Embed Size (px)
Citation preview

i
SOLID DISPERSION – AN APPROACH TO ENHANCE THE DISSOLUTION RATE OF
ACECLOFENAC
By M.A.ALEEM
Reg. No. 03PU254
Dissertation Submitted to the Rajiv Gandhi University of Health Sciences, Karnataka, Bangalore
In partial fulfillment
of the requirements for the degree of
MASTER OF PHARMACY in
PHARMACEUTICS
Under the Guidance of Dr.M.H.DEHGHAN
M.Pharm., Ph.D.
DEPARTMENT OF PHARMACEUTICS
Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com)

ii
LUQMAN COLLEGE OF PHARMACY, GULBARGA-585 102
APRIL 2006
RAJIV GANDHI UNIVERSITY OF HEALTH SCIENCES, KARNATAKA, BANGALORE
DECLARATION BY THE CANDIDATE
I hereby declare that this dissertation/ thesis
entitled “SOLID DISPERSION – AN APPROACH TO
ENHANCE THE DISSOLUTION RATE OF
ACECLOFENAC” is a bonafide and genuine research
work carried out by me under the guidance of
Dr.M.H.Dehghan.
Date:
Place: GULBARGA M.A.ALEEM
Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com)

iii
CERTIFICATE BY THE GUIDE
This is to certify that the dissertation entitled
“SOLID DISPERSION – AN APPROACH TO ENHANCE
THE DISSOLUTION RATE OF ACECLOFENAC” is a
bonafide research work done by Mr.M.A.ALEEM in
partial fulfillment of the requirement for the degree of
MASTER OF PHARMACY in PHARMACEUTICS.
Date:
Place: GULBARGA Dr.M.H.Dehghan M.Pharm. Ph.D.
Hon. Research Guide
Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com)

iv
RAJIV GANDHI UNIVERSITY OF HEALTH SCIENCES, KARNATAKA, BANGALORE
ENDORSEMENT BY THE HOD, PRINCIPAL/ HEAD OF THE INSTITUTION
This is to certify that the dissertation entitled
“SOLID DISPERSION – AN APPROACH TO ENHANCE
THE DISSOLUTION RATE OF ACECLOFENAC” is a
bonafide research work done by Mr.M.A.ALEEM under the
guidance of Dr.M.H.DEHGHAN.
Date:
Place: GULBARGA Prof.Syed Sanaullah Principal, Luqman College of Pharmacy, Gulbarga-585102
Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com)

v
COPYRIGHT
DECLARATION BY THE CANDIDATE
I here by declare that the Rajiv Gandhi University of
Health Sciences, Karnataka shall have the rights to
preserve, use and disseminate this dissertation/ thesis in
print or electronic format for academic/ research purpose.
Date:
Place: GULBARGA Mr.M.A.ALEEM
© Rajiv Gandhi University of Health Sciences, Karnataka
Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com)

vi
ACKNOWLEDGEMENT
I am especially deeply indebted to my Honourable Research Guide Dr.Mohammed Hassan Dehghan, under whose able and valuable guidance, this entire research work was successfully completed. I am wholeheartedly thankful to his constant source of encouragement and support, which provided to impetus and paved the way for the successful completion of the research work. I am highly grateful to Prof.Syed Sanaullah, Principal, Dr.Mujeeb, Treasurer, Luqman College of Pharmacy, Gulbarga, and Dr.Syed Rahmatullah, Secretary, Mr.Abdul Majeed, President, Vocational Education Society, Gulbarga, for providing me all facilities and encouragement throughout the research work. My sincere thanks to all the teaching staff of my college especially Mr.M.A.Saleem, Prof.Satyanandam Sade, Mr.S.S.Bushetti, Mrs.Syeda Humera, Mr.Mohd.Najmuddin, Mr.Sadat Ali, Mr.Adil Shareef, Mr.Asgar Ali, Mr.Omar Khan, Mr.Ashfaq A.Mohsin for their timely guidance in enriching my knowledge and encouragement during the course of my work. A special thanks to M/s.Suyash Laboratories, M.I.D.C. Tarapur for providing the drug sample of Aceclofenac. I also thanks to Mr.Sridhar, M/s.Sipra Labs, Hyderabad for analyzing the IR and DSC Samples. I place on record a respectful thanks to Dr.M.G.Purohit, Department of Pharmaceutical Chemistry, Gulbarga University, Gulbarga for his guidance in my analytical work. I am extremely thankful to my seniors Mr.Md.Jafar, Mr.Abdullah Khan, Mr.Ismail Mouzam for helping me in carrying out this work. It gives me immense pleasure to record my sincere thanks to my colleagues and juniors Anant Kulkarni, Mohan VK, Md.Asif Hussain, Vinod Singh, Tahir Ansari, Muqtadar Ahmed, Imran Ahmed, Nagasesha Reddy, Sarim Imam, Abhishek Bansal, Fazeel Ahmed, Vijay Kumar, Shahidullah, Abdul Rauf, Abdul Razzaq, Maqbool Ahmed, Shravan Kumar for their cooperation.
Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com)

vii
I am thankful to my friends who gave boost in all my achievements Saeed Khan, Dildar Ali Mushtaq, Shahzad Khan, Syed Imran Ali, Md.Sadiq Khan, Syed Ilyas, Md.Haris and Waliuddin. I could never forget Mrs.Farha and Ms.Samreen for the inspiration, encouragement and moral support during the course of my studies. I am thankful to Micro Computers, Gulbarga for their cooperation during the time of typing of this work. I express my thanks to Mr.Asadullah, Mr.Ismail, Mr.Peer Pasha, Mr.Narendra and other non-teaching staff and Librarian Ms.Rubina Anjum and Ms.Pratibha for their cooperation. Above all, words fail to express my feelings to my beloved parents and my family whose initiation, constant source of inspiration and encouragement throughout my career.
Date: Place: Gulbarga M.A.ALEEM
Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com)

viii
LIST OF ABBREVIATIONS USED
%w/v .............. Percent weight / volume %v/v ............... Percent volume/ volume
BP................... British Pharmacopoeia COX ............... Cyclooxygenase
cps .................. centipoise DSC................ Differential scanning calorimetry
GIT................. Gastrointestinal tract gm .................. gram(s)
IP .................... Indian Pharmacopoeia IR.................... Infrared
KBr................. Potassium bromide L ..................... Liter(s)
MCC............... Microcrystalline cellulose
mcg/g............ Microgram(s)
mg .................. Milligram(s) ml ................... Milliliter(s) M.................... Molar
nm .................. Nanometer NSAID............ Non-steroidal antiinflammatory drug
PEG ................ Polyethylene glycol PG .................. Prostaglandin
ppm................. parts per million PVP ................ Polyvinyl pyrrolidone
rpm ................. rotations per minutes SLS................. Sodium lauryl sulphate
UV.................. Ultraviolet
Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com)

ix
FORMULATION CODE
APG Solid dispersions of aceclofenac containing PEG6000 prepared by physical mixture
ASG Solid dispersions of aceclofenac containing PEG6000 prepared by solvent evaporation method
AFG Solid dispersions of aceclofenac containing PEG6000 prepared by fusion method
APV Solid dispersions of aceclofenac containing PVP prepared by physical mixture
ASV Solid dispersions of aceclofenac containing PVP prepared by solvent evaporation method
APS Solid dispersions of aceclofenac containing SLS prepared by physical mixture
ASS Solid dispersions of aceclofenac containing SLS prepared by solvent evaporation method
AFS Solid dispersions of aceclofenac containing SLS prepared by fusion method
APK Solid dispersions of aceclofenac containing PVP and PEG6000 prepared by physical mixture
ASK Solid dispersions of aceclofenac containing PVP and PEG6000 prepared by solvent evaporation method
APGS Solid dispersions of aceclofenac containing PEG6000 and SLS prepared by physical mixture
ASGS Solid dispersions of aceclofenac containing PEG 6000 and SLS prepared by solvent evaporation method
AFGS Solid dispersions of aceclofenac containing PEG 6000 and SLS prepared by fusion method
APVS Solid dispersions of aceclofenac containing PVP and SLS prepared by physical mixture
ASVS Solid dispersions of aceclofenac containing PVP and SLS prepared by solvent evaporation method
Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com)

x
ABSTRACT
Aceclofenac, an analgesic and anti-inflammatory agent used in the management of osteoarthritis, rheumatoid arthritis and ankylosing spondylitis. The objective of the present work was to investigate the effect of different types of carriers such as polyvinyl pyrrolidone (PVP), polyethylene glycol (PEG) 6000 and sodium lauryl sulphate (SLS) as solubilizer on in vitro dissolution of aceclofenac. The effect of change in polymer, polymer composition and drug polymer ratio on dissolution of aceclofenac was also determined. Various carriers, viz., PVP, PEG 6000, PVP-SLS (80:20), PEG 6000-SLS (80:20) have been studied with respect to their effect in increasing the solubility of aceclofenac, among the carriers used PVP-SLS (80:20) showed highest solubility. Aceclofenac solid dispersions were prepared using 3² factorial design by fusion and solvent evaporation method with PEG 6000, PVP, SLS, PVP-PEG 6000, PEG 6000-SLS and PVP-SLS. Prepared aceclofenac solid dispersion were evaluated for physical appearance, drug content uniformity, and in vitro dissolution studies. The dissolution was determined by USP XXIII apparatus using phosphate buffer pH 7.4. The highest aceclofenac dissolution rate, 99.87% in 60 minutes, was obtained from solid dispersion containing SLS (ASS7) prepared by solvent evaporation method. The general trend indicated that there was an increase in dissolution rate for solid dispersions prepared in following order SLS>PVP-SLS>PEG 6000-SLS>PVP-PEG 6000> PVP>PEG 6000. IR and DSC studies showed no chemical change between drug and polymer and aceclofenac is homogeneously distributed in an amorphous state within the carrier and no aceclofenac crystallized out of the dispersions. The formulations studied were found to be stable. Finally it may be concluded that, dissolution rate of aceclofenac can be increased by solid dispersion technique, which may be due to increased hydrophilic nature of carrier and also possibly due to reduction in drug crystallinity.
Keywords: Aceclofenac; Solid dispersion; Solubility; In vitro dissolution; Stability.
Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com)

xi
TABLE OF CONTENTS LIST OF TABLES........................................................... xiii-xv LIST OF FIGURES....................................................... xvi-xvii CHAPTER-1 INTRODUCTION ............................................................ 01-24
1.1 Introduction to Solubilization....................................... 01
1.2 Solubilization Techniques ............................................ 03 1.3 Introduction to Solid Dispersion Technology ............... 08
1.4 Historical Background ................................................. 10 1.5 Definition and Types of Solid Dispersions ................... 12
1.6 Mechanism of Increased Dissolution Rate.................... 13 1.7 Selection of a Carrier ................................................... 15
1.8 Polymers Used in Solid Dispersion .............................. 16 1.9 Methods of Preparing Solid Dispersions....................... 18 1.10 Methods of Determination of Types of Solid
Dispersions .................................................................. 21
1.11 Advantages and Disadvantages of Solid Dispersions .... 22
1.12 Future Prospects........................................................... 23
CHAPTER-2 OBJECTIVES .................................................................. 25-28
2.1 Need for the Study ....................................................... 25
2.2 Objectives of the Study ................................................ 26 2.3 Scheme of Work .......................................................... 27
CHAPTER-3 REVIEW OF LITERATURE .......................................... 29-41 3.1 Review of Literature .................................................... 29 3.2 Drug Profile ................................................................. 38
Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com)

xii
CHAPTER-4 MATERIALS AND METHODS...................................... 42-57 4.1 Raw Material Characterization ..................................... 44
4.2 Methods of Preparation of Aceclofenac Solid Dispersion Systems...................................................... 46
4.3 Evaluation of Aceclofenac Solid Dispersion Systems ....................................................................... 53
CHAPTER-5 RESULTS ....................................................................... 58-132 5.1 Standardization of Materials......................................... 58
5.2 Preparation of Aceclofenac Solid Dispersion Systems ....................................................................... 75
5.3 Evaluation of Aceclofenac Solid Dispersion Systems ....................................................................... 75
CHAPTER-6 DISCUSSION ............................................................... 133-140
CHAPTER-7 CONCLUSIONS........................................................... 141-142
CHAPTER-8 SUMMARY .................................................................. 143-144
CHAPTER-9 BIBLIOGRAPHY......................................................... 145-156
ANNEXURES
Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com)
xiii
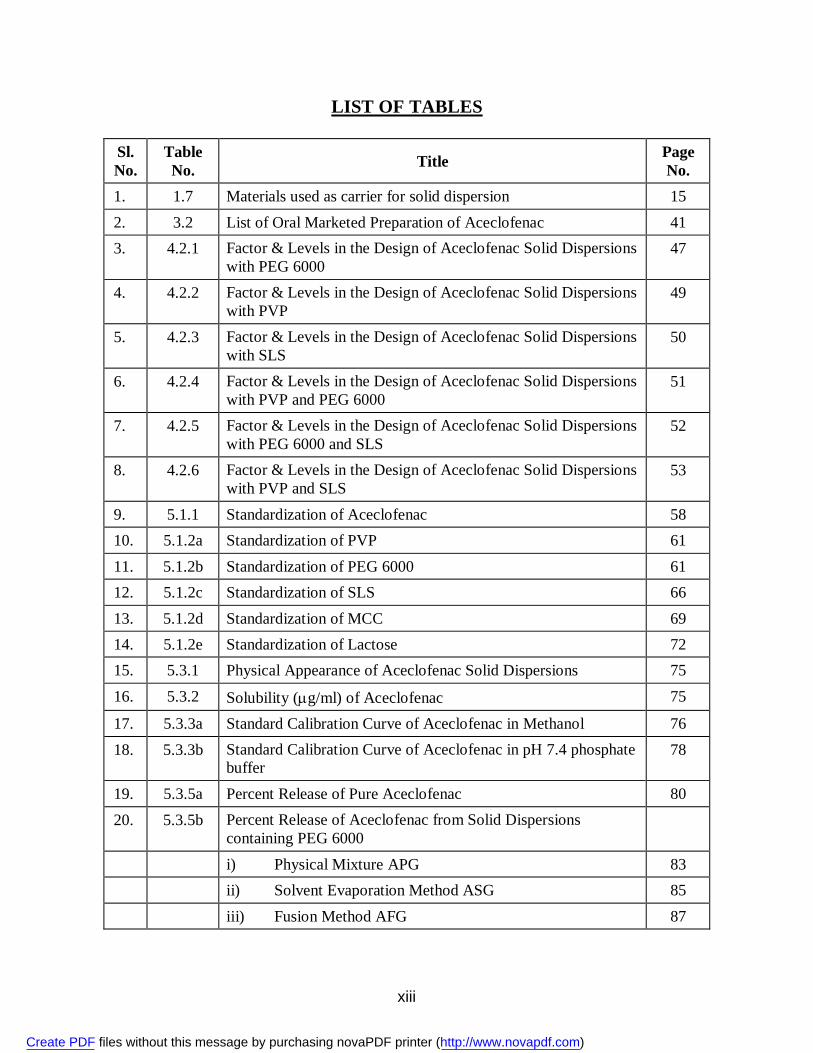
LIST OF TABLES
Sl. No.
Table No. Title Page
No. 1. 1.7 Materials used as carrier for solid dispersion 15 2. 3.2 List of Oral Marketed Preparation of Aceclofenac 41 3. 4.2.1 Factor & Levels in the Design of Aceclofenac Solid Dispersions
with PEG 6000 47
4. 4.2.2 Factor & Levels in the Design of Aceclofenac Solid Dispersions with PVP
49
5. 4.2.3 Factor & Levels in the Design of Aceclofenac Solid Dispersions with SLS
50
6. 4.2.4 Factor & Levels in the Design of Aceclofenac Solid Dispersions with PVP and PEG 6000
51
7. 4.2.5 Factor & Levels in the Design of Aceclofenac Solid Dispersions with PEG 6000 and SLS
52
8. 4.2.6 Factor & Levels in the Design of Aceclofenac Solid Dispersions with PVP and SLS
53
9. 5.1.1 Standardization of Aceclofenac 58 10. 5.1.2a Standardization of PVP 61 11. 5.1.2b Standardization of PEG 6000 61 12. 5.1.2c Standardization of SLS 66 13. 5.1.2d Standardization of MCC 69 14. 5.1.2e Standardization of Lactose 72 15. 5.3.1 Physical Appearance of Aceclofenac Solid Dispersions 75 16. 5.3.2 Solubility (g/ml) of Aceclofenac 75
17. 5.3.3a Standard Calibration Curve of Aceclofenac in Methanol 76 18. 5.3.3b Standard Calibration Curve of Aceclofenac in pH 7.4 phosphate
buffer 78
19. 5.3.5a Percent Release of Pure Aceclofenac 80 20. 5.3.5b Percent Release of Aceclofenac from Solid Dispersions
containing PEG 6000
i) Physical Mixture APG 83 ii) Solvent Evaporation Method ASG 85 iii) Fusion Method AFG 87
Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com)
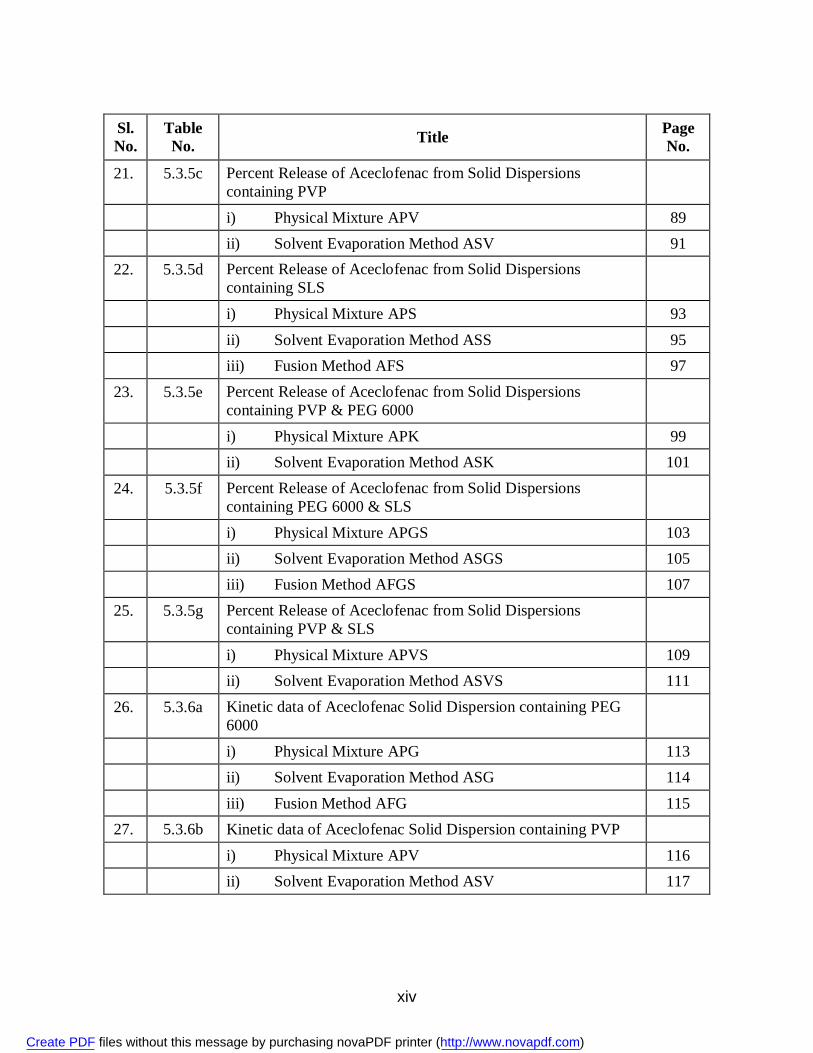
xiv
Sl. No.
Table No. Title Page
No.
21. 5.3.5c Percent Release of Aceclofenac from Solid Dispersions containing PVP
i) Physical Mixture APV 89 ii) Solvent Evaporation Method ASV 91 22. 5.3.5d Percent Release of Aceclofenac from Solid Dispersions
containing SLS
i) Physical Mixture APS 93 ii) Solvent Evaporation Method ASS 95 iii) Fusion Method AFS 97 23. 5.3.5e Percent Release of Aceclofenac from Solid Dispersions
containing PVP & PEG 6000
i) Physical Mixture APK 99 ii) Solvent Evaporation Method ASK 101 24. 5.3.5f Percent Release of Aceclofenac from Solid Dispersions
containing PEG 6000 & SLS
i) Physical Mixture APGS 103 ii) Solvent Evaporation Method ASGS 105 iii) Fusion Method AFGS 107 25. 5.3.5g Percent Release of Aceclofenac from Solid Dispersions
containing PVP & SLS
i) Physical Mixture APVS 109 ii) Solvent Evaporation Method ASVS 111 26. 5.3.6a Kinetic data of Aceclofenac Solid Dispersion containing PEG
6000
i) Physical Mixture APG 113 ii) Solvent Evaporation Method ASG 114 iii) Fusion Method AFG 115 27. 5.3.6b Kinetic data of Aceclofenac Solid Dispersion containing PVP i) Physical Mixture APV 116 ii) Solvent Evaporation Method ASV 117
Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com)
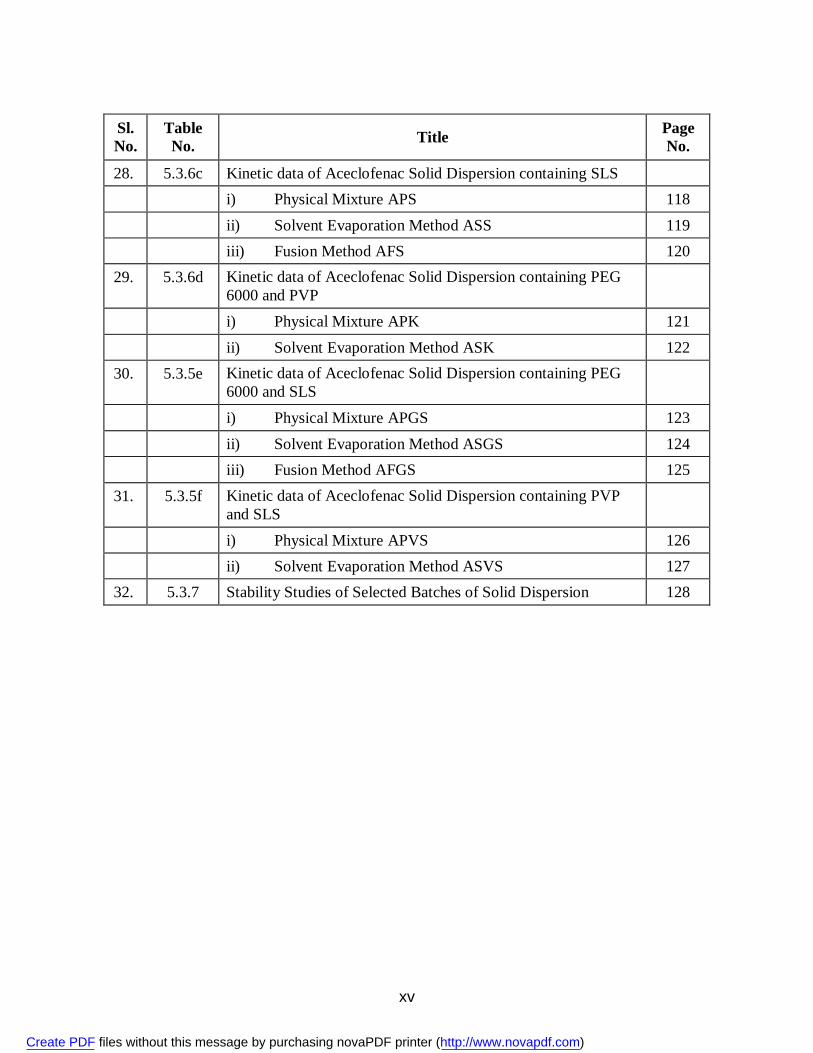
xv
Sl. No.
Table No. Title Page
No.
28. 5.3.6c Kinetic data of Aceclofenac Solid Dispersion containing SLS i) Physical Mixture APS 118 ii) Solvent Evaporation Method ASS 119 iii) Fusion Method AFS 120 29. 5.3.6d Kinetic data of Aceclofenac Solid Dispersion containing PEG
6000 and PVP
i) Physical Mixture APK 121 ii) Solvent Evaporation Method ASK 122 30. 5.3.5e Kinetic data of Aceclofenac Solid Dispersion containing PEG
6000 and SLS
i) Physical Mixture APGS 123 ii) Solvent Evaporation Method ASGS 124 iii) Fusion Method AFGS 125 31. 5.3.5f Kinetic data of Aceclofenac Solid Dispersion containing PVP
and SLS
i) Physical Mixture APVS 126 ii) Solvent Evaporation Method ASVS 127 32. 5.3.7 Stability Studies of Selected Batches of Solid Dispersion 128
Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com)
xvi
LIST OF FIGURES Sl. No.
Figure No. Title Page
No. 1. 1.3 A schematic representation of the bioavailability enhancement
of a poorly water-soluble drug by solid dispersion compared with conventional tablet or capsule
9
2. 5.1.1a IR Spectra of Aceclofenac 59 3. 5.1.1b DSC Thermogram of Aceclofenac 60 4. 5.1.2ai IR Spectra of Polyvinyl Pyrrolidone 62 5. 5.1.2.aii DSC Thermogram of Polyvinyl Pyrrolidone 63 6. 5.1.2bi IR Spectra of PEG 6000 64 7. 5.1.2bii DSC Thermogram of PEG 6000 65 8. 5.1.2ci IR Spectra of Sodium Lauryl Sulphate 67 9. 5.1.2cii DSC Thermogram of Sodium Lauryl Sulphate 68 10. 5.1.2di IR Spectra of Microcrystalline Cellulose 70 11. 5.1.2dii DSC Thermogram of Microcrystalline Cellulose 71 12. 5.1.2ei IR Spectra of Lactose 73 13. 5.1.2eii DSC Thermogram of Lactose 74 14. 5.3.3a Standard Calibration Curve of Aceclofenac in Methanol 77 15. 5.3.3b Standard Calibration Curve of Aceclofenac in pH 7.4 phosphate
buffer 79
16. 5.3.5a Percent Release of Pure Aceclofenac 81 17. 5.3.5b Percent Release of Aceclofenac from Solid Dispersions
containing PEG 6000
i) Physical Mixture APG 84 ii) Solvent Evaporation Method ASG 86 iii) Fusion Method AFG 88 18. 5.3.5c Percent Release of Aceclofenac from Solid Dispersions
containing PVP
i) Physical Mixture APV 90 ii) Solvent Evaporation Method ASV 92
Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com)
xvii
Sl. No.
Figure No. Title Page
No. 19. 5.3.5d Percent Release of Aceclofenac from Solid Dispersions
containing SLS
i) Physical Mixture APS 94 ii) Solvent Evaporation Method ASS 96 iii) Fusion Method AFS 98 20. 5.3.5e Percent Release of Aceclofenac from Solid Dispersions
containing PVP & PEG 6000
i) Physical Mixture APK 100 ii) Solvent Evaporation Method ASK 102 21. 5.3.5f Percent Release of Aceclofenac from Solid Dispersions
containing PEG 6000 & SLS
i) Physical Mixture APGS 104 ii) Solvent Evaporation Method ASGS 106 iii) Fusion Method AFGS 108 22. 5.3.5g Percent Release of Aceclofenac from Solid Dispersions
containing PVP & SLS
i) Physical Mixture APVS 110 ii) Solvent Evaporation Method ASVS 112 23. 5.3.8a DSC Thermogram of Aceclofenac Solid Dispersion containing
PVP and PEG 6000 (ASK9) 129
24. 5.3.8b DSC Thermogram of Aceclofenac Solid Dispersion containing PVP and SLS (APVS9)
130
25. 5.3.9a IR Spectra of Aceclofenac Solid Dispersion containing PVP and PEG 6000 (ASK9)
131
26. 5.3.9b IR Spectra of Aceclofenac Solid Dispersion containing PVP and SLS (APVS9)
132
Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com)

1
CHAPTER–1
INTRODUCTION
1.1 INTRODUCTION TO SOLUBILIZATION:
The solubility is defined as the concentration of the undissolved solid in a
solvent under a given set of conditions. The solution becomes saturated and the
dissolved solute is in equilibrium with the excess undissolved solute1.
Poorly water-soluble drugs are increasingly becoming a problem in terms of
obtaining the satisfactory dissolution within the gastrointestinal tract that is necessary
for good bioavailability. It is not only existing drugs that cause problems but it is the
challenge of medicinal chemists to ensure that new drugs are not only active
pharmacologically but have enough solubility to ensure fast enough dissolution at the
site of administration, often gastrointestinal tract2.
Dissolution of solid dosage forms in gastrointestinal fluids is a prerequisite to
the delivery of the drug to the systemic circulation following oral administration.
Dissolution depends in parts on the solubility of the drug substance in the surrounding
medium. Surface area of drug particle is another parameter that influences drug
dissolution, and in turn drug absorption, particle size is a determinant of surface area3.
The dissolution of a substance may be described by the modified Noye’s-
Whitney equation;
Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com)

2
dc KDS
dt = (Cs-C)
Vh …1
Where dtdc is the rate of increase in C, the concentration of drug in a bulk solution in
which dissolution of the solid particles is taking place; K is a proportionality constant;
D is the diffusion coefficient of the drug in the solvent; S is the surface area of
undissolved solid; V is the volume of the solution; h is the thickness of the diffusion
layer around a particle; and Cs is the solubility of the drug in the solvent. If we
consider a given drug under well-defined conditions (such as controlled liquid
intake), we may assume that D, V and h are relatively constant values. Thus we can
reduce equation (1) to:
dtdc = KS (Cs-C) …2
Equation (2) shows that the two variables, which may be controlled by the
formulation, are the surface area and the solubility of the drug. These two variables
can be altered by the following techniques:
1. Control the solubility of a weak acid or base by buffering the entire dissolution
medium, the “microenvironment”, or the diffusion layer surrounding a particle.
2. Control the solubility of the drug through choice of the physical state, such as
crystal form, its hydrate and its amorphous form.
3. Determine the surface area of the drug through control of particle size4.
1.2 SOLUBILIZATION TECHNIQUES:
Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com)

3
Solubilization is the process by which the apparent solubility of a poorly water
soluble substance is increaed. Solubilization techniques include addition of a
cosolvent, salt formation, prodrug design, complexation, particle size reduction, and
the use of surface active agents (Micellization)5.
Use of solvate and hydrates6, polymorphs7,8, hydrotrophy9,10, use of
absorbents11,12, pH adjustment, solubilizing vehicles, etc. are the some other physico-
chemical approaches to enhancing oral absorption of poorly water soluble drugs.
Use of Surfactants:
Surface active agents (surfactants) are substances which at low concentrations,
adsorb onto the surfaces or interfaces of a system and alter the surface or interfacial
free energy and the surface or interfacial tension. Surface active agents have a
characteristic structure, possessing both polar (hydrophilic) and non-polar
(hydrophobic) regions in the same molecule. Thus, surfactants are said to be
amphipathic in nature.
Classification of Surfactants:
Surfactant molecules may be classified based on the nature of the hydrophilic
group within the molecule. The four main groups of surfactants are defined as
follows:
1. Anionic surfactants, where the hydrophilic group carries a negative charge, such
as carboxyl (RCOO–), sulphonate (RS 3O ) or sulphate (ROS
3O ). Examples of
Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com)

4
pharmaceutical importance include potassium laurate, CH3(CH2)10 COO–K+, and
sodium lauryl sulphate, CH3(CH2)11S 4O Na+.
2. Cationic surfactants, where the hydrophilic group carries a positive charge (e.g.,
quaternary ammonium halides, R4N+Cl–). Examples of pharmaceutical
importance include cetrimide, a mixture consisting mainly of tetradecyl (Ca 68%),
dodecyl (ca 22%), and hexadecyl trimethyl ammonium bromides (Ca 7%), as well
as benzalkonium chloride, a mixture of aklylbenzyl dimethyl ammonium
chlorides of the general formula [C6H5CH2N+(CH3)2R] Cl-, where R represents a
mixture of the alkyls from C8H17 to C18H37.
3. Ampholytic surfactants (also called Zwitter ionic surfactants), where the molecule
contains, or can potentially contain, both a negative and a positive charge (e.g.,
the sulfobetaines, RN+ (CH3)2 CH2 CH2 SO3). Examples of pharmaceutical
importance include N-dodecyl-N, N-dimethyl betaine, C12 H25 N+ (CH3)2 CH2
COO–.
4. Non-ionic surfactants, where the hydrophile carries no charge but derives its
water solubility from highly polar groups such as hydroxyl or polyoxyethylene
(OCH2 CH2O-) groups. Examples of pharmaceutical importance include polyoxy
ethylated glycol mono-ethers (e.g., etomacrogol), sorbitan esters (spans) and
polysorbates (Tweens).
Surfactants as Solubilizing Agents:
Solubilization can be defined as “the preparation of a thermodynamically
stable isotropic solution of a substance normally insoluble or very slightly soluble in a
Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com)

5
given solvent by the introduction of an additional amphiphilic component or
components. The amphiphilic components (surfactants) must be introduced at a
concentration at or above their critical micelle concentrations. Simple micellar
systems (and reverse micellar) as well as liquid crystalline phases and vesicles
referred to above are all capable of solubilization.
Surfactant uses in Pharmaceutical Preparations:
1. They are used in the formulation of liquid dosage forms like solutions,
suspensions, emulsions, etc.
2. They are used in the preparation of aerosols and liposomes.
3. They are also employed in the preparation of some semisolid dosage forms.
4. They are used in the preparation of tablet dosage forms.
Use of Surfactants in Solid Dispersion Systems:
The bioavailability of hydrophobic drugs can be increased by strategies
designed to enhance the dissolution rate of the drug. This has been achieved in many
cases by forming a solid dispersion of the drug in a suitable carrier, often a
hydrophilic polymer such as polyethylene glycol (PEG) or polyvinyl pyrrolidone.
The drug is dispersed in the carrier by coprecipitation from a suitable solution
containing both drug and carrier, by melting both components together, or by some
other process involving a phase change. By using relatively high concentrations of
carrier and a rapid precipitating process, the drug may form as an amorphous or
molecularly dispersed high energy phase in the carrier. A number of workers have
used surfactants as the carrier material to achieve this enhanced dissolution effect.
Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com)

6
Among the surfactants employed are poly oxyethylene stearate, Renex 650,
poloxamer 188, texafor AIP deoxycholic acid, and tweens and spans. Surfactants
have also been added to conventional drug-polymer solid dispersions to further
improve drug release properties13.
Wetting agents speed up the penetration of gastric fluid in the tablets and
hence, tablet disintegration. The effect, which is due to the lowering of contact
angles and of surface and interfacial tensions of the aqueous medium by the
surfactant, has been well documented14.
Aqueous solutions of surfactants exhibit a more or less abrupt change in their
physical properties over a narrow concentration range. This distinct change in
properties is generally accepted to be due to the formation of oriented aggregates or
micelles. The narrow surfactant concentration range at which micelles begin to form
is referred to as the critical concentration for micelle formation or CMC. Among the
more interesting properties of micellar solutions is their ability to solubilize water-
insoluble materials.
Micellar solubilization has been defined by McBain as “the spontaneous
passage of solute molecules of a substance, insoluble in water, into an aqueous
solution of a surfactant in which a thermodynamically stable solution is formed”.
Micellar solubilization of a poorly water-soluble material can be treated as a
process in which the poorly water-soluble material is partitioned between an aqueous
phase and a micellar phase formed by the surfactant above its CMC15.
Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com)

7
Hamid et al16 studied the solubilization of benzocaine with polyxoyethylene
lauryl ether and polysorbate 80 was determined as a function of temperature. The
enhancement of stability of benzocaine and homatropine solubilized with 3 to 15%
polyxoyethylene lauryl ether and polysorbate 80 was quantitatively determined at 30,
50 and 70ºC. For the alkaline hydrolysis of benzocaine and homatropine, the
logarithm of the specific rate constant is linearly related to the percent of the surface-
active agent. The specific rate constant for the hydrolysis of benzocaine is linearly
related to the reciprocal of the apparent solubility of benzocaine in poly oxyethylene
lauryl ether and poly sorbate 80.
Chowdary KPR et al17 studied the effect of two surfactants, sodium lauryl
sulphate (SLS) and tween 80, on the aqueous solubility and dissolution rate of
nimesulide from tablets formulated employing starch paste and PVP as binders. A
marked increase in the solubility as well as dissolution rate of nimesulide was
observed with both the surfactants.
Sreenivasa Rao et al18 studied the release of rifampicin from a matrix
compressed from a physical mixture of rifampicin, gaur gum and SLS. When SLS
was incorporated in the matrix, the release of rifampicin was found to be linearly
related to the square root of time, however, the release depended on the concentration
of SLS. As the concentration of SLS increased up to 15% the release progressively
slowed to a minimum, which could be due to the formation of a poorly soluble
Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com)

8
complex. As the concentration increased further, the release increased as the complex
was micellarly solubilized.
1.3 INTRODUCTION TO SOLID DISPERSION TECHNOLOGY:
The enhancement of oral bioavailability of poorly water soluble drugs remains
one of the most challenging aspects of drug development. Although salt formation,
solubilization and particle size reduction have commonly been used to increase
dissolution rate and thereby oral absorption and bioavailability of such drugs19, there
are practical limitations of these techniques. The salt formation is not feasible for
neutral compounds and the synthesis of appropriate salt forms of drugs that are
weakly acidic or weakly basic may often not be practical. Even when salts can be
prepared, an increased dissolution rate in the GIT may not be achieved in many cases
because of the reconversion of salts into aggregates of their respective acid or base
forms. The solubilization of drugs in organic solvents or in aqueous media by the use
of surfactants and cosolvents leads to liquid formulations that are usually undesirable
from the viewpoints of patient acceptability and commercialization. Although
particle size reduction is commonly used to increase dissolution rate, there is a
practical limit to how much size reduction can be achieved by such commonly used
methods as controlled crystallization, grinding, etc. The use of very fine powders in a
dosage form may also be problematic because of handling difficulties and poor
wettability.
Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com)

9
In 1961, Sekiguchi and Obi20 developed a practical method whereby many of
the limitations with the bioavailability enhancement of poorly water-soluble drugs
can be overcome, which was termed as “Solid Dispersion”21.
POORLY WATER-SOLUBLE DRUG
Tablet/ capsule Dosage form Solid dispersion/ solution
Disintegration Disintegration
Large solid particle (usually 5-100
microns) Drug in GI tract
Colloidal particles/ fine oily globules
(usually <1 microns)
Lower dissolution rate
ABSORPTION INTO BODY
SYSTEM Higher dissolution
rate
Figure-1.3: A schematic representation of the bioavailability enhancement of a
poorly water-soluble drug by solid dispersion compared with conventional tablet or capsule
The advantage of solid dispersion compared with conventional capsule or
tablet formulations is shown schematically in figure-1.322. From conventional
capsules and tablets, the dissolution rate is limited by the size of the primary particles
formed after the disintegration of dosage forms. In this case, an average particle size
of 5m is usually the lower limit, although higher particle sizes are preferred for ease
of handling, formulation and manufacturing. On the other hand, if a solid dispersion
Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com)

10
or a solid solution is used, a portion of the drug dissolves immediately to saturate the
gastrointestinal fluid, and the excess drug precipitates out as fine colloidal particle or
oily globules of submicron size.
Because of such easily promises in the bioavailability enhancement of poorly
water-soluble drugs, solid dispersion has become one of the most active areas of
research in the pharmaceutical field.
1.4 HISTORICAL BACKGROUND:
The effect of the particle size of the drugs on their dissolution rates and
biological availability was reviewed comprehensively by Fincher. For drugs whose
gastrointestinal absorption is rate limited by dissolution, reduction of the particle size
generally increases the rate of absorption and or total bioavailability. This commonly
occurs for drugs with poor water-solubility. For example, the therapeutic dose of
griseofulvin was reduced to 50% by micronization and it also produced a more
constant and reliable blood level. The commercial dose of spironolactone was also
decreased to half by just a slight reduction of particle size. Such enhancement of drug
absorption could further be increased several fold if a micronized product was used.
In 1961, a unique approach of solid dispersion to reduce the particle size and
increase rates of dissolution and absorption was first demonstrated by Sekiguchi and
Obi. They proposed the formation of a eutectic mixture of a poorly soluble drug such
as sulfathiazole with a physiologically inert, easily soluble carrier such as urea. The
eutectic mixture was prepared by melting the physical mixture of the drug and the
Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com)

11
carrier, followed by a rapid solidification process. Upon exposure to aqueous fluids,
the active drug was expected to be released into the fluids as fine, dispersed particles
because of the fine dispersion of the drug in the solid eutectic mixture and the rapid
dissolution of the soluble matrix.
Levy and Kanig subsequently noted the possibility of using a solid solution
approach in which a drug is dispersed molecularly in a soluble carrier. In a series of
reports in 1965-66, Goldberg et al presented a detailed experimental and theoretical
discussion of advantages of solid solution over the eutectic mixture.
In 1965, Tachibana and Nakamaru reported a novel method for preparing
aqueous colloidal dispersions of -carotene by using water-soluble polymers such as
polyvinyl pyrrolidone. They dissolved the drug and the polymer carrier in a common
solvent and then evaporated the solvent completely. A colloidal dispersion was
obtained when the coprecipitate was exposed to water.
In 1966, Mayersohn and Gibaldi demonstrated that the dissolution rate of
griseofulvin could be markedly enhanced when dispersed in polyvinyl pyrrolidone by
the same solvent method.
Chiou and Riegelman recently advocated the application of glass solution to
increase dissolution rates. They used PEG 6000 as a dispersion carrier. It is believed
that this relatively new field of pharmaceutical technique and principles will play an
important role in increasing dissolution, absorption and therapeutic efficacy of drugs
in future dosage forms. Therefore, a thorough understanding of its fast release
Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com)

12
principles, methods of preparation, selection of suitable carriers, determination of
physical properties, limitations and disadvantages will be essential in the practical and
effective application of this approach.
In addition to absorption enhancement, the solid dispersion technique may
have numerous pharmaceutical applications which remain to be further explored. It is
possible that such a technique can be used to obtain a homogeneous distribution of a
small amount of drug at solid state, to stabilize unstable drugs, to dispense liquid or
gaseous compounds, to formulate a fast release priming dose in a sustained release
dosage form, and to formulate sustained release regimens of soluble drugs by using
poorly soluble or insoluble carriers21.
1.5 DEFINITION AND TYPES OF SOLID DISPERSIONS:
1.5.1 Definition:
Solid dispersion technology is the science of dispersing one or more active
ingredients in an inert matrix in the solid stage in order to achieve increased
dissolution rate, sustained release of drugs, altered solid state properties, enhanced
release of drugs from ointment and suppository bases, and improved solubility and
stability.
1.5.2 Types of solid dispersions:
a) Simple eutectic mixture: An eutectic mixture of a sparingly water soluble drug
and a highly water soluble carrier may be regarded thermodynamically as an
intimately blended physical mixture of its two crystalline component. The
Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com)

13
increase in surface area is mainly responsible for increased rate of dissolution.
This led to a conclusion that the increase in dissolution was mainly due to
decreased particle size.
b) Solid solutions: Solid solutions consist of a solid solute dissolved in a solid
solvent. A mixed crystal is formed because the two components crystallize
together in a homogenous one-phase system. Hence, this system would be
expected to yield much higher rates of dissolution than simple eutectic systems.
c) Glass solution of suspension: A glass solution is a homogenous system in which
a glassy or a vitreous of the carrier solubilizer drug molecules in its matrix. PVP
dissolved in organic solvents undergoes a transition to a glassy state upon
evaporation of the solvent.
d) Compound or complex formation: This system is characterized by
complexation of two components in a binary system during solid dispersion
preparation. The availability of the drug from the complex is dependent on the
solubility dissociation constant and the intrinsic absorption rate of the complex.
e) Amorphous precipitation: Amorphous precipitation occurs when drug
precipitates as an amorphous form in the inert carrier. The higher energy state of
the drug in this system generally produces much greater dissolution rates than the
corresponding crystalline forms of the drug23.
1.6 MECHANISM OF INCREASED DISSOLUTION RATE:
The enhancement in dissolution rate as a result of solid dispersion
formulation, relative to pure drug varies from as high as 400 folds to less than two-
Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com)

14
fold. Corrigan reviewed the current understanding of the mechanism of release from
solid dispersion. The increase in dissolution rate for solid dispersion can be attributed
to a number of factors. It is very difficult to show experimentally that any one
particular factor is more important than another. The main reasons postulated for the
observed improvements in dissolution of these systems are as follows:
a) Reduction of particle size: In case of glass, solid solution and amorphous
dispersions, particle size is reduced to a minimum level. This can result in an
enhanced dissolution rate due to an increase in both the surface area
solubilization.
b) Solubilization effect: The carrier material, as it dissolves may have a
solubilization effect on the drug. This was shown to be the case for
acetaminophen and chlorpropamide in urea as well as for numerous other drugs.
c) Wettability and dispersibility: The carrier material may also have an enhancing
effect on the wettability and dispersibility of the drug in the dissolution media.
This should retard any agglomeration or aggregation of the particles, which can
slow the dissolution process.
d) Metastable Forms: Formation of metastable dispersions with reduced lattice
energy would result in faster dissolution rates. It was found that the activation
energies for dissolution for furosemide was 17 K Cal per mol, whereas that for
1:2 furosemide: PVP coprecipitate was only 7.3 K Cal per mol24.
Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com)

15
1.7 SELECTION OF A CARRIER:
The properties of the carrier have a major influence on the dissolution
characteristics of the dispersed drug. A carrier should meet the following criteria to
be suitable for increasing the dissolution rate of a drug.
1. Be freely water-soluble with intrinsic rapid dissolution properties.
2. Be non-toxic and pharmacologically inert.
3. Be heat stable with a low melting point for the melt method.
4. Be soluble in a variety of solvents and pass through a vitreous state upon solvent
evaporation for the solvent method.
5. Be able to preferably increase the aqueous solubility of the drug and
6. Be chemically compatible with the drug and not form a strongly bonded complex
with the drug24.
Table-1.7: Materials used as carrier for solid dispersion
1. Sugars Dextrose, sucrose, galactose, sorbitol, maltose, xylitol, mannitol, lactose.
2. Acids Citric acid, succinic acid
3. Polymeric materials Povidone (PVP), polyethylene glycol (PEG), hydroxypropyl methyl cellulose, methyl cellulose, hydroxy ethyl cellulose, cyclodextrin, hydroxy propyl cellulose, pectin, galactomannan.
4. Insoluble or enteric polymer
Hydroxy propyl methyl cellulose phthalate, eudragit L100, eudragit S100, Eudragit RL, Eudragit RS.
5. Surfactants Polyoxyethylene stearate, renex, poloxamer 188, texafor AIP, deoxycholic acid, tweens, spans.
6. Miscellaneous Pentaerythritol, pentaerythrityl tetraacetate, urea, urethane, hydroxy alkyl xanthins
Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com)

16
1.8 POLYMERS USED IN SOLID DISPERSIONS:
Polymers used in solid dispersions are as follows:
a) Polyethylene glycols (PEG): The term polyethylene glycols refers to compounds
that are obtained by reacting ethylene glycol with ethylene oxide. PEGs whose
molecular weight is above 300000 are commonly termed as polyethylene oxides.
Effect of PEG molecular weight: The dissolution rate of pure PEG decreases with
increasing molecular weight. The dissolution rate of the drug in solid dispersion can
be increased with an increase in molecular weight of PEG. In these cases, the rate at
which the polymer dissolved dictated the rate at which the drug dissolved. Lower
molecular weight PEGs melt at 37ºC in the dissolution medium prior to dissolution,
further increasing the rate of dissolution. In some drug-PEG solid dispersion systems,
the rate dissolution decreases with molecular weight upto a certain composition of the
drug above which the trend becomes irregular.
b) Polyvinyl pyrrolidone (PVP): PVP has a molecular weight ranging from 10,000
to 700,000. It is soluble in solvents like water, ethanol, chloroform and isopropyl
alcohol. PVP is not suitable for preparation of solid dispersions prepared by melt
method because of it melts at a very high temperature above 275ºC, where it
becomes decomposed.
Effect of PVP molecular weight: The effect of molecular weight of PVP on the rate
of dissolution of a drug is more consistent than for PEG. An increase in molecular
weight of PVP will decrease the dissolution rate of most drugs. An increase in
Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com)

17
viscosity of PVP solution due to an increase in molecular weight decreases diffusion
of drug molecules from the surface of viscous material into the dissolution medium.
Lower molecular weight PVP has a short swelling time prior to dissolution resulting
in an increase in dissolution rate of the polymer and drug.
c) Polymers and surface active agent combinations: The addition of surfactants to
dissolution medium lowers the interfacial tension between the drug and the
dissolution medium and promote the wetting of the drug thereby they enhance the
solubility and dissolution of drugs. Ternary dispersion systems have higher
dissolution rates than binary dispersion systems.
d) Cyclodextrins: Cyclodextrins are primarily used to enhance solubility, chemical
protection, taste masking and improved handling by the conversion of liquids into
solids by entrapment.
Oral administration of cyclodextrins: Cyclodextrins play an important role in the
bioavailability of poorly water soluble drugs by increasing the rate and extent of
dissolution of drug.
Cyclodextrins also have the advantage of:
Increasing the stability of the drug.
Release profile during gastrointestinal transit through modification of drug release
site and time profile.
Decreasing local tissue irritation.
Masking unpleasant taste.
Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com)

18
e) Phospholipids: Phospholipids are major structural components of cell
membranes. Phosphotidylcholine was first isolated from egg yolk and brain. Its
chemical name is 1,2-diacyl-in-glycero-3-phosphocholine. In phosphatidyl
ethanolamine and phosphatidyl serine, the choline moiety is replaced by
ethanolamine and serine respectively. Other phospholipids that occur in tissues
include phosphotidyl ethanolamide (PE), phosphotidyl serine (PS), and
phosphotidyl glycerol (PG). Naturally occuring lecithins contain both a saturated
fatty acid and an unsaturated fatty acids with some exceptions23.
1.9 METHODS OF PREPARING SOLID DISPERSIONS:
a) Fusion Process:
The fusion process is technically the less difficult method of preparing
dispersions provided the drug and carrier are miscible in the molten state.
Fusion was used by Sekiguchi and Obi, who melted a sulphathiazole-urea
mixture of eutectic composition at above its eutectic temperature, solidified the
dispersion on an ice bath and pulverized it, to a powder, since a super saturation of
the drug can be obtained by quenching the melt rapidly (when the solute molecules
are arrested in a solvent matrix by instantaneous solidification), rapid congealing is
favoured. Consequently the solidification process is often effected on stainless-steel
plates to favour rapid heat loss. A modification of the process involves spray
congealing from a modified spray drier onto cold metal surfaces and has been used
for dispersions containing mannitol or phenyl butazone urea.
Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com)

19
Decomposition should be avoided during fusion but is often composition
dependent, and affected by fusion time and the rate of cooling. Therefore, to
maintain decomposition at an acceptable level, fusion may be effected at a
temperature only just in excess of that which completely melts both drug and carrier.
b) Solvent Process:
Solid dispersion prepared by solvent removal process were termed by Bates et
al as “coprecipitates”. They should more correctly, be designated as “coevaporates”,
a term that has been recently adopted.
The solvent process used organic solvents, the agent to intimately mix the
drug and carrier molecules and was initially used by Tachibana and Nakamura when
chloroform codissolved –carotene and polyvinylpyrrolidone.
The choice of solvent and its removal rate are critical to the quality of the
dispersion. Since the chosen carriers are generally hydrophilic and the drugs are
hydrophobic, the selection of a common solvent is difficult and its complete removal,
necessitated by its toxic nature, is imperative. Vacuo-evaporation may be used for
solvent removal at low temperature and controlled rate. More rapid removal of the
solvent may be accomplished by freeze-drying. Polyvinylpyrrolidone dispersions of
Ketorpofen or dicoumarol were freeze-dried from their ammonical solutions. The
difficulties in selecting a solvent common to both drug and carrier may be overcome
by using an azeotropic mixture of solvent in water.
Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com)

20
The bioavailability and stability of Nifedipine-enteric coating agents solid
dispersion were studied, using hydroxy propylmethyl cellulosephthalate (HP-55) and
methacrylic methylester copolymer (Eudragit-L) as carriers. These result suggested
that these solid dispersion systems might be useful for bioavailabiltiy enhancement
and development of a sustained release preparation of nifedipine. The solid dispersion
system were prepared by the solvent method. Nifedipine (3g) and a polymer (9g.)
were dissolved in about 90ml of mixed solvent (ethanol: dichloromethane 1:1) and
then the solvent was evaporated off under reduced pressure. The residual solid was
pulverized and the 32-80 mesh fraction was used25.
Solid dispersions of Griseofulvin-PVP26, Sulfathiazole-PVP27, have been
obtained by this method.
c) Fusion Solvent Method:
This method consists of dissolving the drug in a suitable liquid solvent and
incorporating the solution directly in the melt of PEG28. If the carrier is capable of
holding a certain proportion of liquid yet maintaining its solid properties and if the
liquid is innocuous, the need for solvent removal is eliminated. This method is
particularly useful for drugs that have high melting points or that are thermolabile.
Although there are advantages and disadvantages associated with all these
methods, the choice of a method of preparation could affect the intended purpose of
solid dispersion formulations23.
Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com)

21
d) Supercritical Fluid Process:
Supercritical CO2 is a good solvent for water insoluble as well as water
soluble compounds under suitable conditions of temperature and pressure. Therefore,
supercritical CO2 has potential as an alternative for conventional organic solvents
used in solvent based processes for forming solid dispersions due to its favourable
properties of being nontoxic and inexpensive. The process developed by Ferro
Corporation consists of the following steps:
Charging the bioactive material and suitable polymer into the autoclave.
Addition of supercritical CO2 under precise conditions of temperature and
pressure, that causes polymer to swell;
Mechanical stirring in the autoclave; and
Rapid depressurization of the autoclave vessel through a computer controlled
orifice to obtain desire particle size. The temperature conditions used in this
process are fairly mild (35–75°C), which allows handling of heat sensitive
biomolecules, such as enzymes and proteins24.
Solid dispersion of cabamazepine-PEG800029 has been obtained by this method.
1.10 METHODS OF DETERMINATION OF TYPES OF SOLID
DISPERSION:
Various methods, which can contribute information regarding the physical
nature of the solid dispersions, are thermo analytical methods (Thermal Analysis19,
DSC30, X-ray Diffraction Methods31,32, Spectroscopic Methods and Microscopic
Methods19).
Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com)

22
1.11 ADVANTAGES AND DISADVANTAGES OF SOLID DISPERSIONS:
Among the advantages of solid dispersions are the rapid dissolution rates that
result in an increase in the rate and extent of the absorption of the drug, and a
reduction in presystemic metabolism. This latter advantage may occur due to
saturation of the enzyme responsible for biotransformation of the drug, as in the case
of 17 estradiol; or inhibition of the enzyme by the carrier, as in the case of
morphine-tristearin dispersion. Both can lead to the need for lower doses of the drug.
Other advantages include transformation of the liquid form of the drug into a solid
form (e.g., clofibrate and benzoyl benzoate can be incorporated into PEG 6000 to
give a solid, avoidance of polymorphic changes and thereby bio-availability
problems), as in the case of nabilone and PVP dispersion, and protection of certain
drugs by PEGs (e.g., cardiac glycosides) against decomposition by saliva to allow
buccal absorption.
The major disadvantages of solid dispersion are related to their instability.
Several systems have shown changes in crystallinity and a decrease in dissolution rate
with aging. The crystallization of ritonavir from the supersaturated solution in a solid
dispersion system was responsible for the withdrawal of the ritonavir capsule (Norvir,
Abboft) from the market. Moisture and temperature have more of a deteriorating
effect on solid dispersions than on physical mixtures. Some solid dispersions may not
lend themselves to easy handling because of tackiness24.
Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com)

23
1.12 FUTURE PROSPECTS:
Despite many advantages of solid dispersion, issues related to preparation,
reproducibility, formulation, scale up and stability limited its use in commercial
dosage forms for poorly water-soluble drugs. Successful development of solid
dispersion systems for preclinical, clinical and commercial use have been feasible in
recent years due to the availability of surface-active and self-emulsifying carriers with
relatively low melting points. The preparation of dosage forms involves the
dissolving of drug in melted carriers and the filling of the hot solutions into hard
gelatin capsules because of the simplicity of manufacturing and scale up processes,
the physico-chemical properties and, as a result, the bioavailability of solid
dispersions are not expected to change significantly during the scale up. For this
reason, the popularity of the solid dispersion system to solve difficult bioavailability
issues with respect to poorly water-soluble drugs will grow rapidly. Because the
dosage form can be developed and prepared using small amounts of drug substances
in early stages of the drug development process, the system might have an advantage
over such other commonly used bioavailability enhancement techniques as
micronization of drugs and soft gelatin encapsulation.
One major focus of future research will be the identification of new surface-
active and self-emulsifying carriers for solid dispersion. Only a small number of such
carriers are currently available for oral use. Some carriers that are used for topical
application of drug only may be qualified for oral use by conducting appropriate
toxicological testing. One limitation in the development of solid dispersion systems
Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com)

24
may the inadequate drug solubility in carrier, so a wider choice of carriers will
increase the success of dosage form development. Research should also be directed
toward identification of vehicles or excipients that would retard or prevent
crystallization of drugs from super-saturated systems. Attention must be given to any
physiological and pharmacological effects of carriers used. Many of the surface-
active and self-emulsifying carriers are lipidic in nature, so potential roles of such
carriers on drug absorption, especially on their inhibitory effects on CYP-3 based
drug metabolism and p-glycoprotein-mediated drug efflux will require careful
consideration.
In addition to bioavailability enhancement, much recent research on solid
dispersion systems was directed toward the development of extended-release dosage
forms.
Physical and chemical stability of both the drug and the carrier in a solid
dispersion are major developmental issues, an exemplified by the recent withdrawal
of ritonavir capsules from the market, so future research needs to be directed to
address various stability issues. The semisolid and waxy nature of solid dispersions
poses unique stability problems that might not be seen in other types of solid dosage
forms. Predictive methods will be necessary for the investigation of any potential
crystallization of drugs and its impact on dissolution and bioavailability, possible
drug-carrier interactions must also be investigated33.
Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com)

25
CHAPTER–2
OBJECTIVES
2.1 NEED FOR THE STUDY:
By many estimates up to 40 percent of new chemical entities discovered by
the pharmaceutical industry today are poorly soluble or lipophilic compounds. The
solubility issues complicating the delivery of these new drugs also affect the delivery
of many existing drugs.
Aceclofenac is aceclofenacum (O–(2,6-dichloroaniline) phenyl] acetate
glycolic acid ester, 2–(2,6-dichloraniline) phenyl acetoxyacetic acid. Aceclofenac is
a NSAID. It is used in the management of osteoarthritis, rheumatoid arthritis and
ankylozing spondylitis. Aceclofenac when taken orally shows gastrointestinal
disturbances such as GI discomfort, nausea, diarrhea. In some patients peptic
ulceration and severe gastrointestinal bleeding may also occur34.
Solid dispersion technology can be used to improve the in vitro and in vivo
dissolution properties of dissolution dependent poorly water soluble drugs25. PEG’s,
PVP35 and surfactant like SLS36 have been reported to be used for increasing the
solubility of a poorly soluble drugs.
Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com)

26
The usual dose of aceclofenac is 100 mg given twice daily by mouth. The
initial dose should be reduced to 100 mg daily in patients with hepatic impairment34.
Its low solubility makes it a suitable candidate for solid dispersion systems.
2.2 OBJECTIVES OF THE STUDY:
The objectives of the present study include:
Evaluate the potential of polyvinyl pyrolidone, polyethylene glycol 6000
and sodium lauryl sulphate as suitable drug carrier systems for delivery of
aceclofenac.
Determine the effect of change in polymer and polymer composition and
drug-polymer ratio on solubility of aceclofenac.
Study of in vitro dissolution kinetics of aceclofenac from the formulated
solid dispersion systems.
Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com)

27
2.3 SCHEME OF WORK:
PART-I:
1. Extensive literature survey.
2. Procurement of raw materials and drug
3. Standardization of raw materials and drugs.
PART-II:
Preparation of solid dispersions employing 3² factorial design, using different
carrier systems by physical mixture, solvent evaporation method and fusion method.
Carrier Systems Used:
1. Polyethylene glycol 6000
2. Polyvinyl pyrrolidone.
3. Sodium lauryl sulphate
4. PVP and PEG 6000
5. PEG 6000 and SLS
6. PVP and SLS.
PART-III: Evaluation of Aceclofenac Solid Dispersions:
1. Physical appearance
2. Solubility study
3. Construction of standard calibration curve of aceclofenac in methanol and
pH 7.4 phosphate buffer.
4. Drug-content uniformity.
Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com)

28
PART-IV:
1. In vitro drug release studies.
2. Stability study.
PART-V: Statistical Analysis, Data Interpretation and Conclusions.
Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com)

29
CHAPTER–3
REVIEW OF LITERATURE
3.1 REVIEW OF LITERATURE:
Mohamed MS et al studied the solid dispersions containing ibuprofen in
varying amounts of polyethylene glycol 6000 were prepared by the melt method, and
dissolution characteristics of the drug were studied37.
Nakagami H designed a method of increasing the dissolution rates of glass-
forming, poorly water-soluble drugs. It is based on the concept of preparing the
powder form of a glassy drug dispersed in an inert carrier by melting. Indomethacin
and griseofulvin were used as a model drugs, and fumed silicon dioxide was used as
the carrier38.
Chowdary KPR et al evaluated the application of water-soluble cellulose
polymers, HPC-SL, HPMC and HEC in solid dispersions of a poorly soluble drug
naproxen39.
Fernandez M et al prepared the solid dispersions were used to increase the
solubility of active ingredients with the ultimate goal of optimizing their
bioavailability when incorporated into pharmaceuticals. The studies described were
designed to improve the dissolution kinetics of piroxicam by using solid dispersions
in polyethylene glycol 4000. The results showed that PEG 4000 increased the
amount of piroxicam dissolved in both physical mixtures and solid dispersion40.
Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com)

30
Kale SN et al studied the solid dispersions of piroxicam prepared by fusion
and solvent methods using polyethylene glycol 6000 as carrier. All the solid
dispersions showed increased dissolution rate as compared to the dissolution rate of
pure piroxicam41.
Kuchekar BS et al prepared and evaluated -cyclodextrin (-CD) and dextrin
as carriers for solid dispersions of paracetamol, a poorly soluble drug. Marked
increase in the dissolution rate of paracetamol was observed in case of all the solid
dispersions. Among the two carriers used, dextrin was found to increase the
dissolution rate faster than -cyclodextrin42.
Chowdary KPR et al studied nimesulide suspensions formulated by
employing its solid dispersions in PVP, PEG and pregelatinized starch (PGS) and
studied suspensions formulated with dispersions in PGS gave highest dissolution rate
of nimesulide43.
Sreenivasa Rao B et al studied the flurbiprofen, a non-steriodal anti-
inflammatory, analgesic and antipyretic drug. Solid dispersions by common solvent
method was used to enhance the solubility of the poorly soluble flurbiprofen44.
Rama Rao N et al formulated the piroxicam dispersions in pregelatinized
starch prepared in different drug and carrier ratios and were characterized by X-ray
diffractograms (XRD), differential scanning calorimetry (DSC), differential thermal
analysis (DTA) and dissolution studies45.
Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com)

31
Sanjula Baboota et al evaluated the inclusion complexes of aceclofenac with
-cyclodextrin (-CD) prepared by various methods like grinding, kneading, solid
dispersion and freeze drying. The in vitro dissolution rate of drug--CD complex was
faster compared to the drug alone46.
Gowthamarajan K et al studied the -cyclodextrin complexes of aceclofenac
were prepared by solvent evaporation technique in different ratios to enhance the
solubility of the drug47.
Saha RN et al, attempted to enhance solubility and dissolution of nimesulide
and ibuprofen by solid dispersion techniques and complexation using various
hydrophilic excipients48.
Patil CC et al studied the enhancement of dissolution efficiency of naproxen
using solid dispersions. Solid dispersions were prepared by melting method as well
as common solvent method using three hydrophilic carrier viz., PEG 6000, PEG 4000
and polyvinyl pyrrolidone, employing chloroform as a common solvent. Naproxen
showed only 56.51% dissolution at the end of 90 minutes in pH 7.4 phosphate buffer,
whereas solid dispersions showed enhanced drug release. The increase in carrier ratio
in the formulation increased the drug release. Dispersion prepared by melting
methods showed faster dissolution rate than common solvent method49.
Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com)

32
Kusum Devi et al presented a study for three primary objectives. Firstly, in
view of the low aqueous solubility of celecoxib, solid dispersions of the drug were
prepared and evaluated50.
Sanghavi NM et al studied the solid dispersions of methaqualone in
polyethylene glycol (PEG) 6000 were obtained by using melting and solvent method.
Dissolution rate studies of these prepared dispersion were studied in detail and
compared with the pure drug51.
Guangxi Zhai et al investigated and evaluated the bioavailability in rats after
oral administration of puerarin or puerarin-phospholid solid dispersion52.
Pawar SP et al studied the dissolution of trimethoprim by solid dispersion
technique53.
Sheen PC et al attempted to improve the bioavailability of poorly water-
soluble drug, RP 69698, solid dispersion were prepared by melting method and
investigated in beagle dogs showed marked increase in bioavailability54.
Anguiano Igea S et al investigated the effect of clofibrate concentration and
molecular weight of polyethylene glycols on the structure and dissolution rates of
solid dispersions55.
Betageri GV et al evaluated the preparation of a solid dispersion of tolazamide
in polyethylene glycol 8000 by solvent and melt methods is reported. The rate of
Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com)

33
dissolution of tolazamide was faster in the solid dispersions than in physical mixtures
and pure tolazamide56.
Suzuki H et al formulated to improve the poor organic solubility of benidipine
hydrochloride, 2 kinds of solvent systems, an organic solution of Eudragit E-100 and
binary solvent mixtures were used for preparing solid dispersions by the solvent
removal process, the dissolution profile of the drug from these solid dispersions was
investigated57.
Palmieri GE et al prepared solid dispersions of fenofibrate in polyethylene
glycol 4000 (PEG 4000). The solid dispersions were easily prepared by the fusion or
coevaporation methods. Both preparation methods gave very similar results in the
formation of solid solutions and in the improvement of fenofibrate water solubility58.
Torrado S et al prepared and evaluated solid dispersion systems of the
sparingly water soluble drug, albendazole were mixed with varying concentrations of
polyvinyl pyrrolidone (PVP K12) in an attempt to improve the solubility dissolution
rate of albendazole59.
Ruckmani K et al studied carbamazepine, an anti-epileptic drug which is
water insoluble was formulated as solid dispersion using PEG 6000 as carrier to
improve its solubility and dissolution behaviour60.
Dhanaraju MD et al studied griseofulvin solid dispersions were prepared using
polyethylene glycol 6000 (PEG) and polysorbate 80 (Tween 80) mixture using fusion
Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com)

34
technique. The in vitro dissolution release studies indicated that drug release from
PEG with Tween 80 provided dissolution rates faster than dispersion prepared with
PEG alone. The incorporation of surface active agent such as tween 80 has a
considerable effect in the in vitro dissolution profile of griseofulvin61.
Himasankar et al studied solid dispersion of glipizide were prepared using
water soluble carriers such as polyvinyl pyrrolidone and polyethylene glycol by
common solvent method in an attempt to increase the dissolution rate of glipizide62.
Mummaneni V et al studied the aqueous solubility and dissolution of
famotidine from solid glass dispersion of xylitol prepared by the fusion method were
investigated63.
Rabasco AM et al studied a method for the elaboration of solid dispersions,
the weight ratios of diazepam to polyethylene glycol 6000, and the particle size of
drug in the solid dispersion have been investigated64.
Sjokvist E et al studied the non-ionic surfactants polysorbate 80 and
polyethylene dodecyl ether (Brij 35), the anionic surfactant sodium dodecyl sulphate
(SDS) and the cationic surfactant, dodecyltrimethyl ammonium bromide (DTAB)
were incorporated in dispersions of 10% w/w griseofulvin with PEG 3000 as a
carrier. An almost instant and complete dissolution was obtained for dispersions with
1 and 2% w/w SDS65.
Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com)

35
Craig DQM et al investigated the dissolution characteristics of nortriptyline
hydrochloride dispersions in a range of different molecular weight polyethylene
glycol carriers have been investigated. The release rate was found to be higher from
dispersions in PEG 3400 than from the drug alone, while a logarithmic decrease was
seen with increasing carrier molecular weight66.
Ahmed SM et al studied an inclusion complex of bropirimine (ABPP) with -
cyclodextrin (-CD) and its solid dispersion with polyethylene glycol 6000 (PEG
6000) were prepared by the coprecipitation method). Comparative dissolution studies
revealed that the solid complex exhibited a markedly faster dissolution rate compared
to the PEG 6000 solid dispersions and physical mixtures in water and phosphate
buffer (pH 7.4)67.
Kerc J et al studied solid dispersions containing different proportions of
felodipine to urea and relodipine to mannitol have been prepared and studied in water
dissolution media. Enhanced dissolution rate as a result of both surface area increase
and solubilization was noticed68.
Popli H et al investigated solid dispersion systems developed as a drug
delivery system for sulfamethoxazole and nitrofurantoin were evaluated X-ray
diffraction data revealed an increase in the dissolution profile for solid dispersions69.
Betageri GV designed the preparation of solid dispersions and lyophilization
of the dispersions to increase the solubility of glyburide using polyethylene glycol
Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com)

36
(PEG 4000), PEG 6000, and a mixture of the two, are described. Dissolution studies
indicated a significant increase in dissolution of glyburide when dispersed in PEGs70.
Chowdary KPR et al conducted a study to improve the dissolution rate and
efficiency of nimodipine by solid dispersion in individual and combined carrier and to
study the physicochemical nature of the dispersions71.
Khidr SH et al studied the effect of polomaxer 407 (pluronic F-127) on the
dissolution rate of water-insoluble drugs in solid dispersions of poloxamer and
povidone K-30 (polyvinyl pyrrolidone K-30; PVP K-30) at different drug-polymer
ratio is described using nifedipine as a model drug. The solid dispersion technique
using both polymers dramatically increased the dissolution rates of nifedipine72.
Mishra B et al evaluated the solid dispersions of poorly water soluble drug
nimesulide using polyvinyl pyrrolidone. Microcrystalline cellulose (PVP-MCC) and
hydroxy propyl methyl cellulose–microcrystalline cellulose (HPMC-MCC)
combinations were prepared to enhance the dissolution rate of nimesulide73.
Gopal Rao M et al conducted a study to improve the dissolution rate of
naproxen using carrier such as PVP, PEG 4000, PEG 6000, PEG 20000, methyl
cellulose and -cyclodextrin with a view to develop fast release formulations of
naproxen. Solid dispersions of naproxen were prepared by solvent evaporation
method and the dispersions were evaluated for drug content uniformity, dissolution
rates, moisture absorption, thin layer chromatography and X-ray diffraction analysis.
A marked increase in dissolution rate was observed with all solid dispersions74.
Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com)

37
Soniwala MM et al attempted to improve the solubility and dissolution rate of
rofecoxib by formulating its solid dispersions with various hydrophilic carriers (PEG
6000, PVP K-30, Eudragit E-100) and inclusion complex with -cyclodextrin75.
Ahmed M et al studied preparation and in vitro evaluation of solid dispersion
of halofantrine. The low aqueous solubility of halofantrine and its low bioavailability
suggested the formulation of solid dispersions to reduce its particle size and improve
its wettability and aqueous solubility76.
Sanjula Baboota et al attempted to enhance solubility and dissolution of
rofecoxib by complexation using dimethyl -cyclodextrin complexes were prepared
by physical mixture, kneading and spray drying methods77.
Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com)

38
3.2 DRUG PROFILE:
Drug: Aceclofenac
Chemical Name78: 2-[(2,6–dichlorophenylamino) phenyl] acetoxy acetic acid.
Molecular formula: C16H13Cl2NO4
Molecular weight: 354.2
Chemical structure:
Description: A white or almost white crystalline powder
Physico-chemical properties:
Melting point: 149º to 150º
Solubility: Practically insoluble in water, soluble in alcohol and methyl alcohol,
freely soluble in acetone and dimethyl formamide.
Standard79: It contains not less than 99.0 percent and not more than the equivalent of
101.0 percent of 2-[[2–[2–[(2,6–dichloro phenyl) amino] phenyl] acetyl] oxy] acetic
acid, calculated with reference to the dried substance.
Heavy metals: 10 ppm
Loss on drying: Not more than 0.5%
Sulphated ash: Not more than 0.1%
Storage: Storage in a well-closed container, protected from light.
Therapeutic category80: Anti-inflammatory, analgesic.
O
NH
Cl Cl
COOH
O
Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com)

39
Pharmacological Profile81: Aceclofenac is a non-steroidal agent with marked anti-
inflammatory and analgesic properties. The mode of action of aceclofenac is largely
based on the inhibition of prostaglandin synthesis. Aceclofenac is a potent inhibitor
of the enzyme cyclo-oxygenase (COX), which is involved in the production of
prostaglandins.
Pharmacokinetics: After oral administration, aceclofenac is rapidly and completely
absorbed as unchanged drug. Peak plasma concentrations are reached approximately
1.25 to 3.00 hours following ingestion. Aceclofenac penetrates into the synovial
fluid, where the concentration reach approximately 57% of those in plasma. The
volume of distribution is approximately 25 L.
The mean plasma elimination half-life is around 4 hours. Aceclofenac is
highly protein bound >99%, aceclofenac circulates mainly as unchanged drug. 4-
hydroxy aceclofenac is the main metabolite detected in plasma. Approximately two-
thirds of the administered dose is excreted via the urine, mainly as hydroxy
metabolite. No changes in the pharmacokinetics of aceclofenac have been detected in
the elderly.
Adverse Effects34: Aceclofenac when taken orally shows gastrointestinal
disturbances such as GI discomfort, nausea and diarrhea. In some patients, peptic
ulceration and severe gastrointestinal bleeding may also occur.
Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com)

40
Hypersensitivity: Leukocytoelastic vasculitis, a type-III hypersensitivity reaction with
lung hemoptysis has been reported in patients following therapy with aceclofenac.
Drug Interactions:
Enhancement of effects of oral anticoagulants.
Increased plasma concentrations of lithium, methotrexate and cardiac
glycosides.
The risk of nephrotoxicity may be increased if given with ACE inhibitors,
ciclosporin, tacrolimus or diuretics.
Convulsions may occur due to an interaction with quinolones.
The risk of gastrointestinal bleeding and ulceration associated with
NSAIDs is increased when used with corticosteroids, the antiplatelets
clopidogrel and ticlopidine.
There may be increased risk of hemotoxicity during concomitant use of
zidovudine and NSAIDs.
Uses and Administration:
Aceclofenac, a phenyl acetic acid derivative is an NSAID. It is used in the
management of osteoarthritis rheumatoid arthritis and ankylosing
spondylitis.
The usual dose of aceclofenac is 100 mg given twice daily by mouth. The
initial dose should be reduced to 100 mg daily in patients with hepatic
impairment34.
Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com)
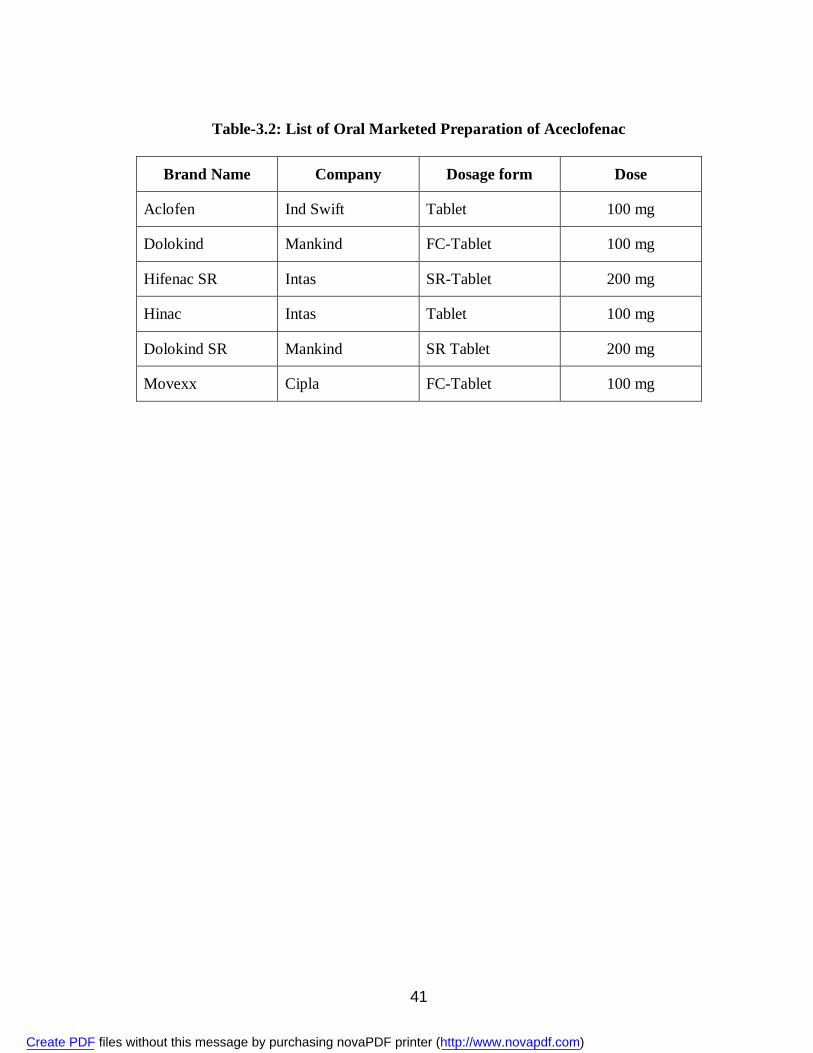
41
Table-3.2: List of Oral Marketed Preparation of Aceclofenac
Brand Name Company Dosage form Dose
Aclofen Ind Swift Tablet 100 mg
Dolokind Mankind FC-Tablet 100 mg
Hifenac SR Intas SR-Tablet 200 mg
Hinac Intas Tablet 100 mg
Dolokind SR Mankind SR Tablet 200 mg
Movexx Cipla FC-Tablet 100 mg
Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com)

42
CHAPTER–4
METHODOLOGY
Materials and Equipment:
Materials:
Sl. No. Materials/ Chemicals Source
1. AceclofenacBP Gift sample from Suyash Laboratories Ltd., MIDC Tarapur
2. Polyvinyl pyrrolidone Loba Chemie Pvt. Ltd., Mumbai
3. Polyethylene Glycol 6000 Loba Chemie Pvt. Ltd., Mumbai
4. Sodium lauryl sulphate Sd Fine Chemicals Ltd., Mumbai
5. Microcrystalline cellulose Loba Chemie Pvt. Ltd., Mumbai
6. Lactose Sd Fine Chemicals Pvt. Ltd., Mumbai
7. Potassium dihydrogen orthophosphate
Sd Fine Chemicals Pvt.Ltd., Mumbai
8. Sodium hydroxide Sd Fine Chemicals Pvt.Ltd., Mumbai
9. Methanol Qualigens Fine Chemicals, Mumbai
Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com)

43
Equipments:
Sl. No. Equipment Name Source
1. UV/Visible spectrophotometer Shimadzu-1700
2. Electronic balance Shimadzu Corporation BL-220H
3. pH meter Elico LI-122
4. Tablet dissolution tester Electrolab USP (XXIII) TDT-06T 9911090
5. Digital Controlled Speed Stirrer Remi Motors RQ 121/D
6. Brookfield DV-II + Programmable Viscometer
M/97-164-E0102
7. Ultrasonicator Flexit Jour
8. Mechanical Shaker Remi Motor Instruments
9. IR-Spectrophotometer Perkin Elmer 1600-Series FTIR
10. Differential Scanning Colorimeter
Pyris-6 DSC
Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com)

44
4.1 RAW MATERIAL CHARACTERIZATION:
4.1.1 Standardization of Drug:
Tests were carried out on the sample of the drug to establish its identity and
purity as per BP 2001 specifications79.
4.1.2 Standardization of Polymers82:
The polymers were tested as per pharmacopoeial or official manufacturers
standards.
4.1.2a Polyvinyl pyrrolidone: It was tested for compliance with IP 1996
specifications.
4.1.2b Polyethylene glycol 6000: It was tested for compliance with IP 1996
specifications.
4.1.2c Sodium lauryl sulphate: It was tested for compliance with IP 1996
specifications.
4.1.2d Microcrystalline cellulose: It was tested for compliance with IP 1996
specification.
4.1.2e Lactose: It was tested for compliance with IP 1996 specification.
Factorial Design83:
It is well known that traditional experimentation involves a good deal of
efforts and time especially when complex formulations are to be developed. It is
desirable to develop an acceptable pharmaceutical formulation in the shortest period
of time using minimum number of man-hours and raw materials. In addition to the
art of formulation, factorial design is an efficient method of indicating the relative
Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com)

45
significance of a number of variables and their interactions. Factorial design
approach shows interactions between factors that a “one factor at a time” model
cannot reveal.
Following are the terms used in factorial:
1. Factors: It is a variable, which has to be assigned such as rpm, drug-polymer
ratio. The choice of factors to be included in an experiment depends on
experimental objective and is predetermined.
2. Level: It takes into account the value beyond or below which a batch cannot
be made effectively and are preselected high and low values of the variables.
3. Runs/trials: These compromises of factorial experiments, which consist of
different combinations of all levels of all factors.
4. Effect of a factor: Is the change in response caused by varying the levels of
the factor.
5. Interaction: Lack of additivity is known as interaction, either an antagonist or
synergistic effect is observed.
Advantages of factorial design:
1. In absence of interaction, factorial designs have maximum efficiency in
estimating main effects.
2. If interaction exists, factorial designs are necessary to reveal and identify
interactions.
3. Maximum use of all data since main effects and interactions are calculated
from all the data.
Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com)

46
4. Factorial designs are orthogonal and all estimated effects and interactions are
independent of the effects of other forms.
Batches were made with the aid of factorial design.
4.2 METHODS OF PREPARATION OF ACECLOFENAC SOLID
DISPERSION SYSTEMS.
4.2.1 Preparation of Aceclofenac Solid Dispersions with PEG 6000
Solid dispersions of aceclofenac in PEG 6000 were prepared using 3² factorial
design with PEG 6000 and aceclofenac as variables and maintaining the amount of
lactose and MCC (4:1) as constant (table-4.2.1). The methods used for the
preparation of these solid dispersions were physical mixtures, solvent evaporation
method and fusion method.
i) Physical Mixture: The physical mixtures were prepared by weighing the
calculated amount of aceclofenac and the carriers and then mixing them in a
glass mortar by triturating. The resultant physical mixtures was passed
through 44-mesh sieve and stored in dessicator until used for further studies.
ii) Solvent Evaporation Method84: The required amount of aceclofenac and the
carrier were dissolved in sufficient volume of methanol with continuous
stirring. The solvent was then completely evaporated at 45º with continuous
stirring to obtain dry mass. The dried mass was pulverized passed through 44
mesh sieve and stored in dessicator until used for further studies.
Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com)

47
iii) Fusion Method85: Accurately weighed amount of carrier was melted in a
porcelain dish at 80-85º and to this calculated amount of aceclofenac was
added with thorough mixing for 1-2 minutes followed by quick cooling. The
dried mass was then pulverized passed through 44-mesh sieve and stored in a
dessicator until used for further studies. PVP containing solid dispersions
were not prepared by the melt method, because PVP melts above 250ºC and
degrades before its melting point23,86.
Table-4.2.1: Factor & Levels in the Design of Aceclofenac Solid Dispersions with
PEG 6000
Levels Independent variables
(–1) Lower (0) Middle (+1) Upper
PEG 6000 (X1) mg 250 300 350
Aceclofenac (X2) mg 50 75 100
(Amount of other additives – lactose and MCC (4:1) was maintained constant in all the preparations)
Experimental Batches for Factorial Design:
Physical mixture (APG)
Solvent evaporation (ASG)
Fusion method (AFG).
Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com)

48
Run No. X1 X2
1 –1 –1
2 –1 0
3 –1 +1
4 0 –1
5 0 0
6 0 +1
7 +1 –1
8 +1 0
9 +1 +1
4.2.2 Preparation of Aceclofenac Solid Dispersions with PVP:
Solid dispersions of aceclofenac in PVP were prepared using 3² factorial
design with PVP and aceclofenac as variables and maintaining the amount of lactose
and MCC(4:1) as constant (table-4.2.2). The methods used for the preparation of
these solid dispersions were physical mixture and solvent evaporation method, as
described for the preparation of aceclofenac solid dispersions with PEG 6000.
Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com)
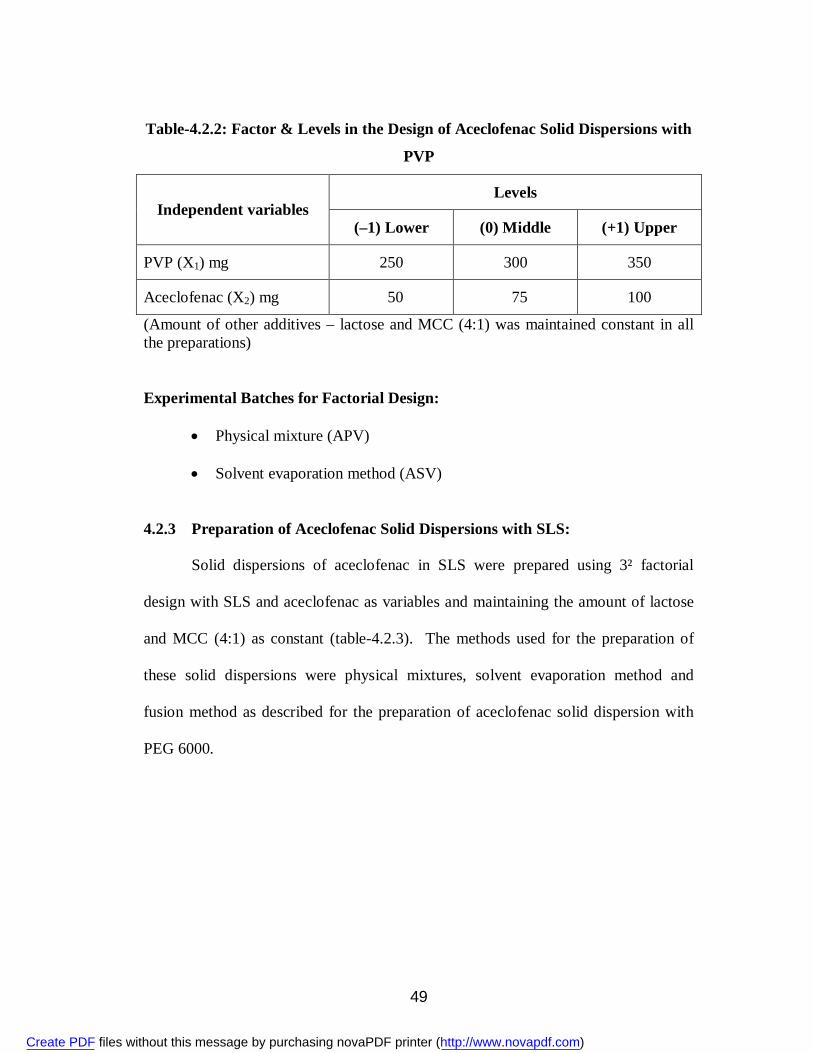
49
Table-4.2.2: Factor & Levels in the Design of Aceclofenac Solid Dispersions with
PVP
Levels Independent variables
(–1) Lower (0) Middle (+1) Upper
PVP (X1) mg 250 300 350
Aceclofenac (X2) mg 50 75 100
(Amount of other additives – lactose and MCC (4:1) was maintained constant in all the preparations)
Experimental Batches for Factorial Design:
Physical mixture (APV)
Solvent evaporation method (ASV)
4.2.3 Preparation of Aceclofenac Solid Dispersions with SLS:
Solid dispersions of aceclofenac in SLS were prepared using 3² factorial
design with SLS and aceclofenac as variables and maintaining the amount of lactose
and MCC (4:1) as constant (table-4.2.3). The methods used for the preparation of
these solid dispersions were physical mixtures, solvent evaporation method and
fusion method as described for the preparation of aceclofenac solid dispersion with
PEG 6000.
Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com)
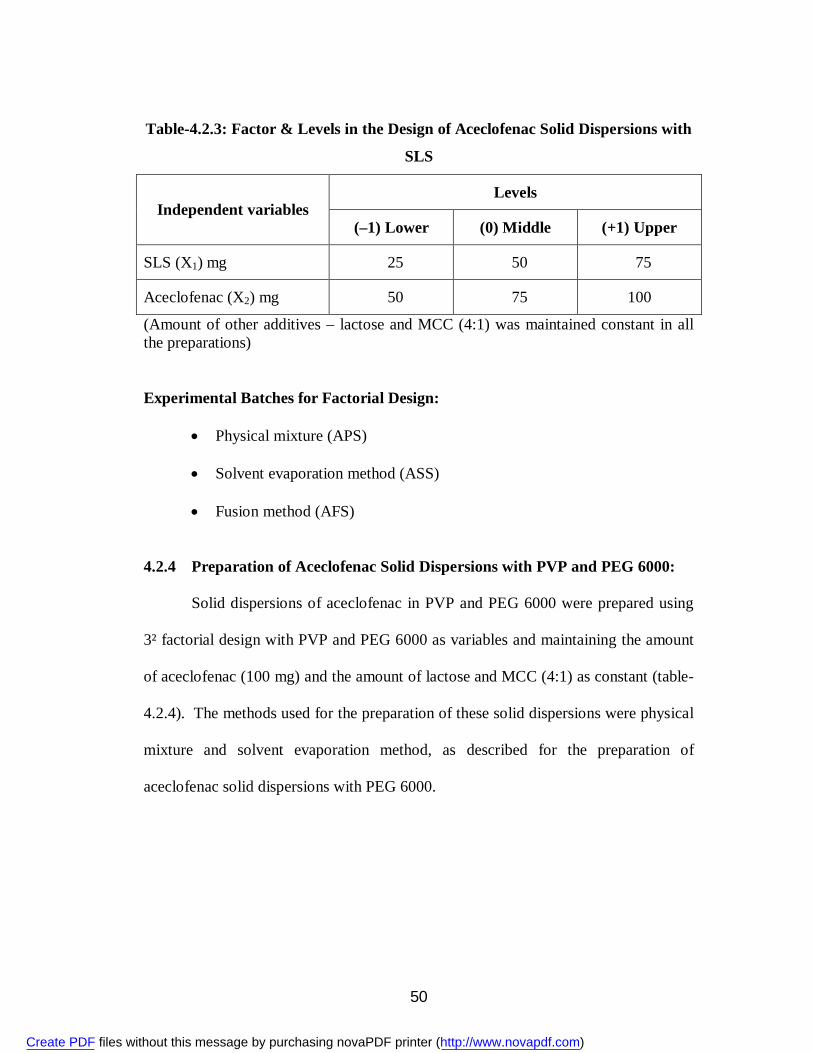
50
Table-4.2.3: Factor & Levels in the Design of Aceclofenac Solid Dispersions with
SLS
Levels Independent variables
(–1) Lower (0) Middle (+1) Upper
SLS (X1) mg 25 50 75
Aceclofenac (X2) mg 50 75 100
(Amount of other additives – lactose and MCC (4:1) was maintained constant in all the preparations)
Experimental Batches for Factorial Design:
Physical mixture (APS)
Solvent evaporation method (ASS)
Fusion method (AFS)
4.2.4 Preparation of Aceclofenac Solid Dispersions with PVP and PEG 6000:
Solid dispersions of aceclofenac in PVP and PEG 6000 were prepared using
3² factorial design with PVP and PEG 6000 as variables and maintaining the amount
of aceclofenac (100 mg) and the amount of lactose and MCC (4:1) as constant (table-
4.2.4). The methods used for the preparation of these solid dispersions were physical
mixture and solvent evaporation method, as described for the preparation of
aceclofenac solid dispersions with PEG 6000.
Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com)
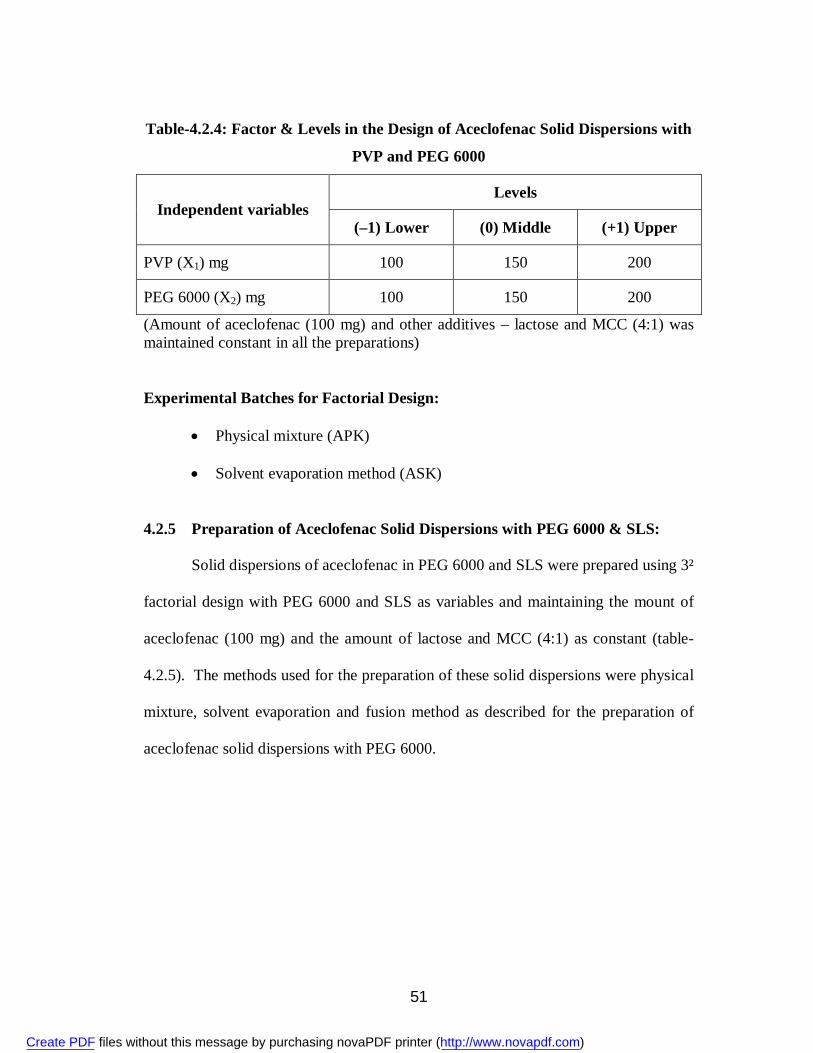
51
Table-4.2.4: Factor & Levels in the Design of Aceclofenac Solid Dispersions with
PVP and PEG 6000
Levels Independent variables
(–1) Lower (0) Middle (+1) Upper
PVP (X1) mg 100 150 200
PEG 6000 (X2) mg 100 150 200
(Amount of aceclofenac (100 mg) and other additives – lactose and MCC (4:1) was maintained constant in all the preparations)
Experimental Batches for Factorial Design:
Physical mixture (APK)
Solvent evaporation method (ASK)
4.2.5 Preparation of Aceclofenac Solid Dispersions with PEG 6000 & SLS:
Solid dispersions of aceclofenac in PEG 6000 and SLS were prepared using 3²
factorial design with PEG 6000 and SLS as variables and maintaining the mount of
aceclofenac (100 mg) and the amount of lactose and MCC (4:1) as constant (table-
4.2.5). The methods used for the preparation of these solid dispersions were physical
mixture, solvent evaporation and fusion method as described for the preparation of
aceclofenac solid dispersions with PEG 6000.
Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com)
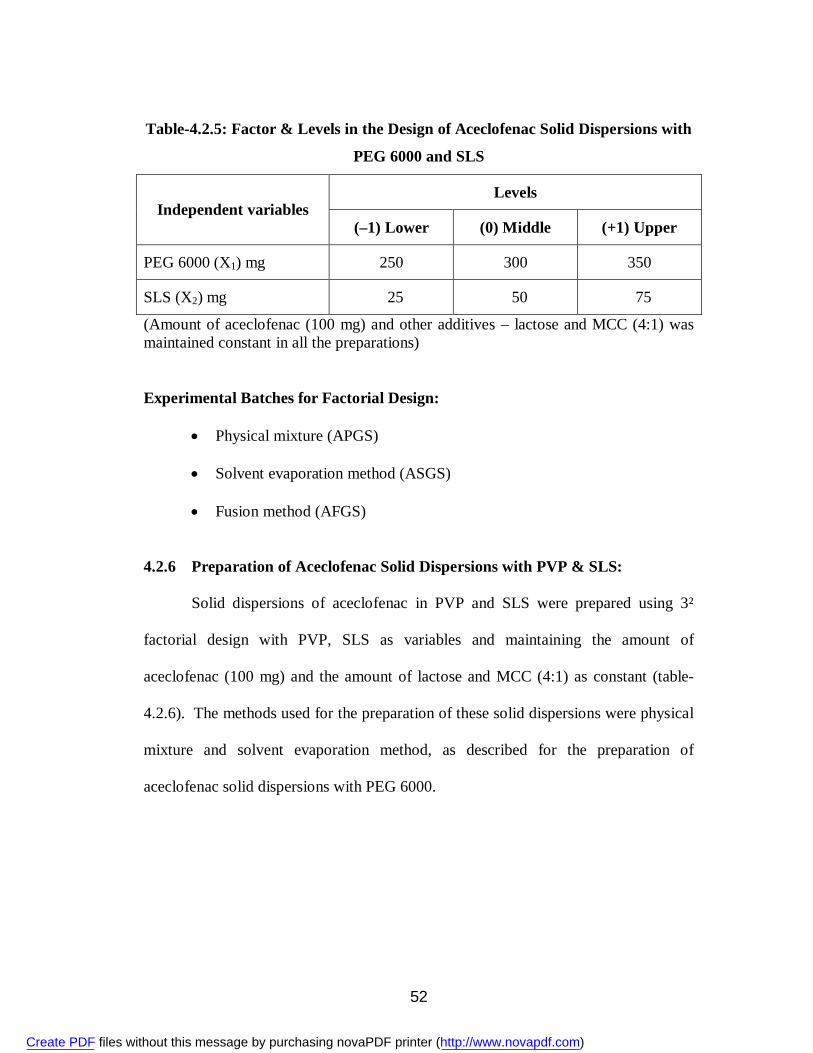
52
Table-4.2.5: Factor & Levels in the Design of Aceclofenac Solid Dispersions with
PEG 6000 and SLS
Levels Independent variables
(–1) Lower (0) Middle (+1) Upper
PEG 6000 (X1) mg 250 300 350
SLS (X2) mg 25 50 75
(Amount of aceclofenac (100 mg) and other additives – lactose and MCC (4:1) was maintained constant in all the preparations)
Experimental Batches for Factorial Design:
Physical mixture (APGS)
Solvent evaporation method (ASGS)
Fusion method (AFGS)
4.2.6 Preparation of Aceclofenac Solid Dispersions with PVP & SLS:
Solid dispersions of aceclofenac in PVP and SLS were prepared using 3²
factorial design with PVP, SLS as variables and maintaining the amount of
aceclofenac (100 mg) and the amount of lactose and MCC (4:1) as constant (table-
4.2.6). The methods used for the preparation of these solid dispersions were physical
mixture and solvent evaporation method, as described for the preparation of
aceclofenac solid dispersions with PEG 6000.
Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com)
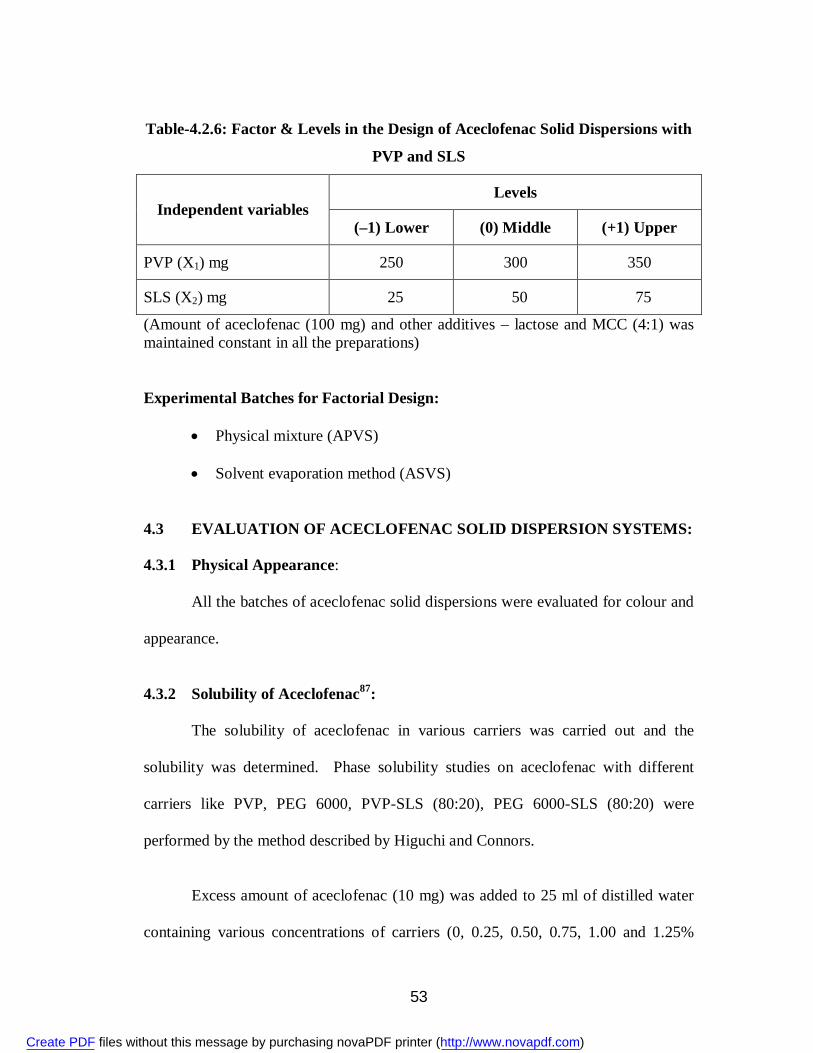
53
Table-4.2.6: Factor & Levels in the Design of Aceclofenac Solid Dispersions with
PVP and SLS
Levels Independent variables
(–1) Lower (0) Middle (+1) Upper
PVP (X1) mg 250 300 350
SLS (X2) mg 25 50 75
(Amount of aceclofenac (100 mg) and other additives – lactose and MCC (4:1) was maintained constant in all the preparations)
Experimental Batches for Factorial Design:
Physical mixture (APVS)
Solvent evaporation method (ASVS)
4.3 EVALUATION OF ACECLOFENAC SOLID DISPERSION SYSTEMS:
4.3.1 Physical Appearance:
All the batches of aceclofenac solid dispersions were evaluated for colour and
appearance.
4.3.2 Solubility of Aceclofenac87:
The solubility of aceclofenac in various carriers was carried out and the
solubility was determined. Phase solubility studies on aceclofenac with different
carriers like PVP, PEG 6000, PVP-SLS (80:20), PEG 6000-SLS (80:20) were
performed by the method described by Higuchi and Connors.
Excess amount of aceclofenac (10 mg) was added to 25 ml of distilled water
containing various concentrations of carriers (0, 0.25, 0.50, 0.75, 1.00 and 1.25%
Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com)

54
w/v). The suspension were shaken for 3 hours on a rotary flask at 37±1ºC and filtered
through a whatman No. 1 filter paper. The filtrate so obtained were analyzed
spectrophotometrically at 275 nm and corresponding concentrations of the drug were
computed from the standard curve.
4.3.3 Construction of Calibration Curve:
a) Calibration Curve of Aceclofenac in Methanol88:
A standard solution containing 1 mg/ ml of aceclofenac was prepared in
methanol by dissolving 50 mg of pure aceclofenac in 50 ml of methanol. From this
solution, working standard solutions of concentrations 0 to 20 g/ml of aceclofenac
was prepared by dilution with methanol. The absorbance of the solutions was
measured at 275 nm against reagent blank. Calibration curve was prepared.
b) Calibration Curve of Aceclofenac in Phosphate Buffer pH 7.4:
An accurately weighed amount of aceclofenac equivalent to 100 mg was
dissolved in small volume of methanol, in 100 ml volumetric flask and the volume
was adjusted to 100 ml with 7.4 phosphate buffer and further dilutions were made
with 7.4 pH phosphate buffer. A series of standard solution containing 2 to 20 g/ml
of aceclofenac were prepared and absorbance was measured at 275 nm against
reagent blank. All spectral absorbance measurements were made on Shimadzu-1700
UV-visible spectrophotometer.
Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com)

55
4.3.4 Determination of Aceclofenac Content:
An accurately weighed amount of each preparation was dissolved in small
volume of methanol and further diluted with methanol. The content of aceclofenac
was determined spectrophotometrically at 275 nm using Shimadzu UV-visible
spectrophotometer.
4.3.5 In Vitro Dissolution:
The dissolution study was carried out using USP XXIII apparatus type-II
(electrolab TDT-OCT). The dissolution medium was 900 ml 7.4 pH phosphate buffer
kept at 37±1ºC. The drug or physical mixture or solid dispersions was taken in a
muslin cloth and tied to the rotating paddle kept in the basket of dissolution
apparatus, the basket was rotated at 50 rpm. Samples of 5 ml were withdrawn at
specified time intervals and analyzed spectrophotometrically at 275 nm88 using
Shimadzu-1700 UV-visible spectrophotometer, the samples withdrawn were replaced
by fresh buffer solutions. Each preparation was tested in triplicate and then mean
values were calculated.
4.3.6 Statistical Comparison:
The dissolution release kinetics and result of best fit model among the
preparations were also compared.
Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com)

56
4.3.7 Stability Studies:
The purpose of stability testing is to provide evidence on how the quality of a
drug substance or drug product varies with time under the influence of a variety of
environmental factors such as temperature, humidity and light, and to establish a
retest period for the drug substance or a shelf-life for the drug product and
recommended storage conditions. Stress testing of the drug substance can help to
identify the likely degradation products, which can in-turn help to establish the
degradation pathways and the intrinsic stability of the molecule and validated the
stability indicating power of analytical procedure used. The nature of the stress
testing will depend upon the individual drug substance and the type of drug-product
involved.
The stability studies should be conducted on the drug substance packaged in a
container closure systems is the same as or simulates the packing proposed for
storage and distribution.
Stability studies on various batches like ASG7, ASV7, ASS7, ASK9, ASGS9
and ASVS9 were carried out by storing 1 gm of solid dispersions in an amber
coloured screw capped bottle at different temperatures for a period of 3 months. The
solid dispersions were visually examined for any physical change and drug content
was estimated at the end of 3 months89.
Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com)

57
4.3.8 Differential Scanning Calorimetry:
Differential scanning calorimetry (DSC) of some selected preparations were
compared with plain aceclofenac, PVP, PEG 6000 and SLS was carried out. The
DSC (pyris-6) thermograms were recorded at a heating of 10ºC/ min from 100ºC to
300ºC.
4.3.9 Infrared Spectroscopy:
The infrared spectra (IR) of aceclofenac, PVP, PEG 6000 and SLS and some
selected preparations was obtained using FTIR (Perkin Elmer 1600 Series). The IR
spectra was carried by KBr pellet method.
Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com)
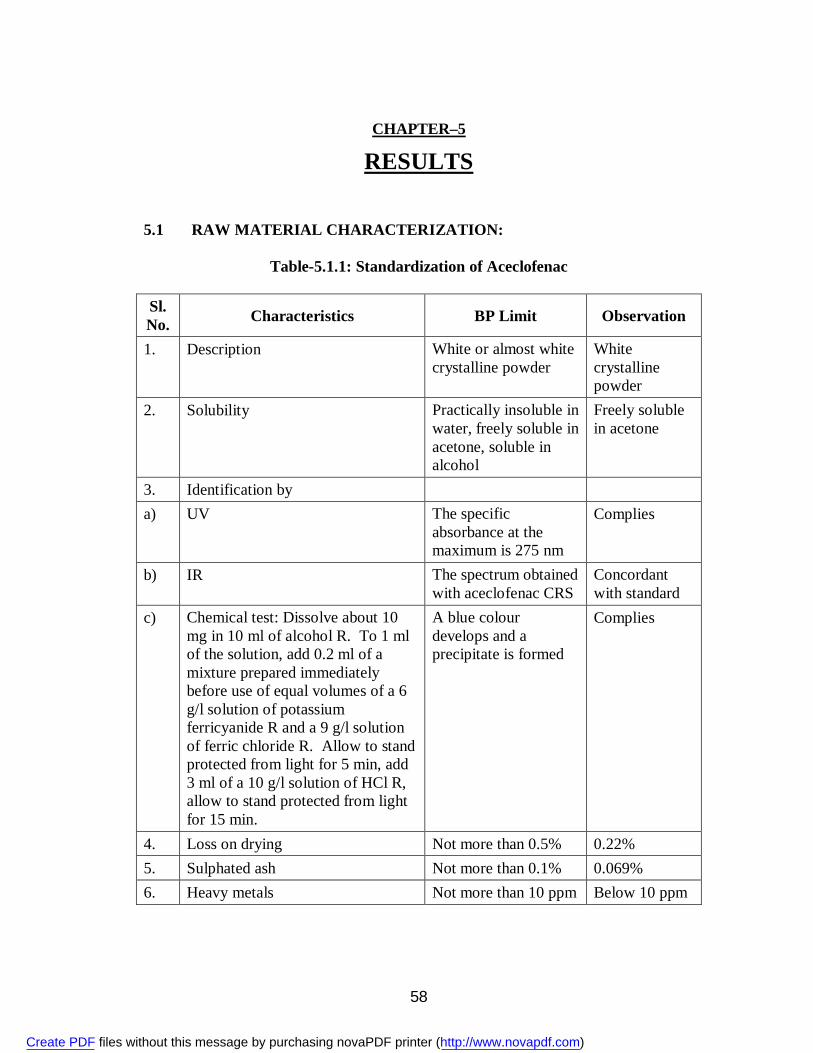
58
CHAPTER–5
RESULTS
5.1 RAW MATERIAL CHARACTERIZATION:
Table-5.1.1: Standardization of Aceclofenac
Sl. No. Characteristics BP Limit Observation
1. Description White or almost white crystalline powder
White crystalline powder
2. Solubility Practically insoluble in water, freely soluble in acetone, soluble in alcohol
Freely soluble in acetone
3. Identification by a) UV The specific
absorbance at the maximum is 275 nm
Complies
b) IR The spectrum obtained with aceclofenac CRS
Concordant with standard
c) Chemical test: Dissolve about 10 mg in 10 ml of alcohol R. To 1 ml of the solution, add 0.2 ml of a mixture prepared immediately before use of equal volumes of a 6 g/l solution of potassium ferricyanide R and a 9 g/l solution of ferric chloride R. Allow to stand protected from light for 5 min, add 3 ml of a 10 g/l solution of HCl R, allow to stand protected from light for 15 min.
A blue colour develops and a precipitate is formed
Complies
4. Loss on drying Not more than 0.5% 0.22% 5. Sulphated ash Not more than 0.1% 0.069% 6. Heavy metals Not more than 10 ppm Below 10 ppm
Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com)
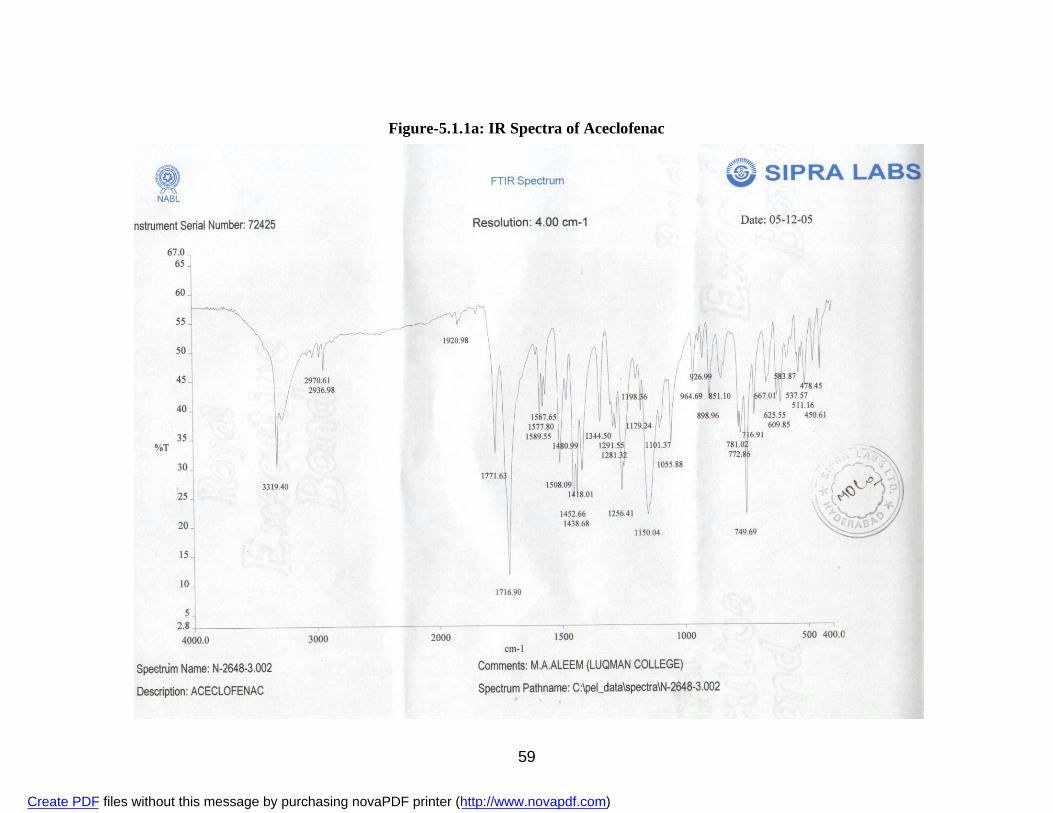
59
Figure-5.1.1a: IR Spectra of Aceclofenac
Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com)
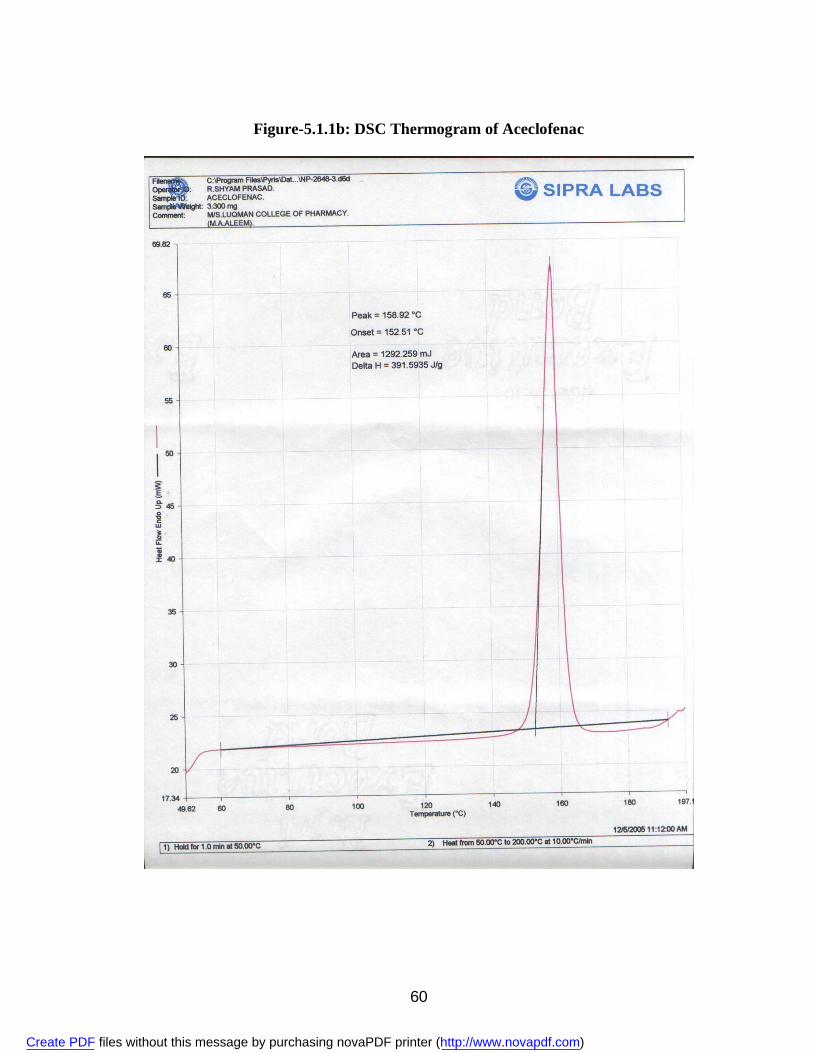
60
Figure-5.1.1b: DSC Thermogram of Aceclofenac
Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com)
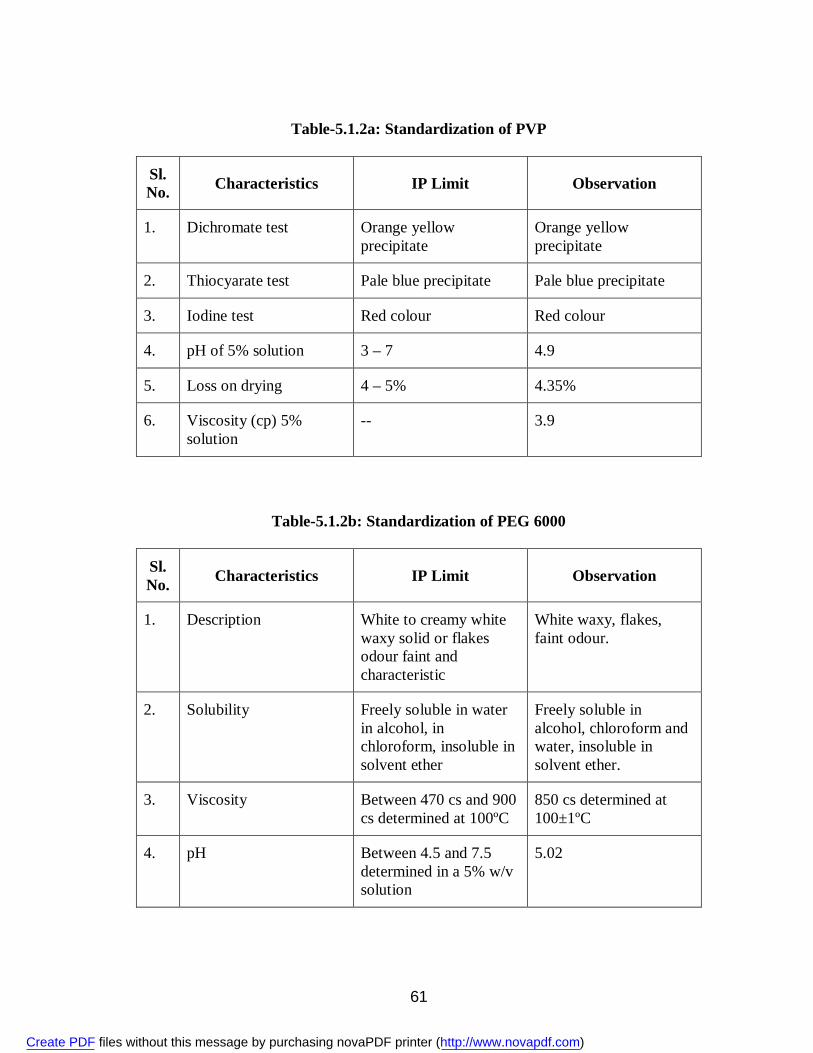
61
Table-5.1.2a: Standardization of PVP
Sl. No. Characteristics IP Limit Observation
1. Dichromate test Orange yellow precipitate
Orange yellow precipitate
2. Thiocyarate test Pale blue precipitate Pale blue precipitate
3. Iodine test Red colour Red colour
4. pH of 5% solution 3 – 7 4.9
5. Loss on drying 4 – 5% 4.35%
6. Viscosity (cp) 5% solution
-- 3.9
Table-5.1.2b: Standardization of PEG 6000
Sl. No. Characteristics IP Limit Observation
1. Description White to creamy white waxy solid or flakes odour faint and characteristic
White waxy, flakes, faint odour.
2. Solubility Freely soluble in water in alcohol, in chloroform, insoluble in solvent ether
Freely soluble in alcohol, chloroform and water, insoluble in solvent ether.
3. Viscosity Between 470 cs and 900 cs determined at 100ºC
850 cs determined at 100±1ºC
4. pH Between 4.5 and 7.5 determined in a 5% w/v solution
5.02
Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com)
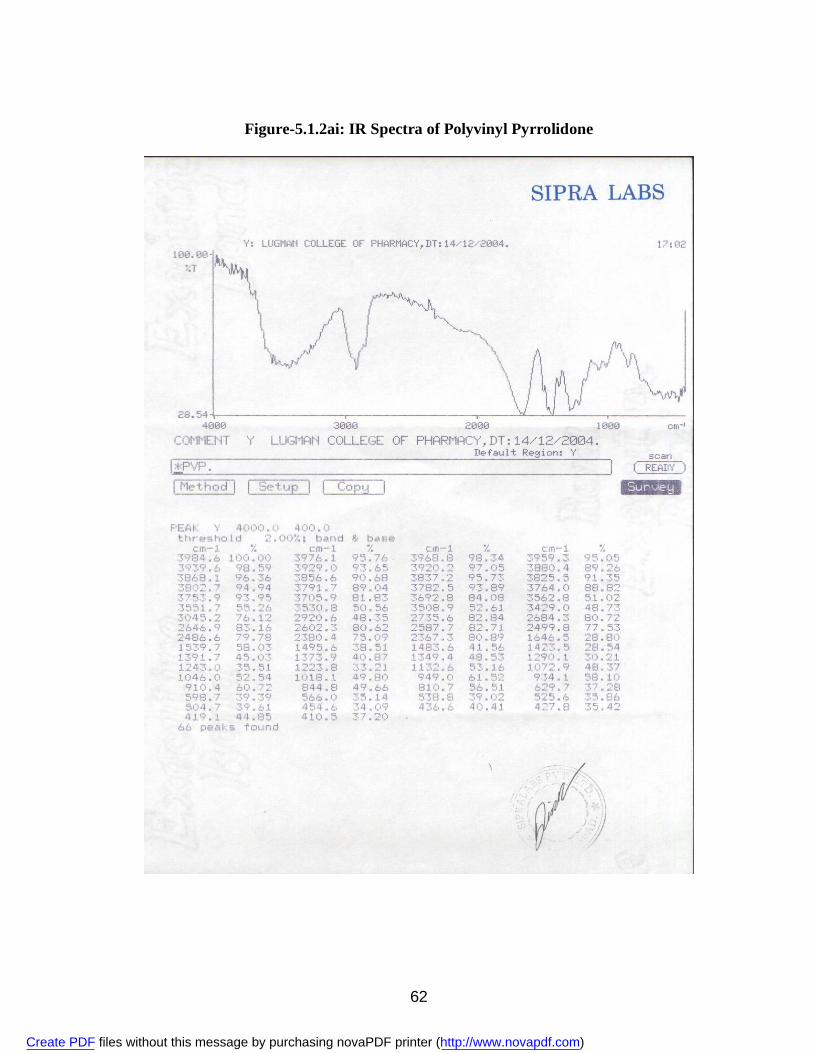
62
Figure-5.1.2ai: IR Spectra of Polyvinyl Pyrrolidone
Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com)
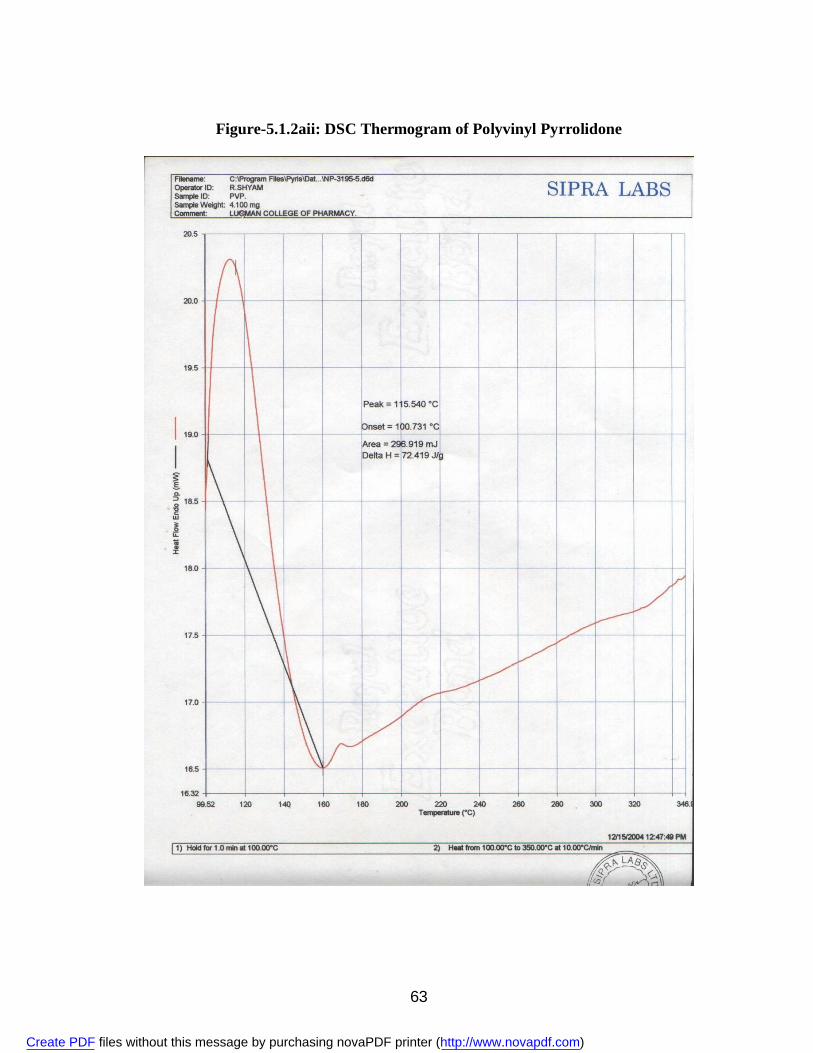
63
Figure-5.1.2aii: DSC Thermogram of Polyvinyl Pyrrolidone
Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com)
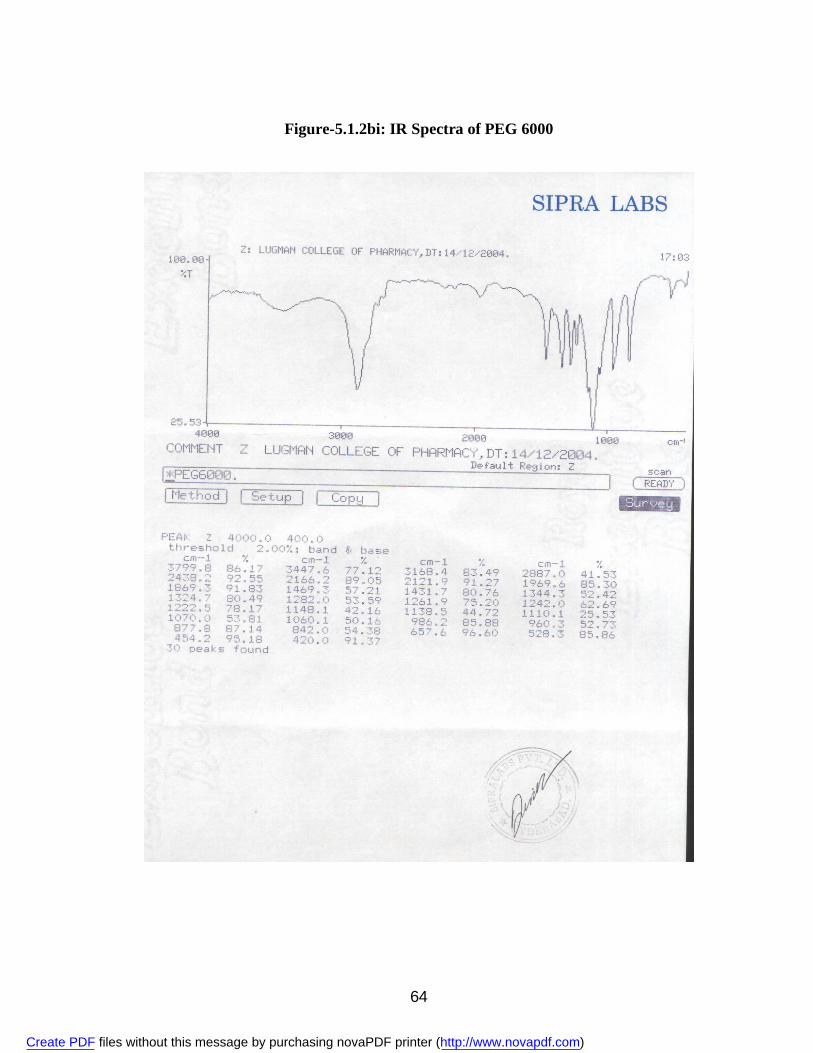
64
Figure-5.1.2bi: IR Spectra of PEG 6000
Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com)
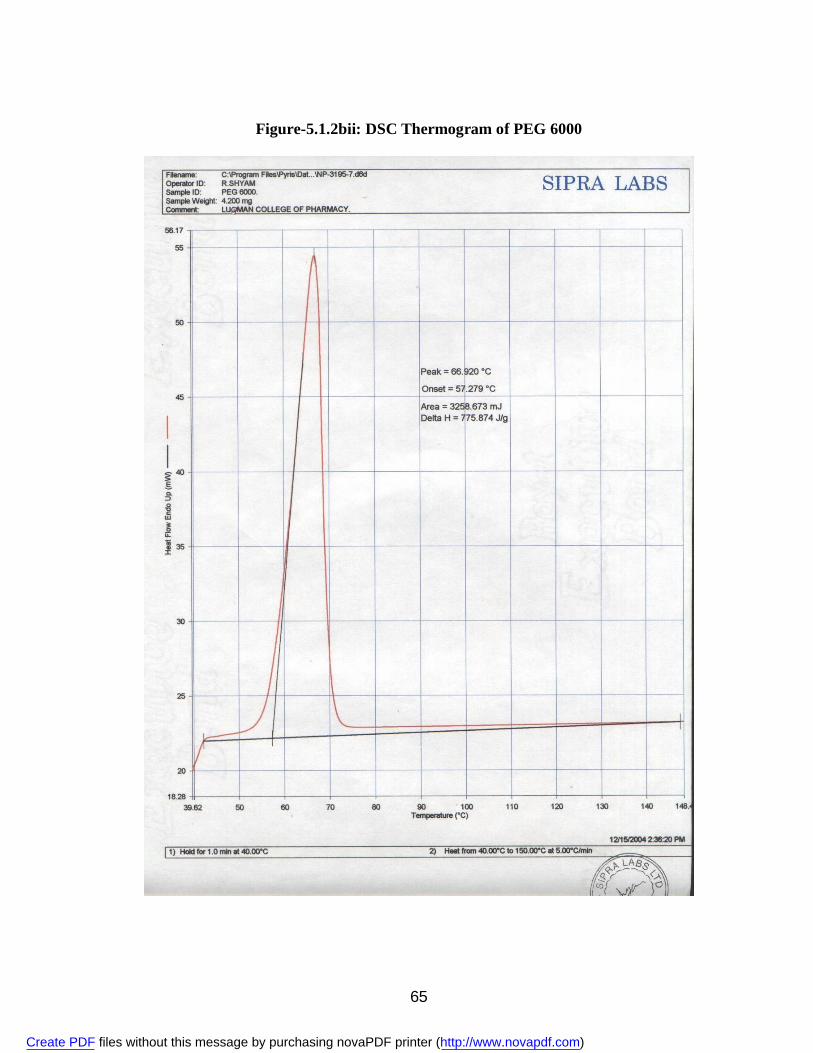
65
Figure-5.1.2bii: DSC Thermogram of PEG 6000
Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com)
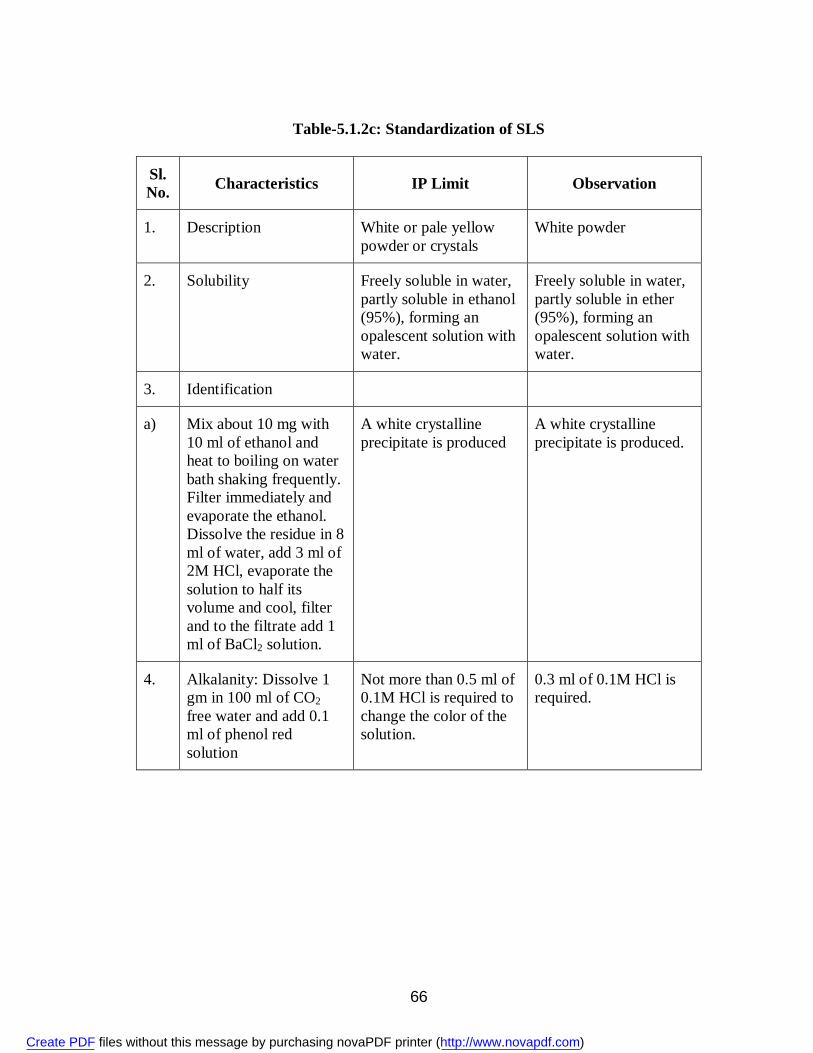
66
Table-5.1.2c: Standardization of SLS
Sl. No. Characteristics IP Limit Observation
1. Description White or pale yellow powder or crystals
White powder
2. Solubility Freely soluble in water, partly soluble in ethanol (95%), forming an opalescent solution with water.
Freely soluble in water, partly soluble in ether (95%), forming an opalescent solution with water.
3. Identification
a) Mix about 10 mg with 10 ml of ethanol and heat to boiling on water bath shaking frequently. Filter immediately and evaporate the ethanol. Dissolve the residue in 8 ml of water, add 3 ml of 2M HCl, evaporate the solution to half its volume and cool, filter and to the filtrate add 1 ml of BaCl2 solution.
A white crystalline precipitate is produced
A white crystalline precipitate is produced.
4. Alkalanity: Dissolve 1 gm in 100 ml of CO2 free water and add 0.1 ml of phenol red solution
Not more than 0.5 ml of 0.1M HCl is required to change the color of the solution.
0.3 ml of 0.1M HCl is required.
Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com)

67
Figure-5.1.2ci: IR Spectra of Sodium Lauryl Sulphate
Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com)
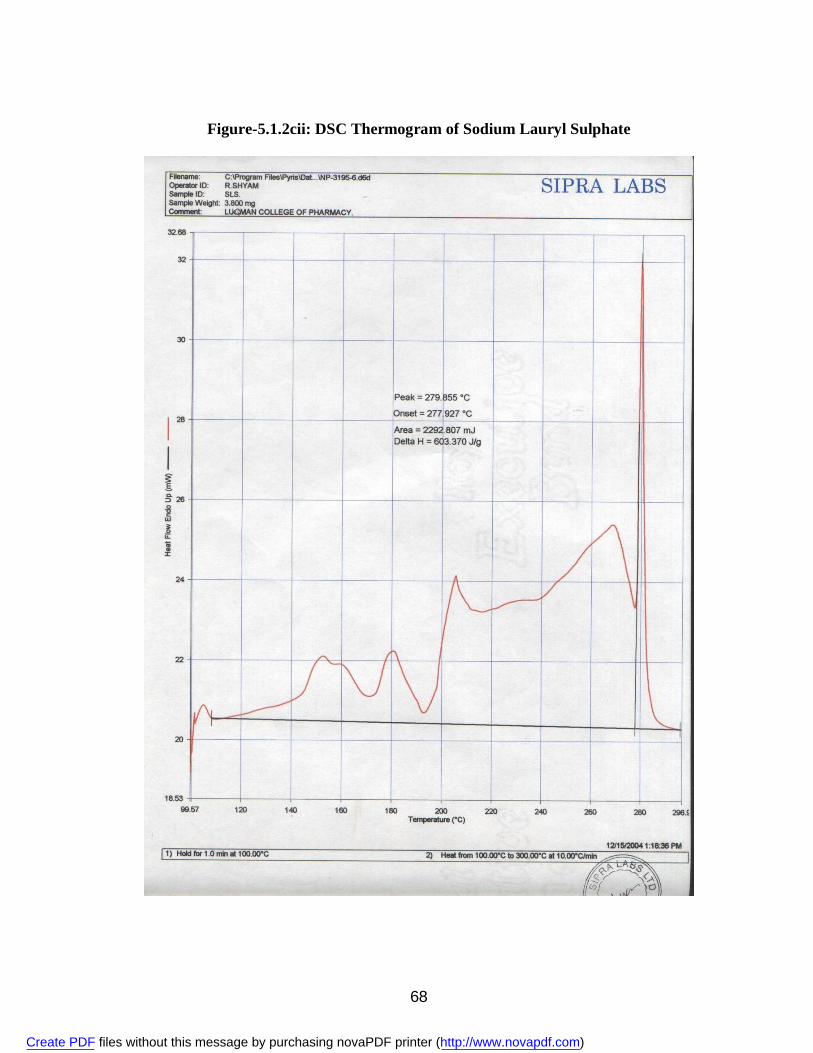
68
Figure-5.1.2cii: DSC Thermogram of Sodium Lauryl Sulphate
Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com)
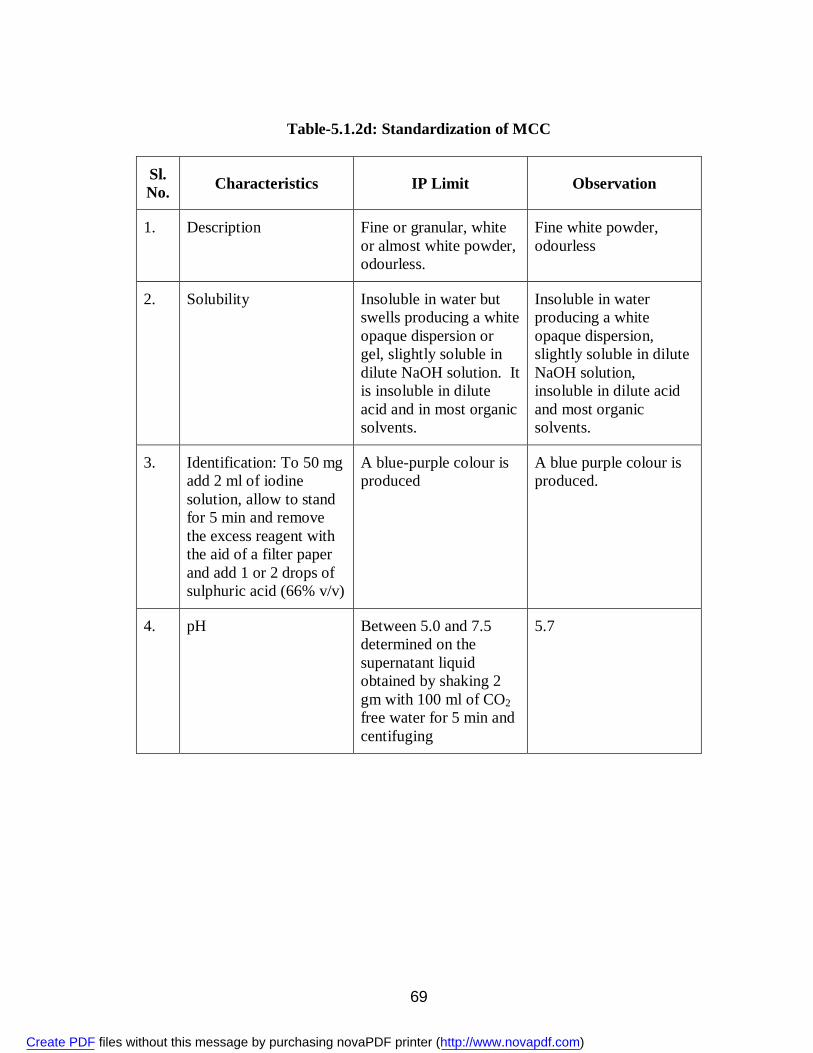
69
Table-5.1.2d: Standardization of MCC
Sl. No. Characteristics IP Limit Observation
1. Description Fine or granular, white or almost white powder, odourless.
Fine white powder, odourless
2. Solubility Insoluble in water but swells producing a white opaque dispersion or gel, slightly soluble in dilute NaOH solution. It is insoluble in dilute acid and in most organic solvents.
Insoluble in water producing a white opaque dispersion, slightly soluble in dilute NaOH solution, insoluble in dilute acid and most organic solvents.
3. Identification: To 50 mg add 2 ml of iodine solution, allow to stand for 5 min and remove the excess reagent with the aid of a filter paper and add 1 or 2 drops of sulphuric acid (66% v/v)
A blue-purple colour is produced
A blue purple colour is produced.
4. pH Between 5.0 and 7.5 determined on the supernatant liquid obtained by shaking 2 gm with 100 ml of CO2 free water for 5 min and centifuging
5.7
Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com)
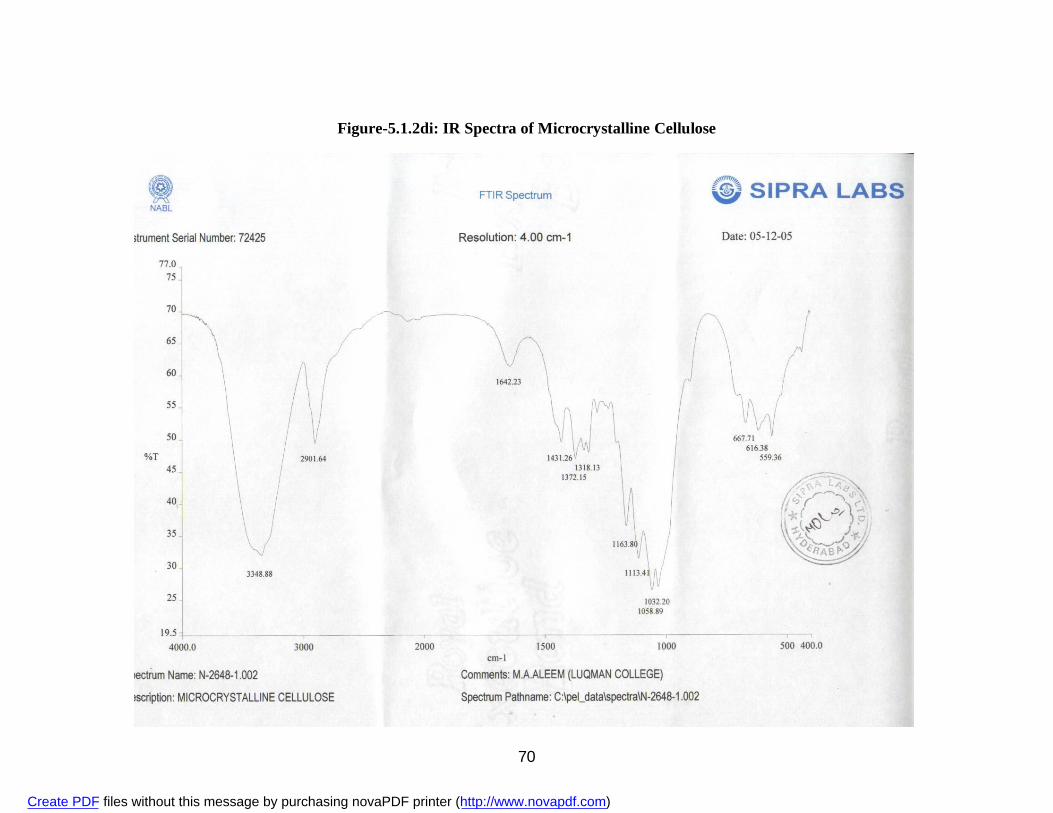
70
Figure-5.1.2di: IR Spectra of Microcrystalline Cellulose
Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com)
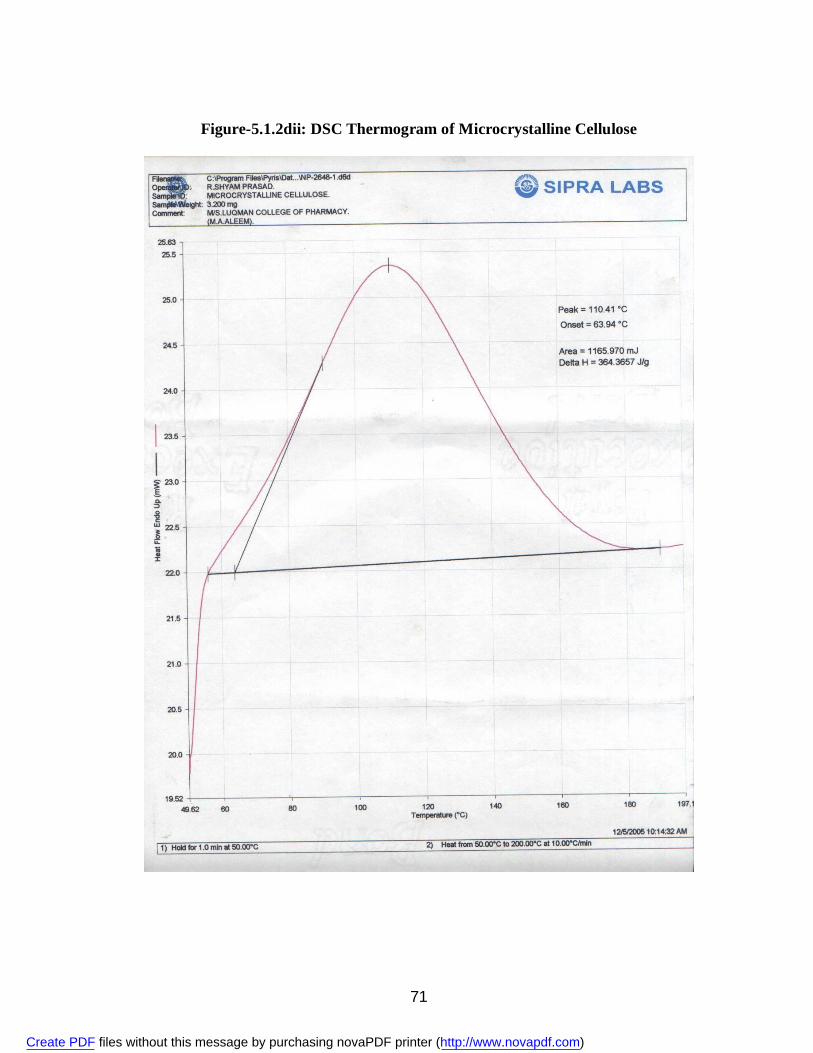
71
Figure-5.1.2dii: DSC Thermogram of Microcrystalline Cellulose
Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com)
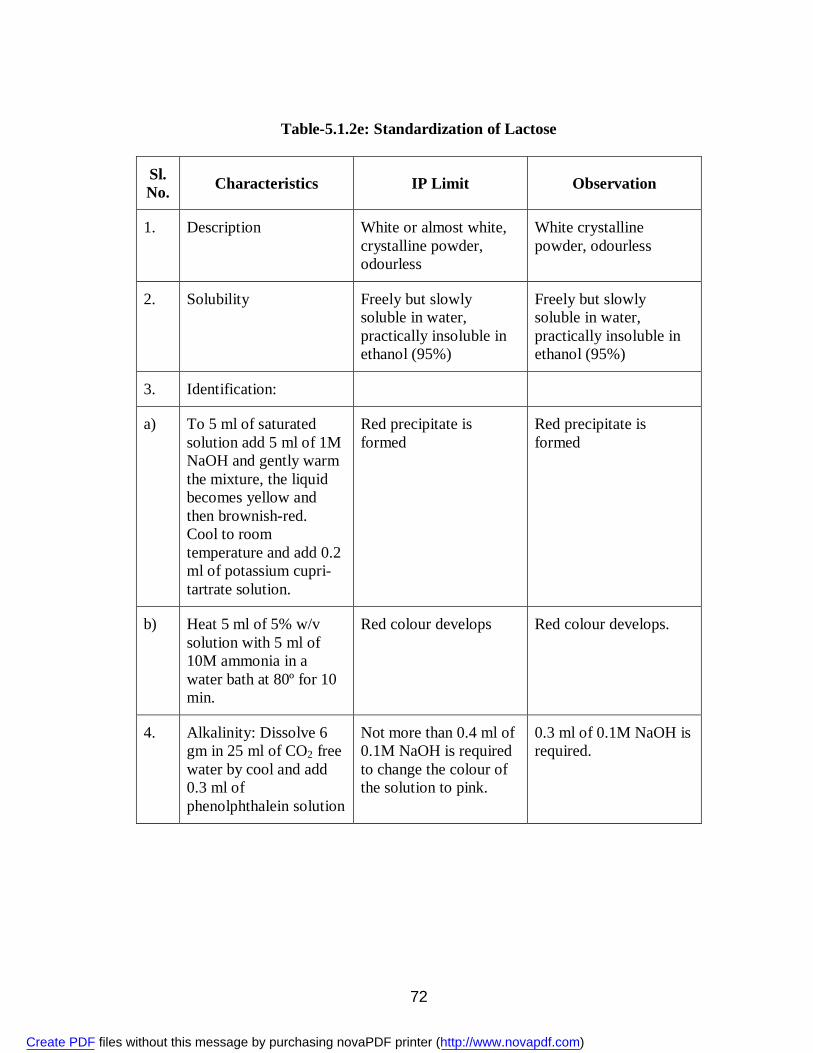
72
Table-5.1.2e: Standardization of Lactose
Sl. No. Characteristics IP Limit Observation
1. Description White or almost white, crystalline powder, odourless
White crystalline powder, odourless
2. Solubility Freely but slowly soluble in water, practically insoluble in ethanol (95%)
Freely but slowly soluble in water, practically insoluble in ethanol (95%)
3. Identification:
a) To 5 ml of saturated solution add 5 ml of 1M NaOH and gently warm the mixture, the liquid becomes yellow and then brownish-red. Cool to room temperature and add 0.2 ml of potassium cupri-tartrate solution.
Red precipitate is formed
Red precipitate is formed
b) Heat 5 ml of 5% w/v solution with 5 ml of 10M ammonia in a water bath at 80º for 10 min.
Red colour develops Red colour develops.
4. Alkalinity: Dissolve 6 gm in 25 ml of CO2 free water by cool and add 0.3 ml of phenolphthalein solution
Not more than 0.4 ml of 0.1M NaOH is required to change the colour of the solution to pink.
0.3 ml of 0.1M NaOH is required.
Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com)

73
Figure-5.1.2ei: IR Spectra of Lactose
Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com)

74
Figure-5.1.2eii: DSC Thermogram of Lactose
Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com)
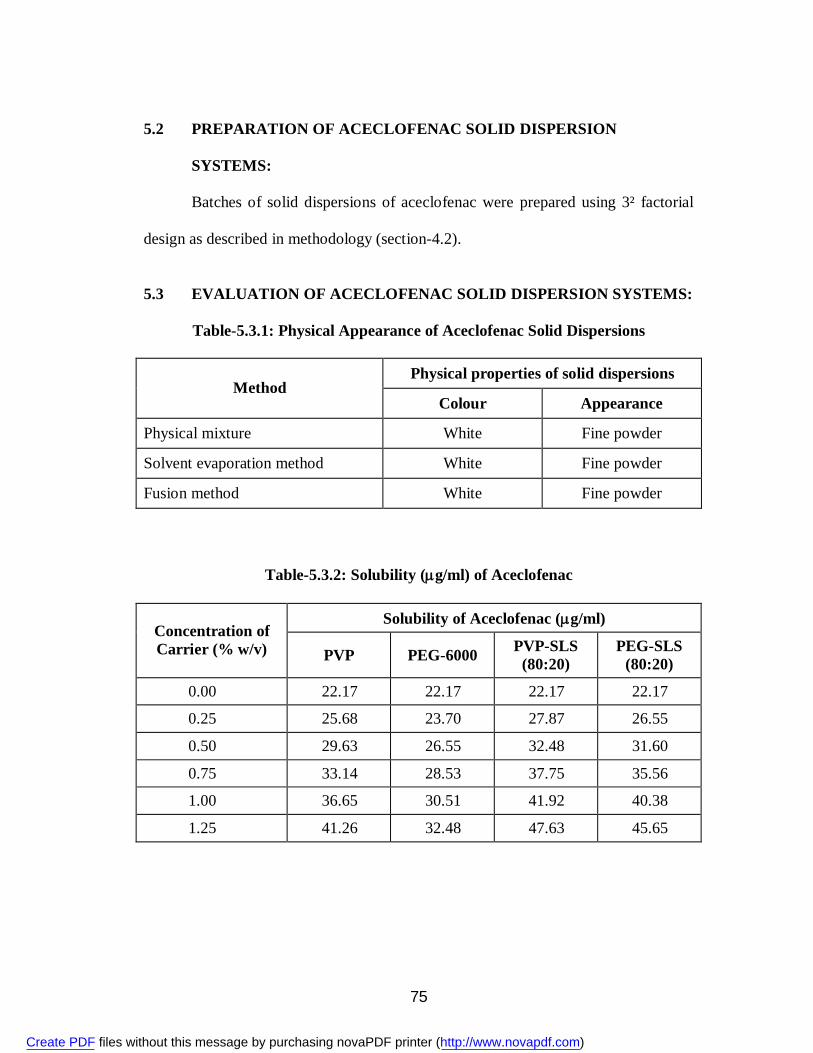
75
5.2 PREPARATION OF ACECLOFENAC SOLID DISPERSION
SYSTEMS:
Batches of solid dispersions of aceclofenac were prepared using 3² factorial
design as described in methodology (section-4.2).
5.3 EVALUATION OF ACECLOFENAC SOLID DISPERSION SYSTEMS:
Table-5.3.1: Physical Appearance of Aceclofenac Solid Dispersions
Physical properties of solid dispersions Method
Colour Appearance
Physical mixture White Fine powder
Solvent evaporation method White Fine powder
Fusion method White Fine powder
Table-5.3.2: Solubility (g/ml) of Aceclofenac
Solubility of Aceclofenac (g/ml) Concentration of Carrier (% w/v) PVP PEG-6000 PVP-SLS
(80:20) PEG-SLS
(80:20)
0.00 22.17 22.17 22.17 22.17
0.25 25.68 23.70 27.87 26.55
0.50 29.63 26.55 32.48 31.60
0.75 33.14 28.53 37.75 35.56
1.00 36.65 30.51 41.92 40.38
1.25 41.26 32.48 47.63 45.65
Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com)
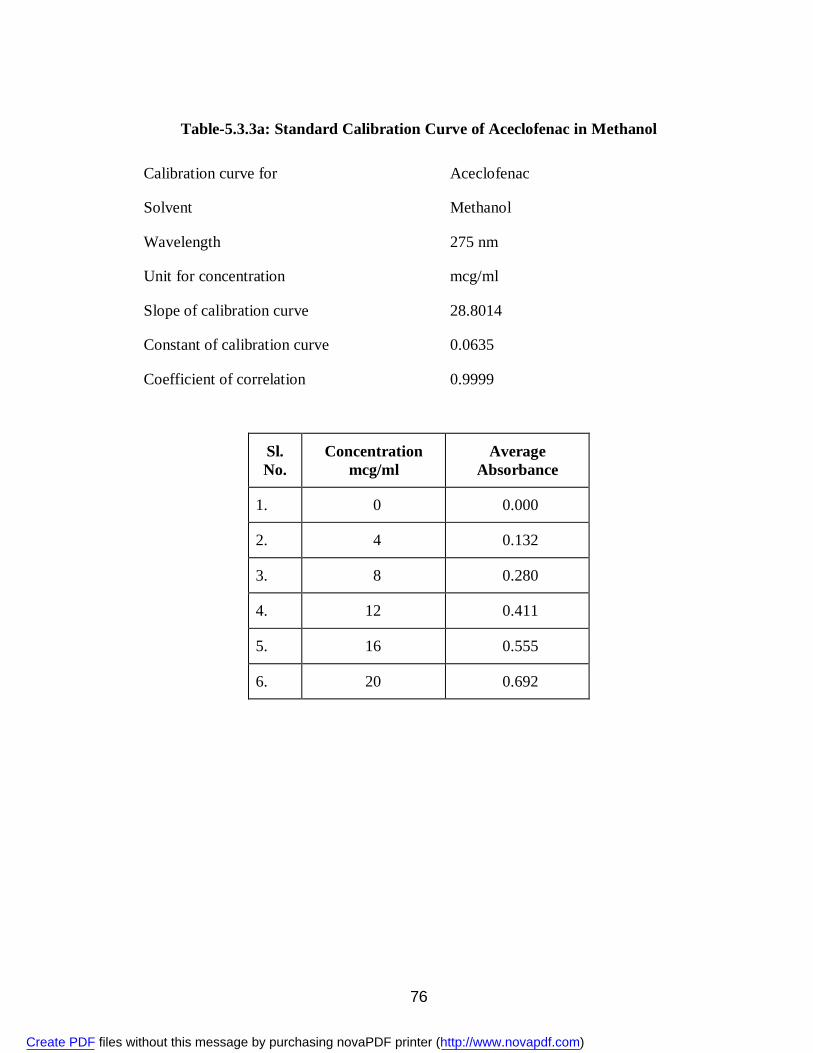
76
Table-5.3.3a: Standard Calibration Curve of Aceclofenac in Methanol
Calibration curve for Aceclofenac
Solvent Methanol
Wavelength 275 nm
Unit for concentration mcg/ml
Slope of calibration curve 28.8014
Constant of calibration curve 0.0635
Coefficient of correlation 0.9999
Sl. No.
Concentration mcg/ml
Average Absorbance
1. 0 0.000
2. 4 0.132
3. 8 0.280
4. 12 0.411
5. 16 0.555
6. 20 0.692
Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com)
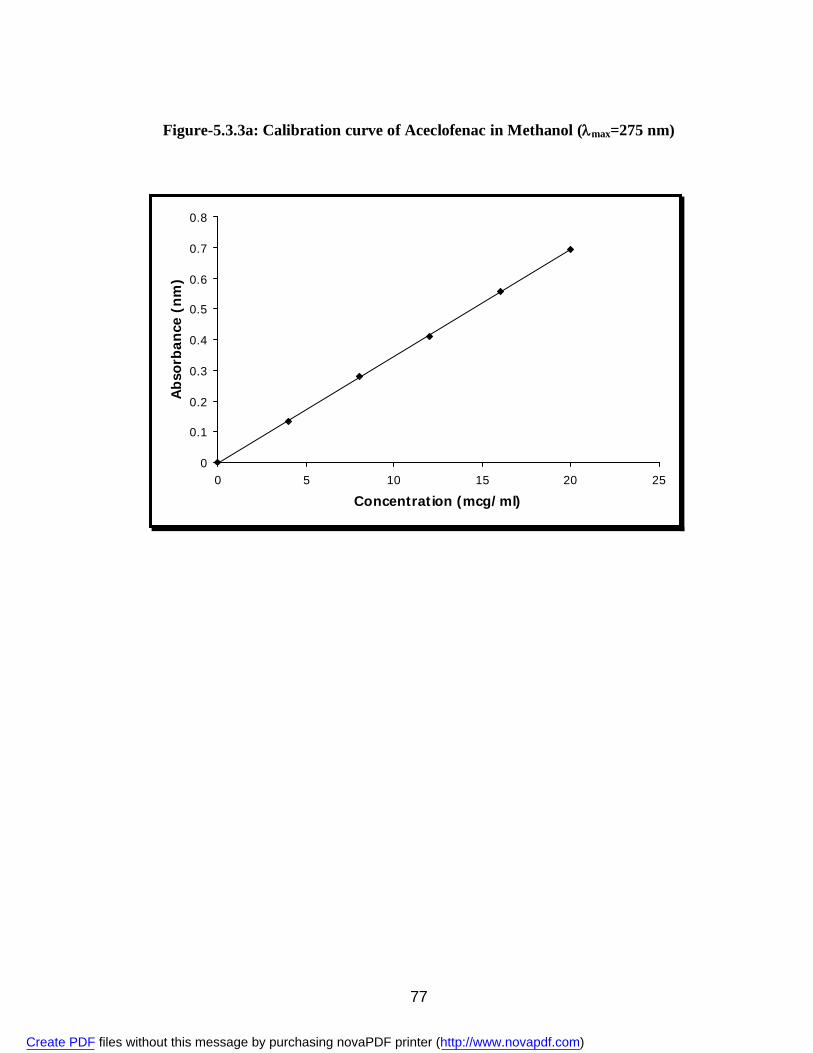
77
Figure-5.3.3a: Calibration curve of Aceclofenac in Methanol (max=275 nm)
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0 5 10 15 20 25
Concentration (mcg/ml)
Ab
sorb
ance
(n
m)
Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com)
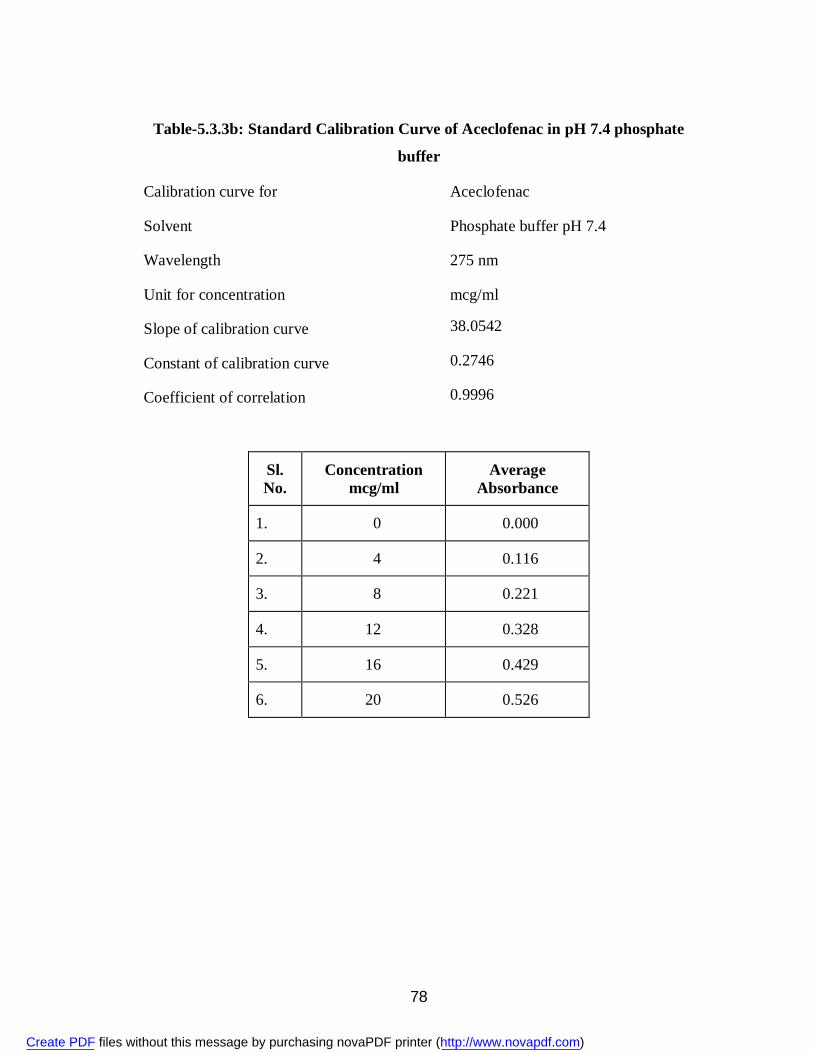
78
Table-5.3.3b: Standard Calibration Curve of Aceclofenac in pH 7.4 phosphate
buffer
Calibration curve for Aceclofenac
Solvent Phosphate buffer pH 7.4
Wavelength 275 nm
Unit for concentration mcg/ml
Slope of calibration curve 38.0542
Constant of calibration curve 0.2746
Coefficient of correlation 0.9996
Sl. No.
Concentration mcg/ml
Average Absorbance
1. 0 0.000
2. 4 0.116
3. 8 0.221
4. 12 0.328
5. 16 0.429
6. 20 0.526
Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com)
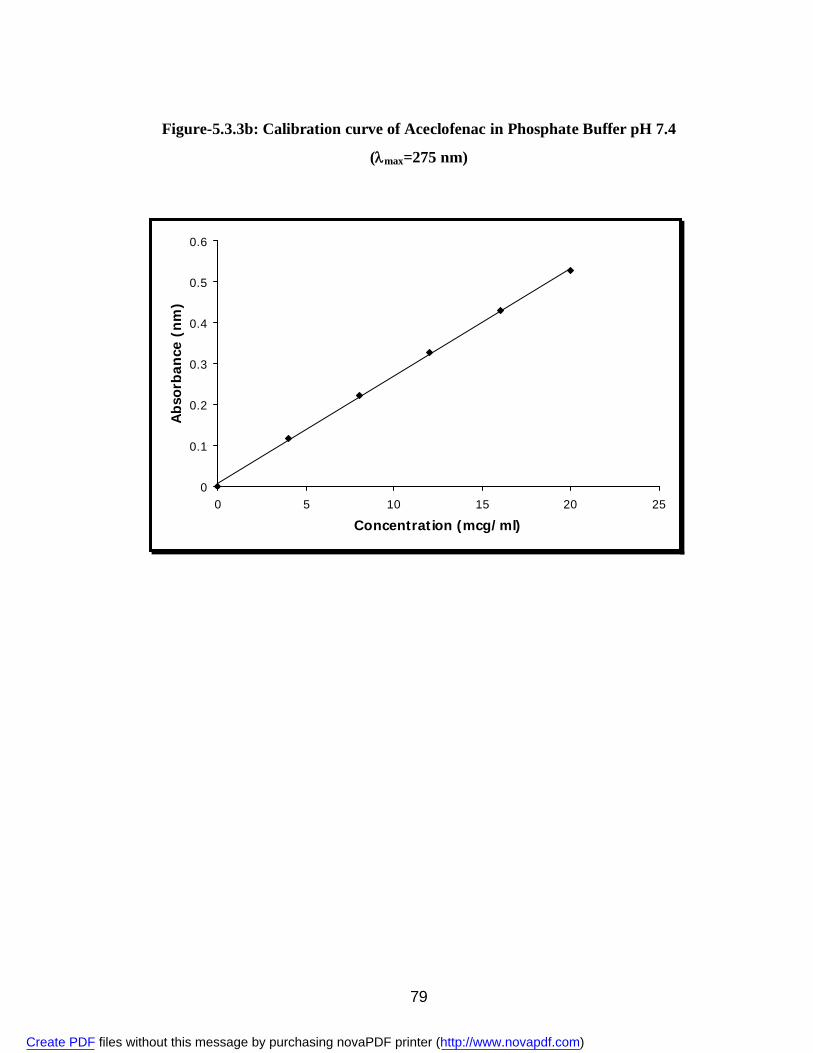
79
Figure-5.3.3b: Calibration curve of Aceclofenac in Phosphate Buffer pH 7.4
(max=275 nm)
0
0.1
0.2
0.3
0.4
0.5
0.6
0 5 10 15 20 25
Concentration (mcg/ml)
Ab
sorb
ance
(n
m)
Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com)
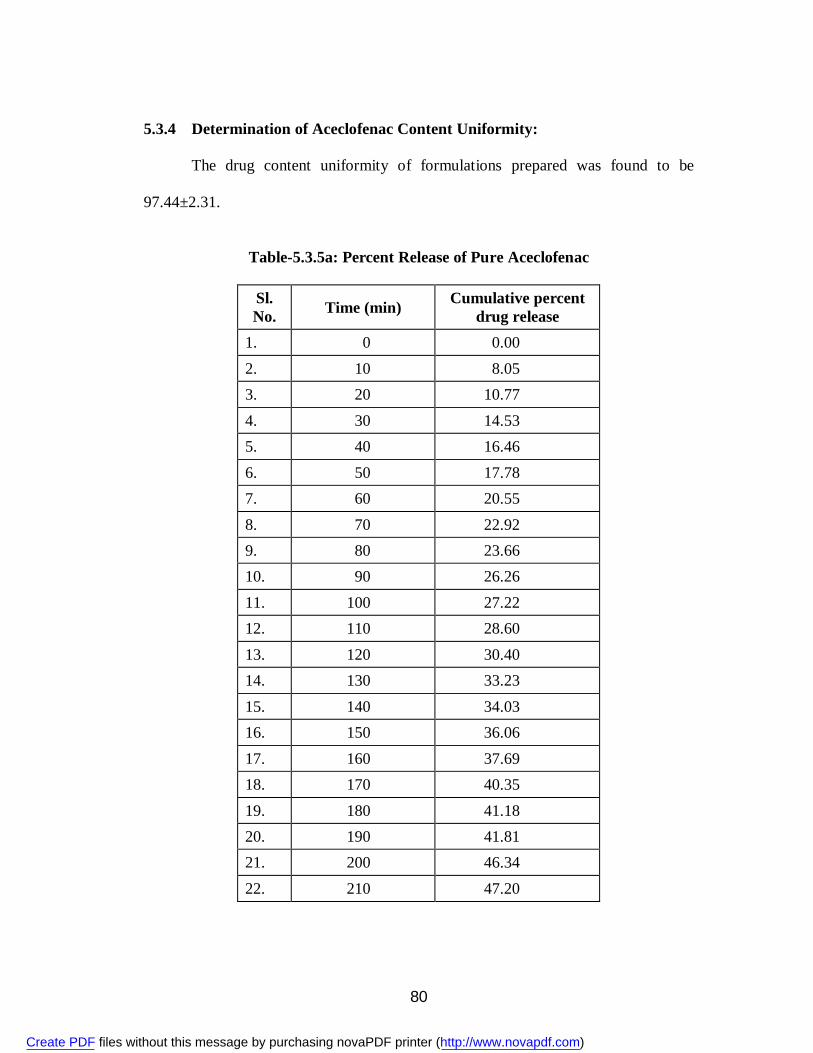
80
5.3.4 Determination of Aceclofenac Content Uniformity:
The drug content uniformity of formulations prepared was found to be
97.44±2.31.
Table-5.3.5a: Percent Release of Pure Aceclofenac
Sl. No. Time (min) Cumulative percent
drug release 1. 0 0.00 2. 10 8.05 3. 20 10.77 4. 30 14.53 5. 40 16.46 6. 50 17.78 7. 60 20.55 8. 70 22.92 9. 80 23.66 10. 90 26.26 11. 100 27.22 12. 110 28.60 13. 120 30.40 14. 130 33.23 15. 140 34.03 16. 150 36.06 17. 160 37.69 18. 170 40.35 19. 180 41.18 20. 190 41.81 21. 200 46.34 22. 210 47.20
Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com)
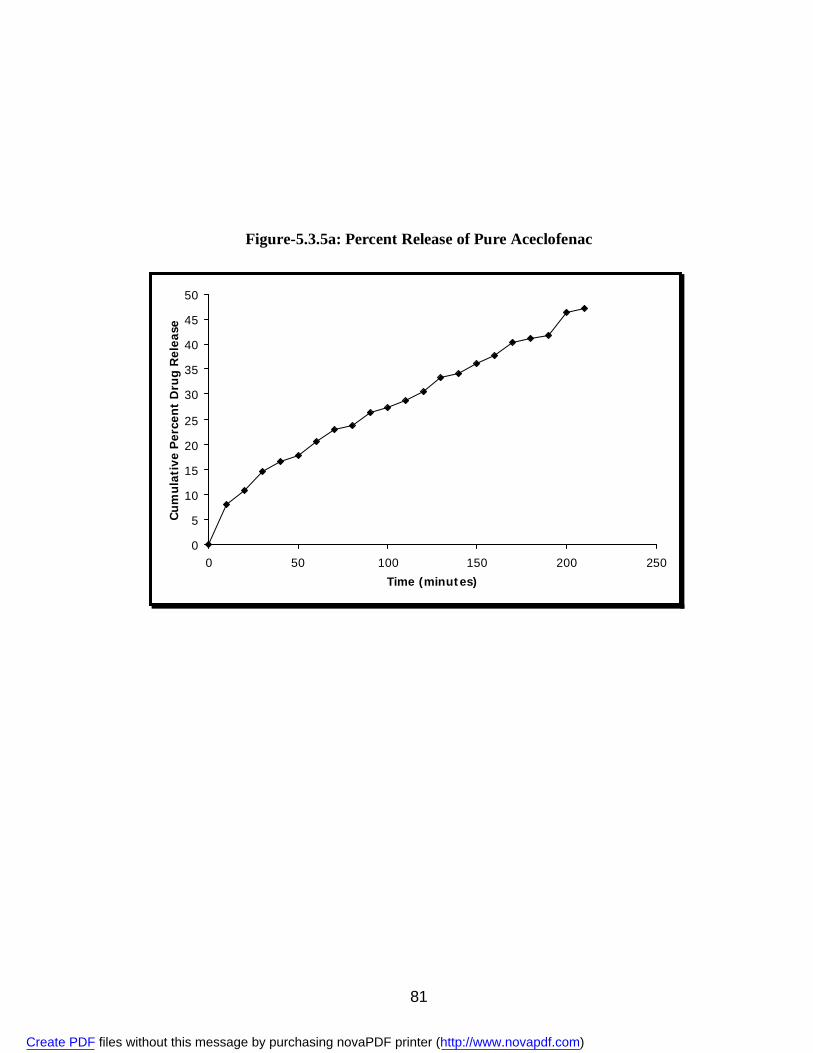
81
Figure-5.3.5a: Percent Release of Pure Aceclofenac
0
5
10
15
20
25
30
35
40
45
50
0 50 100 150 200 250
Time (minutes)
Cu
mu
lati
ve P
erce
nt
Dru
g R
elea
se
Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com)

82
Results of in vitro release of aceclofenac from solid dispersion systems of
various batches prepared by physical mixture, fusion method and solvent
evaporation methods.
Name of the drug Aceclofenac
Loading dose in mg 100
Total No. of readings including zero-time reading 7
Dissolution medium Phosphate buffer pH 7.4
RPM 50
Volume of dissolution medium (ml) 900
Volume of sample removed (ml) 5
Dilution factor 6
Slope of calibration curve 38.0542
Constant of calibration curve 0.2746
R of calibration curve 0.9996
Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com)
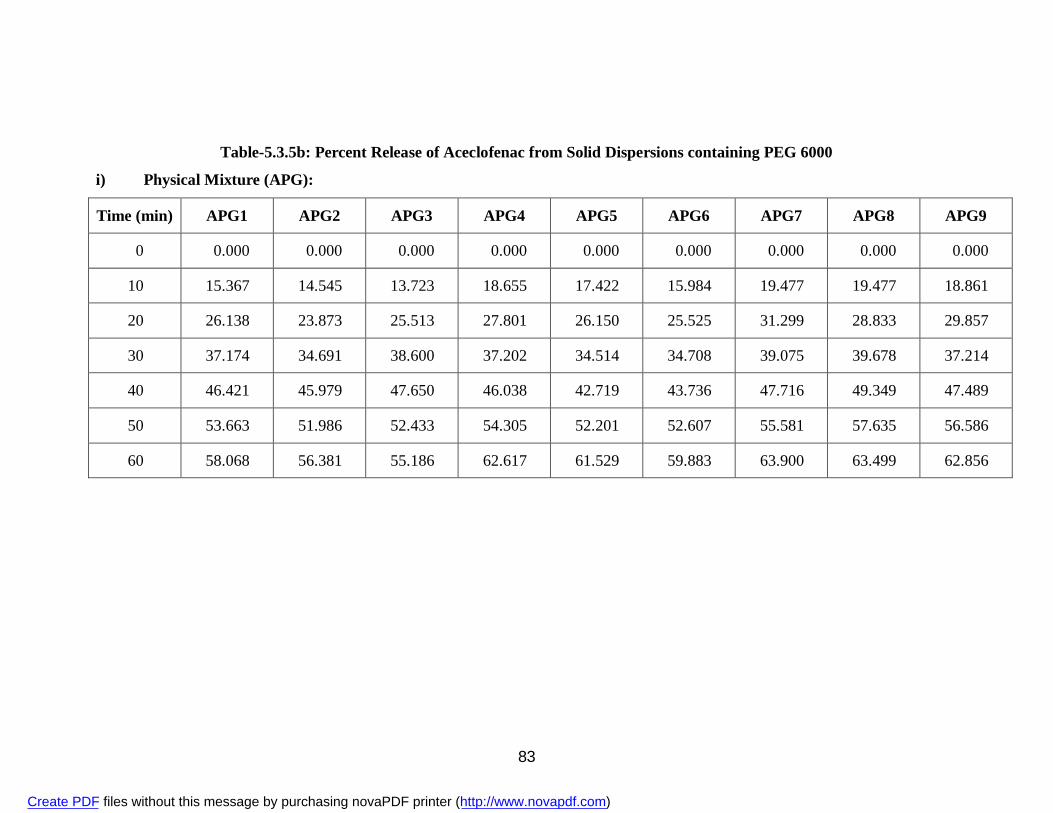
83
Table-5.3.5b: Percent Release of Aceclofenac from Solid Dispersions containing PEG 6000
i) Physical Mixture (APG):
Time (min) APG1 APG2 APG3 APG4 APG5 APG6 APG7 APG8 APG9
0 0.000 0.000 0.000 0.000 0.000 0.000 0.000 0.000 0.000
10 15.367 14.545 13.723 18.655 17.422 15.984 19.477 19.477 18.861
20 26.138 23.873 25.513 27.801 26.150 25.525 31.299 28.833 29.857
30 37.174 34.691 38.600 37.202 34.514 34.708 39.075 39.678 37.214
40 46.421 45.979 47.650 46.038 42.719 43.736 47.716 49.349 47.489
50 53.663 51.986 52.433 54.305 52.201 52.607 55.581 57.635 56.586
60 58.068 56.381 55.186 62.617 61.529 59.883 63.900 63.499 62.856
Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com)
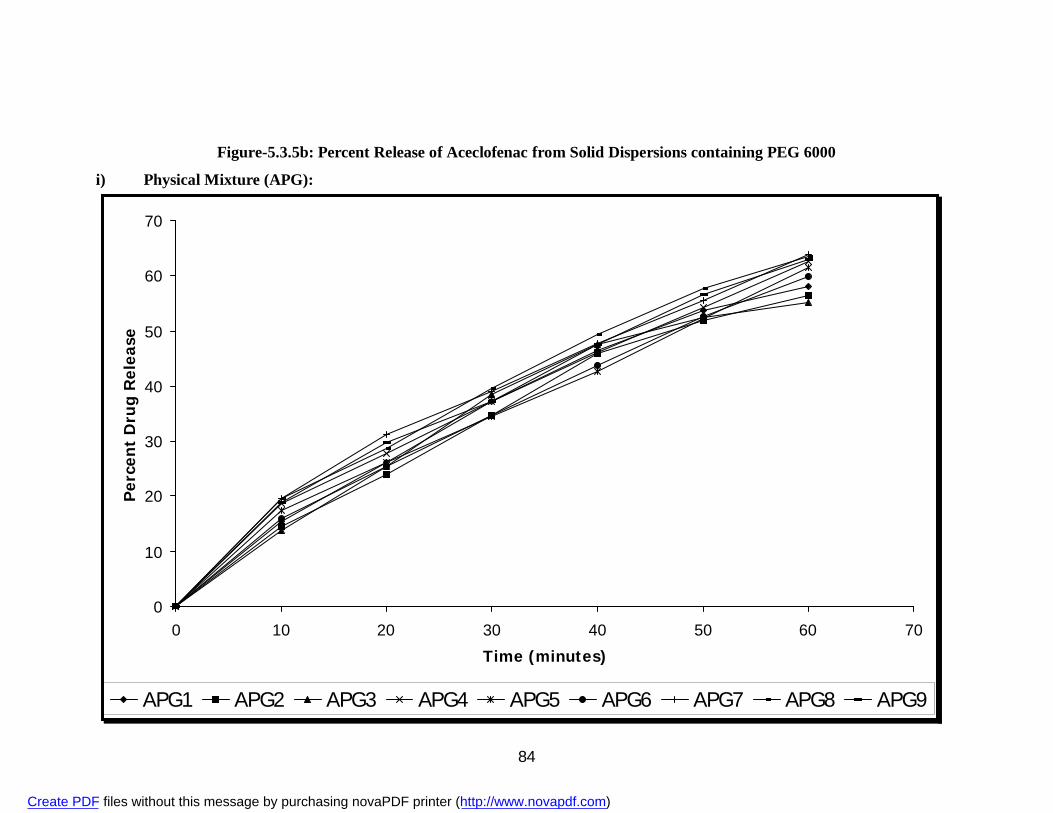
84
Figure-5.3.5b: Percent Release of Aceclofenac from Solid Dispersions containing PEG 6000
i) Physical Mixture (APG):
0
10
20
30
40
50
60
70
0 10 20 30 40 50 60 70
Time (minutes)
Per
cen
t D
rug
Rel
ease
APG1 APG2 APG3 APG4 APG5 APG6 APG7 APG8 APG9
Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com)
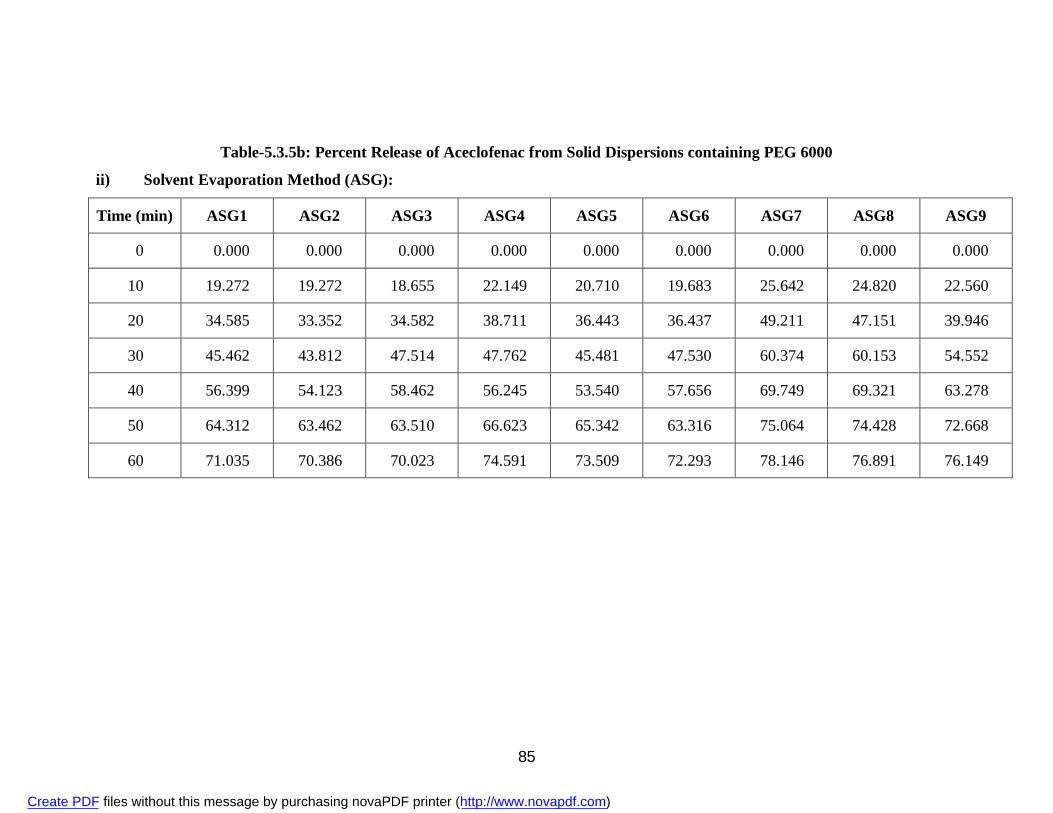
85
Table-5.3.5b: Percent Release of Aceclofenac from Solid Dispersions containing PEG 6000
ii) Solvent Evaporation Method (ASG):
Time (min) ASG1 ASG2 ASG3 ASG4 ASG5 ASG6 ASG7 ASG8 ASG9
0 0.000 0.000 0.000 0.000 0.000 0.000 0.000 0.000 0.000
10 19.272 19.272 18.655 22.149 20.710 19.683 25.642 24.820 22.560
20 34.585 33.352 34.582 38.711 36.443 36.437 49.211 47.151 39.946
30 45.462 43.812 47.514 47.762 45.481 47.530 60.374 60.153 54.552
40 56.399 54.123 58.462 56.245 53.540 57.656 69.749 69.321 63.278
50 64.312 63.462 63.510 66.623 65.342 63.316 75.064 74.428 72.668
60 71.035 70.386 70.023 74.591 73.509 72.293 78.146 76.891 76.149
Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com)
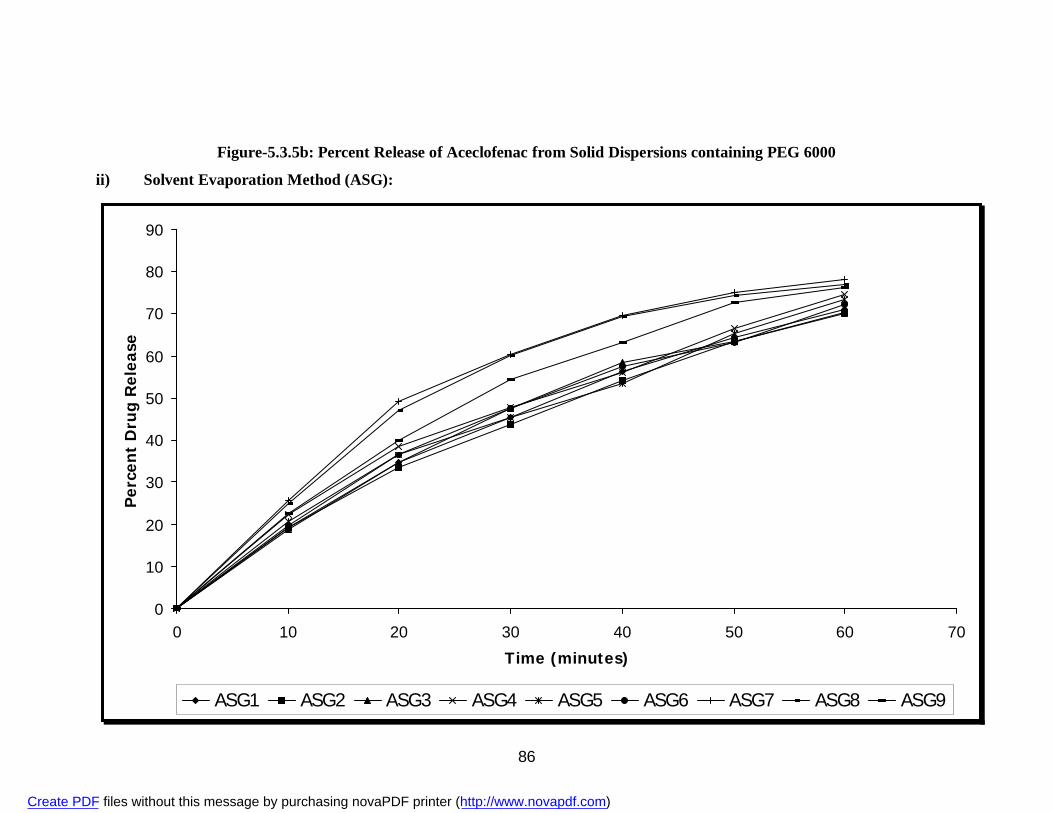
86
Figure-5.3.5b: Percent Release of Aceclofenac from Solid Dispersions containing PEG 6000
ii) Solvent Evaporation Method (ASG):
0
10
20
30
40
50
60
70
80
90
0 10 20 30 40 50 60 70
Time (minutes)
Per
cen
t D
rug
Rel
ease
ASG1 ASG2 ASG3 ASG4 ASG5 ASG6 ASG7 ASG8 ASG9
Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com)
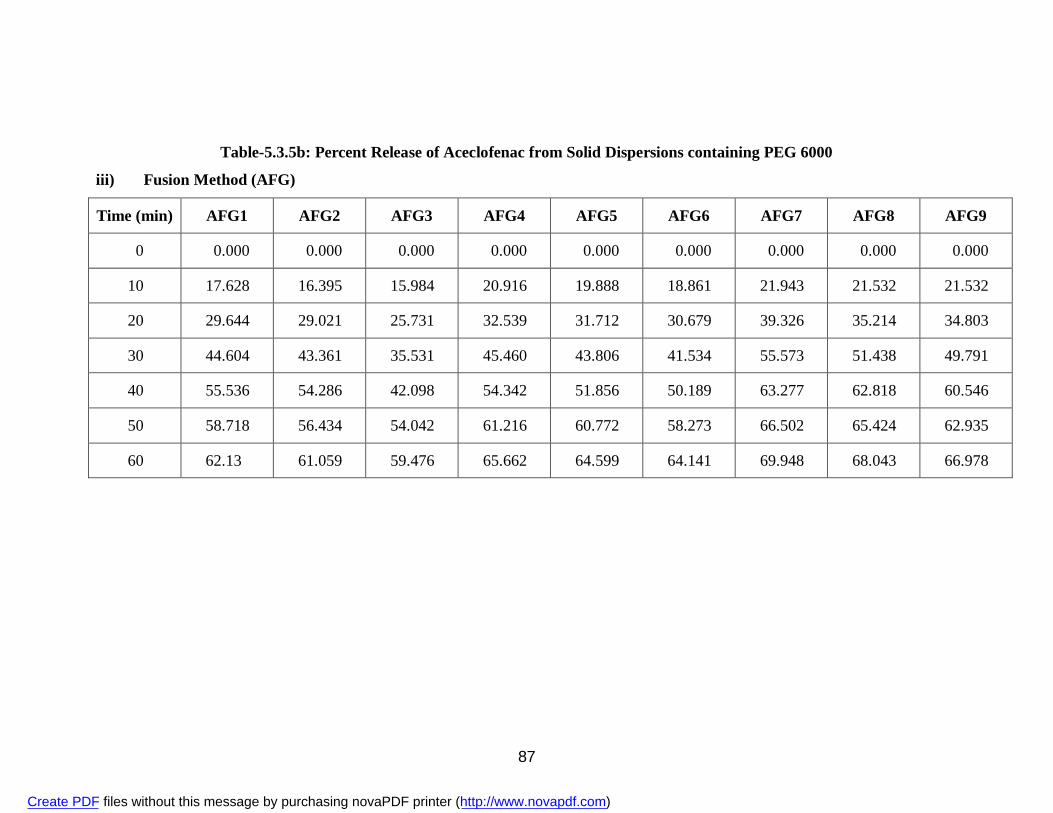
87
Table-5.3.5b: Percent Release of Aceclofenac from Solid Dispersions containing PEG 6000
iii) Fusion Method (AFG)
Time (min) AFG1 AFG2 AFG3 AFG4 AFG5 AFG6 AFG7 AFG8 AFG9
0 0.000 0.000 0.000 0.000 0.000 0.000 0.000 0.000 0.000
10 17.628 16.395 15.984 20.916 19.888 18.861 21.943 21.532 21.532
20 29.644 29.021 25.731 32.539 31.712 30.679 39.326 35.214 34.803
30 44.604 43.361 35.531 45.460 43.806 41.534 55.573 51.438 49.791
40 55.536 54.286 42.098 54.342 51.856 50.189 63.277 62.818 60.546
50 58.718 56.434 54.042 61.216 60.772 58.273 66.502 65.424 62.935
60 62.13 61.059 59.476 65.662 64.599 64.141 69.948 68.043 66.978
Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com)
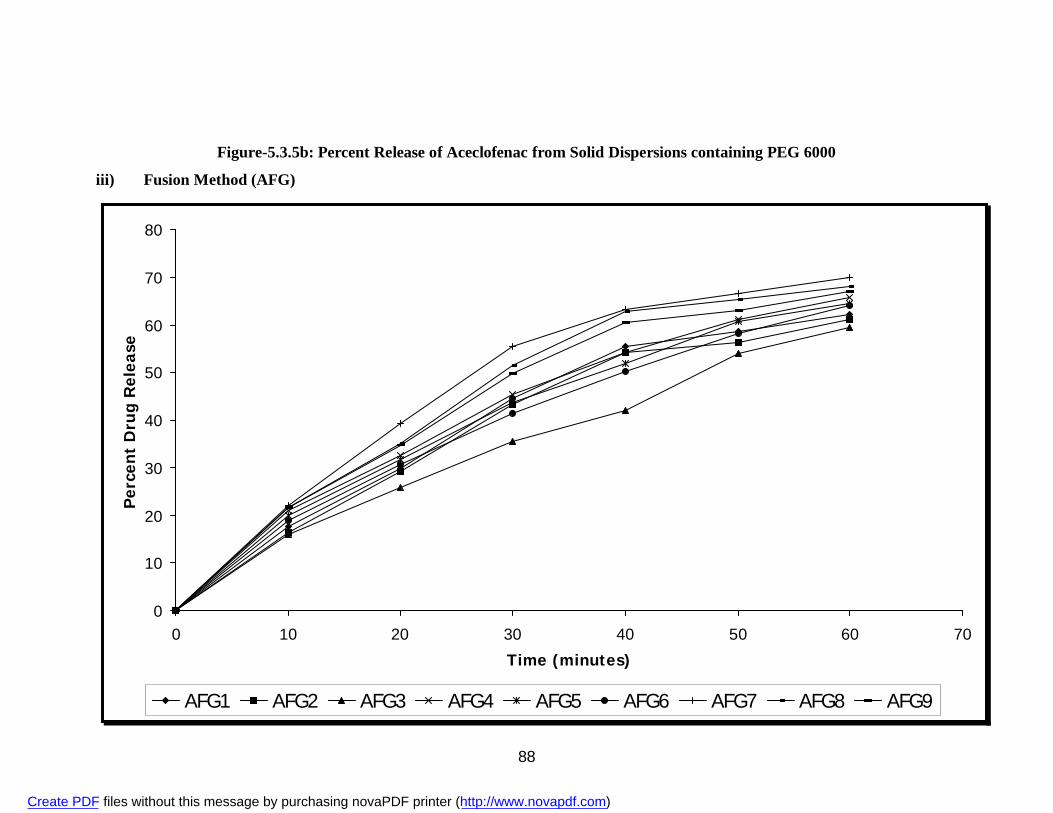
88
Figure-5.3.5b: Percent Release of Aceclofenac from Solid Dispersions containing PEG 6000
iii) Fusion Method (AFG)
0
10
20
30
40
50
60
70
80
0 10 20 30 40 50 60 70
Time (minutes)
Per
cen
t D
rug
Rel
ease
AFG1 AFG2 AFG3 AFG4 AFG5 AFG6 AFG7 AFG8 AFG9
Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com)
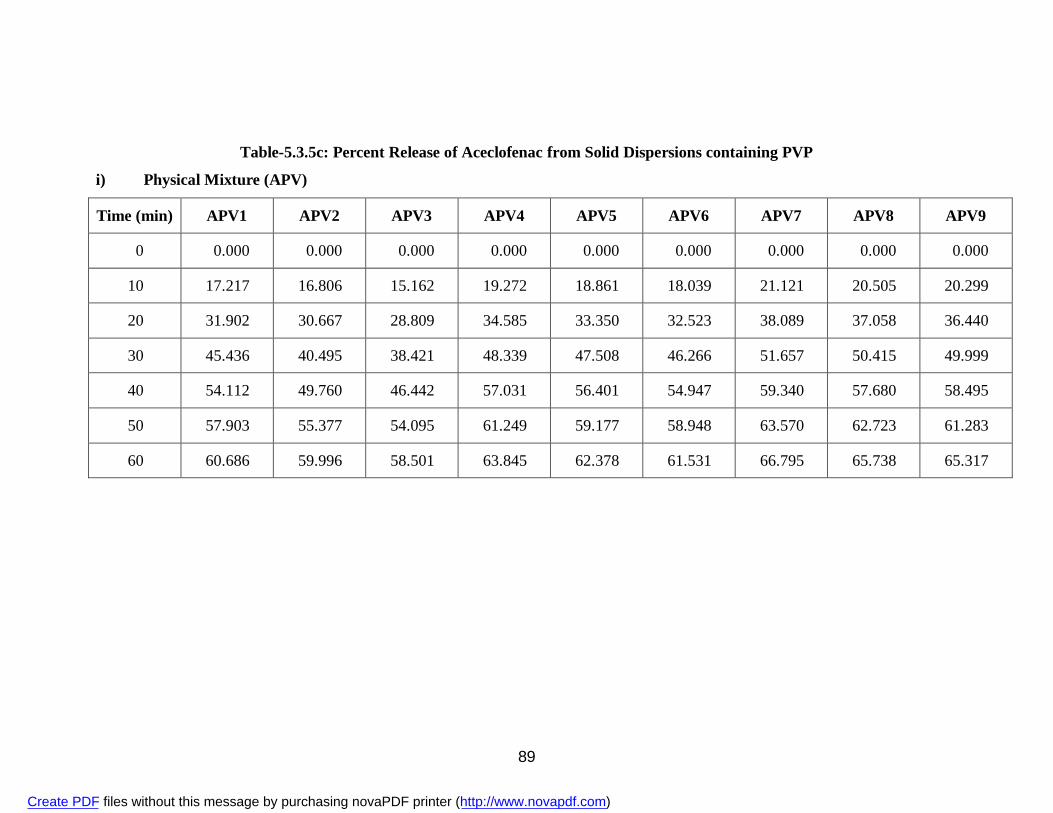
89
Table-5.3.5c: Percent Release of Aceclofenac from Solid Dispersions containing PVP
i) Physical Mixture (APV)
Time (min) APV1 APV2 APV3 APV4 APV5 APV6 APV7 APV8 APV9
0 0.000 0.000 0.000 0.000 0.000 0.000 0.000 0.000 0.000
10 17.217 16.806 15.162 19.272 18.861 18.039 21.121 20.505 20.299
20 31.902 30.667 28.809 34.585 33.350 32.523 38.089 37.058 36.440
30 45.436 40.495 38.421 48.339 47.508 46.266 51.657 50.415 49.999
40 54.112 49.760 46.442 57.031 56.401 54.947 59.340 57.680 58.495
50 57.903 55.377 54.095 61.249 59.177 58.948 63.570 62.723 61.283
60 60.686 59.996 58.501 63.845 62.378 61.531 66.795 65.738 65.317
Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com)
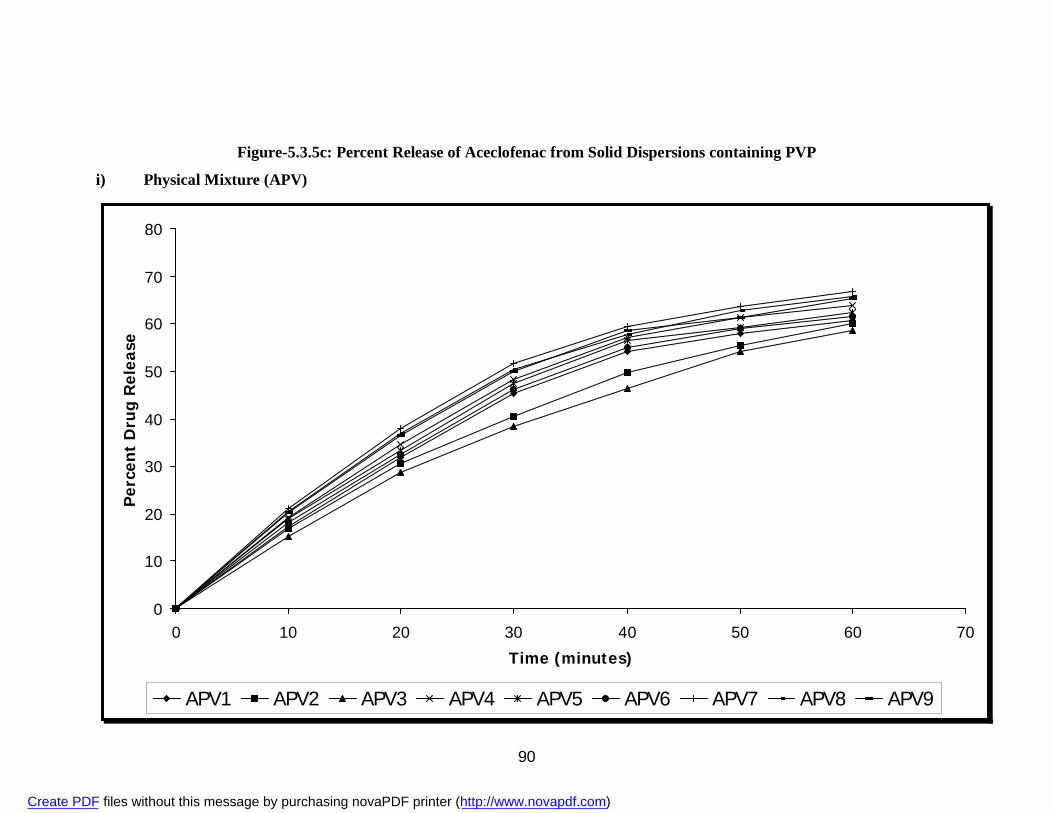
90
Figure-5.3.5c: Percent Release of Aceclofenac from Solid Dispersions containing PVP
i) Physical Mixture (APV)
0
10
20
30
40
50
60
70
80
0 10 20 30 40 50 60 70
Time (minutes)
Per
cen
t D
rug
Rel
ease
APV1 APV2 APV3 APV4 APV5 APV6 APV7 APV8 APV9
Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com)
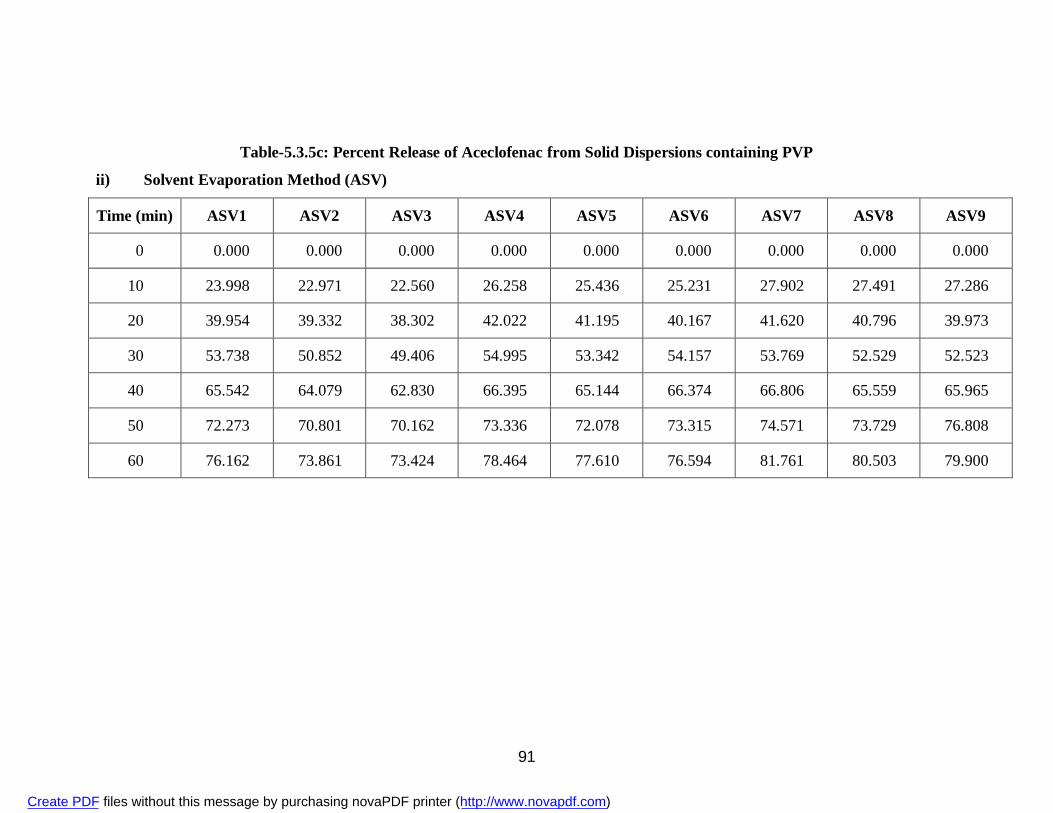
91
Table-5.3.5c: Percent Release of Aceclofenac from Solid Dispersions containing PVP
ii) Solvent Evaporation Method (ASV)
Time (min) ASV1 ASV2 ASV3 ASV4 ASV5 ASV6 ASV7 ASV8 ASV9
0 0.000 0.000 0.000 0.000 0.000 0.000 0.000 0.000 0.000
10 23.998 22.971 22.560 26.258 25.436 25.231 27.902 27.491 27.286
20 39.954 39.332 38.302 42.022 41.195 40.167 41.620 40.796 39.973
30 53.738 50.852 49.406 54.995 53.342 54.157 53.769 52.529 52.523
40 65.542 64.079 62.830 66.395 65.144 66.374 66.806 65.559 65.965
50 72.273 70.801 70.162 73.336 72.078 73.315 74.571 73.729 76.808
60 76.162 73.861 73.424 78.464 77.610 76.594 81.761 80.503 79.900
Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com)
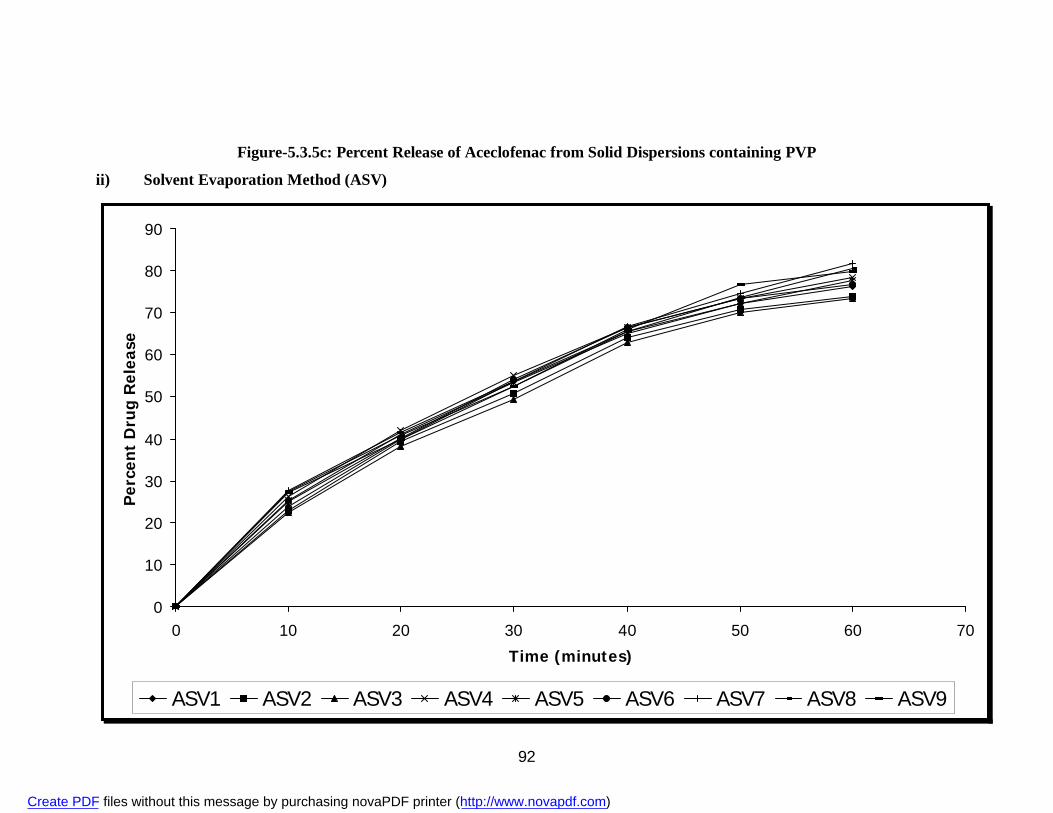
92
Figure-5.3.5c: Percent Release of Aceclofenac from Solid Dispersions containing PVP
ii) Solvent Evaporation Method (ASV)
0
10
20
30
40
50
60
70
80
90
0 10 20 30 40 50 60 70
Time (minutes)
Per
cen
t D
rug
Rel
ease
ASV1 ASV2 ASV3 ASV4 ASV5 ASV6 ASV7 ASV8 ASV9
Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com)
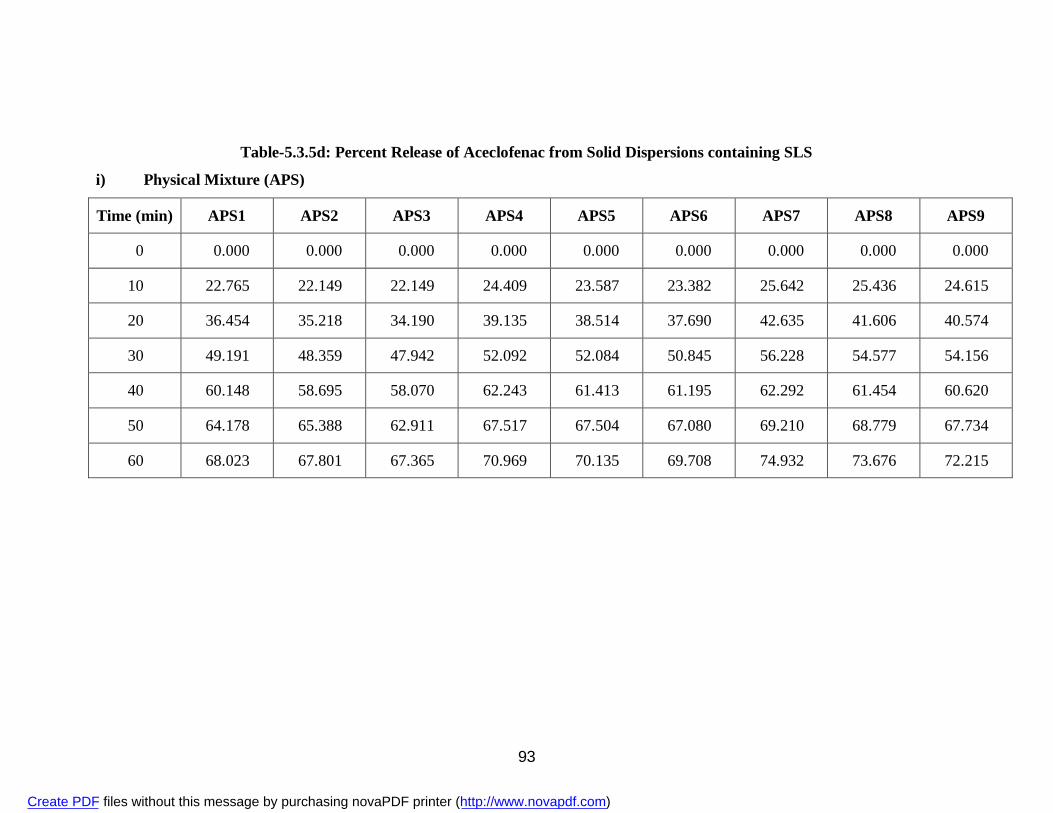
93
Table-5.3.5d: Percent Release of Aceclofenac from Solid Dispersions containing SLS
i) Physical Mixture (APS)
Time (min) APS1 APS2 APS3 APS4 APS5 APS6 APS7 APS8 APS9
0 0.000 0.000 0.000 0.000 0.000 0.000 0.000 0.000 0.000
10 22.765 22.149 22.149 24.409 23.587 23.382 25.642 25.436 24.615
20 36.454 35.218 34.190 39.135 38.514 37.690 42.635 41.606 40.574
30 49.191 48.359 47.942 52.092 52.084 50.845 56.228 54.577 54.156
40 60.148 58.695 58.070 62.243 61.413 61.195 62.292 61.454 60.620
50 64.178 65.388 62.911 67.517 67.504 67.080 69.210 68.779 67.734
60 68.023 67.801 67.365 70.969 70.135 69.708 74.932 73.676 72.215
Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com)