Embed Size (px)
Citation preview

J. Sep. Sci. 2013, 36, 3571–3578 3571
Mir Ali FarajzadehElnaz Marzi KhosrowshahiParisa Khorram
Department of AnalyticalChemistry, Faculty of Chemistry,University of Tabriz, Tabriz, Iran
Received April 24, 2013Revised July 31, 2013Accepted August 18, 2013
Research Article
Simultaneous derivatization and air-assistedliquid–liquid microextraction of someparabens in personal care productsand their determination by GC with flameionization detection
A simultaneous derivatization/air-assisted liquid–liquid microextraction technique has beendeveloped for the sample pretreatment of some parabens in aqueous samples. The analyteswere derivatized and extracted simultaneously by a fast reaction/extraction with butylchlo-roformate (derivatization agent/extraction solvent) from the aqueous samples and thenanalyzed by GC with flame ionization detection. The effect of catalyst type and volume,derivatization agent/extraction solvent volume, ionic strength of aqueous solution, pH,numbers of extraction, aqueous sample volume, etc. on the method efficiency was investi-gated. Calibration graphs were linear in the range of 2–5000 �g/L with squared correlationcoefficients >0.990. Enhancement factors and enrichment factors ranged from 1535 to 1941and 268 to 343, respectively. Detection limits were obtained in the range of 0.41–0.62 �g/L.The RSDs for the extraction and determination of 250 �g/L of each paraben were <4.9%(n = 6). In this method, the derivatization agent and extraction solvent were the sameand there is no need for a dispersive solvent, which is common in a traditional dispersiveliquid–liquid microextraction technique. Furthermore, the sample preparation time is veryshort.
Keywords: Derivatization / Microextraction / Parabens / Personal care productsDOI 10.1002/jssc.201300436
1 Introduction
p-Hydroxybenzoates (parabens), such as methylparaben(MP), ethylparaben (EP), and propylparaben (PP), are effec-tive antibacterial and antifungal agents used in cosmetics andoral and pharmaceutical formulations. Parabens have beenalso used as food additives and preservatives in injections andophthalmic preparations. But they are now generally regardedas being unsuitable for these types of formulations owing totheir irritant potential. These experiences may depend on im-mune responses to enzymatically formed metabolites of theparabens in the skin. Systemically, no adverse reactions toparabens have been reported, although they have been asso-ciated with hypersensitivity reactions [1]. Some studies showthat parabens have been found in breast cancer tumors (an
Correspondence: Dr. M. A. Farajzadeh, Department of AnalyticalChemistry, Faculty of Chemistry, University of Tabriz, 5166614766Tabriz, IranE-mail: [email protected]: +98-411-3340191
Abbreviations: AALLME, air-assisted liquid–liquid microex-traction; BCF, butylchloroformate; DLLME, dispersive liquid–liquid microextraction; EF, enrichment factor; EnF, enhance-ment factor; EP, ethylparaben; ER, extraction recovery; FID,flame ionization detection; MP, methylparaben; PP, propyl-paraben; SDME, single drop microextraction
average of 20 ng/g of tissue). They have also displayed theability to slightly mimic estrogen [2]. Another concern is thatthe estrogen-mimic aspect of parabens may be a factor in theincreasing prevalence of early puberty in girls. Because of thepresence of parabens in the environment and their negativeeffects on human health, there is an increasing interest intheir trace analysis and the monitoring of parabens to protecthuman health.
Due to the polar nature of parabens, they are oftenderivatized prior to GC analysis to reduce their adsorp-tion in the chromatographic system and to improve sensi-tivity, peak separation, and peak symmetry [3]. By this ac-tion, parabens are converted to less polar and more volatilespecies. In addition, the derivatization accomplished beforethe extraction step can increase the extractability of the an-alytes [4]. Therefore, to obtain the optimal analysis condi-tions, an appropriate sample preparation including a clean-upstep or concentration and a derivatization procedure, is usu-ally mandatory for parabens. So far, several derivatizationstrategies have been described using alkylation with dia-zomethane [5], or acetic anhydride under alkaline condi-tions maintained using dipotassium hydrogen phosphate[4], silylation with bis(trimethylsilyl)trifluoroacetamide orN-methyl-N-(tert-butyldimethylsilyl)trifluoroacetamide [6, 7],with silyl [8] or fluoroacetyl groups [9], and derivatizationwith trimethylsilyl ester [10] before analysis. However, these
C© 2013 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.jss-journal.com

3572 M. A. Farajzadeh et al. J. Sep. Sci. 2013, 36, 3571–3578
derivatization procedures involve additional steps that in-crease the time required for sample preparation.
The most common techniques for the determination ofparabens and their derivatives in samples are GC [6, 11–15],CE [16,17], HPLC [18,19], MEKC [20], and spectrophotometricmethods [21–25].
Sample pretreatment is the first and the most importantstep of an analytical procedure. Isolation of analytes from thematrix and their preconcentration and sample clean-up areimportant aspects of this process. Several sample preparationmethods have been developed for the analysis of parabens,such as dispersive liquid–liquid microextraction (DLLME) us-ing an extraction solvent lighter than water [8], supercriti-cal fluid extraction [26], supercritical fluid extraction/on-lineheadspace solid-phase microextraction [27], solid-phase mi-croextraction [6], single drop microextraction (SDME) [15],combination of off-line SPE and on-column sample stack-ing [16], stir-bar sorptive extraction [28,29], and in situ deriva-tization, and DLLME [4]. Each of these techniques has someadvantages, but a number of disadvantages are also outlinedfor them. The difficulty of working with solvents lighter thanwater in DLLME, limitations in using supercritical fluid as anextractant, drop instability in SDME, and long analysis timein SDME and SBSE are some of their disadvantages. In 2012,a novel microextraction technique named air-assisted liquid–liquid microextraction (AALLME) was developed in our lab-oratory [30], which is a new version of the DLLME method.In AALLME, no dispersive solvent is used, which reducesorganic solvent consumption. To disperse the extraction sol-vent into the aqueous phase, it is repeatedly withdrawn intoa syringe and then injected into a tube several times.
The aim of this study is to develop a simultaneous deriva-tization and AALLME method followed by GC with flameionization detection (FID) for the analysis of parabens inaqueous samples. The derivatization step is performed usingbutylchloroformate (BCF) to form butylated analytes. BCF is astrong derivatizing reagent and its reaction with parabens oc-curs directly in aqueous media without heating. The formedderivatives of parabens are extracted and concentrated simul-taneously, because the derivatization agent also acts as anextraction solvent. This method needs only a small volumeof BCF as a derivatization reagent and an extraction solvent.Other advantages of the method are: absence of a dispersersolvent, simplicity, rapidity, less hazardous for the environ-ment, and high extraction recoveries (ERs), enrichment fac-tors (EFs), and enhancement factors (EnFs).
2 Materials and methods
2.1 Chemicals and solutions
All parabens (MP, EP, and PP) had a purity of >98%and were obtained from Sigma-Aldrich (St. Louis, MO,USA). BCF, 3-methylpyridine (picoline), methanol, sodiumchloride, hydrochloric acid, and sodium hydroxide werepurchased from Merck (Darmstadt, Germany). Deionized
water (Ghazi, Tabriz, Iran) was used throughout the studyfor the preparation of aqueous solutions. Sodium hydroxideand hydrochloric acid solutions (1 M) were used in the pHadjustment of samples. Solutions of parabens were preparedin methanol at concentrations of 200 and 2000 mg/L (eachpreservative). A standard solution of derivatized parabenswas prepared by dissolving the analytes in a mixture of 4.9mL of BCF and 0.1 mL of picoline at a concentration of 200mg/L (each paraben) and was injected into the separationsystem each day (three times) for quality control and theobtained peaks areas were used in the calculation of EFs,EnFs, and ERs. Working standard solutions were preparedby appropriate dilutions with deionized water.
2.2 Instrumentation
The analysis was performed on a gas chromatograph (GC-2014, Shimadzu, Japan) equipped with an FID and asplit/splitless injector for the separation and determinationof the selected analytes. Helium (99.999%, Gulf Cryo, UnitedArab Emirates) was used as the carrier gas at a constant linearvelocity of 30 cm/s. The injection port was held at 290�C andused in the splitless mode with a sampling time of 1 min.Separation was carried out on a CP-Sil 8 CB (Chrompack,Milan, Italy) capillary column (30 m × 0.25 mm id, andfilm thickness of 0.25 �m). The oven temperature was pro-grammed as follows: initial temperature 100�C (held for 1min), from 100 to 175�C at a rate of 10�C/min, from 175 to200�C at a rate of 2�C/min, from 200 to 260�C at a rate of30�C/min and held at 260�C for 1 min. The FID temper-ature was maintained at 290�C. Hydrogen gas for the FIDwas generated with a hydrogen generator (OPGU-1500S, Shi-madzu) at a flow rate of 30 mL/min. The flow rate of air forFID was 300 mL/min. A Hettich centrifuge model D-7200(Kirchlengern, Germany) was used for accelerating the phaseseparation in AALLME. pH measurements were carried outwith an Metrohm pH meter model 654 (Herisau, Switzer-land). GC–MS analysis was carried out by an Agilent 6890Ngas chromatograph with a 5973 mass-selective detector (Agi-lent Technologies, CA, USA). The separation was carried outon an HP-5 (Hewlett-Packard, Santa Clara, CA, USA) MScapillary column (30 m × 0.25 mm ID, and film thicknessof 0.25 �m). Helium was used as carrier gas at a flow rateof 1.0 mL/min. The injector temperature and oven tempera-ture programming were the same as that of GC–FID analysismentioned above.
2.3 Real samples
All samples were purchased from local pharmacies (Tabriz,Iran). Ophthalmic preparation, lens rinsing solution, andmouthwash solution were diluted with deionized water atratios of 1:10, 1:10, and 1:6, respectively. In order to extractanalytes from toothpaste, 2 g of the sample was mixed with10 mL methanol and sonicated for 10 min. Then a portion of
C© 2013 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.jss-journal.com

J. Sep. Sci. 2013, 36, 3571–3578 Sample Preparation 3573
Figure 1. Scheme of AALLME procedure.
the supernatant phase was removed and diluted with deion-ized water at a ratio of 1:5. A shampoo sample was dilutedwith deionized water at a concentration of 5% w/v.
2.4 Derivatization/microextraction procedure
5.0 mL of standard or sample solution containing1% w/v sodium chloride with its pH adjusted to 10 ± 0.2,was transferred into a 10 mL glass test tube with a con-ical bottom. Twenty-five microliters BCF (as derivatizationagent/extraction solvent) and 15 �L picoline (as a catalyst forthe derivatization reaction) were added to the solution. Bysucking and expelling the mixture of aqueous sample and or-ganic phase in a 10 mL glass syringe (equipped with a needle)in the conical test tube five times, a cloudy solution consist-ing of very fine droplets of the organic phase dispersed intothe aqueous phase was formed. The mixture was then cen-trifuged for 5 min at 1200 × g, which led to the disperseddroplets of BCF to settle at the bottom of the test tube. Thevolume of the sedimented phase was determined by a 10 �Lmicrosyringe to be 7 ± 1 �L. Finally, 1 �L of the sedimentedphase was removed using a 1 �L GC microsyringe (zero deadvolume, Hamilton, Switzerland) and injected into the GC–FID for analysis. Figure 1 shows a scheme of the AALLMEprocedure.
2.5 Analytical parameters
Three main parameters, namely, EF, EnF, and ER, have beenemployed for evaluation of the proposed procedure. EF is de-fined as the ratio between the analyte concentration in thesedimented phase (Csed) and the initial concentration of ana-lyte (C0) in the sample.
EF = Csed/C0 (1)
Csed was obtained from the comparison of the analytespeak areas in two cases: direct injection of the standardsolution (200 mg/L of each analyte) prepared in the mix-ture of BCF and picoline; and injection of the sedimentedphase after performing the derivatization/microextractionprocedure.
EnF is defined as the ratio between the slope of the cal-ibration graph obtained after performing the derivatization/
microextraction procedure (slopeder) and the slope of the di-rect calibration graph obtained by the direct injection withoutderivatization of paraben solution (slopedir).
EnF = slopeder/slopedir (2)
ER is defined as the percentage of the total analyteamount (n0) extracted into the sedimented phase (nsed).
ER = (nsed/n0) × 100 = [(Csed × Vsed)/(C0 × Vaq)] × 100
ER = (Vsed/Vaq) × EF × 100(3)
where Vsed and Vaq are the volumes of the sedimented phaseand aqueous solution, respectively.
3 Results and discussion
Various parameters such as catalyst type and volume, deriva-tization agent/extraction solvent volume, sample size, saltaddition (ionic strength of aqueous phase), and pH were in-vestigated to select the optimum conditions in order to obtainhigh EFs, improve analytical signals, and to achieve the high-est possible sensitivity for the determination of analytes usingthe proposed method.
3.1 Optimization of the catalyst type and volume
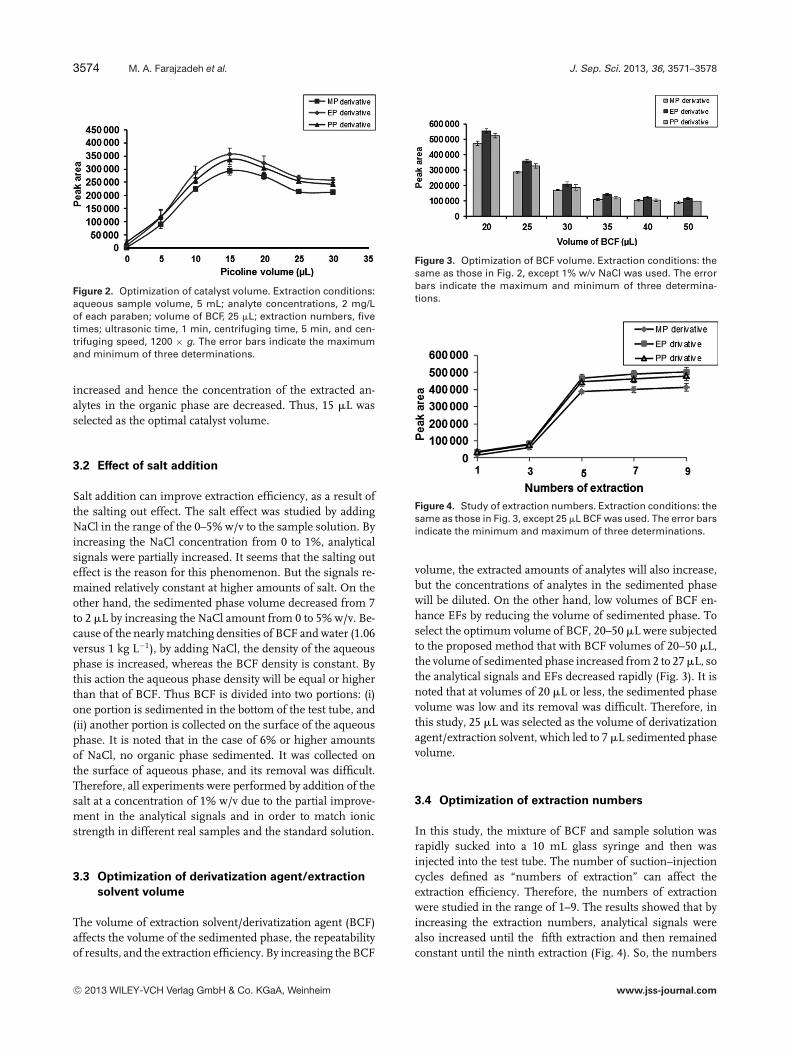
It is predictable that an alkaline catalyst can affect the derivati-zation reaction of parabens. Because the phenolic hydroxidegroups of the parabens are weak acids, therefore, when analkaline catalyst is added to the solution it acts as a protontrap. It takes the paraben’s proton and thus the residual oxy-gen atom that has a negative charge is activated. In order toinvestigate the effect of catalyst type on the obtained results,two alkaline catalysts (picoline and pyridine) were tested. Theresults showed that analytical signals in the presence of bothcatalysts did not differ much, but picoline was slightly bet-ter. Therefore, picoline was selected for further studies. Tostudy the effect of the catalyst volume, different amounts ofpicoline (0–30 �L at 5 �L intervals) were tested. As shown inFig. 2, by increasing the volume of picoline from 0 to 15 �L,the analytical signals increased rapidly and at the volumes>15 �L, peaks areas decreased. It is noted that by increasingthe picoline volume, the sedimented phase volume is also
C© 2013 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.jss-journal.com
3574 M. A. Farajzadeh et al. J. Sep. Sci. 2013, 36, 3571–3578
Figure 2. Optimization of catalyst volume. Extraction conditions:aqueous sample volume, 5 mL; analyte concentrations, 2 mg/Lof each paraben; volume of BCF, 25 �L; extraction numbers, fivetimes; ultrasonic time, 1 min, centrifuging time, 5 min, and cen-trifuging speed, 1200 × g. The error bars indicate the maximumand minimum of three determinations.
increased and hence the concentration of the extracted an-alytes in the organic phase are decreased. Thus, 15 �L wasselected as the optimal catalyst volume.
3.2 Effect of salt addition
Salt addition can improve extraction efficiency, as a result ofthe salting out effect. The salt effect was studied by addingNaCl in the range of the 0–5% w/v to the sample solution. Byincreasing the NaCl concentration from 0 to 1%, analyticalsignals were partially increased. It seems that the salting outeffect is the reason for this phenomenon. But the signals re-mained relatively constant at higher amounts of salt. On theother hand, the sedimented phase volume decreased from 7to 2 �L by increasing the NaCl amount from 0 to 5% w/v. Be-cause of the nearly matching densities of BCF and water (1.06versus 1 kg L−1), by adding NaCl, the density of the aqueousphase is increased, whereas the BCF density is constant. Bythis action the aqueous phase density will be equal or higherthan that of BCF. Thus BCF is divided into two portions: (i)one portion is sedimented in the bottom of the test tube, and(ii) another portion is collected on the surface of the aqueousphase. It is noted that in the case of 6% or higher amountsof NaCl, no organic phase sedimented. It was collected onthe surface of aqueous phase, and its removal was difficult.Therefore, all experiments were performed by addition of thesalt at a concentration of 1% w/v due to the partial improve-ment in the analytical signals and in order to match ionicstrength in different real samples and the standard solution.
3.3 Optimization of derivatization agent/extraction
solvent volume
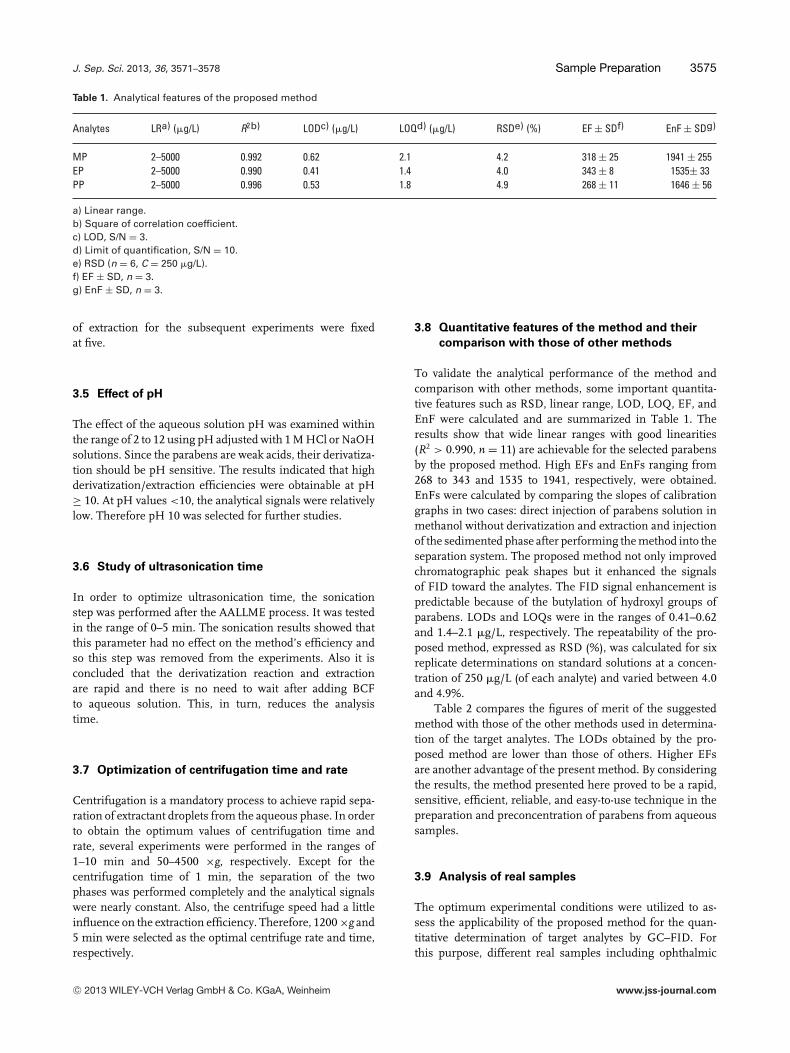
The volume of extraction solvent/derivatization agent (BCF)affects the volume of the sedimented phase, the repeatabilityof results, and the extraction efficiency. By increasing the BCF
Figure 3. Optimization of BCF volume. Extraction conditions: thesame as those in Fig. 2, except 1% w/v NaCl was used. The errorbars indicate the maximum and minimum of three determina-tions.
Figure 4. Study of extraction numbers. Extraction conditions: thesame as those in Fig. 3, except 25 �L BCF was used. The error barsindicate the minimum and maximum of three determinations.
volume, the extracted amounts of analytes will also increase,but the concentrations of analytes in the sedimented phasewill be diluted. On the other hand, low volumes of BCF en-hance EFs by reducing the volume of sedimented phase. Toselect the optimum volume of BCF, 20–50 �L were subjectedto the proposed method that with BCF volumes of 20–50 �L,the volume of sedimented phase increased from 2 to 27 �L, sothe analytical signals and EFs decreased rapidly (Fig. 3). It isnoted that at volumes of 20 �L or less, the sedimented phasevolume was low and its removal was difficult. Therefore, inthis study, 25 �L was selected as the volume of derivatizationagent/extraction solvent, which led to 7 �L sedimented phasevolume.
3.4 Optimization of extraction numbers
In this study, the mixture of BCF and sample solution wasrapidly sucked into a 10 mL glass syringe and then wasinjected into the test tube. The number of suction–injectioncycles defined as “numbers of extraction” can affect theextraction efficiency. Therefore, the numbers of extractionwere studied in the range of 1–9. The results showed that byincreasing the extraction numbers, analytical signals werealso increased until the fifth extraction and then remainedconstant until the ninth extraction (Fig. 4). So, the numbers
C© 2013 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.jss-journal.com
J. Sep. Sci. 2013, 36, 3571–3578 Sample Preparation 3575
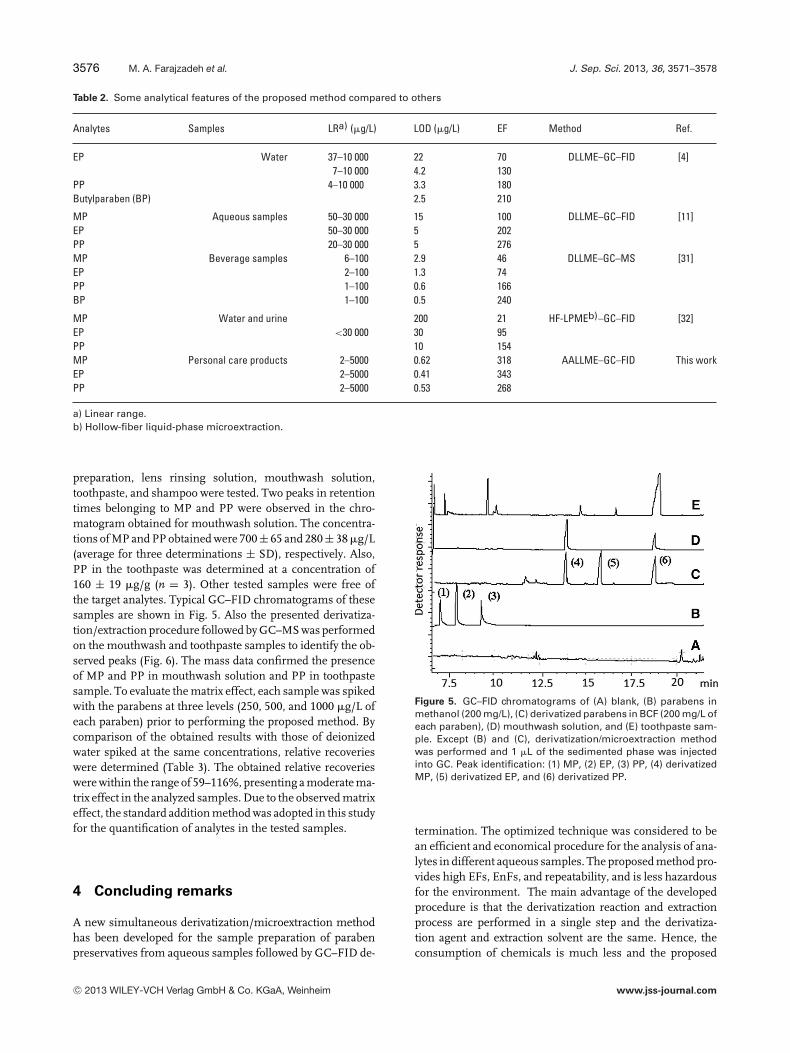
Table 1. Analytical features of the proposed method
Analytes LRa) (�g/L) R2b) LODc) (�g/L) LOQd) (�g/L) RSDe) (%) EF ± SDf) EnF ± SDg)
MP 2–5000 0.992 0.62 2.1 4.2 318 ± 25 1941 ± 255EP 2–5000 0.990 0.41 1.4 4.0 343 ± 8 1535± 33PP 2–5000 0.996 0.53 1.8 4.9 268 ± 11 1646 ± 56
a) Linear range.b) Square of correlation coefficient.c) LOD, S/N = 3.d) Limit of quantification, S/N = 10.e) RSD (n = 6, C = 250 �g/L).f) EF ± SD, n = 3.g) EnF ± SD, n = 3.
of extraction for the subsequent experiments were fixedat five.
3.5 Effect of pH
The effect of the aqueous solution pH was examined withinthe range of 2 to 12 using pH adjusted with 1 M HCl or NaOHsolutions. Since the parabens are weak acids, their derivatiza-tion should be pH sensitive. The results indicated that highderivatization/extraction efficiencies were obtainable at pH≥ 10. At pH values <10, the analytical signals were relativelylow. Therefore pH 10 was selected for further studies.
3.6 Study of ultrasonication time
In order to optimize ultrasonication time, the sonicationstep was performed after the AALLME process. It was testedin the range of 0–5 min. The sonication results showed thatthis parameter had no effect on the method’s efficiency andso this step was removed from the experiments. Also it isconcluded that the derivatization reaction and extractionare rapid and there is no need to wait after adding BCFto aqueous solution. This, in turn, reduces the analysistime.
3.7 Optimization of centrifugation time and rate
Centrifugation is a mandatory process to achieve rapid sepa-ration of extractant droplets from the aqueous phase. In orderto obtain the optimum values of centrifugation time andrate, several experiments were performed in the ranges of1–10 min and 50–4500 ×g, respectively. Except for thecentrifugation time of 1 min, the separation of the twophases was performed completely and the analytical signalswere nearly constant. Also, the centrifuge speed had a littleinfluence on the extraction efficiency. Therefore, 1200 ×g and5 min were selected as the optimal centrifuge rate and time,respectively.
3.8 Quantitative features of the method and their
comparison with those of other methods
To validate the analytical performance of the method andcomparison with other methods, some important quantita-tive features such as RSD, linear range, LOD, LOQ, EF, andEnF were calculated and are summarized in Table 1. Theresults show that wide linear ranges with good linearities(R2 > 0.990, n = 11) are achievable for the selected parabensby the proposed method. High EFs and EnFs ranging from268 to 343 and 1535 to 1941, respectively, were obtained.EnFs were calculated by comparing the slopes of calibrationgraphs in two cases: direct injection of parabens solution inmethanol without derivatization and extraction and injectionof the sedimented phase after performing the method into theseparation system. The proposed method not only improvedchromatographic peak shapes but it enhanced the signalsof FID toward the analytes. The FID signal enhancement ispredictable because of the butylation of hydroxyl groups ofparabens. LODs and LOQs were in the ranges of 0.41–0.62and 1.4–2.1 �g/L, respectively. The repeatability of the pro-posed method, expressed as RSD (%), was calculated for sixreplicate determinations on standard solutions at a concen-tration of 250 �g/L (of each analyte) and varied between 4.0and 4.9%.
Table 2 compares the figures of merit of the suggestedmethod with those of the other methods used in determina-tion of the target analytes. The LODs obtained by the pro-posed method are lower than those of others. Higher EFsare another advantage of the present method. By consideringthe results, the method presented here proved to be a rapid,sensitive, efficient, reliable, and easy-to-use technique in thepreparation and preconcentration of parabens from aqueoussamples.
3.9 Analysis of real samples
The optimum experimental conditions were utilized to as-sess the applicability of the proposed method for the quan-titative determination of target analytes by GC–FID. Forthis purpose, different real samples including ophthalmic
C© 2013 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.jss-journal.com
3576 M. A. Farajzadeh et al. J. Sep. Sci. 2013, 36, 3571–3578
Table 2. Some analytical features of the proposed method compared to others
Analytes Samples LRa) (�g/L) LOD (�g/L) EF Method Ref.
EP Water 37–10 000 22 70 DLLME–GC–FID [4]7–10 000 4.2 130
PP 4–10 000 3.3 180Butylparaben (BP) 2.5 210
MP Aqueous samples 50–30 000 15 100 DLLME–GC–FID [11]EP 50–30 000 5 202PP 20–30 000 5 276MP Beverage samples 6–100 2.9 46 DLLME–GC–MS [31]EP 2–100 1.3 74PP 1–100 0.6 166BP 1–100 0.5 240
MP Water and urine 200 21 HF-LPMEb)–GC–FID [32]EP <30 000 30 95PP 10 154MP Personal care products 2–5000 0.62 318 AALLME–GC–FID This workEP 2–5000 0.41 343PP 2–5000 0.53 268
a) Linear range.b) Hollow-fiber liquid-phase microextraction.
preparation, lens rinsing solution, mouthwash solution,toothpaste, and shampoo were tested. Two peaks in retentiontimes belonging to MP and PP were observed in the chro-matogram obtained for mouthwash solution. The concentra-tions of MP and PP obtained were 700 ± 65 and 280 ± 38 �g/L(average for three determinations ± SD), respectively. Also,PP in the toothpaste was determined at a concentration of160 ± 19 �g/g (n = 3). Other tested samples were free ofthe target analytes. Typical GC–FID chromatograms of thesesamples are shown in Fig. 5. Also the presented derivatiza-tion/extraction procedure followed by GC–MS was performedon the mouthwash and toothpaste samples to identify the ob-served peaks (Fig. 6). The mass data confirmed the presenceof MP and PP in mouthwash solution and PP in toothpastesample. To evaluate the matrix effect, each sample was spikedwith the parabens at three levels (250, 500, and 1000 �g/L ofeach paraben) prior to performing the proposed method. Bycomparison of the obtained results with those of deionizedwater spiked at the same concentrations, relative recoverieswere determined (Table 3). The obtained relative recoverieswere within the range of 59–116%, presenting a moderate ma-trix effect in the analyzed samples. Due to the observed matrixeffect, the standard addition method was adopted in this studyfor the quantification of analytes in the tested samples.
4 Concluding remarks
A new simultaneous derivatization/microextraction methodhas been developed for the sample preparation of parabenpreservatives from aqueous samples followed by GC–FID de-
Figure 5. GC–FID chromatograms of (A) blank, (B) parabens inmethanol (200 mg/L), (C) derivatized parabens in BCF (200 mg/L ofeach paraben), (D) mouthwash solution, and (E) toothpaste sam-ple. Except (B) and (C), derivatization/microextraction methodwas performed and 1 �L of the sedimented phase was injectedinto GC. Peak identification: (1) MP, (2) EP, (3) PP, (4) derivatizedMP, (5) derivatized EP, and (6) derivatized PP.
termination. The optimized technique was considered to bean efficient and economical procedure for the analysis of ana-lytes in different aqueous samples. The proposed method pro-vides high EFs, EnFs, and repeatability, and is less hazardousfor the environment. The main advantage of the developedprocedure is that the derivatization reaction and extractionprocess are performed in a single step and the derivatiza-tion agent and extraction solvent are the same. Hence, theconsumption of chemicals is much less and the proposed
C© 2013 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.jss-journal.com

J. Sep. Sci. 2013, 36, 3571–3578 Sample Preparation 3577
Figure 6. Total ion current chromatograms of(A) toothpaste sample and (B) mouthwash solu-tion after performing the proposed method, andmass spectra of (C) derivatized MP, (D) deriva-tized PP, (E) scan 854, retention time 14.295 min(mouthwash solution), (F) scan 1179, retentiontime 18.927 min (toothpaste), and (G) scan 1177,retention time 18.898 min (toothpaste).
Table 3. Study of the matrix effect in samples spiked at different concentrations
Mean relative recoverya) (%) ± SD (n = 3)
Analytes Lenses rinsing solution Ophthalmic preparations Mouthwash solution Toothpaste Shampoo
All samples were spiked with each analyte at a concentration of 250 �g/LMP 59 ± 1 68 ± 4 88 ± 2 90 ± 2 74 ± 2EP 71 ± 4 73 ± 3 102 ± 6 75 ± 1 100 ± 3PP 70 ± 5 72 ± 3 85 ± 6 80 ± 3 80 ± 2
All samples were spiked with each analyte at a concentration of 500 �g/LMP 70 ± 2 73 ± 2 106 ± 1 116 ± 1 79 ± 3EP 62 ± 2 76 ± 5 100 ± 2 97 ± 3 91 ± 2PP 85 ± 4 72 ± 4 87 ± 5 94 ± 1 87 ± 3
All samples were spiked with each analyte at a concentration of 1000 �g/LMP 79 ± 1 80 ± 1 104 ± 2 80 ± 4 84 ± 5EP 80 ± 2 100 ± 1 115 ± 5 85 ± 2 99 ± 1PP 78 ± 3 97 ± 2 85 ± 3 110 ± 6 103 ± 4
a)Analyte contents of the samples were subtracted.
method is a time-saving procedure. Also the derivatizationreaction has been carried out under mild conditions. So, thismethod is easy to use and fast, and there is no need for heatingor a long time for derivatization.
The authors thank the Research Council of Tabriz Universityfor financial support.
The authors have declared no conflict of interest.
C© 2013 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.jss-journal.com

3578 M. A. Farajzadeh et al. J. Sep. Sci. 2013, 36, 3571–3578
5 References
[1] Rowe, R. C., Handbook of Pharmaceutical Excipients, 5thEdition, Royal Pharmaceutical Society of Great Britain,London 2006.
[2] Harvey, P. W., Everett D. J., J. Appl. Toxicol, 2004, 24 (1),1–4.
[3] Regueiro, J., Llompart, M., Psillakis, E., Monteagudo, J.C. G., Jares, C. G., Talanta 2009, 79, 1387–1397.
[4] Prichodko, A., Janenatte, E., Smittene, V., Vickackaite, V.,J. Acta Chromatogr. 2012, 24, 589–601.
[5] Okumura, T., Nishikawa, Y., Anal. Chim. Acta 1996, 325,175–184.
[6] Canosa, P., Rodriguez, I., Rubi, E., Bollain, M. H., Cela, R.,J. Chromatogr. A 2006, 1124, 3–10.
[7] Canosa, P., Rodriguez, I., Rubi, E. Cela, R., J. Chromatogr.A 2005, 1072, 107–115.
[8] Hajkova, P., Solich, P., Pospisbra, M., Sicha, J., Anal.Chim. Acta 2002, 467, 91–96.
[9] Croo, F. D., Schutter, J. D., Bossche, W. V., Moerloose, P.D., Chromatographia 1984, 18, 260–264.
[10] Conzulez, M., Gallege, M., Valcarcel, M., J. Chromatogr.A 1998, 823, 321–329.
[11] Farajzadeh, M. A., Djozan, Dj., Bakhtiyari, R. F., Talanta2010, 81, 1360–1367.
[12] Yang, T. J., Tsai, F. J., Chen, C. Y., Yang, T. C. C., Lee, M.R., Anal. Chim. Acta 2010, 668, 188–194.
[13] Regueiro, J., Becerril, E., Jares, C. G., Llompart, M., J.Chromatogr. A 2009, 1216, 4693–4702.
[14] Canosa, P., Palacios, D. P., Lopez, A. G., Tena, M. T.,Rodrıguez, I., Rubı, E., Cela, R., J. Chromatogr. A 2007,1161, 105–112.
[15] Saraji, M., Mirmahdieh, S., J. Sep. Sci. 2009, 32, 988–995.
[16] Blanco, E., Casais, M. D. C., Mejuto, M. D. C., Cela, R.,Anal. Chim. Acta 2009, 647, 104–111.
[17] Wang, S. P., Chang, C. L., Anal. Chim. Acta 1998, 377,85–93.
[18] Ye, X., Tao, L. J., Needham, L. L., Calafat, A. M., Talanta2008, 76, 865–871.
[19] Shen, H. Y., Jiang, H. L., Mao, H. L., Pan,G., Zhou, L., Cao, Y. F., J. Sep. Sci. 2007, 30,48–54.
[20] Han, F., He, Y. Z., Yu, C. Z., Talanta 2008, 74,1371–1377.
[21] Lokhnauth, J. K., Snow, N. H., Anal. Chem. 2005, 77,5938–5946.
[22] Claver, J. B., Valencia, M. C., Vallvey, L. F. C., Talanta2009, 79, 499–506.
[23] Basan, H., Dinner, Z., Gozer, N. G., Chem. Anal. 2005, 50,465–473.
[24] Myint, A., Zhang, Q., Liu, L., Cui, H., Anal. Chim. Acta2004, 517, 119–124.
[25] Trabelsi, H., Raouafi, F., Limam, M., Bouzouita,K., J. Pharm. Biomed. Anal. 2002, 29,239–245.
[26] Wang, S. P., Chang, C. L., Anal. Chim. Acta 1998, 377,85–93.
[27] Yang, T. J., Tsai, F. J., Chen, C. Y., Yang, T.C. C., Lee, M. R., Anal. Chim. Acta 2010, 668,188–194.
[28] Melo, L. P., Queiroz, M. E. C., J. Sep. Sci. 2010, 33,1849–1855.
[29] Ramirez, N., Borrull, F., Marce, R. M., J. Sep. Sci. 2010,35, 580–588.
[30] Farajzadeh, M. A., Mogaddam, M. R. A., Anal. Chim. Acta2012, 728, 31–38.
[31] Han, Y., Jia, X., Iu, X., Uan, T., Hen, H., Chromatographia2010, 72, 351–355.
[32] Prichodko, A., Jonusaite, K., Vickackaite, V., Cent. Eur. J.Chem. 2009, 7, 285–290.
C© 2013 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.jss-journal.com



















![Analysis of Steroids using Solid Phase Microextraction-Gas Chromatography-Mass … · 2018-03-29 · spectrometry and tandem mass spectrometry[12], liquid chromatography-mass spectrometry](https://img.pdfslide.us/doc/110x75/5f47f27e21a760452d67e4a6/analysis-of-steroids-using-solid-phase-microextraction-gas-chromatography-mass-2018-03-29.jpg)









