Embed Size (px)
Citation preview

Spin and charge distribution in iron porphyrin models: A coupled cluster and density-functional studyMikael P. Johansson and Dage Sundholm Citation: The Journal of Chemical Physics 120, 3229 (2004); doi: 10.1063/1.1640343 View online: http://dx.doi.org/10.1063/1.1640343 View Table of Contents: http://scitation.aip.org/content/aip/journal/jcp/120/7?ver=pdfcov Published by the AIP Publishing
This article is copyrighted as indicated in the abstract. Reuse of AIP content is subject to the terms at: http://scitation.aip.org/termsconditions. Downloaded to IP:
195.19.233.81 On: Mon, 11 Nov 2013 21:35:20

Spin and charge distribution in iron porphyrin models: A coupled clusterand density-functional study
Mikael P. Johanssona) and Dage Sundholmb)
Department of Chemistry, P.O. Box 55 (A. I. Virtanens Plats 1), FIN-00014 University of Helsinki, Finland
~Received 18 August 2003; accepted 19 November 2003!
We recently performed detailed analyses of the electronic structure of low-spin iron porphyrinsusing density-functional theory~DFT!. Both the spin-density distributions of the oxidized, ferricforms, as well as the changes in total charge density upon reduction to the ferrous forms have beenexplored. Here, we compare the DFT results with wave-function theory, more specifically, with theapproximate singles and doubles coupled-cluster method~CC2!. Different spin states are consideredby studying representative models of low spin, intermediate spin, and high spin species. The CC2calculations corroborate the DFT results; the spin density exhibits the same amount of molecularspin polarization, and the charge delocalization is of comparable magnitude. Slight differences in thedescriptions are noted and discussed. ©2004 American Institute of Physics.@DOI: 10.1063/1.1640343#
I. INTRODUCTION
Density-functional theory~DFT! ~Refs. 1 and 2! is thequantum chemical method of choice for the study of manysystems. A special domain for DFT studies is very large mol-ecules and assemblies. The reason is simple: DFT is still theonly correlation including quantum chemical method capableof routinely treating systems of hundreds of atoms. Corre-lated wave-function methods are still horribly time consum-ing for such entities, due to the much steeper scaling withrespect to system size. Present day density functionals suffer,however, from the lack of a systematic method of checkingthe accuracy of the calculation. Therefore it is desirable tocheck the results of DFT calculations using traditional wave-function methods, especially when new combinations ofproperties and systems are of interest. In this work, two prop-erties are considered. First, the molecular spin-polarizationeffect of oxidized iron porphyrins, and second, the charge-delocalization effect upon reduction of the same.
We recently performed a thorough DFT examination ofthe molecular spin polarization for low-spin ironporphyrins.3 In the work, we showed that the gross spin den-sity, i.e., the sum of unpaireda and b electrons, is signifi-cantly larger than unity, about 1.3. This reconciled two seem-ingly contradictory experimental views of the spin-densitydistribution. The g tensors obtained from electron-spin-resonance spectra~ESR! seem to require practically onewhole unpaired electron in thedp orbitals of iron, whilenuclear magnetic resonance~NMR! and electron nucleardouble resonance~ENDOR! experiments indicate that up to0.2 unpaired electrons reside in the porphyrin ring and itssubstituents.4–7 The contradiction arose from attempts to fitonly one unpaired electron to these data.
In another work,8 the charge delocalization upon reduc-tion of low-spin hemes was studied, and shown to be quite
extensive. Hemes are ligated iron-porphyrins with importantelectron-transfer duties in biological systems. Reduction ofthe Coulomb repulsion by an extensive ‘‘dilution’’ of theadded unit charge is crucial for the protein in its hydrophobicenvironment. Otherwise it would not be able to house theheme during its oxidation/reduction cycle. Our findings alsosupport the edge-to-edge electron-transfer mechanism.9 Ac-cording to this mechanism, the electron transfer rate is re-lated to the distances between the edges of the electron donorand acceptor. There is no need for the electron to go via theiron.
Neither property has previously been studied in greatdetail using quantum chemical methods. Also, both proper-ties are potentially of the sort that current DFT methodscould have difficulties in describing correctly. The large mo-lecular spin polarization might perhaps be a manifestation ofthe recently discussed caveats in the definition of spin DFT.Capelle and Vignale showed,10 with beautiful simplicity, theexternal potential within SDFTnot to be unambiguously de-fined, an assumption made for decades. Argaman and Makovargued on how to deal with this,11 but even so, anothersource of possible problems within SDFT remains. Perdew,Savin, and Burke have proposed an alternative interpretationof the meaning ofra and rb .12 According to this idea, inapproximate DFT it might in certain cases be energeticallyfavorable to usera2rb for a better description of the on-toppair density instead of letting it represent the true spin den-sity at a certain point. Later works discuss the idea further,and provide plausible example systems.13,14Also the chargedelocalization might perhaps be an artifact of the densityfunctionals used; some concern about the ability of moderndensity functionals to describe electron delocalization incharged species exists.15–17
For these reasons, a validation, or rather, a second opin-ion of the results provided by DFT calculations are of con-siderable interest. To this end, we present coupled-clustercalculations on small models of four-, five-, and six-
a!Electronic mail: [email protected]!Electronic mail: [email protected]
JOURNAL OF CHEMICAL PHYSICS VOLUME 120, NUMBER 7 15 FEBRUARY 2004
32290021-9606/2004/120(7)/3229/8/$22.00 © 2004 American Institute of Physics
This article is copyrighted as indicated in the abstract. Reuse of AIP content is subject to the terms at: http://scitation.aip.org/termsconditions. Downloaded to IP:
195.19.233.81 On: Mon, 11 Nov 2013 21:35:20

coordinated iron porphyrins. The models are quite small, andthe ring structure representing the porphyrin plane is morestrained than in a real porphyrin. The models do, however,reproduce both the spin polarization and the charge-delocalization effects. In addition to the iron atom, the mod-els contain two@NHCHNH#2 groups, representing the por-phyrin plane. The intermediate-spin, four-coordinated modelthen has the molecular formula@Fe(NHCHNH)2#q, q50,11. Other models include either one or two axial ligands tothe metal, representing high-spin, five-coordinated and low-spin, six-coordinated systems, respectively. The axial ligandsare either ammonia, giving@Fe(NHCHNH)2(NH3)n#q,n51,2, q50,11, or cyanide, giving@Fe(NHCHNH)2(CN)2#2. This choice of ligands allows usto study both cationic and anionic systems. The axially li-gated model structures are shown in Fig. 1.
II. METHODS
The molecular structures were optimized at the spin-unrestricted density-functional theory~UDFT! level. Thesemilocal gradient-corrected functional BLYP, with Becke’s1988 exchange18 and the Lee, Yang, Parr correlation19 servesas the main functional in this work. The more recent Perdew,Burke, Ernzerhof generalized gradient approximation~GGA!functional from 1996, PBE,20–24 was also tested. The PBEfunctional has some appeal due to its simple formulation andits lack of empirical parameters. As our earlier works3,8,25
have shown no clear advantage in using hybrid functionalscompared with GGA functionals, for describing the molecu-lar spin polarization and charge delocalization, hybrids werenot employed. Instead, the local spin-density functional de-noted SVWN, with Slater-Dirac exchange20–22 in connectionwith the Vosko, Wilk, Nusair correlation functionalVWN-5,26 was tested. The resolution of the identity~RI!approximation of the Coulomb interaction27,28 was used inthe calculations. The RI-DFT calculations are considerablyfaster than ordinary DFT calculations, without any signifi-cant loss of accuracy.
C2v symmetry was imposed on the six-coordinated mod-els, the five-coordinated models usedCs symmetry, and thefour-coordinated systemD2h symmetry. For the six-coordinated ammonia-ligated model, a slight relaxation ofthe structure was noted when going toC1 symmetry. Theammonia groups rotated slightly along the Fe–N axis. Ener-getically, the difference was found to be small. As the spinpolarization and charge delocalization are quite structureinsensitive,3,8,25 the symmetrical geometries were used.
In addition to the various density functionals, the spinand charge densities were calculated with the approximate
coupled-cluster singles and doubles method, CC2,29 employ-ing the resolution of the identity approximation. The RI-CC2methodology developed by Ha¨ttig and Weigend30 signifi-cantly reduces the computer time, disk, and memory require-ments. The electrons of the deep core orbitals of the heavyatoms (1s for C and N, as well as 1s, 2s and 2p for Fe!were not correlated.
All quantum chemical calculations were performed withthe TURBOMOLE program package,31 version 5.6. For hydro-gen, carbon, and nitrogen, Dunning’s augmented correlationconsistent triple-zeta basis sets, aug-cc-pVTZ,32,33were used.For iron, TURBOMOLE’s standard triple-zeta valence basis setaugmented with double polarization functions, TZVPP,34–36
was extended: One set of diffuse functions were added foreach existing angular momenta by dividing the highest ex-ponent by three (s: 0.013 989 409 25,p: 0.044 971 666 70,d: 0.092 897 514 70, andf : 0.833 333 333 3!. The resultingbasis set is here called aug-TZVPP.
In connection with the DFT calculations, the standardTZVPP auxiliary density-fitting~RI! basis set28 was used forall atoms. In the CC2 calculations, iron was designated aTZVPP auxiliary basis set,36 whereas for H, C, and N theoptimized auxiliary aug-cc-pVTZ basis sets37 were used. Inall DFT calculations, TURBOMOLE’s standard grid, denotedm4, was used.
The spin density is obtained as the difference betweenthe a and b spin contributions to the total electron density.We follow the normal convention that the number ofa elec-trons is defined to be greater than the number ofb electrons.The charge-density difference is obtained analogously, nowas the difference in total electron density between the twooxidation states. For the difference in total electron density,the structure of the oxidized form was used for both oxida-tion states. A small structural difference between the oxi-dized and reduced systems exists, but the properties understudy are quite insensitive to the precise structure; theslightly suboptimal structures for the reduced species do notintroduce any computational caveats. In what follows below,electron density refers to both spin and charge density.
The electron density was studied by evaluating it in adiscrete distribution of equidistant Cartesian grid points. Aspacing of 0.2 bohr was used; test calculations with finergrids showed this to be dense enough. The radial distributionof the electron density was explicitly obtained by performingnumerical integration of the density in the discrete represen-tation. The iron atom was placed at the origo. The total ac-cumulated electron density inside a sphere with radiusraround iron, denoted byrel(r ), can then be obtained as
rel~r !51
N~r !
4
3pr 3(
i
N(r )
rel,i , ~1!
where N(r ) is the number of integration points inside thesphere andrel,i is the calculated electron density at grid pointi . To improve the accuracy, the deep core orbitals of theheavy atoms were not considered in the numerical integra-tion. The CC2 electron densities were obtained from the CC2natural orbitals and their occupation numbers.
FIG. 1. Model systems studied in this work. From left to right:Fe(R)2(NH3)2 , Fe(R)2(NH3), Fe(R)2(CN)2 , R5NHCHNH. The figurewas made withXMAKEMOL ~Ref. 54!.
3230 J. Chem. Phys., Vol. 120, No. 7, 15 February 2004 M. P. Johansson and D. Sundholm
This article is copyrighted as indicated in the abstract. Reuse of AIP content is subject to the terms at: http://scitation.aip.org/termsconditions. Downloaded to IP:
195.19.233.81 On: Mon, 11 Nov 2013 21:35:20

III. RESULT AND DISCUSSION
A. Spin densities of the low-spin model
Figure 2 shows the spatial spin-density distribution forthe cationic model system, as calculated with various meth-ods. The local spin-density functional SVWN and thesemilocal BLYP give very similar results. The coupled clus-ter CC2 results differ somewhat. In the CC2 calculation, thecarbon atoms are surrounded by a diffuseb density of pcharacter. The nitrogens of the (NHCHNH)2 plane also havenotably morea spin around them, compared with the DFTspin density. Figure 3 shows the integrated, accumulated netspin density around the central iron. It is immediately seenthat CC2 concentrates the spin density at the iron atom to amuch lesser degree. At the covalent radius of iron, 1.2 Å, theDFT methods have recovered over 85% of the net spin den-sity, whereas CC2 has reached only 60%. Considering onlya-spin density does not change this, only traces ofb spin arefound this near the iron. The gross spin densities obtainedwith various methods are tabulated in Table I.
The spin distribution in the anionic model,@Fe(NHCHNH)2(CN)2#2, turned out to be very similar tothe one of the cationic model. The gross spin density wasslightly higher: 1.23 and 1.28 electrons for BLYP and CC2,respectively. Also, the vicinity of iron was found to have aslightly higher spin density. The CC2 spin density at the ironwas again significantly lower.
For the low-spin iron porphyrin models, then, CC2 con-firms the existence of significant regions with excessb elec-tron spin. The largest discrepancy between DFT and CC2 isseen in the spin density around the iron. It seems that theligating in-plane nitrogens are a tad too fond of spin densityat the CC2 level. To further investigate the matter, we per-formed a Mulliken population analysis38 of the spin andcharge densities on the central iron. In contrast to the grossspin density and the net spin density integration curves, theresults obtained with various population analyses are more ofa qualitative nature. They can, however, provide informationthat otherwise would be difficult to obtain. Table II shows thetotal spin densities,d-orbital spin densities, totald-orbitaloccupation~a plusb!, as well as the charge for the iron atomin the different low-spin compounds, obtained with the Mul-liken analysis. Both of the oxidized (S5 1
2) models have astrikingly larged occupation at the CC2 level, much higherthan obtained with the BLYP functional. It is also higher thanfor any of the higher-spin models, discussed later. The Mul-liken analysis also shows that, compared to the BLYP results,CC2 gives a lower spin density at iron, as well as ahighertotal d population. These facts combined indicates that thed-typeb occupation is too high at the CC2 level. This inter-pretation is further supported by the charge differencebetween the oxidized and reduced model, discussed inSec. III C.
For the cationic model system, a smaller basis set con-sisting of aug-cc-pVDZ for H, C, and N, and TZVPP for Fe,actually gave a very pronounced spin density at iron, reach-ing a neta spin well over one electron within a radius of 1.2.At the same time, thed-orbital occupation at the iron flippedfrom thedp orbital to an in-planedd . This orbital occupationis not the expected one for iron porphyrins,39 which indicatesthat the basis set is too small, or perhaps too unbalanced, fora proper CC2 description. For a good description of the CC2spin density~which in itself might or might not be a gooddescription of thetrue spin density!, an augmented triple-
FIG. 2. Contour plots of the spin-density distribution for the cationic low-spin model as obtained with~a! SVWN, ~b! BLYP, and~c! CC2. Dark areasrepresent excessa density, and light areas excessb density. An isocontourvalue of 0.003e has been used. The plot was created withGOPENMOL ~Refs.55–57!.
FIG. 3. The integrated spin density of@Fe(NHCHNH)2(NH3)2#1 (S512)
inside a sphere with a radius 0.5–3.5 Å, obtained with BLYP~whole!,SVWN ~dashed!, and CC2~dotted!. The graph was created withGNUPLOT
~Ref. 58!.
TABLE I. Gross spin density for the low-spin, ferric@Fe(NHCHNH)2(NH3)2#1 calculated with different methods. HF and CC2used the BLYP structure.
DFT ab initio
SVWN 1.18 HF 1.56BLYP 1.20 CC2 1.24PBE 1.20
TABLE II. Summary of Mulliken total spin densities~SD!, d-orbital spindensities, totald occupation, and charge at the iron atom for the low-spinmodels. R5NHCHNH.
Molecule S SD~Fe! d-SD~Fe! d-tot~Fe! q(Fe)
@Fe(R)2(NH3)2# 0 BLYP 6.30 10.81CC2 6.30 10.73
@Fe(R)2(NH3)2#1 12
BLYP 1.00 0.89 6.08 10.86
CC2 0.63 0.58 6.56 10.31@Fe(R)2(CN)2#2 1
2BLYP 0.95 1.00 6.36 10.75
CC2 0.70 0.62 6.74 10.61
3231J. Chem. Phys., Vol. 120, No. 7, 15 February 2004 Iron porphyrin models
This article is copyrighted as indicated in the abstract. Reuse of AIP content is subject to the terms at: http://scitation.aip.org/termsconditions. Downloaded to IP:
195.19.233.81 On: Mon, 11 Nov 2013 21:35:20

zeta quality basis set seems to be the minimum requirement.Larger basis sets could possibly improve the results further.In contrast, DFT succeeds with much smaller basis sets.3,25
Furthermore, the poor quality of the Hartree–Fock~HF!reference wave function is likely to degrade the CC2 descrip-tion. The low-spin models have several important configura-tions, leading to a multiconfiguration~MC! system. The mul-ticonfiguration character becomes apparent from the highspin-contamination of the HF wave function. The^S2& val-ues for the cationic and anionic low-spin models are 1.07 and0.87, respectively, when the ideal value,S(S11), is 0.75.Apparently HF would prefer the system to be in a higher spinstate, and artificially creates additional spin density to com-pensate for this. The HF gross spin densities of 1.56 and1.69, for the cationic and anionic systems, respectively, aremuch larger than for the other methods.
Another indicator of a multiconfigurational systemcomes from the occupation numbers of the CC2 natural or-bitals. The occupation numbers for the orbitals correspond-ing to the Hartree–Fock frontier orbitals of a given symme-try reveal MC character if they differ much from unity andzero. Table III lists the biggest deviations for botha andbspin. The low-spin models show the highest deviations.
Due to the MC character, DFT most likely gives a betterdescription of the low-spin system, whereas CC2 provides anindependent view of the qualitative effects of molecular spinpolarization. Commonly used exchange functionals accountfor certain terms that in wave-function formalism is consid-ered correlation effects, namely, part of the nondynamic orleft–right correlation.40–42 In passing we note that recentwork actually attributes this effect to the self-interaction er-ror present in most DFT functionals; self-interaction cor-rected exchange functionals seem to includeno electron cor-relation, whereas uncorrected exchange functionals introducenondynamic as well as some dynamic correlation via theelectronic self-interaction present.43,44 All exchange func-tionals used in this work do suffer from self-interaction, andthus mimic nondynamic correlation. Low-order coupled-cluster methods, on the other hand, do not describe multicon-figuration systems very well.45
B. Spin densities for the intermediate-and high-spin models
For the intermediate-spin model without axial ligands,Fe(NHCHNH)2 , and the high spin, five-coordinated model,
Fe(NHCHNH)2(NH3), the spin densities of both the oxi-dized and the reduced species were studied at BLYP andCC2 levels.
BLYP and CC2 described the spin density of theintermediate-spin model very similarly. No notable differ-ence in the graphical representation, shown in Fig. 4, can bediscerned. Therefore only CC2 results are shown. In bothoxidized and reduced intermediate-spin models,b-spin den-sities along the Fe–N bonds, similar to what is observed forthe low-spin models, and iron porphyrins,3 are seen. Theeffect can also be observed as a shallow dip in the net spindensity curve in Fig. 5. In this case the BLYP and CC2curves are very similar, in contrast to the low-spin case,where CC2 apparently underestimated the spin density at theiron ~or rather, overestimated theb-spin contribution!. Now,for the intermediate-spin models, the curves for especiallythe oxidized models almost overlap. The formally two un-pairedd orbitals of the reduced form are of perpendiculardp
type, i.e.,dxz anddyz . This gives the spin density its distinctp-doughnut form.
At reduction of the oxidized model, the additional elec-tron is added to adp-type orbital, rotated 45° in the cut planebetween the two@NHCHNH#2 groups. This is seen in thedifference between the absolute spin densities~i.e., bothaandb spin densities are taken as positive! between the oxi-dized and reduced model, shown in the rightmost picture ofFig. 4. At the same time, the already occupied, correspondingunrotateddp orbital loses some spin density. This only ac-counts for about half of the added unpaired electron density,whereas another half becomesp-spin density at the nitro-gens. The oxidized model thus proportionally delocalizes thespin density more compared to the reduced form; almost halfan unpaired electron is found beyond the ligating nitrogens.The gross spin densities were again slightly higher at theCC2 level compared to the BLYP calculations, see Table IV.The results of the Mulliken population analysis are shown inTable V.
It has to be noted that, at the CC2 level, the oxidizedmodel gave one naturalb orbital with anegativeoccupationnumber of20.043. Efforts to remedy this with stricter con-vergence criteria for both the HF reference function as wellas for the CC2 iterations were unsuccessful. This system alsoshows some multiconfiguration character according to the
TABLE III. CC2 natural occupation numbers for the HF frontier orbitalpairs of a specific symmetry, deviating most from unity and zero for thedifferent models. The S2& expectation value of the HF reference wavefunction is also shown. R5NHCHNH. HOMO5Highest Occupied Molecu-lar Orbital, LUMO5Lowest Unoccupied Molecular orbital.
Molecule
a b
^S2&HOMO LUMO HOMO LUMO
@Fe(R)2(NH3)2#1 0.912 0.088 0.886 0.106 1.07@Fe(R)2(CN)2#2 0.904 0.100 0.894 0.095 0.87@Fe(R)2# 0.960 0.008 0.957 0.016 2.01@Fe(R)2#1 0.931 0.083 0.926 0.068 3.80@Fe(R)2(NH3)# 0.960 0.037 0.959 0.039 6.01@Fe(R)2(NH3)#1 0.961 0.035 0.933 0.049 8.76
FIG. 4. Contour plots of the spin density for the intermediate-spin model,@Fe(NHCHNH)2#11/0. Left: C2/ferrous; middle: CC2/ferric; right: differ-ence in absolute spin density between ferric and ferrous forms. The ferricBLYP structure was used. The spin densities are plotted with an isocontourvalue of 0.003e, the difference-density plot with 0.005e. For the spindensities, dark areas again represent regions with excessa-spin density. Forthe difference in absolute spin density, dark areas represent higher spindensity in the oxidized, ferric form, and light areas show regions with higher
spin density in the reduced form: Upon oxidation fromS51 to S532, the
spin density increases in the dark regions and decreases in the light regions.The three-dimensional distribution at the BLYP level~not shown! is indis-tinguishable from the CC2 distribution.
3232 J. Chem. Phys., Vol. 120, No. 7, 15 February 2004 M. P. Johansson and D. Sundholm
This article is copyrighted as indicated in the abstract. Reuse of AIP content is subject to the terms at: http://scitation.aip.org/termsconditions. Downloaded to IP:
195.19.233.81 On: Mon, 11 Nov 2013 21:35:20
values of Table III, although not as pronounced as for thelow-spin models. The occurrence of negative occupationnumbers for nonvariational wave-function methods has beendiscussed by Gordonet al.,46 and taken as an indicator ofMC character in a system.
At the BLYP level, the lowest energy spin state of theoxidized five-coordinated model was actually found to haveintermediate spin. The intermediate-spin state (S5 3
2) wasfavored over the high spin state (S5 5
2) by 7.5 kcal/mol. Theorder of the states obtained with the hybrid functionalsB3LYP ~Refs. 19, 26, and 47! and PBE0~Refs. 20–24 and48! differed; B3LYP gave identical energies for both con-figurations, whereas PBE0 slightly favored the high-spinstate, giving an energy splitting of 2.9 kcal/mol. In any case,even if obtaining the right energy splittings between spinstates for iron complexes is problematic, property calcula-tions on the high-spin state are not likely to be affected, sincelow-spin configurations cannot intrude into the description ofa higher spin-state.
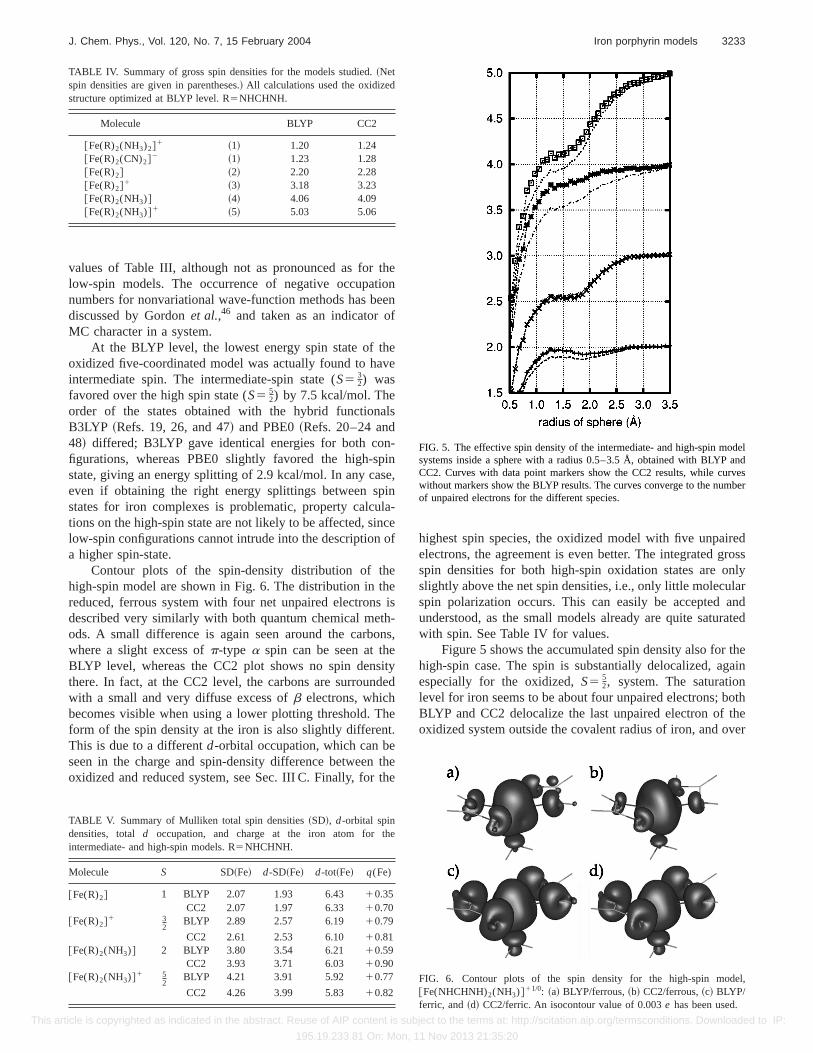
Contour plots of the spin-density distribution of thehigh-spin model are shown in Fig. 6. The distribution in thereduced, ferrous system with four net unpaired electrons isdescribed very similarly with both quantum chemical meth-ods. A small difference is again seen around the carbons,where a slight excess ofp-type a spin can be seen at theBLYP level, whereas the CC2 plot shows no spin densitythere. In fact, at the CC2 level, the carbons are surroundedwith a small and very diffuse excess ofb electrons, whichbecomes visible when using a lower plotting threshold. Theform of the spin density at the iron is also slightly different.This is due to a differentd-orbital occupation, which can beseen in the charge and spin-density difference between theoxidized and reduced system, see Sec. III C. Finally, for the
highest spin species, the oxidized model with five unpairedelectrons, the agreement is even better. The integrated grossspin densities for both high-spin oxidation states are onlyslightly above the net spin densities, i.e., only little molecularspin polarization occurs. This can easily be accepted andunderstood, as the small models already are quite saturatedwith spin. See Table IV for values.
Figure 5 shows the accumulated spin density also for thehigh-spin case. The spin is substantially delocalized, againespecially for the oxidized,S5 5
2, system. The saturationlevel for iron seems to be about four unpaired electrons; bothBLYP and CC2 delocalize the last unpaired electron of theoxidized system outside the covalent radius of iron, and over
TABLE IV. Summary of gross spin densities for the models studied.~Netspin densities are given in parentheses.! All calculations used the oxidizedstructure optimized at BLYP level. R5NHCHNH.
Molecule BLYP CC2
@Fe(R)2(NH3)2#1 ~1! 1.20 1.24@Fe(R)2(CN)2#2 ~1! 1.23 1.28@Fe(R)2# ~2! 2.20 2.28@Fe(R)2#1 ~3! 3.18 3.23@Fe(R)2(NH3)# ~4! 4.06 4.09@Fe(R)2(NH3)#1 ~5! 5.03 5.06
TABLE V. Summary of Mulliken total spin densities~SD!, d-orbital spindensities, totald occupation, and charge at the iron atom for theintermediate- and high-spin models. R5NHCHNH.
Molecule S SD~Fe! d-SD~Fe! d-tot~Fe! q(Fe)
@Fe(R)2# 1 BLYP 2.07 1.93 6.43 10.35CC2 2.07 1.97 6.33 10.70
@Fe(R)2#1 32
BLYP 2.89 2.57 6.19 10.79
CC2 2.61 2.53 6.10 10.81@Fe(R)2(NH3)# 2 BLYP 3.80 3.54 6.21 10.59
CC2 3.93 3.71 6.03 10.90@Fe(R)2(NH3)#1 5
2BLYP 4.21 3.91 5.92 10.77
CC2 4.26 3.99 5.83 10.82
FIG. 5. The effective spin density of the intermediate- and high-spin modelsystems inside a sphere with a radius 0.5–3.5 Å, obtained with BLYP andCC2. Curves with data point markers show the CC2 results, while curveswithout markers show the BLYP results. The curves converge to the numberof unpaired electrons for the different species.
FIG. 6. Contour plots of the spin density for the high-spin model,@Fe(NHCHNH)2(NH3)#11/0: ~a! BLYP/ferrous,~b! CC2/ferrous,~c! BLYP/ferric, and~d! CC2/ferric. An isocontour value of 0.003e has been used.
3233J. Chem. Phys., Vol. 120, No. 7, 15 February 2004 Iron porphyrin models
This article is copyrighted as indicated in the abstract. Reuse of AIP content is subject to the terms at: http://scitation.aip.org/termsconditions. Downloaded to IP:
195.19.233.81 On: Mon, 11 Nov 2013 21:35:20

half of the electron is delocalized further beyond the ligatingnitrogens. This has been noted earlier by Blomberg and Sieg-bahn in a study on high-spin Mn and Fe complexes,49 wherethe effect was attributed to a preference for a 3d occupationof six at the iron. The same preference has been noted also inrecent works.50,51 With six d electrons, two have to bepaired, resulting in a total spin of four at the metal.
In an NMR study of high-spin myogolobin, Maet al.discussed the appearance of negative~b! p spin density.52
They concluded that the higher spin density at the ironshould entice significantly higher spin density inp-type eg
orbitals for the high-spin case compared to a low-spin sys-tem. This does not contradict the findings here, where thelow-spin and intermediate-spin models show higher spin po-larization than the high-spin model. The largest contributionto b-spin density in the models with12<S< 3
2 comes fromorbitals that correspond to theeu-type orbitals inD4h sym-metry. These orbitals show the characteristic, non-p-type,pacifier-shape along the Fe–N bonds in the plane. In thehigh-spin model, the delocalizeda-spin density around ironalready engulfs these regions of the molecules, so nob spincan appear there. A computational study of full iron-porphyrin models of different spin states should be able todiscern the amount of onlyp-type spin density, as this is themajor component of spin density from thea carbons out-ward.
An experimental detection of the negative spin densityalong the Fe–N bonds remains a challenge for EPR andNMR spectroscopists.
C. Change in electron density upon reduction
In a previous work, we discussed the extensive chargedelocalization that takes place upon reduction of heme,8
based on DFT calculations. Here, we study the same effectfor both the low-spin, six-coordinated, and the high-spin,five-coordinated model systems.
When the low-spin system, Fe(NHCHNH)2(NH3)2 , re-ceives an electron and is reduced to its ferrous form, all spinsget paired, which leads to a closed-shell system. But even ifthe main portion of the spin density is found in the vicinityof the iron, the total electron density around the iron changesto a much smaller extent. In other words, the charge of theiron atom itself stays almost unchanged on reduction. Thesame is true for the high-spin model, except that the systemstill possesses four unpaired electrons after reduction. Spinand charge densities are, if not totally independent observ-ables, quite different in nature. The manifestation of thequantum-mechanical spin density in the three-dimensionalCartesian space can move and change over large distanceswith little effort. In contrast, the movement of the more clas-sical charge density is energetically much more constrained.
Figure 7 shows the electron density differences betweenthe oxidized and the reduced models. Very prominent chargereorganization can be noted. Thed orbital getting ‘‘filled’’upon reduction is seen as a region of electron gain~darkareas!. But at the same time, regions in the immediate vicin-ity of the filled d orbital experience a loss of electron density~light areas!. For the low-spin case, both CC2 and BLYP pairthe singly occupiedd orbital seen in Fig. 2. Judging by the
picture~Fig. 7, top!, the addition of electron density to thedorbital is less at the CC2 level than at the BLYP level, inaccordance with the smaller spin density at the CC2 level.For the high-spin model, the difference in the BLYP and CC2spin density distributions around iron in the reduced formgets an explanation; at the CC2 level, adp orbital is filled,whereas BLYP fills add orbital. A direct investigation of thespin density difference between the oxidized and reducedsystems reveals the same. Figure 8 shows the difference inabsolute spin density between the oxidized and reducedhigh-spin model. Again, in analogy with the addition of totalcharge density, the increase in spin density in one type ofdorbital at the iron is partly cancelled by a general decrease inspin density in the immediate surroundings. The correlationbetween changes in spin density and changes in total electrondensity upon oxidation is quite pronounced. Generally, at theiron, electrons are gained in regions where the oxidized formhas higher spin density, while the correlation in the peripheryis lower.
It is, however, impossible to judge the extent of thecharge reorganization by just looking at the contour plots. Adirect integration of the electron density difference aroundiron, Fig. 9, reveals the large delocalization of the added
FIG. 7. Contour plots of the difference in electron-density distribution be-tween the reduced and oxidized forms of Fe(R)2(NH3)2 ~top! andFe(R)2(NH3) ~bottom!, R5NHCHNH. Plots for BLYP ~left! and CC2~right! are shown. Dark areas show regions with higher electron density inthe reduced form, light areas show regions with higher electron density inthe oxidized form. Upon reduction, then, electron density increases in thedark areas, and decreases in the light. An isocontour value of 0.005e hasbeen used.
FIG. 8. Contour plots of the difference in absolute spin density between theoxidized and reduced high-spin model. Dark areas show regions with higherspin density in the oxidized form, light areas show regions with higher spin
density in the reduced form. Upon oxidation fromS52 to S552, the spin
density increases in the dark regions and decreases in the light regions. Plotsfor ~a! BLYP and ~b! CC2 are shown. An isocontour value of 0.005e hasbeen used.
3234 J. Chem. Phys., Vol. 120, No. 7, 15 February 2004 M. P. Johansson and D. Sundholm
This article is copyrighted as indicated in the abstract. Reuse of AIP content is subject to the terms at: http://scitation.aip.org/termsconditions. Downloaded to IP:
195.19.233.81 On: Mon, 11 Nov 2013 21:35:20
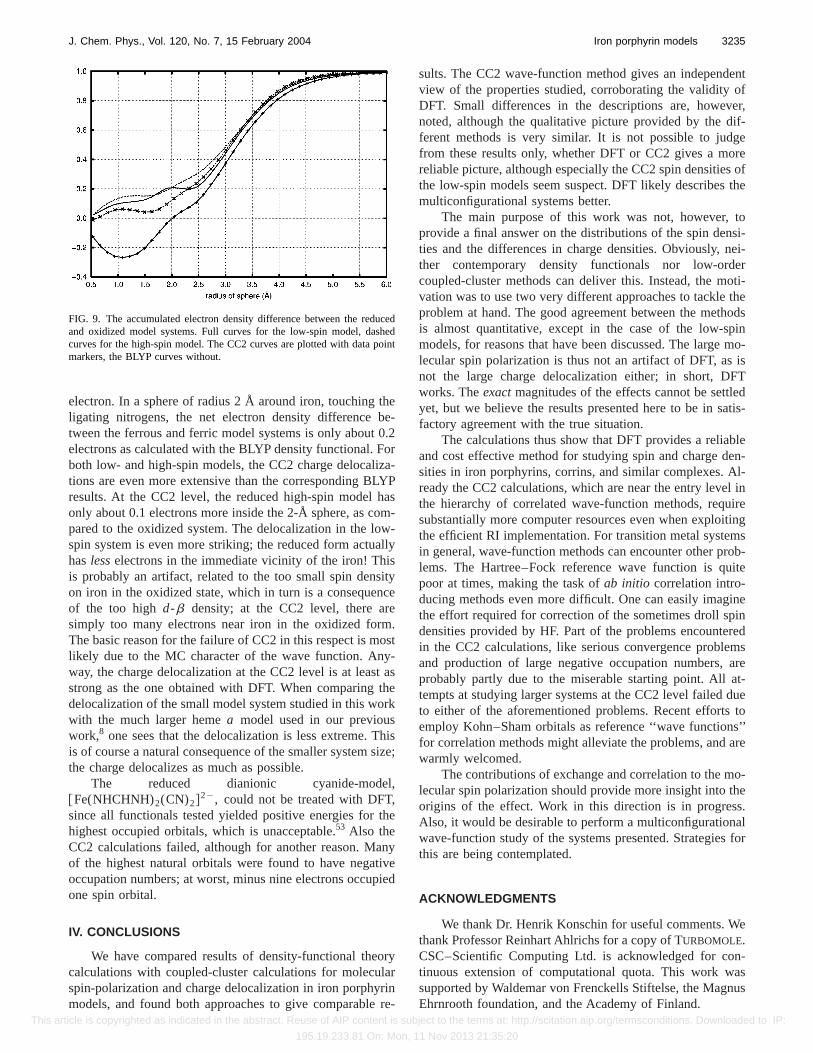
electron. In a sphere of radius 2 Å around iron, touching theligating nitrogens, the net electron density difference be-tween the ferrous and ferric model systems is only about 0.2electrons as calculated with the BLYP density functional. Forboth low- and high-spin models, the CC2 charge delocaliza-tions are even more extensive than the corresponding BLYPresults. At the CC2 level, the reduced high-spin model hasonly about 0.1 electrons more inside the 2-Å sphere, as com-pared to the oxidized system. The delocalization in the low-spin system is even more striking; the reduced form actuallyhaslesselectrons in the immediate vicinity of the iron! Thisis probably an artifact, related to the too small spin densityon iron in the oxidized state, which in turn is a consequenceof the too highd-b density; at the CC2 level, there aresimply too many electrons near iron in the oxidized form.The basic reason for the failure of CC2 in this respect is mostlikely due to the MC character of the wave function. Any-way, the charge delocalization at the CC2 level is at least asstrong as the one obtained with DFT. When comparing thedelocalization of the small model system studied in this workwith the much larger hemea model used in our previouswork,8 one sees that the delocalization is less extreme. Thisis of course a natural consequence of the smaller system size;the charge delocalizes as much as possible.
The reduced dianionic cyanide-model,@Fe(NHCHNH)2(CN)2#22, could not be treated with DFT,since all functionals tested yielded positive energies for thehighest occupied orbitals, which is unacceptable.53 Also theCC2 calculations failed, although for another reason. Manyof the highest natural orbitals were found to have negativeoccupation numbers; at worst, minus nine electrons occupiedone spin orbital.
IV. CONCLUSIONS
We have compared results of density-functional theorycalculations with coupled-cluster calculations for molecularspin-polarization and charge delocalization in iron porphyrinmodels, and found both approaches to give comparable re-
sults. The CC2 wave-function method gives an independentview of the properties studied, corroborating the validity ofDFT. Small differences in the descriptions are, however,noted, although the qualitative picture provided by the dif-ferent methods is very similar. It is not possible to judgefrom these results only, whether DFT or CC2 gives a morereliable picture, although especially the CC2 spin densities ofthe low-spin models seem suspect. DFT likely describes themulticonfigurational systems better.
The main purpose of this work was not, however, toprovide a final answer on the distributions of the spin densi-ties and the differences in charge densities. Obviously, nei-ther contemporary density functionals nor low-ordercoupled-cluster methods can deliver this. Instead, the moti-vation was to use two very different approaches to tackle theproblem at hand. The good agreement between the methodsis almost quantitative, except in the case of the low-spinmodels, for reasons that have been discussed. The large mo-lecular spin polarization is thus not an artifact of DFT, as isnot the large charge delocalization either; in short, DFTworks. Theexactmagnitudes of the effects cannot be settledyet, but we believe the results presented here to be in satis-factory agreement with the true situation.
The calculations thus show that DFT provides a reliableand cost effective method for studying spin and charge den-sities in iron porphyrins, corrins, and similar complexes. Al-ready the CC2 calculations, which are near the entry level inthe hierarchy of correlated wave-function methods, requiresubstantially more computer resources even when exploitingthe efficient RI implementation. For transition metal systemsin general, wave-function methods can encounter other prob-lems. The Hartree–Fock reference wave function is quitepoor at times, making the task ofab initio correlation intro-ducing methods even more difficult. One can easily imaginethe effort required for correction of the sometimes droll spindensities provided by HF. Part of the problems encounteredin the CC2 calculations, like serious convergence problemsand production of large negative occupation numbers, areprobably partly due to the miserable starting point. All at-tempts at studying larger systems at the CC2 level failed dueto either of the aforementioned problems. Recent efforts toemploy Kohn–Sham orbitals as reference ‘‘wave functions’’for correlation methods might alleviate the problems, and arewarmly welcomed.
The contributions of exchange and correlation to the mo-lecular spin polarization should provide more insight into theorigins of the effect. Work in this direction is in progress.Also, it would be desirable to perform a multiconfigurationalwave-function study of the systems presented. Strategies forthis are being contemplated.
ACKNOWLEDGMENTS
We thank Dr. Henrik Konschin for useful comments. Wethank Professor Reinhart Ahlrichs for a copy of TURBOMOLE.CSC–Scientific Computing Ltd. is acknowledged for con-tinuous extension of computational quota. This work wassupported by Waldemar von Frenckells Stiftelse, the MagnusEhrnrooth foundation, and the Academy of Finland.
FIG. 9. The accumulated electron density difference between the reducedand oxidized model systems. Full curves for the low-spin model, dashedcurves for the high-spin model. The CC2 curves are plotted with data pointmarkers, the BLYP curves without.
3235J. Chem. Phys., Vol. 120, No. 7, 15 February 2004 Iron porphyrin models
This article is copyrighted as indicated in the abstract. Reuse of AIP content is subject to the terms at: http://scitation.aip.org/termsconditions. Downloaded to IP:
195.19.233.81 On: Mon, 11 Nov 2013 21:35:20

1P. Hohenberg and W. Kohn, Phys. Rev.136, B864 ~1964!.2W. Kohn and L. J. Sham, Phys. Rev.140, A1133 ~1965!.3M. P. Johansson, D. Sundholm, G. Gerfen, and M. Wikstro¨m, J. Am.Chem. Soc.124, 11 771~2002!.
4W. D. Horrocks and E. S. Greenberg, Biochim. Biophys. Acta322, 38~1973!.
5H. M. Goff, J. Am. Chem. Soc.103, 3714~1981!.6H. Tan, U. Simonis, N. V. Shokhirev, and F. A. Walker, J. Am. Chem. Soc.116, 5784~1994!.
7N. V. Shokhirev and F. A. Walker, J. Phys. Chem.99, 17 795~1995!.8M. P. Johansson, M. R. A. Blomberg, D. Sundholm, and M. Wikstro¨m,Biochim. Biophys. Acta1553, 183 ~2002!.
9C. C. Page, C. C. Moser, X. Chen, and P. L. Dutton, Nature~London! 402,47 ~1999!.
10K. Capelle and G. Vignale, Phys. Rev. Lett.86, 5546~2001!.11N. Argaman and G. Makov, Phys. Rev. B66, 052413~2002!.12J. P. Perdew, A. Savin, and K. Burke, Phys. Rev. A51, 4531~1995!.13J. P. Perdew, M. Ernzerhof, K. Burke, and A. Savin, Int. J. Quantum
Chem.61, 197 ~1997!.14J. Grafenstein, A. M. Hjerpe, E. Kraka, and D. Cremer, J. Phys. Chem. A
104, 1748~2000!.15L. Noodleman, D. Post, and E. J. Baerends, Chem. Phys.64, 159 ~1982!.16T. Bally and G. N. Sastry, J. Phys. Chem. A101, 7923~1997!.17M. Sodupe, J. Bertran, L. Rodrı´guez-Santiago, and E. J. Baerends, J. Phys.
Chem. A103, 166 ~1999!.18A. D. Becke, Phys. Rev. A38, 3098~1988!.19C. Lee, W. Yang, and R. G. Parr, Phys. Rev. B37, 785 ~1988!.20P. A. M. Dirac, Proc. R. Soc. London, Ser. A123, 714 ~1929!.21P. A. M. Dirac, Proc. Cambridge Philos. Soc.26, 376 ~1930!.22J. C. Slater, Phys. Rev.81, 385 ~1951!.23J. P. Perdew and Y. Wang, Phys. Rev. B45, 13 244~1992!.24J. P. Perdew, K. Burke, and M. Ernzerhof, Phys. Rev. Lett.78, 1396
~1996!.25M. Johansson, Master’s thesis, 2002, University of Helsinki.26S. H. Vosko, L. Wilk, and M. Nusair, Can. J. Phys.58, 1200~1980!.27K. Eichkorn, O. Treutler, H. O¨ hm, M. Haser, and R. Ahlrichs, Chem.
Phys. Lett.240, 283 ~1995!.28K. Eichkorn, F. Weigend, O. Treutler, and R. Ahlrichs, Theor. Chem. Acc.
97, 119 ~1997!.29O. Christiansen, H. Koch, and P. Jørgensen, Chem. Phys. Lett.243, 409
~1995!.30C. Hattig and F. Weigend, J. Chem. Phys.113, 5154~2000!.31R. Ahlrichs, M. Bar, M. Haser, H. Horn, and C. Ko¨lmel, Chem. Phys. Lett.
162, 165 ~1989!.
32T. H. Dunning, Jr., J. Chem. Phys.90, 1007~1989!.33R. Kendall, T. H. Dunning, Jr., and R. J. Harrison, J. Chem. Phys.96,
6796 ~1992!.34A. Schafer, C. Huber, and R. Ahlrichs, J. Chem. Phys.100, 5829~1994!.35A. J. H. Wachters, J. Chem. Phys.52, 1033~1970!.36F. Weigend, M. Ha¨ser, H. Patzelt, and R. Ahlrichs, Chem. Phys. Lett.294,
143 ~1998!.37F. Weigend, A. Ko¨hn, and C. Ha¨ttig, J. Chem. Phys.116, 3175~2002!.38R. S. Mulliken, J. Chem. Phys.23, 1833~1955!.39F. A. Walker, Coord. Chem. Rev.185–186, 471 ~1999!.40O. V. Gritsenko, R. van Leeuwen, and E. J. Baerends, Int. J. Quantum
Chem.60, 1375~1996!.41O. V. Gritsenko, P. R. T. Schipper, and E. J. Baerends, J. Chem. Phys.107,
5007 ~1997!.42N. C. Handy and A. J. Cohen, Mol. Phys.99, 403 ~2001!.43V. Polo, E. Kraka, and D. Cremer, Mol. Phys.100, 1771~2002!.44V. Polo, J. Gra¨fenstein, E. Kraka, and D. Cremer, Theor. Chem. Acc.109,
22 ~2003!.45T. Helgaker, P. Jørgensen, and J. Olsen,Molecular Electronic-Structure
Theory~John Wiley & Sons, Chichester, 2000!.46M. S. Gordon, M. W. Schmidt, G. M. Chaban, K. R. Glaesemann, W. J.
Stevens, and C. Gonzalez, J. Chem. Phys.110, 4199~1999!.47A. D. Becke, J. Chem. Phys.98, 5648~1993!.48J. P. Perdew, M. Ernzerhof, and K. Burke, J. Chem. Phys.105, 9982
~1996!.49M. R. A. Blomberg and P. E. M. Siegbahn, Theor. Chem. Acc.97, 72
~1997!.50K. P. Jensen and U. Ryde, ChemPhysChem4, 413 ~2003!.51D. M. A. Smith, M. Dupuis, E. R. Vorpagel, and T. P. Straatsma, J. Am.
Chem. Soc.125, 2711~2003!.52D. Ma, R. Musto, K. M. Smith, and G. N. La Mar, J. Am. Chem. Soc.125,
8494 ~2003!.53N. Rosch and S. B. Trickey, J. Chem. Phys.106, 8940~1997!.54M. P. Hodges,XMAKEMOL : A program for visualizing atomic and molecu-
lar systems, version 5~2001!, http://savannah.nongnu.org/projects/xmakemol/
55L. Laaksonen, J. Mol. Graphics10, 33 ~1992!.56D. L. Bergman, L. Laaksonen, and A. Laaksonen, J. Mol. Graphics Mod-
ell. 15, 301 ~1997!.57http://www.csc.fi/gopenmol/58http://www.gnuplot.info/
3236 J. Chem. Phys., Vol. 120, No. 7, 15 February 2004 M. P. Johansson and D. Sundholm
This article is copyrighted as indicated in the abstract. Reuse of AIP content is subject to the terms at: http://scitation.aip.org/termsconditions. Downloaded to IP:
195.19.233.81 On: Mon, 11 Nov 2013 21:35:20





























