Embed Size (px)
Citation preview

Síndrome de Sézary Criterios diagnósticos y conceptos actuales
Sebastián Colombo\a Wehbe^ y Mercedes L. Hassan^
RESUMEN: Los linfomas cutáneos primarios son procesos linfoproliferativos malignos cuyo órgano diana es la piel y pueden ser de estirpe T o B. El síndrome de Sézary es una rara forma de linfoma cutáneo de células T (LCCT), caracterizado por eritrodermia, con compromiso he-matológico precoz , curso clínico agresivo y principalmente mal pronóstico.
El diagnóstico inicial sigue siendo muy dificultoso; sin embargo los adelantos en el campo de la inmunogenética han hecho posible un avance extraordinario en el conocimiento de esta enfermedad y a partir de ellos se han propuesto criterios concretos que nos acercan al diagnóstico con certeza, a saber: más de 1000 células de Sézary circulantes por mm^ relación CD4/ CD8 > 10, expresión aberrante de marcadores pan T, evidencias de células T clónales o un clon de células T con anormalidades cromosómicas.
Palabras clave: síndrome de Sézary - eritrodermia - leucemia.
ABSTRACT: Primary cutaneous lymphomas are lymphoproliferative disorders that develop pri-marily in the skin. They derive either from T- cell or B- cell. Sézary syndrome is a rare form of cutaneous T- cell lymphoma, characterized by erythroderma, early hematologic involvement, aggressive clinical course and poor prognosis.
The diagnosis is not easy but the current advances in Immunogenetic have increased our knowledge of this disease. The following criteria have been proposed for its' diagnosis: an absolute Sézary cell count of 1000 cells/ mm^ or more; a CD4/CD8 ratio of 10 or higher; aberrant loss or expression of pan- T cell markers; evidence of a T- cell clone in the blood or a chromo-somally abnormal T- cell clone.
Key words: Sézary Syndrome - Erythroderma - Leukemia.
Arch. Argent. Dermatol. 54:147-152, 2004
INTRODUCCION
El síndrome de Sézary (SS), expresión leucémica de un l infoma cutáneo de células T eritrodérmico, es una entidad rara de curso clínico agresivo y mal pronóstico. Su diagnóst ico, dif icultoso y excepcional , se basa en la demostración de compromiso hematológico neoplásico por el hal lazgo en sangre periférica de linfocitos T con rasgos morfológicos distintivos y evidencia de clonali-dad mediante técnicas inmunohistoquímicas e inmuno-genéticas.
El objetivo de este trabajo es destacar los nuevos criterios diagnóst icos que definen al síndrome de Sézary y remarcar las diferencias clínico-evolutivas entre los distintos l infomas cutáneos eri trodérmicos.
COMENTARIOS
Si bien Hal lopeau en 1892 y Leredde en 1894 fueron los primeros en describir la eri trodermia en el contexto
' Médico especialista en Dermatología. ^ Médica Residente de 4- año.
Jefe de División Dermatología. Prof. Adjunta U.B.A. División Dermatología. Hospital Gral. de Agudos J. M Ramos Me-jía. Urquiza 609, 2° piso. (1221). Buenos Aires.
de los linfomas cutáneos, y que Leo von Zumbusch en 1919 relata un paciente con eritrodermia, leucemia y lin-focitosis (leucemia cutis)', la denominación actual de esta entidad hace referencia a Sézary y Bouvrain, quienes en 1938 identifican por primera vez células mononuclea-res atípicas ("células monstruosas") tanto en sangre como en piel de un paciente con eritrodermia crónica^ Posteriormente, en 1939 Baccaredda hizo un exhaustivo estudio del tema, a propósito de un caso correspondiente a una mujer de 67 años con eritrodermia, adeno-patías múltiples, hepatoesplenomegal ia y hallazgo de células de núcleo voluminoso en sangre periférica. El autor consideró el cuadro como una reticuiohistiocitosis hiperplásica que da lugar a una eritrodermia pigmentada con hipertrofia ganglionar En 1949, Sézary efectuó una revisión sobre esta patología basándose en sus dos observaciones de 1938, agrega otra de 1943 e incorpora la de Baccaredda. Denominó descript ivamente a la enfermedad como una reticuiosis maiigna ieucémica a tiistiocitos - monocitos monstruosos ya forma de eritrodermia edematosa pigmentada^.
En 1959, Bureau, Barriere y Guénel señalaron la posible participación de la médula ósea, hasta ese momento no mencionada". TasweII y Winkelmann, dos años más tarde, destacaron la naturaleza linfocitaria de las células
Recibido: 21-8-2003. Aceptado para pubiicación: 16-3-2004. 147

Sebastián Colombo y colaboradores
Fig. 1: Eritrodermia rojo intenso. FIg. 2: Eritrodermia rojo intenso.
neoplásicas y designaron a esta entidad como síndrome de Sézary. Mas adelante, en 1968, Lutzner y Jordán describieron al microscopio electrónico las características morfológicas ultraestrucurales de dichas células, proveyendo un nuevo criterio para su identificación^.
En 1978, la División Tratamiento del Cáncer del Instituto Nacional del Cáncer de los EEUU, propuso englobar bajo el término Linfomas cutáneos a linfocitos Ta la MF y SS".
En Argent ina Pierini, Abulaf ia y Carvalho en 1958 aportaron la pr imera publicación sobre el tema, t i tulando el trabajo Eritrodermia reticulósicá^. Le siguieron luego los trabajos de Mosto^ en 1959 y de Mazzini y col.^^ en 1961 , destacándose por ser las investigaciones iniciales de la enfermedad en nuestro país.
Se manif iesta como una eri trodermia de curso crónico, usualmente acompañada de pol iadenopatías, alopecia difusa, distrofias ungulares, queratodermia palmo-plantar y prurito intenso (Figs. 1-2-3). Clásicamente se describe la progresión hacia una melanodermia, localizada o difusa, adquir iendo el paciente una tonalidad "gris acerada". Esta condición podría estar relacionada con factores raciales, ya que la mayoría de las comunicaciones en nuestro medio mencionan el oscurecimiento cu-
Fig. 3: Queratodermia plantar
148 Arch. Argent. Dermatol.
Síndrome de Sézary
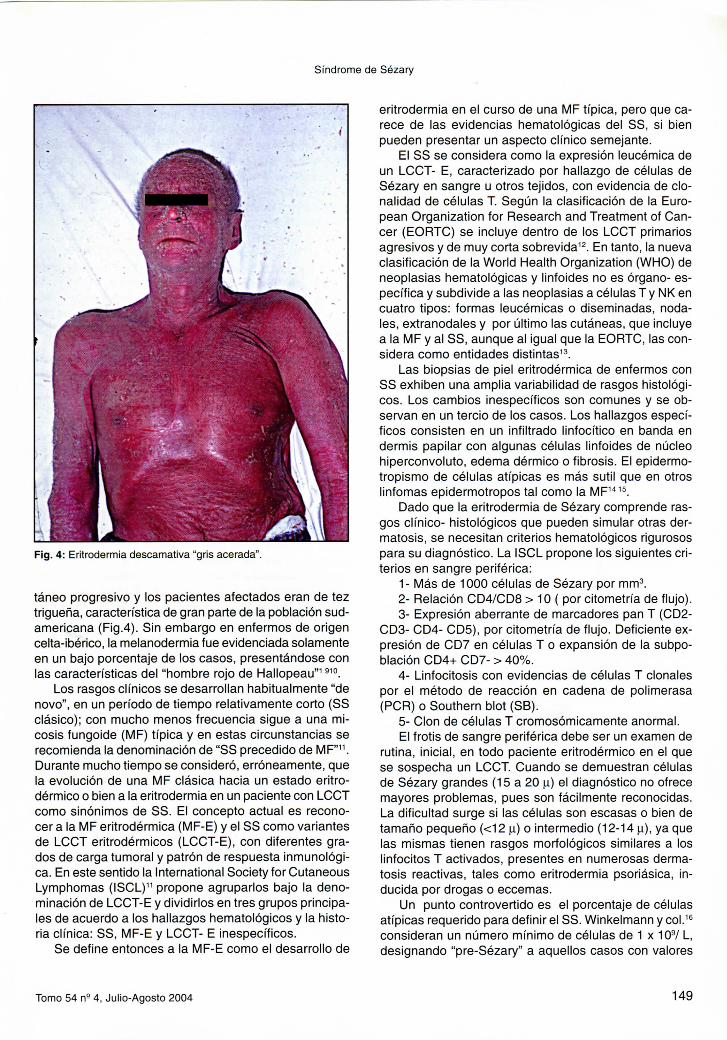
Fig. 4: Eritrodermia descamativa "gris acerada".
táneo progresivo y los pacientes afectados eran de tez trigueña, característica de gran parte de la población sudamericana (Fig.4). Sin embargo en enfermos de origen celta-ibérico, la melanodermia fue evidenciada solamente en un bajo porcentaje de los casos, presentándose con las características del "hombre rojo de Ha l lopeau" ' ' ' ° .
Los rasgos clínicos se desarrol lan habitualmente "de novo", en un período de t iempo relat ivamente corto (SS clásico); con mucho menos frecuencia sigue a una micosis fungoide (IVIF) típica y en estas circunstancias se recomienda la denominación de "SS precedido de M F " " . Durante mucho t iempo se consideró, erróneamente, que la evolución de una MF clásica hacia un estado eritrodérmico o bien a la eritrodermia en un paciente con LCCT como sinónimos de SS. El concepto actual es reconocer a la MF eri trodérmica (MF-E) y el SS como variantes de LCCT eri trodérmicos (LCCT-E), con diferentes grados de carga tumoral y patrón de respuesta inmunológi-ca. En este sentido la International Society for Cutaneous Lymphomas ( ISCL) " propone agruparlos bajo la denominación de LCCT-E y dividirlos en tres grupos principales de acuerdo a los hal lazgos hematológicos y la historia clínica: SS, MF-E y LCCT- E inespecíf icos.
Se define entonces a la MF-E como el desarrol lo de
eri trodermia en el curso de una MF típica, pero que carece de las evidencias hematológicas del SS, si bien pueden presentar un aspecto clínico semejante.
El SS se considera como la expresión leucémica de un LCCT- E, caracterizado por hallazgo de células de Sézary en sangre u otros tejidos, con evidencia de clo-nalidad de células T. Según la clasificación de la Euro-pean Organizat ion for Research and Treatment of Cáncer (EORTC) se incluye dentro de los LCCT primarios agresivos y de muy corta sobrevida'^. En tanto, la nueva clasif icación de la World Health Organization (WHO) de neoplasias hematológicas y linfoides no es órgano- específ ica y subdivide a las neoplasias a células T y NK en cuatro t ipos: formas leucémicas o diseminadas, nodales, extranodales y por último las cutáneas, que incluye a la MF y al SS, aunque al igual que la EORTC, las considera como entidades d is t in tas" .
Las biopsias de piel eritrodérmica de enfermos con SS exhiben una amplia variabil idad de rasgos histológicos. Los cambios inespecíf icos son comunes y se observan en un tercio de los casos. Los hallazgos específ icos consisten en un infiltrado linfocítico en banda en dermis papilar con algunas células linfoides de núcleo hiperconvoluto, edema dérmico o fibrosis. El epidermo-tropismo de células atípicas es más sutil que en otros l infomas epidermotropos tal como la M F " " ^ .
Dado que la eri trodermia de Sézary comprende rasgos clínico- histológicos que pueden simular otras dermatosis, se necesitan criterios hematológicos rigurosos para su diagnóst ico. La ISCL propone los siguientes criterios en sangre periférica:
1- Más de 1000 células de Sézary por mm^. 2- Relación CD4/CD8 > 10 ( por citometría de flujo). 3- Expresión aberrante de marcadores pan T (CD2-
CD3- CD4- CD5), por citometría de flujo. Deficiente expresión de CD7 en células T o expansión de la subpo-blación CD4+ CD7- > 40%.
4- Linfocitosis con evidencias de células T clónales por el método de reacción en cadena de polimerasa (PCR) o Southern blot (SB).
5- Clon de células T cromosómicamente anormal. El frotis de sangre periférica debe ser un examen de
rutina, inicial, en todo paciente eritrodérmico en el que se sospecha un LCCT. Cuando se demuestran células de Sézary grandes (15 a 20 |^) el diagnóstico no ofrece mayores problemas, pues son fáci lmente reconocidas. La dificultad surge si las células son escasas o bien de tamaño pequeño (<12 \i) o intermedio (12-14 | i ) , ya que las mismas t ienen rasgos morfológicos similares a los linfocitos T act ivados, presentes en numerosas dermatosis reactivas, tales como eritrodermia psoriásica, inducida por drogas o eccemas.
Un punto controvert ido es el porcentaje de células atípicas requerido para definir el SS. Winkelmann y col.'^ consideran un número mínimo de células de 1 x 10^/ L, designando "pre-Sézary" a aquellos casos con valores
Tomo 54 n** 4, Julio-Agosto 2004 149

Sebastián Colombo y colaboradores
inferiores, lo cual no signif ica una etapa previa a la verdadera ent idad tal como lo demostró el seguimiento de esos pacientes. Otros autores sugieren un porcentaje necesario de células de Sézary en un rango entre el 5% al 2 0 % del total de linfocitos en el conteo. La ISCL recomienda utilizar como valor de referencia un conteo absoluto de linfocitos de 1000 células por mm^, porque tendría implicancias pronost icas" . De todos modos se debe tener en cuenta que tales criterios son arbitrarios y no diferencian entre una población clonal o policlonal de células T; además son hal lazgos dependientes del observador.
La EORTC enuncia que la evidencia de una expansión de la población T CD4+, resultante en un significativo incremento de la relación CD4 / CD8 (>10), debe considerarse como criterio diagnóst ico ya que suele observarse en un 8 0 % de los enfermos con SS. Los pacientes incluidos en este grupo t ienen alta carga tumo-ral y pobre pronóst ico, con una sobrevida promedio de 24 meses. La ISCL avala dichas considerac iones"
Actualmente se ha desarrol lado un amplio panel de anticuerpos monoclonales para determinar el inmunofe-notipo de los l infomas tanto en tejido fijado en formol e incluido en parafina como en sangre periférica. En ésta, la técnica de ci tometría de flujo permite trabajar con un número importante de anticuerpos y se puede detectar la existencia de una proliferación de linfocitos T cooperadores (CD4-I-), maduros (CD2-I-, CD3-I-, CD5+) , de memor ia (CD45-RO-I-, CDw29-i-). La pérdida de marcadores pan-T es sugestiva de mal ignidad'^
El antígeno CD7 es una molécula expresada en un 9 0 % de los l infocitos T CD4+ normales'^. La ausencia de CD7 permite del inear un subgrupo de células de memoria (CD4-(-, CD45-RO-(-, CD45-RA-) , el cual es pequeño en pacientes normales, pero se expande marcadamente en enfermos con SS^". Sin embargo, se ha demostrado un incremento de esta subpoblación en dermatosis benignas, como así también no todas las células de Sézary son CD7 negativas. En conclusión, los estudios por citometría de flujo ayudan al diagnóstico si demuestran una expansión CD4-I- / CD7- (> 40%), junto con una pérdida parcial o completa de marcadores T'^. Bernengo y col. determinaron que un incremento del fenotipo celular T CD4+ / CD26- mayor al 3 0 % de los linfocitos de sangre periférica, evidenciaría el compromiso leucémico del SS^'.
Recientemente, Scala y col. demostraron que el in-munofenotipo CD4-I- CD60-h CD26- CD49d- está fuertemente asociado con clonal idad de células T en sangre de pacientes con SS^^.
Para cumplir con su función en el reconocimiento an-tigénico, los linfocitos reordenan el ADN de los genes que codif ican las regiones variables de los receptores de superficie responsables de dicha interacción, o sea las inmunoglobul inas de superficie en los linfocitos B y el receptor T (TCR) en las células T. En una población
monoclonal todos los linfocitos poseen el mismo reordenamiento. Su utilidad radica, por un lado, en determinar la naturaleza T o B de la población linfoide en estudio en función de los genes que reordena, y además permite el diagnóstico de policlonalidad (y presumible benignidad) o monoclonal idad (y presumible malignidad) de una población l infoide". El análisis inmunogenético de los LCCT puede efectuarse por técnicas moleculares tales como Southern blotting o el método de reacción en cadena de polimerasa (PCR) que miden genes de la cadena (3 del TCR (TCR-P) o genes de la cadena (3 del TCR (TCRP). Ambas son altamente específicas, ya que muy raramente se reportan hallazgos de rearreglo clonal de TCR en pacientes con dermatosis benignas. Sin embargo la técnica por PCR es mucho más sensible, porque al basarse en la amplif icación del ADN pueden detectar poblaciones que representan entre el 0 , 0 0 1 % al 0 , 1 % de las células en estudio. Por otra parte son más rápidas, sencillas y no emplean sondas radioactivas, siendo las técnicas de elección en la actual idad'^
Dada la alta sensibil idad de este método se han detectado células T clónales en parapsoriasis u otros LCCT incluso no eritrodérmicos. Por esta razón la ISCL sugiere que la determinación de clonalidad debe estar acompañada de otros criterios que expresen la carga tumo-ral, como el conteo absoluto de células de Sézary o la relación C D 4 / C D 8 " .
Los estudios citogenéticos sobre células malignas circulantes en sangre periférica tienen como objetivo identificar anormalidades cromosómicas y la correlación de las mismas con los estadios clínicos y la sobrevida. Las anormalidades cromosómicas no son específicas y pueden ser tanto numéricas como estructurales. La alteración más frecuente resulta de la pérdida de material a nivel de los brazos cortos de los cromosomas 1 y 8. En el Cr. 1, la región ubicada entre 1p 22 y 1p 36 requiere un análisis detallado, ya que las aberraciones de dicha zona estarían involucradas en procesos de transformación maligna y/o progresión de MF / SS. Se describen, también alteraciones en los cromosomas 2, 4, 5, 6, 9, 10, 15, 17, 1 9 y 202''25.
Recientemente Karenko y col.^"^ demostraron que las aberraciones en los cromosomas 8 y 17 están especialmente asociadas con la actividad y progresión de la enfermedad, ya que podrían reflejar mutaciones a nivel del gen supresor tumoral p53 (17p 13) o del oncogén c- myc (8q 24.12- 24.13). Si bien en los LCCT primarios no existe una correlación concreta entre la expresión de p53 y el pronóstico, en este estudio seis de siete pacientes con aberraciones clónales evidenciaron progreso continuo del proceso y cuatro de ellos fallecieron dentro de los 30 meses de la detección del clon.
La demostración de un clon celular cromosómicamente anormal debiera ser adoptado como criterio independiente para el diagnóstico de SS, que si bien es detectable en pocos casos su hallazgo es altamente específ ico".
150 Arch. Argent. Dermatol.

Síndrome de Sézary
Por últ imo queda definir aquel los casos de LCCT-E que no alcanzan a cumplir los criterios diagnósticos de MF-E o SS. Se recomienda categorizarlos como "LCCT-E inespecíf icos o no bien definidos" hasta tanto el curso evolutivo de la enfermedad permita o no un acercamiento con mayor certeza al d iagnóst ico" .
CONCLUSIONES
Con los criterios actuales la eritrodermia de Sézary podría homologarse a la " leucemia cutis" de von Zumbusch y el "hombre rojo" de Hal lopeau, en tanto que el color gris acerado sería común a otras eritrodermias ines-pecíf icas de curso crónico en pacientes con tendencia a la h iperpigmentación, por lo tanto es difícil confirmar la presunción diagnóst ica sólo por el color de la piel.
Predecir la evolución del enfermo eritrodérmico con células de Sézary circulantes parece basarse en criterios laboratoriales bien definidos, mediante técnicas especial izadas ( inmunohistoquímicas e inmunogenéticas), que permiten delimitar poblaciones de pacientes con distinta sobrevida. Esta es del 11 % en 5 años, promedio 24 meses si se toma en cuenta un incremento de la relación C D 4 / C D 8 ( > 1 0 ) " ' 2 .
Russel l -Jones y Wh i t take r " demostraron que 57 de 65 pacientes con diagnóstico de SS y más de 10% de células atípicas circulantes exhibieron clonalidad en sangre periférica (por SB y/o PCR), y que 23 de las 40 muertes de este grupo fueron atribuidas al l infoma vs. ninguna del grupo no clonal. La sobrevida media del grupo clonal fue de 45 meses.
A su vez, en enfermos en los que se detectó un clon con aberraciones cromosómicas, la supervivencia no fue superior a los 30 meses
Considerando que es necesario distinguir dos cuadros de muy diferente pronóstico y sobrevida, estos criterios parecen ser los más vál idos a la hora de diagnosticar una er i t rodermia de Sézary (l infoma cutáneo de células T agresivo), diagnóst ico presuntivo excepcional-mente conf i rmado sólo por el aspecto clínico de la erit rodermia.
Los adelantos en el campo de la inmunogenética y la biología molecular han hecho posible un avance extraordinario para el diagnóst ico, clasif icación y diferenciación del SS, y han ampl iado el conocimiento para el desarrollo de nuevas modal idades terapéuticas de esta compleja enfermedad.
BIBLIOGRAFIA
1. Burg, G.: Cutaneous lymphomas: A practical approach to erythrodermic CTCL (E-CTCL), AAD Lymphoma 2002, Sym-p o s i u m 3 1 1 ; F r iday ; F e b r u a r y 22 , 2 0 0 2 , www.cutaneouslymphoma.org
2. Sézary A.; Bouvrain, Y.: Erythrodermie avec présence de cel-lules monstrueuses dans le derme et le sang circulant. Bul l Soc Fr Derm Syph 1938; 45: 254-260.
3. P ie r in i , L.E.; Abu la f i a , J . ; Ca rva lho , A. : Er i t rodermia reticulósica. A rch Argent Dermatol 1958, 8: 171-189. 4. Cardama, J.E.; Olivares, L.M.; Gil, J.; Duarte, A.M.; Bianchi, O.: A propósito de aspectos clínicos y nosológicos de la eritrodermia reticulósica de Sézary-Baccaredda. Rev Argent Dermatol 1983, 64: 448-454.
5. Lutzner, IVI.A.; Jordán, H.W.: The ultraestructure ot an abnormal cell in Sézary's syndrome. B lood 1968, 3 1 : 719-726.
6. Mosto, S.J.: Eritrodermia reticulósica (Sézary- Baccaredda). Arch Argent Dermatol 1959, 9: 113-121.
7. Mazzini, M.A.; Scaletzky, A.; Doreski, R: Eritrodermia con melanodermia y eosinof i l ia de evoluc ión fatal (Sézary-Baccaredda frustro). Rev Argent Dermatol 1961: 45: 55-57.
8. Mazzini, M.A.: Scaletzky, A.: Eritrodermia reticulósica de Sézary-Baccaredda. Rev Argent Dermatol 1961: 45: 51-54.
9. Jonquiéres, E.D.: Síndrome de Sézary. Arch Argent Dermatol 1983: 33: 263-281.
10. Scaletzky, A.: A propósito de las eritrodermias melanóticas. Dermatol ILA 1966: 8: 433-437.
11. Vonderheid, E.C.; Bernengo, M.G.: Burg, G.: Duvic. M .e ta l : Update on erythrodermic cutaneous T- cell lymphoma: report of the International Society for Cutaneous Lymphomas. J Am Acad Dermatol 2002: 46: 95-106.
12. Willemze, R.: KerI, H.: Sterry, W.: Berti, E. et.al: EORTC clas-sification for primary cutaneous lymphomas: A proposal from the cutaneous lymphoma study group of the European Organization for Research and Treatment of Cáncer B lood 1997; 90: 354-371.
13. Russell-Jones, R.: World Health Organization classitication of hematopoietic and lymphoid tissues: Implications for der-matology J Am Acad Dermatol 2003: 48: 93-102.
14. Trotter, M.J.; Whittaker, S.J.: Orchard, G.E.; Smith, N.P.: Cutaneous histopathology of Sézary syndrome: a study of 41 cases with a proven circulating T- cell clone. J Cutan Pathol 1997: 24: 286-291.
15. Kamarashev, J.; Burg, G.: Kempf, W. et al: Comparativa analy-sis of histological and immunohistological features in mycosis fungoides and Sézary syndrome. J Cutan Pathol 1998: 25: 407-412.
16. Winkelmann, R.K.: Buechner, S.A.; Díaz Pérez, J.L.: Pre-Sézary syndrome. J Am Acad Dermatol 1984: 10: 992-999.
17. Martí Laborda, R.M.: Estracht, T : Linfomas cutáneos. Med C u t I L A 1998: 26: 113-136.
18. Russell-Jones, R.: Whittaker, S.: T-cell receptor gene analy-sis in the diagnosis of Sézary syndrome. J Am Acad Dermatol 1999: 4 1 : 254-259.
19. Vonderheid, E.C.: Bigler, R.D.: Kotecha. A.: Boselli, C.M.: Lessin, S.R.: Bernengo, M.G.: Polansky, M.: Variable CD7 expression on T cells in the leukemic phase of cutaneous T cell lymphoma (Sézary syndrome). J Invest Dermatol 2001: 117: 654-662.
20. Rappl, G.: Muche, J.M.: Abken, H.: Sterry W. et al: CD4(-^) CD7(-) T cells compose the dominant T- cell clone in the peripheral blood of patients with Sézary syndrome. J Am Acad Dermatol 2001; 44: 456-461.
2 1 . Bernengo, M.G.: Novelli, M.: Quaglino, P. e ta l : The relevance of the CD4+ CD26- subset in the identification of circulating Sézary cells. Br J Dermatol 2001: 144: 125-135.
22. Scala, E.: Narducci, M.G.: Amerio, R: Saliva. G. et al: T cell receptor- V beta analysis identifies a dominant 0060-»- CD26-CD49d- T cell clone in the peripheral blood of Sézary syndrome patients. J Invest Dermatol 2002; 119: 193-196.
23. Cordel, N.; Lenormand, B.: Courville. P.: Laurel, P: Joly. P: Detection of clonal T- cell receptor gamma gene rearrange-
Tomo 54 n- 4, Julio-Agosto 2004 151

Sebastián Colombo y colaboradores
ment with the use of PCR- DGGE for diagnosis of erythroderma. A n n Dermato l Venereol 2001; 128: 220-223.
24. Thangavelu, M.; Finn, W.; Yelavarthi, K.; Roenigk, H. et al: Recurring structural chromosome abnormalities in peripheral blood lymphocytes of patients with mycosis fungoides / Sézary syndrome. B lood 1997, 89: 3371-3377.
25. Mao, X.: Lillington, D.; Scarisbrick, J.J.; Mitchel, T. et al: Molecular cytogenetic analysis of cutaneous T-cell lymphomas: identification of common genetic alterations in Sézary syndrome and mycosis fungoides. Br J Dermatol 2002; 147: 464-475.
26. Karenko, L.; Sarna, S.; Káhkónen, M.; Ranki, A.: Chromosomal
abnormalities in relation to clinical disease in patients with cutaneous T- cell lymphoma: a 5- year follow-up study. Br J Dermatol 2003: 148: 55-64.
27. Russell-Jones, R.; Whittaker, S.: Sézary syndrome: Diagnos-tic criteria and therapeutic options. Semin Cutan Med Surg 2000; 19: 100-108.
Direcc ión posta l : S. Colombo Arenales 2347 5^ C 1124. Buenos Aires E- mail: [email protected]
152 Arch. Argent. Dermatol.






![Expression and Role of Integrin Receptors in Sézary Syndrome · lymphadenopathy, and atypical mononuclear cells (Sezary cells, SC) in the peripheral blood [1,2]. A dense ~ t bandlike](https://img.pdfslide.us/doc/110x75/5e46a965b623cd2b5a646ae7/expression-and-role-of-integrin-receptors-in-szary-syndrome-lymphadenopathy-and.jpg)