Embed Size (px)
Citation preview

Available online at www.sciencedirect.com
www.elsevier.com/locate/jocn
Journal of Clinical Neuroscience 16 (2009) 169–177
Review
Spinal tumors in neurofibromatosis-2: Managementconsiderations – a review
Katherine Holland *, Andrew H. Kaye
Department of Neurosurgery, Department of Surgery, University of Melbourne, Royal Melbourne Hospital, Parkville 3052, Victoria, Australia
Received 19 March 2008; accepted 21 March 2008
Abstract
Neurofibromatosis Type 2 (NF-2) is a distinct clinical entity, characterized by multiple intracranial and spinal tumors. While bilateralvestibular schwannomas are the pathological hallmark of the disease, significant morbidity in NF-2 is attributable to the presence of bothintramedullary and extramedullary spinal tumors. With the advent of MRI as a screening modality, multiple, extensive spinal tumors inthe NF-2 population are often seen, which may be clinically quiescent at the time of initial diagnosis. All NF-2 patients should haveroutine screening with full spinal MRI at the time of diagnosis, regardless of symptoms. Early surgical intervention is indicated in caseswhere a neurological deficit is attributable to a focal expanding spinal lesion. In asymptomatic patients, the decision to operate is tailoredto the individual patient, with the ultimate goal of preserving function. In these cases, surgery should be considered where there is evi-dence of progressive tumor growth, with attendant risk to the patient of functional deterioration.� 2008 Elsevier Ltd. All rights reserved.
Keywords: Extramedullary tumors; Intramedullary tumors; Neurofibromatosis type 2; Spinal tumors
1. Introduction
Neurofibromatosis Type 2 (NF-2) is an autosomal dom-inant disorder, characterized by the development of benigntumors of the neural crest. NF-2 is much less prevalentthan NF-1, occurring at a rate of 1 in 40,000 people.1 LikeNF-1, NF-2 tumors derive from Schwann cells, the neuro-ectodermal cells that produce myelin. However, NF-2 is aunique clinical and pathological entity, entirely distinctboth phenotypically and genotypically from NF-1. Previ-ous authors have acknowledged that the designation ‘‘neu-rofibromatosis 2” is a misnomer, as there are noneurofibromas within NF-2, and most neural tumors areschwannomas.1 While the diagnosis of NF-2 requires thepresence of bilateral ‘‘acoustic neuromas”, or vestibularschwannomas, many other nervous system tumors arepresent within the syndrome.1 Molecular genetics has pro-gressed our understanding of the molecular basis of NF-2,
0967-5868/$ - see front matter � 2008 Elsevier Ltd. All rights reserved.
doi:10.1016/j.jocn.2008.03.005
* Corresponding author. Tel.: +61 3 9342 7000; fax: +61 3 9342 7273.E-mail address: [email protected] (K. Holland).
and the introduction of MRI has reshaped our understand-ing of the disease, as one typified by variegated, often com-plex spinal tumors. Neuroimaging studies reveal that, withthorough evaluation with MRI, tumors of the spine andother cranial nerves are almost as common as vestibularschwannomas in NF-2 patients.4 As distinct from sporadicextramedullary spinal tumors, spinal schwannomas in NF-2 tend to be multiple, and grow as expanding, multicentricmasses. Intramedullary spinal tumors (IMSTs) are also fre-quent, and are usually ependymomas.1,2 The challenge forthe surgeon is to rationalize and tailor intervention to theindividual patient, who may or may not have symptomsattributable to an often alarming array of spinal tumors,present at multiple levels.
As the genetics of NF-2 are better understood, so too isour appreciation of the unique behavior of IMSTs withinthe spectrum. While NF-2 patients represent only 0.03%of the population, they account for about 2.5% of patientswith IMSTs.2 Likewise, NF-2 related mutations have beenidentified in DNA from isolated, sporadic IMSTs.2,5 Spe-cifically, an association between NF-2 and intramedullary

170 K. Holland, A.H. Kaye / Journal of Clinical Neuroscience 16 (2009) 169–177
ependymoma has been demonstrated.2–4 However, as yetthere is no specific correlation between the underlyingmutation in NF-2 and specific patterns of disease or recur-rence of IMSTs after surgical resection.5,6 Demonstrating alink between this rare, gene-based disorder and a specifictumor type, with unique natural history, highlights a po-tential ‘‘cause and effect” relationship between disease phe-notype and underlying gene mutation. The potential is foremerging treatment modalities, such as gene therapy, to bedeveloped based on specific genetic mutations detected inindividual tumors.2
The preponderance of spinal and cranial tumors in NF-2 accounts for its greater overall morbidity and mortality,compared to NF-1.7 Patients with NF-2 may present withan overwhelming array of complex cranial and spinaltumors. This may pose a challenge to the treating surgeonin prioritizing surgical intervention.8 A patient may presentwith a combination of threatened hearing or enlargingtumors of the cerebellopontine angle, in addition to subtleneurological symptoms of an expanding spinal tumor.Alternatively, they may be asymptomatic with respect tospinal pathology, but with known extensive intracranialdisease. In a recent series of 41 NF-2 patients, routinespinal imaging studies were normal in only 7 patients.1 In12 patients, imaging of the spine unmasked a multitudeof spinal lesions. This paper reviews the current literatureon spinal tumors in NF-2, and aims to clarify managementissues relating to this complex clinical entity.
2. Discussion
2.1. Genetics of NF-2
NF-2 is characterized by autosomal dominant inheri-tance, with high penetrance.8 Located on chromosome22q12, the NF-2 gene was first identified by Trofatterand Rouleau in 1993.9 About half of the reported casesof NF-2 represent new mutations.8 The NF-2 gene productencodes the 595-amino acid protein schwannomin, or‘‘merlin”, an acronym for ‘‘moesin–ezrin–radixin-like pro-tein”. Merlin is part of a family of proteins that link thecytoskeleton to the plasma membrane.10,11 Outside theneurofibromatosis spectrum, NF-2 gene mutations havebeen associated with sporadic central nervous system(CNS) tumors. The NF-2 gene has been linked to the devel-opment of sporadic acoustic neuromas, meningiomas,astrocytomas and other tumor types. Earlier literature sup-ports an association between sporadic meningiomas andacoustic neuromas occurring in patients unaffected byNF-2, with mutations of the NF-2 gene and itstranscript.12,13 Previous genetic research has identified thatabout 50% of sporadic acoustic neuromas and 30%of intracranial meningiomas are associated with NF-2mutations.14
Most tumors demonstrate loss of heterozygosity forchromosome 22q, suggesting that the NF-2 locus functionsas a tumor suppressor site.8 At a cellular level, the primary
inherited mutation in NF-2 is recessive to the normal allele,and tumors only occur when a second mutation eliminatesthe normal allele.15 However, NF-2 is dominant at the levelof the organism, due to a high likelihood of secondarymutation and associated tumorigenesis.15 This correspondsto loss or inactivation of a tumor-suppressor gene in NF-2.The clinical spectrum is variable: with single aminoacid abnormality, disease may be mild, whereas severedisease is associated with frameshift deletions or nonsensemutations.1
2.2. Spinal tumors in NF-2
2.2.1. Extramedullary spinal tumorsResearch has identified that NF-2 patients have a predi-
lection for multiple spinal nerve sheath schwannomas.15,16
Halliday and colleagues examined the clinical and patho-logical features of 86 benign spinal nerve sheath tumors(SNSTs) removed surgically from 65 patients, as well as 5autopsy specimens.15 Seven out of 86 patients met the diag-nostic criteria for NF-2.15 These tumors were intradural,extramedullary neoplasms, arising from dorsal nerve roots.With one exception, all SNSTs in both surgically treatedand autopsied NF-2 patients were schwannomas,15 withmost occurring at the cervical or thoracic level.15 OneNF-2 patient demonstrated a ‘‘mixed-type” tumor.15 Theauthors concluded that while there may be a degree of phe-notypic heterogeneity, ‘‘if a patient with NF-2 has a spinalnerve sheath tumor, it is very likely to be a schwannoma ormixed tumor”.15
Clinical studies support the preponderance, and associ-ated morbidity, of extramedullary spinal tumors in NF-2.In his personal series of 41 NF-2 patients, treated over a30-year period, Malis found that tumors of the spine andcranial nerves were nearly as common as vestibular sch-wannomas when patients were thoroughly evaluated withMRI.1 Among 41 NF-2 patients, all of whom where ini-tially referred due to bilateral vestibular schwannomas, atotal of 99 extramedullary spinal tumors were treated sur-gically. Of these, 58 were schwannomas and 41 were menin-giomas.1 In the same group, there were 14 intramedullarytumors, of which 9 were ependymomas, 2 central schwan-nomas and 3 astrocytomas. Malis’s series highlights theclinical burden of meningiomas in NF-2. Every patientdeath from NF-2-related disease was attributable to over-whelmingly rapid growth of multiple meningiomas.1 Thisis in contrast to vestibular schwannomas, the clinical ‘‘hall-mark” of NF-2, from which no patient died.1
The incidence of spinal meningiomas and schwannomain NF-2 is comparable, and the two tumors may occursimultaneously.1,4 A common pathogenetic mechanismrelated to derangements in chromosome 22 may help toexplain why meningiomas, a tumor of mesodermal origin,should occur in association with schwannomas, which de-rive from neuroectoderm.17 Dorizzi and colleagues postu-lated that meningiomas and schwannoma cells derivefrom the same mesenchymal cell,18 a common origin that

K. Holland, A.H. Kaye / Journal of Clinical Neuroscience 16 (2009) 169–177 171
could explain the occurrence of meningiomas in associationwith schwannomas in NF-2.
While marred by selection bias through recruitment ofsubjects through specialist referrals, several neuroimagingstudies have detected a high incidence of extramedullaryspinal tumors in NF-2 patients, suggesting that spinal tu-mors are more common among these patients than previ-ously reported. Mautner and colleagues attempted todefine the frequency of asymptomatic spinal tumors inNF-2 patients, also characterizing the incidence of cerebraltumors and ocular abnormalities.4 Forty-eight patientswith NF-2 underwent complete neuroradiological, neuro-logical and ophthalmological examinations. Most patientswere referred by treating specialists, resulting in a selectionbias towards those with severe phenotype. Patients werepredominantly young (mean age 28 years), presented earlywith symptoms (mean age of onset 17 years, range 2–36)and were on average 22 years old at time of diagnosis.4
All patients met the National Institute of Health NF-2diagnostic criteria. All patients underwent gadolinium-enhanced MRI of the brain and entire spine, in additionto compete neurological examination.
With respect to presenting symptoms, spinal neurolog-ical symptoms and neck or back pain were rare (4% and2% respectively).4 The most common presenting symptomwas related to hearing loss or tinnitus in 31% of patients,followed by skin tumors (25%), ocular abnormalities(13%) and polyneuropathy (6%).4 Paucity of localizingsymptoms notwithstanding, paraspinal tumors were iden-tified in 43 of 48 patients (90%). Forty-one patients (85%)had cervical tumors, while 38 (79%) had thoracic tumorsand 36 (75%) had lumbar tumors. These tumors weremultiple in 56% patients.4 Tumors were defined histolog-ically in three patients with meningiomas and four withschwannomas. Only 14 of 43 patients (33%) withspinal tumors had symptoms. Fifteen patients (31%) hadIMSTs.4
Previous neuroimaging studies support the high preva-lence of extramedullary spinal tumors in NF-2. Evansand colleagues detected asymptomatic spinal tumors in26% of NF-2 patients, who were investigated prior toundergoing surgery for vestibular schwannomas.19 How-ever, spinal MRI was limited to the cervical spine. Parryand colleagues detected spinal tumors in 67% of NF-2 pa-tients, but MRI was also mainly limited to the cervical re-gion.20 More caudal spinal lesions almost certainly mayhave been missed by these studies, as Mautner and col-leagues noted two-thirds of tumors occurred in the thora-columbar region.4 In that series, three of 43 patients withspinal tumors had lesions only in the lumbar spine.4 How-ever, the two earlier studies by Evans and Parry may haveincluded a greater proportion of patients with mild pheno-type, possibly correlating with lower incidence of spinallesions among their sample populations. Several authorshave described a severe phenotype characterized by earlysymptom onset, rapid progression of hearing loss, and mul-tiple intracranial and spinal tumors.1,21 Such patients may
be more likely to exhibit multiple extramedullary spinaltumors, which may be occult at time of diagnosis.
2.2.2. Intramedullary spinal tumors
Various authors have reported an association betweenNF-2 and intramedullary ependymoma.2,8 In an early liter-ature review, Rodriguez and Berthrong identified 16 re-ported cases of patients with NF-2 and IMSTs, eight ofwhich were classified as ependymoma, in addition to threeastrocytomas and three ‘‘gliomas”.22 More recently, Leeand colleagues described nine patients with NF, including3 with NF-1, 5 with NF-2 and 1 with ‘‘type uncertain” dis-ease.2 Ependymoma was associated with NF-2 in four outof five patients. Conversely, out of a total of three patientswith NF-1, the predominant pathology was astrocytoma(two with ‘‘low-grade” and one with ‘‘anaplastic”).2 Leeand colleagues reported the incidence of IMSTs in a popu-lation of NF-1 and NF-2 patients as 19% overall (threepatients had NF-1, and five patients had NF-2), a high inci-dence that may reflect referral patterns associated with spe-cialized neurosurgical services.2
While generally regarded as rarities in the spectrum ofspinal pathology, spinal ependymomas account for 60%of IMSTs,8,14 and represent the most frequent IMSTs inadults,3–5,7 with a reported rate of 53% in a recent interna-tional series.23 Almost all are histologically benign tumors,which although unencapsulated, are well-circumscribed,and do not infiltrate adjacent spinal cord tissue (Figs 1and 2).3 Spinal ependymomas present with variable clinicalfeatures; often non-specific in their early stages, with a po-tential time-lag of three to four years between symptom on-set and diagnosis.3
The available, albeit limited literature supports a pre-sumption that solitary intramedullary spinal tumors inNF-1 patients will most likely be an astrocytoma, and anNF-2 patient presenting with an intramedullary tumor willmost likely have an ependymoma.2,5,8 However, this pre-sumption is based on small series and selected case reports.NF-2 gene mutations have been identified in sporadicIMSTs.5,8,24,25 Within sporadic IMSTs, there is a geneticdistinction between astrocytomas and ependymomas. Lossof chromosome 22q has been implicated in the first transi-tion from astrocyte to astrocytoma, along with a mixtureof p53 mutations and chromosome 17q loss.5 However,when Rubio and colleagues examined 30 astrocytomas theyfound that the NF-2 gene was not mutated in these cases.26
Differences in pathogenetic mechanisms seem to exist notonly between ependymomas in the brain and spine, butalso between different IMSTs. Although chromosome 22qabnormalities and NF-2/merlin mutations occur in spinalependymomas, they are not exclusive to ependymoma.27
Recent studies have demonstrated a more compellinglink between sporadic intramedullary ependymomas andthe NF-2 gene mutation. Birch and colleagues examined se-ven intramedullary spinal ependymomas from unrelatedpatients who did not have NF-2.14 They demonstrated that71% of patients with sporadic intramedullary ependymoma
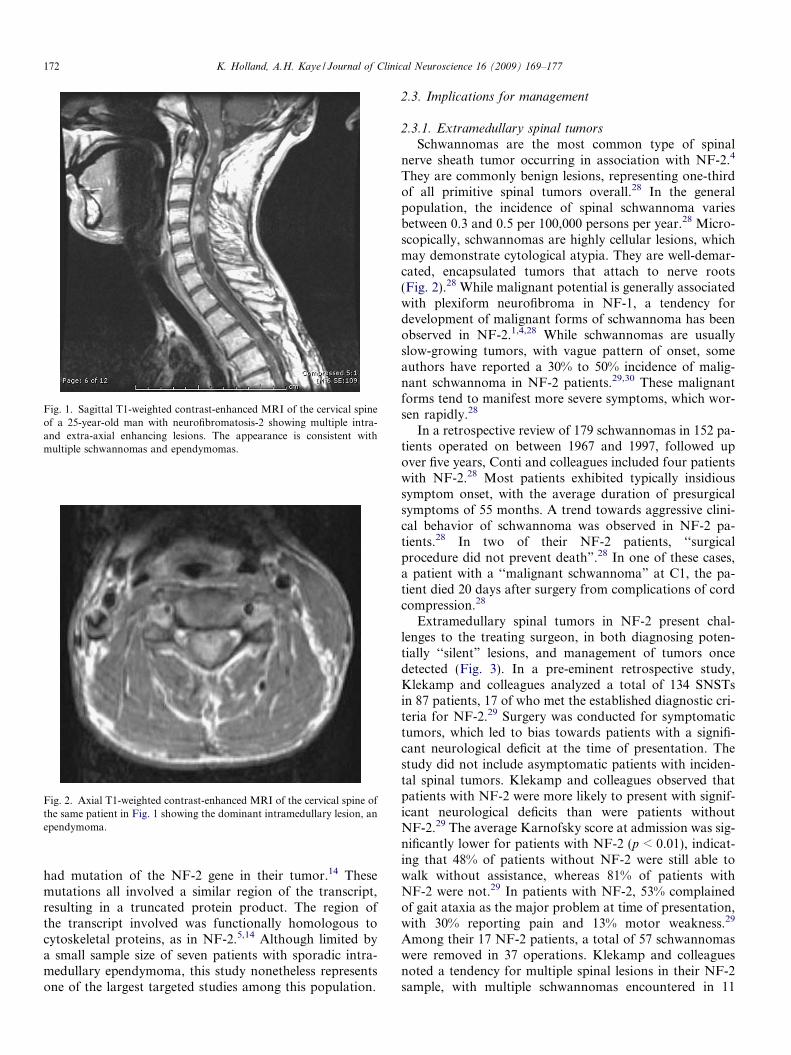
Fig. 1. Sagittal T1-weighted contrast-enhanced MRI of the cervical spineof a 25-year-old man with neurofibromatosis-2 showing multiple intra-and extra-axial enhancing lesions. The appearance is consistent withmultiple schwannomas and ependymomas.
Fig. 2. Axial T1-weighted contrast-enhanced MRI of the cervical spine ofthe same patient in Fig. 1 showing the dominant intramedullary lesion, anependymoma.
172 K. Holland, A.H. Kaye / Journal of Clinical Neuroscience 16 (2009) 169–177
had mutation of the NF-2 gene in their tumor.14 Thesemutations all involved a similar region of the transcript,resulting in a truncated protein product. The region ofthe transcript involved was functionally homologous tocytoskeletal proteins, as in NF-2.5,14 Although limited bya small sample size of seven patients with sporadic intra-medullary ependymoma, this study nonetheless representsone of the largest targeted studies among this population.
2.3. Implications for management
2.3.1. Extramedullary spinal tumors
Schwannomas are the most common type of spinalnerve sheath tumor occurring in association with NF-2.4
They are commonly benign lesions, representing one-thirdof all primitive spinal tumors overall.28 In the generalpopulation, the incidence of spinal schwannoma variesbetween 0.3 and 0.5 per 100,000 persons per year.28 Micro-scopically, schwannomas are highly cellular lesions, whichmay demonstrate cytological atypia. They are well-demar-cated, encapsulated tumors that attach to nerve roots(Fig. 2).28 While malignant potential is generally associatedwith plexiform neurofibroma in NF-1, a tendency fordevelopment of malignant forms of schwannoma has beenobserved in NF-2.1,4,28 While schwannomas are usuallyslow-growing tumors, with vague pattern of onset, someauthors have reported a 30% to 50% incidence of malig-nant schwannoma in NF-2 patients.29,30 These malignantforms tend to manifest more severe symptoms, which wor-sen rapidly.28
In a retrospective review of 179 schwannomas in 152 pa-tients operated on between 1967 and 1997, followed upover five years, Conti and colleagues included four patientswith NF-2.28 Most patients exhibited typically insidioussymptom onset, with the average duration of presurgicalsymptoms of 55 months. A trend towards aggressive clini-cal behavior of schwannoma was observed in NF-2 pa-tients.28 In two of their NF-2 patients, ‘‘surgicalprocedure did not prevent death”.28 In one of these cases,a patient with a ‘‘malignant schwannoma” at C1, the pa-tient died 20 days after surgery from complications of cordcompression.28
Extramedullary spinal tumors in NF-2 present chal-lenges to the treating surgeon, in both diagnosing poten-tially ‘‘silent” lesions, and management of tumors oncedetected (Fig. 3). In a pre-eminent retrospective study,Klekamp and colleagues analyzed a total of 134 SNSTsin 87 patients, 17 of who met the established diagnostic cri-teria for NF-2.29 Surgery was conducted for symptomatictumors, which led to bias towards patients with a signifi-cant neurological deficit at the time of presentation. Thestudy did not include asymptomatic patients with inciden-tal spinal tumors. Klekamp and colleagues observed thatpatients with NF-2 were more likely to present with signif-icant neurological deficits than were patients withoutNF-2.29 The average Karnofsky score at admission was sig-nificantly lower for patients with NF-2 (p < 0.01), indicat-ing that 48% of patients without NF-2 were still able towalk without assistance, whereas 81% of patients withNF-2 were not.29 In patients with NF-2, 53% complainedof gait ataxia as the major problem at time of presentation,with 30% reporting pain and 13% motor weakness.29
Among their 17 NF-2 patients, a total of 57 schwannomaswere removed in 37 operations. Klekamp and colleaguesnoted a tendency for multiple spinal lesions in their NF-2sample, with multiple schwannomas encountered in 11
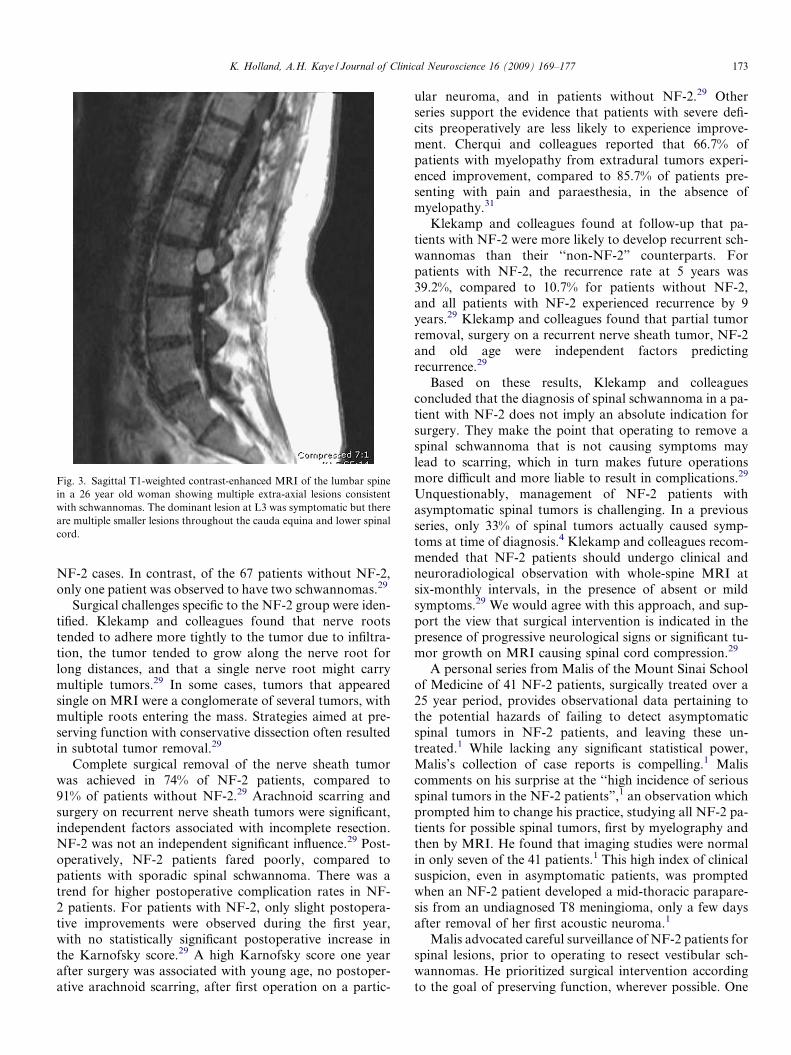
Fig. 3. Sagittal T1-weighted contrast-enhanced MRI of the lumbar spinein a 26 year old woman showing multiple extra-axial lesions consistentwith schwannomas. The dominant lesion at L3 was symptomatic but thereare multiple smaller lesions throughout the cauda equina and lower spinalcord.
K. Holland, A.H. Kaye / Journal of Clinical Neuroscience 16 (2009) 169–177 173
NF-2 cases. In contrast, of the 67 patients without NF-2,only one patient was observed to have two schwannomas.29
Surgical challenges specific to the NF-2 group were iden-tified. Klekamp and colleagues found that nerve rootstended to adhere more tightly to the tumor due to infiltra-tion, the tumor tended to grow along the nerve root forlong distances, and that a single nerve root might carrymultiple tumors.29 In some cases, tumors that appearedsingle on MRI were a conglomerate of several tumors, withmultiple roots entering the mass. Strategies aimed at pre-serving function with conservative dissection often resultedin subtotal tumor removal.29
Complete surgical removal of the nerve sheath tumorwas achieved in 74% of NF-2 patients, compared to91% of patients without NF-2.29 Arachnoid scarring andsurgery on recurrent nerve sheath tumors were significant,independent factors associated with incomplete resection.NF-2 was not an independent significant influence.29 Post-operatively, NF-2 patients fared poorly, compared topatients with sporadic spinal schwannoma. There was atrend for higher postoperative complication rates in NF-2 patients. For patients with NF-2, only slight postopera-tive improvements were observed during the first year,with no statistically significant postoperative increase inthe Karnofsky score.29 A high Karnofsky score one yearafter surgery was associated with young age, no postoper-ative arachnoid scarring, after first operation on a partic-
ular neuroma, and in patients without NF-2.29 Otherseries support the evidence that patients with severe defi-cits preoperatively are less likely to experience improve-ment. Cherqui and colleagues reported that 66.7% ofpatients with myelopathy from extradural tumors experi-enced improvement, compared to 85.7% of patients pre-senting with pain and paraesthesia, in the absence ofmyelopathy.31
Klekamp and colleagues found at follow-up that pa-tients with NF-2 were more likely to develop recurrent sch-wannomas than their ‘‘non-NF-2” counterparts. Forpatients with NF-2, the recurrence rate at 5 years was39.2%, compared to 10.7% for patients without NF-2,and all patients with NF-2 experienced recurrence by 9years.29 Klekamp and colleagues found that partial tumorremoval, surgery on a recurrent nerve sheath tumor, NF-2and old age were independent factors predictingrecurrence.29
Based on these results, Klekamp and colleaguesconcluded that the diagnosis of spinal schwannoma in a pa-tient with NF-2 does not imply an absolute indication forsurgery. They make the point that operating to remove aspinal schwannoma that is not causing symptoms maylead to scarring, which in turn makes future operationsmore difficult and more liable to result in complications.29
Unquestionably, management of NF-2 patients withasymptomatic spinal tumors is challenging. In a previousseries, only 33% of spinal tumors actually caused symp-toms at time of diagnosis.4 Klekamp and colleagues recom-mended that NF-2 patients should undergo clinical andneuroradiological observation with whole-spine MRI atsix-monthly intervals, in the presence of absent or mildsymptoms.29 We would agree with this approach, and sup-port the view that surgical intervention is indicated in thepresence of progressive neurological signs or significant tu-mor growth on MRI causing spinal cord compression.29
A personal series from Malis of the Mount Sinai Schoolof Medicine of 41 NF-2 patients, surgically treated over a25 year period, provides observational data pertaining tothe potential hazards of failing to detect asymptomaticspinal tumors in NF-2 patients, and leaving these un-treated.1 While lacking any significant statistical power,Malis’s collection of case reports is compelling.1 Maliscomments on his surprise at the ‘‘high incidence of seriousspinal tumors in the NF-2 patients”,1 an observation whichprompted him to change his practice, studying all NF-2 pa-tients for possible spinal tumors, first by myelography andthen by MRI. He found that imaging studies were normalin only seven of the 41 patients.1 This high index of clinicalsuspicion, even in asymptomatic patients, was promptedwhen an NF-2 patient developed a mid-thoracic parapare-sis from an undiagnosed T8 meningioma, only a few daysafter removal of her first acoustic neuroma.1
Malis advocated careful surveillance of NF-2 patients forspinal lesions, prior to operating to resect vestibular sch-wannomas. He prioritized surgical intervention accordingto the goal of preserving function, wherever possible. One

174 K. Holland, A.H. Kaye / Journal of Clinical Neuroscience 16 (2009) 169–177
case of a 22-year-old woman with known bilateral vestibu-lar schwannomas, with unilateral deafness, presented withsubacute gait deterioration, and was found to have a mid-cervical lesion with a block at C5 on myelography. Malisopted to treat the spinal lesion first, conducting a C2–C7decompressive laminectomy for an extensive intramedul-lary ependymoma. The patient subsequently underwentstepwise removal of her acoustic schwannomas, in additionto intracranial meningiomas, with preservation of mobilityand hand function. She was subsequently able to completeschooling and gain employment, despite hearing impair-ment.1 In another case, a 14-year-old female with similarpresentation underwent diagnostic imaging of her thoraco-lumbar spine, after developing unilateral foot drop.Myelography showed a complete block at T8 from an intra-dural, extramedullary mass. After a multilevel thoraciclaminectomy, the patient postoperatively developed com-plete lower limb paralysis, with loss of hand power. Imagingof her cervical spine demonstrated a lesion at C1, which hadnot been previously diagnosed. Despite an urgent cervicallaminectomy, the patient failed to regain fine finger move-ment, and retained flaccid lower extremities.1 In eight cases,Malis describes NF-2 patients who initially presented withdeafness and bilateral vestibular schwannomas, who onsubsequent imaging had clinically occult compressive spinallesions. Anecdotally, failing to detect these lesions, or alter-natively, not operating on them early, may result in devas-tating functional outcomes.1
The available literature suggests that once symptomatic,spinal schwannomas in association with NF-2 tend to growfaster, are more likely to infiltrate nerve roots and thereforeprogress to serious deficits sooner.29 Schwannomas in NF-2 tend to be larger and are often multiple, and are thereforemore likely to cause complications, with the potential forpostoperative instability.1,29 Multiple tumors may beencountered throughout the cord, and may be quiescent,without associated neurological deficit. Observational datawould suggest that waiting to operate until such tumors be-come symptomatic results in potential significant neurolog-ical deficit, with poor prospect of recovery. Klekamp andcolleagues found that even with successful surgical treat-ment, maintaining preoperative clinical status is all thatcan be achieved in most cases.29 Furthermore, where a pa-tient has preoperative motor deficit attributable to thenerve root carrying an extradural tumor, the likelihoodof permanent deficit with complete resection, necessitatingen bloc resection of the nerve root, is high. Preservation offunction must be sacrificed for complete resection of thetumor.29
In a patient with NF-2, preservation of function is offundamental importance. Such patients typically have mul-tiple neurological problems associated with other varied tu-mors, including significant visual and hearing deficits.1 Theprospect of further deficits from aggressive surgical resec-tion may be unacceptable in this population. However,Klekamp and colleagues make the valid point that futurerecurrence of the tumor with further nerve root damage
is an inevitable consequence of incomplete tumor removal,and chances of radical resection are significantly reduced ata second operation, due to postoperative arachnoid scar-ring and cord tethering.29 Nonetheless, the ‘‘watch andwait” approach advocated by Klekamp may be unaccept-able to an NF-2 patient with severe phenotype, who facesthe real possibility of complete deafness. Such patients relyon manual dexterity and mobility for communication, andbasic independence.
2.3.2. Intramedullary spinal tumors
Spinal ependymomas arise from ependymal cells liningthe central canal and are slow-growing tumors, with propen-sity to cause symmetric expansion of the spinal cord.32 Incontrast to intramedullary astrocytomas, which tend topresent with neuraxis pain and progressive motor dysfunc-tion over a shorter time course, spinal ependymoma tendto be indolent, with motor impairment usually occurringlate in disease progression. This gradual tumor growth hasimplications for diagnosis, as even a large tumor may exhibitfew neurological deficits. Chang and colleagues retrospec-tively reviewed 31 cases of spinal ependymoma, attemptingto assess the rate of disease progression and identify contrib-utory factors to neurological outcome after treatment. Theyreported a protracted period of 31.9 months between initialsymptom presentation and time to surgery.32
Even with occult symptoms at presentation, a delay indiagnosis of intramedullary ependymoma, associated withan expanding tumor, may affect the normal functional plas-ticity of the cord, with serious implications for the patient’spostoperative outcome. Preoperative functional ability isoverwhelmingly regarded as the strongest predictor ofpostoperative outcome in spinal ependymoma.33–38 Thecohort of patients reviewed by Chang and colleaguesunderwent variable surgical resection, using a mid linemyelotomy with internal debulking and dissection of thelateral tumor margin, followed by ventral dissection. Post-operative outcome was assessed clinically, and by serialMRI performed at six months after surgery and then re-peated annually in asymptomatic patients. The median fol-low-up period was 33 months. Complete surgical removalwas achieved in 74% of patients, with subtotal removal in16%, and partial removal in 10%. Postoperative functionalstatus assessed by Nurick’s grade showed no change in64%, with improvement in 26% and worsened status in10%.32 All cases of postoperative functional improvementoccurred in cases with a preoperative functional status bet-ter than Nurick’s grade 4, while no patients with poorerpreoperative status achieved improvement. There was nostatistical difference in terms of neurological deteriorationbetween patients with complete versus incomplete removalof tumor.32 The correlation between poorer preoperativefunctional status and absence of postoperative neurologicalimprovement suggests that once normal functional plastic-ity is impaired, even with marginal associated neurologicaldeficit, surgery has reduced benefits in terms of functionaloutcome. In patients with better preoperative functional

K. Holland, A.H. Kaye / Journal of Clinical Neuroscience 16 (2009) 169–177 175
status, however, gross-total removal of intramedullaryependymoma can be associated with absence of furtherneurological deficit.32,35
Similarly, complete surgical removal of spinal ependy-moma is associated with limited tumor progression. Changand colleagues reported that disease progression occurred insix of 31 cases during follow-up, with an overall 70% 5-yearprogression-free rate. Radiological recurrence was found inonly two out of 23 cases where the tumor was completely re-moved. In contrast, radiological regrowth was observed infour out of eight cases in the incomplete removal group.There was a statistically significant difference in progres-sion-free rate between these two groups (p = 0.008). Whenthe factors influencing disease progression were analyzed,only the extent of surgical removal was found to correlatewith prognosis.32 In a larger series, Raco and colleagues re-viewed 202 consecutive patients who underwent surgery forintramedullary tumors between 1972 and 2003.33 Of the 68ependymomas, 55 (81%) were completely removed, andmean follow-up was 7.1 years. In patients with ependymo-mas, the outcome depended strictly on the completeness oftumor removal, with tumor regrowth developing in 6 ofthe 13 patients who underwent incomplete removal.33 Thepathology of spinal ependymoma would support the goalof gross-total removal as a viable surgical aim. Ependymo-mas are often well-encapsulated, non-infiltrative lesions dis-playing a distinct plane, with well-defined interface betweentumor mass and surrounding normal cord tissue.3 We wouldsuggest that it is reasonable, in the vast majority of non-NF-2, sporadic cases, to attempt a gross total resection of epen-dymoma, without risk of permanent spinal cord injury. Thisview is well supported in the literature.8,39 However, cautionmust be exercised in applying results of studies based onsporadic ependymoma to the management of NF-2 relatedtumors. In NF-2, IMSTs tend to be multiple and moreextensive than sporadic tumors, with multicentric, lobulatedlesions all along the neuraxis. It may, therefore, be more dif-ficult to obtain complete resection without risking damageto neural tissue. In such cases, the risk of neurological deficitmay preclude an attempt at complete resection.
Even with gross total resection there remains a risk ofrecurrence with benign intramedullary tumors, resultingin the need for long-term clinical and radiographicfollow-up. Several authors have reported that risk ofrecurrence depends mainly on completeness of the originalresection.8,39 When compared to subtotal resection andradiotherapy, gross-total removal of benign intramedullaryependymoma is better at ensuring long-term tumor re-moval.36,37,39 Gross-total resection may still be associatedwith up to a 5% to 10% recurrence rate, with some authorsreporting late recurrence in these slow-growing tumors,even up to 12 years after surgery.40 There is consensus,however, that radiation therapy is unnecessary if completeremoval has been accomplished.36,41 Conversely, subtotalresection is associated with high recurrence rates, with even99% removal leading to recurrence in up to 30% of patientsin spite of postoperative radiation therapy.41
3. Conclusion
Controversy in management of NF-2 patients has lar-gely centered around timing and indications for surgeryin the setting of bilateral acoustic neuromas, where hearinghas already been lost on one side.1 Difficulties arise in pa-tients with NF-2 in whom hearing loss is the dominant pre-senting complaint, but who have additional spinal lesions,single or multiple, which may be incidentally discovered orassociated with subtle or subacute neurological distur-bance. To date, there have been few studies devoted to con-sideration of management of spinal lesions in NF-2patients. In such a vulnerable patient group, who are al-ready seriously impaired by their hearing loss, it is impera-tive to ensure that mobility is preserved. In additionfunctional ability hinges on use of sign-language and writ-ing for communication. There is a case for operating pre-emptively on expanding and compressive spinal lesions,even in the absence of focal neurology.
All patients with bilateral acoustic neuromas should bescreened at the time of diagnosis with full spinal MRI todetect occult spinal lesions. The literature would suggestthat these lesions might present insidiously, and cause pre-cipitous neurological deterioration. In many patients withNF-2, ‘‘serious morbidity and death are caused by . . .spinal and paraspinal tumors of various types, includingependymomas, schwannomas, astrocytomas and meningio-mas. The clinicians attending patients with NF-2 mustconstantly be on guard for problems beyond those ofcompromise of the VIIIth cranial nerve”.42
All patients with NF-2, regardless of symptoms, shouldundergo serial follow-up radiological screening by spinalMRI to detect occult spinal lesions. Patients known to haveclinically quiescent tumors should undergo regular radio-logical surveillance to monitor for tumor growth. It hasbeen established that early diagnosis and surgical removalof spinal tumors, both intramedullary and extramedullary,confers to patients the best opportunity for functionalimprovement.32 Clearly, patients who exhibit minimal orno neurological deficit at the time of surgery stand to ob-tain the greatest benefit from surgical resection.33,35,41,43
However, the presence of spinal cord atrophy and arach-noid scarring, indicating a degree of chronic cord compres-sion with associated loss of plasticity, may be anindependent predictor of poor functional outcome,3,34 evenin the absence of overt neurological symptoms. Waiting forpatients to present once symptomatic clearly confers a de-lay in time to surgery, which may inevitably reduce thechances of achieving a favorable outcome.
The management of spinal tumors in NF-2 demandsthat treatment is tailored to the individual patient. Thegoal of preserving function must be paramount in anymanagement decision. NF-2 patients often display multi-ple, extensive spinal tumors on MRI, which may be associ-ated with spinal cord compression at more than one level.In a symptomatic patient, the surgeon must carefully inter-pret the available imaging, which will often demonstrate a

176 K. Holland, A.H. Kaye / Journal of Clinical Neuroscience 16 (2009) 169–177
distracting multiplicity of spinal lesions. The lesion respon-sible for the patient’s clinical presentation should be iden-tified, and surgical intervention should be targeted at thatoffending lesion. In asymptomatic patients, the decisionto operate is taken when there is radiological evidence ofprogressive tumor growth, with real or impending compro-mise to neural structures. Here, it is imperative to balancethe risk of iatrogenic nerve injury against that of anexpanding spinal tumor, which may cause precipitous neu-ral compression, and corresponding deficit, if left un-treated. The anatomical location of the tumor must beconsidered, in assessing potential loss of function.
Genetic research will no doubt facilitate greater under-standing of the molecular behavior of spinal tumors in NF-2.Thismayinturnpavethewayforpotentialgene-basedther-apy for both NF-2-linked and sporadic intramedullary epen-dymoma.3 While advancement in microsurgical techniqueshas decreased the morbidity of surgical resection,5,44 thereare clear limitations in our ability to arrest neurological dete-rioration, and eradicate risk of recurrence. The need for long-term clinical and radiological follow-up, even after successfulsurgical resection, places a significant burden on the individ-ual, and on community resources. If indeed ‘‘the frontier ofspinal cord tumor therapy now lies in the realm of molecularbiology and genetics”,5 further exploration of a link betweentheNF-2mutationandspinal tumors isof interest tosurgeonsand scientists alike.
References
1. Malis L. Neurofibromatosis type 2 and central neurofibromatosis.Neurosurg Focus 1998;4:Article 1.
2. Lee M, Rezai A, Freed D, et al. Intramedullary spinal cord tumors inneurofibromatosis. Neurosurgery 1996;38:32–7.
3. Martuza RL, Eldridge R. Neurofibromatosis 2 (bilateral acousticneurofibromatosis). N Engl J Med 1988;318:684–8.
4. Mautner VF, Lindenau M. The neuroimaging and clinical spectrumof neurofibromatosis 2. Neurosurgery 1996;38:880–6.
5. Parsa A, Fiore A, et al. Genetic basis of intramedullary spinalcord tumors and therapeutic implications. J Neuro-Onc 2000;47:239.
6. Schwartz T, McCormick P. Intramedullary ependymomas: Clinicalpresentation, surgical treatment strategies and prognosis. J Neuro-Onc
2000;47:211.7. Huson S. What level of care for the neurofibromatoese? The Lancet
1999;353:1114–6.8. Chi J, Cachola K, Parsa A. Genetics and molecular biology of
intramedullary spinal cord tumors. Neurosurg Clin N Am 2006;17:1–5.
9. Trofatter JA, MacCollin MM. A novel moesin-, ezrin-, radixin-likegene is a candidate for the neurofibromatosis 2 tumor suppressor. Cell
1993;72:791–800.10. Hovens CM, Kaye AH. The tumor suppressor protein NF2/merlin:
The puzzle continues. J Clin Neurosci 2001;8:4–7.11. Wickremasekera A, Hovens CM, Kaye AH. Expression of ERB-1 and
2 in vestibular schwannomas. J Clin Neurosci 2007;14:1199–206.12. Bianchi AB, Hara T. Mutations in transcript isoforms of the
neurofibromatosis 2 gene in multiple human tumour types. Nat Genet
1994;6:185–92.13. Ng H, Lau K, Tse J, et al. Combined molecular genetic studies of
chromosome 22q and the neurofibromatosis type 2 gene in centralnervous system tumors. Neurosurgery 1995;37:764–73.
14. Birch B, Johnson J, Parsa A, et al. Frequent type 2 neurofibromatosisgene transcript mutations in sporadic intramedullary spinal cordependymomas. Neurosurgery 1996;39:135–40.
15. Halliday A, Sobel R, et al. Benign spinal nerve sheath tumors: theiroccurrence sporadically and in neurofibromatosis types 1 and 2. J
Neurosurg 1991;74:248–53.16. Rubenstein LJ. The malformative central nervous system lesions
in the central and peripheral forms of neurofibromatosis. Aneuropathological study of 22 cases. Ann NY Acad Sci 1986;468:14–29.
17. Glasauer FE, Castiglia GJ. Coexistence of two different intraspinaltumors. Case report and review of the literature. Neurosurg Focus
1999;6:Article 7.18. Dorizzi A, Crivelli G. Associated cervical schwannoma and dorsal
meningioma. Case report and review of the literature. J Neurosurg Sci
1992;36:173–6.19. Evans DGR, Huson SM. A clinical study of type 2 neurofibromatosis.
Q J Med 1992;304:603–18.20. Parry DM, Eldridge R. Neurofibromatosis 2: Clinical characteristics
of 63 affected individuals and clinical evidence for heterogeneity. Am J
Med Genet 1994;52:450–61.21. Wishart JH. Cases of tumors of the skull, dura mater and brain.
Edinburgh Med Surg J 1822;18:393–7.22. Rodriguez H, Berthrong M. Multiple primary intracranial tumors in
von Recklinghausen’s neurofibromatosis. Arch Neurol 1966;14:467–75.
23. Miller D. Surgical pathology of intramedullary spinal cord neoplasms.J Neuro-Onc 2000;47:189.
24. Ebert C, von Haken M. Molecular genetic analysis of ependymaltumors. NF2 mutations and chromosome 22q loss occur preferentiallyin intramedullary spinal ependymomas. Am J Pathol 1999;155:627–32.
25. Lamszus K, Lachenmayer L. Molecular genetic alterations onchromosomes 11 and 22 in ependymomas. Int J Cancer
2001;91:803–8.26. Rubio MP, Correa KM. Analysis of the neurofibromatosis 2 gene
in human ependymomas and astrocytomas. Cancer Res 1994;54:45–7.
27. Huang B, Starostik P. Human ependymomas reveal frequentdeletions on chromosomes 6 and 9. Acta Neuropathol (Berl)
2003;106:357–62.28. Conti P, Pansini G, Mouchaty H, et al. Spinal neurinomas:
retrospective analysis and long-term outcome of 179 consecutivelyoperated cases and review of the literature. Surg Neurol
2004;61:34–43.29. Klekamp J, Samii M. Surgery of spinal nerve sheath tumors with
special reference to neurofibromatosis. Neurosurgery 1998;42:279–89.
30. Matti ST, Matti HJ, Risto SJ, et al. Long-term outcome after removalof spinal schwannoma; a clinicopathological study of 187 cases. J
Neurosurg 1995;83:621–6.31. Cherqui A, Kim DH, Se-Hoon K, et al. Surgical approaches
to paraspinal nerve sheath tumors. Neurosurg Focus 2007;22:E9.
32. Chang U, Choe W, Chung SK, et al. Surgical outcome and prognosticfactors of spinal intramedullary ependymoma in adults. J Neuro-Onc
2002;57:133.33. Raco A, Esposito V, Lenzi J, et al. Long-term follow-up of
intramedullary spinal cord tumors: A series of 202 cases. Neurosurgery
2005;56:972–81.34. Hoshimaru M, Koyamua T, et al. Results of microsurgical treatment
for intramedullary spinal cord ependymomas: Analysis of 36 cases.Neurosurgery 1999;44:264–9.
35. Hanibali F, Fourney D, Marmor E, et al. Spinal cord ependymoma:Radical surgical resection and outcome. Neurosurgery 2002;51:1162–74.
36. McCormick PC, Torres R. Intramedullary ependymoma of the spinalcord. J Neurosurg 1990;72:523–32.

K. Holland, A.H. Kaye / Journal of Clinical Neuroscience 16 (2009) 169–177 177
37. Epstein F, Farmer J, Freed D, et al. Adult intramedullary spinal cordependymomas: the result of surgery in 38 patients. J Neurosurg
1993;79:204–9.38. Woodworth G, Chaichana K, McGirt MJ, et al. Predictors of
ambulatory function after surgical resection of intramedullary spinalcord tumors. Neurosurgery 2007;61:99–106.
39. Crisante L, Hermann H. Surgical management of intramedullaryspinal cord tumors: Functional outcome and sources of morbidity.Neurosurgery 1994;35:69–76.
40. Lindstat D, Wara W. Postoperative radiotherapy of primary spinal cordtumors. Int J Radiat Oncol Biol Phys 1989;16:1397–403.
41. Cooper P. Outcome after operative treatment of intramedullary spinalcord tumors in adults: intermediate and long-term results in 51patients. Neurosurgery 1989;25:855–9.
42. Riccardi VM. Von Recklinghausen neurofibromatosis. N Engl J Med
1981;305:1617–27.43. Yagi O, Haque M. Intramedullary spinal cord tumour associ-
ated with neurofibromatosis type 1. Acta Neurochir 1997;139:1055–60.
44. Briggs RJ, Fabinyi G, Kaye AH. Current management of acousticneuroma: A review of surgical approaches and outcomes. J Clin
Neurosci 2000;7:521–6.





























