Embed Size (px)
Citation preview

Simple Synthesisof F-18-Labeled 2-Fluoro-2-Deoxy-D-Glucose:
Concise Communication
RichardE. Ehrenkaufer,Jan F. Potocki, and Douglas M.Jewett
Universityof MichiganMedicalSchool,AnnArbor,Michigan
Fluorine-iS acetyl hypofluorite,producedby a gas/solid-phasereaction, wasreacted with D-glucal in water, followed by treatment with HCI and serial passagethroughcolumnsof charcoal, ion-retardationresin,and alumina.The productwasan injectablesolutionof 2-[18Fjfluoro-2-deoxy-D-giucose.The radiochemicalpuritywas greaterthan95% andthe radiochemicalyield was about40%. The synthesiswas completed in about 15 mm. Acetyl hypofluoritewas shownto be a stable,highly stereoselective fluorinatingagent In aqueous systems over a wide pHrange.
J Nuci Med 25: 333—337,1984
New methods are continuously being sought for thesynthesis of 2-fluoro-2-deoxy-D-glucose(FDG), the mostwidely useful radiopharmaceutical for metabolic PETimaging. In addition to the methods involving electrophilic fluorination now in widespread use, nucleophilicreactions of ‘8F are also finding increasing application
(1 ). Both approaches involve the reaction of protectedprecursors in organic solvents, with subsequent deblocking, chromatographic purification, and removal oforganic solvents—operationsthat are difficult to performin remote, automated systems.
Acetyl hypofluorite [AcOF (2)] has recently beenshown to react with protected I ,2-unsaturated sugarswith high stereoselectivity to give the corresponding2-fluoro-2-deoxy sugars in good yield (3,4). This reactionhas been applied to the radiosynthesis of [F-18}FDG(5—8),resulting in higher yields and simpler proceduresthan for methods involving direct fluorination with[I8F]F2.Recentlywedeveloped,forthesynthesisofAcOF (9), a simple gas/solid-phase method that isreadily applicable to the synthesis of AcO'8F. Thismethod allowed considerable flexibility in the choice of
Received July 21, 1983; revision accepted Oct. 24, 1983.For reprints contact: Richard E. Ehrenkaufer, PhD, Cyclotron/PET
Facility, 3480 Kresge I Bldg., Box056, University of Michigan, AnnArbor, Ml 48109.
solvent for the subsequent reaction of AcOF with anunsaturated compound. We therefore considered thepossibility of reacting ACOFdirectly with an unprotectedsugar in water.
When AcO'8F in neon was bubbled into a dilute solution of D-glucal in water, there resulted a mixture ofF-18-labeled FDG and a radiolabeled acid-labile precursor of FDG. Addition of HCI followed by briefheating and serial passage through columns of charcoal,ion-retardation resin, and alumina converted the precursor to FDG and, at the same time, removed the excessstarting materials, HCI and HF. The resulting solutionof [F-I8JFDG with high radiochemical purity was readyfor injection after passage through a 0.2-s filter.
Because the procedure requires no organic solvents,evaporations, or chromatographic separations, a verysimple automatic system is adequate for remote synthesis. Since dry reagents or glassware are unnecessary,the system is ready for reuse after automatic washingwith sterile, nonpyrogenic water and replacement of thecolumns.
MATERIALS AND METHODS
Potassiumacetate —acetic acidcomplexfor generatingacetyl hypofluorite. Anhydrous KOAc (1 .0 mole) wasmixed with glacial acetic acid (1.5 mole) and stored in
Volume 25, Number 3 333
by on February 27, 2019. For personal use only. jnm.snmjournals.org Downloaded from

LHRENKAUFER. POTOCKI. AND JEWEIT
a sealed container for 48 hr. The resulting solid cake waspulverized with a spoon and placed in a desiccator undervacuum over P2O5 for 24 hr. The product was stored intightly sealed bottles to exclude moisture.
D-Clucal (10). Amberlyte CG 4ØØ*,OH—form, waswashed several times with anhydrous methanol. Theresin was added to a solution of 27.2 g 3,4,6-tri-O-acetyl-D-glucal in 200 ml anhydrous MeOH. After stirringat 60°for 30 mm, the solvent was removed on a rotaryevaporator. A second 200 ml MeOH was added, and thesuspension was stirred and heated for 30 mm more. Theresin was then removed by suction filtration and thesolvent was removed on a rotary evaporator. The resuiting syrup was maintained under a high vacuum for24 hr. The resulting solid, mp 54—57°(58—60°Lit.) gavea single spot by TLC [MeCH/H2O (95:5); silica gel;detected with 121.
Preparation of the columns. Powdered activatedcharcoal, USP grade, was mixed with diatomaceousearth I :4 w/w. This material, 0.5 g, was packed looselyin a 7-mm column over a 10 @zmsintered polyethylenefrit. A small plug ofcotton was placed above the charcoalto trap polymer particles that would otherwise clog thecolumn bed. Before use, the column was washed with 20ml sterile, pyrogen-free water. These columns were usedonce and discarded.
The ion-retardation resin and alumina were packedin a single column, which was regenerated after use.Neutral alumina, 100-200 mesh, was washed with waterto remove fines, and slurry-packed to a depth of 70 mmin a glass column (7 mm i.d. X 300 mm) fitted withI0-zm sintered polyethylene frits at both ends. The remainder of the column was slurry-packed with AG I IA8resint. The column was regenerated by washing with 100ml N NaOH followed by I00 ml sterile, pyrogen-freewater. The upper frits become clogged with charcoalparticles after several uses and are periodically replaced.
Synthesis of[F-I8JFDG. [‘8F]F2,nominally 0.1% inneon, was produced by the 20Ne(d,a)'8F reaction. AI 5.0-MeV deuteron beam from a CS-30 cyclotron wasused ( I 3.3 MeV D@on the target gas at I0 @Afor 0.5hr), with a nickel target (70 cc volume) containing 0.1%F2in neon at 80 psig. The total activity recovered fromthe target was 20 mCi after 15 mm irradiation, or 40mCi after 30 mm.
After irradiation, the target gas was passed at 100mI/mm through a stainless steel tube, 4 mm X 100 mm,packed with KOAc(HOAc)1.5. The effluent was passedinto a reaction vessel similar to that described by Ido etal. (ii) containing 30 mg D-glucal dissolved in 2.5 mlH2O. The effluent gas from this vessel was passedthrough a small column of soda lime followed by acharcoal trap maintained at —70°.
After 5 mm, 0.6 ml 37% HCI was added. The solutionwas transferred to a tube maintained at 100°. Heating
was continued for 3 mm, during which the excess Dglucal was converted to a green polymeric precipitate.The resulting suspension was drawn by a vacuumthrough the charcoal, resin, and alumina columns connected in series. The two reaction vessels and columnswere washed with two 3.5-ml portions of sterile, pyrogen-free water. The combined fractions were passedthrough a 0.2 ,um Millipore filter into a multidosevial.
Analysis of the product.The radiochemical purity ofthe product was determined by high performance liquidchromatography (HPLC) and thin layer chromatography (TLC), and the chemical purity by gas-liquidchromatography (GLC) and TLC. For HPLC the radioactivity was measured directly with a detector consisting ofa cell containing a solid glass scintillator coupled to two photomultiplier tubes. For TLC the chromatograms were cut into strips for gamma counting.
For HPLC we used a HPX-87C Ca@-exchangedpolystyrene sulfonate resint with water as the mobilephase at 85°and at a pressure of900 psig (12). Retention time for [F-18]FDG was 6.4 mm.
For GLCt (/3), a l00-jzl aliquot was evaporated todryness, the residue was dissolved in pyridine, and 25 @zlhexamethyldisilazane and I5 jzl of a 1.5 M solution oftrimethylsilylimidazole in pyridine were added. Thesolution was kept at 60°for I .5 hr. The resulting tnmethylsilyl derivative, I .5 @l,was injected on a 2-m SE3Ocolumn, temperature-programmed from I30°to 240°at the rate of I0°/mm.Retention times for the alpha andbeta anomers of FDG were I2.5 and I3.4 mm, respectively (6).
For TLC, silica-gel coated strips were used with twosolvent systems: MeCN/H2O (95:5), RfOf FDG 0.46(11); and butanol/AcOH/H2O (5:1:1), Rf of FDG0.52 (14). The stripsweresprayedwith alkaline AgNO3for visualization. Unreacted starting material was specifically detected by 12.
RESULTS AND DISCUSSION
Using the above method, [F-l8]FDG was produced
F2+CH,[email protected]@H @CH3CO@F+ CH3CO2H+KF (I)
CH1OH
cH,0H NOKl@;)@ @H,O,4
H@K@)+CH3CO2F@i9•@F @0K@.;>;-1@@(2)(I) (n) cH2014
@:@
FIG.1. Synthesisof2-fluoro-2-deoxy-D-glucosefromacetylhypofluorite and D-glucal.
334 THE JOURNAL OF NUCLEAR MEDICINE
by on February 27, 2019. For personal use only. jnm.snmjournals.org Downloaded from
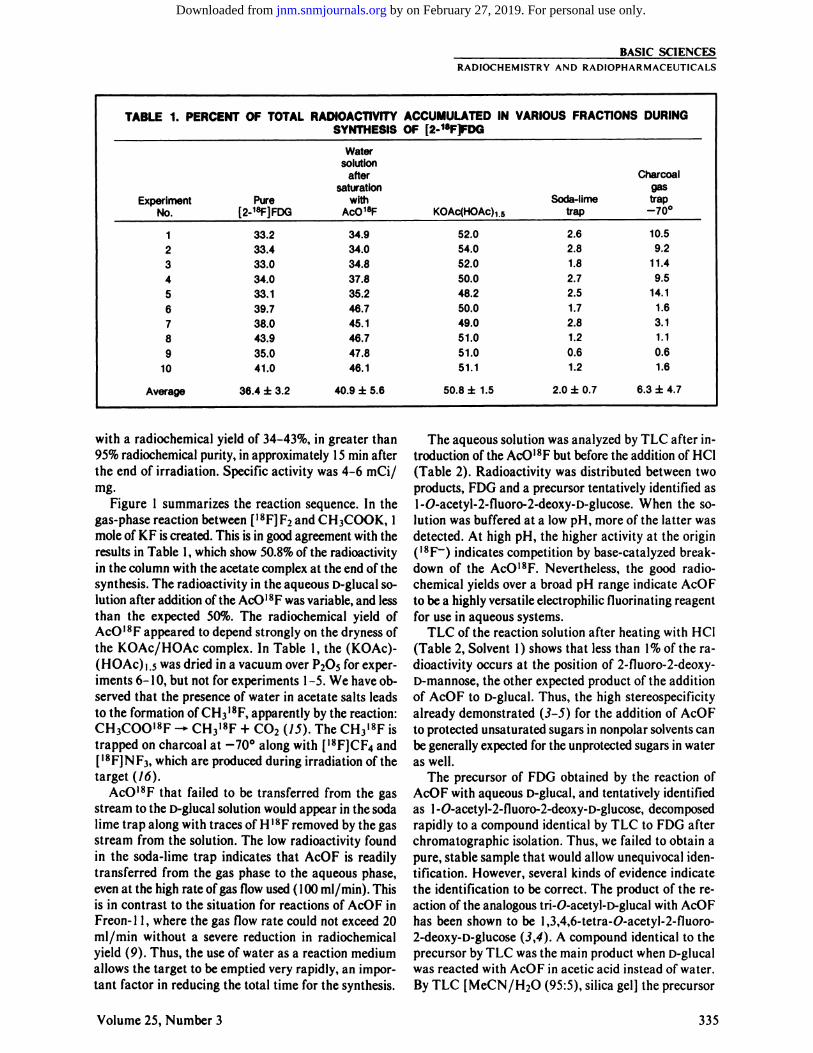
TABLE1. PERCENTOF TOTALRADIOACTIVITYSYNTHESISACCUMULATED
INOF[2-1F1F00VARIOUSFRACTIONSDURINGWatersolutionafterCharcoalsaturationgasExperiment
No.Pure [2-1@F]FDGwith ACO18FKOAc(HOAc)1.5Soda-lime traptrap —70°
BASIC SCIENCESRADIOCHEMISTRYAND RADIOPHARMACEUTICALS
1 33.2 34.9 52.0 2.6 10.52 33.4 34.0 54.0 2.8 9.23 33.0 34.8 52.0 1.8 11.44 34.0 37.8 50.0 2.7 9.55 33.1 35.2 48.2 2.5 14.16 39.7 46.7 50.0 1.7 1.67 38.0 45.1 49.0 2.8 3.18 43.9 46.7 51.0 1.2 1.19 35.0 47.8 51.0 0.6 0.6
10 41.0 46.1 51.1 1.2 1.6
Average 36.4 ±3.2 40.9 ±5.6 50.8 ±1.5 2.0 ±0.7 6.3 ±4.7
with a radiochemical yield of 34—43%,in greaten than95% radiochemical purity, in approximately 15 mm afterthe end of irradiation. Specific activity was 4-6 mCi/mg.
Figure 1 summarizes the reaction sequence. In thegas-phase reaction between [‘8FJF2 and CH3COOK, Imole of KF is created. This is in good agreement with theresults in Table 1, which show 50.8% of the radioactivityin the column with the acetate complex at the end of thesynthesis. The radioactivity in the aqueous D-glucal solution after addition of the ACO'8F was variable, and lessthan the expected 50%. The radiochemical yield ofAcO'8F appeared to depend strongly on the dryness ofthe KOAc/HOAc complex. In Table I , the (KOAc)
I .s was dried in a vacuum over P205 for exper
iments 6—10, but not for experiments I —5.We have observed that the presence of water in acetate salts leadsto the formation of CH318F, apparently by the reaction:CH3COO'8F —‘CH318F+ CO2 (15). The CH318Fistrapped on charcoal at —70°along with [‘8F]CF4and[‘8F]NF3,which are produced during irradiation of thetarget (16).
AcO'8F that failed to be transferred from the gasstream to the D-giucal solution would appear in the sodalime trap along with traces of H'8F removed by the gasstream from the solution. The low radioactivity foundin the soda-lime trap indicates that AcOF is readilytransferred from the gas phase to the aqueous phase,even at the high rate ofgas flow used (100 mi/mm). Thisis in contrast to the situation for reactions of AcOF inFreon- I I , where the gas flow rate could not exceed 20mi/mm without a severe reduction in radiochemicalyield (9). Thus, the use of water as a reaction mediumallows the target to be emptied very rapidly, an important factor in reducing the total time for the synthesis.
The aqueous solution was analyzed by TLC after introduction of the AcO'8F but before the addition of HC1(Table 2). Radioactivity was distributed between twoproducts, FDG and a precursor tentatively identified asl-O-acetyl-2-fluoro-2-deoxy-D-glucose. When the solution was buffered at a low pH, more of the latter wasdetected. At high pH, the higher activity at the origin(I 8F—) indicates competition by base-catalyzed break
down of the AcO18F. Nevertheless, the good radiochemical yields over a broad pH range indicate AcOFto be a highly versatileelectrophilicfluorinatingreagentfor use in aqueous systems.
TLC of the reaction solution after heating with HCI(Table 2, Solvent 1) shows that less than 1%of the radioactivity occurs at the position of 2-fluoro-2-deoxyD-mannose, the other expected product of the additionof AcOF to D-glucal. Thus, the high stereospecificity
already demonstrated (3—5) for the addition of AcOFto protected unsaturated sugars in nonpolar solvents canbe generally expected for the unprotected sugars in wateras well.
The precursor of FDG obtained by the reaction ofACOFwith aqueous D-glucal, and tentatively identifiedas 1-O-acetyl-2-fluoro-2-deoxy-D-glucose,decomposedrapidly to a compound identical by TLC to FDG afterchromatographic isolation. Thus, we failed to obtain apure, stable sample that would allow unequivocal identification. However, several kinds of evidence indicatethe identification to be correct. The product of the reaction of the analogous tni-O-acetyl-D-glucalwith ACOFhas been shown to be 1,3,4,6-tetra-O-acetyl-2-fluoro2-deoxy-D-glucose (3,4). A compound identical to theprecursor by TLC was the main product when D-glucalwas reacted with AcOF in acetic acid instead of water.By TLC [MeCN/H2O (95:5), silica gel] the precursor
Volume 25, Number 3 335
by on February 27, 2019. For personal use only. jnm.snmjournals.org Downloaded from
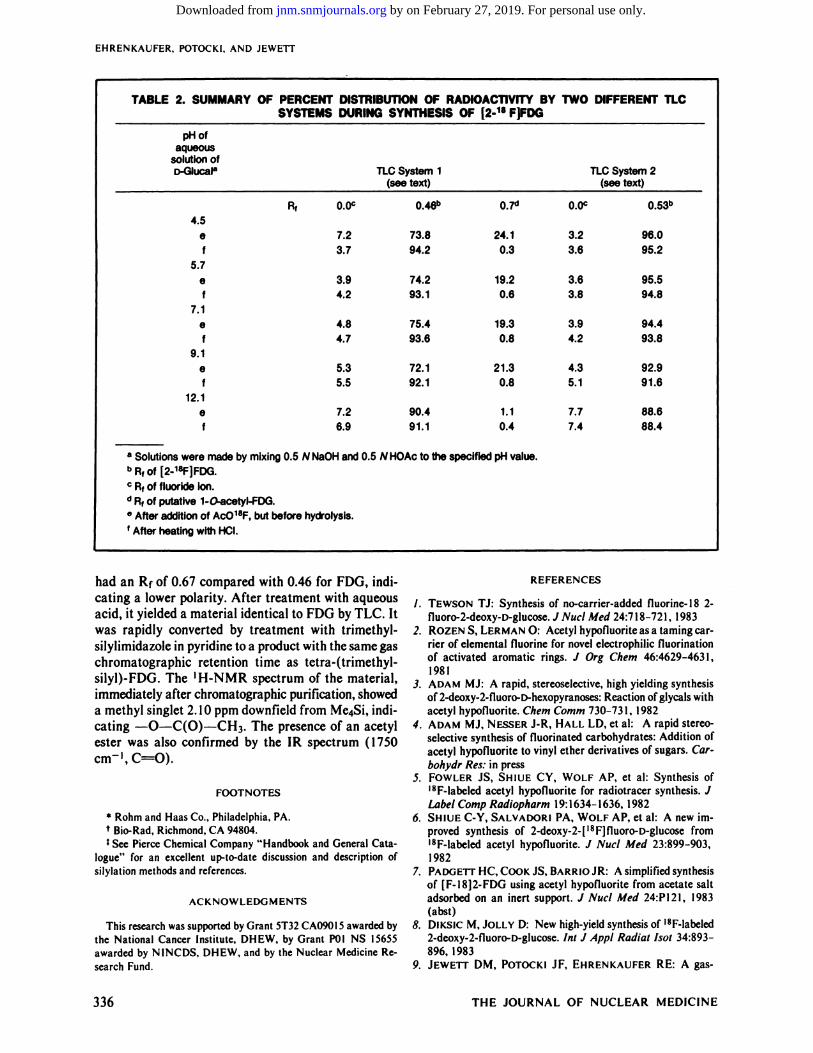
pHofaqueoussolution
ofD-GIucaI8TICSystem 1
(see text)TLCSystem 2
(see text)
EHRENKAUFER, POTOCKI, AND JEWETT
TABLE2. SUMMARYOF PERCENTDISTRIBUTIONOF RADIOACTIVITYBY TWO DIFFERENTTLCSYSTEMS DURING SYNTHESIS OF [2@18F]FDG
A1 0.0c 0.0c4.5
ef
7.23.7
73.894.2
24.10.3
3.23.6
96.095.2
5.7ef
3.94.2
74.293.1
19.20.6
3.63.8
95.594.8
7.1ef
4.84.7
75.493.6
19.30.8
3.94.2
94.493.8
9.1ef
5.35.5
72.192.1
21.30.8
4.35.1
92.991.6
12.10 7.2
6.990.491.1
1.1 7.70.4 7.4
88.688.4
a Solutions were made by mixing 0.5 N NaOH and 0.5 N HOAc to the specified pH value.
b Rf of [2-1@]FDG.
C Rf of fluoride ion.
d Rf of putative 1-O-acetyl-FDG.
a After addition of ACO18F, but before hy&olysis.
f After heating with HCI.
had an Rf of 0.67 compared with 0.46 for FDG, mdicating a lower polarity. After treatment with aqueousacid, it yielded a material identical to FDG by TLC. Itwas rapidly converted by treatment with trimethylsilylimidazole in pyridine to a product with the same gaschromatographic retention time as tetra-(trimethylsilyl)-FDG. The â€H̃-NMR spectrum of the material,immediately after chromatographic purification, showeda methyl singlet 2. 10 ppm downfield from Me4Si, mdieating —O—C(O)—CH3. The presence of an acetylester was also confirmed by the IR spectrum (1750cm1, C=O).
FOOTNOTES
* Rohm and Haas Co., Philadelphia, PA.
t Bio-Rad, Richmond, CA 94804.
I Sec Pierce Chemical Company “Handbookand General Catalogue―for an excellent up.to-date discussionand description ofsilylation methods and references.
ACKNOWLEDGMENTS
This researchwassupportedbyGrant 5132 CAO9OI5 awardedbythe National Cancer Institute, DHEW, by Grant P01 NS 15655awarded by NINCDS, DHEW, and by the Nuclear MedicineResearch Fund.
REFERENCES
1. TEWSONTJ: Synthesisof no-carrier-addedfluorine-182-fluoro-2-deoxy-D-glucose. J Nuc! Med 24:7 18-721 , 1983
2. ROZEN S, LERMAN 0: Acetyl hypofluorite as a taming carncr of elemental fluorine for novel electrophilic fluorinationof activated aromatic rings. J Org Chem 46:4629-4631,I981
3. ADAM Mi: A rapid, stereoselective, high yielding synthesisof 2-deoxy-2-fluoro-D-hexopyranoses:Reactionofglycals withacetylhypofluorite.ChemComm730-731,1982
4. ADAMMi, NESSERJ-R,HALLLD,etal: A rapidstereoselective synthesis of fluorinated carbohydrates: Addition ofacetyl hypofluorite to vinyl ether derivatives of sugars. Carbohydr Res: in press
5. FOWLER iS, SHIUE CY, WOLF AP, et al: Synthesis of‘8F-labeledacetyl hypofluorite for radiotracer synthesis. JLabelComp Radiopharm19:1634-1636,1982
6. SHIUEC-Y,SALVADORIPA,WOLFAP,Ctal: A newimproved synthesisof 2-deoxy-2-['8F]fluoro-D-glucosefrom‘8F-labeledacetyl hypofluorite. I Nuci Med 23:899-903,I982
7. PADGETFHC,CooKiS, BARRIOiR: A simplifiedsynthesisof [F-18]2.FDG using acetyl hypofluorite from acetate saltadsorbed on an inert support. I Nuci Med 24:Pl2l, 1983(abst)
8. DIKSICM,JOLLYD: Newhigh-yieldsynthesisof ‘8F-labeled2-deoxy-2-fluoro-D-glucose.ml J App! Radial Isot 34:893-896, 1983
9. JEWETTDM, PolocKI iF, EHRENKAUFERRE:A gas
336 THE JOURNAL OF NUCLEAR MEDICINE
by on February 27, 2019. For personal use only. jnm.snmjournals.org Downloaded from

BASIC SCIENCESRADIOCHEMISTRY AND RADIOPHARMACEUTICALS
13. SWEELEY CC, BENTLEY R, MAKITA M, et al: Gas-liquidchromatography of trimethylsilyl derivatives of sugars andrelated substances. J Am Chem Soc 85:2497-2507, 1963
14. BARRIO JR, MACDONALD NS, ROBINSON GD, et al:Remote, semiautomated production of F-I 8-labeled 2-deoxy-2-fluoro-D-glucose. J Nucl Med 22:372-375, 1981
15. GRAKAUSKAS V: Aqueous fluorination ofcarboxylic acidsalts.J Org(‘hem34:2446-2451,1969
16. BIDA GT, EHRENKAUFER RL, WOLF AP, et al: The effectof target-gas purity on the chemical form of F-I 8 during‘8F-F2productionusingthe neon/fluorinetarget. J NucI Med21:758-762,1980
solid-phase microchemical method for the synthesis of acetylhypofluorite.J FluorineChem:in press
10. OVEREND WG, STACEY M, STANEK J: Deoxy-sugars. PartVII. A study of the reactions of somederivativesof 2-deoxyD-glucose.J Chen,Soc 2841-2845,I949
II. IDo T, WAN C-N, CASELLA V, et al: Labeled 2-deoxy-D-glucose analogs. I@ F-labeled 2-deoxy-2-fluoro-D-glucose,2-deoxy-2-fluoro-D-mannose, and I4C-2-deoxy-2-fluoro-D-glucose.J LabelComp Radiopharm14:175-183,1978
12. PALMER AJ: Liquid chromatography of (‘8F)-2-deoxy-2-fluoro-D-glucose on cationic exchange resins. J Label CompRadiopharm 18:264-266, 1981
The next examination of the American Board of Science in Nuclear Medicine will be held June 4, 1984, in conjunctionwith the 31st Annual Meeting of the Society of Nuclear Medicine.
Specialty areas which may be chosen for the examination include:. NuclearMedicinePhysicsandInstrumentation. Radiation Protection. RadiopharmaceuticalandRadiochemistryScience. NuclearMagneticResonance. NuclearMedicineComputerScience. NuclearMedicineLaboratoryScience
For further information contact:Dr. Eugene Vinciguerra, Secretary
American Board of Science in Nuclear Medicine145 W. 58th St., New York, NY 10019
Tel: (212)757-0520Completedapplicationsmustbe receivedby May15,1984.
Volume 25, Number 3 337
American Board of Science in Nuclear MedicineLos Angeles, CaliforniaJune 4, 1984
HawaiiChapterSociety of Nuclear Medicine
7th Annual MeetingMay 26—28,1984 Kahuku, Oahu, Hawaii
The Annual Meeting of the HawaiiChapter,SNM, will be held May26—28,1984at the Hilton TurtleBay Resort Hotel locatedon Oahu's beautiful north shore.
Topics to be addressed at this Memorial Day Weekend Conference include NMR, SPECT,monoclonal antibodies, andcorrelativeimaging.
Continuing Education and VOICE Credits will be available for participants.
For further informationcontact:Patrick McGuigan
The HonoluluMedicalGroupDept. of Nuclear Medicine
550 S. Beretania StreetHonolulu, Hawaii 96813
by on February 27, 2019. For personal use only. jnm.snmjournals.org Downloaded from

1984;25:333-337.J Nucl Med. Richard E. Ehrenkaufer, Jan F. Potocki and Douglas M. Jewett CommunicationSimple Synthesis of F-18-Labeled 2-Fluoro-2-Deoxy-¢-Glucose: Concise
http://jnm.snmjournals.org/content/25/3/333This article and updated information are available at:
http://jnm.snmjournals.org/site/subscriptions/online.xhtml
Information about subscriptions to JNM can be found at:
http://jnm.snmjournals.org/site/misc/permission.xhtmlInformation about reproducing figures, tables, or other portions of this article can be found online at:
(Print ISSN: 0161-5505, Online ISSN: 2159-662X)1850 Samuel Morse Drive, Reston, VA 20190.SNMMI | Society of Nuclear Medicine and Molecular Imaging
is published monthly.The Journal of Nuclear Medicine
© Copyright 1984 SNMMI; all rights reserved.
by on February 27, 2019. For personal use only. jnm.snmjournals.org Downloaded from




![ORIGINAL RESEARCH Open Access F]FDG-PET imaging is an ... · Background: Positron emission tomography (PET) with [2-18 F]-2-fluoro-2-deoxy-D-glucose ([18 F]FDG-PET) was acquired at](https://img.pdfslide.us/doc/110x75/5fffd04346622d24ad6dffc9/original-research-open-access-ffdg-pet-imaging-is-an-background-positron-emission.jpg)


![Ivyspring International Publisher Thheeranoossttiiccss · [5,6-3H]-2-Fluoro-2-deoxy-D-glucose (3H-FDG; catalog number: ART0105; SA, 2.22 TBq/mmol; RCP, >99%) was purchased from American](https://img.pdfslide.us/doc/110x75/5f4847d1b7e252723751ee38/ivyspring-international-publisher-thheeranoossttiiccss-56-3h-2-fluoro-2-deoxy-d-glucose.jpg)

![EfficientStereospecificSynthesis ofNo-Carrier-Added2@[18F ...jnm.snmjournals.org/content/27/2/235.full.pdf · ofNo-Carrier-Added2@[18F]-Fluoro-2-Deoxy D-GlucoseUsingAminopolyether](https://img.pdfslide.us/doc/110x75/5af4e91e7f8b9a190c8da917/efficientstereospecificsynthesis-ofno-carrier-added218f-jnm-18f-fluoro-2-deoxy.jpg)
![Imaging, Diagnosis, Prognosis Cancer Research Preclinical and … · Imaging, Diagnosis, Prognosis Preclinical and Clinical Evidence that Deoxy-2-[18F]fluoro-D-glucose Positron Emission](https://img.pdfslide.us/doc/110x75/5e9a6009a0a8a60ac52aaf27/imaging-diagnosis-prognosis-cancer-research-preclinical-and-imaging-diagnosis.jpg)
![Potential of Early [18F]-2-Fluoro-2-Deoxy-D-Glucose ...stroke.ahajournals.org/content/strokeaha/43/1/193.full.pdf · ent diffusion coefficient as predictors for final tissue outcome](https://img.pdfslide.us/doc/110x75/5ab2f0ba7f8b9a00728db1b6/potential-of-early-18f-2-fluoro-2-deoxy-d-glucose-diffusion-coefficient-as.jpg)
![O-[3-18F-fluoropropyl]-𝛼-methyl Tyrosine in Mesothelioma ... · for imaging the metabolic activity of cancers is positron emissiontomography(PET)using18F-2-fluoro-2-deoxy-D-glucose](https://img.pdfslide.us/doc/110x75/5e7ae8d894f8f8158a33608d/o-3-18f-fluoropropyl-methyl-tyrosine-in-mesothelioma-for-imaging-the.jpg)



![MT-1769 - Moravek, Incproducts.moravek.com/DataSheets/MT1769DS.pdfMT-1769 . 1-(2-Deoxy-2-fluoro-ß-D-arabinofuranosyl)-5-methyluracil, [methyl-3H(N)]-. Lot 237-001-0032-A-20080416-TN](https://img.pdfslide.us/doc/110x75/5f3de3458996c66c5709948f/mt-1769-moravek-mt-1769-1-2-deoxy-2-fluoro-d-arabinofuranosyl-5-methyluracil.jpg)












