Embed Size (px)
Citation preview

1
SOLIDARITY TRIAL PLUS Standard Operating Procedures
-
SOLIDARITY TRIAL PLUS
World Health Organization
An international randomised trial of additional
treatments for COVID-19 in hospitalised patients
who are all receiving the local standard of care
Standard Operating Procedures and
Appendixes
Version 1.0
April 5, 2021
This SOPs are confidential to trial investigators. It should not be disclosed to others
without permission from the WHO, except to seek the consent of collaborators or
participants.

2
SOLIDARITY TRIAL PLUS Standard Operating Procedures
Contents
SOP-1 APPROVAL OF HOSPITALS TO JOIN THE TRIAL AND ACCESS TRIAL DRUGS ....................... 3
SOP-2 ACCESS TO GLOBAL ENROLMENT AND RANDOMIZATION CENTER ...................................... 4
SOP-3 INFORMED CONSENT FORM ......................................................................................................... 5
SOP-4 PHARMACY MANUAL FOR ARTESUNATE .................................................................................... 9
SOP-5 PHARMACY MANUAL FOR IMATINIB ........................................................................................... 11
SOP-6 PHARMACY MANUAL FOR INFLIXIMAB ...................................................................................... 13
SOP-7 REPORTING OF MAIN OUTCOMES WHILE A PATIENT IS IN HOSPITAL ................................. 16
SOP-8 REPORTING SAES AND SUSARS ................................................................................................ 17
SOP-9 REPORTING OF PROTOCOL DEVIATIONS AND VIOLATIONS ................................................. 19
APPENDIX 1. GLOBAL DATA AND SAFETY MONITORING COMMITTEE ............................................. 20
APPENDIX 2. SOLIDARITY TRIAL GOVERNANCE ................................................................................. 21
APPENDIX 3. PROTOCOL AMENDMENT HISTORY ............................................................................... 22
3
SOLIDARITY TRIAL PLUS Standard Operating Procedures
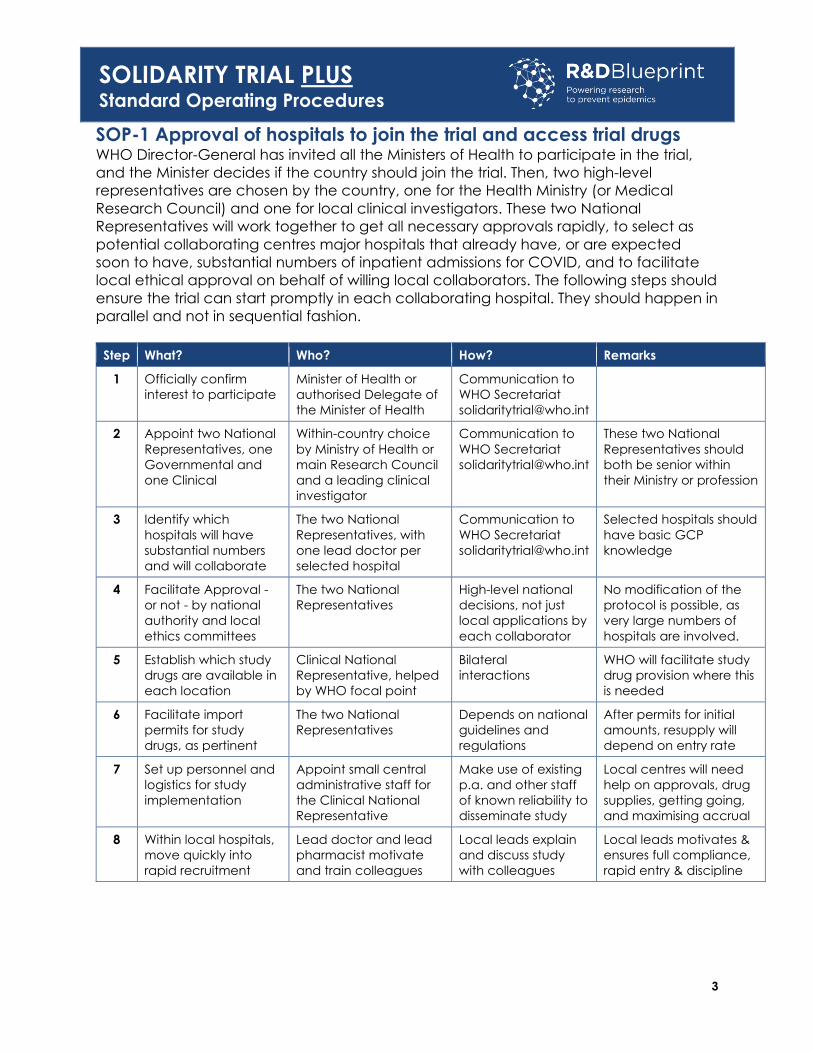
SOP-1 Approval of hospitals to join the trial and access trial drugs WHO Director-General has invited all the Ministers of Health to participate in the trial,
and the Minister decides if the country should join the trial. Then, two high-level
representatives are chosen by the country, one for the Health Ministry (or Medical
Research Council) and one for local clinical investigators. These two National
Representatives will work together to get all necessary approvals rapidly, to select as
potential collaborating centres major hospitals that already have, or are expected
soon to have, substantial numbers of inpatient admissions for COVID, and to facilitate
local ethical approval on behalf of willing local collaborators. The following steps should
ensure the trial can start promptly in each collaborating hospital. They should happen in
parallel and not in sequential fashion.
Step What? Who? How? Remarks
1 Officially confirm
interest to participate
Minister of Health or
authorised Delegate of
the Minister of Health
Communication to
WHO Secretariat
2 Appoint two National
Representatives, one
Governmental and
one Clinical
Within-country choice
by Ministry of Health or
main Research Council
and a leading clinical
investigator
Communication to
WHO Secretariat
These two National
Representatives should
both be senior within
their Ministry or profession
3 Identify which
hospitals will have
substantial numbers
and will collaborate
The two National
Representatives, with
one lead doctor per
selected hospital
Communication to
WHO Secretariat
Selected hospitals should
have basic GCP
knowledge
4 Facilitate Approval -
or not - by national
authority and local
ethics committees
The two National
Representatives
High-level national
decisions, not just
local applications by
each collaborator
No modification of the
protocol is possible, as
very large numbers of
hospitals are involved.
5 Establish which study
drugs are available in
each location
Clinical National
Representative, helped
by WHO focal point
Bilateral
interactions
WHO will facilitate study
drug provision where this
is needed
6 Facilitate import
permits for study
drugs, as pertinent
The two National
Representatives
Depends on national
guidelines and
regulations
After permits for initial
amounts, resupply will
depend on entry rate
7 Set up personnel and
logistics for study
implementation
Appoint small central
administrative staff for
the Clinical National
Representative
Make use of existing
p.a. and other staff
of known reliability to
disseminate study
Local centres will need
help on approvals, drug
supplies, getting going,
and maximising accrual
8 Within local hospitals,
move quickly into
rapid recruitment
Lead doctor and lead
pharmacist motivate
and train colleagues
Local leads explain
and discuss study
with colleagues
Local leads motivates &
ensures full compliance,
rapid entry & discipline

4
SOLIDARITY TRIAL PLUS Standard Operating Procedures
SOP-2 Access to global enrolment and randomization center To facilitate collaboration even in overloaded hospitals, patient enrolment and
randomisation (via a cloud-based GCP-compliant platform) and all other trial
procedures are greatly simplified.
Within each country, the national co-ordinator invites selected hospitals and
helps them get ethical and regulatory approval and study drugs then patient
entry can begin. Access is via the WHO study website
https://data.castoredc.com/.
Each doctor has access to the platform using an individual secured password. A
study ID for the patient is then generated and displayed.
Eligibility: Adults (age ≥ 18 years), hospitalised with laboratory-confirmed COVID,
not expecting transfer within 72 hours, and, in the view of their doctors, with no
contra-indication to any potentially relevant study drug.
Consent: The study website https://data.castoredc.com/studies has printable
patient information in some UN official languages. Once the information has
been explained to patients, obtaining consent takes only a few minutes. An
electronic image of the signature page is kept (or, if national regulations forbid
this, a note to file), and the printed information and original consent stays with
the patient or legal representative.
Data collected electronically immediately before randomisation:
• Country, hospital (from a list of approved hospitals) and randomising doctor
• Confirmation that informed consent was obtained from the patient (or a
surrogate/ representative)
Age, sex and (yes/no): smoking, diabetes, heart disease, chronic liver
disease, chronic lung disease, asthma, HIV, obesity
• Date of onset of symptoms, date of hospitalization, and (yes/no)
current/planned use of a few drugs
• Respiratory support (highest level currently being given): A. No oxygen, B.
Low-flow oxygen, C. High-flow nasal oxygen, D. Non-invasive ventilation, E.
Invasive ventilation, F. ECMO
• SpO2 (%) and, if not ventilated, respiratory rate
• Any major bilateral lung abnormality? (not imaged/no major
abnormality/infiltrations/patchy shadowing)
• Which relevant study drugs are locally available? (yes/no: artesunate,
infliximab, imatinib; confirm that none of these locally available drug(s) is, in
the doctor’s view, definitely contra-indicated)
Trial entry; randomization: Once electronic data collection has been completed
the patient automatically enters the trial and a random allocation of their trial
treatment is generated (by an algorithm that ensures eventual balance in the
characteristics just recorded between each study drug and its controls) and
displayed. The patients will be randomly allocated either to Standard of Care
(SoC) or to one of the study drugs.

5
SOLIDARITY TRIAL PLUS Standard Operating Procedures
SOP-3 Informed consent form
Coronavirus-induced disease (COVID-19, or COVID)
Informed consent to join the WHO Solidarity randomised trial
To the health worker: We are inviting hospitalised adults 18 years or older to
consent to join a research study. All who participate will receive the usual care
offered in this hospital, and in addition some but not all will be given one extra
treatment chosen at random from a few untested treatments or a placebo. The
aim is to discover whether any of these treatments slows disease progression or
improves survival.
To the patient: You have a lung disease called COVID. Several medicines for it have
been proposed. These medicines may have no effect or a moderate effect on the
disease, but none is expected to have a big effect. This hospital is collaborating through
the World Health Organization and hospitals in many other countries in a study to help
discover whether any of these treatments can help.
Whether or not you choose to join, you will still receive the usual standard of care at this
hospital. If you do not join the study, you will receive whatever your doctor thinks is best.
If you decide to join, you may receive one untested treatment chosen by chance, as if
on the roll of a dice. This form has two parts:
1. To share information about the treatments being tested.
2. For your signature, if you do decide to take part
PART 1: Information Sheet
Introduction
I am _____________________________, working for the _________________________.
You have a lung disease called COVID that recently spread across the world. Most
people recover fully from COVID, but a few die. This hospital is collaborating in an
international research study to help discover whether some untested treatments could
help treat COVID. I am going to tell you about the study, then invite you to join it.
Before you decide, you can talk to anyone about it. If there are any words you don’t
understand, please ask me to stop and explain. If you have questions later, I’ll be
available to answer them, and so will the study doctors.
Principal Investigator
Study Sponsor Ministry of Health and WHO
Name of protocol and version Solidarity Trial PLUS

6
SOLIDARITY TRIAL PLUS Standard Operating Procedures
Purpose of the research
The disease is called COVID-19 because it was first discovered in 2019, but I’ll just call it
COVID. It’s caused by a new virus that can be passed between people touching or
passed through the air when an infected person coughs or sneezes, so the hospital staff
wears protective clothing. Most people with COVID get better without coming to the
hospital, and most who come to the hospital also get better, although they may get
worse before they get better. But, a few of those in the hospital die from the illness. The
Ministry of Health and the World Health Organization are organizing a study in many
countries in which various treatments are studied, to see whether they are of any use for
treating COVID. The study treatments are listed briefly below.
During the study, some treatments may get removed from this list, and others may get
added to it. Each patient will receive at most one of the treatments.
Invitation to participate
Adults admitted to this hospital with COVID can join this study. If you join, you will be
asked to sign that you understand that there are possible risks and benefits and consent
to join the study. Your doctor will check whether you are eligible to join, and whether
any of the study treatments would definitely be unsuitable for you.
After those checks, brief details identifying you and any other conditions you have are
put into the computer, and you are then randomly allocated to receive the care you
normally would receive in this hospital alone or with one of the drugs being evaluated
in the study. Neither you nor the medical staff can choose which of the study options
you will receive, as the computer makes this allocation at random, as if on the roll of a
dice.
Any study treatment should start promptly unless you or your doctor decide for any
reason that it should stop. In addition, you will still receive what is already being done
for COVID patients in this hospital. No additional visits after you leave the clinic/hospital
are required. You or our doctor can stop the untested treatment at any time before it
has been completed, and you are free at any time to change your mind and stop
participating.
Joining is voluntary and will not affect the care you receive at this hospital. You have
the right to withdraw consent to participate at any time without reprisal. but could still
remain in the study, with the outcome of your disease being reported by the study
team. You will not be paid for your participation, and neither will the medical staff. Your
identifying details will be shared confidentially with international researchers, along with
information about the course of your illness, and we would like to use your identifying
details to link your treatment to your future medical records for long enough to know
whether you are properly cured. The findings will be made freely available worldwide
to help future patients, but your details will not be identifiable.

7
SOLIDARITY TRIAL PLUS Standard Operating Procedures
I’ve got a list describing the possible side-effects of each of the study drugs briefly. We
could either read it now, or you could join the study, find out immediately whether
you’d be taking one of these drugs, and then just learn about that one and decide
whether or not you want to take it.
Risks and benefits
Any study treatment you receive may or may not help you personally, but this study
could help future patients. This study has been reviewed and approved by
__________________________________________________, a committee (contact details) set
up to make sure research participants are protected.
Some of the study treatments are given as infusions in the vein and some orally.
Artesunate is a medication used in the treatment of malaria. Artesunate is part of the
standard treatment for malaria globally. The dose that will be administered is the
standard dose recommended for the treatment of severe malaria. It will involve an
initial infusion and then additional injections after 12 hours, and 24 hours and thereafter
every 24 hours for maximum 7 days.
Infliximab is a medication used to treat a number of diseases of the immune system.
The dose that will be administered is the standard dose that is given repeatedly for the
treatment of psoriasis (a skin disease that causes red, itchy scaly patches, most
commonly on the knees, elbows, trunk and scalp). This will be given as an infusion in the
vein given only once only. The administration takes about 2 hours.
Imatinib is an oral medication used to treat cancer. The dose that will be given is the
standard maintenance dose administered for several years to patients with cancers of
the blood. However, in this study it will be given orally, once daily, for a maximum 14
days.
Before administering you any of these drugs your doctor will review if any of these drugs
is not appropriate for you. There are a few specific contraindications:
Artesunate will not be given to you if you have a history of allergic reaction to it or other
drugs of the same family (called artemisinins), or to any of the components of the drug.
In people with malaria, the doctor will monitor the red blood cell counts after about 2
weeks to evaluate if you have developed anemia.
Infliximab will not be administered if you have a history of allergic reaction to it or to any
of the components of this drug. The doctor may also decide not to give this drug to you
if you have moderate or severe heart failure, tuberculosis or certain severe infections. A
test to determine if you may have latent tuberculosis or hepatitis B may be necessary. If
one of them is positive, the doctor will refer you for treatment if necessary. Receiving
the treatment of the study drug does not have to wait for these results.
Imatinib will not be administered if you have a history of allergic reaction to it or to any
of the components of the drug. This drug may affect the functioning of your liver and
therefore the doctor will not administer it to you if you have certain liver diseases.
There is limited data on its use during pregnancy and breastfeeding, so the doctor will
discuss with you to decide if the potential benefits are greater than the unknown risks.
8
SOLIDARITY TRIAL PLUS Standard Operating Procedures
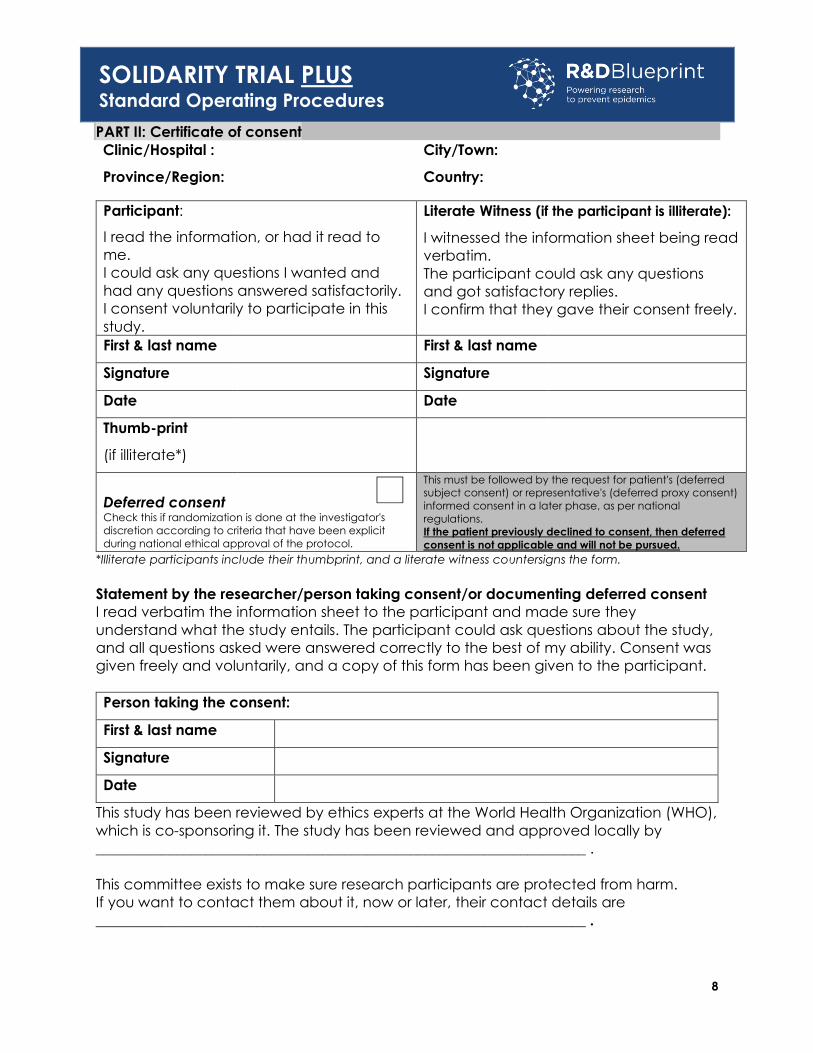
PART II: Certificate of consent
Clinic/Hospital : City/Town:
Province/Region: Country:
Participant:
I read the information, or had it read to
me.
I could ask any questions I wanted and
had any questions answered satisfactorily.
I consent voluntarily to participate in this
study.
Literate Witness (if the participant is illiterate):
I witnessed the information sheet being read
verbatim.
The participant could ask any questions
and got satisfactory replies.
I confirm that they gave their consent freely.
First & last name First & last name
Signature Signature
Date Date
Thumb-print
(if illiterate*)
Deferred consent Check this if randomization is done at the investigator's
discretion according to criteria that have been explicit
during national ethical approval of the protocol.
This must be followed by the request for patient's (deferred
subject consent) or representative's (deferred proxy consent)
informed consent in a later phase, as per national
regulations.
If the patient previously declined to consent, then deferred
consent is not applicable and will not be pursued. *Illiterate participants include their thumbprint, and a literate witness countersigns the form.
Statement by the researcher/person taking consent/or documenting deferred consent
I read verbatim the information sheet to the participant and made sure they
understand what the study entails. The participant could ask questions about the study,
and all questions asked were answered correctly to the best of my ability. Consent was
given freely and voluntarily, and a copy of this form has been given to the participant.
Person taking the consent:
First & last name
Signature
Date
This study has been reviewed by ethics experts at the World Health Organization (WHO),
which is co-sponsoring it. The study has been reviewed and approved locally by
___________________________________________________________________ .
This committee exists to make sure research participants are protected from harm.
If you want to contact them about it, now or later, their contact details are
___________________________________________________________________ .

9
SOLIDARITY TRIAL PLUS Standard Operating Procedures
SOP-4 Pharmacy manual for Artesunate
1. Drug product
Artesunate for injection is a sterile white crystalline powder, in clear, transparent, Type 1
(5 ml) vial. In addition to the Artesunate vial, the package contains: 1 ml type 1
ampoule with 50mg/ml sodium bicarbonate as a sterile clear colourless liquid and a 5
ml type1 ampoule with 9 mg/ml sodium chloride as a sterile clear colourless liquid.
2. Drug receipt
Artesunate must be shipped at temperature below 30˚C. Upon receipt, the recipient
should check all vials and ensure no vials are broken. If vials are broken, the vials should
be discarded. Receipt and accountability of the product on site should be
documented according to the study procedures and the appropriate persons should
be notified.
3. Storage and handling
Store vials of Artesunate for Injection and sterile diluent in the carton at 20°C to 25°C
(68°F to 77°F); excursions permitted to 15°C to 30°C (59°F to 86°F). Do not freeze. Avoid
exposure to heat. Keep protected from light. Do not use beyond the expiration date.
4. Dose preparation
The Artesunate white powder needs to be reconstituted with the sodium bicarbonate
solution and diluted with the sodium chloride solution. The reconstituted Artesunate
solution should always be used immediately and discarded if not used within 1.5 hours.
Artesunate is to be administered at a dose of 2.4 mg/kg body weight, by intravenous
(IV) at 0, 12 and 24 hours and then once daily. The total duration of the treatment is 7
days. The required dose in mg of Artesunate should be calculated and the number of
vials of Artesunate needed should be determined prior to reconstituting the Artesunate
powder.
Preparation of the solution of Artesunate for injection:
1. Artesunate for Injection must be constituted with the supplied diluent prior to
administration. A diluent consisting of 12 mL of sterile 0.3 M pH 8.0 sodium phosphate
buffer is provided with Artesunate for Injection.
2. To constitute Artesunate for Injection, withdraw 11 mL of this diluent with a needle
and syringe and inject into the artesunate vial (when constituted the final
concentration of artesunate is 10 mg/mL).

10
SOLIDARITY TRIAL PLUS Standard Operating Procedures
3. Swirl gently (do not shake) for up to 5 to 6 minutes until the powder is fully dissolved
and no visible particles remain.
4. Parenteral drug products should be inspected visually for particulate matter and
discoloration prior to administration, whenever solution and container permit. Do not
administer Artesunate for Injection if particulate matter and/or discoloration is
observed.
5. After constitution, inject the constituted solution intravenously (through an
established intravenous line or needle) as a slow bolus over 1 to 2 minutes. Discard
the vial and any unused portion of the drug product after use.
6. The volume of the solution required (mL) will be: Volume(mL)= (dose(mg)):10.
7. The Artesunate solution should NOT be administrated as an intravenous drip.
5. Disposition of unused product
Records of doses dispensed should be kept and provided to study staff on request. If
any unopened study packages remain at the end of the study, the local study PI
should be contacted for further instructions regarding the disposition of these vials.
Opened, partially unused packages should be destroyed at study end in consultation
with study staff after proper accountability has been performed.
6. Maintenance of inventory logs
The use of doses should be tracked and recorded on drug disposition logs for use
during accountability procedures by study staff. Records of when doses were
dispensed or destroyed must be kept in these logs.
7. Emergency contact
In the event of an emergency or the need for immediate information regarding
product preparation, handling, storage or administration, contact the study’s local
investigator.

11
SOLIDARITY TRIAL PLUS Standard Operating Procedures
SOP-5 Pharmacy manual for Imatinib
1. Drug product
Imatinib is available as brown-orange film coated Imatinib Mesylate tablets of 400 mg in
PVD C/ aluminium blisters to be administered orally. The tablet is breakable to get 200
mg. It contains the following main excipients: cellulose microcrystalline, crospovidone,
hypomellose, magnesium stearate, silicia colloidal anhydrous, Iron oxide red and
yellow, macrogol, talc.
2. Drug receipt
Imatinib must be shipped at temperature below 30˚C. Upon receipt, the recipient
should check the integrity of the package. If packages are broken, they should be
discarded. Receipt and accountability of the product on site should be documented
according to the study procedures and the appropriate persons should be notified.
3. Storage and handling
Store at 25°C (77°F); excursions permitted to 15-30°C (59-86°F). Protect from moisture.
4. Dose preparation
The recommended dosage for the study is 400mg to be administered orally once daily
for a total duration of 14 days. The dose should be taken orally with a meal and a large
glass of water. For patients unable to swallow, the tablets may be dispersed in a glass of
still water or apple juice.
The required number of tablets should be placed in the appropriate volume (200 mL for
400 mg tablet). The suspension should be administered immediately after the complete
disintegration of the tablets.
5. Disposition of unused product
Records of doses dispensed should be kept and provided to study staff on request. If any
unopened study packages remain at the end of the study, the local study PI should be
contacted for further instructions regarding the disposition of these vials.
Opened, partially unused packages should be destroyed at study end in consultation
with study staff after proper accountability has been performed.

12
SOLIDARITY TRIAL PLUS Standard Operating Procedures
6. Maintenance of inventory logs
The use of doses should be tracked and recorded on drug disposition logs for use during
accountability procedures by study staff. Records of when doses were dispensed or
destroyed must be kept in these logs.
7. Emergency Contact
In the event of an emergency or the need for immediate information regarding
product preparation, handling, storage or administration, contact the study’s local
investigator.

13
SOLIDARITY TRIAL PLUS Standard Operating Procedures
SOP-6 Pharmacy manual for Infliximab
1. Drug product
Infliximab is available as a freeze-dried white pellet powder in a type 1 glass vial with
rubber stopper and aluminium crimp protected by a plastic bag. Each vial contains
100 mg of infliximab and the following main excipients: sucrose, polysorbate 80,
monobasic sodium phosphate, dibasic sodium phosphate.
2. Drug receipt
Infliximab could be shipped at temperature up to a maximum of 25˚C (recommended
storage temperature 2˚C - 8˚C). The product may be at a temperature up to 25˚C for a
period of 6 months, but not exceeding the original expiry date. Upon removal from
refrigerated storage, Infliximab must not be returned to refrigerated storage.
Upon receipt, the recipient should check the integrity of the package. If packages are
broken, they should be discarded. Receipt and accountability of the product on site
should be documented according to the study procedures and the appropriate
persons should be notified.
3. Storage and handling
Before reconstitution, the recommended storage temperature is between 2˚C - 8˚C.
However, the product may be stored up to 25˚C (refer to section 2).
After reconstitution, from a microbiological point of view, the infusion solution should be
administered immediately. If not possible, it should not be kept no longer than 24 hours
at 2˚C - 8˚C. Unless, if the reconstitution/ dilution is done in a controlled and validated
aseptic conditions. From a physical and chemical point of view, the infusion solution is
stable for up to 28 days at 2˚C - 8˚C and for an additional 24 hours at 25˚C after
removal from the refrigerator.
4. Dose preparation
The proposed dosing schedule of this study is 5mg/Kg/dose to be administered as a
single IV infusion.
Infliximab powder reconstitution:
The reconstituted infusion solution should be prepared by a trained medical
professional using aseptic technique by the following procedure:

14
SOLIDARITY TRIAL PLUS Standard Operating Procedures
1. Calculate the dose, total volume of reconstituted INFLIXIMAB solution
required and the number of INFLIXIMAB vials needed. Each INFLIXIMAB vial
contains 100 mg of the infliximab antibody.
2. Reconstitute each INFLIXIMAB vial with 10 mL of Sterile Water for Injection,
USP, using a syringe equipped with a 21-gauge or smaller needle as follows:
o Remove the flip-top from the vial and wipe the top with an alcohol swab.
Insert the syringe needle into the vial through the center of the rubber
stopper and direct the stream of Sterile Water for Injection, USP, to the
glass wall of the vial.
o Gently swirl the solution by rotating the vial to dissolve the lyophilized
powder. Avoid prolonged or vigorous agitation.
o DO NOT SHAKE.
o Foaming of the solution on reconstitution is not unusual.
o Allow the reconstituted solution to stand for 5 minutes. The solution should
be colorless to light yellow and opalescent, and the solution may develop
a few translucent particles as infliximab is a protein.
o Do not use if the lyophilized cake has not fully dissolved or if opaque
particles, discoloration, or other foreign particles are present.
Infusion solution preparation:
3. Dilute the total volume of the reconstituted INFLIXIMAB solution dose to 250
mL with sterile 0.9% Sodium Chloride Injection, USP, by withdrawing a volume
equal to the volume of reconstituted INFLIXIMAB from the 0.9% Sodium
Chloride Injection, USP, 250 mL bottle or bag.
4. Slowly add the total volume of reconstituted INFLIXIMAB solution to the 250
mL infusion bottle or bag.
5. Gently mix.
6. The resulting infusion concentration should range between 0.4 mg/mL and 4
mg/mL.
Infusion solution administration:
7. The INFLIXIMAB infusion should begin within 3 hours of reconstitution and
dilution.
8. The infusion must be administered over a period of not less than 2 hours and
must use an infusion set with an in-line, sterile, non-pyrogenic, low-protein-
binding filter (pore size of 6 of 58 Reference ID: 3403013 1.2 µm or less).
9. The vials do not contain antibacterial preservatives. Therefore, any unused
portion of the infusion solution should not be stored for reuse.
10. No physical biochemical compatibility studies have been conducted to
evaluate the coadministration of INFLIXIMAB with other agents. INFLIXIMAB

15
SOLIDARITY TRIAL PLUS Standard Operating Procedures
should not be infused concomitantly in the same intravenous line with other
agents.
11. Parenteral drug products should be inspected visually before and after
reconstitution for particulate matter and discoloration prior to administration,
whenever solution and container permit. If visibly opaque particles,
discoloration or other foreign particulates are observed, the solution should
not be used.
12. Observe patients for at least 1-2 hours post infusion for acute infusions related
reactions.
13. Emergency equipment must be available: adrenaline, antihistamine, corticosteroids,
and artificial airway.
14. Pre-treatment with an antihistamine, hydrocortisone and/ or paracetamol is
possible. The infusion rate may be slowed in order to decrease the risk of infusion
related reactions.
5. Disposition of unused product
Records of doses dispensed should be kept and provided to study staff on request. If any
unopened study packages remain at the end of the study, the local study PI should be
contacted for further instructions regarding the disposition of these vials.
Opened, partially unused packages should be destroyed at study end in consultation
with study staff after proper accountability has been performed.
6. Maintenance of inventory logs
The use of doses should be tracked and recorded on drug disposition logs for use during
accountability procedures by study staff. Records of when doses were dispensed or
destroyed must be kept in these logs.
7. Emergency Contact
In the event of an emergency or the need for immediate information regarding
product preparation, handling, storage or administration, contact the study’s local
investigator.

16
SOLIDARITY TRIAL PLUS Standard Operating Procedures
SOP-7 Reporting of main outcomes while a patient is in hospital
To facilitate collaboration even in hospitals that have become overloaded, patient
enrolment and randomization (via the internet) and all other trial procedures are
greatly simplified, and no paperwork at all is required.
Withdrawal from the treatment that was randomly allocated at study entry need not
imply withdrawal from information on outcome in hospital being reported to the WHO
at the end of the hospital stay. But, if the patient (or a legal representative) decides the
patient will withdraw and that no further data will be sent to the WHO study office, then
only the date of withdrawal will be reported; no further information will be given, unless
an adverse drug reaction report is legally required.
At discharge or death, the study doctor will log into the study website
https://data.castoredc.com/and enter:
When patients die or are discharged, follow-up ceases and it is reported:
• Which study drug was given (and for how long?)
• Which of a few selected other drugs were given (and for how long?)
• What respiratory support was given (and first and last dates used?) A No oxygen,
B Low-flow supplemental oxygen, C High-flow nasal oxygen, D Non-invasive
ventilation, E Invasive ventilation, F ECMO upplemental oxygen, high-flow nasal
oxygen, non-invasive ventilation, invasive ventilation, ECMO
• Date and cause of death in hospital or date discharged alive (and, if discharged
alive but still at appreciable risk of death from COVID, give details)
• Pregnant? Yes/No/unknown
Patient information will be encrypted and held securely by the at the cloud database.
Those analysing it will use only anonymized data, and no identifiable patient details will
be released to any unauthorized third party. Patient confidentiality will be maintained
when study results are disseminated.

17
SOLIDARITY TRIAL PLUS Standard Operating Procedures
SOP-8 Reporting SAEs and SUSARS
An adverse event is any undesirable experience associated with the use of a medical
product in a patient. Serious Adverse Events (SAEs) associated with the study drug
(possibly related to the treatment), and any suspected unexpected serious adverse
reactions (SUSARs) that are life-threatening must be reported within 24 hours.
The event is a SAE and should be reported when the patient outcome is:
o Death : Report if you suspect that the death was an outcome of the adverse event,
and include the date if known.
o Life-threatening : Report if suspected that the patient was at substantial risk of dying
at the time of the adverse event or use or continued use of the device or other
medical product might have resulted in the death of the patient.
o Hospitalization (prolonged) : Report if admission to the hospital or prolongation of
hospitalization was a result of the adverse event.
o Disability or Permanent Damage : Report if the adverse event resulted in a
substantial disruption of a person's ability to conduct normal life functions, i.e., the
adverse event resulted in a significant, persistent or permanent change, impairment,
damage or disruption in the patient's body function/structure, physical activities
and/or quality of life.
o Congenital Anomaly/Birth Defect : Report if you suspect that exposure to a medical
product prior to conception or during pregnancy may have resulted in an adverse
outcome in the child.
o Required Intervention to Prevent Permanent Impairment or Damage (Devices) :
Report if you believe that medical or surgical intervention was necessary to
preclude permanent impairment of a body function, or prevent permanent
damage to a body structure, either situation suspected to be due to the use of a
medical product.
o Other Serious (Important Medical Events) : Report when the event does not fit the
other outcomes, but the event may jeopardize the patient and may require
medical or surgical intervention (treatment) to prevent one of the other outcomes.
Serious Adverse Events possibly related to the study drug with causality assessment must
be reported. Report includes only SAEs possibly related to the treatment. SAEs must be
reported promptly by logging into the study website https://data.castoredc.com/.
The local doctor reporting the event enters a narrative description of the event and its
seriousness that includes the patient’s trial identification number. Include in the report
results of relevant lab tests and other examinations performed outside protocol
scheduled visits.

18
SOLIDARITY TRIAL PLUS Standard Operating Procedures
SUSARS are Suspected Unexpected Serious Adverse Reactions that are fatal or life-
threatening, such as Stevens-Johnson syndrome, anaphylaxis, aplastic anemia, or
anything comparably uncommon.
Suspected Unexpected Serious Adverse Reaction (SUSAR) includes:
o serious adverse reactions not listed in the Investigator’s Brochure (IB), or
o not listed in the IB at the specificity or severity observed, or
o mentioned in IB as anticipated due to pharmacokinetic properties of the drug, or
o occurred with other drugs in this class, but not with the trial study drug.
SUSARS should be uncommon but must be reported promptly by logging into the study
website https://data.castoredc.com/.
The local doctor reporting the event then enters a narrative description of the event
and its seriousness that includes the patient’s trial identification number.
SAEs (only those possibly related to the treatment)and SUSARs entered onto the study
website are reviewed promptly and, unless refuted, will be reported by the trial center
to the Global Data and Safety Monitoring Committee (Appendix 1), the appropriate
National Regulatory Agency, and the local Ethics Review Committee, the WHO Ethics
Review Committee and the National Regulatory Agency holding the investigational
new drug application via the drug developer within 24 hours of having been entered.
The trial center may need to seek further clarification of the nature and outcome of the
event, and any new information on it that is considered significant on the central review
will be sent as a follow-up report within 15 days.

19
SOLIDARITY TRIAL PLUS Standard Operating Procedures
SOP-9 Reporting of protocol deviations and violations
A protocol deviation is generally considered to be a departure from the study protocol,
or other study-related documents, that has not been approved by the Ethics Review
Committee.
The investigator should not implement any deviation from, or changes of the protocol
without agreement by the sponsor and prior review and documented approval/
favourable opinion from the Ethics Review Committee of such amendment, except
where:
o it is necessary to eliminate an immediate hazard(s) to trial subjects (because the
protocol leaves the local doctor fully responsible for all decisions about patient
care, including discontinuing study medication if considered appropriate), or
o when the change(s) involves only logistical or administrative aspects of the trial
(e.g., change in monitor(s), change of telephone number(s)
A protocol violation is a divergence from the protocol that materially (a) reduces the
quality or completeness of the data, (b) makes the Informed Consent Form inaccurate,
or (c) impacts a subject's safety, rights, or welfare.
When investigators are aware that a deviation is likely to take place, they may take a
prior approval of the sponsor in the form of a waiver for the deviation, but when the
deviation is discovered after it has taken place, and then the incident is a deviation
and not a waiver.
The local monitor may grant waivers (in writing) for deviations that are not likely to
impact either the data quality or safety of the subject. Since waivers are preapproved
deviations, no action is necessary to be taken.
Major protocol deviations and protocol violation should be uncommon but must be
reported promptly by logging into the study website https://data.castoredc.com/.
The investigator reporting the event then enters a narrative description of the event and
its seriousness that includes the trial identification number patient’s affected and the
corrective actions taken.

20
SOLIDARITY TRIAL PLUS Standard Operating Procedures
Appendix 1. Global Data and Safety Monitoring Committee
Global Data and Safety Monitoring Committee (DSMC): This independent committee
will not include representatives of the trial sponsors, trial committees or trial centers, and
will not include any doctors who are directly responsible for the treatment of individual
COVID patients.
Interim trial analyses will be monitored by the DSMC. Otherwise, the WHO, collaborators,
and administrative staff (except those who produce the confidential analyses) will
remain ignorant of the interim results.
The DSMC will, at intervals decided by itself, examine confidential interim analyses of
safety and efficacy, reporting them to the executive group only if the DSMC considers
them likely to require publication or a change in the conduct of the trial. Otherwise, the
trial sponsors, trial committees, and trial centers will remain blind to the interim findings.
The protocol states that the evidence on mortality must be strong enough, and the range
of uncertainty around the results must be narrow enough to affect national and global
treatment strategies. It also states that realistic, appropriate sample sizes could not be
estimated at the start of the trial; that it may be possible to enter several thousand
hospitalized patients with the relatively mild disease when admitted and a few thousand
with severe disease, yielding results that are separately reliable for each; and that the
response to certain treatments may differ substantially between different populations or
sub-populations (e.g., patients with particular prior conditions, older adults, patients in
one or another large country).
In the light of this, the DSMC will independently evaluate the interim analyses and will
inform the Executive Group of the Steering Committee if at any stage the results are, in
the judgement of the DSMC, sufficiently robust for general release (ie, strong enough
and with a sufficiently narrow range of uncertainty around the findings to affect
national and global treatment strategies). The DMSC will decide independently how
best to respond to interim analyses of safety and apparent efficacy, and what further
such analyses to require.
Although the DSMC will be informed of each such SAE and SUSAR and major protocol
violations as the trial office deals with reporting it, the DSMC may chiefly be concerned
not with each individual event, but with the confidential analyses of the accumulated
evidence on all such events. In light of this, the DMSC will decide independently how
best to respond to the evidence on adverse reactions.

21
SOLIDARITY TRIAL PLUS Standard Operating Procedures
Appendix 2. Solidarity Trial Governance
The Director-General of the WHO will invite the Ministries of Health of selected member
states to join the trial. Within each country, the WHO and the Ministry of Health would
act as co-sponsors of the trial.
International Steering Committee: This will govern the conduct of the trial in accordance
with the agreed international protocol, amended as necessary during the study.
Governments that do decide to join will, in consultation with WHO, appoint two members
to the international steering committee. One will be a Governmental representative of
the Ministry of Health (or national Medical Research Council, or equivalent), and the
other will be a leading clinician who would act as the national PI of the trial, and who
would seek the collaboration of appropriate local clinicians at major hospitals. These two
members from each country would be jointly responsible for obtaining rapidly, at national
and at the local hospital level, ethical approval and any other relevant permissions, and
for encouraging whole-hearted participation by the collaborating hospitals.
Executive group of the steering committee: For practicality, a smaller executive group
of about 5-9 members of this committee will be set up in consultation with WHO. They
will confer electronically at frequent intervals with WHO to ensure the study is being
conducted appropriately by the team within WHO and to ensure the trial steering
committee is appropriately informed and consulted.
WHO trial center (Geneva): This will be responsible for the conduct of the trial. It will
establish and maintain a web-based service for randomization, for receiving electronic
reports of any SAEs and SUSARs for entry of follow-up information, and for sending
reminders when follow-up is overdue. The trial center will help deal with obtaining all
necessary approvals, recruiting and motivating centers, ensuring drug supplies, and
reporting SUSARS. It will monitor recruitment rates, follow-up rates, data plausibility, and
overdue follow-up, describing and regularly discussing with the executive group all
aspects of the progress of the trial, but not conducting interim analyses of trial results.
Global Data and Safety Monitoring Committee (DSMC): This independent committee
will examine confidential interim analyses of safety and efficacy, reporting them to the
executive group only if the DSMC considers them likely to require publication or a
change in the conduct of the trial. Otherwise, the trial sponsors, trial committees, and
trial centers will remain blind to the interim findings.
Add-on studies: The trial involves only a simple characterization of patients and outcomes.
So, within particular countries, some hospitals participating in the trial may choose to
collaborate with each other in more detailed studies. Such add-on studies should not
analyze trial treatment allocation in relation to the main trial outcomes of the duration of
hospital stay, ventilation, and mortality until after the main trial findings have been
published in the names of all collaborators. Apart from that, the planning, conduct, and
reporting of any such studies are wholly independent of WHO trial governance.
22
SOLIDARITY TRIAL PLUS Standard Operating Procedures
Appendix 3. Protocol amendment history
Version Date Description of main amendments o
o
o
o
















![Inter Ceptor NC-81 plus rE6300B] 1 CHEMICAL TRIAL REPORT](https://img.pdfslide.us/doc/110x75/61e126f41140c340693b55da/inter-ceptor-nc-81-plus-re6300b-1-chemical-trial-report-.jpg)











