Embed Size (px)
Citation preview

SPECTRAL-KINETIC PATTERNS OF THE PHOTOISOMERIZATION OF
o-AMINOAZOBENZENES
Ya. N. Malkin, N. O. Pirogov, L. S. Tul'chinskaya, and V. A. Kuz'min
UDC 541.127:543.422:541.14: 542.952.1:547.566.33
The study of isomerization of aromatic azo compounds is of interest since they are used as photochromic agents for light filters of varying optical density, which automatically alter light transmission depending on the level of illumination.
In this study, we investigated the o-aminoazo compounds (I)-(III), obtained according to [i], and o-dimethylaminobenzenes (IV)-(V), obtained by the method described in [2, 3]. In condensation with barbituric acid, such azo compounds form tricyclic condensed alloxazines and isoalloxazines [3]; riboflavin (vitamin Ba) belongs to these latter. The study of photo- isomerization of compounds (I)-(V) [particularly (1)-(Ill)] is thus essential to elucidate the role of such structures in the production process of riboflavin and its analogs
Rs / Compound R, R, R ~ m R'.
N _ R ~ R 3 / / (I) H CH~ H H }I
_/2--~_N =N_/2--~_ R2 (lI) CHs CH3 H H H ~ k_/ (Ill) CH3 CH3 CH3 H H
/ R \ I (IV) CHs CH~ H CH3 H IV) CH3 CH3 H CH, CH~
EXPERIMENTAL
The absorption spectra of compounds (I)-(V) were recorded on a Specord UV-VIS spectro- photometer; the absorption spectra and the kinetics of disappearance of short-lived intermed- iate products were observed on a pulsed photolysis instrument [4].
DISCUSSION OF RESULTS
Pulsed irradiation of solutions of compounds (1)-(V) in polar (alcohol, DMFA) or non- polar (toluene, hexane) solvents by UV or visible light will induce the formation of an intermediate product with an absorption spectrum which, compared to that of the initial azo compound, has shifted towards the long-wave region (Fig. i).
Ar' /
N=N +h~-~ N=N / / \
Ar Ar Ar'
The d i f f e r e n t i a l a b s o r p t i o n s p e c t r a o f compounds ( I ) - ( V ) a r e c l o s e t o t h o s e o f t h e t r a n s and c i s i s o m e r s o f a z o b e n z e n e , b u t s h i f t e d t o w a r d s t h e l o n g - w a v e r e g i o n [ 5 ] . S i n c e u n d e r irradiation the absorption spectrum of the short-lived product will retain that of the initial compound in various parts, it has been assumed that at about 20~ the o-aminoazo compound in solution is present only in the form of a single stereoisomer (trans); the pro- cess observed constitutes a photochemical trans--cis-photolsomerization.
The kinetics of the decay of the short-lived cis isomer of (1)-(V), measured from ad- sorption and fading (Fig. 2), is subject to a first-order law with rate constants weakly depending on the substituent on the benzene ring and on the nature of the solvent (Table I). Nor do the rate constants of thermal cis--trans isomerization of (1)-(V) depend on the o- aminoazo compound concentration in the i0-~-i0 -4 M range.
The fact that the rate constants of thermal isomerization are independent of the nature of the substituents has been interpreted in [6, 7] as proof of an inversion mechanism of iso-
Institute of Chemical Physics, Academy of Sciences of the USSR, Moscow. All-Union Scien- tific Research Institute for Vitamins, Moscow. Translated from Izvestiya Akademii Nauk SSSR, Seriya Khimicheskaya, No. 3, pp. 533-539, March, 1980. Original article submitted January 18, 1979.
354 0568-5230/80/2903-0354507.50 �9 1980 Plenum Publishing Corporation

TABLE i. Rate Constants and Activation Energy of Cis-Trans Iso- merization of o-Aminoazo Compounds
Com- pound
(I) (n) (iit) (iv) (v)
~max, ILrn
trans- I ci~- isomer i~omer
440 430 441 440 443 445
lEa, kcal / h, sec'* Imble
toluene
0,tt 4,6 0,t2 4,8 0,t7 4,4
0,072 5,0
I k, sec "I IE~ kcal/mole
propanol
0,080 5,7 0,065 5,0 0,t0 5,4
merization, i.e., a mechanism whereby change in the molecular structure proceeds through in- version of part of the molecule in the plane [8] rather than by its rotation around the N=N bond.
It has been found, via pulsed photolysis, that irradiation of the trans form will yield not only the cis isomers of (I)-(III) but also a short-lived intermediate product with a life of 0.1-0.2 sec (see Fig. 2). Such intermediate products were recorded for the above compounds in benzene, toluene, hexane, methanol, ethanol, propanol, and CCI~. Prolonged irradiation of solutions of compounds (I)-(III) in any of the solvents used failed to induce irreversible changes in the absorption spectra of the initial products; this indicates that formation of the intermediate product is not accompanied by irreversible side processes. According to [9], 4-aminoazobenzene will not form any intermediate products under pulsed photoactivation. The formation of a short-lived intermediate product from 2-aminoazaobenzene (whose amino group is adjacent to the azo group) is apparently determined by a reversible transfer of an H atom from the amino to the azo group. Subutitution of H atoms in the amino group of com- pounds (I)-(I!I) by the CHa group (compound (V)) will exclude the possibility of intramolecu- far hydrogen transfer according to the equation
NH2 NH H / / J 1 /
/ \ / \
The absence of an intermediate product in this case indicates that for (I)-(III), formation of the product is related to the reversible transfer of an H atom.
The reaction of the H atom transfer apparently proceeds as a result of its attraction by the azo group due to the latter's excited state. We may exclude the possibility of an inter- molecular nature of this transfer in a solution of (I)-(III) in CC14, where this H atom trans- fer was also observed. The detachment of the H atom by the excited electrons of the azo group has been assumed in [i0], based on calculation of the properties of the azo group in the excited state, but this reaction had not been recorded in the experiment, although photo- reduction of azobenzene to hydrazobenzene [ii] had been observed in other studies: this pro- cess has certainly started with an initial split-off of an H atom from the solvent molecule.
No intermediate product formation was observed in photoactivation of solutions of com- pounds (I)-(III) in alcohol in the presence of AcOH il0 -5 M). On the oscillogram we observed only the process of cis--trans isomerization of azo compounds around the N=~I bond. The quantum yield of photoisomerization increases 2-3-fold under these conditions, and the rate constant of thermal izomerization also increases by the same order of magnitude. We believe that the absence of a reversible transfer of the H atom in acid solutions is due to conversion of com- pounds (I)-(III) into quaternary salts:
NH3 + / /
--@--N=N-.2-AS._ / \
Transfer of an H atom from the positively charged ammonium ion is thereby rendered more dif-
ficult.
For the direct proof that the hydrogen atom can be split-off by the azo group in its excited state, we used the reaction of o-aminoazo compounds [compounds (llI)] with the hydrogen
355
o z ~ t , s ec O Z # ~,sec
0
.-#,oo
-#,aa
=#,tz
I q50
/ / / /
i// ,
~ m .4.V
0,02
f # !
-#,, 0r
~50 / 5o0 i
,t
,, / / / ,,, '//
\ ~ /
Fig. i Fig. 2
Fig. I. Differential absorption spectra for compounds (I)-(III) taken in toluene 10 -3 sec after the activating pulse (UFS-5 filter): i) (I); 2) (II) ; 3) ( i x I ) .
Fig. 2. Differential absorption spectra of compound (II) (a) and oscil- lograms of the decay of cis isomers (b = 440, c = 520 nm) in 2-propanol under activation by UV light (UFS-5 filter), i) After i0 -s sec; 2) after 0.I sec; 3) 7 sec after the flash.
donor 2,6-diphenylstearoxyphenol (Fig. 3). Under irradiation of a solution of (III) in hexane in the presence of this electron donor with light of ~ > 400 nm (i.e., in the region where there is no absorption of the phenol itself), we recorded the absorption spectrum of the well-known phenoxy radical [12]. The phenoxy radicals are formed as a result of the split- off of hydrogen by o-aminoazobenzene (III) according to the reaction
A*-+-POH . -+AH + PO"
where A* is the excited o-aminoazobenzene, POH is the phenol, and PO" is the phenoxy radical.
This thus explains the increased yield of the intermediate product of H transfer in al- cohols since this proceeds due to an intermolecular reaction involving detachment of the H atom from the alcohol by the azo compound. We imagine the overall scheme of the processes leading to the reversible transfer of an H atom in the o-aminoazo compound molecule from the amino to the azo group as follows:
-S* cis - H
cis -S*
tran~-So q- Av ~ tram - 8 "
tran~-S* ~ z+ trans -So
tmns_~* --+ cis..-S.
cis ~'So + hv --~ d s - S *
-+ e .~ -H
-+ :cis-S o
-+ t~aaS -3 o
CO) (1)
(2)
(3)
(4)
(5)
(6)
where cisiSo, and trans-So are the ground, and cis-S* and trans-S* the excited, states of o- aminoazobenzene, and cis-H is the product of transfer of the H atom to the azo group.
It is assumed that separation of the H atom proceeds only in the excited state of the cis form of compounds (I)-(III); this occurs as a result of absorption of a second light
356

//
J
CI81t3 7
qgg 5g# ##g h, nmTgg
Fig. 3. Spectrum of the 2,6-di- phenyl-4-stearoxyphenol radical in hexane.
quantum by the cis form [the course of the curve expressing optical density from c to d in Fig. 2 is determined by the process (5) of cis formation as a result of the reversible H transfer]. The participation of the cis form in detaching the H atom agrees with data in [ii] in that the photoreduction of azobenzene to hydrazobenzene occurs only via the cis form. To confirm the mechanism (1)-(6) of the reaction involving the intramolecular transfer of the H atom in the excited state of the cis form of (I)-(III), we investigated the dependence of the yield in the cis-H form on the intensity of light absorption (Fig. 4) (change in the yield of the cis-H form was estimated as a change in the distance from c to d on the oscillograms) o The quadratic dependence obtained supports the assumption that detachment of the H atom re- quires absorption of a second light quantum by the photoinduced form.
To determine the nature of the electron-induced state responsible for photoisomerizatlon of o-aminoazo compounds, we investigated the photoinduction of solutions (I)-(III) in the presence of quenchers with a heavy atom belonging to various classes of organic compounds, i.e., 2,3-dibromobutane, 2-bromofluorene, KBr, and KI. With an increase of the quencher con- centration, the quantum yield of photoisomerization was found to rise significantly (Fig. 5)9 due to acceleration of the intercombinational conversion into the triplet state from which the photoisomerization process arises.
Based on the experiments with quenchers, we are proposing the following scheme for the process of photoisomerization of compounds (1)-(V):
tram-So + h~, --+ tmns -S 1
tram-S 1 -+ t~ans_So tram-S~ -+ ~ans-T~
t~ans.T~ --+ cis-So
trans-T 1 ._>. t~ans.S o
trans-S1 + Q -+ tram-T 1
tram-T1 + Q -+- tram-S o
(0)
(7)
(8)
(9)
(i0)
(11)
(12)
where trans-T~ and trans-S~ are the triplet and singlet states of the trans isomers (1)-(V), respectively; Q is the quencher containing the heavy atom. In this case, the quantum yields of photoisomerization of (1)-(V) will be as follows in the presence and the absence of the quencher:
k8 k9 (P ----- k7 ~- kS " k9 ~- k10 (13a)
k8 + h'11 [Q] k9 (13b) q~o = k 7 + ks + kll[Q] k9 +klo+ k~[Ql
Assuming that at the concentrations used the quencher will insignificantly change the life of the trans-T~ triplet state, i.e., that [Q]kta << k, + k~o, we obtain
ks { 1 + k, (14) - - k T + k 8 k s + k , , [ Q l J
357
Z,5
2,0
0,5 I I
g. 75 ~g U 40
Fig. 4
! I I o s ~z
Fig. 5
I Y
/0
,F
O,J Q,#/
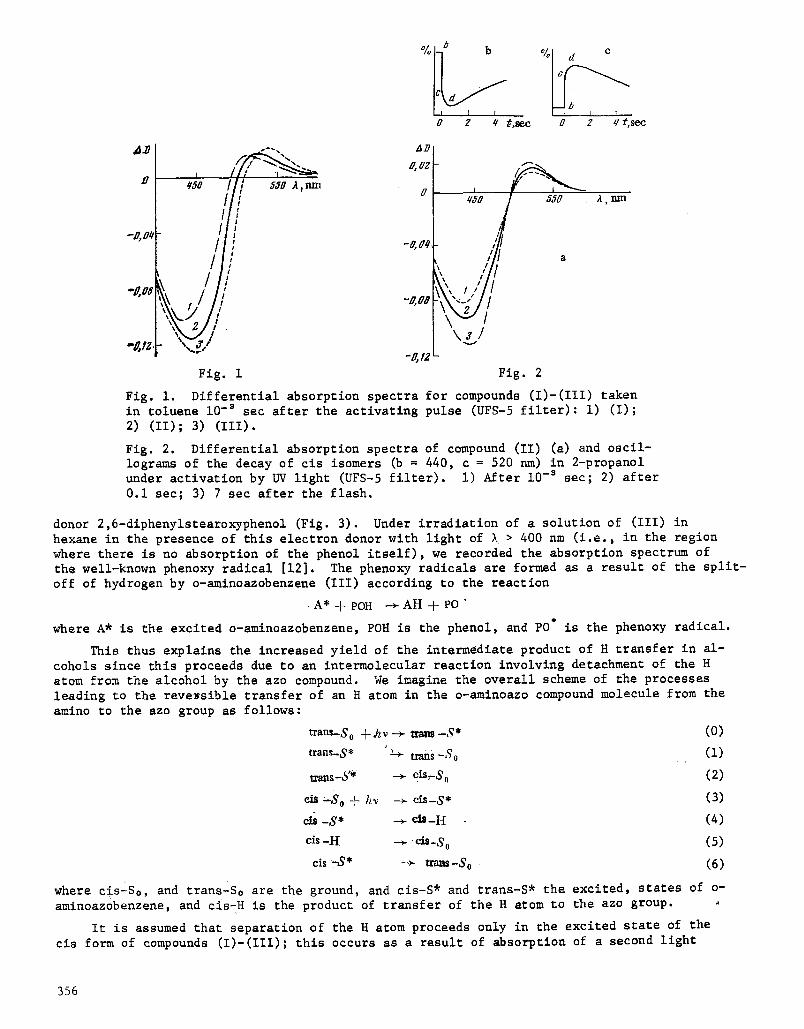
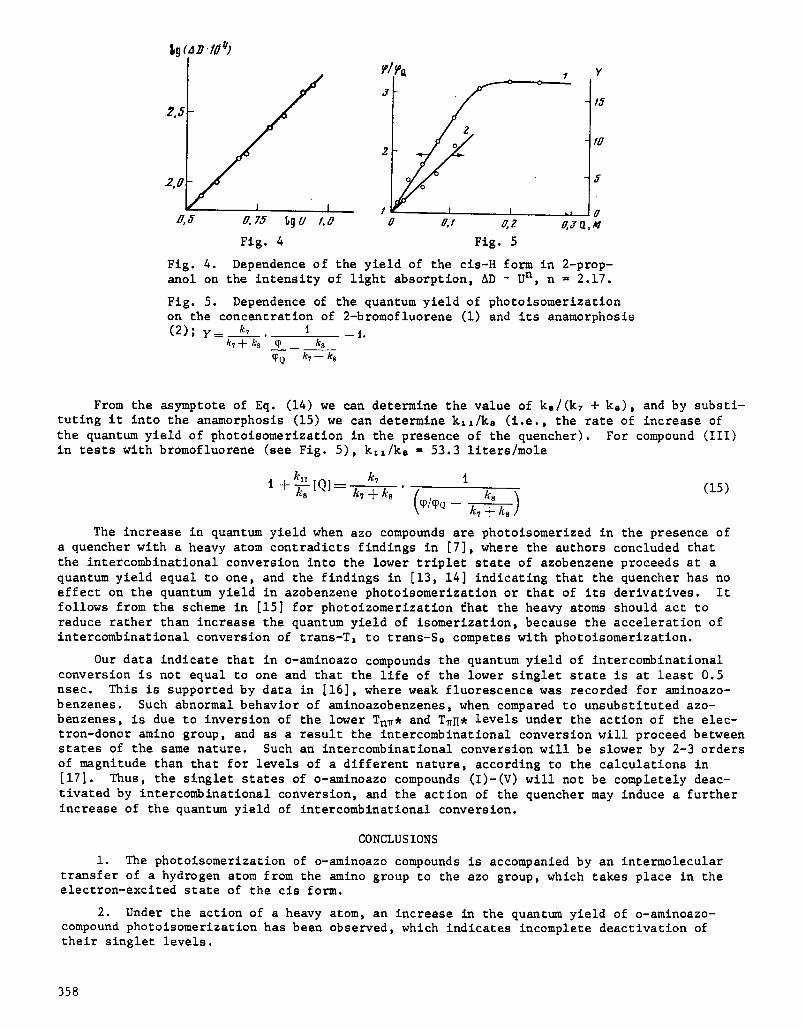
Fig. 4. Dependence of the yield of the cls-H form in 2-prop- anol on the intensity of light absorption, AD ~ U n, n = 2.17.
Fig. 5. Dependence of the quantum yield of photoisomerization on the concentration of 2-bromofluorene (i) and its anamorphosls (2) ; y = k, I I.
k7 + ks " ~ ks ~ O - - k7 - - ks
From the asymptote of Eq. (14) we can determine the value of k./(k7 + k,), and by substi- tuting it into the anamorphosis (15) we can determine k**/k. (i.e., the rate of increase of the quantum yield of photolsomerlzatlon in the presence of the quencher). For compound (III) in tests with bromofluorene (see Fig. 5), k:,/k, ffi 53.3 liters/mole
k . k7 i k8 (15) t + ~-8 [Q]___-- k:+ks (~l~e k,+ks)
The increase in quantum yield when azo compounds are photoisomerized in the presence of a quencher with a heavy atom contradicts findings in [7], where the authors concluded that the intercombinational conversion into the lower triplet state of azobenzene proceeds at a quantum yield equal to one, and the findings in [13, 14] indicating that the quencher has no effect on the quantum yield in azobenzene photoisomerization or that of its derivatives. It follows from the scheme in [15] for photoizomerlzation Chat the heavy atoms should act to reduce rather than increase the quantum yield of isomerization, because the acceleration of intercombinational conversion of trans-T, to trans-So competes with photoisomerizatlon.
Our data indicate that in o-aminoazo compounds the quantum yield of intercombinational conversion is not equal to one and that the llfe of the lower slnglet state is at least 0.5 nsec. This is supported by data in [16], where weak fluorescence was recorded for amlnoazo- benzenes. Such abnormal behavior of amlnoazobenzenes, when compared to unsubstituted azo- benzenes, is due to inversion of the lower Tnz, and T~H* levels under the action of the elec- tron-donor amino group, and as a result the intercomblnatlonal conversion will proceed between states of the same nature. Such an intercombinational conversion will be slower by 2-3 orders of magnitude than that for levels of a different nature, according to the calculations in [17]. Thus, the singlet states of o-amlnoazo compounds (1)-(V) will not be completely deac- tivated by intercombinational conversion, and the action of the quencher may induce a further increase of the quantum yield of intercombinatlonal conversion.
CONCLUSIONS
i. The photoisomerlzation of o-amlnoazo compounds is accompanied by an intermolecular transfer of a hydrogen atom from the amino group to the azo group, which takes place in the electron-excited state of the cis form.
2. Under the action of a heavy atom, an increase in the quantum yield of o-aminoazo- compound photoisomerizatlon has been observed, which indicates incomplete deactivation of their singlet levels.
358

LITERATURE CITED
i. V. M. Berezovskii and L. S. Tul'chinskaya, Zh. Org. Khim., 31, 2779 (1961). 2. V. M. Berezovskii, L. I. Gurko, and E. P. Rodionova, Zh. Org. Khim., 32, 2951 (1962). 3. L. S. Tul'chinskaya, Dissertation, Moscow (1963). 4. Yu. E. Borisevich, A. S. Tatikolov, and V. A. Kuz'min, Khim. Vys. Energ., 12, 474 (1978). 5. G. Zimmerman, L. Phow, and U. Paik, J. Am. Chem. Soc., 80, 3528 (1958). 6. P. Haberluld, P. M. Block, and M. S. Lux, J. Am. Chem. Soc., 97, 5804 (1975). 7. R. Camp, J. Epstein, and C. Stell, J. Am. Chem. Soc., 99, 2453 (1977). 8. N. Nishimara, T. Suegoshi, H. Yamanaka, E. Imu, S. Yamamoto, and S. Haseguwa, Bull. Chem.
Soc., Jpn., 49, 1381 (1976). 9. P. D. Wilds, J. Am. Chem. Soc., 93, 2004 (1971).
i0. S. J. Formosinho, J. Chem. Soc. Faraday Trans. II, 72, 1332 (1976). ii. B. E. Blaisdell, J. Soc. Dyers Colour, 65, 618 (1949). 12. I. V. Khudyakov, P. P. Levin, Vo A. Kuz'min, and G. R. H. I. de Jonge, Izv. Akad. Nauk
SSSR, Ser. Khim., 1303 (1977). 13. J. Ronayette, R. Arnaud, P. Lebourgeois, and J. Lemaire, Can. J. Chem., 52, 1848 (1974), 14. J. Ronayette, R. Arnaud, and J. Lemaire, Can. J. Chem., 52, 1858 (1974). 15. J. L. Gardette, G. Gayot, R. Arnaud, and J. Lemaire, Nouv. J. Chem., ~, 283 (1977). 16. H. Bisle, M. Romer, and H. Rou, Bet. Bunsenges, Phys. Chem., 80, 301 (1976)o 17. S. Louer and M. EI-Saied, Usp. Fiz. Nauk, 94, 289 (1968).
OPTICALLY ACTIVE MIXED CHELATE COMPLEXES.
6, MIXED CHELATES OF Co(ll) AND Co(Ill) WITH ACETYLACETONE
AND AROMATIC AMINO ACIDS
V. A. Pavlov, S. R. Piloyan, and E. I. Klabunovskil
UDC 541.65:541.49:546.73:547. 422.3:547.583.5
We have attempted to clarify the stereochemistry of the intermediate ternary catalyst-- modifier--substrate complexes in enantloselective hydrogenation catalysts [i] in work on models of these ternary complexes -- mixed chelates of Cu and Ni with acetylacetone (acac) and aromatic amino acids (AA) in solution and in the solid state -- by circular dichroism (CD), optical rotatory dispersion (ORD), magnetooptical rotatory dispersion (MORD), IR and UV spec- troscopy, and x-ray diffraction [2-6].
Here we have used CD, MOP, D, and UV spectroscopy to examine the Co(acac)~--Co(AA)~ system, where AA is D-phenylalanine (D-Phe), D-tyrosine (D-Tyr), and D-tryptophan (D-Trp), in DM_F
solution.
EXPERIMENTAL
The amino acid complexes of Co(ll), Co(acac)2, and Co(acac)2(AA) were prepared by the methods of [7-9]. The CD spectra were measured with a Jobln--Yvon III dichrograph in the 250- 750 nm region with solution concentrations of 0.652-3.73 mmole/liter. The UV spectra were recorded on a Specord UV-VIS spectrophotomater in the 200-800 nm region in the concentration range 0.1-1.55 mmole/liter. The MORD curves were measured in DMF solution on a Spectropol-i spectropolarimeter in a magnetic field of 7490 • 30 Oe in the 320-580 nm region in a cell with layer thickness 2 mm in the concentration range 1-12 mmole/liter.
RESULTS AND DISCUSSION
Unstable mixed chelate complexes of Co(ll) with acetylacetone and aromatic AA's can be formed in solution by ligand exchange between the simple chelates in the same way as the Ni
complexes [6]
Co(acac)~ + Co(AA)~ ~ 2 Co(acac) (AA) (1)
....... N.D. Zelinskii Institute of Organic Chemistry, Academy of Sciences of the usSR, Moscow. Translated from Izvestiya Akademii Nauk SSSR, Seriya Khimicheskaya, No. 3, pp. 539-545, March, 1980. Original article submitted January 2, 1979.
0568-5230/80/2903-0359507.50 �9 1980 Plenum Publishing Corporation 359





























