Embed Size (px)
Citation preview

SYSTEMIC VASCULITIDES
Small vessel vasculitidesLorraine Harper
Frauke Weidanz
AbstractSmall vessel vasculitis may be associated with immune complex disease,
such as Henoch Schonlein purpura or systemic lupus erythematosus, or
primary such as Wegener’s granulomatosis, microscopic polyangiitis, and
renal limited vasculitis. These primary conditions are associated with cir-
culating anti-neutrophil cytoplasm antibodies and are the most common
cause of rapidly progressive glomerulonephritis. This review describes
recent advances in the understanding of the pathogenesis underlying
these conditions and reviews common presentations. Consideration is
given to recent clinical trials in the management of anti-neutrophil cyto-
plasm antibodies (ANCA)-associated vasculitides.
Keywords anti-neutrophil cytoplasm antibodies (ANCA); microscopic
polyangiitis; therapy; vasculitis; Wegener’s granulomatosis
Primary small vessel vasculitis includes Wegener’s granulo-matosis, microscopic polyangiitis and Churg–Strauss syndrome, and is commonly associated with anti-neutrophil cytoplasm antibodies (ANCA). The incidence of these diseases is increasing; more than 20/million population are affected. They can occur at any age, including in children, but are most common in elderly patients (peak age 55–70 years). They predominantly affect Caucasians, with no difference between the sexes. Management of these diseases is challenging. Rapid diagnosis is essential to reduce the permanent scarring caused by vasculitis and death from pulmonary haemorrhage and renal failure, but there is often considerable delay because of the multiple and nonspecific manifestations. If untreated, these conditions are fatal.
Henoch–Schönlein purpura is a related disorder that should be differentiated from ANCA-associated vasculitis (see below). It is the commonest vasculitis in children.
Lorraine Harper PhD MRCP is Senior Lecturer and Honorary Consultant
Physician in Nephrology at the University of Birmingham and Queen
Elizabeth Hospital, Birmingham, UK. She qualified from the University
of Edinburgh, and trained in renal medicine in Birmingham. Her
research interests include the pathogenesis and management of
primary small vessel vasculitides. Competing interests: none declared.
Frauke Weidanz MBBS Dr med is an Academic Foundation Year Two
trainee at the University Hospital Birmingham NHS Trust. She qualified
from Freiburg University in Germany. Competing interests: none
declared.
MEDICINE 34:11 456
Secondary vasculitides include leucocytoclastic vasculitis, rheumatoid vasculitis and systemic lupus erythematosus (SLE) vasculitis.
Primary small vessel vasculitis
Aetiology and pathogenesisThe aetiology of primary small vessel vasculitis is unknown. However, significant advances have been made in understanding disease pathogenesis. The strong clinical association with ANCA suggests an autoimmune disease and that these antibodies are directly pathogenic.1 ANCA correlate closely with clinical activity. The autoantibodies activate cytokine primed neutrophils and monocytes, which express the ANCA antigens proteinase 3 (PR3) and myeloperoxidase (MPO) on the cell surface. Neutrophils respond by generating a respiratory burst, degranulation and secreting pro-inflammatory cytokines. Endothelial cells are also important in localizing inflammation. These cells are important for maintaining an anticoagulant and anti-inflammatory envi-ronment. In response to cytokines, endothelial cells take on an activated phenotype with enhanced expression of adhesion molecules that allow interaction with circulating leukocytes and release factors promoting thrombosis. ANCA activation of neutrophils in this environment promotes stationary adhesion to endothelial cells and transmigration, releasing inflammatory factors in the wrong place and, therefore, promoting endothelial damage.
Animal models support the view that these autoantibodies are pathogenic.2,3 In one model, MPO knockout mice were immu-nized with murine MPO. Anti-MPO antibodies, purified from the serum of immunized animals, were transferred to wild-type mice that express MPO. The recipient mice developed pauci-immune focal necrotizing crescentic nephritis, demonstrating that anti-MPO antibodies alone were sufficient to cause disease. No good model of granulomatous inflammation exists but passive transfer of anti-PR3 antibodies into a mouse exacerbates local cutaneous inflammation following tumour necrosis factor (TNF) administration.4
Activation of neutrophils and endothelial cells is important for the early development of vasculitic lesions and progression is
• Recent evidence has shown that ANCA tested by immunofluorescence and ELISA are sensitive and specific markers of primary systemic vasculitis
• In the management of primary systemic vasculitis, azathioprine is as effective as cyclophosphamide in maintaining remission, with fewer side-effects
• Etanercept is not useful in the treatment of ANCA- associated vasculitis
• Methotrexate may be used for induction therapy in patients with limited ANCA associated vasculitis but is associated with more relapses than cyclophosphamide
What’s new?
© 2006 Elsevier Ltd. All rights reserved.

SYSTEMIC VASCULITIDES
accompanied by recruitment of T cells and monocytes. Several studies have suggested that T cells can proliferate to MPO or PR3 and that they remain activated despite disease remission, sug-gesting that T cells contribute to the relapsing–remitting nature of ANCA-associated vasculitis.
Environmental and genetic factors also influence disease pathogenesis. Autoimmune diseases are often associated with major histocompatibility antigens; however, no consistent asso-ciations are reported with primary small vessel vasculitis. There is an association between a1-antitrypsin deficiency (the main inhibitor of proteinase 3) and Wegener’s granulomatosis. Patients carrying the main deficiency allele, PiZ, have more severe dis-ease and a poor prognosis. However, a1-antitrypsin deficiency per se is not sufficient to induce vasculitis, suggesting that it acts as a ‘second-hit’ amplifier of inflammation. Neutrophil surface expression of PR3 is also genetically determined and individuals expressing large amounts of proteinase 3 on the neutrophil sur-face are also more likely to develop disease.5
Drug exposure may also precipitate ANCA-associated vascu-litis, including propylthiouracil, minocycline and penicillamine. Infectious agents have often been implicated as initiators of vas-culitis and nasal carriage of Staphylococcus aureus has been asso-ciated with relapse in Wegener’s granulomatosis. Silica exposure, which may also result in granuloma formation, is associated with an increased risk of ANCA-associated vasculitis.
PathologySmall vessel ANCA-associated vasculitides are characterized by inflammation and necrosis of capillaries, arterioles and venules, but may also affect larger vessels. In the kidney, the process primarily affects the glomeruli, leading to focal segmen-tal necrotizing glomerulonephritis with crescent formation with-out immunoglobulin deposition; this is termed ‘pauci-immune glomerulonephritis’. There is often an associated interstitial inflammation. Within the kidney there are no absolute differences in pathology between microscopic polyangiitis and Wegener’s granulomatosis, although patients with microscopic polyangiitis tend to have more chronic lesions. In the lung, the findings are usually of capillaritis, often associated with lung haemorrhage.
Granulomatous lesions occur in Wegener’s granulomatosis and Churg–Strauss syndrome but not in microscopic polyangiitis. In the lung, there are often large, ill-defined collections of inflam-matory cells near affected vessels; these may present as cavitating nodules. In the upper airways, this granulomatous reaction may present as ulceration. Renal granulomata are probably whole necrotic glomeruli.
Clinical featuresClinical differentiation between Wegener’s granulomatosis and microscopic polyangiitis is often difficult. However, this distinction is not important clinically, because their treatment and prognosis are similar. Systemic non-specific symptoms such as malaise, flu-like symptoms and weight loss are common and may pre-date other symptoms.6
Wegener’s granulomatosisLimited disease – respiratory tract symptoms are common and
may be present in 90% of patients at the time of diagnosis. Upper respiratory tract symptoms include sinusitis, epistaxis, otitis
MEDICINE 34:11 45
media and hoarseness. Complications of granulomatous inflam-mation may cause mucosal ulceration and nasal septal perfora-tion with a saddle nose. Subglottic stenosis occurs in up to 16% of adults and 48% of children. It often becomes fixed and irre-versible and can cause life-threatening upper airways obstruction. This so-called limited disease may have an indolent course for many years before transforming into systemic disease.
Systemic Wegener’s granulomatosis is characterized by: • upper respiratory tract involvement • pulmonary disease • renal involvement with glomerulonephritis • ANCA directed against the neutrophil enzyme proteinase 3. Typically, there are granulomata and small vessel vasculitis. Patients often present initially with limited upper airways disease with granulomatous inflammation, though presentation with a fulminating life-threatening illness is not uncommon.
Microscopic polyangiitis is characterized by : • Renal disease • Pulmonary disease • ANCA directed against myeloperoxidase.
Specific organ involvement may occur in Wegener’s granulomatosis or microscopic polyangiitis
Pulmonary involvement is less common at presentation (45–70%), but occurs in 85% of patients with Wegener’s granu-lomatosis at some stage. Presentation may be with asymptomatic pulmonary infiltrates or cough, haemoptysis, pleuritis or dys-pnoea. Life-threatening alveolar haemorrhage may occur and is associated with poor prognosis. Pulmonary disease is less com-mon in microscopic polyangiitis but 30% of patients will have pulmonary haemorrhage.
Renal disease is characterized by necrotizing small vessel vasculitis associated with no significant immune deposits (pauci-immune) and can range from an active urinary sediment to rap-idly progressive glomerulonephritis with severe damage. Renal involvement in microscopic polyangiitis is invariable. Hyper-tension occurs in 30% of patients. So-called idiopathic rapidly progressive glomerulonephritis, without immune deposits and with no systemic features of vasculitis, is now considered part of the spectrum of microscopic polyangiitis. Renal involvement is not a common presentation (18%) in patients with Wegener’s granulomatosis, but up to 77% of patients will develop glomerulonephritis.
Other organs – ocular involvement is common and may pres-ent as conjunctivitis, scleritis or uveitis. In those with granulo-matous inflammation, proptosis may occur and is commonly associated with extensive sinus disease. Optic nerve vasculitis and retinal artery thrombosis are rare but important complica-tions. Loss of sight has been reported in 8% of patients. Myalgia and arthralgia are common. Non-erosive arthritis occurs in up to 28% of patients. Skin disease may manifest as palpable purpura, ulcers and subcutaneous nodules, and occurs in up to 50% of patients. The heart (pericarditis, coronary arteritis), nervous sys-tem (mononeuritis multiplex, peripheral neuropathy) and gastro-intestinal tract (haemorrhagic ulceration, bowel perforation) may also be involved.
Churg–Strauss syndrome is characterized by hypereosinophilia with tissue infiltration, formation of granuloma and vasculitis.
7 © 2006 Elsevier Ltd. All rights reserved.
SYSTEMIC VASCULITIDES
It is less common than Wegener’s granulomatosis and micro-scopic polyangiitis; the estimated incidence is 2.4/million. Aeti-ology is unknown although there have been several reports of the syndrome occurring during leukotriene antagonist therapy for chronic asthma.
Allergic rhinitis and/or asthma precede the development of vasculitis, often associated with nonspecific symptoms. Asthma is often more severe in the weeks preceding vasculi-tis. A rash and mononeuritis multiplex are common presenting features. Pulmonary involvement includes dyspnoea, alveolar haemorrhage or pleurisy and is associated with nonspecific pulmonary infiltrates on chest radiography. Cardiac involve-ment, manifest by myocarditis with eosinophilic infiltration, coronary artery vasculitis and myocardial infarct, carries a poor prognosis and is often a late manifestation. Gastrointesti-nal involvement can present in various ways with pain, diar-rhoea and ascites. Renal involvement is not uncommon (up to 50%); it is usually mild.
InvestigationsRoutine laboratory investigations are generally nonspecific.
Common abnormalities include leucocytosis, thrombocytosis (>400,000/mm3), normochromic, normocytic anaemia and eleva-tion of inflammatory markers including erythrocyte sedimentation rate (ESR) and C-reactive protein (CRP). Churg–Strauss syndrome is usually associated with blood eosinophilia (>1.5 × 109/litre) and elevated IgE levels.
Urinalysis is a sensitive marker of renal involvement. An active urinary sediment with red blood cells and casts indicates glomerular disease. Proteinuria is often present. Elevated urea and creatinine levels are common, though active renal disease may be present even if biochemical indices are normal.
ANCA are a useful marker for diagnosis. Wegener’s granulo-matosis is associated with ANCA directed against proteinase 3 (PR3-ANCA), giving a granular cytoplasmic staining pattern on immunofluorescence. This test is highly sensitive (>95%) and specific (>99%) in patients with active systemic disease. MPO-ANCA are found in microscopic polyangiitis (70%), but PR3-ANCA can also occur. MPO-ANCA give a perinuclear staining pattern on immunofluorescence. Of patients with Churg–Strauss syndrome, 50% are positive for MPO-ANCA. The positive predic-tive value of a positive test for ANCA is, however, very depen-dent on the clinical situation.
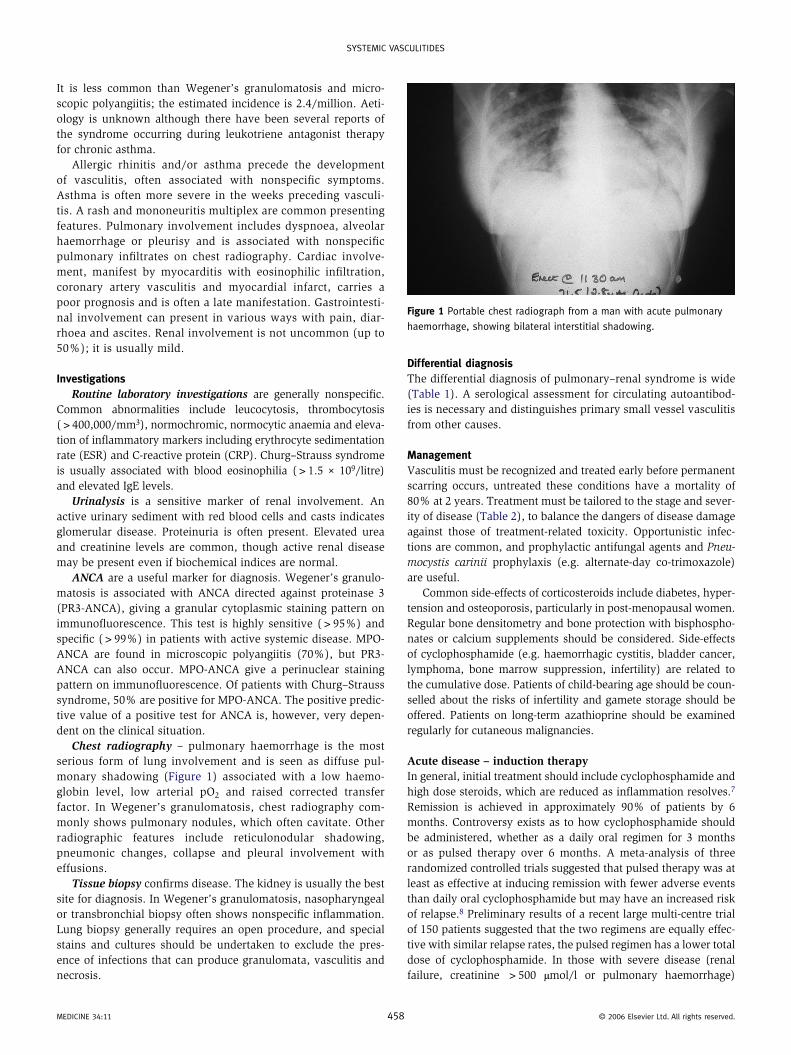
Chest radiography – pulmonary haemorrhage is the most serious form of lung involvement and is seen as diffuse pul-monary shadowing (Figure 1) associated with a low haemo-globin level, low arterial pO2 and raised corrected transfer factor. In Wegener’s granulomatosis, chest radiography com-monly shows pulmonary nodules, which often cavitate. Other radiographic features include reticulonodular shadowing, pneumonic changes, collapse and pleural involvement with effusions.
Tissue biopsy confirms disease. The kidney is usually the best site for diagnosis. In Wegener’s granulomatosis, nasopharyngeal or transbronchial biopsy often shows nonspecific inflammation. Lung biopsy generally requires an open procedure, and special stains and cultures should be undertaken to exclude the pres-ence of infections that can produce granulomata, vasculitis and necrosis.
MEDICINE 34:11 45
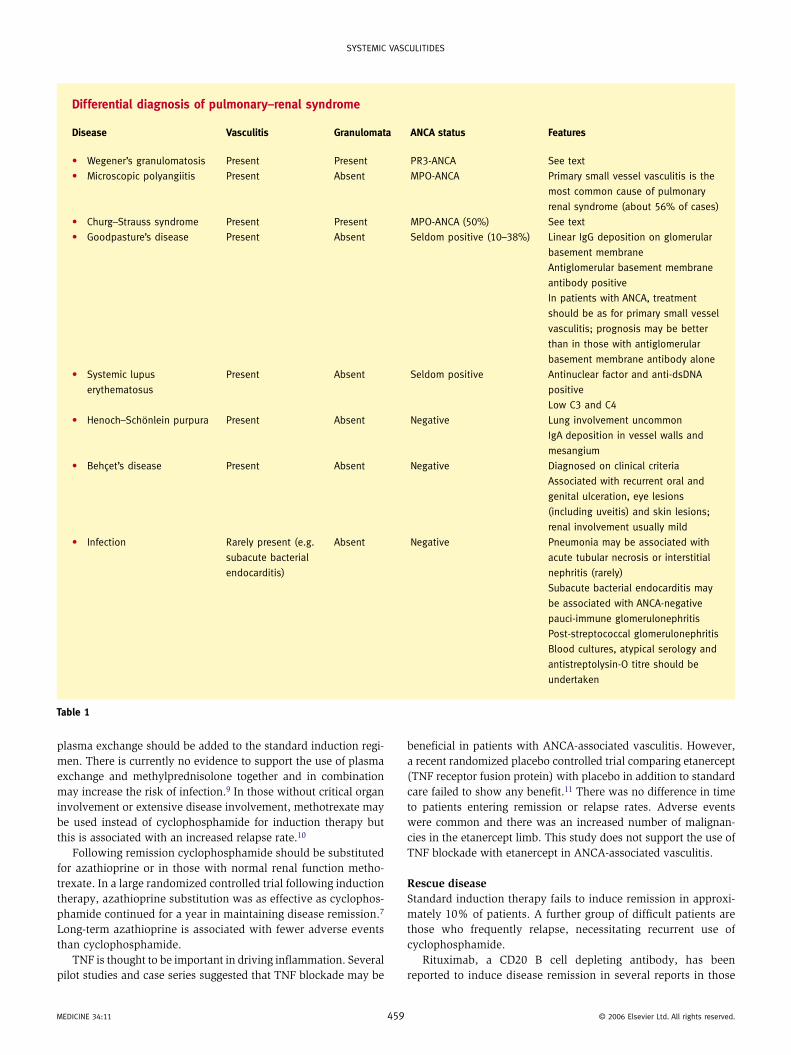
Differential diagnosisThe differential diagnosis of pulmonary–renal syndrome is wide (Table 1). A serological assessment for circulating autoantibod-ies is necessary and distinguishes primary small vessel vasculitis from other causes.
ManagementVasculitis must be recognized and treated early before permanent scarring occurs, untreated these conditions have a mortality of 80% at 2 years. Treatment must be tailored to the stage and sever-ity of disease (Table 2), to balance the dangers of disease damage against those of treatment-related toxicity. Opportunistic infec-tions are common, and prophylactic antifungal agents and Pneu-mocystis carinii prophylaxis (e.g. alternate-day co-trimoxazole) are useful.
Common side-effects of corticosteroids include diabetes, hyper-tension and osteoporosis, particularly in post-menopausal women. Regular bone densitometry and bone protection with bisphospho-nates or calcium supplements should be considered. Side-effects of cyclophosphamide (e.g. haemorrhagic cystitis, bladder cancer, lymphoma, bone marrow suppression, infertility) are related to the cumulative dose. Patients of child-bearing age should be coun-selled about the risks of infertility and gamete storage should be offered. Patients on long-term azathioprine should be examined regularly for cutaneous malignancies.
Acute disease – induction therapyIn general, initial treatment should include cyclophosphamide and high dose steroids, which are reduced as inflammation resolves.7 Remission is achieved in approximately 90% of patients by 6 months. Controversy exists as to how cyclophosphamide should be administered, whether as a daily oral regimen for 3 months or as pulsed therapy over 6 months. A meta-analysis of three randomized controlled trials suggested that pulsed therapy was at least as effective at inducing remission with fewer adverse events than daily oral cyclophosphamide but may have an increased risk of relapse.8 Preliminary results of a recent large multi-centre trial of 150 patients suggested that the two regimens are equally effec-tive with similar relapse rates, the pulsed regimen has a lower total dose of cyclophosphamide. In those with severe disease (renal failure, creatinine >500 μmol/l or pulmonary haemorrhage)
Figure 1 Portable chest radiograph from a man with acute pulmonary
haemorrhage, showing bilateral interstitial shadowing.
8 © 2006 Elsevier Ltd. All rights reserved.
SYSTEMIC VASCULITIDES
Differential diagnosis of pulmonary–renal syndrome
Disease Vasculitis Granulomata ANCA status Features
• Wegener’s granulomatosis Present Present PR3-ANCA See text
• Microscopic polyangiitis Present Absent MPO-ANCA Primary small vessel vasculitis is the
most common cause of pulmonary
renal syndrome (about 56% of cases)
• Churg–Strauss syndrome Present Present MPO-ANCA (50%) See text
• Goodpasture’s disease Present Absent Seldom positive (10–38%) Linear IgG deposition on glomerular
basement membrane
Antiglomerular basement membrane
antibody positive
In patients with ANCA, treatment
should be as for primary small vessel
vasculitis; prognosis may be better
than in those with antiglomerular
basement membrane antibody alone
• Systemic lupus
erythematosus
Present Absent Seldom positive Antinuclear factor and anti-dsDNA
positive
Low C3 and C4
• Henoch–Schönlein purpura Present Absent Negative Lung involvement uncommon
IgA deposition in vessel walls and
mesangium
• Behçet’s disease Present Absent Negative Diagnosed on clinical criteria
Associated with recurrent oral and
genital ulceration, eye lesions
(including uveitis) and skin lesions;
renal involvement usually mild
• Infection Rarely present (e.g.
subacute bacterial
endocarditis)
Absent Negative Pneumonia may be associated with
acute tubular necrosis or interstitial
nephritis (rarely)
Subacute bacterial endocarditis may
be associated with ANCA-negative
pauci-immune glomerulonephritis
Post-streptococcal glomerulonephritis
Blood cultures, atypical serology and
antistreptolysin-O titre should be
undertaken
Table 1
plasma exchange should be added to the standard induction regi-men. There is currently no evidence to support the use of plasma exchange and methylprednisolone together and in combination may increase the risk of infection.9 In those without critical organ involvement or extensive disease involvement, methotrexate may be used instead of cyclophosphamide for induction therapy but this is associated with an increased relapse rate.10
Following remission cyclophosphamide should be substituted for azathioprine or in those with normal renal function metho-trexate. In a large randomized controlled trial following induction therapy, azathioprine substitution was as effective as cyclophos-phamide continued for a year in maintaining disease remission.7 Long-term azathioprine is associated with fewer adverse events than cyclophosphamide.
TNF is thought to be important in driving inflammation. Several pilot studies and case series suggested that TNF blockade may be
MEDICINE 34:11 45
beneficial in patients with ANCA-associated vasculitis. However, a recent randomized placebo controlled trial comparing etanercept (TNF receptor fusion protein) with placebo in addition to standard care failed to show any benefit.11 There was no difference in time to patients entering remission or relapse rates. Adverse events were common and there was an increased number of malignan-cies in the etanercept limb. This study does not support the use of TNF blockade with etanercept in ANCA-associated vasculitis.
Rescue diseaseStandard induction therapy fails to induce remission in approxi-mately 10% of patients. A further group of difficult patients are those who frequently relapse, necessitating recurrent use of cyclophosphamide.
Rituximab, a CD20 B cell depleting antibody, has been reported to induce disease remission in several reports in those
9 © 2006 Elsevier Ltd. All rights reserved.

SYSTEMIC VASCULITIDES
Management of primary small vessel vasculitis
Induction therapy • Continued for 3 months following remission
• Prednisolone, 1 mg/kg, maximum dose 80 mg; rapid reduction in corticosteroid dose –
50% over 2 weeks and to a dose of 0.25 mg/kg at week 8
• Cyclophosphamide, 2 mg/kg/day p.o., maximum dose 200 mg; age > 60 years reduce by
25%, age > 70 years reduce by 50%
• Pulsed cyclophosphamide can also be given – 10 pulses over 25 weeks, 15 mg/kg i.v.;
dose reductions must be made for age and creatinine
• WBC count must be checked between day 10 and day 14 following pulse because of
decrease in leucocyte count; if < 3 × 109/litre, a suitable dose reduction must be made
for the next pulse
Adjuvant therapy for life-threatening disease • Life-threatening disease includes creatinine > 500 μmol/litre and/or pulmonary haemorrhage
• Plasma exchange should be considered
Maintenance therapy • Prednisolone, 5–10 mg/day
• Azathioprine, 1.5 mg/kg/day, maximum dose 200 mg
• Methotrexate 20–25mg/week (contraindicated in patients with creatinine > 170 μmol/litre)
• Alternatives to azathioprine include mycophenolate mofetil, 1g b.d.,
• Consider addition of co-trimoxazole
Relapse therapy • Major relapse – return to initial induction therapy
• Minor relapse – increase corticosteroid dose
Rescue therapy for relapsing and refractory
disease
• Standard induction therapy fails to induce remission in 10% of patients; patients who
frequently relapse necessitating recurrent use of cyclophosphamide are also a difficult
group
• Newer therapies that may prove beneficial include B cell depletion with rituximab,
polyclonal antithymocyte globulin, deoxyspergualin and other newer immunosuppressant
drugs that have shown promise in organ transplantation
Table 2
with refractory disease. In some series, there was a reduction in ANCA without a fall in total immunoglobulin levels although the mechanism of this is controversial. Disease may recur with B cell repletion and some subsets of disease may be less responsive (e.g. chronic granulomatous disease). Rituximab was licensed for treatment of non-Hodgkins lymphoma and has a good safety and tolerability profile. It has shown promise in many autoim-mune diseases and a randomized controlled study in patients with ANCA-vasculitis has been commenced.
Other therapies in small series have also suggested benefit in refractory patients. Anti-thymocyte globulin is a T cell depleting polyclonal antibody directed against surface antigens of activated T cells and deoxyspergualin (an immunosuppressant success-fully used in renal allograft rejection) have both shown benefit including in patients with granulomatous disease. Side effects are not uncommon with these treatments and should not be used routinely.
Churg–StraussSteroids are the mainstay of treatment in Churg–Strauss syn-drome; however, cyclophosphamide should be added in those with severe disease. Disease severity can be assessed using the five factor score (1 point for each: creatinine >140 μmol/l, pro-teinuria >1 g, severe GI involvement, cardiomyopathy and CNS signs). Cyclophosphamide improves survival in those with a five factor score of more than 2.12
MEDICINE 34:11 46
Monitoring during treatment – regular white blood cell (WBC) monitoring is essential. Patients should undergo blood tests twice weekly in the first month, on alternate weeks in the second month and monthly thereafter. If the WBC count falls below 4 x 109/litre, cyclophosphamide or azathioprine should be discontinued and restarted at a lower dose once the count has recovered. An urgent WBC count should be performed in patients with signs of infection. Allopurinol (a xanthine oxidase inhibitor) is contraindicated in those taking azathioprine, because of the high risk of bone marrow suppression.
Relapse – it is unclear how long maintenance immuno- suppressive therapy should be continued; most physicians rec-ommend at least 2 years. Relapse (30–50% over 3–5 years) can occur at any time, and patients should be reviewed regularly (3- monthly) to detect early signs of disease recurrence and drug toxicity. Relapse occurs more commonly in Wegener’s granu-lomatosis and those who remain ANCA positive. ANCA test-ing should be performed at each review. Indicators of relapse include: • recurrence of symptoms (these may differ from the original
presentation) • rising inflammatory markers (CRP, ESR) • recurrence of or rising ANCA titre (patients who remain ANCA
positive may be at greater risk of relapse) • reappearance of haematuria and/or granular casts in urine • increased serum creatinine.
0 © 2006 Elsevier Ltd. All rights reserved.

SYSTEMIC VASCULITIDES
Treatment depends on the severity of the relapse. Induction ther-apy as described above should be re-started with a major relapse and an increase in steroids with a minor relapse. Escalation of therapy should not be based solely on a rising ANCA titre. ANCA levels generally correlate with clinical activity, but the positive predictive value of an increasing ANCA level varies (20–80%) depending on the study.
PrognosisSurvival is 70–80% at 5 years. Early mortality often results from opportunistic infection. Poor prognostic indicators include greater age, pulmonary haemorrhage and severe renal disease. End-stage renal disease occurs in 20–25% of patients. With improved survival, long-term damage from disease and drug toxicity is a major problem.
Henoch–Schönlein purpura
Henoch–Schönlein purpura is a systemic vasculitis characterized by IgA deposition. It is most common in children, often occurring before the age of 5 years, though it can occur at any age. The aetiology is unknown, but the condition commonly follows an upper respiratory infection.
Henoch–Schönlein purpura is characterized by: • rash, usually affecting the buttocks and lower limbs (100%)
(Figure 2) • arthralgia (75%) • gastrointestinal involvement, with abdominal pain and bloody
diarrhoea (30–40%) • glomerulonephritis (50%), often more severe with increasing
age. Other organs, including the CNS (seizures) and lung (pulmonary haemorrhage), may also be involved.
Diagnosis is based on clinical symptoms and confirmed by tissue biopsy. Skin biopsy reveals leucocytoclastic vasculitis with IgA deposition in the blood vessels or dermo-epidermal junction. Renal histology may vary from mild mesangial proliferation to
Figure 2 This purpuric rash has coalesced to form necrotic patches.
The differential diagnosis includes primary small vessel vasculitis and
Henoch-Schönlein purpura. The rash associated with Henoch-Schönlein
purpura usually involves the buttocks and extensor aspects of the
lower limbs. It usually resolves within 1 month, but fresh crops often
appear.
MEDICINE 34:11 461
focal segmental necrotizing glomerulonephritis, but always shows IgA deposition in the mesangium. Renal biopsy should be performed in patients with proteinuria and haematuria, because the severity of renal involvement (particularly the number of crescents) is important for prognosis.
Management – Henoch–Schönlein purpura is usually self- limiting and requires only symptomatic relief. Immunosuppres-sants should be considered in patients with renal involvement, particularly those with severe disease. Uncontrolled studies have suggested benefit in patients with poor prognostic indicators, though no randomized controlled studies have been performed.
Prognosis – in most patients, the prognosis is excellent. In those with renal involvement, however, damage may persist and progress without signs of active disease. The course of the renal lesion is often worse in older patients. Relapses are common, particularly in patients with nephritis.
Secondary vasculitides
Leucocytoclastic vasculitisLeucocytoclastic vasculitis is also known as cutaneous vas-culitis or hypersensitivity vasculitis, and is often part of a systemic disorder (Table 3). The most common cause is drug related, the penicillins, cephalosporins, sulphonamides (including most loop and thiazide-type diuretics), phenytoin and allopurinol have been most often implicated. The most common cutaneous lesions include palpable purpura and atypical urticarial lesions. Livedo reticularis, ulcerations and necrosis also occur.
Pathology – leucocytoclastic vasculitis is characterized by fibrinoid necrosis of the vessel walls with infiltration of neutrophils, some of which are disrupted, resulting in the presence of nuclear debris within the tissue. With repair mechanisms, a mononuclear infiltrate may develop and become evident when biopsies are taken late.
Investigations – patients who present with leucocytoclastic vasculitis should be investigated to find a cause and to assess the presence and severity of systemic disease. The diagnosis is often suggested by the clinical findings and the history of an offend-ing drug or infection, and will influence other investigations. Patients in whom there is no obvious cause should be screened for systemic disease (Table 4).
Systemic disease associated with leucocytoclastic vasculitis
• Other vasculitic syndromes – Wegener’s granulomatosis,
microscopic polyangiitis, Henoch–Schönlein purpura
• Autoimmune rheumatic disorders – rheumatoid arthritis,
systemic lupus erythematosus, dermatomyositis
• Infections – hepatitis B and C (often from cryoglobulins),
HIV, streptococci, subacute bacterial endocarditis
• Paraproteins – cryoglobulinaemia, Waldenström’s
macroglobulinaemia
• Others – inflammatory bowel disease, neoplasia,
a1-antitrypsin deficiency
Table 3
© 2006 Elsevier Ltd. All rights reserved.

SYSTEMIC VASCULITIDES
Management depends on the presence of systemic involve-ment and the diagnosis. Symptomatic relief may be adequate in patients with isolated cutaneous involvement, though oth-ers may require immunosuppression. The need for treatment should be weighed against the toxicity of immunosuppres-sive therapies. Antihistamines and colchicine have also been successful.
Rheumatoid vasculitisRheumatoid vasculitis is a small vessel vasculitis, though medium-sized vessels may also be involved. It typically presents in patients with severe destructive rheumatoid arthritis (RA) and rheumatoid nodules, and is associated with high rheumatoid fac-tor levels. Clinically evident vasculitis occurs in 2–5% of patients; subclinical disease is more common.
Clinical features – cutaneous disease manifested as purpura is the most common symptom of rheumatoid vasculitis. Vascu-litic ulceration and, less commonly, pyoderma gangrenosum may occur. Isolated nail-fold infarcts are not predictive of rheumatoid vasculitis. Peripheral neuropathy is common; 40% of patients have sensory neuropathy, and mononeuritis multiplex may also be present. CNS involvement is rare. Eye disease manifests as necrotizing scleritis, which may result in scleral ulceration. These patients often require surgical intervention in addition to cytotoxic therapy. Necrotizing scleritis must be distinguished from other ocular conditions such as non-necrotizing scleritis and episcleritis or keratoconjunctivitis sicca, which are not as threatening to the eye and do not herald the onset of vasculitis.
Renal disease in RA is often associated with nephrotic syn-drome caused by membranous nephropathy following gold or penicillamine therapy or amyloid deposition. About one-quarter of patients with rheumatoid vasculitis have renal involvement; focal segmental necrotizing glomerulonephritis without immuno-globulin deposition has most often been reported. Presentation is with microscopic haematuria, proteinuria and renal impairment.
Diagnosis and investigations – rheumatoid vasculitis should be suspected in any patient with a long history of RA or juvenile arthritis who develops new extra-articular or constitutional symp-toms such as weight loss, fever and/or lethargy. Tissue biopsy of the affected organ should be undertaken whenever possible because of the poor prognosis of untreated systemic vasculitis and toxicity of treatment.
Laboratory findings include raised inflammatory markers (CRP and ESR), high levels of IgM and IgG rheumatoid factor, though
Patient evaluation in leucocytoclastic vasculitis
• History – drugs, infection, previous history of systemic
disease, systemic symptoms
• Examination – physical examination, urinalysis
• Skin biopsy – light microscopy, immunofluorescence for IgA
• Laboratory tests – full blood count, urea and electrolytes,
inflammatory markers (C-reactive protein and ESR),
autoantibody screen (antinuclear antobodies, dsDNA,
ANCA, rheumatoid factor, complement), hepatitis B and C,
cryoglobulins and protein electrophoresis, blood cultures
Table 4
MEDICINE 34:11 462
these are not good markers of clinical activity. Antinuclear anti-body is present in most cases of rheumatoid vasculitis. Atypical ANCA (commonly against lactoferrin) may be present, but it is unclear whether they are involved in the pathogenesis.
Management – isolated skin vasculitis may be treated with corticosteroids. Biopsy-proven systemic vasculitis must be treated aggressively. Regimens similar to those for primary systemic vas-culitis should be used. Relapses occur in up to 25% of patients.
SLE and vasculitisVasculitis is uncommon in patients with SLE occurring in approx-imately 10% of patients.
Clinical features (Table 5) – cutaneous vasculitis is the most common feature (90% of those with vasculitis). Other organ involvement is uncommon. Many patients have constitutional symptoms and other features of active SLE when they develop vas-culitis. Those patients with vasculitis are more likely to have livedo reticularis have higher scores of disease activity and have anti-La/SS-B antibodies.13 The CNS is commonly affected in patients with SLE, but true CNS vasculitis is uncommon. Microvascular
Systemic lupus erythematosus and vasculitis – organ involvement
Skin • Purpuric lesions, digital pulp infarcts, livedo reticularis;may
be associated with Raynaud’s syndrome
• Note that patients with SLE are at increased risk of drug
allergies; development of new rashes should raise the
possibility of drug reactions
CNS • True CNS vasculitis occurs in < 10% of patients; symptoms
range from mild cognitive dysfunction to psychosis, seizures
and ischaemia
• MRI is often unhelpful; it may reveal periventricular white
matter lesions but is not diagnostic
Cardiovascular system • Extramural arteries may be involved, resulting in myocardial
infarct (rare)
• Myocardial infarction is common as a result of accelerated
atherosclerosis
Mesenteric vasculitis • With or without infarction, most serious complication
• Presents with acute abdomen, fever and vomiting; may have
an insidious onset
• High mortality
• Often associated with renal disease and other internal organ
vasculitis
Kidney • Focal segmental necrotizing glomerulonephritis with fibrinoid
necrosis (rare)
• Distinguished from primary systemic vasculitis by deposition
of immunoglobulin and complement
Table 5
© 2006 Elsevier Ltd. All rights reserved.

SYSTEMIC VASCULITIDES
capillaritis may occur in the kidneys, leading to abnormal urinaly-sis and rapidly deteriorating renal function, and less commonly in the lungs, where it may manifest as pulmonary haemorrhage.
Investigations – vasculitis occurs most often in patients with active disease. Complement levels are often low and dsDNA antibody levels high. ESR is usually elevated and CRP is low. Antiphospholipid antibodies may be present. Regular urinalysis is essential. The threshold for renal biopsy should be low in any patient with cutaneous vasculitis.
Management – vasculitis in SLE requires early and aggres-sive treatment with corticosteroids and cyclophosphamide. Intra-venous pulse cyclophosphamide may be less toxic than daily oral therapy. Anticoagulation should be considered if anti- phospholipid antibodies are present or the patient has neurologi-cal vasculitis. ◆
REFERENCES
1 Morgan M D, Harper L, Williams J, Savage C. Anti-neutrophil
cytoplasm-associated glomerulonephritis. J Am Soc Nephrol 2006;
17: 1224–34.
2 Xiao H, Heeringa P, Liu Z et al. The role of neutrophils in the
induction of glomerulonephritis by anti-myeloperoxidase antibodies.
Am J Pathol 2005; 167: 39–45.
3 Little M A, Smyth C L, Yadav R et al. Antineutrophil cytoplasm
antibodies directed against myeloperoxidase augment leukocyte-
microvascular interactions in vivo. Blood 2005; 106: 2050–8.
4 Pfister H, Ollert M, Frohlich L F et al. Antineutrophil cytoplasmic
autoantibodies against the murine homolog of proteinase 3
(Wegener autoantigen) are pathogenic in vivo. Blood 2004; 104:
1411–8.
5 Jagiello P, Gross W L, Epplen J T. Complex genetics of Wegener
granulomatosis. Autoimmun Rev 2005; 4: 42–7.
6 Savage C O. ANCA-associated renal vasculitis. Kidney Int 2001;
60: 1614–27.
7 Jayne D, Rasmussen N, Andrassy K et al. A randomized trial of
maintenance therapy for vasculitis associated with antineutrophil
cytoplasmic autoantibodies. N Engl J Med 2003; 349: 36–44.
8 de Groot K, Adu D, Savage C. The value of pulse cyclophosphamide
in ANCA-associated vasculitis: meta-analysis and critical review.
Nephrol Dial Transplant 2001; 16: 2018–27.
9 Jayne D R W, Gaskin G, Rasmussen N et al. Randomised trial of
plasma exchange versus methyl prednisolone for severe renal
vasculitis. N Engl J Med; in press.
10 De Groot K, Rasmussen N, Bacon P A et al. Randomized trial of
cyclophosphamide versus methotrexate for induction of remission
in early systemic antineutrophil cytoplasmic antibody-associated
vasculitis. Arthritis Rheum 2005; 52: 2461–9.
11 Etanercept plus standard therapy for Wegener’s granulomatosis.
N Engl J Med 2005; 352: 351–61.
12 Guillevin L, Cohen P, Mahr A et al. Treatment of polyarteritis
nodosa and microscopic polyangiitis with poor prognosis factors:
a prospective trial comparing glucocorticoids and six or twelve
cyclophosphamide pulses in sixty-five patients. Arthritis Rheum
2003; 49: 93–100.
13 Ramos-Casals M, Nardi N. Lagrutta M et al. Vasculitis in systemic
lupus erythematosus: prevalence and clinical characteristics in 670
patients. Medicine (Baltimore) 2006; 85: 95–104.
MEDICINE 34:11 4
FuRTHER READING
Adu D, Tse W Y. Rheumatoid arthritis, mixed connective tissue
disease and polymyositis. In: Adu A, Emery P, Madaio M, eds.
Rheumatology and the kidney. Oxford: Oxford University Press,
2001: 293–304.
(This chapter focuses on rheumatoid vasculitis.)
Ball E, Knight J, Bridges Jr. S L, eds. Vasculitis. Oxford: Oxford
University Press, 2001.
(A comprehensive overview of many forms of vasculitis, including
Wegener’s granulomatosis and Henoch–Schönlein nephritis.)
Buhaescu I, Covic A, Levy J. Systemic vasculitis: still a challenging
disease. Am J Kidney Dis 2005; 46: 173–85.
(A recent review of treatment, well referenced for further reading.)
Danning C L, Illei G G, Boumpas D T. Vasculitis associated with primary
rheumatologic diseases. Curr Opin Rheum 1998; 10: 58–65.
(A good overview of vascultis in RA and SLE.)
D’Cruz D. Vasculitis in systemic lupus erythematosus. Lupus 1998; 7:
270–4.
(Includes additional information on treatment and pathogenesis.)
Levy J, Pusey C D. Systemic Vasculitis. In: Davison A M, Cameron J S,
Grünfeld J-P, Kerr D S, Ritz E, Winearls C G, eds. Oxford Textbook
of Clinical Nephrology. Oxford: Oxford University Press, 2005:
559–78.
(A review of renal involvement in primary small vessel vasculitis.)
Practice points
• Perform an ANCA test (as part of a full autoantibody
screen) in any patient with an undiagnosed multisystem
illness, particularly when there is involvement of the upper
respiratory tract, lungs or kidneys
• A negative ANCA test does not exclude primary small vessel
vasculitis
• Consider the diagnosis of primary small vessel vasculitis
in any patient with multisystem disease, particularly when
urinalysis reveals microscopic haematuria and proteinuria;
early diagnosis and rapid treatment improve the prognosis
• Manage the patient with help from a specialist centre; careful
monitoring of clinical and laboratory markers of disease
activity guides treatment and allows early detection of
relapse
• Any vasculitic rash should be taken seriously and efforts
made to find a cause; a systemic cause should be considered
if there is no obvious drug or infectious cause
• The threshold for renal investigation, including biopsy, should
be low in patients with cutaneous vasculitis
• Non-cutaneous vasculitis should be treated early and
aggressively
• Necrotizing scleritis in RA must be detected early and
differentiated from other eye lesions to preserve sight
• Vasculitis is uncommon but should be considered in patients
with SLE or RA with active disease and atypical symptoms
63 © 2006 Elsevier Ltd. All rights reserved.





























