Embed Size (px)
Citation preview

Sonochemical Oxidation of Carbon Disulfide in Aqueous Solutions:Reaction Kinetics and Pathways
Yusuf G. Adewuyi* and Collins Appaw
Department of Chemical Engineering, North Carolina A and T State University,Greensboro, North Carolina 27411
The kinetics of sonochemical oxidation of CS2 and the effects of process parameters (e.g.,concentration, pH, temperature, ultrasonic intensity, irradiation medium, dissolved gas and time,etc.) on the degradation rates and product distributions were studied in a batch reactor at 20kHz. Sonochemical oxidation was found to decrease with an increase in the solution temperature,and the reaction rate order was dependent on the temperature (T) range studied: zero-order atT g 10 °C [with rate constants of (0.66-3.68) × 10-5 M min-1] and first-order at T e 5 °C (withrate constants of 0.037-0.266 min-1). From Arrhenius law, k ) A exp(EA/RT), the activationenergy, EA, for the zero-order degradation of CS2 in the presence of air as the irradiating gaswas found to be 7.2 kJ/mol at the higher temperatures compared to 28.7 kJ/mol for the first-order degradation at the lower temperatures. Sonochemical oxidation pathways leading to sulfateformation are discussed. The results of this study suggest that the ultrasonic degradation ofCS2 might provide an environmentally conscious method for the control of this hazardouspollutant in industrial wastewater.
Introduction
Biogenic sulfur (or reduced sulfur) compounds are themain causes of odor in natural waters and processwastewaters, and control of their emissions is a keyenvironmental concern in the purification of natural gas,viscose rayon manufacture, tanneries, and the kraftpulp and petroleum refining industries.1 These com-pounds include carbon disulfide (CS2) and its hydrolysisproducts in an aquatic environment, hydrogen sulfide(H2S), and carbonyl sulfide (OCS).2,3 Carbon disulfide(CS2) is a poisonous, volatile, and pungent-smellingliquid with wide industrial applications as an excellentsolvent. It is also toxic to animals and aquatic organ-isms, and its aqueous hydrolysis products, H2S andOCS, are also malodorous and corrosive. When releasedinto the atmosphere, CS2 is mostly converted to SO2 andOCS in the lower atmosphere, with the latter being oneof the greenhouse gases with a lifetime of more than 1year in the atmosphere.4 It is also classified as ahazardous air pollutant under Title III of the 1990 CleanAir Act Amendment of the United States.5 The develop-ment of cost-effective and environmentally conscioustechnologies for its control is therefore desirable and ofan increased interest.4-6
The kinetics and mechanisms of oxidation of carbondisulfide in aqueous solutions by hydrogen peroxide andother chemical oxidants (e.g., O3, Cl2) at different pHsand temperatures were studied in detail by previousinvestigators.1-3 However, today’s treatment processesmust meet more stringent requirements of being envi-ronmentally responsible, and innovative technologiessuch as sonochemical oxidation, requiring little to nochemical additions for effective remediation, are moredesirable.7,8 Sonochemical techniques utilize ultrasoundto produce an oxidative environment via acoustic cavi-tation due to the formation and subsequent collapse of
microbubbles from acoustical wave-induced compres-sion/rarefaction. The collapse of these bubbles leads tolocalized transient high temperatures (g5000 K) andpressures (g1000 atm), resulting in the generation ofhighly reactive species including hydroxyl (•OH), hy-drogen (H•), and hydroperoxyl (HO2
•) radicals andhydrogen peroxide. The reactive species are capable ofinitiating or promoting many reduction-oxidation reac-tions. Adewuyi8 provides a comprehensive review of thefundamentals of environmental sonochemistry discuss-ing applications to contaminant remediation and chal-lenges for scale-up and commercialization.
Entezari et al.9 studied the sonochemical degradationof pure liquid carbon disulfide and the effects of fre-quency, temperature, intensity, and gases on the rateof its dissociation. They found that ultrasonic irradiationof the CS2 liquid at 20 kHz resulted in the formation ofa heterogeneous mixture of black particles (amorphouscarbon) in a yellow solution (monoclinic sulfur). Weinvestigated the kinetics of the sonochemical oxidationof unbuffered aqueous carbon disulfide earlier at 20 kHzand 20 °C in a batch reactor.10 With an initial CS2concentration of (13.2-13.6) × 10-4 M, we found thereaction rate to be zero-order, and the rate constant forthe degradation at 20 °C and 14 W (11.04 W/m2) in airwas 21.1 µM/min compared with 46.7 µM/min at 50 W(39.47 W/m2). The formation of sulfate as the mainreaction product was enhanced in the presence ofhydrogen peroxide but inhibited in the presence of1-butanol. To further understand the kinetics of thesonochemical oxidation of CS2 and the reaction path-ways, we have extended this study to include reactionsat different temperatures (1-50 °C), unbuffered solu-tions, and solutions with pHs of 8-11. The results ofthese studies are reported here.
Experimental Procedures
The experimental setup for this study consisted of a20 kHz sonifer capable of a maximum power output of
* Corresponding author. Phone: (336) 334-7564. Fax: (336)334-7904. E-mail: [email protected].
4957Ind. Eng. Chem. Res. 2002, 41, 4957-4964
10.1021/ie020069a CCC: $22.00 © 2002 American Chemical SocietyPublished on Web 08/31/2002
400 W (Branson model 450 sonifer), jacketed glassreactor, sound abatement enclosure box, and circulatorywater bath described elsewhere in detail.10 Kinetic runswere carried out by ultrasonically irradiating the result-ing solutions for a desired length of time, using intensi-ties of 14-50 W, and at temperatures of 1-50 °C. Atthe start of each experiment, a prepared stock solution(50 mL) of CS2 with known concentration (e.g., 0.02,0.03, or 0.04 M) was poured into the reactor. The reactorwas immediately sealed to prevent any contaminantsfrom volatizing from the stock solution. A gas dispersiontube was inserted in the reactor below the surface ofthe solution. An irradiating gas, air, helium, argon, ornitrous oxide was bubbled into the sample for a periodof 30 min to saturate the solution. The bubbling processresulted in the reduction of the initial concentrationsof the stock CS2 solutions of 0.02, 0.03, and 0.04 M totypically (6.4-7.0) × 10-4, 10.5 × 10-4, and (13.2-13.9)× 10-4 M, respectively, due to volatilization. Theseresulting concentrations were recorded as the initial CS2concentrations at the start of ultrasonic irradiations.Degradation of the gas-saturated CS2 solutions wasmonitored at 314 nm at different time intervals usinga Beckman DU-7000 spectrophotometer. The productsof the reaction were analyzed using a DIONEX ionchromatograph (with an IonPac AS11 2 mm analyticalcolumn and an EG 40 eluent generator using 1 Mpotassium hydroxide as the eluent) at the end of eachirradiation period. The experimental procedure wasrepeated for different sets of process parameters (e.g.,pH, temperature, solute concentration, power or inten-sity, dissolved gases). Experiments presented here wereconducted with unbuffered solutions, and at pH valuesof 11, 10, 9, and 8 buffered with NaOH-glycol-NaCl,potassium carbonate-potassium borate-KOH, boricacid-KCl-NaOH, and potassium phosphate monoba-sic-NaOH buffer systems, respectively. These bufferswere concentrates, and appropriate amounts were addedto Milli-Q water and CS2 dissolved in the solutions atthe start of the experiments. The solution pH wasmeasured with a Fisher Scientific pH/ion conductivitymeter (model 50). All of the buffer solutions were ACSreagent grade obtained from Fisher Scientific Co. Ul-trapure-grade air, nitrous oxide (N2O), helium, or argonused as the nucleating or saturating gas was obtainedfrom Air Products Co., Ltd.
Results and Discussion
In our earlier study, which was carried out in a batchreactor at a frequency of 20 kHz and 20 °C, thesonochemical oxidation of CS2 was shown to follow zero-order with the production of sulfate as the product.10
To further understand the kinetics of the sonochemicaloxidation of CS2 and the reaction pathways, experi-ments were conducted at different process parameters(e.g., intensities, initial aqueous CS2 concentrations,irradiation time, and medium), temperatures rangingfrom 1 to 50 °C, unbuffered solutions, and solutions withpHs of 8-11, and the reaction products were analyzed.The experimental conditions and results are sum-marized in Table 1 and Figures 1-11. Table 1 alsoincludes two data points at 20 °C (P ) 14 W) with airas the irradiating gas: [CS2]0 ) 6.90 × 10-4 and 13.36× 10-4 from our earlier study10 duplicated here for thebenefit of comparison.
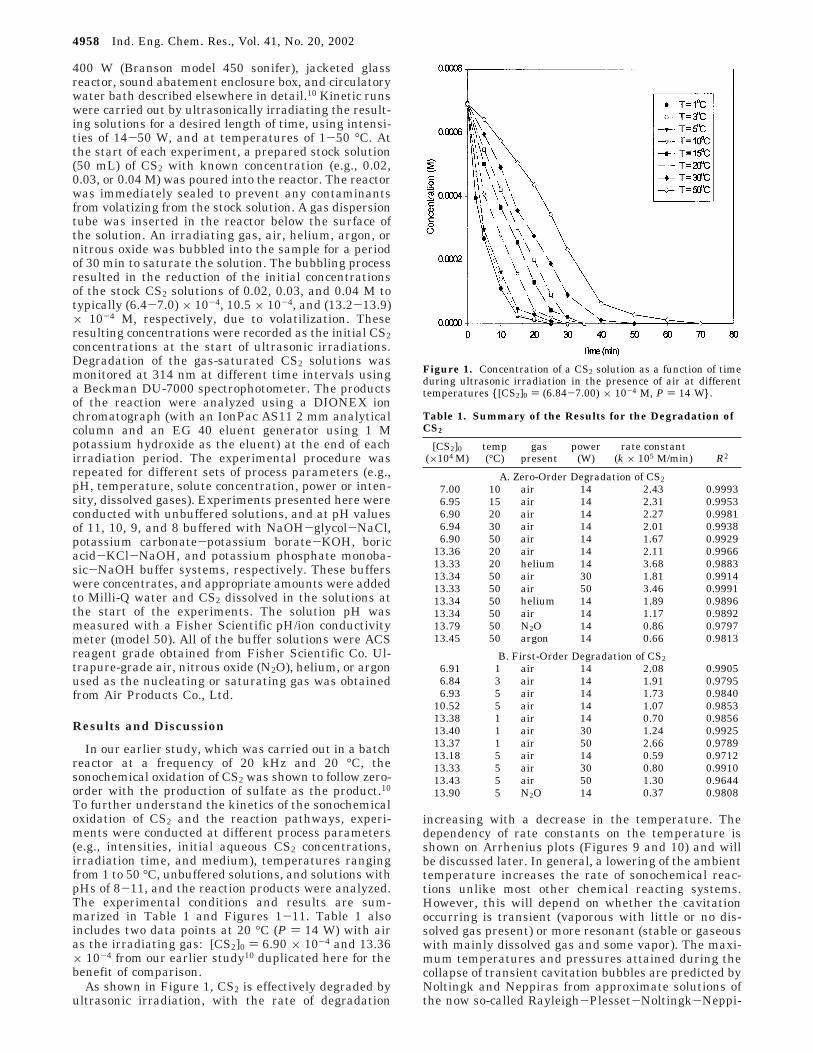
As shown in Figure 1, CS2 is effectively degraded byultrasonic irradiation, with the rate of degradation
increasing with a decrease in the temperature. Thedependency of rate constants on the temperature isshown on Arrhenius plots (Figures 9 and 10) and willbe discussed later. In general, a lowering of the ambienttemperature increases the rate of sonochemical reac-tions unlike most other chemical reacting systems.However, this will depend on whether the cavitationoccurring is transient (vaporous with little or no dis-solved gas present) or more resonant (stable or gaseouswith mainly dissolved gas and some vapor). The maxi-mum temperatures and pressures attained during thecollapse of transient cavitation bubbles are predicted byNoltingk and Neppiras from approximate solutions ofthe now so-called Rayleigh-Plesset-Noltingk-Neppi-
Figure 1. Concentration of a CS2 solution as a function of timeduring ultrasonic irradiation in the presence of air at differenttemperatures {[CS2]0 ) (6.84-7.00) × 10-4 M, P ) 14 W}.
Table 1. Summary of the Results for the Degradation ofCS2
[CS2]0(×104 M)
temp(°C)
gaspresent
power(W)
rate constant(k × 105 M/min) R2
A. Zero-Order Degradation of CS2
7.00 10 air 14 2.43 0.99936.95 15 air 14 2.31 0.99536.90 20 air 14 2.27 0.99816.94 30 air 14 2.01 0.99386.90 50 air 14 1.67 0.9929
13.36 20 air 14 2.11 0.996613.33 20 helium 14 3.68 0.988313.34 50 air 30 1.81 0.991413.33 50 air 50 3.46 0.999113.34 50 helium 14 1.89 0.989613.34 50 air 14 1.17 0.989213.79 50 N2O 14 0.86 0.979713.45 50 argon 14 0.66 0.9813
B. First-Order Degradation of CS2
6.91 1 air 14 2.08 0.99056.84 3 air 14 1.91 0.97956.93 5 air 14 1.73 0.9840
10.52 5 air 14 1.07 0.985313.38 1 air 14 0.70 0.985613.40 1 air 30 1.24 0.992513.37 1 air 50 2.66 0.978913.18 5 air 14 0.59 0.971213.33 5 air 30 0.80 0.991013.43 5 air 50 1.30 0.964413.90 5 N2O 14 0.37 0.9808
4958 Ind. Eng. Chem. Res., Vol. 41, No. 20, 2002

ras-Poritsky (RPNNP) bubble dynamic equation as-suming adiabatic collapse and are given in eqs 1 and2,11-14 where T0 ) temperature of the bulk solution, Pv
) pressure in the bubble at its maximum size, Pa )pressure in the bubble at the moment of transientcollapse (i.e., acoustic pressure), and γ ) polytropic
index or heat capacity ratio of the cavity medium.8,11
For vaporous cavitation, increasing the ambient tem-perature, T0, will raise the equilibrium vapor pressureof the medium and so lower both Tmax and Pmax. As thetemperature of the liquid, T0, is increased, its vapor
Figure 2. Concentration of a CS2 solution as a function of timeduring ultrasonic irradiation in the presence of different gases {-[CS2]0 ) (13.2-13.9) × 10-4 M, T ) 5 °C, P ) 14 W}.
Figure 3. Concentration of a CS2 solution as a function of timeduring ultrasonic irradiation in the presence of different gases {-[CS2]0 ) (13.4-13.8) × 10-4 M, T ) 50 °C, P ) 14 W}.
Tmax ) T0[Pa(γ - 1)Pv
] (1)
Pmax ) Pv[Pa(γ - 1)Pv
]γ/(γ-1)
(2)
Figure 4. Zero- and first-order kinetic degradations of CS2 at 5and 20 °C with air as the irradiating gas {[CS2]0 ) (13.2-13.4) ×10-4 M, P ) 14 W}.
Figure 5. Zero- and first-order kinetic degradations of CS2 at 5and 50 °C with N2O as the irradiating gas {[CS2]0 ) (13.8-13.9)× 10-4 M, P ) 14 W}.
Figure 6. Zero-order kinetic degradations of CS2 at the highertemperatures (T ) 10, 15, 20, 30, and 50 °C) with air as theirradiating gas {[CS2]0 ) (6.9-7.0) × 10-4 M, P ) 14 W}.
Ind. Eng. Chem. Res., Vol. 41, No. 20, 2002 4959

pressure, Pv, is also increased, but much more dramati-cally than the temperature. The vapor, which enters thebubble during its formation, cushions the collapse of thebubble and dampens the dissipation of ultrasonic en-ergy. This predicts that, as the bulk temperatureincreases, the temperature of the “hot spot” formed bythe collapsing cavity decreases, resulting in a decreasein the reaction rate as indicated by the results of thisstudy.
It should be noted that, in certain reaction systems,more favorable results are attained at an optimumreaction temperature. In such systems, an increase inthe ambient temperature is found to increase the kineticreaction to a point before the cushioning effect of thevapor in the bubble begins to dominate, resulting in adecrease in the reaction rate upon further temperatureincrease.11 In the investigation of the degradation ofthymine, Sehgal and Wang15 also found that the rate
may even reach a plateau with an increase in thetemperature before decreasing upon a further increasein temperature. Studies also indicate that the natureof cavitation prevailing is determined to a large extentby the ultrasonic frequency of the system. Entezari andKruss16,17 studied the sonochemical reaction rate ofiodide oxidation at different temperatures (0-50 °C) andat 20 and 900 kHz. At 20 kHz the reaction ratedecreased with an increase in the temperature at allpower levels, but at 900 kHz the rate showed a maxi-mum at a temperature intermediate between 5 and 50°C, dependent on the power level. They observed thatthe noise caused by the collapse of the cavitation bubbleswas very noticeable at 20 kHz compared with that at900 kHz and concluded that transient cavitation wasmore likely occurring at the 20 kHz frequency andresonant cavitation at the higher frequency of 900kHz.17 The effect of temperature on degradation ratesobserved at 20 kHz in this study is consistent with theresults of our studies. However, using a model whichlinked bubble dynamics with the production of free
Figure 7. First-order kinetic degradations of CS2 at differentinitial concentrations, [CS2]0 (T ) 5 °C, P ) 14 W).
Figure 8. First-order kinetic degradation of CS2 at the lowertemperatures (T ) 1, 3, and 5 °C) with air as the irradiating gas{[CS2]0 ) (6.90-7.00) × 10-4 M, P ) 14 W}.
Figure 9. Arrhenius plot of CS2 degradation rate constants atthe higher temperatures (T ) 10, 15, 20, 30, and 50 °C) with airas the irradiating gas {[CS2]0 ) (6.9-7.0) × 10-4 M, P ) 14 W}.
Figure 10. Arrhenius plot of CS2 degradation rate constants atthe lower temperature (T ) 1, 3, and 5 °C) with air as theirradiating gas {[CS2]0 ) (6.84- 6.93) × 10-4 M, P ) 14 W}.
4960 Ind. Eng. Chem. Res., Vol. 41, No. 20, 2002

radicals, Sochard et al.18,19 predicted a maximum forboth •OH and H• radicals with an increase in the liquidbulk temperature. They attributed these results to twocompetitive but opposite effects: (1) an increasing liquidtemperature leading to an increased amount of watervapor in the bubble, which can promote formation offree radicals from dissociation of water molecules; (2)increasing liquid temperature leading to less violentcollapse, which can result in lower internal tempera-tures at the end of the collapse phase.
As shown in Table 1, the sonochemical reaction ratesare greater at higher power and hence higher intensity.The acoustic power (W) represents the intensity emittedby a given surface. For example, with approximately thesame [CS2]0 ) 13.3 × 10-4 M and temperature (5 °C)and with air as the irradiating gas, the first-orderdegradation rate constant, k, of CS2 at 50 W (39.47W/m2) was about twice that at 14 W (11.04 W/m2), 0.13vs 0.06 min-1. Similarly, at a temperature of 50 °C, thezero-order degradation rate constant at 30 W (23.68W/m2) was 50% greater than that at 14 W, 1.8 × 10-5
vs 1.2 × 10-5 M min-1. The power (or acoustic) intensityI, which is proportional to the applied power density, isa function of the acoustic amplitude or pressure, Pa. Inthe case of a progressive planar or spherical wave, I (inW/m2) is directly related to Pa by eq 3, where F is the
density of the fluid (e.g., water) and c is the speed ofsound in the fluid (1500 m/s in water), and the term Fcrepresents the acoustic impedance (Z) of the medium.12
An increase in the ultrasound intensity results in anincrease in the acoustic amplitude, which favors moreviolent cavitation bubble collapse because the bubblecollapse time, the transient temperature, and the in-ternal pressure in the cavitation bubble during collapseare all dependent on the acoustic amplitude, Pa. Thatis, high enough acoustic power results in transientcavitation.20 Hence, the results of an increase in thesound intensity are greater sonochemical effects, result-ing in higher CS2 degradation rates. However, Vichare
et al.21 in their theoretical study based on the model ofsingle cavity dynamics using the Rayleigh-Plessetequation discussed the effect of intensity and thefrequency of ultrasound on the quantum of energydissipation and rate of dissipation. They noted that inlowering the intensity, though it resulted in an increasein the estimated energy associated with the individualcavity, the number of cavities could also be substantiallyless. They also noted that the energy dissipation ratefor complete adiabatic collapse conditions decreasesinitially but increases at higher intensities, althoughmarginally. They concluded that the net effect of thiscould be an optimal intensity, giving the maximumsonochemical effect. Thus, it could also be concludedthat the higher CS2 degradation at higher intensitiesresulted from an increased number of cavitation events.
As illustrated in Figures 2 and 3, the rate of CS2sonochemical degradation in the presence of the differ-ent irradiating gases was in the order He > air > N2O> Ar, at both low temperature (5 °C) and high temper-ature (50 °C). These results are consistent with ourearlier studies10 at 20 °C and are well explained byprevious investigators.8-10
Reaction Kinetics
As shown in Figures 4 and 5, zero- and first-orderplots of CS2 degradation at 5 and 20 °C (air as theirradiating gas) and 5 and 50 °C (N2O as the irradiatinggas) both indicate a change in the kinetic order of thereaction with a change in the temperature of thesolution. We observed that the reaction rate order isdependent on the temperature (T) range studied: zero-order at T g 10 °C and first-order at T e 5 °C. The zero-and first-order rate constants for CS2 degradation underthe conditions of these experiments are summarized inTable 1. The zero-order plots for the degradation at T) 10, 15, 20, 30, and 50 °C are shown in Figure 6. Thefirst-order plots for the degradation of CS2 at 5 °C butdifferent initial CS2 concentrations, [CS2]0, and atapproximately the same [CS2]0 but different tempera-tures (i.e., T ) 1, 3, and 5 °C) are shown respectively inFigures 7 and 8. As expected, sonochemical degradationdecreases with an increase in the temperature. Hence,the appropriate form of the Arrhenius law11 is appliedto the data to obtain the activation energy, EA, for thereactions, and the results are shown in Figures 9 and10 respectively for the zero- and first-order reaction rateconstants. As indicated in Figures 9 and 10, the activa-tion energies for the zero- and first-order degradationof CS2 with air as the irradiating gas were foundrespectively to be 7.2 kJ/mol at the higher temperaturesand 28.7 kJ/mol at the lower temperatures. Because EArepresents the potential energy of activation, the higheractivation observed for the reactions at the lowertemperature range suggests that the higher amount isassociated with the cavitation event at the lower tem-peratures compared with that at the higher tempera-tures. That is, the factor exp[(bond energy)/RT] that canbe achieved at the lower temperature is more signifi-cant, and the fraction of the molecules with sufficientenergy for dissociation will be higher.16
The kinetics of the sonochemical degradation ofpollutants is either first- or zero-order as observed bymost investigators. According to the “structured hotspot” model used to explain the results of most studiesin environmental sonochemistry, the three regions for
Figure 11. Sulfate formation as a function of time for solutionsof different initial temperatures, T, in the presence of air as theirradiating gas {[CS2]0 ) (13.60-13.8) × 10-4 M, pH ) 11, P ) 50W}.
I ) Pa2/2Fc (3)
Ind. Eng. Chem. Res., Vol. 41, No. 20, 2002 4961

the occurrence of chemical reactions in a cavitationevent are a hot gaseous cavity, an interfacial regionsurrounding the inner cavity, and the bulk liquidmedium at ambient temperature.8,15 The changes insonochemical activities resulting from temperaturechanges are believed to be due to the phase in whichthe reaction leading to chemical degradation occurs andto the physical and chemical properties of the medium(e.g., concentration of the solute).15 The availability andthe relative rates of diffusion of free radicals (e.g., •OH)to the reaction zone might then determine the rate-limiting step and the overall order of the reaction.Sehgal and Wang15 in the sonochemical degradation ofthymine also observed a change in the reaction orderfrom first to zero as the temperature increased: thereaction rate was zero-order in the temperature rangeof 10-50 °C and first-order at lower temperatures. Theyexplained the variation of the reaction order with thehelp of a cavitation-diffusion model proposed by Mar-gulis.22 They noted that the concentration and temper-ature gradients inside a bubble and in the solution forcethe free radicals and thymine to diffuse from opposingdirections into the bubble-liquid interface, where theyreact almost instantaneously. As the reaction temper-ature increased, they suggested that the rate of diffusionof thymine from the bulk liquid to the reaction zone wasaccelerated. However, the increase in the temperaturewas simultaneously accompanied by a decrease in thecavitation intensity, reducing the amount of free radi-cals produced within the bubble. On the other hand,they indicated that, at low solution temperatures,intense cavitation resulted in high intracavity temper-atures and high concentrations of radicals, which ledto a rapid diffusion of radicals into the interface. Hence,they concluded that the rate-limiting step for thereaction was the diffusion of the substrate (thymine)into the interface at the low temperatures, and at thehigh temperatures, it was the diffusion of the freeradicals into the interface. In general, the results fromthese studies support the theory that there is anabundance of free radicals, which rapidly diffuse intothe interface at low solution temperatures. On the otherhand, the diffusion of the substrate (here CS2) in theopposite direction into the interface is slow, and it is,therefore, the rate-determining step. It is, therefore,expected that CS2 would be used up as soon as itdiffused into the reaction zone, resulting in a first-orderdegradation as observed experimentally at the lowertemperatures. At the higher solution temperature,cavitation could be inhibited to the extent that thediffusion of free radicals would become the rate-determining step. The sonochemical degradation reac-tion is then zero-order with respect to CS2, as illustratedexperimentally at the higher temperatures.
Reaction Products
Analysis of the reaction products indicates thatultrasonic irradiation of CS2 in the presence of air, Ar,and N2O results in its oxidation to mainly sulfate. Theformation of sulfate as a function of temperature isillustrated in Figure 11. As expected, the lower thetemperatures are, the higher the sulfate production ratedue to more severe cavitation events, resulting in theenhanced availability and diffusion of •OH radicals. Theformation of sulfate was also enhanced at the lower pHvalues. With an initial CS2 concentration of (13.60-13.85) × 10-4 M and at 20 °C and 50 W, the amount of
sulfate formed after 16 h was about 1.8 × 10-3 M at pH9 compared with 9.0 × 10-4 M at pH 11 using air asthe dissolved gas. For the same conditions but at pH 9and after 6 h, the amount of sulfate formed was 1.1 ×10-3, 8.3 × 10-4, and 9.1 × 10-4 M respectively indissolved air, Ar, and N2O. The decrease in the sulfateproduction rate with an increase in the solution pH canbe partly explained by the rapid dissociation of •OH inalkaline solutions as illustrated by eq 4, where kf ) 1.2
× 1010 M-1 s-1 for the forward reaction and kb ) 9.3 ×107 s-2 for the backward reaction and the oxide radicalion (O•-) is known to react more slowly with the samesubstrate than •OH.22 Results of previous sonochemicalstudies of other reduced sulfur compounds showedsimilar observations.23,24 Kotronarou et al.23 found thesonochemical oxidation of H2S or S(II-) {[S(-II) ) [H2S]+ [HS-] + [S2-]} solutions to proceed rapidly with azero-order rate, resulting in the formation of sulfate(SO4
2-) and sulfite (SO32-) as the main products and
thiosulfate (S2O32-) as the minor product at pH g 10.
They suggested that the apparent zero-order depen-dence on [S(II-)] was the result of the reaction of thesubstrate with a •OH radical as the main pathway. Theyproposed that the rate-determining step in the overallreaction was the reaction of HS- and the oxidationintermediates with a •OH radical in the liquid phase asit diffused out of the cavitation bubble (HS- + •OH fHSOH-). They also observed a decrease in the zero-order rate constant at pH > 10 and also attributed theirobservation partly to the dissociation of •OH in thealkaline solutions.
In our earlier study we investigated the effects ofoxidants, such as H2O2, and •OH radical scavengers,such as 1-butanol, on the product (i.e., sulfate) produc-tion rate during ultrasonic irradiation in the presenceof air as the irradiating gas.10 We found that theformation of sulfate was enhanced by the addition ofH2O2 and inhibited by the addition of 1-butanol, sug-gesting that the •OH radical played a major role in thesulfate formation mechanism. It is, therefore, conceiv-able that the rate of formation of an activated complexinvolving CS2 and •OH determines the overall reaction.
Reaction Pathways
On the basis of the results discussed, the rate-determining step in the overall sonochemical oxidationof CS2 in the presence of air (i.e., oxygenated aqueoussolutions) to produce sulfate appears to be dependenton the availability of •OH radicals for reactions in theinterface of the bubble. The following reaction pathwaysare proposed:
•OH + OH - y\zkf
kbO•- + H2O (4)
CS2 + •OH f CS2OH• (5)
CS2OH• f OCS + HS• (6)
OCS + H2O f H2S + CO2 (7)
H2S + •OH f HS• + H2O (8)
HS• + O2 f HSO2• (9)
HSO2• h SO2
•- + H+ (10)
4962 Ind. Eng. Chem. Res., Vol. 41, No. 20, 2002

This mechanism indicates that the sonochemicaloxidation of CS2 to sulfate proceeds mainly throughoxidation by the •OH radical and H2O2 produced fromits recombination reactions. In addition, the low EAvalues (<42 kJ/mol) in both the low- and high-temper-ature range in this study suggest that diffusion-controlled transport processes dictate the overall reac-tion.24,25 During ultrasound-induced cavitation, thedecomposition of water vapor present in the cavities toproduce H• and •OH radicals during the compressionphase is well-known.8,26-28 The •OH radical is a powerfuland efficient chemical oxidant in both the gas and liquidphase, and its reactions with inorganic and organicsubstrates are often near the diffusion-controlled rate.29
The sonolysis of water to produce H2O2 and hydrogengas via hydroxyl radicals and hydrogen atoms is well-known and occurs in the presence of any gas, O2, or puregases (e.g., Ar).8,26 It has also been demonstratedexperimentally by numerous investigators.26,30,31 In theabsence of a scavenger, the hydroxyl radicals willrecombine to form H2O2 (2•OH f H2O2) and hydrogenradicals to form hydrogen (2H• f H2). The rate constantfor the recombination of •OH to form H2O2 (i.e., in theabsence of a substrate) is reported to be 5.5 × 109 M-1
s-1.29 However, these radicals will react preferentiallywith scavengers (e.g., H• with O2 in the inner cavitiesand •OH with substrate (here, CS2) in the interface ofthe cavitation bubbles). The reaction between H• andO2 at high temperatures is believed to produce oxygenatoms and •OH radical (H• + O2 f •OH + •O).32 In thepresence of O2 in the vapor phase of the bubble, thehydrogen radicals also react with O2, and this resultsin the rapid formation of a hydroperoxyl (or peroxy)radical, HO2
• (H• + O2 f HO2•), suppressing the
recombination of H• and •OH. The hydroperoxyl radicalssubsequently decay to produce hydrogen peroxide inaerated solutions via HO2
• + HO2• f H2O2 + O2 or in
the presence of hydrogen atom via HO2• + H• f H2O2.
The rate constant for the recombination of HO2• to form
H2O2 is reported to be 8.6 ( 0.6 × 105 M-1 s-1.33-35 Thespontaneous disproportionation of the peroxy radicalwith generation of H2O2 reduces the effectiveness of itsdirect attack on the substrate (e.g., CS2) but enhancesthe oxidation process because of further formation ofH2O2 by its recombination reaction. According to thesereactions, both hydrogen atoms and hydroperoxyl radi-cals are converted to other species before they can reachthe interface and react with CS2. In addition, thedisproportionation reaction of HO2
•/O2•- is expected to
contribute to H2O2 regeneration via33-36 HO2• + O2
•- +H2O f H2O2 + O2 + OH-.
Hence, the main pathway in the sonochemical oxida-tion of CS2 is the initial reaction of the •OH radical inthe interface of the bubble with CS2 to produce the
intermediate, dithiocarbonate ion (CS2OH•), as in eq 5.The rate constant at 23 °C for this reaction is reportedto be 8.0 ( 2.0 × 109 M-1 s-1.37,38 The intermediate CS2-OH• so formed decomposes instantaneously to producethe hydrogen sulfide radical (HS•) as in eqs 6-8, whichis then oxidized to sulfate as the main final product bya variety of radical reactions, and H2O2 as indicated ineqs 9-16. The subsequent intermediate steps after theformation of HS• in the pathway to sulfate productionare discussed further elsewhere.23,24 In the presence ofpure gas (e.g., Ar), the formation of H2O2 and O2 afterthe initial dissociation of H2O to produce H• and •OHproceeds via reactions8,39 2•OH + M f H2O2, 2•OH f•O + H2O, and 2•O + M f O2 + M (where M is an inertthird molecule), and sulfate production should proceedvia the same mechanism. However, the presence ofoxygen might improve sonochemical activities by pro-viding additional reactions such as O2 f 2•O, O2 + H•
f HO2•, and 2HO2
• f H2O2 + O2, as discussed earlier.Also, most investigators have observed that the kineticsof sonochemical degradation of pollutants is first- orzero-order irrespective of the dissolved gas medium (air,O2, or Ar), as demonstrated in the results of our studies(Table 1) and reviewed extensively elsewhere.8 However,the nature of the cavitating gas (Ar and O2) mightinfluence the overall degradation rate constant and theresulting product distribution as observed in ourstudies.10,39-41 For example, Beckett and Hua39 observedthat the first-order kinetic rate constants for 1,4-dioxanein an aqueous solution at 358 kHz were highest with asparge gas ratio of 75% Ar/25% O2 (k ) 4.32 ( 0.31 ×10-4 s-1) and lowest in the presence of pure argon (k )8.67 ( 0.47 × 10-5 s-1) and attributed the difference tothe production of additional radical species during thedecomposition of O2 compensating for the lower internalcavitation temperature arising from the use of O2 overAr. Also, Hua et al.40 found that the rate constant forn-nitrophenol at 20 kHz in the presence of pure O2, kO2
) 5.19 × 10-4 s-1, is lower than that in the presence ofpure Ar, kAr ) 7.94 × 10-4 s-1, and a 4:1 (v/v) Ar/O2mixture yielded the highest degradation rate, kAr/O2 )1.2 × 10-3 s-1. These results suggest that, among otherfactors, an optimum balance between higher tempera-tures generated during acoustic cavitation and genera-tion of active radical species promotes the most effectiveconditions for compound destruction.
The results from this research clearly demonstratethe utility of sonochemical oxidation for CS2 control.However, to develop the engineering data that areessential for scale-up, pilot-scale studies are needed.Furthermore, appropriate treatment methods such asprecipitation need to be developed for spent solutionsthat may contain high levels of sulfate (SO4
2-).
Conclusions
The rate of sonochemical degradation in the presenceof the different irradiating gases was in the order He >air > N2O > Ar, at both low and high temperatures.The sonochemical oxidation decreases with an increasein the temperature, and the reaction rate order istemperature dependent: zero-order at T g 10 °C andfirst-order at T e 5 °C. With air as the irradiating gas,the activation energy, EA, for the zero-order degradationof CS2 was 7.2 kJ/mol at the higher temperaturescompared to 28.7 kJ/mol for the first-order degradationat the lower temperatures. The results suggest that theavailability and the relative rates of diffusion of free
SO2•- + O2 f SO2 + O2
•- (11)
SO2 + H2O h SO2‚H2O (12)
SO2‚H2O h H+ + HSO3- (13)
HSO3- h H+ + SO3
2- (14)
2•OH f H2O2 (15)
HSO3- + H2O2 f HSO4
- + H2O (16)
Ind. Eng. Chem. Res., Vol. 41, No. 20, 2002 4963

radicals (e.g., •OH) to the interfacial reaction zonedetermine the rate-limiting step and the overall orderof the reaction. The product of the reaction in thepresence of air (and other dissolved gases, e.g., Ar, N2O)was mainly sulfate, whose formation was enhanced atlower pH values and also at lower temperatures whensolutions from experiments in the pH range of 8-11 (T) 20 °C) and temperatures of 5-20 °C (pH ) 11) wereanalyzed.
Acknowledgment
The authors are grateful to Air Force Office ofScientific Research for financial assistance (GrantF49620-95-1-0541) and Department of Energy (GrantDE-FC04-90AL66158) and the equipment support(through Title III) of the Chemical Engineering Depart-ment at North Carolina Agricultural and TechnicalState University.
Literature Cited
(1) Adewuyi, Y. G. Oxidation of Biogenic Sulfur Compounds inAqueous Media In Biogenic Sulfur in the Environment. ACS Symp.Ser. 1989, 393, 529.
(2) Adewuyi, Y. G.; Carmichael, G. R. Kinetics of Hydrolysisand Oxidation of Carbon Disulfide by Hydrogen Peroxide inAlkaline Medium and Application to Carbonyl Sulfide. Environ.Sci. Technol. 1987, 21, 170.
(3) Elliot, S. Effect of Hydrogen Peroxide on Alkaline Hydrolysisof Carbon Disulfide. Environ. Sci. Technol. 1990, 24, 264.
(4) Hartikainen, T.; Ruuskanen, J.; Martikainen, P. J. CarbonDisulfide and Hydrogen Sulfide Removal with a Peat Biofilter. J.Air Waste Manage. Assoc. 2001, 51, 387.
(5) Hugler, W.; Acosta, C. Biological Removal of Carbon Dis-ulfide from Waste Air Streams. Environ. Prog. 1999, 18, 173.
(6) Tsai, C.-H.; Lee, W.-J.; Chen, C.-Y.; Liao, W.-T.; Shih, M.Formation of Solid Sulfur by Decomposition of Carbon Disulfidein the Oxygen-Lean Cold Plasma Environment. Ind. Eng. Chem.Res. 2002, 41, 1412.
(7) Suslick, K. Sonochemistry. Science 1990, 247, 1439.(8) Adewuyi, Y. G. Sonochemistry: Environmental Science and
Engineering Applications. Ind. Eng. Chem. Res. 2001, 40, 4681.(9) Entezari, M. H.; Peeter, K.; Otson, R. The Effect of Fre-
quency on Sonochemical Reactions III: Dissociation of CarbonDisulfide. Ultrason. Sonochem. 1997, 4, 49.
(10) Appaw, C.; Adewuyi, Y. G. Destruction of Carbon Disulfidein Aqueous Solutions by Sonochemical Oxidation. J. Hazard.Mater. 2001, B90, 237.
(11) Thompson, L. H.; Doraiswamy, L. K. Sonochemistry:Science and Engineering. Ind. Eng. Chem. Res. 1999, 38, 1215.
(12) Luche, J.-L. Synthetic Organic Sonochemistry; PlenumPress: New York, 1998.
(13) Noltingk, B. E.; Neppiras, E. A. Cavitation Produced byUltrasonics. Proc. Phys. Soc. London, Ser. B 1950, 63, 674.
(14) Neppiras, E. A. Acoust. Cavitation Phys. Rep. 1980, 61,159.
(15) Sehgal, C. M.; Wang, S. Y. Threshold Intensities andKinetics of Sonoreaction of Thymine in Aqueous Solutions at LowUltrasonic Intensities. J. Am. Chem. Soc. 1981, 103, 6606.
(16) Entezari, M. H.; Kruus, P. Effect of Frequency on Sonochem-ical Reactions. I: Oxidation of Iodide. Ultrason. Sonochem. 1994,1, S75.
(17) Entezari, M. H.; Kruus, P. Effect of Frequency on theSonochemical Reactions II: Temperature and Intensity Effects.Ultrason. Sonochem. 1996, 3, 19.
(18) Sochard, S.; Wilhelm, A. M.; Delmas, H. Modeling of FreeRadicals Production in a Collapsing Gas-Vapor Bubble. Ultrason.Sonochem. 1997, 4, 77.
(19) Sochard, S.; Wilhelm, A. M.; Delmas, H. Gas-VaporBubble Dynamics and Homogeneous Sonochemistry. Chem. Eng.Sci. 1998, 53, 239.
(20) Monnier, H.; Wilhelm, A. M.; Delmas, H. The Influence ofUltrasound on Micromixing in a Semi-batch Reactor. Chem. Eng.Sci. 1999, 54, 2953.
(21) Vichare, N. P.; Senthikumar, P.; Moholkar, V. S.; Gogate,P. R. Energy Analysis in Acoustic Cavitation. Ind. Eng. Chem.2000, 39, 1480.
(22) Margulis, M. Sonochemistry and Cavitation; OPA (Am-sterdam) B. V. Gordon and Breach Science Publ.: New York, 1995.
(23) Kotronarou, A.; Mills, G.; Hoffmann, M. R. Oxidation ofHydrogen Sulfide in Aqueous Solution. Environ. Sci. Technol.1992, 26, 2420.
(24) Kotronarou, A.; Hoffmann, M. R. The Chemical Effects ofCollapsing Cavitation Bubbles: Mathematical Modeling. Adv.Chem Ser. 1995, 244, 233.
(25) Sparks, D. L. Environmental Soil Chemistry; AcademicPress: New York, 1995.
(26) Kang, J.-W.; Hung, H.-M.; Lin, A.; Hoffmann, M. SonolyticDestruction of Methyl tert-Butyl Ether by Ultrasonic Irradiation:The Role of O3, H2O2, Frequency, and Power Density. Environ.Sci. Technol. 1999, 33, 3199.
(27) Serpone, N.; Colarusso, P. Sonochemistry I. Effects ofUltrasound on Heterogeneous Chemical ReactionssA Useful Toolto Generate Radicals and to Examine Reaction Mechanisms. Res.Chem. Intermed. 1994, 20, 635.
(28) Riesz, P.; Berdahl, D.; Christman, C. L. Free RadicalGeneration by Ultrasound in Aqueous and Nonaqueous Solutions.Environ. Health Perspect. 1985, 64, 233.
(29) Buxton, G. V.; Greenstock, C. L.; Helman, W. P.; Ross, A.B. Critical Review of Rate Constants for Reactions of HydratedElectrons, Hydrogen Atoms and Hydroxyl Radicals (•OH/•O-) inAqueous Solution. J. Phys. Chem. Ref. Data 1988, 17, 513.
(30) Margulis, M. A.; Didenko, Yu. T. Energetics and Mecha-nism of Acoustochemical Reactions. Yields of Hydrogen andHydrogen Peroxide in Different Aqueous Systems. Russ. J. Phys.Chem. 1984, 58, 848.
(31) Hart, E. J.; Henglein, A. Free Radical and Free AtomReactions in the Sonolysis of Aqueous Iodide and Formate Solu-tions. J. Phys. Chem. 1985, 89, 4342.
(32) Kotronarou, A.; Mills, G.; Hoffmann, M. R. UltrasonicIrradiation of p-Nitrophenol in Aqueous Solution. J. Phys. Chem.1991, 95, 3630.
(33) Bielski, B. H. J.; Cabelli, D. E.; Arudi, R. L.; Ross, A. B.Reactivity of HO2/O2
- Radicals in Aqueous Solution. J. Phys.Chem. Ref. 1985, 14, 1041.
(34) Bielski, B. H. J.; Allen, A. O. J. Mechanism of theDisproportionation of Superoxide Radicals. Phys. Chem. 1977, 81,1048.
(35) Bielski, B. H. J. Reevaluation of the Spectral and KineticProperties of HO2 and O2
- Free Radicals. Photochem. Photobiol.1978, 28, 645.
(36) Liao, C.-H.; Gurol, M. D. Chemical Oxidation by PhotolyticDecomposition of Hydrogen Peroxide. Environ. Sci. Technol. 1995,29, 3007.
(37) Farhataziz, R. A. B. Selected Specific Rates of Reactionsof Transients from Water in Aqueous Solutions. III. HydroxylRadicals and Perhydroxyl and Their Radical Ions; Report No.NSRDS-NBS59; National Bureau of Standards: Washington, DC,1977.
(38) Graedel, T. E.; Wechsler, C. J. Chemistry Within AqueousAerosols and Raindrops. Rev. Geophys. 1981, 19, 505.
(39) Beckett, M. A.; Hua, I. Elucidation of the 1,4-DioxaneDecomposition Pathway at Discrete Ultrasonic Frequencies. En-viron. Sci. Technol. 2000, 34, 3944.
(40) Hua, I.; Hochemer, R. H.; Hoffmann, M. R. SonochemicalDegradation of p-Nitrophenol in a Parallel-Plate Near-FieldAcoustical Processor. Environ. Sci. Technol. 1995, 29, 2790.
(41) Hoffmann, M. R.; Hua, I.; Hochemer, R. Applications ofUltrasonic Irradiation for the Degradation of Chemical Contami-nants in Water. Ultrason. Sonochem. 1996, 3, S163.
Received for review January 22, 2002Revised manuscript received July 24, 2002
Accepted July 29, 2002
IE020069A
4964 Ind. Eng. Chem. Res., Vol. 41, No. 20, 2002





























