Embed Size (px)
Citation preview

1
Synthesis and Biological Testing of Novel Pharmacological Agents: Dihydropyrimidinone Derivatives
By Santiago Gonzalez
Presented to The Honors Collegein partial fulfillment of the requirements for Honors Senior Thesis
Arkansas State University
_________________ __________________________________
Dr. John Hershberger, Advisor
_________________ _________________________________
Dr. Allyn Ontko
_________________ ________________________________
Dr. Mohammad Alam
_________________ __________________________________
Ms. Rebecca Oliver, Director of The
Honors College
May 2016

2
TABLE OF CONTENTS
TITLE PAGE…………………………………………………………………………………… .1
TABLE OF CONTENTS……………………………………………………………….………. 2
LIST OF FIGURES……………………………………………………………………………... 3
ACKNOWLEDGEMENTS………………………………………………………….…………. .4
ABSTRACT…………………………………………………………………….………………...5
INTRODUCTION/BACKGROUNG…………………………………………………………….6
DIHYDROPYRIMIDINONE BIOACTIVITY..............................................................................6
REASONS FOR DIHYDROPYRIMIDINONE ACTIVITY…………………………….……....7
DIHYDROPYRIMIDINONE SYNTHESIS…………………………………………………......8
PHENETHYLAMINE BIOACTIVITY………………………………………………………….9
APPLICATION OF THE RESEARCH…………………………………………………………10
SYNTHETIC PATHWAY OUTLINE..........................................................................................11
DIHYDROPYRIMIDINONE WITH DIFFERENT FUNCTIONAL GROUPS……………......12
BROMINATION OF THE ALLYLIC SITES IN DIHYDROPYRIMIDINONES………….….14
NBS VS. TRIMETHYLAMMONIUM TRIBROMIDE………………………………...............14
CYCLIZATION METHODOLOGY AND MECHANISM…………………………………. ...15
SUBSTITUTED PHENETHYLAMINES USED IN THE PROJECT.........................................16
PHENETHYLAMINE...................................................................................................................16
INCREASED TEMPERATURES…………………………………………….……………........18
DECREASED STERIC HINDRANCE…………….…………………………………………....19
SYNTHESIZED STRUCTURES..................................................................................................19
EXPERIMENTAL SECTION...................................................................................................... 21
EXPERIMENTAL DATA FROM SYNTHESIZED PRODUCTS..............................................21
ANTIBIOTIC TESTING...............................................................................................................31
ANTIBIOTIC TESTING RESULTS.............................................................................................31
CONCLUSIONS ……………………………………………………………………….……….32
REFERENCES………………………………………………………………….……………….34
APPENDICES (1-10) ...................................................................................................................37

3
List of Figures FIGURE 1: DIHYDROPYRIMIDINONES IN PHARMACOLOGICAL AGENTS
FIGURE 2: DNA INTERCALATION
FIGURE 3: BIGINELLI REACTION
FIGURE 4: BASIC PHENETHYLAMINE SCAFFOLD
FIGURE 5: NOREPINEPHRINE
FIGURE 6: DOPAMINE
FIGURE 7: ALDEHYDE AND PHENETHYLAMINE VARIANTS
FIGURE 8: SYNTHETIC PATHWAY OUTLINE
FIGURE 9: DIHYDROPYRIMIDINONE SCAFFOLDS
FIGURE 10: BROMINATION REACTION
FIGURE 11: CYCLIZATION
FIGURE 12: CYCLIZED PRODUCTS MADE WITH REGULAR PHENETHYLAMINE
FIGURE 13: INTERMEDIATE PRODUCTS
FIGURE 14: CYCLIZED PRODUCTS AT 100° CELSIUS
FIGURE 15: ETHYL ESTERS VS. METHYL ESTERS
FIGURE 16: SYNTHESIZED PRODUCTS

4
Acknowledgements This thesis could not have been completed without the support of my mentor and committee
members. I would like to thank Dr. John Hershberger for his mentorship for the past two years in
the process of gathering data, presenting, and writing of the thesis. I am also grateful with Dr.
Hershberger for his guidance in the process of making career decisions, applying to medical
schools, and pursuing various academic opportunities. My experience working in your lab and
having you as a mentor have made me grow as a person and as a student of science, and for all of
this, I am deeply grateful. I would like to thank my committee members Dr. Allyn Ontko, and
Dr. Mohammad Abrar Alam for their input throughout the research process. I would like to
thank Dr. David Gilmore for his assistance in the antibiotic testing of the synthesized
compounds. Lastly, I would like to thank my parents, grandmother, and sister for their trust and
support throughout my entire college career. I could not have done it if it were not for your
unconditional love, for your bold decision of sacrificing everything to seek a better future for all
of us, and for the fact that you all are living role models of the importance of education and the
pursuit of one’s dreams.

5
Abstract The goal of this project is to synthesize novel pharmacological agents by combining two
privileged structures; dihydropyrimidinones and phenethylamines. Dihydropyrimidinones are
significant building blocks and versatile synthons that are frequently seen in medicinal chemistry
due to their many pharmacological properties, including calcium channel blocking, bacterial
growth inhibiting, HIV inhibiting, and antitumor activity. Phenethylamines form the scaffold for
catecholamine adrenergic hormones such as norepinephrine and dopamine. By combining these
two privileged bioactive scaffolds, we predict that the resulting structure will also be bioactive.
After various experimental trials, we have developed a reaction pathway that encompasses three
straightforward steps to synthesize the desired products. We have characterized and tested the
synthesized products for bioactivity using the Kirby-Bauer disk diffusion assay.

6
Introduction/Background Dihydropyrimidinone bioactivity
Dihydropyrimidinones are significant building blocks and versatile synthons that are frequently
seen in medicinal chemistry and are used in pharmacological agents as calcium channel blockers,
HIV inhibitors, antitumor agents, HIV gp-120-CD4 inhibitors and neuropeptide Y antagonists
(1). One of the most widely used cancer drugs is a dihydropyrimidinone derivative named
Monastrol, which is an inhibitor of human kinesin Eg5 that leads to mitotic arrest and apoptotic
cell death (Figure 1) (2). Another example of a bioactive dihydropyrimidinone is Nitractin,
which is a highly effective antiviral agent. (Figure 1) (3).
Figure 1: Dihydropyrimidinones in Pharmacological agents; Monastrol and Nitractin

7
Reasons for Dihydropyrimidinone Bioactivity
Dihydropyrimidinones have the ability to interact with DNA. A planar polycyclic aromatic
portion of the drug is inserted between adjacent stacked base pairs of a double stranded DNA in
a process named intercalation, which results in helix extension and unwinding of the DNA
(Figure 2) (4). The insertion of an intercalating molecule occurs in the minor groove of DNA and
is held together by hydrogen bonds, charge transfer bonds, and van der Waals forces (5). The
most effective compounds are ligands capable of structure and sequence selective binding to
DNA, since they are able to unwind a specific target sequence that causes the inhibition of a
particular gene or protein that produces a disease (5). The unwinding of the DNA molecule
prevents transcription, which blocks the replication process of the cell containing the intercalated
DNA (6). There is not a consensus on how the unwinding disrupts the transcription process, but
one hypothesis is that it inhibits topoisomerases, which are necessary for uncoiling a DNA
molecule prior to replication (6). Inhibiting cell replication leads to cell death, which can cause a
decrease in tumor size in cancer patients (6). By being able to stop transcription, and thus
causing death of specific cells, dihydropyrimidinones are able to act as a powerful cancer
treatment.
Figure 2: DNA Intercalation

8
Dihydropyrimidinone Synthesis
The first dihydropyrimidinone was first synthesized by Pietro Biginelli in 1893 by an acid-
catalyzed cyclocondensation reaction of ethyl acetoacetate, urea, and benzaldehyde (Figure 3)
(7). The reaction was performed by heating a mixture of the three components dissolved in
ethanol with hydrochloric acid as a catalyst (7). The product of this synthesis was identified by
Pietro Biginelli as 3, 4- dihydropyrimidine- 2 (1H) – one (Figure 3) (7). Although this reaction is
simple, it has poor yield. Many catalytic processes have been discovered (8). A method to
catalyze the cyclization of the Biginelli product is to use a Bronsted or Lewis acid with methods
based on metal salts with non-nucleophilic anions (8). The most effective catalysts use reagents
which have dehydrating properties as well as protic or Lewis acidic behavior. Some of these
compounds are: ethyl polyphosphate, acetic anhydride, and trimethylsilyl chloride (TMSCl) (8).
Synthetic studies also show that triethylorthoformate (TEOF), associated with citric acid or
oxalic acid acts as a system that highly increases yields of dihydropyrimidinones, especially
when weak acids are used (8).
Figure 3: Biginelli Reaction. Cyclocondensation of ethyl acetoacetate, benzaldehyde, and urea

9
Phenethylamine Bioactivity
Phenethylamines (Figure 4) are a basic scaffold for a series of compounds that have high
biological activity. Some of these compounds are neurotransmitters such as norepinephrine,
sympathomimetics such as ephedrine and amphetamine, and hallucinogens such as mescaline
(9). Many phenethylamines are also ubiquitous in hallucinogenic designer drugs such as alpha-
methyltryptamine (AMT), dimethyltryptamine (DMT), and psilocybin (10).
Figure 4: Phenethylamine
The reason for the neurologic activity of phenethylamine scaffolds has to do with the structural
similarity between the tryptamines and serotonin, and phenethylamines and dopamine, which
makes these compounds act as 5 hydroxytryptamine (5HT) receptor agonists in the central
nervous system (10). Norepinephrine is an example of one of the most significant
phenethylamines, which can be seen by the many crucial effects that it has on the body (Figure
5) (11). Norepinephrine controls the flight-or fight response by acting as an alpha-adrenergic
receptor activator, which increases heart rate, dilates bronchioles, increases gluconeogenesis and
sugar breakdown, and it increases blood flow to muscles (11).
Figure 5: Norepinephrine

10
Another important example of a bioactive phenethylamine is dopamine (Figure 6) (12).
Dopamine regulates hormonal, cardiovascular, retinal, renal, and immune system functions
among others (12). Proper release and functioning of dopamine and its receptors is so crucial in
the body, that malfunctions in these systems have been linked to Parkinson’s disease, ADHD,
and Tourette’s syndrome (12).
Figure 6: Dopamine
As it can be seen by the information above, phenethylamines and its derivatives have lots of
bioactive properties in the body, which is expected to also occur in the compounds that were
synthesized in this project.
Applications to the Research
The main idea of the project is to combine two privileged structures; dihydropyrimidinones and
phenethylamines in order to form a highly bioactive chimeric compound. The strategy of
combining two or more bioactive structures with the goal of enhancing pharmacological benefits,
and decreasing side effects has been seen in literature. The name of this strategy is the synthesis
of polyfunctional drugs (13). These polyfunctional drugs are made by combining two or more
bioactive agents in order to produce a single compound with multiple biological activities (13).
The advantages of using polyfunctional drugs include the possibility of synergistic drug effects,

11
and more predictable pharmacokinetics and pharmacodynamics than drug cocktails which use
various drugs to achieve the same effect (13). As a result of combining dihydropyrimidinones
and phenethylamines, it is expected to synthesize a chimeric molecule with unique
pharmacological properties, which will be locally tested for antibiotic activity with the use of the
Kirby-Bauer disk diffusion assay. Pharmacological Screening will also be performed by sending
the synthesized products to the High Throughput Screening (HTS) services of the National
Institutes of Health (NIH) (14).
Synthetic pathway outline
The synthesis of the target molecules is split into three separate steps. The first step is to
synthesize the Biginelli products with various functional groups. Different functional groups are
added to the molecule by changing the aldehyde at the beginning of the reaction, which end up
carrying over to the final synthesized products (Figure 7).
Figure 7: Aldehyde and Phenethylamine
The second step of the synthetic pathway is to brominate the allylic site in the Biginelli product.
The third step is to perform a novel cyclization of a lactam ring by using a phenethylamine as a
nucleophile. Various functional groups can be added to the target product by changing the
functional groups in the phenethylamines used for cyclization (Figure 7). The various

12
modifications to the reaction conditions in each of these steps will be explored in depth
throughout the remainder of the thesis. The outline can be seen below (Figure 8):
Figure 8: Synthetic Pathway Outline. Biginelli Synthesis, Bromination, and Cyclization
Synthesis of dihydropyrimidinone scaffolds with various functional groups
In this section of the project, Nozomi Arai and Kei Ohgo helped with the synthesis of some
dihydropyrimidinones. As mentioned earlier in the paper, the synthesis of dihydropyrimidinones
is done through the standard Biginelli reaction. The reaction is done by an acid-catalyzed
cyclocondensation reaction of ethyl acetoacetate, urea, and benzaldehyde (2). The way in which
we added functional groups to the dihydropyrimidinones is by using different commercially
available aldehydes at the beginning of the cyclocondensation reaction. The aldehydes used in
the project include: benzaldehyde, 1-napthaldehyde, 2- naphthaldehyde, 4-nitrobenzaldehyde, 4-
trifluoromethylbenzaldehyde, 2-nitrobenzaldehyde, 3-chloro benzaldehyde, 2-
bromobenzaldehyde, 3-bromobenzaldehyde, 2-furaldehyde, 3-furaldehyde, and 4-

13
trifluoromethoxy benzaldehyde. The raw synthesized products were recrystallized with ethanol
in order to increase purity. The synthesized dihydropyrimidinone scaffolds can be seen below
(Figure 9).
Figure 9: Synthesized Dihydropyrimidinone Scaffolds

14
Bromination of the allylic sites in dihydropyrimidinones
The next step in the project was to brominate the allylic site in the synthesized
dihydropyrimidinone structures (Figure 10). The two brominating agents we used for this step in
the process are N-bromosuccinimide (NBS) and phenyltrimethylammonium tribromide. The
NBS reacts with the allylic site through a radical reaction that involves the homolytic cleavage of
the Br2 with light, followed by the extraction of the allylic H and the reaction of this radical with
another equivalent of Br2 to give the desired product (15). The preferred method due to
experimental observations is the bromination with phenyltrimethylammonium tribromide due to
its tendency to cause only a single bromination (16).
Figure 10: Bromination Reaction
N-bromosuccinimide (NBS) vs. Phenyltrimethylammonium Tribromide
The procedure for the Bromination reaction with NBS involved 1.1 NBS equivalent, and 1
equivalent of the Biginelli product dissolved in dichloromethane (DCM), and reacted at room
temperature. As predicted by literature, NBS was a strong brominating agent (15), and the NMR
spectra showed dibrominations at the allylic site. Phenyltrimethylammonium tribromide (PMTB)
was used due to its behavior as a milder brominating agent (16). The methodology for the
Bromination reaction with PMTB involved 1 PMTB equivalent, and 1 equivalent of the Biginelli

15
product dissolved in DCM, and reacted at room temperature. The 1H Nuclear Magnetic
Resonance (NMR) Spectra showed a single Bromination at the allylic site. In order to increase
the purity of the brominated Biginelli product, recrystallization with ethanol was performed. The
desired goal of recrystallizing the molecule was not reached however, as the NMR spectra after
the recrystallization showed the dissociation of the bromine from the molecule. Due to the
repeated debromination after various recrystallization procedures, we decided to perform a one
pot reaction mechanism. The one pot reaction mechanism involves the evaporation of DCM from
the reaction flask by rotavaping, followed by the addition of methanol in order to re-dissolve the
brominated Biginelli product. Lastly, the phenethylamine is added to perform the cyclization.
Cyclization Methodology and Mechanism The third step in the synthesis of the desired product is the addition of the primary
phenylethylamine group to perform a second order nucleophilic substitution (SN2) intermolecular
cyclization. The cyclization reaction was done by adding the 5 equivalents of the phenethylamine
to the round bottom flask with the brominated Biginelli dissolved in methanol. The reaction was
stirred at room temperature overnight. The hypothesized mechanism involves the nucleophilic
attack from the lone pair on the nitrogen of the primary amine on the electrophilic carbon
attached to the bromine. The bromine will be knocked off the molecule as a leaving group. The
amino group from the extra equivalents of the phenethylamine will pull a proton off of the
nitrogen, and the remaining lone pair in the nitrogen will then nucleophilically attack the
electrophilic carbonyl carbon in the ester group, the O-R group will act as a leaving group, and
an intra-molecular cyclization will occur as a result (Figure 11). A similar mechanism is seen in
literature in a reaction called the Pictet-Spengler cyclization in which the primary amine acts as a

16
nucleophile by attacking an electrophilic carbonyl group that subsequently undergoes an
intramolecular cyclization (17).
Figure 11: SN2 reaction followed by an intramolecular cyclization
Substituted Phenethylamines used in the project
In order to add variety to the functional groups present in the final cyclized products, pre-
manufactured phenethylamines with different functional groups were used to perform the
cyclization. The phenethylamines that were used include: phenethylamine, tyramine, 2-
methoxyphenethylamine, 3-methoxyphenethylamine, 4-methoxyphenethylamine, 3,4-
dimethoxyphenethylamine, 2-(3-chlorophenyl) ethylamine, 4-trifluoromethylphenethylamine, 2-
(3-trifluoromethylphenyl) ethylamine, 3-fluorophenethylamine, and 4-fluorophenethylamine.
Phenethylamine
The phenethylamine that worked the best for the cyclization step is the unsubstituted
phenethylamine with no groups attached to it. As soon as the phenethylamine was added to the
reaction, there was a visible color change from light yellow to clear, and soon thereafter a white

17
precipitate was formed. The product of this reaction was then easily filtered, and recrystallized
with ethanol. The following products were formed with phenethylamine (Figure 12).
Figure 12: Cyclized products made with phenethylamine
Not all of the phenethylamine variants precipitated in the cyclization step. We assume most
phenethylamines worked to perform the first SN2 reaction in which the lone pair on the
phenethylamine’s nitrogen nucleophilically attacks the brominated site on the molecule. Most of
the phenethylamine variants ended up forming an intermediate in which the cyclization did not
yet occur. The intermediate product of the SN2 prior to the cyclization, which was precipitated
from methanol can be seen below (Figure 13).
Figure 13: Intermediate product from the SN2 reaction

18
In order to increase the chances for cyclization, various changes to the reaction conditions were
performed. The modifications made to the conditions were focused on increasing the entropy of
the system by raising the temperature in order to increase chances for favorable molecular
collisions, and a decrease in the steric hindrance in areas of the molecule that are crucial for
proper cyclization.
Increased Temperature The first attempt at increasing the entropy of the system was done by increasing the temperature
of the reaction from room temperature to 60° Celsius with methanol as a solvent. As indicated by
NMR spectra, there were no intramolecular cyclizations that occurred at this temperature. In
order to further increase the entropy of the system the solvent was changed to propanol, and the
temperature was brought up to 100° Celsius. Favorable results were encountered at this
temperature, and two molecules that were unable to cyclize prior to the increase in temperature
showed peaks on the NMR that indicated full cyclization. The structures that were synthesized
after the increase in temperature are seen below (Figure 14).
Figure 14: Cyclized structures at 100° Celsius

19
Decreased steric hindrance In order to decrease the steric hindrance at the site of cyclization, a different acetoacetate was
used in the cyclocondensation reaction of the starting dihydropyrimidinone (Biginelli) product.
The acetoacetate used at the beginning of the project was ethyl acetoacetate. In order to decrease
the hindrance of the molecule, methyl acetoacetate was used instead. By taking away a carbon
from the R group in the ester group, steric hindrance was decreased. The difference in the
structures made with the ethyl acetoacetate vs. the methyl acetoacetate are seen below (Figure
15). By decreasing the hindrance, there is an increased chance of a favorable collision between
the nitrogen and the carbonyl carbon from the ester group in the molecule.
Figure 15: Structures made with ethyl acetoacetate vs. methyl acetoacetate Synthesized Structures After the methyl acetoacetate modification was done to decrease hindrance, the success in the
making of the cyclized final products increased dramatically. Multiple phenethylamines were
able to trigger a full inner molecular cyclization at room temperature. The synthesized structures
are seen below (Figure 16).
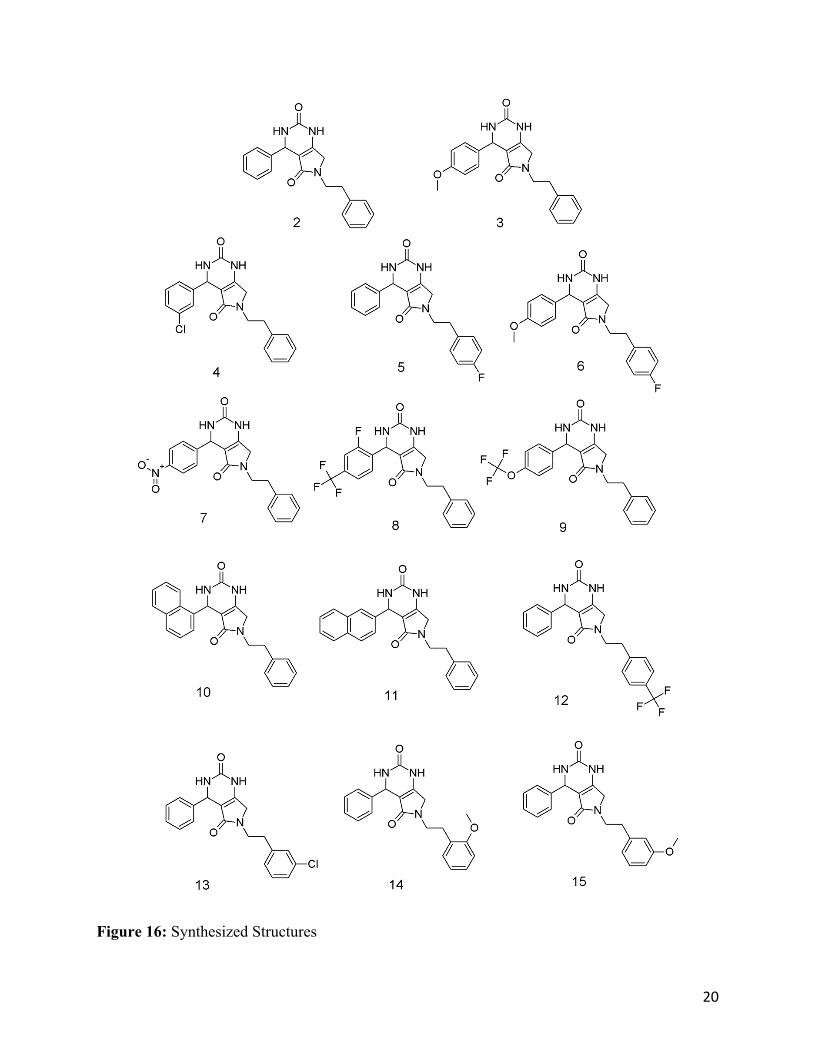
20
Figure 16: Synthesized Structures

21
Experimental Section The reagents were used as received and no precautions were taken to exclude air or moisture.
Experimental Data from Synthesized Products
Product 2 (APPENDIX 1,2): Dihydropyrimidinone 1A (2 mmol) was dissolved in 40 ml of
DCM, and mixed with Phenyltrimethylammonium tribromide (2 mmol) at room temperature and
the reaction was stirred for 2 hours. The DCM was rotavaped, and 40 ml methanol was added as
a solvent. Five equivalents of Phenethylamine (10 mmol) was added to the reaction flask, and it
was stirred overnight. The precipitate was filtered, and later recrystallized with ethanol. A yield
of 356.8 mg was obtained, with a percent yield of 52.5%. 1H NMR (300 MHz, DMSO-d6) δ 2.74
(t, 2H, J= 7.3 Hz), 3.44 (m, 7H), 3.84 (s, 2H), 5.14 (s, 1H), 7.16-7.35 (m, 10H), 7.54 (d, 7H, J=
2.1 Hz), 9.52 (s, 2H); 13C NMR 126.7, 126.8, 128.0, 128.8(2), 129.0, 139.4, 143.7, 150.2, 152.5,
168.7.

22
Product 3: Dihydropyrimidinone 1K (4.1 mmol) was dissolved in 40 ml of DCM, and mixed
with Phenyltrimethylammonium tribromide (4.1mmol) at room temperature and the reaction was
stirred for 2 hours. The DCM was rotavaped, and 30 ml methanol was added as a solvent. Five
equivalents of Phenethylamine (20.5 mmol) was added to the reaction flask, and it was stirred
overnight. The precipitate was filtered, and later recrystallized with ethanol. A yield of 31.0 mg
was obtained, with a 2.7% yield.
Product 4: Dihydropyrimidinone 1G (1.7 mmol) was dissolved in 40 ml of DCM, and mixed
with Phenyltrimethylammonium tribromide (1.7 mmol) at room temperature and the reaction
was stirred for 2 hours. The DCM was rotavaped, and 30 ml methanol was added as a solvent.

23
Five equivalents of Phenethylamine (8.5 mmol) was added to the reaction flask, and it was
stirred overnight. The precipitate was filtered, and later recrystallized with ethanol. The yield
was small, and it was used in its entirety for 1H NMR analysis.
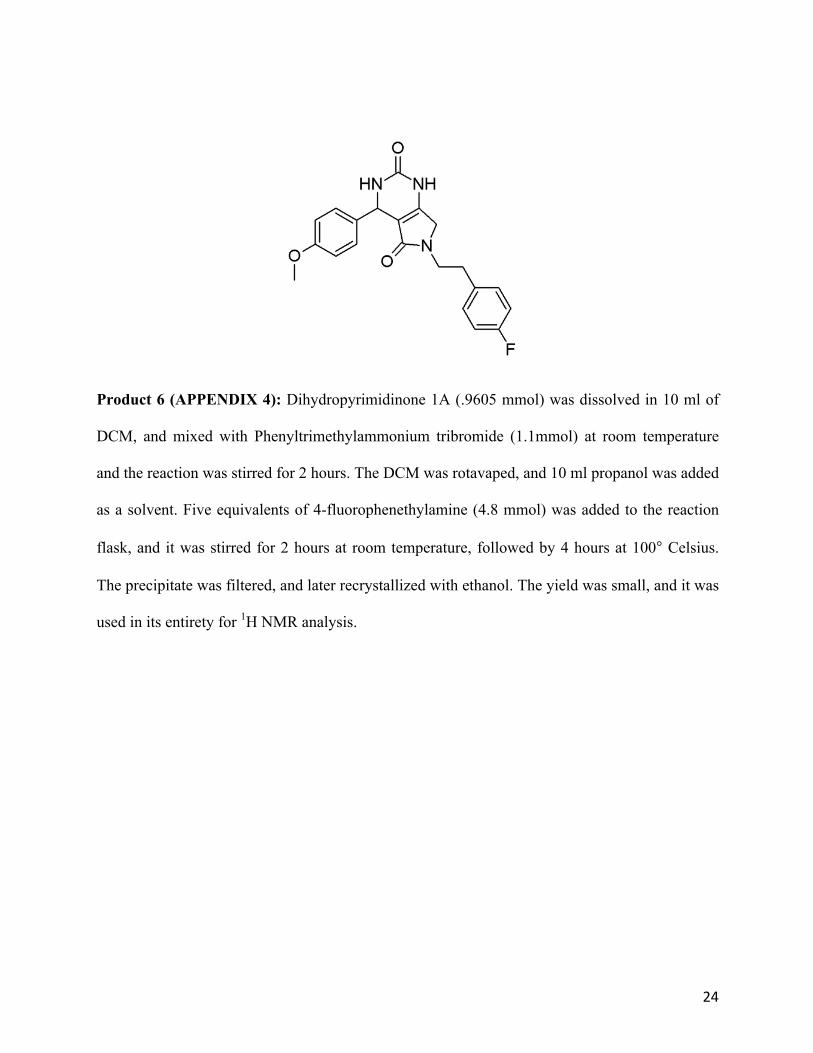
Product 5 (APPENDIX 3): Dihydropyrimidinone 1A (.9605 mmol) was dissolved in 10 ml of
DCM, and mixed with Phenyltrimethylammonium tribromide (1.1mmol) at room temperature
and the reaction was stirred for 2 hours. The DCM was rotavaped, and 10 ml propanol was added
as a solvent. Five equivalents of 4-fluorophenethylamine (4.8 mmol) was added to the reaction
flask, and it was stirred for 2 hours at room temperature, followed by 4 hours at 100° Celsius.
The precipitate was filtered, and later recrystallized with ethanol. The yield was small, and it was
used in its entirety for 1H NMR analysis.
24
Product 6 (APPENDIX 4): Dihydropyrimidinone 1A (.9605 mmol) was dissolved in 10 ml of
DCM, and mixed with Phenyltrimethylammonium tribromide (1.1mmol) at room temperature
and the reaction was stirred for 2 hours. The DCM was rotavaped, and 10 ml propanol was added
as a solvent. Five equivalents of 4-fluorophenethylamine (4.8 mmol) was added to the reaction
flask, and it was stirred for 2 hours at room temperature, followed by 4 hours at 100° Celsius.
The precipitate was filtered, and later recrystallized with ethanol. The yield was small, and it was
used in its entirety for 1H NMR analysis.
25
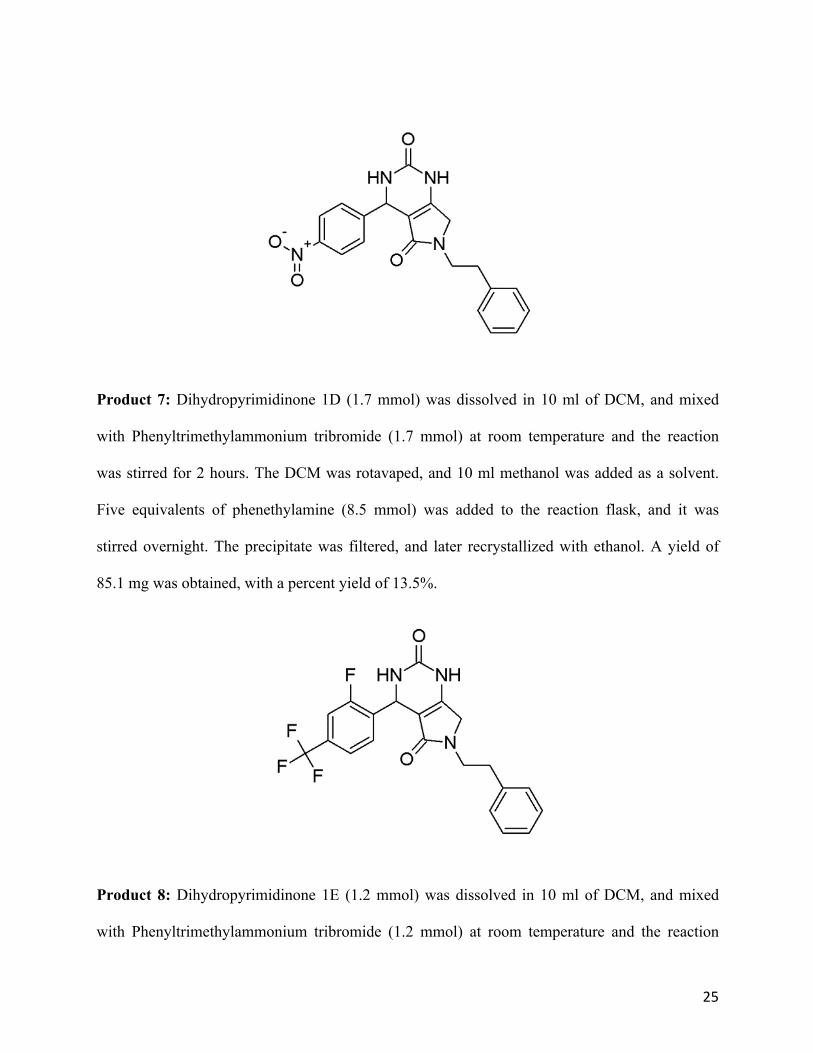
Product 7: Dihydropyrimidinone 1D (1.7 mmol) was dissolved in 10 ml of DCM, and mixed
with Phenyltrimethylammonium tribromide (1.7 mmol) at room temperature and the reaction
was stirred for 2 hours. The DCM was rotavaped, and 10 ml methanol was added as a solvent.
Five equivalents of phenethylamine (8.5 mmol) was added to the reaction flask, and it was
stirred overnight. The precipitate was filtered, and later recrystallized with ethanol. A yield of
85.1 mg was obtained, with a percent yield of 13.5%.
Product 8: Dihydropyrimidinone 1E (1.2 mmol) was dissolved in 10 ml of DCM, and mixed
with Phenyltrimethylammonium tribromide (1.2 mmol) at room temperature and the reaction

26
was stirred for 2 hours. The DCM was rotavaped, and 10 ml methanol was added as a solvent.
Five equivalents of phenethylamine (6 mmol) was added to the reaction flask, and it was stirred
overnight. The precipitate was filtered, and later recrystallized with ethanol. A yield of 224.4 mg
was obtained, with a percent yield of 35.4%.
Product 9 (APPENDIX 5): Dihydropyrimidinone 1L (.314 mmol) was dissolved in 10 ml of
DCM, and mixed with Phenyltrimethylammonium tribromide (.314 mmol) at room temperature
and the reaction was stirred for 2 hours. The DCM was rotavaped, and 10 ml methanol was
added as a solvent. Five equivalents of phenethylamine (1.57 mmol) was added to the reaction
flask, and it was stirred overnight. The precipitate was filtered, and later recrystallized with
ethanol. A yield of 26.4 mg was obtained, with a percent yield of 14.7%.
27
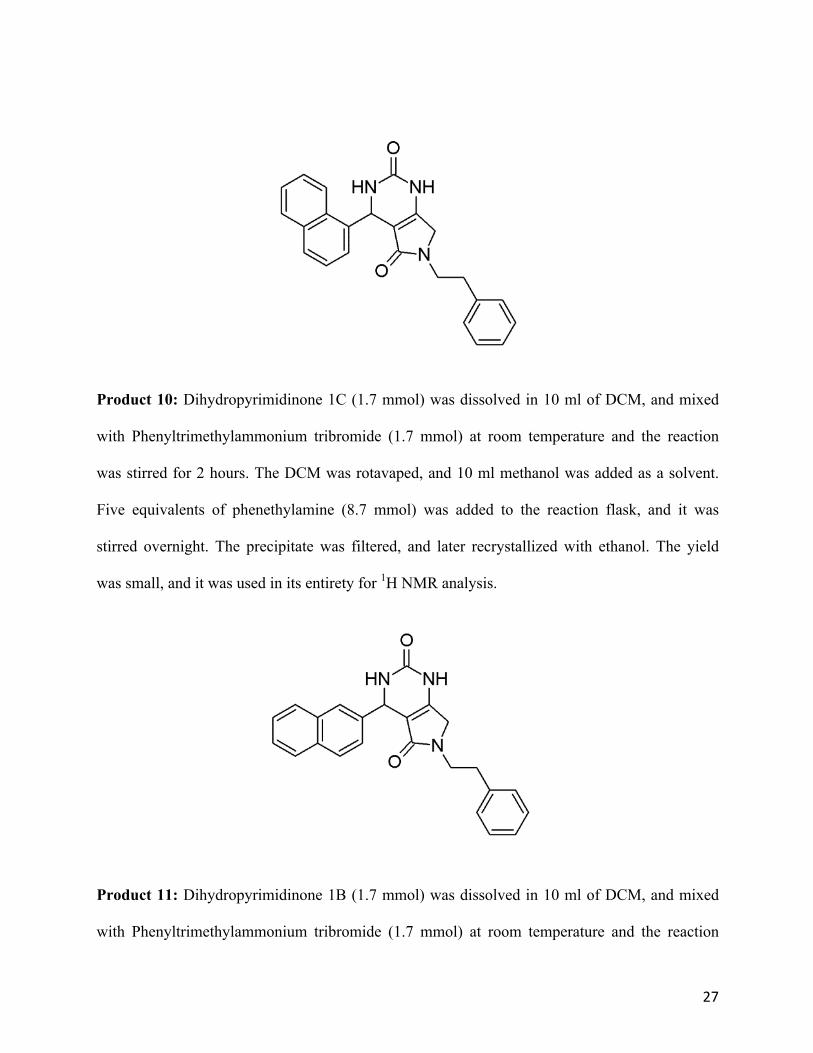
Product 10: Dihydropyrimidinone 1C (1.7 mmol) was dissolved in 10 ml of DCM, and mixed
with Phenyltrimethylammonium tribromide (1.7 mmol) at room temperature and the reaction
was stirred for 2 hours. The DCM was rotavaped, and 10 ml methanol was added as a solvent.
Five equivalents of phenethylamine (8.7 mmol) was added to the reaction flask, and it was
stirred overnight. The precipitate was filtered, and later recrystallized with ethanol. The yield
was small, and it was used in its entirety for 1H NMR analysis.
Product 11: Dihydropyrimidinone 1B (1.7 mmol) was dissolved in 10 ml of DCM, and mixed
with Phenyltrimethylammonium tribromide (1.7 mmol) at room temperature and the reaction

28
was stirred for 2 hours. The DCM was rotavaped, and 10 ml methanol was added as a solvent.
Five equivalents of phenethylamine (8.7 mmol) was added to the reaction flask, and it was
stirred overnight. The precipitate was filtered, and later recrystallized with ethanol. A yield of
69.3 mg was obtained, with a percent yield of 10.6%.
Product 12 (APPENDIX 6, 7): Dihydropyrimidinone 1A (1 mmol) was dissolved in 10 ml of
DCM, and mixed with Phenyltrimethylammonium tribromide (1 mmol) at room temperature and
the reaction was stirred for 2 hours. The DCM was rotavaped, and 10 ml methanol was added as
a solvent. Five equivalents of 4 trifluoromethylphenylethylamine (5 mmol) was added to the
reaction flask, and it was stirred overnight. The precipitate was filtered, and later recrystallized
with ethanol. A yield of 135.4 mg was obtained, with a percent yield of 35.6%.

29
Product 13 (APPENDIX 8): Dihydropyrimidinone 1A (1 mmol) was dissolved in 10 ml of
DCM, and mixed with Phenyltrimethylammonium tribromide (1 mmol) at room temperature and
the reaction was stirred for 2 hours. The DCM was rotavaped, and 10 ml methanol was added as
a solvent. Five equivalents of 2-(3-chlorophenylthylamine) (5 mmol) was added to the reaction
flask, and it was stirred overnight. The precipitate was filtered, and later recrystallized with
ethanol. A yield of 88.9 mg was obtained, with a percent yield of 24.7%.
Product 14 (APPENDIX 9): Dihydropyrimidinone 1A (1 mmol) was dissolved in 10 ml of
DCM, and mixed with Phenyltrimethylammonium tribromide (1 mmol) at room temperature and
the reaction was stirred for 2 hours. The DCM was rotavaped, and 10 ml methanol was added as

30
a solvent. Five equivalents of 2-methoxyphenethylamine (5 mmol) was added to the reaction
flask, and it was stirred overnight. The precipitate was filtered, and later recrystallized with
ethanol. A yield of 67.0 mg was obtained, with a percent yield of 18.9%.
Product 15 (APPENDIX 10): Dihydropyrimidinone 1A (1 mmol) was dissolved in 10 ml of
DCM, and mixed with Phenyltrimethylammonium tribromide (1 mmol) at room temperature and
the reaction was stirred for 2 hours. The DCM was rotavaped, and 10 ml methanol was added as
a solvent. Five equivalents of 3-methoxyphenethylamine (5 mmol) was added to the reaction
flask, and it was stirred overnight. The precipitate was filtered, and later recrystallized with
ethanol. A yield of 152.2 mg was obtained, with a percent yield of 40.9%.

31
Antibiotic testing Local antibiotic testing was done with the use of the Kirby Bauer Disk Diffusion Assay. The
synthesized compounds were dissolved in dimethylsulfoxide (DMSO) to a concentration of
0.1M, and were tested against a strain of gram positive bacteria (Staphylococcus aureus), and a
strain of gram negative bacteria (Enterobacter aerogenes). The extent of the antibiotic activity is
qualitatively seen by the diameter of the zone of inhibition produced by the compound (18). The
larger the zone of inhibition, the higher the antibacterial activity of the compound (18). The
positive control substance was chloramphenicol, which is always expected to have a relatively
large zone of inhibition with both gram positive and gram negative bacterial strains.
Antibiotic testing results
The final cyclized scaffolds, the intermediate uncycled products, and some of the starting
dihydropyrimidinone scaffolds were tested. The results of the experiment indicate that none of
the compounds synthesized thus far possess antibacterial properties as indicated by the lack of a
zone of inhibition.

32
Conclusion
The goal of the experiment was to synthesize novel pharmacological agents by combining two
privileged structures; dihydropyrimidinones and phenethylamines. The project was separated
into four parts; making dihydropyrimidinone scaffolds with different functional groups,
brominating the allylic site in the dihydropyrimidinone, performing a cyclization with various
phenethylamines, and testing for bioactivity of the compounds.
The first part of the project involved the synthesis of dihydropyrimidinones with various
functional groups by using pre-manufactured aldehydes. Twelve dihydropyrimidinone scaffolds
were synthesized, and their purity was increased through recrystallization with ethanol as a
solvent. The second part of the project involved the bromination of the allylic site in the
dihydropyrimidinone scaffolds. It was found that phenytrimethylammonium tribromide was the
preferred brominating agent. Due to the dissociation of the bromine group during the
recrystallization, a one pot reaction was chosen. The goal of the third part of the project was to
perform an intermolecular cyclization which combined pre-manufactured phenethylamines with
different functional groups with the brominated dihydropyrimidinone scaffolds. The temperature
was raised to 60° Celsius, and 100° Celsius respectively, which led to improved results. The
reaction was also maximized by decreasing the steric hindrance of the molecule by altering the
ester group from an ethyl ester to a methyl ester. The increased temperature, and the decreased
hindrance led to the successful synthesis of fourteen cyclized compounds. The fourth part of the
project involved the antibacterial testing of the synthesized compounds using the Kirby-Bauer
disk diffusion assay.

33
Although the compounds did not show any antibacterial activity, it is a possibility that they
possess other pharmacological properties of dihydropyrimidinones such as calcium channel
blockers, anti-cancer drugs, and anti-inflammatory agents. These pharmacological properties will
be tested for by the Developmental Therapeutics Program (DTP) of the NIH. This NIH program
is designed to help the general research community to screen bioactive compounds with the goal
of discovering new chemical leads and biological mechanisms. Another program that we will
send our compounds to the Lilly Open Innovation Drug Discovery (OIDD) Program for
bioactivity screenings (19). Once hits are discovered, we will move to more conventional SAR
studies to determine how to improve the absorption and bioavailability of these compounds in
both in vitro and in vivo.

34
References 1. Vijay, K.; Ganapaty, S.; Rao, A. S. Synthesis And Characterization and Biological Evaluation
of Some Dihydropirimidinones. Asian Journal of Chemistry. 2010, 22, 2518–2528.
2. Soumyanarayanan, U.; Bhat, V. G.; Kar, S. S.; Mathew, J. A. Monastrol Mimic Biginelli
Dihydropyrimidinone Derivatives: Synthesis, Cytotoxicity Screening against HepG2 and HeLa
Cell Lines and Molecular Modeling Study. Organic and Medicinal Chemistry Letters Org Med
Chem Lett. 2012, 2, 1–11.
3. Yadlapalli, R. K.; Chourasia, O.; Vemuri, K.; Sritharan, M.; Perali, R. S. Synthesis And in
Vitro Anticancer and Antitubercular Activity of Diarylpyrazole Ligated Dihydropyrimidines
Possessing Lipophilic Carbamoyl Group. Bioorganic & Medicinal Chemistry Letters. 2012, 22,
2708–2711
4. Gribble, G.; Lopchuck, J. “Compositions and Methods for Treating Cancer.” Publication
number: US20140228392 A1. 2014. (1-7)
5. Wang, G.; Yan, C.; Lu, Y. Exploring DNA Binding Properties and Biological Activities of
Dihydropyrimidinones Derivatives. Colloids and Surfaces B: Biointerfaces. 2013, 106, 28–36.
6. Thomas, G. Medicinal Chemistry. An Introduction; 2nd ed.; 2007, 372-373.
7. Mishra, M. K. Anti-Inflammatory Activity of Some New Dihydropyrimidinines Derivatives.
International Journal of Pharmaceutical Sciences and Research. 2010, 1, 92–95.

35
8. Canto, R. F. S.; Bernardi, A.; Battastini, A. M. O.; Russowsky, D.; Eifler-Lima, V. L.
Synthesis Of Dihydropyrimidin-2-One/Thione Library and Cytotoxic Activity against the
Human U138-MG and Rat C6 Glioma Cell Lines. J. Braz. Chem. Soc. Journal of the Brazilian
Chemical Society. 2011, 22, 1379–1388.
9. Pullman, B.; Coubeils, J. L.; Courriere, P.; Gervois, J. P. Quantum Mechanical Study of the
Conformational Properties of Phenethylamines of Biochemical and Medicinal Interest. J. Med.
Chem. Journal of Medicinal Chemistry. 1972, 15, 17–23.
10. Vorce, S. P.; Sklerov, J. H. A General Screening And Confirmation Approach to the Analysis
of Designer Tryptamines and Phenethylamines in Blood and Urine Using GC-EI-MS and HPLC-
Electrospray-MS. Journal of Analytical Toxicology. 2004, 28, 407–410
11. Norepinephrine. Rice University, http://www.caam.rice.edu/~cox/wrap/norepinephrine.pdf
12. Beaulieu, J.-M.; Gainetdinov, R. R. The Physiology, Signaling, And Pharmacology of
Dopamine Receptors. Pharmacological Reviews. 2011, 63, 182–217.
13. Bansal, Y.; Silakari, O. Synthesis And Pharmacological Evaluation of Polyfunctional
Benzimidazole-NSAID Chimeric Molecules Combining Anti-Inflammatory, Immunomodulatory
and Antioxidant Activities. Arch. Pharm. Res. Archives of Pharmacal Research. 2013, 37, 1426–
1436
14. PAR-13-134: High Throughput Screening (HTS) to Discover Chemical Probes (X01).
http://grants.nih.gov/grants/guide/pa-files/par-13-134.html (accessed Apr 18, 2016).

36
15. F. Von Nussbaum, D. Karthaus, S. Anlauf, M. Klein, V. M-J. Li, D. Meiborn, K. Lustig, J.
Schamberger. “4-(4-Cyano-2-thioaryl)-dihydropyrimidinones as neutrophil elastase inhibitors
and their preparation and use in the treatment of diseases of the lung and cardiovascular system”
US2014/0045802A1 US Patent Application. 2014.
16. Kajigaeshi, S.; Kakinimi, T. Bromination And Oxidation with Benzyltrimethylammonium
Tribromide. Elsevier. 1995, 7, 29–48.
17. Cejka, J. Introduction To Zeolite Molecular Sieves. Elsevier. 2007, 3.
18. Leboffe, M.; Pierce, B. Microbiology: Laboratory Theory and Application; 3rd edition;
Morton Publishing Company, 2010.
19. OIDD Screening. Open Innovation Drug Discovery,
https://openinnovation.lilly.com/dd/what-we-offer/screening.html (accessed Apr 18, 2016)

37
APPENDIX 1: 1H NMR from Product 2

38
APPENDIX 2: 13C NMR from Product 2
39
APPENDIX 3: 1H NMR from Product 5

40
APPENDIX 4: 1H NMR from Product 6

41
APPENDIX 5: 1H NMR from Product 9

42
APPENDIX 6: 1H NMR from Product 12

43
APPENDIX 7: 19F NMR from Product 12

44
APPENDIX 8: 1H NMR from Product 13

45
APPENDIX 9: 1H NMR from Product 14

46
APPENDIX 10: 1H NMR from Product 15





























