrovir 213 ... · CCR5 gene via Zinc finger nucleases [4], cleavage of CCR5 mRNA by multimeric ribozymes [5], inhibition of CCR5 mRNA](https://img.pdfslide.us/doc/110x75/5fd3f8f670db7b30b42beea9/journal-of-falkenhagen-et-al-j-antivir-antiretrovir-213-ccr5-gene-via-zinc.jpg)























