Embed Size (px)
Citation preview

Vol. 173, No. 6JOURNAL OF BACTERIOLOGY, Mar. 1991, p. 1920-19310021-9193/91/061920-12$02.00/0Copyright © 1991, American Society for Microbiology
Cloning and Nucleotide Sequences of Helicobacter pylornGenes Responsible for Urease Activity
AGNtS LABIGNE,* VALEIRIE CUSSAC, AND PASCALE COURCOUXUnite des Enterobacteries, INSERM U199, Institut Pasteur, 75724 Paris Cedex 15, France
Received 17 September 1990/Accepted 9 January 1991
Production of a potent urease has been described as a trait common to all Helicobacter pylori so far isolatedfrom humans with gastritis as well as peptic ulceration. The detection of urease activity from genes cloned fromH. pylod was made possible by use of a shuttle cosmid vector, allowing replication and movement of clonedDNA sequences in either Escherichia coli or Campylobacter jejuni. With this approach, we cloned a 44-kbportion of H. pylorz chromosomal DNA which did not lead to urease activity when introduced into E. coi butpermitted, although temporarily, biosynthesis of the urease when transferred by conjugation to C. jejuni. Therecombinant cosmid (pILL585) expressing the urease phenotype was mapped and used to subclone an 8.1-kbfragment (pILL590) able to confer the same property to C. jejuni recipient strains. By a series of deletions andsubclonings, the urease genes were localized to a 4.2-kb region of DNA and were sequenced by the dideoxymethod. Four open reading frames were found, encoding polypeptides with predicted molecular weights of26,500 (ureA), 61,600 (ureB), 49,200 (ureC), and 15,000 (ureD). The predicted UreA and UreB polypeptidescorrespond to the two structural subunits of the urease enzyme; they exhibit a high degree of homology withthe three structural subunits of Proteus mirabilis (56% exact matches) as well as with the unique structuralsubunit of jack bean urease (55.5% exact matches). Although the UreD-predicted polypeptide has domainsrelevant to transmembrane proteins, no precise role could be attributed to this polypeptide or to the UreCpolypeptide, which both mapped to a DNA sequence shown to be required to confer urease activity to a C. jejunirecipient strain.
Helicobacter pylori (previously designated Campylobac-ter pylori) is a small, curved, gram-negative bacillus found inthe stomach of patients with active chronic gastritis andduodenal ulcers. Since its discovery by Warren and Marshall(49) and successful isolation by Marshall et al. in 1984 (30),clinical, histological, and bacteriological investigations havebeen conducted worldwide in an attempt to determine therole of the bacteria as a causative agent in gastroduodenaldiseases. H. pylorn is now recognized as the etiological agentof active chronic gastritis (5), and there is accumulatingevidence that the organism contributes to peptic ulceration.
Several properties commonly associated with H. pyloriare suspected to play a role in the pathogenic process ofgastritis as well as ulcer formation. These include adhesionto the gastric epithelium layer (17), a property which corre-lates with the expression of hemagglutinins (11, 35), andadhesion to cell lines (10, 36); the production of proteasescapable of degrading mucus glycoproteins (42); and produc-tion of cytotoxins (22). Whether or not the genes expressingthese traits are harbored by all H. pylori isolates is stillunknown. In contrast, the expression of very high ureaseactivity responsible for hydrolysis of urea to ammonia andcarbon dioxide has been described as a common trait to allH. pylori so far isolated (5). Although it is not yet clear howthe urease enzyme acts, it is suspected to play a major rolein the ability of the bacteria to colonize and cause damage tothe gastric mucosa. It has been proposed that the ureaseenzyme might (i) allow the survival of the bacteria in anacidic medium (29), a prerequisite for colonization; and (ii)be responsible for the enhancement of the back-diffusion ofhydrogen ions (16) or stimulate gastrin production (27),resulting in increased acidity leading to gastric mucosal
* Corresponding author.
damage. (iii) More recently, Smoot et al. (45) have demon-strated that H. pylon' was cytotoxic to cultured humangastric epithelial cells and that this toxicity was due in part toammonia produced by hydrolysis of urea.To clarify the role of urease in the pathogenic process, we
attempted to identify the genes responsible for urease activ-ity in H. pylori. Because direct cloning into Escherichia colidid not result in expression of urease activity, we developeda shuttle approach for the cloning and expression of H.pylori genes. A genomic library was prepared from the totalDNA of an H. pylori strain in an E. coli host strain, using acosmid cloning vector derived from the shuttle vectorpILL550 that we designed previously for investigation of thegenetics of Campylobacter species (25). Each recombinantcosmid was then shuttled from E. coli to a Campylobacterjejuni recipient strain to examine for expression of the H.pylori genes in a Campylobacter background. By using thisshuttle approach and constructing a series of plasmid deriv-atives of a hybrid cosmid harboring the H. pylori genesresponsible for the urease activity in C. jejuni, we were ableto identify and sequence the two genes encoding the poly-peptides of the urease enzyme as well as additional se-quences required for the urease expression.
(Parts of this work were presented at the First and theSecond International Meetings of the European Campylo-bacter pylori Study Group Workshop on GastroduodenalPathology and Campylobacter pylori, 1988, Bordeaux,France, and 1989, Ulm, Germany; and at the Annual Meet-ing of the American Society for Microbiology, 1990.)
MATERIALS AND METHODSBacterial strains and plasmids. H. pylori 85P, isolated from
a patient with gastritis, and C. jejuni C31 were kindlyprovided by J. L. Fauchere (Hopital Necker-Enfants
1920
Shuttle
on July 15, 2018 by guesthttp://jb.asm
.org/D
ownloaded from
CLONING OF H. PYLORI UREASE GENE CLUSTER 1921
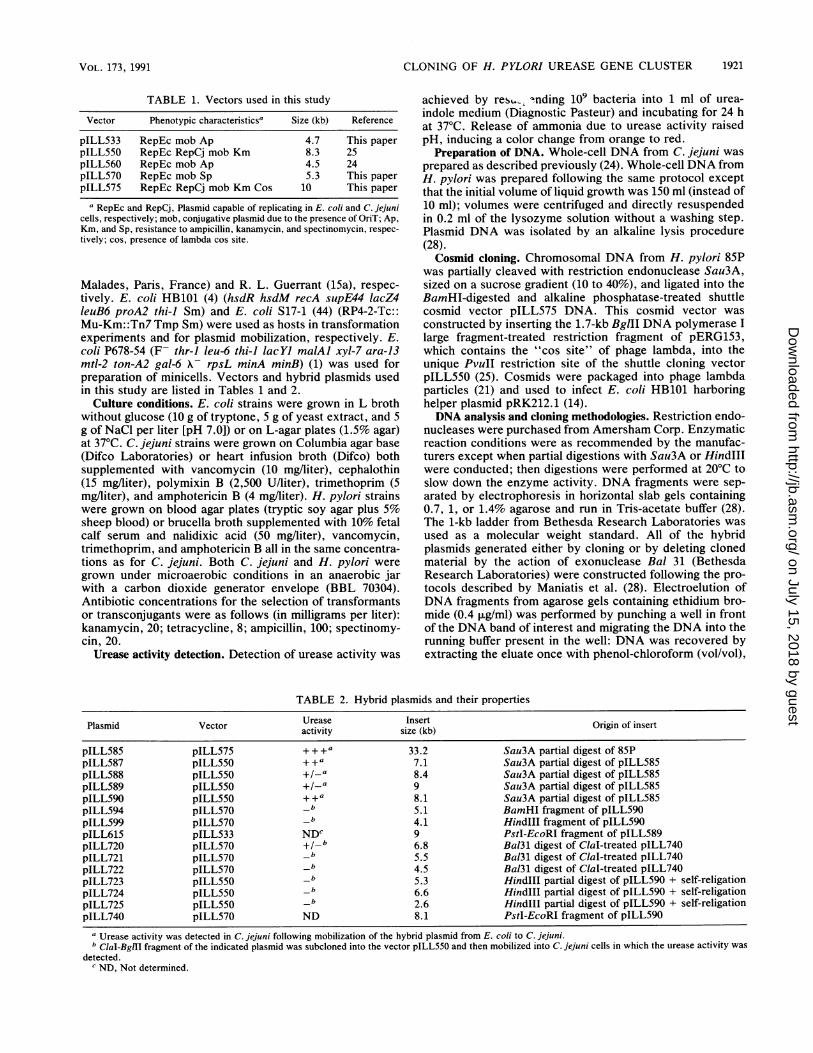
TABLE 1. Vectors used in this study
Vector Phenotypic characteristicsa Size (kb) Reference
pILL533 RepEc mob Ap 4.7 This paperpILL550 RepEc RepCj mob Km 8.3 25pILL560 RepEc mob Ap 4.5 24pILL570 RepEc mob Sp 5.3 This paperpILL575 RepEc RepCj mob Km Cos 10 This paper
a RepEc and RepCj, Plasmid capable of replicating in E. coli and C. jejunicells, respectively; mob, conjugative plasmid due to the presence of OriT; Ap,Km, and Sp, resistance to ampicillin, kanamycin, and spectinomycin, respec-tively; cos, presence of lambda cos site.
Malades, Paris, France) and R. L. Guerrant (1Sa), respec-tively. E. coli HB101 (4) (hsdR hsdM recA supE44 lacZ4leuB6 proA2 thi-J Sm) and E. coli S17-1 (44) (RP4-2-Tc::Mu-Km: :Tn7 Tmp Sm) were used as hosts in transformationexperiments and for plasmid mobilization, respectively. E.coli P678-54 (F- thr-J leu-6 thi-J lacYl malAl xyl-7 ara-13mtl-2 ton-A2 gal-6 A- rpsL minA minB) (1) was used forpreparation of minicells. Vectors and hybrid plasmids usedin this study are listed in Tables 1 and 2.
Culture conditions. E. coli strains were grown in L brothwithout glucose (10 g of tryptone, 5 g of yeast extract, and 5g of NaCl per liter [pH 7.0]) or on L-agar plates (1.5% agar)at 37°C. C. jejuni strains were grown on Columbia agar base(Difco Laboratories) or heart infusion broth (Difco) bothsupplemented with vancomycin (10 mg/liter), cephalothin(15 mg/liter), polymixin B (2,500 U/liter), trimethoprim (5mg/liter), and amphotericin B (4 mg/liter). H. pylori strainswere grown on blood agar plates (tryptic soy agar plus 5%sheep blood) or brucella broth supplemented with 10% fetalcalf serum and nalidixic acid (50 mg/liter), vancomycin,trimethoprim, and amphotericin B all in the same concentra-tions as for C. jejuni. Both C. jejuni and H. pylori weregrown under microaerobic conditions in an anaerobic jarwith a carbon dioxide generator envelope (BBL 70304).Antibiotic concentrations for the selection of transformantsor transconjugants were as follows (in milligrams per liter):kanamycin, 20; tetracycline, 8; ampicillin, 100; spectinomy-cin, 20.
Urease activity detection. Detection of urease activity was
achieved by res, -nding 109 bacteria into 1 ml of urea-indole medium (Diagnostic Pasteur) and incubating for 24 hat 37°C. Release of ammonia due to urease activity raisedpH, inducing a color change from orange to red.
Preparation of DNA. Whole-cell DNA from C. jejuni wasprepared as described previously (24). Whole-cell DNA fromH. pylori was prepared following the same protocol exceptthat the initial volume of liquid growth was 150 ml (instead of10 ml); volumes were centrifuged and directly resuspendedin 0.2 ml of the lysozyme solution without a washing step.Plasmid DNA was isolated by an alkaline lysis procedure(28).Cosmid cloning. Chromosomal DNA from H. pylori 85P
was partially cleaved with restriction endonuclease Sau3A,sized on a sucrose gradient (10 to 40%), and ligated into theBamHI-digested and alkaline phosphatase-treated shuttlecosmid vector pILL575 DNA. This cosmid vector wasconstructed by inserting the 1.7-kb BglII DNA polymerase Ilarge fragment-treated restriction fragment of pERG153,which contains the "cos site" of phage lambda, into theunique PvuII restriction site of the shuttle cloning vectorpILL550 (25). Cosmids were packaged into phage lambdaparticles (21) and used to infect E. coli HB101 harboringhelper plasmid pRK212.1 (14).DNA analysis and cloning methodologies. Restriction endo-
nucleases were purchased from Amersham Corp. Enzymaticreaction conditions were as recommended by the manufac-turers except when partial digestions with Sau3A or HindlIlwere conducted; then digestions were performed at 20°C toslow down the enzyme activity. DNA fragments were sep-arated by electrophoresis in horizontal slab gels containing0.7, 1, or 1.4% agarose and run in Tris-acetate buffer (28).The 1-kb ladder from Bethesda Research Laboratories was
used as a molecular weight standard. All of the hybridplasmids generated either by cloning or by deleting clonedmaterial by the action of exonuclease Bal 31 (BethesdaResearch Laboratories) were constructed following the pro-tocols described by Maniatis et al. (28). Electroelution ofDNA fragments from agarose gels containing ethidium bro-mide (0.4 ,ug/ml) was performed by punching a well in frontof the DNA band of interest and migrating the DNA into therunning buffer present in the well: DNA was recovered byextracting the eluate once with phenol-chloroform (vol/vol),
TABLE 2. Hybrid plasmids and their properties
Plasmid Vector Urease Insert Origin of insert
pILL585 pILL575 +++a 33.2 Sau3A partial digest of 85PpILL587 pILL550 ++a 7.1 Sau3A partial digest of pILL585pILL588 pILL550 +/-a 8.4 Sau3A partial digest of pILL585pILL589 pILL550 +/-a 9 Sau3A partial digest of pILL585pILL590 pILL550 ++a 8.1 Sau3A partial digest of pILL585pILL594 pILL570 _b 5.1 BamHI fragment of pILL590pILL599 pILL570 _b 4.1 HindlIl fragment of pILL590pILL615 pILL533 NDC 9 PstI-EcoRI fragment of pILL589pILL720 pILL570 +/_b 6.8 Bal31 digest of ClaI-treated pILL740pILL721 pILL570 _b 5.5 BaIl3 digest of Clal-treated pILL740pILL722 pILL570 _b 4.5 Bal31 digest of ClaI-treated pILL740pILL723 pILL550 _b 5.3 HindIII partial digest of pILL590 + self-religationpILL724 pILL550 _b 6.6 HindIll partial digest of pILL590 + self-religationpILL725 pILL550 _b 2.6 Hindlll partial digest of pILL590 + self-religationpILL740 pILL570 ND 8.1 PstI-EcoRI fragment of pILL590
aUrease activity was detected in C. jejuni following mobilization of the hybrid plasmid from E. coli to C. jejuni.b ClaI-BglII fragment of the indicated plasmid was subcloned into the vector pILL550 and then mobilized into C. jejuni cells in which the urease activity was
detected.c ND, Not determined.
VOL. 173, 1991
on July 15, 2018 by guesthttp://jb.asm
.org/D
ownloaded from

1922 LABIGNE ET AL.
then several times with butanol to reduce the eluate volumeto 100 ,ul, and finally with diethyl ether. DNA was thenprecipitated with cold ethanol before recovering it by cen-trifugation. DNA polymerase I large fragment, T4 DNApolymerase (used to make blunt-end fragments) and T4 DNAligase were purchased from Amersham, and calf intestinephosphatase was purchased from Pharmacia.
Hybridization. DNA restriction fragments fractionated byagarose gel electrophoresis were transferred to nitrocellu-lose sheets (0.45-tLm pore size; Schleicher & Schuell, Inc.)by the Southern technique (46) and hybridized at 68°C with32P-labeled deoxyribonucleotide probes (Amersham) labeledby random priming (13), using as primers the random hex-amers from Pharmacia. Hybridization was revealed by au-toradiography with Kodak XAR-Omat film.Mating between donor E. coil cells and C. jejuni C31
recipients. Conditions to run series of 24 mating experimentssimultanously were adapted from the previously describedprotocol (25) as follows. E. coli cells harboring the IncPhelper plasmid plus the hybrid plasmid to mobilize weregrown in L broth without antibiotic with gentle shaking to adensity of 10i bacteria per ml. Campylobacter recipient cellswere grown overnight in heart infusion broth (100 ml)supplemented as described above at 37°C with vigorousshaking under microaerobic conditions. A 4-ml portion ofthe overnight recipient cultures (108 bacteria per ml) wascentrifuged, washed in sterile water, pelleted again, andsuspended in 100 ,ul of donor E. coli cells. Each mating mix(100 p1) was spread on the surface of Mueller-Hinton me-dium freshly poured into a 12-well tissue culture plate (eachwell containing 3 ml of Muller-Hinton) and incubated inmicroaerobic conditions for 5 h at 37°C. The bacteria wereharvested by shaking three glass beads per well with a 100-lU1volume of heart infusion broth, and 100 itl of broth contain-ing bacteria was plated on Columbia medium containingvancomycin, polymixin, cephalothin, trimethoprim, ampho-tericin B, and kanamycin. Plates were incubated at 37°C inmicroaerobic conditions for 48 to 60 h.
Analysis of proteins expressed in minicells. Minicells har-boring the appropriate hybrid plasmid were isolated andlabeled with [35S]methionine (50 ,uCi/ml) (15). Approxi-mately 100,000 cpm of acetone-precipitable material wassubjected to sodium dodecyl sulfate-polyacrylamide gel elec-trophoresis in a 12.5% gel (26). Standard proteins withmolecular weights ranging from 94,000 to 14,000 (low-mo-lecular-weight kit from Bio-Rad Laboratories) were run inparallel. The gel was stained and examined by fluorography,using En3Hance (New England Nuclear).DNA sequencing. Appropriate DNA fragments were
cloned into M13mpl9 (31). Plaques containing inserts wereidentified by using X-Gal (5-bromo-4-chloro-3-indolyl-P-D-galactopyranoside) and isopropyl-p-D-thiogalactopyrano-side. Sequential series of overlapping clones were producedby using the Cyclone I Biosystem (I.B.I.). Single-strandedDNA templates were prepared by the polyethylene glycolmethod (41), and the sequence was determined by dideoxy-nucleotide chain termination (40), using a Sequenase kit(United States Biochemical Corp.). Sequence analyses per-formed directly on the product of amplification (polymerasechain reaction) were performed by using dimethyl sulfoxide(1% final concentration) in the annealing mixture (50). Theamplification was carried out from total DNA extracted fromH. pylori 85P for 25 cycles in a DNA Thermal Cycler (PerkinElmer-Cetus), using the two oligonucleotides shown in Fig.4 and a GeneAmp kit (Perkin Elmer-Cetus). The DNA was
denatured at 94°C for 2 min, annealed at 55°C for 2 min, andextended at 75°C for 2 min.
Nucleotide sequence accession number. The nucleotidesequence accession number is X57132.
RESULTS
Cosmid cloning of a DNA sequence responsible for ureaseexpression from H. pylon 85P. Due to unsuccessful attemptsat cloning and expressing the H. pylori genes of interest intoE. coli recipient strains, we designed a shuttle approach foridentifying the genes involved in urease expression in H.pylori strains, using C. jejuni as a final recipient strain. C.jejuni is a Campylobacter species naturally devoid of ureaseactivity and was thought to be a more closely related host forthe expression of H. pylori genes than E. coli. We modifiedthe E. coli-C. jejuni shuttle vector pILL550 that we de-scribed previously (25) by introducing a DNA fragmentcontaining a cos site (see Materials and Methods), so that theshuttle vector could be used as a cosmid vector. Bacterio-phage lambda-transducing particles carrying recombinantcosmid molecules with segments (35 to 48 kb) of the chro-mosomal DNA of H. pylori 85P total DNA were preparedwith pILL575 as a vector and were transduced into E. coliK-12 strain HB101 harboring the IncP helper plasmidpRK212.1. A total of 400 independent tetracycline- andkanamycin-resistant E. coli transductants harboring recom-binant cosmids were frozen as a gene bank. None of the 400E. coli transductants exhibited urease activity. Each recom-binant cosmid was then mobilized from E. coli to C. jejuniC31, and kanamycin transconjugants were tested for theircapacity to hydrolyze urea, a phenotype designated urease+.Of 106 C. jejuni kanamycin-resistant transconjugants tested,1 exhibited urease activity; the enzymatic activity of thetransconjugant was considerably lower than that observedwith the wild-type H. pylori strain (85P). Whereas with H.pylori the reaction was immediate, 4 h of incubation wererequired to detect the activity from the transconjugants. Thepositive colony harbored a recombinant plasmid designatedpILL585, 54 kb in size, which was purified from E. colitransductants as well as from Campylobacter transconju-gants. Comparison of the HindIll restriction profiles ofpILL585 isolated from either E. coli or C. jejuni (Fig. 1)clearly showed that the recombinant cosmid once in C. jejunicells was totally unstable and gave rise to DNA rearrange-ments associated with deletions. As a consequence, in thesubsequent steps, the DNA material used was solely thatprepared from E. coli strains. The BamHI, EcoRI, PstI, andSmaI restriction sites of pILL585 were located by single anddouble digestion, and the resulting restriction map of thecosmid was established as shown in Fig. 2.
Defining the smallest urease-expressing DNA region ofcosmid pIL585. BamHI-generated DNA fragments from the44-kb insert were cloned into shuttle vector pILL550, trans-formed in the mobilizing E. coli S17-1, and mobilized into C.jejuni, and the resulting transconjugants harboring the hy-brid plasmids were assayed for urease activity. None of theBamHI-generated fragments led to a urease+ phenotype inC.jejuni, indicating that the genes involved in the expressionof urease contain at least one BamHI site. In the absence ofsuitable restriction sites and to define the smallest DNAfragment able to confer the urease+ phenotype on C. jejuniC31, pILL585 cosmid DNA (20 ,g) was partially digestedwith endonuclease Sau3A to generate fragments rangingfrom 6 to 12 kb. The fragments were treated with alkalinephosphatase to prevent any rearrangement of the initial
J. BACTERIUOL.
on July 15, 2018 by guesthttp://jb.asm
.org/D
ownloaded from

CLONING OF H. PYLORI UREASE GENE CLUSTER 1923
ino
1.21)..ImX 3
X- Q.
LnCso0, v-X sLf IV
'& ,l (Kb)
Ltn
kf0o Go cO
4- m- LInh6_j
.j j
m4
Lir,(OC
c. Lncr% or, =Ln Ln >-.j IM61.4 .j" -8, -41:.6 .64
-76 _ -41 5 .2-_
.4
-3
-2
15
.1 -1.15.V.
Hind III Hind III BamH I
FIG. 1. Comparison of the urease region when cloned in shuttlecosmid pILL575 (pILL585) or in shuttle plasmid pILL550 (pILL590)following their propagation in E. coli or C. jejuni C31 (pILL585*).Plasmid DNA (0.5 p.g) or H. pylori 85P total DNA (3 ,ug) was
digested with HindlIl or BamHI restriction enzymes. The resultingfragments were separated by electrophoresis through a 1% agarose
gel for 17 h at 2.5 V/cm, transferred to nitrocellulose, and hybridizedwith in vitro 32P-labeled EcoRI-PstI 8.1-kb insert from pILL590.Values on the left correspond to the sizes (in kilobases) of the 1-kbladder used as the standard; those on the right indicate the size ofthe restriction fragments present in chromosomal DNA, hybridcosmid pILL585, and the derivative plasmid (pILL590).
genome and were ligated with BamHI-treated pILL550 (75ng). After transformation into E. coli S17-1, kanamycin-resistant transformants were mated with C. jejuni C31 andkanamycin transconjugants were assayed for urease activ-ity. Of 60 transconjugants tested, 4 were urease positive;they contained plasmids designated pILL587, pILL588,pILL589, and pILL590 with inserts of 7.8, 8.6, 9, and 8.1 kb,respectively. The plasmids exhibited instability in C. jejunicells, however, to a lesser extend than cosmid pILL585.Based on the rapidity of the reaction relative to that ofrecombinant cosmid pILL585 (urease +. ), pILL587 andpILL590 exhibited a phenotype which was designated ure-
ase++, whereas pILL588 and pILL589 were urease+'-. TheBamHI, ClaI, EcoRI, HindlIl, PstI, PvuII, and SmaI rec-
ognition sites were mapped in the four plasmids, and theorientation of the insert relative to the vector was deter-mined as illustrated in Fig. 2: comparison of the restrictionendonuclease-generated maps showed that expression of theurease activity was independent of the orientation of theinsert relative to the vector and that they shared a common
DNA sequence of 4.2 kb, designated the urease region.Plasmid pILL590 with relatively high urease expression was
chosen as the prototype plasmid for further characterizationof the urease region. Using the 8.1-kb EcoRI-PstI restrictionfragment of pILL590 as a probe, we confirmed by Southernhybridization that the sequences cloned did not suffer anyrearrangements through the cloning process, i.e., that theHindIll and BamHI restriction fragments of subclones andcosmid were present at the same size as in the original strain
of H. pylori (Fig. 1). This hybridization allowed us toconclude that a single copy of the urease region was presentin the H. pylori strain. Moreover, hybridizing the 8.1-kbinsert of pILL590 against nondigested total DNA extractedfrom H. pylori 85P did not allow us to visualize a band whichwould migrate as a supercoiled form of plasmid DNA,suggesting that the genetic information that we cloned orig-inated from chromosomal DNA (data not shown).To generate deletions, unique restriction sites were added
on each side of the insert by cloning the 8.1-kb fragment ofpILL590 into vector pILL570 (Spr, 5.3 kb in size). ThepILL570 vector was constructed from pILL560, describedpreviously (24), following the removal of the DNA fragmentcontaining the ampicillin resistance marker (located betweenthe DraI site [position 3232 of pBR322] and the EcoRI site[position 1 of pBR322]) and ligation with the filled ends of theHindIII-generated interposon Ql (37). A series of deletionsstarting at either end of the 8.1-kb insert of the resultingplasmid pILL740 and extending up to 3.6 kb in the insertwere performed by (i) using Bal3l, (ii) subcloning restric-tion fragments, or (iii) partially digesting the plasmid withHindIll. Following these steps, each deleted insert wascloned again into pILL550, introduced into a mobilizing E.coli strain (S17.1) and shuttled into C. jejuni to determineurease activity. The results are summarized in Fig. 2. Anydeletion extending into the previously defined urease region(4.2 kb in length) led to a negative phenotype, suggesting thatthis region was the smallest DNA fragment absolutely re-quired for the urease expression in a C. jejuni host.DNA sequence of the urease region of H. pylori 85P. The
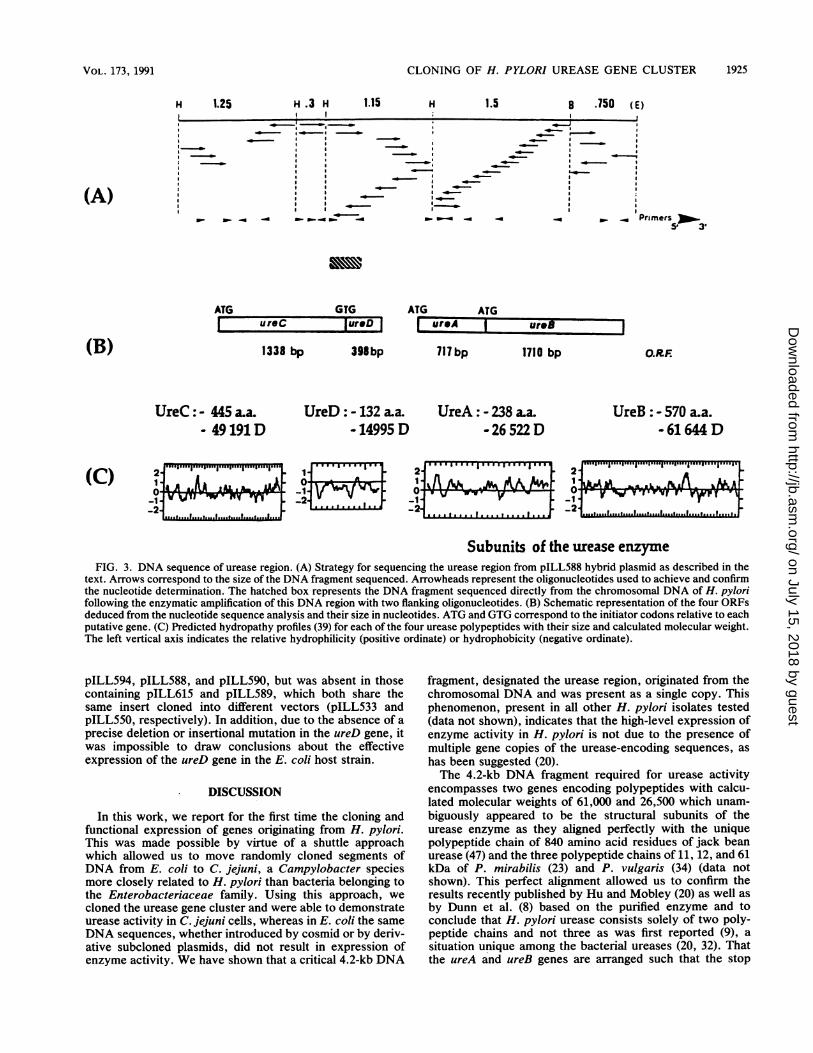
5,100 bp depicted in Fig. 3A were sequenced with thefollowing strategy; the HindIll 0.3-, 1.15-, and 1.25-kbfragments as well as the 1.5-kb HindIII-BamHI and 0.7-kbBamHI-EcoRI fragments originating from pILL588 wereindependently sequenced by creating overlapping deletionson the restriction fragments cloned into M13mpl9 DNAphage. In addition, 16 synthetic oligonucleotide primerswere synthesized to generate sequences overlapping the fiveindependently sequenced fragments and to sequence thecomplementary strand when required.The 5,100 bp spanning the urease region were analyzed for
open reading frames (ORFs): four ORFs of >132 codonswere found encoded by the same strand (Fig. 3B), whereasno ORF of any significant length was found on the reversecomplement of the sequence shown in Fig. 4. These fourORFs were designated ureA, ureB, ureC, and ureD; three ofthem begin with the characteristic ATG start codon, and onebegins with the less frequent GTG start codon (Fig. 3). Thefour ORFs were each preceded by sites similar to the E. coliconsensus ribosome-binding (Shine-Dalgarno) sequence(43): GGAG preceding the ureA and ureB genes and AAGGpreceding the ureC and ureD genes. The precise positionsare indicated in Fig. 4. The atypical GTG start codon of ureDis localized a few nucleotides upstream of the ureC stopcodon, and a unique frameshift is responsible for the sepa-ration of the left end side of the urease region into two ORFs.To be sure that these two ORFs really did exist in H. pylori,we used the polymerase chain reaction to amplify a 450-bpDNA fragment spanning the 3' end of the putative ureC geneand the 5' end of the ureD gene from total DNA extractedfrom H. pylori 85P. The amplified DNA was then directlysequenced and the same nucleotide sequence as the onedetermined from DNA cloned and propagated in E. coli wasfound in the genome of H. pylori, indicating that ureC andureD actually represented two distincts ORFs.From the 5' to the 3' end of the sequenced region, several
VOL. 173, 1991
on July 15, 2018 by guesthttp://jb.asm
.org/D
ownloaded from

1924 LABIGNE ET AL.
UreaseActivity (P) BB
Conjugative cosmid I IJplL1585 (33.2 kb) 4 kb
B BS
I
Sm P E (E)I I Il
(A)
++ pILL587(7.1 kb)
+/- pILLS88(8.4 kb)
+1- pILL589(9 kb)
++ plLU590(8.1 kb)
(P) H BI HIA .a 1.1 I H2 H3 H4
I(P)H H BI HI H2H3 H4
I I I I _ I I
(d,C) H2 H3 H4
JII(E,C)B1 HI i H2H3
I I I I I
I
H4
B2 I
82 (EC)
B2 1 83B2 I B3
I
B3 (E,C)
B2 I B3 HS (P)I a I IL
(C)
pILL720 (6.8 kb)
- pILL721 (5.5 kb)
- pILL594 (5.1 kb)
pILL722 (4.5 kb)
pILL723 (5.3 kb) _ g
pILLS99 (4.1 kb) -
pILL724 (6.6 kb) I -l_
pILL725 (2.6 kb) I -I
...4.2 kilobasesFIG. 2. Linear restriction maps of hybrid cosmid pILL585 (A) and (B) hybrid plasmids pILL587, pILL588, pILL589, and pILL590
resulting from Sau3A partial digestion of pILL585 and the subsequent cloning of the generated fragments into vector pILL550 (heavy line).(C) Boxes represent the extent of the deletions generated with nuclease Bal 31 or restriction endonuclease HindIII performed on plasmidpILL590. Numbers in parentheses correspond to the size of the H. pylori DNA fragment inserted into the cloning vector (pILLS75 orpILL550). B, BamHI; C, ClaI; E, EcoRI; H, HindIII; P, PstI; Sm, SmaI restriction sites; these letters within parentheses indicated that therestriction sites originated from the cloning vector. +++, + +, or +/- refer to the urease activity detected in the C. jejuni kanamycin-resistant transconjugants harboring the indicated recombinant plasmid.
other remarkable features were found at the DNA level (Fig.3B). (i) Upstream of the ureC gene an E. coli consensuspromoterlike sequence (ar70) was found between nucleotides188 and 225, where 10 nucleotides matched the 12 requirednucleotides (TIGACA, -35 and TATAAT, -10) (38). (ii) Astem-loop structure with the features of a typical rho-independent transcriptional stop signal (38) was found down-stream of the ureC gene (Fig. 4). (iii) Exactly 310 bpupstream of the ureD and ureA genes, sequences were foundwhich exhibit a high degree of homology with the nitrogenregulation site specifying the a54 recognition sequence (nifpromoter) (33): 13 and 14 of the 16 nucleotides of theconsensus nifpromoter sequence TGGYAYR ---- YYGCZ,where Y = T or C, R = G or A, and Z = A or T (i.e.,TGGTAgA - - - - TacCT for ureA and TGGCtTG - - - -
gCGCT for ureD), were conserved and aligned in such a waythat the critical spacing between the GG and the GC doubletswas 10 bp. No stem-loop structure was identified down-stream of the ureD gene which might lead to the terminationof the putative transcript initiated at the first nifpromoterlikesequence or downstream of the second ni/ promoterlikesequence mapping within the 480-bp noncoding regionflanked by the ureD and ureA genes.
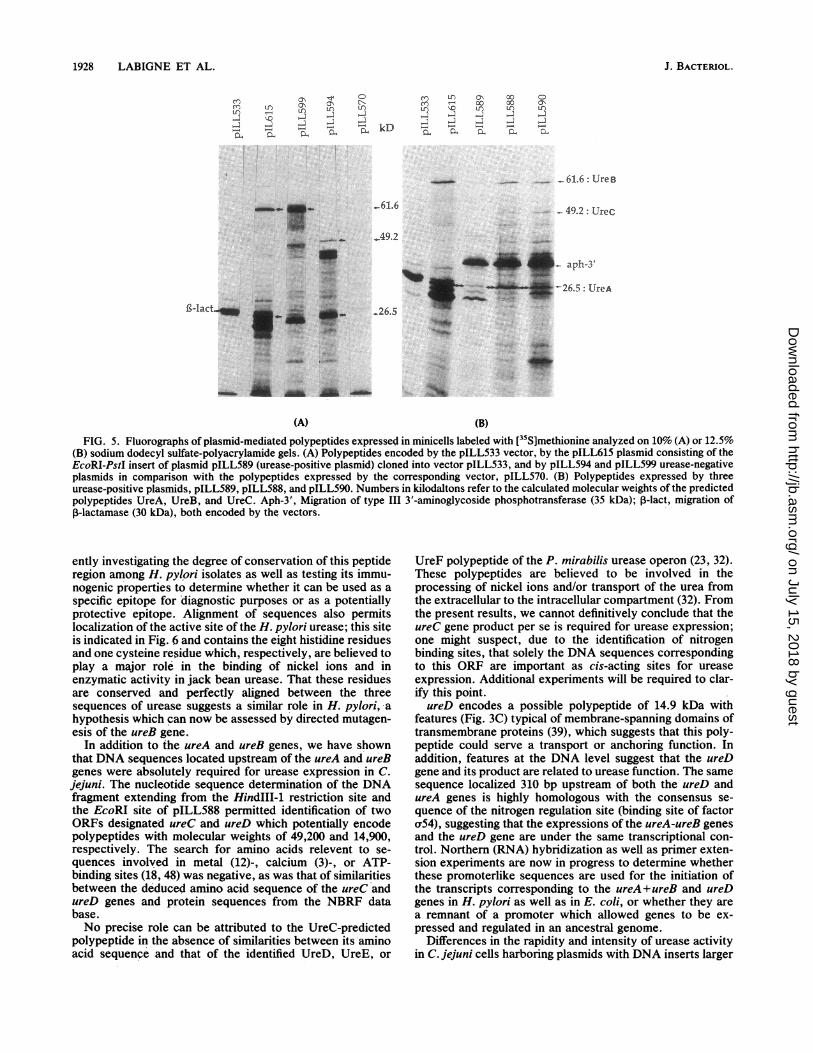
Analysis of polypeptides expressed in minicells. pILL615,pILL589, pILL588, pILL590, pILL594, and pILL599 hybrid
plasmids as well as the corresponding cloning vectors wereintroduced by transformation into E. coli P678-54, a minicell-producing strain. Minicells were isolated and the polypep-tides encoded by the plasmids synthesized in E. coli werelabeled with [35S]methionine and resolved by sodium dode-cyl sulfate-polyacrylamide gel electrophoresis. The twopolypeptides expressed from DNA fragments correspondingto ureA and ureB were clearly detected from these experi-ments (Fig. 5); they migrated with polypeptides havingapparent molecular weights of 66,000 and 30,000 and wereshown to disappear when deletions mapped to the ureA andureB loci encoding the 61.6- and 26.5-kDa predicted poly-peptides: UreA was expressed by pILL615, pILL589,pILL588, and pILL590 as well as by the subcloned pILL599,but not by pILL594; reciprocally, UreB was expressed bypILL594 but not by pILL599, as expected. The intensity ofthe bands corresponding to the expressed polypeptide rela-tive to that of type III 3'-aminoglycoside phosphotransferaseconferring kanamycin resistance (35,000 in molecularweight) was independent of the orientation of the insert inthe cloning vector, suggesting that the ureA and ureB geneswere expressed from promoter sequences located in thecloned sequences; a polypeptide with an apparent electro-phoretic mobility corresponding to the ureC gene predictedproduct (49.2 kDa) was synthetized in minicells harboring
(B)
HS H (P)
a,zI if I - LL I I 1-- 2% s |I
I,
III
gm.I
--------
.M-
J. BACTERIOL.
I#/ Urease region
I-
I 0
on July 15, 2018 by guesthttp://jb.asm
.org/D
ownloaded from
CLONING OF H. PYLORI UREASE GENE CLUSTER
1.15 H 1.5 8 .750 (E)
. . -, | - ._
- ~ ~ ~ _~
.. - .
_ _ _ _ _ _ _Z _~~~~ a -
_ Primers5' 3'
ATG GTGI ureC lureDI
1338 bp 398 bp
ATG ATGureA J urIeB717bp 1710 bp
UreC: - 445 aa.- 49191 D
UreD: - 132 aa.- 14995 D
UreA: - 238 La.- 26 522 D
UreB : - 570 a.a.- 61 644 D
0-1 VVII VVW __IV _ _- __-__2 ,.I,,,s,,,_-21lll.
Subunits of the urease enzymeFIG. 3. DNA sequence of urease region. (A) Strategy for sequencing the urease region from pILL588 hybrid plasmid as described in the
text. Arrows correspond to the size of the DNA fragment sequenced. Arrowheads represent the oligonucleotides used to achieve and confirmthe nucleotide determination. The hatched box represents the DNA fragment sequenced directly from the chromosomal DNA of H. pylorifollowing the enzymatic amplification of this DNA region with two flanking oligonucleotides. (B) Schematic representation of the four ORFsdeduced from the nucleotide sequence analysis and their size in nucleotides. ATG and GTG correspond to the initiator codons relative to eachputative gene. (C) Predicted hydropathy profiles (39) for each of the four urease polypeptides with their size and calculated molecular weight.The left vertical axis indicates the relative hydrophilicity (positive ordinate) or hydrophobicity (negative ordinate).
pILL594, pILL588, and pILL590, but was absent in thosecontaining pILL615 and pILL589, which both share thesame insert cloned into different vectors (pILL533 andpILL550, respectively). In addition, due to the absence of aprecise deletion or insertional mutation in the ureD gene, itwas impossible to draw conclusions about the effectiveexpression of the ureD gene in the E. coli host strain.
DISCUSSION
In this work, we report for the first time the cloning andfunctional expression of genes originating from H. pylori.This was made possible by virtue of a shuttle approachwhich allowed us to move randomly cloned segments ofDNA from E. coli to C. jejuni, a Campylobacter speciesmore closely related to H. pylori than bacteria belonging tothe Enterobacteriaceae family. Using this approach, wecloned the urease gene cluster and were able to demonstrateurease activity in C. jejuni cells, whereas in E. coli the sameDNA sequences, whether introduced by cosmid or by deriv-ative subcloned plasmids, did not result in expression ofenzyme activity. We have shown that a critical 4.2-kb DNA
fragment, designated the urease region, originated from thechromosomal DNA and was present as a single copy. Thisphenomenon, present in all other H. pylori isolates tested(data not shown), indicates that the high-level expression ofenzyme activity in H. pylori is not due to the presence ofmultiple gene copies of the urease-encoding sequences, as
has been suggested (20).The 4.2-kb DNA fragment required for urease activity
encompasses two genes encoding polypeptides with calcu-lated molecular weights of 61,000 and 26,500 which unam-
biguously appeared to be the structural subunits of theurease enzyme as they aligned perfectly with the uniquepolypeptide chain of 840 amino acid residues of jack beanurease (47) and the three polypeptide chains of 11, 12, and 61kDa of P. mirabilis (23) and P. vulgaris (34) (data notshown). This perfect alignment allowed us to confirm theresults recently published by Hu and Mobley (20) as well as
by Dunn et al. (8) based on the purified enzyme and toconclude that H. pylori urease consists solely of two poly-peptide chains and not three as was first reported (9), a
situation unique among the bacterial ureases (20, 32). Thatthe ureA and ureB genes are arranged such that the stop
1.25 H .3 HH
(A)
(B) O.RF
1925VOL. 173, 1991
on July 15, 2018 by guesthttp://jb.asm
.org/D
ownloaded from

31 1680/301 _1710UL1LAM CT? T AOC TMO AM £01 CAT GCM AMA CA C AT? AM AM £20 T0£ AMA AM AMA OW A AM =C CT 22£ AM CM 22£ GM 3MG CIarARC CoC CA! 220 ATC COT TMTgly tyr assn aa IOU inu lye gin inu amp lysevin gin ile erg his len ±l* arg tyr61 9
TCC AMC A12 CTA AAR AMC AC&AA3R C22 AMA AMG AM CCC AAM AM AM AM CCI TTA C2M 1740/411 1770/421ACC GCC ACT aM AC AMA TT£ CCa £20 CT? 22h aM OCT AM CAT aM AMA CT? 22£ aJk
121 151 anr gly tbr gin ass. lye len erg ±1. InU IOU gin ala lye asp gin lye lOU lOn 'ginCC 22£ AMC AMG TTA AMA ICC CCC CIA AM 23£ GM 30£ COO AT? 222 220 ACT aM ow1800/431 1830/441
181 211 200 AM £20 Ca aM 22£ AM 9Mg TTT 222 CJa 000 CAT 220 2CC TA£ AMA CCI CIA AMA22£ ACWI AG TYC CT aM owG0M2TT CTT V3 CC? 220 C20 ACI AMT enr lye set gin gin lOU lye Vlu phe phs gin gly his lOn cye OCR
241 271 50 122£ ?QC AMC ?TC 22£ 22£ 0G AT? 2TC AMh CC£ CTC 220 AMC TC1 TCC TMC £20 AT? CM 222 TGA5M OCA 222 020 CT£ AMA £00 AC? AM AMA
val leu Lye tin tin lye lye301 321A00 CM CCC 2TC ACC CMC 00 CT? AMT CAT AT? TT2 AM? 2T WIT 222 ATA AMA CC? CT? 1862/S8 __________ 1892/16
£00 CRC TIC 0MM 222AT£21 0M02 2211' CT? A T? 222 0CC2 0G2 CII ow AT? AMA361 291 oan len inu val pbs ile gly Val pbs ph* inu Ii. pho, gy Val asp gin ala ±2* lyeow 220 0GM TCT TTT AMTCOT CTO TCT 0M22CG CT? TIC 2CC CCI CT? £20 £22 ?CC CAT
t~~~~~~~~~~~~~~~~~~~~~~L2UttLG 1952/36422 451 ~~~~~~~~~~~~~~~~~TICOCT £22 22£ GM 0002T2 CCC TAT a WI 220TT?MM 2A OAT AT? 02 2T0 020 22CAMC CIT MMT? 22 £20 CC? 220 23£ TOT AM £22 TOO. CAT AM ?C2 £20 23£ AMA220 £22 tXr ala Lle inu gin gly ph. erg tyr gin enr len vel il. a-sp Ile Val 1ion val phs,431 S5 510/1 1902/46 2012/56AlA AM?WI 22£ AlA_? £22T 22£ 23£ CO. MG AO MCAM£222M0C 012000030 AM? AMhO MG=T C00020ow222 C22CM WI T222 22MGMT0000122 AM CTM 2CMset lye ilie pbs gly tin asp gly val arg assn lye gly va.l ala phs, sen InU inu an pbs inu gin gly gly leu lye tyr len gin540/11 570/21 2042/68 2072/78OG? AMk GCA 000 020 AM CMC A0C CCC £20 222 020 £20 COT 22£ CC £22 OCT CC 0GM £20 CT? T20 AT? 22£ 000 CT 222 £20 222 22£ £20 CCC CIA 300 CM CT? 222 AMA AMCgly lye ala gly vat lye len tin pro set pbs vat set arg In gly 12* ala Ala gly 1le Inoleinl2 inu gly len pbse il* phs len set ary gin arg gluinue phs, lye sass
600/31 630/41 2102/S0 2132/55220 TMT 222 AMR AMR CIT TC? CIA 30G AM? AMA AWT?22 £20002 AMAA MC £0CAMAA CAT ow ATh GM 222 00 £20 CM 222f 002 oa ooo 0' TM AM?0MM m CM COO 222inu tyr phs lye lys his snx gin tin ass lye Ilie inu il* gly lye asp tir arg lys his ala ±2* gin phs, gly set vat pbs gly ala gly vat snr ass vat inu asp arg ph*660/51 690/61 2162/10S 2192/116MC G TAT21232 02£ aJ AMACC 21 00CMAC OCT CMC ACT T00 £TA CCC 2A AM 00 0GM CIT 00CC 02£ 0200M2GA TMT0CM 2A2 TAT CA? TM 000 222 CAT 220 CCI 222 22£enr gly tyr set vat gin &ass ala inu vat eno &alai tin enr Ile gly tyr ass' Vat vat his gly gly vat Vat asp tyr vat tyr tyr his tyr gly pho asp len pro phb, len
720/71 750/Si 2222/12S 2252AT?f CIA £2£ GM0CCT £20 CCT A0CC?owT £20AC = 222 22£ £0= GM £20 CCC TOT AC? 200 CS £20G TCI r A201230£20000020 ow 222212TOT?T 23£ M AM? TC? 222Ile gin il* gly pro set pro tin pro ala ile ala pbis len tin gin asp m!et arg cye tin eno Iwonost sen 0£
730/61 310/101 2282/146 2312GM2 ow GM £22 £20 A2A MC ow MC CMC AMC CC? 222 Ca GM AM? 00 AT? AMr 222 22£ AC AMR AMC AMA ACA AMR 22£ A0 CAT AM TCC CC? 222 23£ AMT AMR 300 TCr CO.asp ala gly Ilie set il sen ala eno his assn pro pho, gin asp assn gy Lie lye pbs
2341 2271340/111 370/121 TMC 020 AGITMTI;AiC CT AC 3£ 200T TMG200 CC= CS 022T CAA 020 CMG 20220 AAM T TAT COT TMT AM CMC AM WA aM a aM AGA ow £22 a am £C2022phs ass eno tyr gly tyr lye Ion lye gin gin gin gin arg ala ±le gin gin Ile phs 2401 2431
TOO CCI CCA 22£ ?CA CMC CIAR 22223 220T TCA 22? 222 200 GMt 222 TT0 £2022 £21A900/131 930/141CAT GAT aL 00£ 22£ CS CiaT2CC WIT 2TM AM GM CCC GMS MC 020 CO MC OCT AM 2461 2491his asp gin gly len len his enr eno tyr lye vat gly gin eno vat gly enr ala ly AM 2TM AMR 000 0TA 22£ AMT GrA CMC CCAT3.2 3.00 CT? 22£ TMG CCC 220 AMA AMC £21
960/151 990/161 2521 2551A00 £2£ GM GAT 00 32£ 000 COT TMT £20 1*0 CAT T20 AM CMC TC? 220 CCC AMA CAT AMA CTA £22 CAT 222 AM23£ 23£ 22£ 022 AMAT CCL 12 20223£TA TC? TMa 23£ £20arg ile asp asp vat Ile giy arg tyr I1. ala his len lye his enr pho, pro lye his
2531 26111020/171 1050/iSi AM MAC 220 CIA CMA TC£ CAT CC AC0 220. £22 ow 22£ TOT CT? CM 00£ AMA MAC CT?2T0 AMT 22£ CM WIA 22£ 3032 on CTA 0GM1A=CC?AAM 0WCC o OT TMa AMonMleu &asinIogin snr in arg ile vat len asp tin ala ass gly ala ala tyr Lye val
2641 53 1 2671/51030/191 1110/201 23£ a 2MS&a- AM? Gm £20 AMA CMc £00 CCI am Gm 22£ OAT AM 220 £20 CMTCMCM? CCC 2TCT 222MTAC GMG CTT GM GCC? T1 on T21 on £22 AM? CAT GM CC? AMC set lys len tin pro lye yin inu asp lye inu set inu hisala pro vat vat ph eno gin Inu gly ala asp Vat inu vat ile ass asp gin pro &ass
2701/15 2731/251140/211 1170/221 TMC OCT 00£ aM 22G OCT AMA AMA COO AMA CaR 31£ GCC £22 AMG CT? AMC TMT 02£ aG00 TOT AMC £22 AM JGM CMK TOOG0G OCMTT22 CAC CC? AMC CM TM AMC CAGM aon tyr ala gly gin inu ala lye lye arg lye gin lys gly I*lye ins y a igly cys ass l2o ass gin gin eye giy ala len his pro &ass gin Ion snr gin gin vat
2761/35 2791/451200/231 1230/241 G 02£ GCC 220 £22 WIT GC CAT £22 3.20 aM a ow 30£ OCT GOT AMA AM AC1 owAMA AMA 230 CCC owr OAT CSG CC 222 GM? 22 CAT CCC CIT ow CAT A0 CIA on on &al Vat ala inuil* ana ala his il* eat gin gin ala arg ala gly lye lye tin alalye lye tyr ar'g ala asp in gly phe ala pbs asp gly asp ala asp arg ino vat val
2821/55 2851/651260/251 1290/261 OCT aM 2T £20 CM GAR CS CCC ICT CIT 22£ AMA CCI GMAT CM £2A0 OAT CC onGM GAT AA? 22£ CS AMT £20 on CIT CS GAT AMC CMTM22£ CSMo 22£ CS 0 TMT ala gin inu set gin gin gly arg tin inu Ion lye pro asp asp vat set asp gly vatvat asp &asinIogly ass ilie vat1 his gly asp lye inu In gly vat inu gly Vat tyr-
2831/75 2911/651320/271 1250/261 WCAMC £20 £20 CIT Ga on COT £22GAao £20 222 CCT GA? CS MCT AMA CTC oTCAM AMA 2CT AMR AMC GCC C2T 2C ?2CT CM CGM £2? 0 CT MAC ARC £20 MC AM2 22£ ala ent set Ile his gin vat -gly Ile gin ala set -h pro asp gly tin lye len vatgin lye enr lye ass ala In eno anr gin ala ile Vat &ala tin assn set sne ass InU
2941/95 2971/1051260/291 1410/301 30C CM CAT A00 CC? £22C GM00AMT COT AM T21 GT2 CC?T0GMT 220TIC 22£T AMCIT AMR CA TMC 22£ AMA 2CC CIA GM2 2 aM TIO AM CIT 2CC 50 £22TC CAT tin vat his tlin pro ile gin sIn ass gly lye inu vat pro gly gin len ph* len lye&ala i lys gin tyr inu lye enr gin as in gin inu lye his cye ala I2*gl asp
3001/115 2031/1251440/311 1470/321 AMT a GM £20 ACT £20 AMaC CM AMA AMA CCC 0G2 MC on AMR022 AMA AMT GTTAMC T2' on AGC a 7CC £20 00£ 220 AM AMA 00 AM 222 00£ 00 GMG CMR MCe CS ass gin asp ile tin ±l* ass gin glI lye lye ala viat anvts yatleasralye pbs Vat sen gin cys set arg inu assn lye ala assn phe gly gly gin gin eno gly
3061/135 3091/1451500/331 1520 3 0/3 C GM £01 CCC G2 CM& A20 0CC 2CA CM 212C CAT 22C TT2 Ga Gon AMT AOl TCC CIACA? £20 £22 222 MOC CAT TMACC 35£AAA CCACO=Ta TMCC 220 CM on gy s rg pro val gin lle gly sen his phe his pb phe gin vat &ass Mrg eye leUhis ilie le phbs en asp tyr ala lyeo tfin gly asp IFi1yfl ngi a3121/155 2151/165'1560/351 1590/361 GM TWr GW M3 aM AM AC? 220 0G1 AMA COO 22TM £22owTMCACS MAA 02£AMC owM 22£ CM 22£ CM WIT AMG CTT 02£ MC 2CM GT CGrC 22£ AM CC TTT GCM 22£ asp pho, asp arg gin lyes tin ph, gly iye ar9 inu asp Ile &a sear g'ly tin ala valane ala inu Vat ion gin enc lye InU Vat en enr Vat arg in6 ass pro phe gin inu3151/175 3211/1SS1620/371 1650/31 AM 222 GMA CCT 00 Qa aM AMA 7=CC C M 2W AT? GMC ATn oc GOT AMC aG ATM CC? CMR AM CSG on AM 22G AMT CM CM AMh LAG = CC? 22£ Ca AGC CS0 AM an pi- gin pro gly gin gin lye an val gin in Ile asp Ile gly gly ass arg argtyr pro gin asn Ion Vat assnaoussn Vat gin lys lye pro pro inu gin enr InU lye
FIG. 4. Nucleotide sequence of the H. pylori urease genes. Numbers above the sequence indicate the nucleotide position. Predicted aminoacid sequences, in sequential order, for UreC (bp 510 to 1844), UreD (bp 1841 to 2235), UreA (bp 2659 to 3372), and UreB (bp 3379 to 5085)are shown'below the DNA sequence. Putative ribosome-binding seque'nces (Shine-Dalgarno [SD] sites) are underlined; boxed sequencescorrespond to the promoterlike sequences (a70 as well as cr54), and arrows above the sequence indicate stem-loop structures with'the featuresof a rho-independent transcriptional stop signal.
1926
on July 15, 2018 by guesthttp://jb.asm
.org/D
ownloaded from

CLONING OF H. PYLORI UREASE GENE CLUSTER
3241/195 3271/205ATC TTT GGA TTT AAC 0CA TT7 CTT GAT AGK CAA 0CA CAC AAC OAA ACC AAA AAA ATT OCT
ile phs gly phe asn ala leu val asp erg gln ala asp asn glu smr lys lys ile ala
3301/215 3331/225TTA CAC AGK OCT AAA GAO COT OCT TTT CAT COC OCT AaA ACC OAT CAG AAO TAT GTA AAAleu his arg ala lys glu arg gly pbs his gly ala lys ser asp asp asn tyr val lys
3361/235 MD 1 3391/5ACA AT? Aa_gC TAA OAA ATO AAA AAO ATT AOC AGA AAA GAK TAT 7T. TCT ATG TAT cOT
thr ile lys glu OCR -- met lys lys ile ser arg lys glu tyr val ser met tyr gly
3421/15 3451/25CCT ACT ACA GOC OAT AAA AKA TTG CCC GAT ACA GAC TTG ATC OCT GAA CTA OAK CATpro thr thr gly asp lys val arg leu gly asp thr asp lou Lie ala glu val glu his
3481/35 3511/45GAC TAC ACC ATT TAT 0CC CAA CAG CTT AAA TTC COT GCC 00T AAA ACC CTA AGC OAAKCCasp tyr thr ile tyr gly glu glu leu lys pbs gly gly gly lys thr lou erg glu gly
3541/55 3571/65ATG AGOC CAA TCT AA AAC CCT AOC AAA OAA GAG TT7 GAT TTA AT? ATC ACT AAC OCT TTAset ser gln ser asn asn pro ser lye glu glu lou asp lou Lie ile thr asn ala leu
3601/75 3631/95ATC 0TC GAT TAC ACC OCT AT? TAT AAA CCC OAT ATT G0T ATT AAA GAT aCC AAA ATC OCT
ile val asp tyr thr gly Lie tyr lys ala asp ile gly Lie lys asp gly lys Lie ala
3661/95 3691/105CGC ATT 0OT AAA CGC 00T AAC AAA CAC ATG CAK GA CC GTT AAK AAC AAT CTT AGC G7Agly Lie gly lys gly gly asn lys asp met gln asp gly val lye asn asn leu ser val
3721/115 3751/125GOT CCT OCT ACT OAA 0CC TTA GCC 0CT GAA G0T TTG ATC GTA ACG OCT 0GT CGT ATT CACgly pro ala thr glu ala leu ala gly glu gly lou Lie val thr ala gly gly Lie asp
3781/135 3811/145ACA CAC ATC CAC TTC AT? TCA CCC CAA CAA ATC CCT ACA CCT TTT 0CA AOC 0CT CTA ACA
thr his ile his pbs ile ser pro gln gln ile pro thr ala ph* ala ser gly val thr
3841/155 3871/165ACC ATC ATT 02T 07T 00A ACC G0T CCT OCT CAT 0CC ACT AAT CCC ACT ACT ATC ACT CCAthr met ile gly gly gly thr gly pro ala asp gly thr asn ala thr thr ile thr pro
3901/17S 3931/195GCC ACA AGA AAT TTA AAA TOG ATC CTC AGKA CC OCT CAA OAA TAT TCT ATC AAT TTA G0Tgly arg arg asn leu lys trp net lou erg ala ala glu glu tyr ser met asn leu gly
3961/195 3991/205TTC TT7 OCT AAA GOT AAC OCT TCT AAC CAT CCG AGOC TTA GCC GAT CAA AT? OAA CCC GOTphe leu ala lys gly aen ala ser aen asp ala ser leu ala asp gln ile glu ala gly
4021/215 4051/225CC AT? GCC TST AAA AT? CAC OAA GAC TOO CCC ACC ACT CCT TCT GCA ATC AAT CA? CGOala ile gly phe lys ile his glu asp trp gly thr thr pro ser ala Lie &en his ala
4081/235 4111/245TTA GAT GT? GCC CAC AAA TAC OAT 0T7 CAA OCT ATC CAC ACA GAO ACT TTG AAT OAKleu asp val ala asp lys tyr asp val gln val ala ili his thr asp thr lou asn glu
4201/275 4231/285ACT OAA GGC OCT GGC CCC GGA CAC GCT CCT GAT ATT ATT AAA GTA CCC GOT GAK CAC AAC
thr glu gly ala gly gly gly his ala pro asp Li. ile lys val &la gly glu his asn
4261/295 4291/305ATT CTT CCC CCT TCC ACT AAC CCC ACC ATC CCT TTC ACC CT0 AAT ACA CAA GCA GAC CAC
1ie l-u pro ala ser thr asn pro thr Lie pro phs thr val &an thr glu ala glu his
4321/315 4351/325ATC CAC ATC CTT ATO CTO TOC CAC CAC TTC CAT AAA AGC ATT AAA CAA GAT 0TT CAt TTCnet asp met lou nst val cys his his leu asp lys ser ile lys glu asp val gln phs
4381/335 4411/345OCT GA? TCA AGO ATC CCC CCT CAA ACC ATT CCC CCT GAK CAC ACT TTC CAT CAC ATO 0GGala asp ser arg i11 erg pro gln thr £1. ala ala glu asp thr leu his asp net gly
4441/355 4471/365AT? TTC TCa ATC ACC ACT TCT CAC TCT CAA CCC ATO CCC COT 0T7 COT OAA GTT ATC ACTLi. pbs ser £1. thr s*r ser asp ser gln ala *nt gly arg val gly glu val ile thr
4501/375 4531/395AGK ACT TG0 CAK ACK CCT GAC AAA AAC AAO AAA CAA TTT CCC CCC TTG AAA GAA OAA AAAarg thr trp gln thr ala asp lys asn lys lys glu phb gly erg lu lys glu glu lys
4561/395 4591/4 05CCC CA? AAC cac AAC TTC AGO ATC AAA CGC TAC TTC. TCT AAK TAC ACC ATT AAC CCA GC0gly asp asn asp asn phe erg ile lys erg tyr leu ser lys tyr thr Lie asn pro ala
4621/415 4651/425ATC CCT CAT COO ATT AGC GAO TAT CTA COT TCA GTA OAA 0T7 CGC AAA 0TC CCT GAC TT0ile ala his gly 1ie ser glu tyr val gly ser val glu val gly lys val ala asp leu
4681/435 4711/445OTA TT7. TGO ACT CCA GCA TTC TTT GGC GT0 AAA CCC AAC ATO ATC ATC AAA CGC G0A TTCval leu trp ser pro ala phe phb gly val lys pro asn set Lie ile lys gly gly phs
4741/455 4771/4 65ATT CCC TTA AGC CAA ATC CGC CAT CCC AAC CCT TCT ATC CCT ACC CCA CAA CCO CTT TATile ala lou ser gln net gly asp ala asn ala ser ile pro thr pro gln pro val tyr
4801/475 4631/495TAC AGA GAA ATG TTC CCT CAT CAT COT AAK CCT AAA TAC CAT cca AAC ATC ACT TTT 0T7tyr arg glu net phr ala his his gly lys ala lys tyr asp ala asn Lie thr phb val
4661/495 4891/505TCT CAA CCC OCT TAT CAC AAA CGC ATT AAA OAK OAA TTA GGA CTT OAA AGA CAA GT0. TTCser gln ala ala tyr asp lys gly 1ie lys glu glu leu gly leu glu arg gln val leu
4921/515 4951/525CCO C02 AAA AAT TCC AGA AAT ATC ACT AAK AAA GAC ATC CAA TTC AAC CAC ACT ACT GCTpro val lys asn cys erg asn ile thr lys lys asp met gln phb asn asp thr thr ala
4981/535 5011/545CAC ATT CAA 0TC AAT CCT OAK ACT TAC CAT 0T7 TTC 0T2 OAT CCC AAA OAA GTA ACT TCThis ile glu val asn pro glu thr tyr his val phe val asp gly lys glu val thr ser
5041/555 5071/565AAA CCA CCC AA? AAA 0T0 AOC TTG CCG CAA C?C TTT AGOC ATT TTC TAG GAT TT TTA GAGlys pro ala asn lys val ser leu ala gln leu phe ser ie phe AMB
4141/255 4171/265GCC G0T TGT GTA OAA GAC ACT ATCGC? CC? AT? OCT 00A CCC ACT ATG CAC AC? T7.C CACala gly cys val glu asp thr m*t ala ala ile ala gly arg thr met his thr phe his
FIG. 4-Continued.
codon of the ureA gene is separated from the methionineinitiator codon of the ureB gene by a single codon suggeststhat a single mutation in the stop codon of ureA could lead tothe fusion of the two polypeptides encoded by ureA and ureBand therefore generate a single polypeptide. Based on this,the H. pylori urease appears phylogenetically more closelyrelated to the jack bean urease than to the three-subunitbacterial ureases; this statement is also documented by thehigher degree of conservation observed between the 26.5-kDa subunit of H. pylori with the amino-terminal sequence
ofjack bean urease (48%) compared with that of the homol-ogous urease subunits of P. mirabilis (42%).Although the urease of H. pylori has been described as an
extracellular enzyme (8), i.e., has to be transported to theexternal membrane, no leader peptide sequence was foundfor either of the two polypeptides. In addition, there is an
excellent agreement between the amino acid sequence de-duced from the DNA analyses and that of the N-terminalamino acid of the H. pylori subunits purified and sequenced(8, 20), indicating that no maturation of the N-terminal endsof the urease subunits is required either for the export of thesubunits or to generate enzymatic activity.Clayton et al. (7) were the first to report the cloning of
specific H. pylori antigens reacting with antisera raised
against the purified H. pylori urease. Subsequently, theydemonstrated by DNA sequence analyses (6) that the anti-gens expressed in E. coli by the cloned DNA fragmentcorrespond to two polypeptides of the urease enzyme (i.e.,UreA and UreB); however, there was no expression ofurease activity. The ureA and ureB nucleotide sequencespresented in this work and the sequences reported byClayton et al. (6) for a different H. pylori isolate indicate thatthe urease subunits of H. pylori are highly conserved poly-peptides as >98% of the amino acids of the urease enzymewere conserved, if one excludes from the comparison thecarboxy-terminal sequence on which there is substantialdisagreement. However, the high degree of conservation inprimary structure observed between the urease sequencesoriginating from bacteria (P. mirabilis) or from a plant (jackbean) suggests that the whole urease enzyme cannot be usedas a specific antigen for serological tests since it is likely thatcommon epitopes will be present in the different bacterialurease proteins which might lead to false-positive serologicaltests. Nevertheless, alignment of the three sequences al-lowed us to identify a domain (shown in Fig. 6) of the ureAgene product which is unique to H. pylori; no equivalentsequence exists in the urease subunits of P. mirabilis or
Ureaplasma urealyticum (2) (data not shown). We are pres-
VOL. 173, 1991 1927
on July 15, 2018 by guesthttp://jb.asm
.org/D
ownloaded from
1928 LABIGNE ET AL.
(ONE CDO m ON 0 0
1\ 0I 000 10J 6
.~~~~~~~~61.6:UreB
_61.6- 49.2: Urec___
-........ .. _. __1!.... , .,:t * .:
S-lact_" __= ^_
.= ...
... * ...... .. . , _(A)
_49.2qr:.. ..:....
Am -aph-3'-.*~&- 2 6.526.5: UreA
_26.5
(B)FIG. 5. Fluorographs of plasmid-mediated polypeptides expressed in minicells labeled with [35S]methionine analyzed on 10% (A) or 12.5%
(B) sodium dodecyl sulfate-polyacrylamide gels. (A) Polypeptides encoded by the pILL533 vector, by the pILL615 plasmid consisting of theEcoRI-PstI insert of plasmid pILL589 (urease-positive plasmid) cloned into vector pILL533, and by pILL594 and pILL599 urease-negativeplasmids in comparison with the polypeptides expressed by the corresponding vector, pILL570. (B) Polypeptides expressed by threeurease-positive plasmids, pILL589, pILL588, and pILL590. Numbers in kilodaltons refer to the calculated molecular weights of the predictedpolypeptides UreA, UreB, and UreC. Aph-3', Migration of type III 3'-aminoglycoside phosphotransferase (35 kDa); r-lact, migration ofP-lactamase (30 kDa), both encoded by the vectors.
ently investigating the degree of conservation of this peptideregion among H. pylori isolates as well as testing its immu-nogenic properties to determine whether it can be used as aspecific epitope for 'diagnostic purposes or as a potentiallyprotective epitope. Alignment of sequences also permitslocalization of the active site of the H. pylori urease; this siteis indicated in Fig. 6 and contains the eight histidine residuesand one cysteine residue which, respectively, are believed toplay a major role in the binding of nickel ions and inenzymatic activity in jack bean urease. That these residuesare conserved and perfectly aligned between the threesequences of urease suggests a similar role in H. pylori, ahypothesis which can now be assessed by directed mutagen-esis of the ureB gene.
In addition to the ureA and ureB genes, we have shownthat DNA sequences located upstream of the ureA and ureBgenes were absolutely required for urease expression in C.jejuni. The nucleotide sequence determination of the DNAfragment extending from the HindIII-1 restriction site andthe EcoRI site of pILL588 permitted identification of twoORFs designated ureC and ureD which potentially encodepolypeptides with molecular weights of 49,200 and 14,900,respectively. The search for amino acids relevent to se-quences involved in metal (12)-, calcium (3)-, or ATP-binding sites (18, 48) was negative, as' was that of similaritiesbetween the deduced amino acid sequence of the ureC andureD genes and protein sequences from the NBRF database.No precise role can be attributed to the UreC-predicted
polypeptide in the absence of similarities between its'aminoacid sequence and that of the identified UreD, UreE, or
UreF polypeptide of the P. mirabilis urease operon (23, 32).These polypeptides are believed to be involved in theprocessing of nickel ions and/or transport of the urea fromthe extracellular to the intracellular compartment (32). Fromthe present results, we cannot definitively conclude that theureC gene product per se is required for urease expression;one might suspect, due to the identification of nitrogenbinding sites, that solely the DNA sequences correspondingto this ORF are important as cis-acting sites for ureaseexpression. Additional experiments will be required to clar-ify this point.ureD encodes a possible polypeptide of 14.9 kDa with
features (Fig. 3C) typical of membrane-spanning domains oftransmembrane proteins (39), which suggests that this poly-peptide could serve a transport or anchoring function. Inaddition, features at the DNA level suggest that the ureDgene and its product are related to urease function. The samesequence localized 310 bp upstream of both the ureD andureA genes is highly homologous with the consensus se-quence of the nitrogen regulation site (binding site of factora54), suggesting that the expressions of the ureA-ureB genesand the ureD gene are under the same transcriptional con-trol. Northern (RNA) hybridization as well as primer exten-sion experiments are now in progress to determine whetherthese promoterlike sequences are used for the initiation ofthe transcripts corresponding to the ureA+ureB and ureDgenes in H. pylori as well as in E. coli, or whether they area remnant of a promoter which allowed genes to be ex-pressed and regulated in an ancestral genome.
Differences in the rapidity and intensity of urease activityin C. jejuni cells harboring plasmids with DNA inserts larger
J. BACTERIOL.
WAMOMMI. .& a& jm 40M.
on July 15, 2018 by guesthttp://jb.asm
.org/D
ownloaded from

CLONING OF H. PYLORI UREASE GENE CLUSTER 1929
1
Protous mlrbille NmpJmjaTjAr-LvAziL&lx/ijmAuLss:&aGcc-KTAQIszcRTLTnfSvbc.PDa cDrVFTPDHalicebacter pylorn SLTPLDcIaa"LAIsUHINEAR^IMHIaCGTLLRTDDVWVASvGIuPDCJack boon 18sp9vwuaaLMBrIAQURIAvRLVRTKTAVALIAsQDmM)GKnVKAQIcLCoQm.LQILlUnAVPHLLuAVQwATKPDr.Jeck~~~~ ~ ~ ~ ~ ~ ~AAVP I
100 1P.m. TUVPIIRAVaPctalcDIlReTzsIalatPcs
H p. SE5mimIpl-------------------LVPCUISINWVSITVGIKVaMlPVKGWQ tCSIWYTZV>
J.b. TKLVTVI SIDKTATnILA:XILxLaEG4EVIISPIQVSHHFIrNPL
109
P.m. RrRJr VRDILWaFA;lIY------------------rVG*:-::::::::::::::::: 238
H.p. D IDLTVRDIC QWULVDRNlIA... . . . ..::: : : ::::: :: . . .. :: :: 270
Pro Icue mirabilia M=RhA1FPTRJLDZJLIXFrGZVFGKICWQQSC-VINIIYC
Halicobacter pylort a Y V )Z SY Gt o4t IT . I
Jack boan -MWIWP YYCPT D I YU) Y0K:VrGCGIVI PAISXDMTH&VIIDYCI271 A A
P.m. VIADICIt VCIDVD rI-_ITDTIHFIC CLVSCTICCCSCPIT
HIPp DICICDGIAW P A_DII HFSPMQPTAASGaU4;GA;PADKrr
J.b. tSaDICGsIQXZQZDDN:vsTVIAGAIDctYCPQLVYZWISsITVRTGAocTT
A
P.m. VII_VDcZIN9FGKQCV RIGCI&UMWGATcrcHNaDD4vQvXuHSccrY
J.b. ~~~~CTPTWF LFICTK s KIZMEXKAICIA GSAINO TIAZHHIDIQIIHT0TLNAJC KMSWJ
P.m. VUITAGMIWVIf'EGM CH VKgJGZPNILASMaMW1YTIRrDEHWDKIMJOWLDPSIPEDVAFAES PPE?IAAEDILH
J.b. I~~~~XA&FKGRINI G&GMAIIKVCGIIUWLPSS25WTPLTaIEIDHDKIa.D VCJOUU.RZIPILDLAFAhI BKKT1AAMVLH
P.m. MCXVSSMMG LTQARPQ=GSM*IRIKT&LH;ATClK.LDntPi
H.p. IT _ cr= cm^ v = nc
J.O. C W t S DICTDz.IQN
P.m. vlcFDICAl;;ccwwMKavI^GMS§twgLpzsEQTplj=STINWRIRRIAKcllDWAsIp4LrDAYGs} GKIADL£DIUS
P.m.
H.p.
J.b.
58918I!WMIUDP!FDlxDGVPLVCKvA1ILLFAiQmrFI
:::::: : .. :S69NDTUVWTWlvCMSPNShLSI:::: :::. :840
1CA.IPIITYDPZSgY'IVnDCU..CWVATTVPLUINTFrLVA
FIG. 6. Alignment of the first 270 amino acids of the jack bean urease (bottom line) with that of the two small subunits of Proteus mirabilisand the product of the ureA gene (top) and alignment of the last 550 amino acids of the jack bean urease with that of the large subunit of P.mirabilis and of the product of the ureB gene (bottom). Colons between two lines indicate the presence of the identical amino acid in twopolypeptide chains. Numbers above the letters represent amino acid positions for each subunit. Dashed lines represent gaps introduced tooptimize the alignments. The box in which arrows point out eight histidine residues and one cysteine residue correspond to the jack beanurease active site (47). Triangles point out the discrepancies with the H. pylori urease sequence as published by Clayton et al. (6). Hatch linescorrespond to a peptide sequence unique to H. pylori.
than the 4.2-kb obligately required fragment indicates that,in addition to the four identified genes or loci, other genes orDNA sequences located upstream as well as downstreamappear to be involved in high-level activity.The absence of detectable urease activity in E. coli cells is
not yet understood: in vivo transcription and translation
experiments performed in a minicell-producing strain indi-cated functional transcription as well as translation of atleast the ureA, ureB, and ureC genes. All of our attempts toinduce urease activity in recombinant E. coli cells by chang-ing growth conditions (temperature, media, or addition ofurea) or to detect activity from sonicated bacterial cells in
VOL. 173, 1991
P.m.
H.p.
J.b.
IW"ALIXXGGNVkXKPHMnAAAIPTPQPVUYP.IlbaAClarjmQTsmrmmm.jLxw.vptKwLitsLsLiGRvzGckHiTyAsmilI
FCVKPNNCEI PTPQPVYYRDWAHHGKAKrDANITIFVSQAAYD=IMLLTAIZAQVLPVXN--CPSITMMmW
FCMZWXI=WAWXDIGDPNKSIPTPZPVIMPWGTIA2MArALSXMVSIUUUMORVW=r.IMUCVTAVSN--VPJMTKIDMM
on July 15, 2018 by guesthttp://jb.asm
.org/D
ownloaded from

1930 LABIGNE ET AL.
the presence or absence of nickel ions were unsuccessful.That by shuttling the DNA fragment cloned in E. coli to C.jejuni cells we were capable to detect urease activity indi-cates that C. jejuni cells are capable of complementing afunction that E. coli cells cannot accomplish; this functionshould be investigated. The cloning and expression of theurease genes of H. pylori allow us to think that the shuttleapproach described in this paper might be generalized as apossible route for the successful cloning and expression ofother H. pylori genes of interest.
ACKNOWLEDGMENTS
We thank J. L. Fauchere for providing H. pylori 85P and M.Blaser for help with the writing as well as for critically reading themanuscript.
REFERENCES1. Alder, H. I., D. Fisher, A. Cohen, and A. A. Hardlget. 1967.
Miniature Escherichia coli cells deficient in DNA. Proc. Natl.Acad. Sci. USA 57:321-326.
2. Blanchard, A. 1990. Ureaplasma urealyticum urease genes; useof a UGA tryptophan codon. Mol. Microbiol. 4:669-676.
3. Boehm, D., R. Welch, and I. S. Snyder. 1990. Domains ofEscherichia coli hemolysin (HlyA) involved in binding of cal-cium and erythrocyte membranes. Infect. Immun. 58:1959-1964.
4. Boyer, H. W., and D. Roulland-Dussoix. 1969. A complementa-tion analysis of the restriction and modification of DNA inEscherichia coli. J. Mol. Biol. 41:459-472.
5. Buck, G. E. 1990. Campylobacter pylori and gastroduodenaldisease. Clin. Microbiol. Rev. 3:1-12.
6. Clayton, C. L., M. J. Pallen, H. Kleanthous, B. W. Wren, and S.Tabaqchali. 1990. Nucleotide sequence of two genes fromHelicobacter pylori encoding for urease subunits. Nucleic AcidsRes. 18:362.
7. Clayton, C. L., B. W. Wren, P. Mullany, A. Topping, and S.Tabaqchali. 1989. Molecular cloning and expression of Cam-pylobacter pylori species-specific antigens in Escherichia coliK-12. Infect. Immun. 57:623-629.
8. Dunn, B. E., G. P. Campbell, G. I. Perez-Perez, and M. J.Blaser. 1990. Purification and characterization of urease fromHelicobacter pylori. J. Biol. Chem. 265:9464-469.
9. Dunn, B. E., G. I. Perez-Perez, and M. Blaser. 1989. Two-dimensional gel electrophoresis and immunoblotting of Cam-pylobacter pylori proteins. Infect. Immun. 57:1825-1833.
10. Evans, D. G., D. J. Evans, Jr., and D. Y. Graham. 1989.Receptor-mediated adherence of Campylobacter pylori tomouse Y-1 adrenal cell monolayers. Infect. Immun. 57:2272-2278.
11. Evans, D. J., D. G. Evans, K. E. Smith, and D. Y. Graham. 1989.Serum antibody responses to the N-acetylneuraminyllactose-binding hemagglutinin of Campylobacter pylori. Infect. Immun.57:664-667.
12. Evans, R. M., and S. M. Hollenberg. 1988. Zinc fingers: gilt byassociation. Cell 52:1-3.
13. Feinberg, A., and B. Vogelstein. 1983. A technique for radiola-beling DNA restriction endonuclease fragments to high specificactivity. Anal. Biochem. 132:6-13.
14. Figurski, D. H., and D. R. Helinski. 1979. Replication of anorigin-containing derivative of plasmid RK2 dependent on aplasmid function provided in trans. Proc. Natl. Acad. Sci. USA76:1648-1652.
15. Gill, R. E., F. Heffron, and S. Falkow. 1979. Identification of theprotein encoded by the transposable element Tn3 which isrequired for its transposition. Nature (London) 282:797-801.
15a.Guerrant, R. L., R. A. Pennie, L. J. Barrett, and A. O'Brien.1985. 3rd International Workshop on Campylobacter Infections,abstr. 093.
16. HazeIl, S., and A. Lee. 1986. Campylobacter pyloridis, urease,hydrogen ion back diffusion, and gastric ulcers. Lancet ii:15-17.
17. Hazell, S. L., A. Lee, L. Brady, and W. Hennessy. 1986.
Campylobacterpyloridis and gastritis: association with intercel-lular spaces and adaptation to an environment of mucus asimportant factors in colonization of the gastric epithelium. J.Infect. Dis. 153:658-663.
18. Higgins, C. F. I. D. Hiles, K. Whalley, and D. J. Jamieson.Nucleotide binding by membrane components of bacterial peri-plasmic binding protein-dependent trnasport systems. EMBO J.4:1033-1040.
19. Hopp, T., and K. R. Woods. 1981. Prediction of protein anti-genic determinants from amino acid sequences. Proc. Natl.Acad. Sci. USA 78:3824-382.
20. Hu, L., and H. L. T. Mobley. 1990. Purification and N-terminalanalysis of urease from Helicobacter pylori. Infect. Immun.58:992-998.
21. Hull, R. A., R. E. Gill, P. Hsu, B. H. Minshew, and S. Falkow.1981. Construction and expression of recombinant plasmidsencoding type 1 or D-mannose-resistant pili from a urinary tractinfection Escherichia coli isolate. Infect. Immun. 33:933-938.
22. Hupertz, V., and S. Czinn. 1988. Demonstration of a cytotoxinfrom Campylobacterpylori. Eur. J. Clin. Microbiol. Infect. Dis.7:576-578.
23. Jones, B. D., and H. L. T. Mobley. 1989. Proteus mirabilisurease: nucleotide sequence determination and comparison withjack bean urease. J. Bacteriol. 171:6414-6422.
24. Labigne-Roussel, A., P. Courcoux, and L. Tompkins. 1988. Genedisruption and replacement as a feasible approach for mutagen-esis of Campylobacterjejuni. J. Bacteriol. 170:1704-1708.
25. Labigne-Roussel, A., J. Harel, and L. Tompkins. 1987. Genetransfer from Escherichia coli to Campylobacter species: devel-opment of shuttle vectors for genetic analysis of Campylobacterjejuni. J. Bacteriol. 169:5320-5323.
26. Laemmli, U. K. 1970. Cleavage of structural proteins during theassembly of the head of bacteriophage T4. Nature (London)227:680-685.
27. Levi, S., K. Beardshall, G. Haddad, R. Flayford, P. Ghosh, andJ. Calam. 1989. Campylobacter pylori and duodenal ulcers: thegastrin link. Lancet i:1167-1168.
28. Maniatis, T., E. Fritsch, and J. Sambrook. 1983. Molecularcloning: a laboratory manual. Cold Spring Harbor Laboratory,Cold Spring Harbor, N.Y.
29. Marshall, B. J., L. Barret, C. Prakesh, R. Mc Callum, and R.Guerrant. 1988. Protection of Campylobacter pyloridis but notCampylobacter jejuni against acid susceptibility by urea, p.402-403. In B. Kaijser and E. Falsen (ed.), Campylobacter IV.University of Goteborg, Goteborg, Sweden.
30. Marshall, B. J., H. Royce, D. I. Annear, C. S. Goodwin, J. W.Pearman, J. R. Warren, and J. A. Armstrong. 1984. Originalisolation of Campylobacter pyloridis from human gastric mu-cosa. Microbios Lett. 25:83-88.
31. Meissing, J., and J. Vieira. 1982. A new pair of M13 vectors forselecting either DNA strand of double-digest restriction frag-ments. Gene 19:269-276.
32. Mobley, H. L. T., and R. P. Hausinger. 1989. Microbial ureases:significance, regulation, and molecular characterization. Micro-biol. Rev. 53:85-108.
33. Morett, E., and M. Buck. 1989. In vivo studies on the interactionof RNA polymerase-54 with the Klebsiella pneumoniae andRhizobium meliloti nifH promoters. J. Mol. Biol. 210:65-77.
34. Morsdorf, G., and H. Kaltwasser. 1990. Cloning of the genesencoding urease from Proteus vulgaris and sequencing of thestructural genes. FEMS Microbiol. Lett. 66:67-74.
35. Nakazawa, T., M. Ishibashi, H. Konishi, T. Takemoto, M.Shigeeda, and T. Kochiyama. 1989. Hemagglutination activity ofCampylobacter pylori. Infect. Immun. 57:989-991.
36. Neman-Simha, V., and F. Megraud. 1988. In vitro model forCampylobacter pylori adherence properties. Infect. Immun.56:3329-3333.
37. Prentki, P., and H. M. Krisch. 1984. In vitro insertional muta-genesis with a selectable DNA fragment. Gene 29:303-313.
38. Rosenberg, M., and D. Court. 1979. Regulatory sequencesinvolved in the promotion and termination of RNA transcrip-tion. Annu. Rev. Genet. 13:319-359.
39. Sabatini, D. D., G. Kreibich, T. Morimoto, and M. Adesnik.
J. BACTERIOL.
on July 15, 2018 by guesthttp://jb.asm
.org/D
ownloaded from

CLONING OF H. PYLORI UREASE GENE CLUSTER
1982. Mechanisms for the incorporation of proteins in mem-branes and organelles. J. Cell Biol. 92:1-22.
40. Sanger, F., S., A. R. Coulson, B. G. Barreli, A. J. H. Smith, andB. A. Roe. 1980. Cloning in single-stranded bacteriophage as an
aid to rapid DNA sequencing. J. Mol. Biol. 143:161-178.41. Sanger, F., S. Nicklen, and A. R. Coulson. 1977. DNA sequenc-
ing with chain-terminating inhibitors. Proc. Natl. Acad. Sci.USA 74:5463-5467.
42. Sarosiek, J., A. Slomiany, and B. L. Slomiany. 1988. Evidencefor weakening of gastric mucus integrity by Campylobacterpylori. Scand. J. Gastroenterol. 23:585-590.
43. Shine, J., and L. Dalgarno. 1974. The 3'-terminal sequence ofEscherichia coli 16S ribosomal RNA: complementarity to non-sense triplets and ribosome binding sites. Proc. Natl. Acad. Sci.USA 71:1342-1346.
44. Simon, R., U. Priefer, and A. Puihler. 1983. A broad host range
mobilization system for in vivo genetic engineering: transposonmutagenesis in gram negative bacteria. Biotechnology 1:784-791.
45. Smoot, D. T., H. L. T. Mobley, G. R. Chippendale, J. F.
Lewison, and J. H. Resau. 1990. Helicobacter pylori ureaseactivity is toxic to human gastric epithelial cells. Infect. Immun.58:1992-1994.
46. Southern, E. M. 1975. Detection of specific sequences amongDNA fragments separated by gel electrophoresis. J. Mol. Biol.98:503-517.
47. Takishima, K., T. Suga, and G. Mamiya. 1988. The structure ofjack bean urease. The complete amino acid sequence, limitedproteolysis and reactive cysteine residues. Eur. J. Biochem.175:151-165.
48. Walker, J. E., M. Saraste, M. J. Runswick, and N. J. Gay. 1982.The ATP operon nucleotide sequence of the genes for thegamma sub-unit, beta-subunit, epsilon-subunit of Escherichiacoli ATP synthetase. EMBO J. 1:945-951.
49. Warren, J., and B. Marshal. 1983. Unidentified curved bacilli ongastric epithelium in active chronic gastritis. Lancet i:1273-1275.
50. Winship, P. R. 1989. An improved method for directly sequenc-ing PCR amplified material using dimethyl sulphoxide. NucleicAcids Res. 17:1266.
VOL. 173, 1991 1931
on July 15, 2018 by guesthttp://jb.asm
.org/D
ownloaded from





























