Embed Size (px)
Citation preview

Skeletal Involution by Age-associated Oxidative Stressand Its Acceleration by Loss of Sex Steroids*
Received for publication, April 3, 2007, and in revised form, July 6, 2007 Published, JBC Papers in Press, July 10, 2007, DOI 10.1074/jbc.M702810200
Maria Almeida‡, Li Han‡, Marta Martin-Millan‡, Lilian I. Plotkin‡, Scott A. Stewart‡, Paula K. Roberson§,Stavroula Kousteni‡1, Charles A. O’Brien‡, Teresita Bellido‡, A. Michael Parfitt‡, Robert S. Weinstein‡,Robert L. Jilka‡, and Stavros C. Manolagas‡2
From the ‡Division of Endocrinology and Metabolism, Center for Osteoporosis and Metabolic Bone Diseases,and the §Department of Biostatistics, University of Arkansas for Medical Sciences and the Central ArkansasVeterans Health Care System, Little Rock, Arkansas 72205
Both aging and loss of sex steroids have adverse effects onskeletal homeostasis, but whether and how they may influenceeach others negative impact on bone remains unknown. Wereport herein that both female andmale C57BL/6mice progres-sively lost strength (as determined by load-to-failure measure-ments) and bone mineral density in the spine and femurbetween the ages of 4 and 31 months. These changes were tem-porally associated with decreased rate of remodeling as evi-denced by decreased osteoblast and osteoclast numbers anddecreased bone formation rate; as well as increased osteoblastand osteocyte apoptosis, increased reactive oxygen species lev-els, and decreased glutathione reductase activity and a corre-sponding increase in the phosphorylation of p53 and p66shc, twokey components of a signaling cascade that are activatedby reac-tive oxygen species and influences apoptosis and lifespan.Exactly the same changes in oxidative stress were acutely repro-duced by gonadectomy in 5-month-old females or males andreversed by estrogens or androgens in vivo aswell as in vitro.Weconclude that the oxidative stress that underlies physiologic or-ganismal aging in mice may be a pivotal pathogenetic mecha-nismof the age-related bone loss and strength. Loss of estrogensor androgens accelerates the effects of aging onbone by decreas-ing defense against oxidative stress.
Age-related loss of bone mass and strength is an invariablefeature of human biology, affecting women and men alike.Moreover, population-based studies demonstrate that substan-tial bone loss begins as early as the 20s in young adult womenandmen, long before any hormonal changes (1).3 The extent towhich estrogen deficiency contributes to age-related bone loss
and the slower rate of decline of bonemass and strength duringthe late postmenopausal years, and the molecular and cellularmechanisms of such putative interactions, are unknown.The universality of age-associated bone loss irrespective of
sex steroid status notwithstanding, age is by far a more criticaldeterminant of fracture risk than bonemass in humans indicat-ing that age-related increase in fracture risk reflects a loss ofbone strength that is only partly accounted for by loss of bonemass (2). Whereas an increased propensity to fall due to age-related decline in neuromuscular function is a factor, thereare also age-related changes in the bone itself. Such changesinclude disrupted architecture, altered composition of thebone mineral and matrix, delayed repair of fatigue micro-damage, excessive turnover, and inadequate bone size (3–7).The most recently appreciated qualitative factor is loss ofosteocytes (8, 9), former osteoblasts entombed into the min-eralized matrix. Osteocyte death may influence the signalsnecessary for mechanical adaptation and repair and also leadto long term changes in bone hydration. The anti-apoptoticeffect of sex steroids on osteocytes, which has been well doc-umented in mice, rats, and humans (10–12), may contributeto their anti-fracture efficacy independently of their effecton bone mineral density (BMD)4 (8).We and others had shown earlier that estrogens and andro-
gens protect the adult skeleton against bone loss by suppressingthe rate of bone turnover and maintaining a focal balancebetween bone formation and resorption (13–15). Suppressionof bone turnover results from attenuating effects of sex steroidson the birth rate of osteoblast and osteoclast progenitors.Main-tenance of a focal balance between formation and resorptionresults from opposite effects on the lifespan of osteoblasts/os-teocytes and osteoclasts: an anti-apoptotic effect on the formerand a pro-apoptotic effect on the latter cell type. Conversely,loss of sex steroids increases the rate of remodeling by up-reg-ulating osteoblastogenesis and osteoclastogenesis. Specifically,we have shown that estrogen loss rapidly up-regulates osteo-
* This work was supported by National Institutes of Health Grants P01AG13918, R01 AR51187, R01 AR49794, and R01 AR46191, Department ofVeterans Affairs merit review grants (to R. S. W., R. L. J., and S. C. M.) and aresearch enhancement award program; and tobacco settlement fundsprovided by the University of Arkansas for Medical Sciences. The costs ofpublication of this article were defrayed in part by the payment of pagecharges. This article must therefore be hereby marked “advertisement” inaccordance with 18 U.S.C. Section 1734 solely to indicate this fact.
1 Present address: Dept. of Medicine, Division of Endocrinology, ColumbiaUniversity, New York, NY 10032.
2 To whom correspondence should be addressed: 4301 West Markham, 587,Little Rock, AR 72205-7199. Tel.: 501-686-5130; Fax: 501-686-8148; E-mail:[email protected].
3 B. L. Riggs, L. J. Melton III, A. L. Oberg, E. K. Atkinson, and S. Khosla personalcommunication.
4 The abbreviations used are: BMD, bone mineral density; AFU, arbitrary fluo-rescence units/mg of protein; BSO, L-buthionine-(S,R)-sulfoximine; DEM,diethyl maleate; DHT, dihydrotestosterone; E2, estradiol; ER, estrogenreceptor; NAC, N-acetyl-L-cysteine; ORX, orchidectomy; OVX, ovariectomy;ROS, reactive oxygen species; TNF, tumor necrosis factor; N, newton; ERK,extracellular signal-regulated kinase; AR, androgen receptor; ANOVA, anal-ysis of variance; MSC, mesenchymal stem cell; FOXO, Forkhead box O; MEK,mitogen-activated protein kinase/extracellular signal-regulated kinasekinase.
THE JOURNAL OF BIOLOGICAL CHEMISTRY VOL. 282, NO. 37, pp. 27285–27297, September 14, 2007Printed in the U.S.A.
SEPTEMBER 14, 2007 • VOLUME 282 • NUMBER 37 JOURNAL OF BIOLOGICAL CHEMISTRY 27285
by guest on June 13, 2018http://w
ww
.jbc.org/D
ownloaded from

blastogenesis in mice, and this effect is at least in part cellautonomous, and independent of the increased osteoclasticresorption, as it is manifested in ovariectomized (OVX) micetreated with bisphosphonates, known inhibitors of osteoclasticbone resorption (16). Moreover, the adverse effects of estrogenor androgen withdrawal on bone seem to be mediated, at leastin part, by cells of the osteoblastic lineage and are obviated byconstraints on osteoblastogenesis, whether genetic (17) oracquired (18), most likely because of the critical dependence ofosteoclastogenesis on osteoblastogenesis (19, 20). Nonetheless,the effect of the loss of estrogens on the rate of remodeling andthe up-regulation of osteoblastogenesis and bone formation inanimals and humans wanes with time (5–10 years in womenand 6–8 weeks in mice) to about the same rate as in elderlyeugonadal males, raising the possibility that aging might beoverriding the acute effects of estrogen deficiency.Increased levels of reactive oxygen species (ROS) influence
numerous cellular processes, including the timing of death byapoptosis; and have been linked to aging and the developmentof age-related diseases. Moreover, oxidative stress has beenstrongly correlated with longevity in flies, nematodes, andmammals (21, 22). The p53 tumor suppressor and the adapterprotein p66shc represent key components of a signal transduc-tion pathway that not only is activated by increased intracellu-lar ROS and converts oxidative signals into apoptosis but alsogenerates ROS in themitochondria (23–25). Strikingly, an acti-
vating mutation of p53 causes earlyonset of aging-associated pheno-types inmice (26). Conversely, dele-tion of p66shc increases resistance tooxidative stress, as well as lifespan,by as much as 30% (23). One of themechanisms used by cells to defendagainst oxidative damage involvesthe reduction of peroxides to harm-less alcohols in a reaction in whichglutathione peroxidase oxidizes glu-tathione (GSH) to the disulfideGSSH, and glutathione reductase(GSR) converts it back into GSH(27).The above lines of evidence have
strongly suggested to us the possi-bility that organismal aging per se,rather than an age-associated failureof other organs and tissues, may bethe predominant mechanism of thebone fragility disease, which hasbecome synonymous with osteopo-rosis, just one of the many featuresand risk factors underlying theproblem of fractures. And, that lossof estrogens or androgens mayexaggerate the adverse effects of or-ganismal aging on bone. We reportseveral previously unappreciatedage-related changes in both femaleand male C57BL/6 mice that may
provide critical clues into the mechanisms of the age-relateddecline of bone strength and mass, and the influence of sexsteroid deficiency in the process.
EXPERIMENTAL PROCEDURES
Chemicals and Reagents—L-Buthionine-(S,R)-sulfoximine(BSO), diethyl maleate (DEM),N-acetyl-L-cysteine (NAC), eto-poside, flutamide, H2O2, dihydrotestosterone (DHT), and E2were purchased fromSigma. PD98059was purchased fromCellSignaling Technology, Inc. (Danvers, MA). ICI 182,780 waspurchased fromTocris Cookson Inc. (Ellisville, MO), PP1 fromBiomol International P.A. (PlymouthMeeting, PA), and U0126from Promega (Madison, WI). Tumor necrosis factor (TNF) �recombinant protein was purchased from R&D Systems (Min-neapolis, MN). Sixty-day slow-release pellets containing DHTwere purchased from Innovative Research of America (Sara-sota, FL). Estradiol was assayed with a kit from DiaSorin (Still-water, MN) and testosterone with a kit from MP Biomedicals(CostaMesa, CA). Glutathione reductase was assayedwith a kitfrom Cayman Chemical Co. (Ann Arbor, MI). IntracellularROS were quantified using dichlorodihydrofluorescein diac-etate dye (28), using bone marrow cells flushed from femursand washed with phosphate-buffered saline.Animal Experimentation—Male and female C57BL/6 mice
4–31 months old were purchased from Harlan Inc. from acohort maintained with support from the National Institute of
FIGURE 1. BMD and strength decrease with age in sex steroid sufficient female or male C57BL/6 mice.A, BMD at the spine and femur was assessed by dual energy x-ray absorptiometry in two experiments withfemale and one experiment with male mice. The n was 12 animals per age group in each experiment withfemales, and 10 –12 in the experiment with males. In the two experiments with females the age-depend-ent changes were statistically indistinguishable, hence the data were combined. B, load-to-failure, ameasure of strength, was determined by compression testing of the 6th lumbar vertebra (L6) and by3-point bending of the left femur. Colored horizontal lines in A and B indicate the mean values for each sex.* and † indicate the age after which a time-dependent decline began in females and males, respectively.
Skeletal Involution, Oxidative Stress, and Sex Steroids
27286 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 282 • NUMBER 37 • SEPTEMBER 14, 2007
by guest on June 13, 2018http://w
ww
.jbc.org/D
ownloaded from

Aging. The age-associated changes of intact animals were stud-ied in three separate experiments: two with females (the first at8, 16, 25, and 31 months old; and the second at 4, 8, 16, and 25months old) and one with males (4, 8, 16, 25, and 31 monthsold). For the studies examining the effects of sex steroid defi-ciency, 5 month-old C57BL/6 mice were purchased from Har-lan Sprague-Dawley Inc. Two to 3 days before surgery, all ani-mals were electronically tagged (Biomedic Data System Inc.,Maywood, NJ), BMD measurements were performed on each,and they were then allocated to various experimental groups toachieve equivalent mean femur BMD values. Animals werethen sham-operated, OVX, or orchidectomized (ORX). Sham-operated animals were administered vehicle or BSO (2mmol/kg intraperitoneally) twice a day (n� 12 per group). BSOwas also included in the drinkingwater (20mM).OVXandORXanimals were subcutaneously injected with vehicle or withreplacement doses of E2 (30 ng/g) or NAC (100 mg/kg/day)twice a day or were implanted with 60-day slow-release pelletscontaining DHT (10 mg) (n � 12 per group). After 6 weeks oftreatment, animals were sacrificed and the tissues dissected forfurther analyses. BMD, bone geometry measurements, histo-morphometry, and osteoblast/osteocyte apoptosis were per-formed as previously described (29–31).Biomechanical Testing—The load bearing properties of L6
were measured using a single columnmaterial testing machineand a calibrated tension/compression load cell (model 5542,Instron Corp., Canton, MA). Load cell calibration was verified
in accordance with American Soci-ety for Testing and MaterialsE74-02 standards and traceable tothe National Institute of Standardsand Technology. Data wererecorded and analyzed using theMerlin IX software package(Instron Corp.). The L6 specimenswere cleaned of surrounding softtissue, wrapped in gauze soaked in37 � 0.5 °C normal saline, andtested on the day of sacrifice. Thelength, width, and depth of thebones were recorded with a digitalcaliper at a resolution of 0.01 mm(Mitutoyo number 500-196, AceTools, Ft. Smith, AR). The cross-sectional area was assumed to be anellipse and calculated as A � 0.25 �(width)(depth). Articular and spi-nous processes that would interferewith compression were excisedusing an iris scissors. After pre-seat-ingwith less than 0.5 newtons (N) ofapplied load, vertebrae were com-pressed between screw-driven load-ing platens using a lower-platen,customizedminiature spherical seatthatminimizes shear by adjusting toirregularities in the end plates of thespecimens. Best seating was
obtained with the load applied along the caudocephalad axis ata speed of 0.5 mm/min until failure. Standard materials forcompression were run before each set of determinations.Three-point bending of the femur was also performed at 37 �0.5 °C using a miniature bending apparatus with the posteriorfemoral surface lying on lower supports (7 mm apart) and theleft support immediately proximal to the distal condyles. Loadwas applied to the anterior femoral surface by an actuator mid-way between the two supports moving at a constant rate of 3mm/min to produce a physiological in vivo stain rate of 1% forthe averagemurine femur. The externalmeasurements (length,width, and thickness) of the femorawere recordedwith a digitalcaliper. Measurements of the internal marrow cavity (greaterand lesser diameters) were obtained with a hand-held micro-scope at �100 magnification using a calibrated linear reticuleeyepiece (KlarmannRulings,Manchester, NH).Maximum load(N) and displacement (mm) were recorded. The mechanicalproperties were normalized for bone size and ultimate strengthor stress (N/mm2; in megapascals) was calculated. Standardprecision steel piano wire with stiffness in the same range asmurine femoral bone was evaluated before each set ofdeterminations.Western Blot Analysis—The phosphorylation status of p53,
p66shc, and ERK1/2 was analyzed by immunoblotting in fifthlumbar vertebra lysates, as previously described (32). The anti-bodies used were: a rabbit polyclonal antibody recognizingSer15-phosphorylated p53 (Cell Signaling Technology, Inc.,
FIGURE 2. Bone remodeling and bone formation rate decrease with age in sex steroid sufficient female ormale C57BL/6 mice, whereas osteoblast and osteocyte apoptosis increases. A, static and dynamic histo-morphometric analysis of longitudinal undecalcified sections of L1–L4 vertebrae. Osteoblasts were enumer-ated on sections from the same specimens stained with toluidine blue and bone formation rate was deter-mined from tetracycline-labeled surfaces. B, osteoblast and osteocyte apoptosis were determined by in situend-labeling. Bars indicate mean � S.D.; * or † indicate p � 0.05 versus 8-month-old animals in females and4-month-old in males. In A, only mice from the first experiment with females were analyzed.
Skeletal Involution, Oxidative Stress, and Sex Steroids
SEPTEMBER 14, 2007 • VOLUME 282 • NUMBER 37 JOURNAL OF BIOLOGICAL CHEMISTRY 27287
by guest on June 13, 2018http://w
ww
.jbc.org/D
ownloaded from

Danvers, MA), a mouse monoclonal antibody recognizingSer36-phosphorylated p66shc (Calbiochem, SanDiego, CA), anda mouse monoclonal antibody recognizing tyrosine-phospho-rylated ERK1/2 (Santa Cruz Biotechnology Inc., Santa Cruz,CA). Protein levels of p-53, p66shc, and ERK1/2 were analyzedusing a mouse monoclonal antibody recognizing p53 (Cell Sig-naling), a rabbit polyclonal antibody recognizing p66shc (BDBiosciences, Palo Alto, CA), and a rabbit polyclonal antibodyrecognizing total ERK1/2 (Santa Cruz).Cell Culture—OB-6 cells, an osteoblastic cell line derived in our
laboratory from the murine bone marrow (33), were cultured in�-minimal essential medium (Invitrogen) supplemented with10% fetal calf serum (Hyclone, Logan, UT), penicillin (100units/ml), streptomycin (100 �g/ml), and glutamine (292�g/ml). C2C12 cells were cultured in Dulbecco’s modified
Eagle’s medium supplemented with10% fetal bovine serum, antibioticsas above, and 1% sodium pyruvate.Osteoclasts were derived frombone marrow cells cultured in�-minimal essential medium sup-plemented with 30 ng/ml macroph-age-colony stimulations factor and30 ng/ml soluble RANK ligand. Forthe quantification of osteoblast apo-ptosis, OB-6 cells were treated for1 h with BSO or DEM followed bythe steroids for 1 and 6 h with thepro-apoptotic agent etoposide (5 �10�5 M), TNF� (10�9 M), or H2O2(5 � 10�5 M). Osteoclast apoptosiswas assayed in cells treated withBSO or DEM for 1 h followed by thesteroids for 24 h. Apoptotic cellswere quantified by measuringcaspase 3 activity as described pre-viously (32). For quantification ofosteoclasts, after 4 days in culture,cells were treated for 1 h with BSOor DEM followed by the steroids for24 h. Osteoclasts were enumeratedafter staining for TRAPase; bothmononucleated and multinucleatedcells were counted.Quantitative PCR—Total RNA
was extracted from tibia or calvariausing Ultraspec RNA (Biotecx Lab-oratories, Houston, TX) andreverse-transcribed using the HighCapacity cDNA Archive Kit(Applied Biosystems, Foster City,CA). Primers and probes for estro-gen receptor (ER) �, ER�, androgenreceptor (AR), and glyceraldehyde-3-phosphate dehydrogenase weremanufactured by the TaqMan�Gene Expression Assays service(Applied Biosystems).
Transient Transfections—C2C12 cells were transfected with0.1 �g of green fluorescent protein, 0.1 �g of empty vector orwild-type p66shc plasmid, and 0.2 �g of pcDNA using Lipo-fectamine Plus (Invitrogen).Statistical Analysis—Effects of age and gender on BMD
and strength of the spine and the femur were evaluated bytwo-way ANOVA. The time of onset of the changes was deter-mined by linear regression followed by a test for significant lackof fit or departure from linearity (34). Whenever there was sig-nificant departure from linearity for the entire data set, datafrom one or two earlier time points (4 month or both 4 and 8months) were excluded from the analysis so that linearity couldbe established within the remaining set of data. Subsequently,the earliest time point that was used to establish linearity, i.e.8 or 16 months, was compared with 4 or 4 and 8 months by
FIGURE 3. The external and internal diameters of the femoral diaphysis increase with age in both femaleand male C57BL/6 mice. The geometry of the left femurs from the two experiments with females and thesingle experiment with males was determined after performance of the 3-point bending test at the diaphysealbreakpoint by measuring the diameters depicted in the inset. The anterior-posterior external diameter (AP) andlateral exterior diameter (L) were measured using a digital caliper; and the corresponding internal diameters,(AP�) and (L�), were measured with a hand-held microscope, as described under “Experimental Procedures.”† indicates significant difference only between 6 and 18 months by ANOVA. ‡ indicates a progressive increaseafter 16 months of age. * indicates progressive increase after 4 months of age without significant departurefrom linearity for the entire data set.
Skeletal Involution, Oxidative Stress, and Sex Steroids
27288 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 282 • NUMBER 37 • SEPTEMBER 14, 2007
by guest on June 13, 2018http://w
ww
.jbc.org/D
ownloaded from

Student’s t test or ANOVA. If no different from the precedingtime points, the earliest time point of the linearity plot wasdeclared the time after which a particular change was mani-fested and the change was continuous from that time point on;if, on the other hand, differences were detected, we concludedthat the change began at the earliest time point, but it wasdiscontinuous.ANOVA was used to detect other age-related changes, and
effects of various in vivo and in vitro treatments, after establish-ing that the data were normally distributed and equivalency ofvariances. Bonferroni’s method was used to perform appropri-ate pairwise comparisons of treatment groups. In cases whenone or both of the requirements for performing ANOVA werenot met, Kruskal-Wallis ANOVA on Ranks test was used fol-lowed by Dunn’s method to perform pairwise comparisons oftreatment groups.
RESULTS
Similar Age-associated Changes in BMD and Strength inFemale and Male Mice—Both female and male C57BL/6 miceexhibited loss of bone mass, as determined by the surrogatemeasurement of BMD, and loss of strength in the spine andfemur between ages of 4 and 31 months (Fig. 1, A and B).
Strength in these experiments wasassessed by obtaining load-to-fail-ure estimates of the 6th lumbar ver-tebra (L6) and the left femur, usingcompression and 3-point bending,respectively. The decline in bonestrength occurred earlier as com-paredwith the loss of BMD,with theexception of the spine in females inwhich both changes began simulta-neously. Furthermore, the declineof BMD and strength was progres-sive (i.e. the data fit linear regres-sion) in all instances other than ver-tebral and femoral strength inmales, which were discontinuous.The fall in spinal BMD and femoralstrength occurred earlier in femalesthan in males.Age-associated Changes in Osteo-
blast and Osteoclast Number, BoneFormation, and Osteoblast andOsteocyte Apoptosis—As evidencedfrom histomorphometric analysis(Fig. 2) of the vertebrae of the first ofthe two experiments with femaleC57BL/6 mice, advancing age from 8to 31 months was associated withdecreased cancellous bone area andtrabecular width and increased trabe-cular separation (Fig. 2A). Further-more, advancing age was associatedwith decreased wall width, decreasedosteoblast and osteoclast number, aswell as a decrease in bone formation
rate. All these changes are unmistakable indications of a low turn-over state, with resorption exceeding formation; and are clearlydistinct from the high turnover state caused by loss of estrogensor androgens in rodents and humans. Furthermore, advancingage was associated with an increase in osteoblast and osteocyteapoptosis (Fig. 2B). The increase in osteoblast and osteocyteapoptosis with age in females was confirmed in the experimentwith the males between the ages of 4 and 31 months. As is thecase in humans (35, 36), both female and male mice exhibitedan age-dependent increase in both the external and internaldiameter of the femoral diaphysis, indicating ongoing perios-teal apposition and endocortical resorption, respectively (Fig.3).Age-related Increase of Oxidative Stress in the Bone of
Females andMales—Consistent with the evidence for a criticalrole of oxidative damage in age-related diseases in general, andof the p53/p66shc signaling cascade in apoptosis, the changes inbone mass, strength, osteoblast/osteocyte apoptosis, osteoblastnumber, and bone formation rate with age in the aging murinemodel were associated with age-dependent decreased activityof GSR (Fig. 4A) and increased levels of ROS in the bone mar-row (Fig. 4B). Furthermore, the pattern of all these changes wastemporally associated with a progressive increase in the phos-
FIGURE 4. Oxidative stress increases with age in the bone of female or male C57BL/6 mice. GSR activity (A)and ROS levels (B) were determined in bone marrow aspirates. The results from the two experiments withfemales and the single experiment with males are shown separately (n � 4 animals per group per experiment).Bars indicate mean � S.D.; * or † indicate p � 0.05 versus 8-month-old animals in females and 4-month-old inmales. C, the levels of phosphorylated p53; and D, p66shc were determined by Western blot analyses in verte-bral lysates; each lane represents one animal. The mean ratio of phosphorylated to total protein is depictednumerically at the bottom of the corresponding blots.
Skeletal Involution, Oxidative Stress, and Sex Steroids
SEPTEMBER 14, 2007 • VOLUME 282 • NUMBER 37 JOURNAL OF BIOLOGICAL CHEMISTRY 27289
by guest on June 13, 2018http://w
ww
.jbc.org/D
ownloaded from
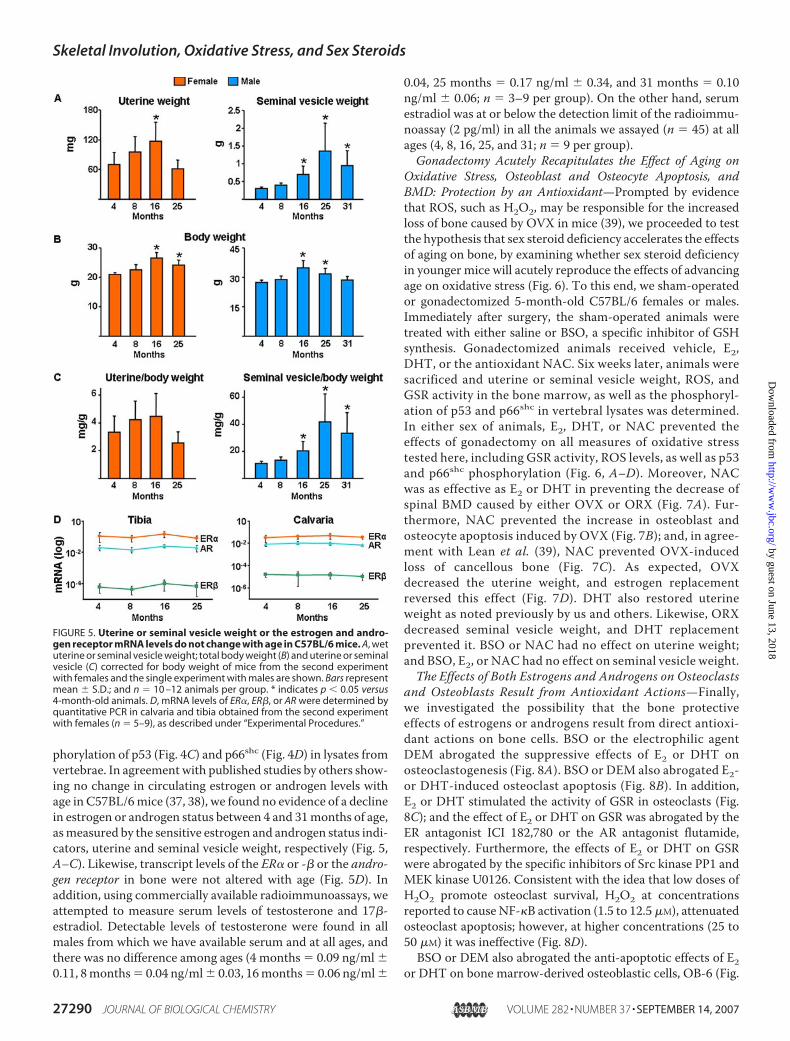
phorylation of p53 (Fig. 4C) and p66shc (Fig. 4D) in lysates fromvertebrae. In agreement with published studies by others show-ing no change in circulating estrogen or androgen levels withage in C57BL/6mice (37, 38), we found no evidence of a declinein estrogen or androgen status between 4 and 31months of age,asmeasured by the sensitive estrogen and androgen status indi-cators, uterine and seminal vesicle weight, respectively (Fig. 5,A–C). Likewise, transcript levels of the ER� or -� or the andro-gen receptor in bone were not altered with age (Fig. 5D). Inaddition, using commercially available radioimmunoassays, weattempted to measure serum levels of testosterone and 17�-estradiol. Detectable levels of testosterone were found in allmales from which we have available serum and at all ages, andthere was no difference among ages (4 months � 0.09 ng/ml �0.11, 8months� 0.04 ng/ml� 0.03, 16months� 0.06 ng/ml�
0.04, 25 months � 0.17 ng/ml � 0.34, and 31 months � 0.10ng/ml � 0.06; n � 3–9 per group). On the other hand, serumestradiol was at or below the detection limit of the radioimmu-noassay (2 pg/ml) in all the animals we assayed (n � 45) at allages (4, 8, 16, 25, and 31; n � 9 per group).Gonadectomy Acutely Recapitulates the Effect of Aging on
Oxidative Stress, Osteoblast and Osteocyte Apoptosis, andBMD: Protection by an Antioxidant—Prompted by evidencethat ROS, such as H2O2, may be responsible for the increasedloss of bone caused by OVX in mice (39), we proceeded to testthe hypothesis that sex steroid deficiency accelerates the effectsof aging on bone, by examining whether sex steroid deficiencyin younger mice will acutely reproduce the effects of advancingage on oxidative stress (Fig. 6). To this end, we sham-operatedor gonadectomized 5-month-old C57BL/6 females or males.Immediately after surgery, the sham-operated animals weretreated with either saline or BSO, a specific inhibitor of GSHsynthesis. Gonadectomized animals received vehicle, E2,DHT, or the antioxidant NAC. Six weeks later, animals weresacrificed and uterine or seminal vesicle weight, ROS, andGSR activity in the bone marrow, as well as the phosphoryl-ation of p53 and p66shc in vertebral lysates was determined.In either sex of animals, E2, DHT, or NAC prevented theeffects of gonadectomy on all measures of oxidative stresstested here, including GSR activity, ROS levels, as well as p53and p66shc phosphorylation (Fig. 6, A–D). Moreover, NACwas as effective as E2 or DHT in preventing the decrease ofspinal BMD caused by either OVX or ORX (Fig. 7A). Fur-thermore, NAC prevented the increase in osteoblast andosteocyte apoptosis induced by OVX (Fig. 7B); and, in agree-ment with Lean et al. (39), NAC prevented OVX-inducedloss of cancellous bone (Fig. 7C). As expected, OVXdecreased the uterine weight, and estrogen replacementreversed this effect (Fig. 7D). DHT also restored uterineweight as noted previously by us and others. Likewise, ORXdecreased seminal vesicle weight, and DHT replacementprevented it. BSO or NAC had no effect on uterine weight;and BSO, E2, or NAC had no effect on seminal vesicle weight.The Effects of Both Estrogens and Androgens on Osteoclasts
and Osteoblasts Result from Antioxidant Actions—Finally,we investigated the possibility that the bone protectiveeffects of estrogens or androgens result from direct antioxi-dant actions on bone cells. BSO or the electrophilic agentDEM abrogated the suppressive effects of E2 or DHT onosteoclastogenesis (Fig. 8A). BSO or DEM also abrogated E2-or DHT-induced osteoclast apoptosis (Fig. 8B). In addition,E2 or DHT stimulated the activity of GSR in osteoclasts (Fig.8C); and the effect of E2 or DHT on GSR was abrogated by theER antagonist ICI 182,780 or the AR antagonist flutamide,respectively. Furthermore, the effects of E2 or DHT on GSRwere abrogated by the specific inhibitors of Src kinase PP1 andMEK kinase U0126. Consistent with the idea that low doses ofH2O2 promote osteoclast survival, H2O2 at concentrationsreported to cause NF-�B activation (1.5 to 12.5�M), attenuatedosteoclast apoptosis; however, at higher concentrations (25 to50 �M) it was ineffective (Fig. 8D).BSO or DEM also abrogated the anti-apoptotic effects of E2
or DHT on bone marrow-derived osteoblastic cells, OB-6 (Fig.
FIGURE 5. Uterine or seminal vesicle weight or the estrogen and andro-gen receptor mRNA levels do not change with age in C57BL/6 mice. A, wetuterine or seminal vesicle weight; total body weight (B) and uterine or seminalvesicle (C) corrected for body weight of mice from the second experimentwith females and the single experiment with males are shown. Bars representmean � S.D.; and n � 10 –12 animals per group. * indicates p � 0.05 versus4-month-old animals. D, mRNA levels of ER�, ER�, or AR were determined byquantitative PCR in calvaria and tibia obtained from the second experimentwith females (n � 5–9), as described under “Experimental Procedures.”
Skeletal Involution, Oxidative Stress, and Sex Steroids
27290 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 282 • NUMBER 37 • SEPTEMBER 14, 2007
by guest on June 13, 2018http://w
ww
.jbc.org/D
ownloaded from

9A), as well as in primary cultures of calvaria cells and theC2C12 osteoblast progenitor cell line (data not shown), irre-spective of whether apoptosis was induced by the topoisomer-ase inhibitor etoposide, TNF�, orH2O2. BSOorDEM, at higherconcentrations than those that blocked the anti-apoptoticeffect of E2, induced apoptosis of calvaria cells, presumably bydepleting GSH (data not shown). This observation raises thepossibility that a threshold level of GSH production by osteo-blasts may be an inhibitor of endogenous ROS production and,thereby, apoptosis.Phosphorylation of serine 36 (Ser36) of p66shc is required for
transduction of oxidant stress signals leading to apoptosis.Consistent with this, overexpression of p66shc in C2C12 cellsinduced apoptosis both under basal conditions and in the pres-ence of H2O2 (Fig. 9B). Therefore, we examined whether E2 orDHT affects p66shc phosphorylation. H2O2 stimulated the
phosphorylation of p66shc as early as2 min and for at least 1 h followingits addition to cultures of OB-6osteoblastic cells but had no effecton the expression of the protein(Fig. 9C). E2 or DHT suppressed theH2O2-induced p66shc phosphoryla-tion as early as 15 min followingtreatment of OB-6 cells; and theeffect of either steroid was reversedin the presence of the MEK inhibi-tor PD98059. These results suggestthat, not only attenuation of oste-oclastogenesis, but also the stimula-tion of osteoclast and attenuation ofosteoblast apoptosis by estrogensinvolve non-protein thiol metabo-lism and are the result of actionsmediated via cytoplasmic kinasesand probably downstream tran-scriptional control (32).
DISCUSSION
The results reported herein dem-onstrate that both female and maleC57BL/6 mice exhibit an age-re-lated decrease in strength, BMD,bone remodeling, and bone forma-tion rate together with an increasein osteoblast and osteocyte apopto-sis. These changes are temporallyassociated with increased ROS lev-els, decreased GSR activity, andincreased phosphorylation of p53and p66shc. The exact same changesin ROS, GSR, and the phosphoryla-tion of p53 and p66shc were acutelyreproduced by gonadectomy in5-month-old females or males; andprevented by the antioxidant N-ace-tyl-L-cysteine. Moreover, estradiolor DHT acted directly in vitro to
attenuate osteoclastogenesis and osteoblast apoptosis, stimu-late osteoclast apoptosis, and suppress p66shc phosphorylationby a kinase-dependent mechanism that involves non-proteinthiol metabolism. This evidence strongly suggests thatincreased ROS is a pivotal mechanism of age-related bone lossand strength; and that estrogen or androgen deficiency accel-erates the adverse effects of aging on bone by decreasingdefense against ROS. To the best of our knowledge, the in-depth characterization of the mice used in our studies, includ-ing the demonstration of increased ROS and p66shc phospho-rylation with physiologic aging and acute loss of estrogens orandrogens, demonstrates for the first time that aging and sexsteroid deficiency exert their adverse effects by similar mecha-nisms in an animal model at the molecular, cellular, and tissuelevel. A model summarizing the results of the present report
FIGURE 6. The antioxidant NAC, as well as estrogens or androgens, prevent gonadectomy-inducedincrease in oxidative stress in females and males. A–D, 5-month-old mice were sham-operated, OVX, orORX. Sham-operated animals were administered vehicle (V) or BSO twice a day. One day after surgery, OVX andORX animals were injected daily with E2 (30 ng/g) or NAC (100 mg/kg) or were implanted with 60-day slow-release pellets containing DHT (10 mg). Animals were sacrificed 6 weeks later. In A and B, n � 4 animals pergroup. The results depicted for males in D were reproduced in a second blot, in which lysates from two moreanimals were assayed. The mean ratio of phosphorylated to total protein is depicted numerically in the bottomof the corresponding blots; and, in the case of the male data, represents the results from all four animals.* indicates p � 0.05 versus vehicle-treated OVX or ORX animals; and ‡ indicates p � 0.05 versus shamoperated animals treated with vehicle.
Skeletal Involution, Oxidative Stress, and Sex Steroids
SEPTEMBER 14, 2007 • VOLUME 282 • NUMBER 37 JOURNAL OF BIOLOGICAL CHEMISTRY 27291
by guest on June 13, 2018http://w
ww
.jbc.org/D
ownloaded from

and findings from earlier studies by us and others on the effectsof ROS and sex steroids on bone cells is provided in Fig. 10.In agreement with the findings of the present report, osteo-
porosis has been noted by us and others in mouse models ofpremature aging (17, 26, 40). Moreover, consistent with ourobservations temporally linking increased oxidative stress withincreased osteoblast apoptosis and decreased osteoblast number
and bone formation rate in aging C57BL/6 mice, in studies byChambers and colleagues (41) both osteoblast numbers and boneformationweredecreased in2-month-oldB6;129SF2mice treatedwith the glutathione inhibitor BSO. Likewise, in agreement withour findingswith the 5-month-old ovariectomizedC57BL/6mice,these workers had shown that the antioxidants, NAC and ascor-bate, or inhibition of H2O2 by pegylated catalase prevents theincreased osteoclastogenesis and loss of bone caused by acute lossof estrogens in 2-month-oldMF-1mice (39, 41).Beneficial effects of estrogens in several other tissues, such as
lens epithelial cells, arteries, central nervous system, fat, liver,and oviducts, are also shown to result from improved defenseagainst oxidative stress (42–53). Importantly, in view of the factthat E2 inhibits oxidized LDL (54) inhibition of 15-lipoxygen-ase, the enzyme responsible for the generation of oxidized lowdensity lipoprotein, partially prevents OVX-induced bone loss(55). Moreover, bone loss and vascular calcification progress inparallel with advancing age, indicating an age-dependent rela-tionship between atherosclerosis and osteoporosis (56). On theother hand, adverse effects of estrogens on breast cancer, theuterus, and spermatogenesis may be due to increased ROS pro-duction or decreased antioxidant defense (57–59).The results of the present study inmice also suggest that loss
of bone strength with age is only partially accounted for by areduction in bone mass. Moreover, even though we did notstudy fractures inmice, our observations are in linewith clinicalevidence from humans that the age-related increase in fracturerisk reflects a loss of bone strength that is only partly accountedfor by loss of bone mass (2). Indeed, consistent with this con-tention, at the lowest values for bone mass for humans, a20-year increase in age is accompanied by a 4-fold increase infracture risk. Whereas an increased propensity to fall due toage-related decline in neuromuscular function is clearly a factorin humans, there are also age-related changes in the bone itself.The importance of non-mass factors is demonstrated by severallines of evidence. First, a fracture at any site increases the risk ofa subsequent fracture at any other site (60). Second, only a smallpart of the reduction in fracture incidence in response to anti-catabolic therapy can be accounted for by the increase in bonemass (61). Third, many of the genetic effects on bone strengthare mediated by factors other than bone mass (62, 63). Theresults reported here support the idea that loss of osteocytes,which make an independent contribution to vertebral bonestrength both in patientswith vertebral fracture (9) and inmice,may be an additional factor (8, 64). Importantly, the age-de-pendent decline in murine bone strength in the present studyoccurred despite the expansion in bone size, exactly as is thecase in humans. Therefore, our findings also support the con-tention that theC57BL/6mouse is a goodmodel for the study ofage-related geometric changes in human bone.In line with the results of the present report suggesting that
sex steroid status was not altered appreciably, at least as far asthe skeletal changes concerned, between the age of 4 and 31months in C57BL/6 mice, Mobbs and Finch (37) had found nodifference in estradiol levels in females up to the age of 18months or androgen in male C57BL/6 mice up to 31 months(38). Nonetheless, inmore detailed studies of the estrus cycle inthe same mice these workers subsequently reported that estro-
FIGURE 7. The antioxidant NAC, as well as estrogens or androgens, pre-vent gonadectomy-induced bone loss and osteoblast and osteocyteapoptosis in females and males. A, spinal BMD was determined by dualenergy x-ray absorptiometry 1 to 3 days before and 6 weeks after surgery inthe mice of the experiments shown in Fig. 4. The mean � S.D. of the percentchange from the pre-surgery measurement is shown (n � 10 –12 per group).B, osteoblast and osteocyte apoptosis were determined in longitudinalundecalcified sections of L1–L4 by in situ end labeling, n � 7–13 per group.* indicates p � 0.05 versus vehicle-treated OVX or ORX animals; and ‡indicates p � 0.05 versus sham operated animals treated with vehicle.C, representative photomicrographs of lumbar vertebrae from the femalemice of the experiment. Note that the loss of central cancellous bone inthe OVX/vehicle (OVX/V), as compared with the Sham/vehicle control(Sham/V), has been prevented in the OVX animals that received NAC (OVX/NAC). Unstained and viewed at �25 with no coverslip. D, wet uterine orseminal vesicle weight of female and male mice. Bars represent mean �S.D.; and n � 10 –12 animals per group. * indicates p � 0.05 versus vehicletreated sham operated animals.
Skeletal Involution, Oxidative Stress, and Sex Steroids
27292 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 282 • NUMBER 37 • SEPTEMBER 14, 2007
by guest on June 13, 2018http://w
ww
.jbc.org/D
ownloaded from

gen levels on day 3 and the preovulatory rise beginning on day 4were decreased at 10–12.5 months as compared with 5.5–7.5months of age (65). Moreover, whereas Nelson et al. (38) didnot show declines in testosterone levels in healthy mice, micewith other diseases of aging did have markedly reduced testos-terone levels. In addition, in a very recent paper (66) the authorsfoundmarked decreases in testosterone levels inmale C57BL/6mice at age 15months comparedwith 4months.We and othershave shown here and elsewhere that seminal vesicle weight hasless than half the variance of the testosterone measurementsand indicates cumulative androgen status in mice better than asingle serum sample (18, 67, 68). Be that as it may, we cannotcategorically exclude the possibility that the age-related find-ings we have demonstrated herein may be accentuated bysuperimposed sex steroid deficiency in aging mice, as they arein humans. That having been said, however, acute loss of sexsteroids causes an increase in the rate of bone remodeling,underlined by an increase in osteoclastogenesis and osteoblas-togenesis and a corresponding increase in both bone resorp-tion and formation, with the former exceeding the latter. Incontrast, our aging C57BL/6 mice (and elderly individualswithout vitamin D deficiency and secondary hyperthyroid-ism) exhibit a low rate of bone remodeling, as well as adecrease in osteoblast number and bone formation, a pheno-type reminiscent also of the osteoporosis associated withglucocorticoid excess. In fact, work, from our group and oth-ers in several animal models, has strongly suggested thatdecreased wall width, the hallmark of age-associated osteo-porosis and decreased bone formation results fromdecreased osteoblastogenesis (13, 17). If both aging and lossof sex steroids exert their adverse effects on bone by oxida-tive damage, how can osteoblast and osteoclast number below in the former and high in the latter?In studies reported in the accompanying manuscript by
Almeida et al. (82) we have determined that ROS antagonizethe skeletal effects of Wnt/�-catenin in vitro by diverting�-catenin from Tcf- to FoxO-mediated transcription. More-over, consistent with the notion that increased ROS productionwith age attenuates Wnt/�-catenin signaling, Axin2 and OpgmRNAwere decreased in old as comparedwith youngC57BL/6mice. Because activation of Wnt signaling enables osteoblasto-genesis, suppresses osteoblastic cell apoptosis, inhibits oste-oclastogenesis and increases bone mass, the cellular changesseen with increased aging and the accompanying oxidativedamage may be due in part to attenuation of Wnt signaling.In work reported elsewhere (69, 70), we have found that the
estrogen-liganded ER� inhibits BMP-2-induced osteoblastprogenitor differentiation, whereas the unliganded ER� orER�, but not the AR, promote the pro-differentiating actionsof BMP-2 by enhancing Smad-mediated transcription. In
FIGURE 8. Estrogens or androgens regulate osteoclastogenesis and thesurvival of osteoclasts via antioxidant actions. A and B, bone marrow-derived osteoclasts were treated for 1 h with BSO (10�6
M) or DEM (10�4M),
followed by E2 or DHT (10�8M) for 24 h. Osteoclasts were enumerated after
staining for TRAPase and apoptosis was quantified by determining caspase 3activity. C, GSR activity in osteoclasts treated with ICI 182,780 (10�7
M), flut-amide (10�7
M), PP1 (10�6M), or U0126 (10�6
M) for 1 h followed by the indi-cated steroids for 24 h. D, apoptosis was quantified by determining caspase 3activity in bone marrow-derived osteoclasts treated for 24 h with 10�8
M E2 orthe indicated doses of H2O2. Bars indicate mean � S.D. of triplicate determi-nations; * p � 0.05 versus vehicle.
Skeletal Involution, Oxidative Stress, and Sex Steroids
SEPTEMBER 14, 2007 • VOLUME 282 • NUMBER 37 JOURNAL OF BIOLOGICAL CHEMISTRY 27293
by guest on June 13, 2018http://w
ww
.jbc.org/D
ownloaded from

addition, we have developed methods of expanding the self-renewal of mesenchymal stem cell (MSC) progenitors ofosteoblasts, while preserving stemness, and found that theirreplication capacity is significantly lower in old versus youngmice (71).Based on our own findings and parallel developments in the
understanding of the role of FoxOs in hematopoietic stem cellresistance to physiologic oxidative stress (72) we hypothesizethat estrogens influence osteoblastogenesis by two distinct butoverlapping cell autonomous mechanisms: (a) antioxidanteffects on MSC osteoblast progenitors and (b) transcriptionalrepression of BMP-induced differentiation of their progeny.Increased levels of ROS have deleterious effects on MSC self-renewal (by increasing apoptosis), but promote MSCs entryinto the cell cycle and terminal differentiation. These effects arecounteracted byROS-enhanced binding of FOXO to�-catenin,leading to increased defense against ROS. Because of theirability to defend against ROS, estrogens favor MSC quies-cence and survival and oppose terminal differentiation. Fol-lowing acute loss of estrogens, the increased ROS levels inMSCs promote their exit from quiescence, replication, anddevelopment of transit amplifying osteoblast progenitors.These mechanisms, together with the unleashing of the tran-scriptional restraint of the liganded ER� on BMP, lead toup-regulation of osteoblast differentiation and therebyincreased bone formation. However, this effect is reversedwith time because the gradual buildup of ROS productionwith aging (accelerated by estrogen deficiency) antagonizesWnt signaling and also leads to a decrease in the size of theMSC compartment by increasing MSC apoptosis.In addition, we have found that as compared with 4-month-
old mice, 25- and 31-month-old mice exhibited a 2-foldincrease in serum corticosterone, adrenal weight, and bonemRNA for 11�-Hsd1, the enzyme that amplifies glucocorticoidaction.5 The age-dependent decrease in bone formation rateand increased osteoblast and osteocyte apoptosis, cardinal fea-tures of the adverse effects of glucocorticoid excess on bone,along with the increase in 11�-Hsd1 mRNA, suggest thatlocal amplification of endogenous glucocorticoids may alsocontribute to decreased bone formation, the decline in bonestrength, and the disparity between bone quantity and qual-ity with aging. Additional mechanisms could well includeincreased production of peroxisome proliferator-activatedreceptor �-activating oxidized lipids derived from thelipoxygenase Alox15 (55, 73) and the skeletal unloading
5 R. S. Weinstein, R. L. Jilka, and S. C. Manolagas unpublished data.
FIGURE 9. Estrogens or androgens regulate the survival of osteoblasts viaantioxidant actions. A, apoptosis was quantified by determining caspase 3activity in OB-6 cells treated for 1 h with BSO or DEM followed by the steroids
for 1 and 6 h with the pro-apoptotic agent etoposide (5 � 10�5M), TNF� (10�9
M), or H2O2 (5 � 10�5M). Bars indicate mean � S.D. of triplicate determina-
tions; * indicates p � 0.05 versus vehicle. B, C2C12 cells transfected with avector control or wild-type p66shc plasmid, along with green fluorescent pro-tein, were treated with or without H2O2 (5 � 10�5
M). The number of apopto-tic cells was determined by examining the nuclear morphology of fluorescentcells 6 h later. Bars indicate mean � S.D. of triplicate determinations; * p �0.05 versus vector control untreated. C, OB-6 cells were treated for 1 h with theMEK inhibitor PD98059 (5 � 10�5
M), then the indicated steroids (10�8M)
were added, and 1 h later the cultures were exposed for 15 min to H2O2 (5 �10�5
M). Phosphorylated p66shc was determined by Western blot analyses incell lysates.
Skeletal Involution, Oxidative Stress, and Sex Steroids
27294 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 282 • NUMBER 37 • SEPTEMBER 14, 2007
by guest on June 13, 2018http://w
ww
.jbc.org/D
ownloaded from

resulting from reduced physical activity with old age (74).Hence, the effects of aging must override the effects of sexsteroid deficiency by several mechanisms, some of whichresult directly from increased oxidative stress. In agreementwith this contention we have shown before that the expectedchanges in bone cell progenitor numbers, histomorphom-etry, and BMD that occur after orchidectomy were eitherabsent or greatly attenuated in a mouse model of defectiveosteoblastogenesis or in Swiss Webster mice treated simul-taneously with glucocorticoids (17, 18).Although much has been learned about age-related bone
loss, it remains unknown whether its mechanisms are the samein all aging persons, but recent evidence indicates that the cel-lular and molecular mechanisms responsible may vary fromsubject to subject (75–77). One reason for such variation couldbe that the increase in responsiveness differs among individu-
als, so that somewould losemore bone than others at, for exam-ple, the same glucocorticoid level. In closing, we submit that theresults presented in this report constitute a major paradigmshift in the understanding of the pathogenesis of osteoporo-sis from the “estrogen-centric” view that has dominated thelast 50 years to a multifactorial one, in which oxidative dam-age is a pivotal mechanism for females and males alike.Appreciation of the facts that loss of bone mass and strengthoccur with aging irrespective of sex steroid loss in animalsand humans, and that many other factors contribute to thepathogenesis of skeletal involution, necessitates reappraisal ofcurrent ideas about the timing of initiation of prevention strat-egies, meaningful re-classification of osteoporosis based on theprevailing pathogenetic factor(s), and perhaps even the need forindividualization of treatment with future pathogenetic factor-tailored therapies.
FIGURE 10. ROS-activated signals affecting the genesis and lifespan of osteoblasts and osteoclasts and the counter-regulatory actions of sex steroids. Effectsof ROS, exemplified here by H2O2, on the genesis and survival of both osteoblasts and osteoclasts are depicted in black, and as shown require the same signalingcascades and factors, i.e. ERKs, NF-�B, and osteoclastogenic cytokines like Rankl, Tnf, and interleukin (IL) 6, used by estrogens to regulate the birth and death ofosteoblasts and osteoclasts, albeit in the exactly opposite manner (13, 14). Like most other cell types, bone cells attempt to counteract the adverse effects of ROS byseveral defense mechanisms (depicted in blue). Such mechanisms include the up-regulation of ROS scavenging enzymes (superoxide dismutases and catalases) aswell as DNA-damage repair genes by Forkhead transcription factors (FoxO; see accompanying article, Almeida et al. (82)). Additionally, enzymes like glutathioneperoxidase use glutathione to reduce ROS to alcohols. Glutathione reductase is a key partner in this cycle because it converts the disulfide (GSSG) back into glutathione(GSH). The pro-apoptotic effects of H2O2 on osteoblasts (and probably their mesenchymal stem cell progenitors) are associated with phosphorylation of p53 andp66shc. ROS decreases osteoblastogenesis by at least two mechanisms: 1) antagonism of Wnt signaling by diversion of �-catenin from Tcf- to FoxO-mediatedtranscription (see accompanying article, Almeida et al. (82)); and 2) direct and sustained activation of ERKs and NF-�B (78). ROS inhibit osteoclast apoptosis andstimulate osteoclastogenesis by increasing RANKL production in cells of the stromal/osteoblastic lineage (79) as well as an ERK/NF-�B/Tnf/interleukin 6-mediatedmechanism (39). E2 or DHT antagonizes the effects of ROS via several mechanisms (depicted in red): (a) up-regulation of GSR (and thioredoxin reductase activity (39, 80);(b) attenuation of p66shc phosphorylation via a Src- and ERK-dependent pathway; and (c) down-regulation of the production of osteoclastogenic cytokines like TNFand interleukin 6 via attenuation of NF-�B. In addition, E2 or DHT antagonizes the effects of ROS by attenuating osteoblast apoptosis and stimulating osteoclastapoptosis via a transient and sustained ERK activation, respectively; and by inhibiting osteoclastogenesis, through a sustained ERK activation (81). For the interplaybetween ROS and estrogens on osteoblastogenesis, see “Discussion.”
Skeletal Involution, Oxidative Stress, and Sex Steroids
SEPTEMBER 14, 2007 • VOLUME 282 • NUMBER 37 JOURNAL OF BIOLOGICAL CHEMISTRY 27295
by guest on June 13, 2018http://w
ww
.jbc.org/D
ownloaded from

Acknowledgments—We thank B. L. Riggs, J. Potts, Jr., and R. Bouil-lon for critical review of the manuscript prior to submission; A.Warren, V. Lowe, R. Shelton, K. Vyas, T. Chambers, S. Berryhill, C.Wicker III, E. Hogan, R. Wynne, J. Crawford, and W. Webb fortechnical assistance; and R. I. DeWall for assistance in the prepa-ration of the manuscript.
REFERENCES1. Looker, A. C., Wahner, H. W., Dunn, W. L., Calvo, M. S., Harris, T. B.,
Heyse, S. P., Johnston, C. C. Jr., and Lindsay, R. (1998)Osteoporosis Int., 8,468-489
2. Hui, S. L., Slemenda, C.W., and Johnston, C. C., Jr. (1988) J. Clin. Investig.81, 1804–1809
3. Kleerekoper, M., Villanueva, A. R., Stanciu, J., Rao, D. S., and Parfitt, A.M.(1985) Calcif. Tissue Int. 37, 594–597
4. Burr, D. B., Turner, C. H., Naick, P., Forwood, M. R., Ambrosius, W.,Hasan, M. S., and Pidaparti, R. (1998) J. Biomech. 31, 337–345
5. Heaney, R. P. (2003) Bone 33, 457–4656. Duan, Y., Parfitt, A., and Seeman, E. (1999) J. Bone Miner. Res. 14,
1796–18027. Silva, M. J., and Gibson, L. J. (1997) Bone 21, 191–1998. O’Brien, C. A., Jia, D., Plotkin, L. I., Bellido, T., Powers, C. C., Stewart, S. A.,
Manolagas, S. C., and Weinstein, R. S. (2004) Endocrinology 145,1835–1841
9. Qiu, S., Rao, D. S., Palnitkar, S., and Parfitt, A.M. (2003) J. BoneMiner. Res.18, 1657–1663
10. Tomkinson, A., Reeve, J., Shaw, R. W., and Noble, B. S. (1997) J. Clin.Endocrinol. Metab. 82, 3128–3135
11. Tomkinson, A., Gevers, E. F., Wit, J. M., Reeve, J., and Noble, B. S. (1998)J. Bone Miner. Res. 13, 1243–1250
12. Kousteni, S., Bellido, T., Plotkin, L. I., O’Brien, C. A., Bodenner, D. L., Han,K., DiGregorio, G., Katzenellenbogen, J. A., Katzenellenbogen, B. S., Rob-erson, P. K., Weinstein, R. S., Jilka, R. L., and Manolagas, S. C. (2001) Cell104, 719–730
13. Manolagas, S. C. (2000) Endocr. Rev. 21, 115–13714. Manolagas, S. C., Kousteni, S., and Jilka, R. L. (2002) Recent Prog. Horm.
Res. 57, 385–40915. Manolagas, S. C., Kousteni, S., Chen, J. R., Schuller, M., Plotkin, L., and
Bellido, T. (2004) Kidney Int. Suppl. 91, S41–S4916. Jilka, R. L., Takahashi, K.,Munshi,M.,Williams,D.C., Roberson, P. K., and
Manolagas, S. C. (1998) J. Clin. Investig. 101, 1942–195017. Jilka, R. L., Weinstein, R. S., Takahashi, K., Parfitt, A. M., and Manolagas,
S. C. (1996) J. Clin. Investig. 97, 1732–174018. Weinstein, R. S., Jia, D., Powers, C. C., Stewart, S. A., Jilka, R. L., Parfitt,
A. M., and Manolagas, S. C. (2004) Endocrinology 145, 1980–198719. Abe, E., Yamamoto, M., Taguchi, Y., Lecka-Czernik, B., O’Brien, C. A.,
Economides, A. N., Stahl, N., Jilka, R. L., and Manolagas, S. C. (2000)J. Bone Miner. Res. 15, 663–673
20. Manolagas, S. C., Jilka, R. L., Bellido, T., O’Brien, C. A., and Parfitt, A. M.(1996) in Principles of Bone Biology, (Bilezikian, J. P., Raisz, L. G., andRodan, G. A., eds) Academic Press, San Diego
21. Quarrie, J. K., and Riabowol, K. T. (2004) Sci. Aging Knowledge Environ.2004, re5
22. Finkel, T., and Holbrook, N. J. (2000) Nature 408, 239–24723. Migliaccio, E., Giorgio, M., Mele, S., Pelicci, G., Reboldi, P., Pandolfi, P. P.,
Lanfrancone, L., and Pelicci, P. G. (1999) Nature 402, 309–31324. Trinei, M., Giorgio, M., Cicalese, A., Barozzi, S., Ventura, A., Migliaccio,
E., Milia, E., Padura, I. M., Raker, V. A., Maccarana, M., Petronilli, V.,Minucci, S., Bernardi, P., Lanfrancone, L., and Pelicci, P. G. (2002) Onco-gene 21, 3872–3878
25. Giorgio, M., Migliaccio, E., Orsini, F., Paolucci, D., Moroni, M., Contursi,C., Pelliccia, G., Luzi, L.,Minucci, S.,Marcaccio,M., Pinton, P., Rizzuto, R.,Bernardi, P., Paolucci, F., and Pelicci, P. G. (2005) Cell 122, 221–233
26. Tyner, S. D., Venkatachalam, S., Choi, J., Jones, S., Ghebranious, N.,Igelmann,H., Lu, X., Soron,G., Cooper, B., Brayton, C., Hee, P. S., Thomp-son, T., Karsenty, G., Bradley, A., and Donehower, L. A. (2002) Nature
415, 45–5327. Dickinson, D. A., and Forman, H. J. (2002) Ann. N. Y. Acad. Sci. 973,
488–50428. Huang, X., Frenkel, K., Klein, C. B., and Costa, M. (1993) Toxicol. Appl.
Pharmacol. 120, 29–3629. Kousteni, S., Chen, J.-R., Bellido, T., Han, L., Ali, A. A., O’Brien, C., Plotkin,
L. I., Fu, Q., Mancino, A. T., Wen, Y., Vertino, A. M., Powers, C. C.,Stewart, S. A., Ebert, R., Parfit, A. M., Weinstein, R. S., Jilka, R. L., andManolagas, S. C. (2002) Science 298, 843–846
30. Weinstein, R. S., Jilka, R. L., Parfitt, A. M., and Manolagas, S. C. (1998)J. Clin. Investig. 102, 274–282
31. Weinstein, R. S., Chen, J. R., Powers, C. C., Stewart, S. A., Landes, R. D.,Bellido, T., Jilka, R. L., Parfitt, A. M., and Manolagas, S. C. (2002) J. Clin.Investig. 109, 1041–1048
32. Kousteni, S., Han, L., Chen, J.-R., Almeida,M., Plotkin, L. I., Bellido, T., andManolagas, S. C. (2003) J. Clin. Investig. 111, 1651–1664
33. Lecka-Czernik, B., Gubrij, I., Moerman, E. J., Kajkenova, O., Lipschitz,D. A., Manolagas, S. C., and Jilka, R. L. (1999) J. Cell Biochem. 74, 357–371
34. Netter, J., Wasserman, W., and Kutner, M. H. (1990) Applied Linear Sta-tistical Models, 3rd ed., Richard D. Irwin, Inc., Homewood, IL
35. Ahlborg, H. G., Johnell, O., Turner, C. H., Rannevik, G., and Karlsson,M. K. (2003) N. Engl. J. Med. 349, 327–334
36. Duan, Y., Beck, T. J.,Wang, X. F., and Seeman, E. (2003) J. BoneMiner. Res.18, 1766–1774
37. Mobbs, C. V., Cheyney, D., Sinha, Y. N., and Finch, C. E. (1985) Endocri-nology 116, 813–820
38. Nelson, J. F., Latham, K. R., and Finch, C. E. (1975) Acta Endocrinol. 80,744–752
39. Lean, J. M., Davies, J. T., Fuller, K., Jagger, C. J., Kirstein, B., Partington,G. A., Urry, Z. L., andChambers, T. J. (2003) J. Clin. Investig. 112, 915–923
40. De Boer, J., Andressoo, J. O., de Wit, J., Huijmans, J., Beems, R. B., vanSteeg, H., Weeda, G., van der Horst, G. T., van Leeuwen, W., Themmen,A. P., Meradji, M., and Hoeijmakers, J. H. (2002) Science 296, 1276–1279
41. Jagger, C. J., Lean, J. M., Davies, J. T., and Chambers, T. J. (2005) Endocri-nology 146, 113–118
42. Moor, A. N., Gottipati, S., Mallet, R. T., Sun, J., Giblin, F. J., Roque, R., andCammarata, P. R. (2004) Exp. Eye Res. 78, 933–944
43. Chiang, K., Parthasarathy, S., and Santanam, N. (2004) Life Sci. 75,2425–2438
44. Darblade, B., Pendaries, C., Krust, A., Dupont, S., Fouque, M. J., Rami, J.,Chambon, P., Bayard, F., and Arnal, J. F. (2002) Circ. Res. 90, 413–419
45. Sack, M. N., Rader, D. J., and Cannon, R. O., III (1994) Lancet 343,269–270
46. Quintanilla, R. A., Munoz, F. J., Metcalfe, M. J., Hitschfeld, M., Olivares,G., Godoy, J. A., and Inestrosa, N. C. (2005) J. Biol. Chem. 280,11615–11625
47. Sudoh, N., Toba, K., Akishita, M., Ako, J., Hashimoto, M., Iijima, K., Kim,S., Liang, Y. Q., Ohike, Y.,Watanabe, T., Yamazaki, I., Yoshizumi, M., Eto,M., and Ouchi, Y. (2001) Circulation 103, 724–729
48. Arnal, J. F., Clamens, S., Pechet, C., Negre-Salvayre, A., Allera, C., Giro-lami, J. P., Salvayre, R., and Bayard, F. (1996) Proc. Natl. Acad. Sci. U. S. A.93, 4108–4113
49. Sawada, H., Ibi, M., Kihara, T., Urushitani, M., Honda, K., Nakanishi, M.,Akaike, A., and Shimohama, S. (2000) FASEB J. 14, 1202–1214
50. Chambliss, K. L., Simon, L., Yuhanna, I. S., Mineo, C., and Shaul, P. W.(2005)Mol. Endocrinol. 19, 277–289
51. Lu, Q., Pallas, D. C., Surks, H. K., Baur, W. E., Mendelsohn, M. E., andKaras, R. H. (2004) Proc. Natl. Acad. Sci. U. S. A. 101, 17126–17131
52. Baba, T., Shimizu, T., Suzuki, Y. I., Ogawara, M., Isono, K. I., Koseki, H.,Kurosawa, H., and Shirasawa, T. (2005) J. Biol. Chem. 280, 16417–16426
53. Lapointe, J., Kimmins, S., Maclaren, L. A., and Bilodeau, J. F. (2005) Endo-crinology 146, 2583–2592
54. Yen, C. H., Hsieh, C. C., Chou, S. Y., and Lau, Y. T. (2001) Life Sci. 70,403–413
55. Klein, R. F., Allard, J., Avnur, Z., Nikolcheva, T., Rotstein, D., Carlos, A. S.,Shea, M., Waters, R. V., Belknap, J. K., Peltz, G., and Orwoll, E. S. (2004)Science 303, 229–232
56. Schulz, E., Arfai, K., Liu, X., Sayre, J., and Gilsanz, V. (2004) J. Clin. Endo-
Skeletal Involution, Oxidative Stress, and Sex Steroids
27296 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 282 • NUMBER 37 • SEPTEMBER 14, 2007
by guest on June 13, 2018http://w
ww
.jbc.org/D
ownloaded from

crinol. Metab. 89, 4246–425357. Hurh, Y. J., Chen, Z. H., Na, H. K., Han, S. Y., and Surh, Y. J. (2004) J.
Toxicol. Environ. Health Part A 67, 1939–195358. Deroo, B. J., Hewitt, S. C., Peddada, S. D., and Korach, K. S. (2004) Endo-
crinology 145, 5485–549259. Mishra, D. P., and Shaha, C. (2005) J. Biol. Chem. 280, 6181–619660. Klotzbuecher, C. M., Ross, P. D., Landsman, P. B., Abbott, T. A., III, and
Berger, M. (2000) J. Bone Miner. Res. 15, 721–73961. Cummings, S. R., Karpf, D. B., Harris, F., Genant, H. K., Ensrud, K.,
LaCroix, A. Z., and Black, D. M. (2002) Am. J. Med. 112, 281–28962. Li, X., Masinde, G., Gu, W., Wergedal, J., Mohan, S., and Baylink, D. J.
(2002) Genomics 79, 734–74063. Wergedal, J. E., Sheng, M. H., Ackert-Bicknell, C. L., Beamer, W. G., and
Baylink, D. J. (2005) Bone 36, 111–12264. Manolagas, S. C. (2006) BoneKey-Osteovision 3, 5–1465. Nelson, J. F., Felicio, L. S., Osterburg, H. H., and Finch, C. E. (1992) Endo-
crinology 130, 805–81066. Lacombe, A., Lelievre, V., Roselli, C. E., Salameh, W., Lue, Y. H., Lawson,
G.,Muller, J. M.,Waschek, J. A., and Vilain, E. (2006) Proc. Natl. Acad. Sci.U. S. A. 103, 3793–3798
67. Bellido, T., Jilka, R. L., Boyce, B. F., Girasole, G., Broxmeyer, H., Dalrymple,S. A., Murray, R., and Manolagas, S. C. (1995) J. Clin. Investig. 95,2886–2895
68. Vanderschueren, D., Boonen, S., Ederveen, A. G., de Coster, R., VanHerck, E., Moermans, K., Vandenput, L., Verstuyf, A., and Bouillon, R.(2000) Bone 27, 611–617
69. Kousteni, S., Almeida, M., Han, L., Bellido, T., Jilka, R. L., and Manolagas,S. C. (2007)Mol. Cell Biol. 27, 1516–1530
70. Almeida, M., Chen, X., Han, L., Martin-Millan, M., Lowe, V., Warren, A.,Stewart, S. A., Kousteni, S., Weinstein, R. S., O’Brien, C. A., Bellido, T.,Jilka, R. L., and Manolagas, S. C. (2006) J. Bone Miner. Res. 21, S95
71. Chen, X.-D., Dusevich, V., Feng, J. Q., Manolagas, S. C., and Jilka, R. L.(2007) J. Bone Miner. Res., in press
72. Tothova, Z., Kollipara, R., Huntly, B. J., Lee, B. H., Castrillon, D. H., Cullen,D. E., McDowell, E. P., Lazo-Kallanian, S., Williams, I. R., Sears, C., Arm-strong, S. A., Passegue, E., DePinho, R. A., and Gilliland, D. G. (2007) Cell128, 325–339
73. Akune, T., Ohba, S., Kamekura, S., Yamaguchi, M., Chung, U. I., Kubota,N., Terauchi, Y., Harada, Y., Azuma, Y., Nakamura, K., Kadowaki, T., andKawaguchi, H. (2004) J. Clin. Investig. 113, 846–855
74. Aguirre, J. I., Plotkin, L. I., Stewart, S. A., Weinstein, R. S., Parfitt, A. M.,Manolagas, S. C., and Bellido, T. (2006) J. Bone Miner. Res. 21, 605–615
75. Smith, R. G., Betancourt, L., and Sun, Y. (2005) Endocr. Rev. 26, 203–25076. Sapolsky, R. M., Romero, L. M., and Munck, A. U. (2000) Endocr. Rev. 21,
55–8977. Seeman, T. E., and Robbins, R. J. (1994) Endocr. Rev. 15, 233–26078. Bai, X. C., Lu, D., Bai, J., Zheng,H., Ke, Z. Y., Li, X.M., and Luo, S.Q. (2004)
Biochem. Biophys. Res. Commun. 314, 197–20779. Bai, X. C., Lu, D., Liu, A. L., Zhang, Z.M., Li, X.M., Zou, Z. P., Zeng,W. S.,
Cheng, B. L., and Luo, S. Q. (2005) J. Biol. Chem. 280, 17497–1750680. Lean, J. M., Jagger, C. J., Kirstein, B., Fuller, K., and Chambers, T. J. (2005)
Endocrinology 146, 728–73581. Chen, J. R., Plotkin, L. I., Aguirre, J. I., Han, L., Jilka, R. L., Kousteni, S.,
Bellido, T., and Manolagas, S. C. (2005) J. Biol. Chem. 280, 4632–463882. Almeida, M., Han, L., Martin-Millan, M., O’Brien, C. A., and Manolagas,
S. C. (2007) J. Biol. Chem. 280, 27298–27305
Skeletal Involution, Oxidative Stress, and Sex Steroids
SEPTEMBER 14, 2007 • VOLUME 282 • NUMBER 37 JOURNAL OF BIOLOGICAL CHEMISTRY 27297
by guest on June 13, 2018http://w
ww
.jbc.org/D
ownloaded from

Parfitt, Robert S. Weinstein, Robert L. Jilka and Stavros C. ManolagasK. Roberson, Stavroula Kousteni, Charles A. O'Brien, Teresita Bellido, A. Michael
Maria Almeida, Li Han, Marta Martin-Millan, Lilian I. Plotkin, Scott A. Stewart, PaulaLoss of Sex Steroids
Skeletal Involution by Age-associated Oxidative Stress and Its Acceleration by
doi: 10.1074/jbc.M702810200 originally published online July 10, 20072007, 282:27285-27297.J. Biol. Chem.
10.1074/jbc.M702810200Access the most updated version of this article at doi:
Alerts:
When a correction for this article is posted•
When this article is cited•
to choose from all of JBC's e-mail alertsClick here
http://www.jbc.org/content/282/37/27285.full.html#ref-list-1
This article cites 79 references, 16 of which can be accessed free at
by guest on June 13, 2018http://w
ww
.jbc.org/D
ownloaded from