Embed Size (px)
Citation preview

Sequence AlignmentsSequence Alignments
Introduction to BioinformaticsIntroduction to Bioinformatics

Intro to Bioinformatics – Sequence Alignment 2
Sequence AlignmentsSequence Alignments Cornerstone of bioinformatics What is a sequence?
• Nucleotide sequence
• Amino acid sequence
Pairwise and multiple sequence alignments• We will focus on pairwise alignments
What alignments can help• Determine function of a newly discovered gene sequence
• Determine evolutionary relationships among genes, proteins, and species
• Predicting structure and function of protein
Acknowledgement: This notes is adapted from lecture notes of both Wright State University’s Bioinformatics Program and Professor Laurie Heyer of Davidson College with permission.

Intro to Bioinformatics – Sequence Alignment 3
DNA ReplicationDNA Replication Prior to cell division, all the
genetic instructions must be “copied” so that each new cell will have a complete set
DNA polymerase is the enzyme that copies DNA• Reads the old strand in the 3´ to 5´
direction

Intro to Bioinformatics – Sequence Alignment 4
Over time, genes accumulate Over time, genes accumulate mutationsmutations Environmental factors
• Radiation
• Oxidation Mistakes in replication or
repair Deletions, Duplications Insertions, Inversions Translocations Point mutations

Intro to Bioinformatics – Sequence Alignment 5
Codon deletion:ACG ATA GCG TAT GTA TAG CCG…• Effect depends on the protein, position, etc.• Almost always deleterious• Sometimes lethal
Frame shift mutation: ACG ATA GCG TAT GTA TAG CCG… ACG ATA GCG ATG TAT AGC CG?…• Almost always lethal
DeletionsDeletions

Intro to Bioinformatics – Sequence Alignment 6
IndelsIndels Comparing two genes it is generally impossible
to tell if an indel is an insertion in one gene, or a deletion in another, unless ancestry is known:
ACGTCTGATACGCCGTATCGTCTATCTACGTCTGAT---CCGTATCGTCTATCT
Intro to Bioinformatics – Sequence Alignment 7
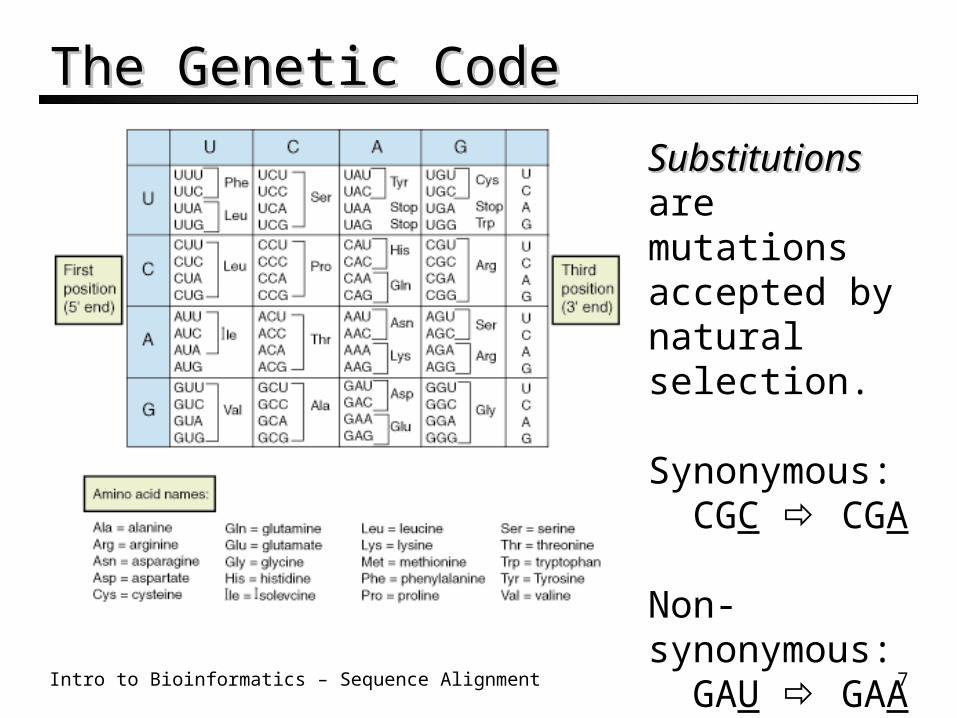
The Genetic CodeThe Genetic Code
SubstitutionsSubstitutions are mutations accepted by natural selection.
Synonymous: CGC CGA
Non-synonymous: GAU GAA

Intro to Bioinformatics – Sequence Alignment 8
Comparing Two SequencesComparing Two Sequences Point mutations, easy:ACGTCTGATACGCCGTATAGTCTATCTACGTCTGATTCGCCCTATCGTCTATCT
Indels are difficult, must align sequences:ACGTCTGATACGCCGTATAGTCTATCTCTGATTCGCATCGTCTATCT
ACGTCTGATACGCCGTATAGTCTATCT----CTGATTCGC---ATCGTCTATCT

Intro to Bioinformatics – Sequence Alignment 9
Why Align Sequences?Why Align Sequences? The draft human genome is available Automated gene finding is possible Gene: AGTACGTATCGTATAGCGTAA
• What does it do?What does it do?
One approach: Is there a similar gene in another species?• Align sequences with known genes• Find the gene with the “best” match

Intro to Bioinformatics – Sequence Alignment 10
Gaps or No GapsGaps or No Gaps Examples
Intro to Bioinformatics – Sequence Alignment 11
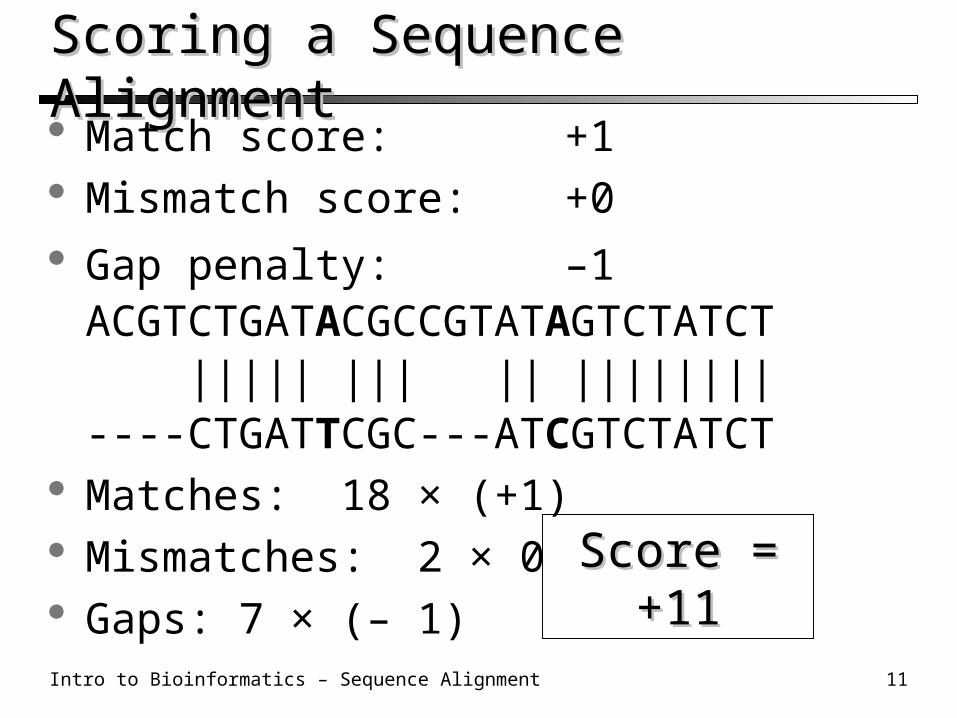
Scoring a Sequence AlignmentScoring a Sequence Alignment Match score: +1 Mismatch score:+0
Gap penalty: –1ACGTCTGATACGCCGTATAGTCTATCT ||||| ||| || ||||||||----CTGATTCGC---ATCGTCTATCT
Matches: 18 × (+1) Mismatches: 2 × 0 Gaps: 7 × (– 1)
Score = +11Score = +11

Intro to Bioinformatics – Sequence Alignment 12
Origination and Length PenaltiesOrigination and Length Penalties We want to find alignments that are
evolutionarily likely. Which of the following alignments seems more
likely to you?ACGTCTGATACGCCGTATAGTCTATCTACGTCTGAT-------ATAGTCTATCT
ACGTCTGATACGCCGTATAGTCTATCTAC-T-TGA--CG-CGT-TA-TCTATCT
We can achieve this by penalizing more for a new gap, than for extending an existing gap
Intro to Bioinformatics – Sequence Alignment 13
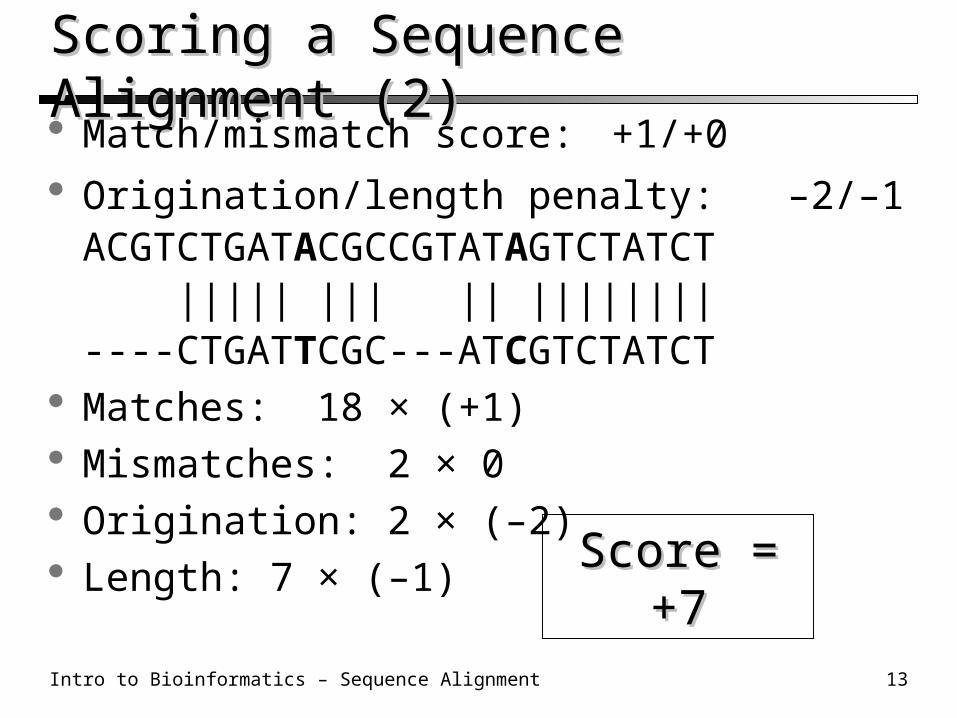
Scoring a Sequence Alignment (2)Scoring a Sequence Alignment (2) Match/mismatch score: +1/+0
Origination/length penalty: –2/–1ACGTCTGATACGCCGTATAGTCTATCT ||||| ||| || ||||||||----CTGATTCGC---ATCGTCTATCT
Matches: 18 × (+1) Mismatches: 2 × 0 Origination: 2 × (–2) Length: 7 × (–1)
Score = +7Score = +7

Intro to Bioinformatics – Sequence Alignment 14
How can we find an optimal alignment?How can we find an optimal alignment? Finding the alignment is computationally hard:ACGTCTGATACGCCGTATAGTCTATCTCTGAT---TCG-CATCGTC--T-ATCT
C(27,7) gap positions = ~888,000 possibilities It’s possible, as long as we don’t repeat our
work! Dynamic programming: The Needleman &
Wunsch algorithm

Intro to Bioinformatics – Sequence Alignment 15
Dynamic ProgrammingDynamic Programming Technique of solving optimization problems
• Find and memorize solutions for subproblems• Use those solutions to build solutions for larger
subproblems• Continue until the final solution is found
Recursive computation of cost function in a non-recursive fashion
Intro to Bioinformatics – Sequence Alignment 16
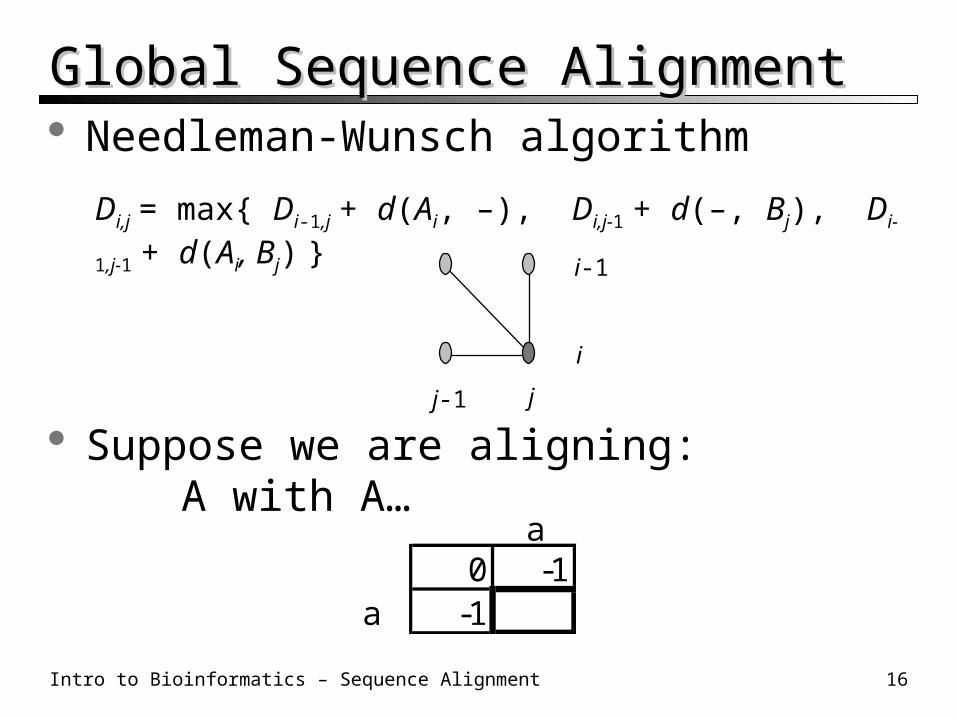
Global Sequence AlignmentGlobal Sequence Alignment Needleman-Wunsch algorithm
Suppose we are aligning:A with A…
a0 -1
a -1
Di,j = max{ Di-1,j + d(Ai, –), Di,j-1 + d(–, Bj), Di-1,j-1 + d(Ai, Bj) }
i-1
j-1 j
i
Intro to Bioinformatics – Sequence Alignment 17
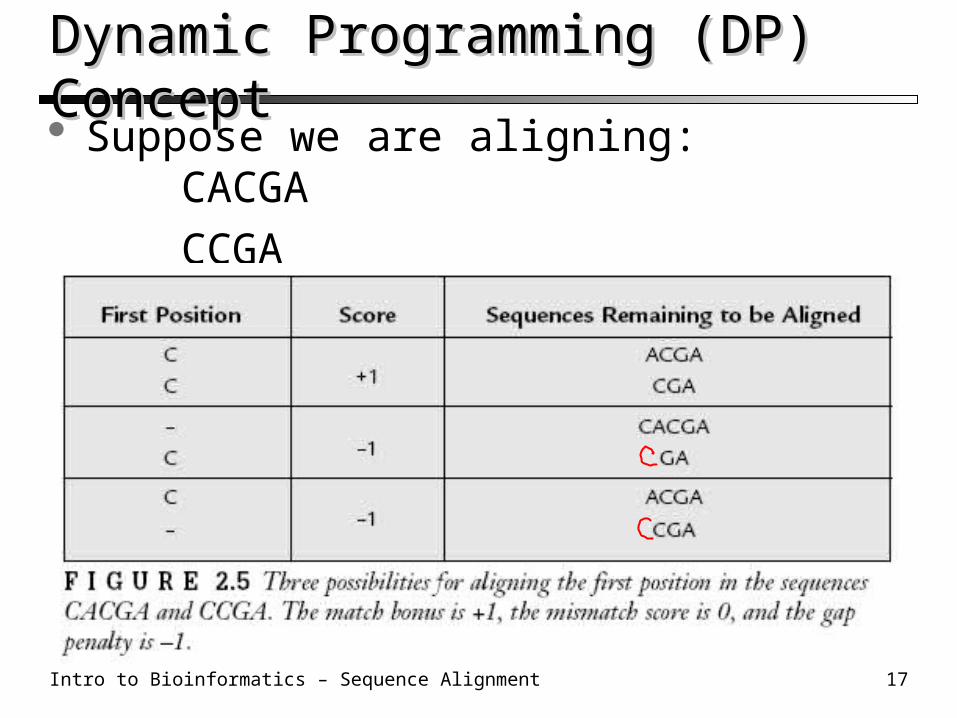
Dynamic Programming (DP) ConceptDynamic Programming (DP) Concept Suppose we are aligning:
CACGA
CCGA

Intro to Bioinformatics – Sequence Alignment 18
DP – Recursion PerspectiveDP – Recursion Perspective Suppose we are aligning:ACTCGACAGTAG
Last position choices:G +1 ACTCG ACAGTA
G -1 ACTC- ACAGTAG
- -1 ACTCGG ACAGTA

Intro to Bioinformatics – Sequence Alignment 19
What is the optimal alignment?What is the optimal alignment? ACTCGACAGTAG
Match: +1 Mismatch: 0 Gap: –1

Intro to Bioinformatics – Sequence Alignment 20
Needleman-Wunsch: Step 1Needleman-Wunsch: Step 1 Each sequence along one axis Mismatch penalty multiples in first row/column 0 in [1,1] (or [0,0] for the CS-minded)
A C T C G0 -1 -2 -3 -4 -5
A -1 1C -2A -3G -4T -5A -6G -7

Intro to Bioinformatics – Sequence Alignment 21
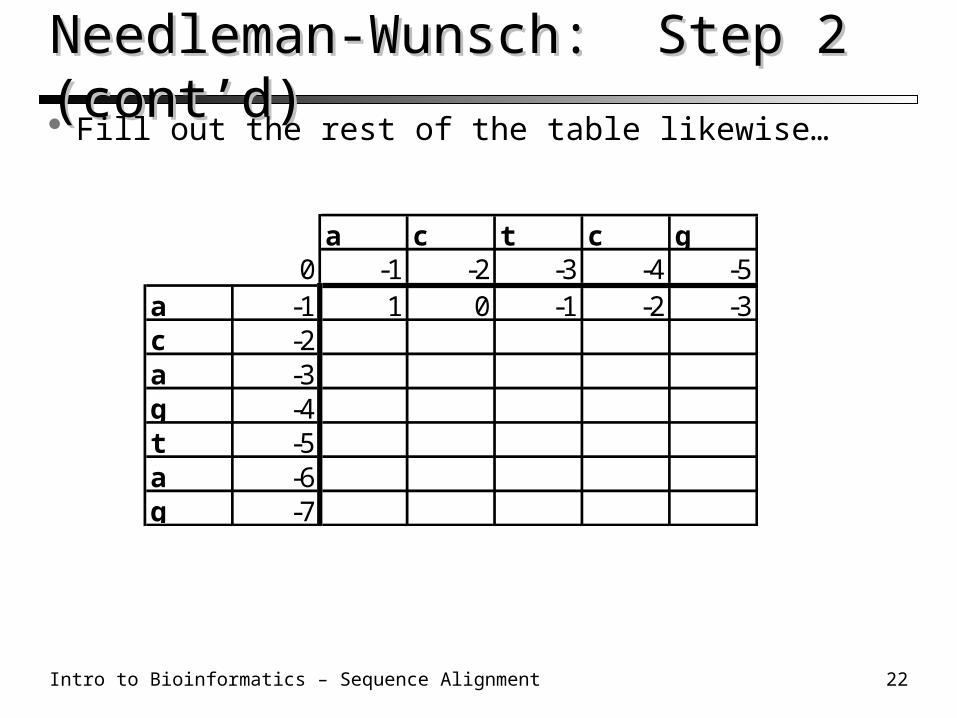
Needleman-Wunsch: Step 2Needleman-Wunsch: Step 2 Vertical/Horiz. move: Score + (simple) gap penalty Diagonal move: Score + match/mismatch score Take the MAX of the three possibilities
A C T C G0 -1 -2 -3 -4 -5
A -1 1C -2A -3G -4T -5A -6G -7
Intro to Bioinformatics – Sequence Alignment 22
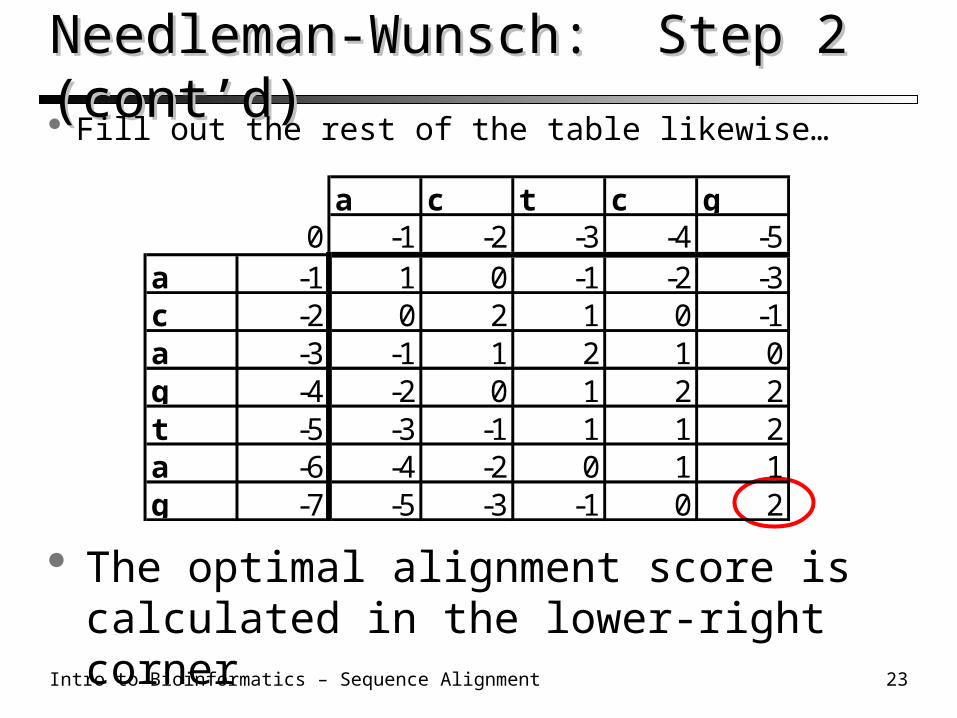
Needleman-Wunsch: Step 2 (cont’d)Needleman-Wunsch: Step 2 (cont’d) Fill out the rest of the table likewise…
a c t c g0 -1 -2 -3 -4 -5
a -1 1 0 -1 -2 -3c -2a -3g -4t -5a -6g -7
Intro to Bioinformatics – Sequence Alignment 23
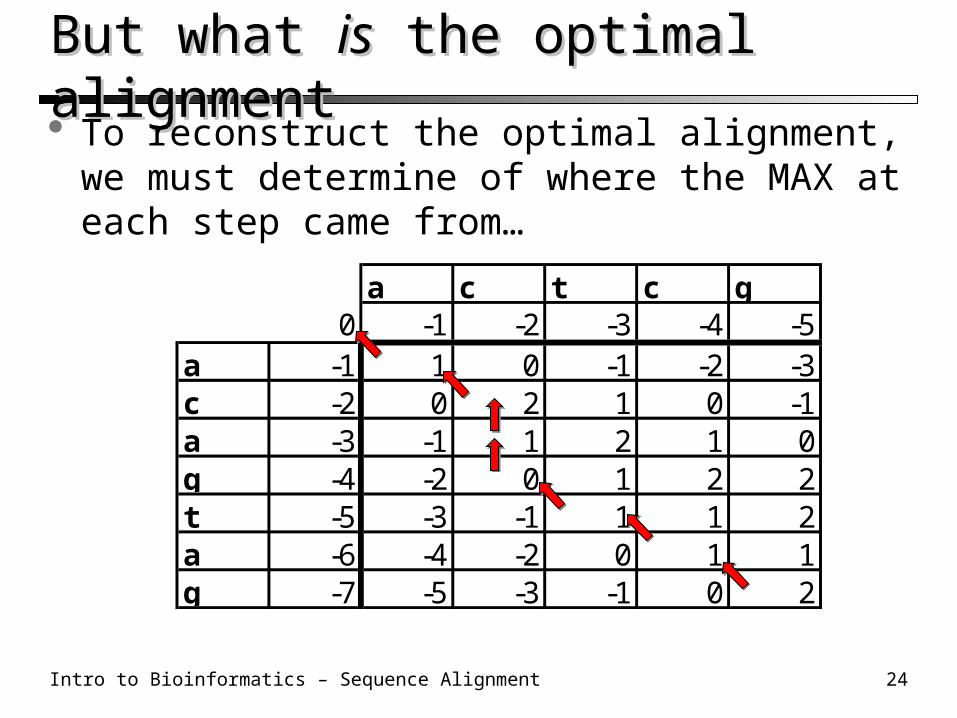
Needleman-Wunsch: Step 2 (cont’d)Needleman-Wunsch: Step 2 (cont’d) Fill out the rest of the table likewise…
The optimal alignment score is calculated in the lower-right corner
a c t c g0 -1 -2 -3 -4 -5
a -1 1 0 -1 -2 -3c -2 0 2 1 0 -1a -3 -1 1 2 1 0g -4 -2 0 1 2 2t -5 -3 -1 1 1 2a -6 -4 -2 0 1 1g -7 -5 -3 -1 0 2
Intro to Bioinformatics – Sequence Alignment 24
a c t c g0 -1 -2 -3 -4 -5
a -1 1 0 -1 -2 -3c -2 0 2 1 0 -1a -3 -1 1 2 1 0g -4 -2 0 1 2 2t -5 -3 -1 1 1 2a -6 -4 -2 0 1 1g -7 -5 -3 -1 0 2
But what But what isis the optimal alignment the optimal alignment To reconstruct the optimal alignment, we must
determine of where the MAX at each step came from…

Intro to Bioinformatics – Sequence Alignment 25
A path corresponds to an alignmentA path corresponds to an alignment = GAP in top sequence = GAP in left sequence = ALIGN both positions One path from the previous table: Corresponding alignment (start at the end):
AC--TCGACAGTAG
Score = +2

Intro to Bioinformatics – Sequence Alignment 26
Algorithm AnalysisAlgorithm Analysis Brute force approach
• If the length of both sequences is n, number of possibility = C(2n, n) = (2n)!/(n!)2 22n / (n)1/2, using Sterling’s approximation of n! = (2n)1/2e-nnn.
• O(4n)
Dynamic programming• O(mn), where the two sequence sizes are m and n,
respectively • O(n2), if m is in the order of n

Intro to Bioinformatics – Sequence Alignment 27
Practice ProblemPractice Problem Find an optimal alignment for these two
sequences:GCGGTTGCGT
Match: +1 Mismatch: 0 Gap: –1
g c g g t t0 -1 -2 -3 -4 -5 -6
g -1c -2g -3t -4

Intro to Bioinformatics – Sequence Alignment 28
Practice ProblemPractice Problem Find an optimal alignment for these two
sequences:GCGGTTGCGT g c g g t t
0 -1 -2 -3 -4 -5 -6g -1 1 0 -1 -2 -3 -4c -2 0 2 1 0 -1 -2g -3 -1 1 3 2 1 0t -4 -2 0 2 3 3 2
GCGGTTGCG-T-
Score = +2

Intro to Bioinformatics – Sequence Alignment 29
g c g0 -1 -2 -3
g -1 1 0 -1g -2 0 1 1c -3 -1 1 1g -4 -2 0 2
Semi-global alignmentSemi-global alignment Suppose we are aligning:GCGGGCG
Which do you prefer?G-CG -GCGGGCG GGCG
Semi-global alignment allows gaps at the ends for free.

Intro to Bioinformatics – Sequence Alignment 30
Semi-global alignmentSemi-global alignment
g c g0 0 0 0
g 0 1 0 1g 0 1 1 1c 0 0 2 1g 0 1 1 3
Semi-global alignment allows gaps at the ends for free.
Initialize first row and column to all 0’s Allow free horizontal/vertical moves in last row
and column

Intro to Bioinformatics – Sequence Alignment 31
Local alignmentLocal alignment Global alignments – score the entire alignment Semi-global alignments – allow unscored gaps
at the beginning or end of either sequence Local alignment – find the best matching
subsequence CGATGAAATGGA
This is achieved by allowing a 4th alternative at each position in the table: zero.

Intro to Bioinformatics – Sequence Alignment 32
Local Sequence Alignment Local Sequence Alignment Why local sequence alignment?
• Subsequence comparison between a DNA sequence and a genome
• Protein function domains• Exons matching
Smith-Waterman algorithm
Di,j = max{ Di-1,j + d(Ai, –), Di,j-1 + d(–,Bj), Di-1,j-1 + d(Ai,Bj), 0 }
Initialization: D1,j = , Di,1 =
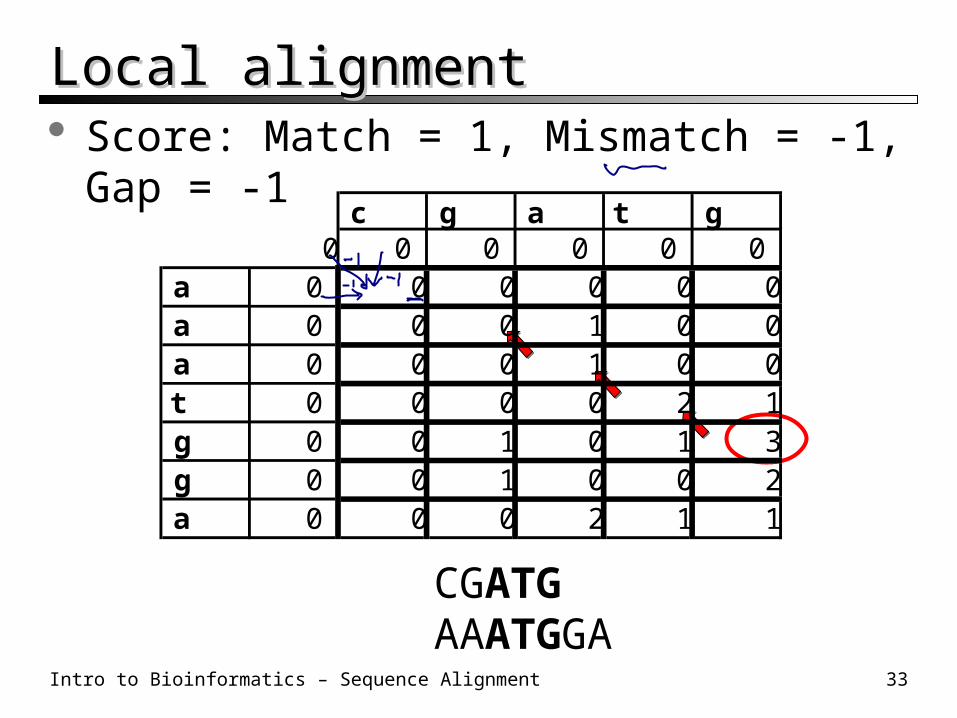
Intro to Bioinformatics – Sequence Alignment 33
Local alignmentLocal alignment Score: Match = 1, Mismatch = -1, Gap = -1
CGATGAAATGGA
c g a t g0 0 0 0 0 0
a 0 0 0 0 0 0a 0 0 0 1 0 0a 0 0 0 1 0 0t 0 0 0 0 2 1g 0 0 1 0 1 3g 0 0 1 0 0 2a 0 0 0 2 1 1
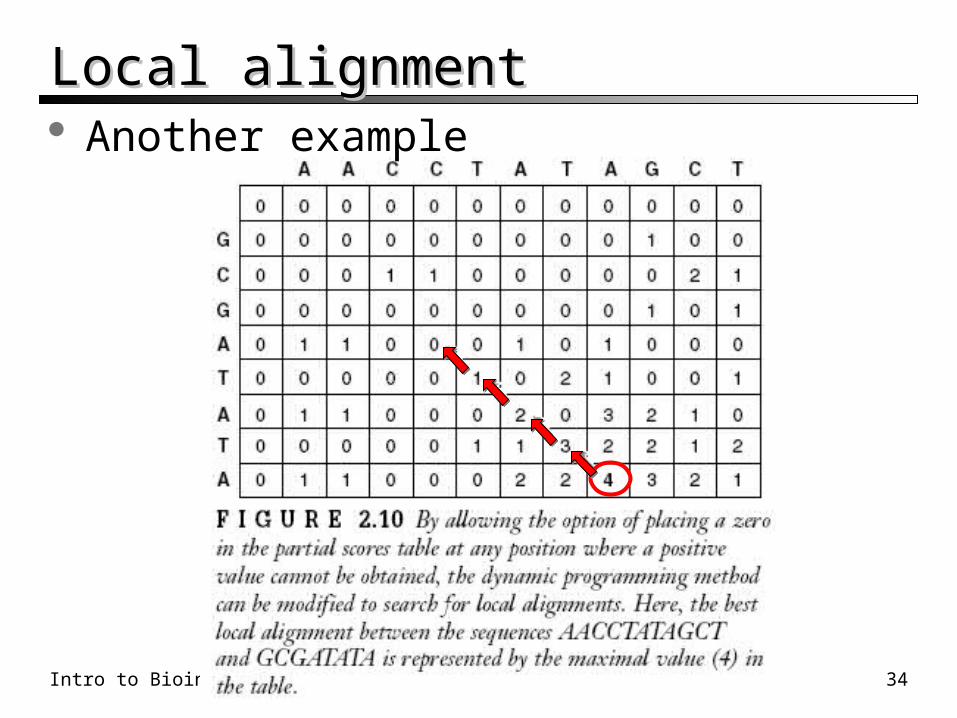
Intro to Bioinformatics – Sequence Alignment 34
Local alignmentLocal alignment Another example
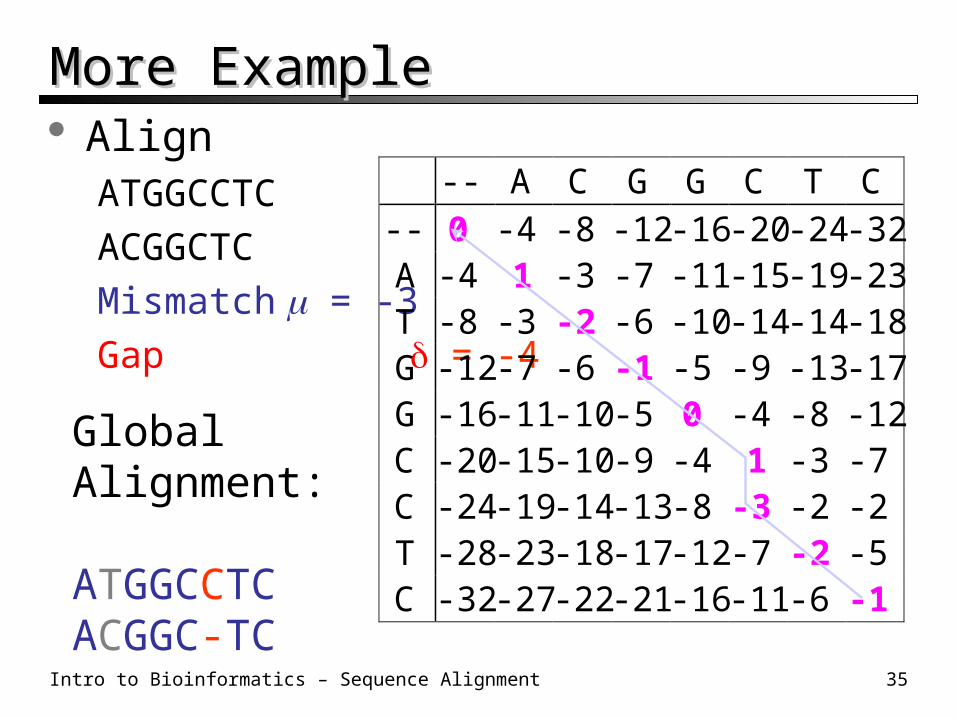
Intro to Bioinformatics – Sequence Alignment 35
More ExampleMore Example Align
ATGGCCTC
ACGGCTC
Mismatch = -3
Gap = -4
-- A C G G C T C
0 -4 -8 -12 -16 -20 -24 -321 -3
-3
--ATGGCCTC
-4-8-12-16-20-24-28-32
-7 -11 -15 -19 -23-2 -6 -10 -14 -14 -18
-7 -6 -1 -5 -9 -13 -17-11 -10 -5 0 -4 -8 -12-15 -10 -9 -4 1 -3 -7-19 -14 -13 -8 -3 -2 -2-23 -18 -17 -12 -7 -2 -5-27 -22 -21 -16 -11 -6 -1
GlobalAlignment:
ATGGCCTCACGGC-TC

Intro to Bioinformatics – Sequence Alignment 36
More ExampleMore Example
LocalAlignment:
ATGGCCTCACGG CTC
or
ATGGCCTCACGGC TC
-- A C G G C T C
0 0 0 0 0 0 0 01 00
--ATGGCCTC
00000000
0 0 0 0 00 0 0 0 1 0
0 0 1 1 0 0 00 0 1 2 0 0 00 1 0 0 3 0 10 1 0 0 1 0 10 0 0 0 0 2 00 1 0 0 1 0 3

Intro to Bioinformatics – Sequence Alignment 37
Scoring Matrices for DNA SequencesScoring Matrices for DNA Sequences Transition: A G C T Transversion: a purine (A or G) is replaced by a
pyrimadine (C or T) or vice versa
Intro to Bioinformatics – Sequence Alignment 38
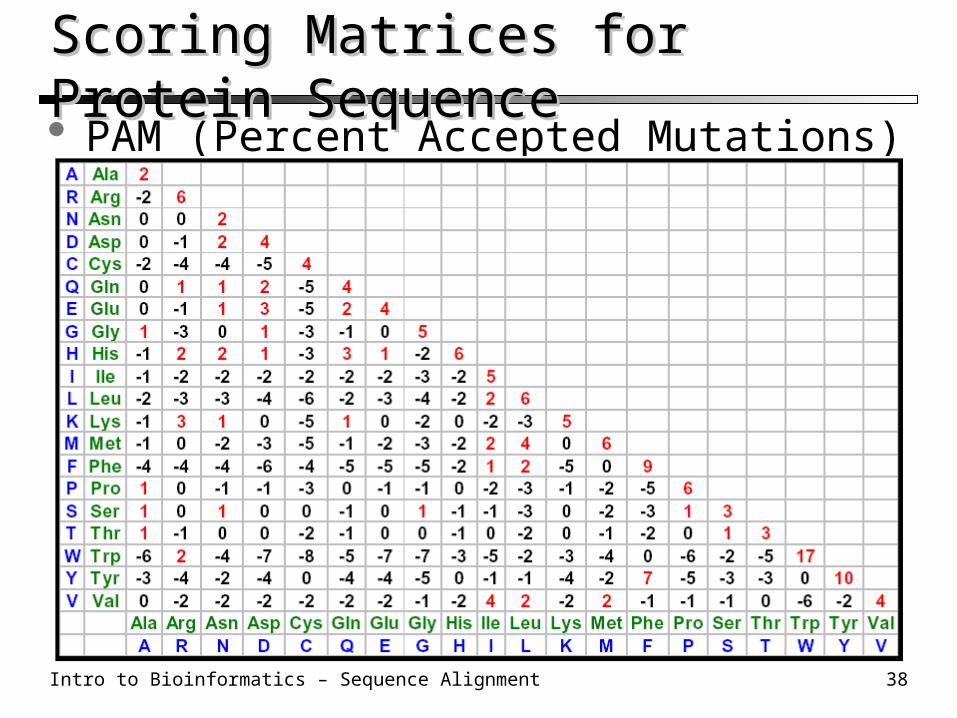
Scoring Matrices for Protein Sequence Scoring Matrices for Protein Sequence PAM (Percent Accepted Mutations) 250
Intro to Bioinformatics – Sequence Alignment 39
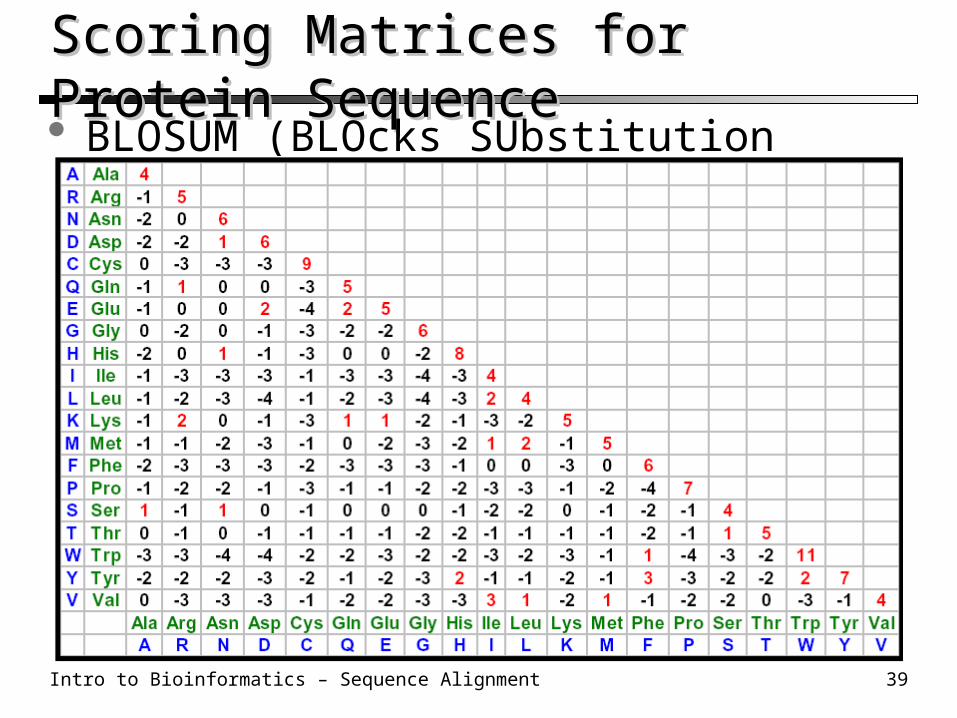
Scoring Matrices for Protein Sequence Scoring Matrices for Protein Sequence BLOSUM (BLOcks SUbstitution Matrix) 62

Intro to Bioinformatics – Sequence Alignment 40
Using Protein Scoring MatricesUsing Protein Scoring Matrices Divergence
BLOSUM 80 BLOSUM 62 BLOSUM 45PAM 1 PAM 120 PAM 250Closely related Distantly relatedLess divergent More divergentLess sensitive More sensitive
Looking for• Short similar sequences → use less sensitive matrix• Long dissimilar sequences → use more sensitive matrix• Unknown → use range of matrices
Comparison• PAM – designed to track evolutionary origin of proteins• BLOSUM – designed to find conserved regions of proteins





























