Embed Size (px)
Citation preview

Sensitivity to DNA Damage Is a Common Componentof Hormone-Based Strategies for Protectionof the Mammary Gland
Yifan Tu,1,3 D. Joseph Jerry,1,2,3 Brooke Pazik,3 and Sallie Smith Schneider1,2,3
1Molecular and Cellular Biology Program and 2Department of Veterinary and Animal Sciences, University ofMassachusetts, Amherst, Massachusetts, and 3Pioneer Valley Life Sciences Institute Baystate MedicalCenter/UMass Biomedical Research Institute, Springfield, Massachusetts
AbstractAn early full-term pregnancy significantly reduces the
risk of getting breast cancer in women. In animals, this
protection can be mimicked by a short-term exposure to
physiologic doses of estrogen plus progesterone.
Sensitization of p53 and up-regulation of transforming
growth factor B are believed to be important aspects of
the mechanism by which protection is imparted. Little is
known, however, about the use of this pathway in
response to other chemopreventive agents. In this
article, we investigated the ability of retinoids, such
as 9-cis retinoic acid, all-trans retinoic acid, and
N-4-hydroxyphenylretinamide (4-HPR), to sensitize the
ductal epithelial cells of virgin mammary glands to DNA
damage responses. Using a whole-organ culture system,
we observed enhanced cell death in response to
;-irradiation in the virgin tissues treated with retinoids
for 72 hours. These retinoids were partially dependent
on p53 and transforming growth factor B to exert their
radiosensitizing effects. However, 4-HPR seemed to
sensitize other cells or activate these pathways in a
different manner as costimulation with ovarian
hormones and 4-HPR was additive, whereas coculture of
ovarian hormones and the natural retinoids did not
increase amount of death. Taken together, these data
suggest that sensitization of the mammary epithelium to
p53-dependent apoptosis is a common pathway, which
is engaged by retinoids as well as ovarian hormones.
(Mol Cancer Res 2005;3(8):435–42)
IntroductionBreast cancer is the most common cancer among women in
the Western countries. Risk factors include hormonal alterations
such as early menarche, late menopause, late first pregnancy, or
prolonged use of hormone replacement therapy (1). However,
the risk of developing breast cancer can be reduced. A full-term
pregnancy before 20 years of age is the most effective natural
protection against breast cancer in women. This protection
occurs universally among women with different ethnic back-
grounds. This indicates that the protective effect does not result
from environmental or socioeconomic factors, but from a
permanent biological change within the breast caused by
pregnancy (2-4). During pregnancy, the mammary gland is
exposed to the highest physiologic concentrations of ovarian
steroids (estradiol and progesterone), pituitary hormone (pro-
lactin and growth hormone), and placental (hCG as well as
placental lactogens) hormone, which cause proliferation and
differentiation of the mammary gland (5-7). In rats and mice,
the protection of pregnancy can be mimicked by short-term
exposure to physiologic doses of estrogen plus progesterone
(8). It has been hypothesized that hormone stimulation at a
critical stage of mammary development causes cells to change
both in the regulatory loops governing proliferation and in the
response to DNA damage (9).
Persistent molecular changes induced by parity or hormones
in mammary gland have been noted in several laboratories. With
the use of DNA microarrays, D’Cruz et al. (2) found that a full-
term pregnancy results in the persistent up-regulation of
transforming growth factor (TGF) h3, a growth-inhibitory
molecule. Ginger et al. (10) noted changes in the chromatin
remodeling gene RbAp46 in response to estrogen and
progesterone. Sivaraman and coworkers found that in Wistar-
Furth rats as well as in BALB/c mice, a 21-day estrogen +
progesterone treatment induced the up-regulation and nuclear
accumulation of p53 protein. The elevation of p53 was persistent
28 days after hormone withdrawal. Pregnancy induced a similar
pattern of expression (11). Minter and colleagues strengthened
these findings. They showed that p53-mediated apoptosis in
response to g-radiation was greater in proliferating or estrogen +
progesterone–treated mammary glands than in glands of virgin
mice (12). Based on these findings, a model has been proposed
that p53 activity is impaired during specific periods of mammary
gland development, causing the mammary epithelium to be
susceptible to cancer. However, prolonged ovarian hormone
stimulation, such as during pregnancy, serves to sensitize p53 to
the effects of DNA damage. The responsiveness of the p53
pathway to the changes of the endocrine environment associated
with pregnancy renders the mammary epithelium refractory to
tumors (13).
The main function of activated p53 is to arrest cell cycle and
induce apoptosis in response to DNA damage. Medina et al.
showed that the deletion of the tumor suppressor gene p53
Received 3/31/05; revised 7/19/05; accepted 7/21/05.Grant support: Rays of Hope at the Comprehensive Breast Center in theBaystate Health System.The costs of publication of this article were defrayed in part by the payment ofpage charges. This article must therefore be hereby marked advertisement inaccordance with 18 U.S.C. Section 1734 solely to indicate this fact.Requests for reprints: Yifan Tu, University of Massachussetts at Amherst,Amherst, MA 01107. Phone: 413-695-1679. E-mail: [email protected] D 2005 American Association for Cancer Research.doi:10.1158/1541-7786.MCR-05-0038
Mol Cancer Res 2005;3(8). August 2005 435
results in enhanced tumorigenic risk. They found that the
mammary gland from the p53-wild-type and p53-null mice
exhibited the same dependence on ovarian hormones for
growth. However, estrogen and progesterone strongly enhanced
carcinogenesis in the p53-null mammary epithelium (14). This
suggests that p53 may be a critical factor for protection of the
mammary gland from further accumulation of mutations.
Furthermore, recent studies have indicated that pregnancy does
not impart protection in p53-null mice, suggesting that p53 is
required, in part, for the protective response imparted by
hormones (15).
Although an early full-term pregnancy can reduce the risk of
women developing breast cancer, it is not a practical approach
to breast cancer protection. Chemoprevention is defined as the
use of specific chemical substances (natural or synthetic) or
their mixtures to suppress, retard, or reverse the process of
carcinogenesis. Among these chemicals, the retinoids (deriva-
tives of vitamin A) have shown great promise in the area of
cancer chemotherapy and chemoprevention (16). The most
extensively studied native retinoids in cancer medicine are all-
trans retinoic acid (RA) and 9-cis RA. In humans, all-trans RA
has been used to treat acute promyelocytic leukemia with great
success. All-trans retinoids, alone or in combination with other
drugs, also showed activity in juvenile chronic myeloid
leukemia (17). Retinoids inhibit breast cancer growth and
augment the action of other breast cancer cell growth inhibitors
both in vivo and in vitro (18). However, retinoids cause serious
side effects at high doses (19), and thus more selective retinoids
have been designed (20). Among these is N-4-hydroxyphenyl-
retinamide (4-HPR), a synthetic retinoid that has been
examined as a cancer chemopreventive agent (21). A phase
III clinical breast cancer trial found a beneficial trend with 4-
HPR in the protection of premenopausal women from
contralateral and ipsilateral breast cancer (22).
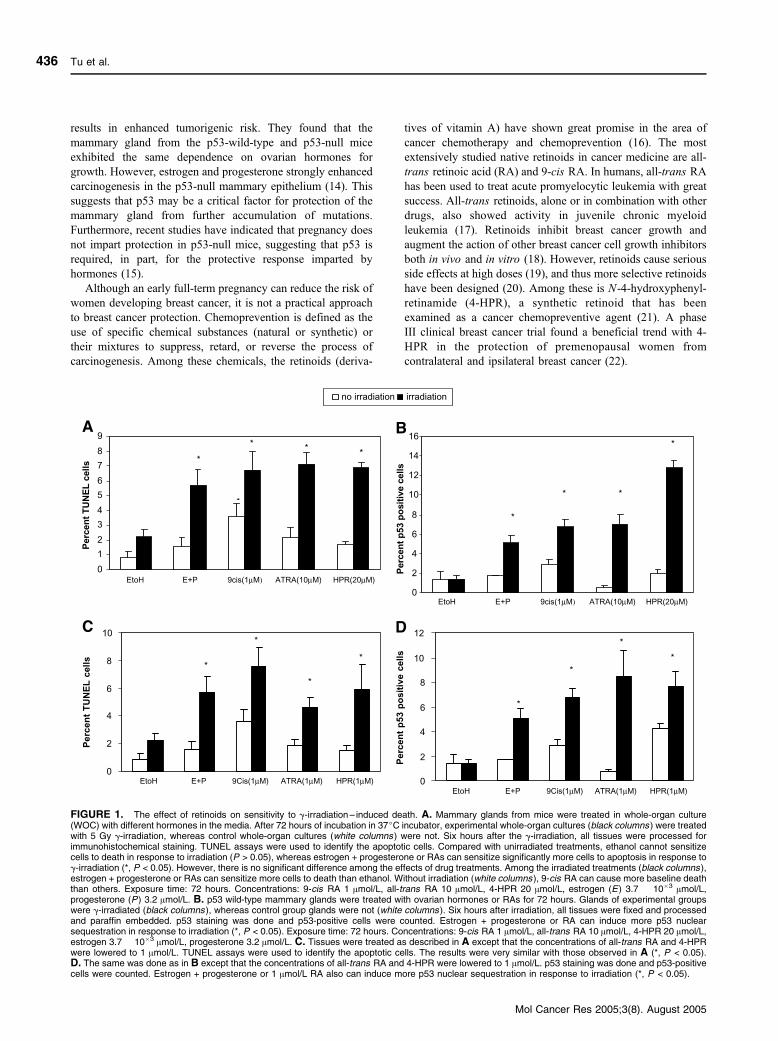
FIGURE 1. The effect of retinoids on sensitivity to g-irradiation – induced death. A. Mammary glands from mice were treated in whole-organ culture(WOC) with different hormones in the media. After 72 hours of incubation in 37jC incubator, experimental whole-organ cultures (black columns ) were treatedwith 5 Gy g-irradiation, whereas control whole-organ cultures (white columns ) were not. Six hours after the g-irradiation, all tissues were processed forimmunohistochemical staining. TUNEL assays were used to identify the apoptotic cells. Compared with unirradiated treatments, ethanol cannot sensitizecells to death in response to irradiation (P > 0.05), whereas estrogen + progesterone or RAs can sensitize significantly more cells to apoptosis in response tog-irradiation (*, P < 0.05). However, there is no significant difference among the effects of drug treatments. Among the irradiated treatments (black columns ),estrogen + progesterone or RAs can sensitize more cells to death than ethanol. Without irradiation (white columns ), 9-cis RA can cause more baseline deaththan others. Exposure time: 72 hours. Concentrations: 9-cis RA 1 Amol/L, all-trans RA 10 Amol/L, 4-HPR 20 Amol/L, estrogen (E) 3.7 � 10�3 Amol/L,progesterone (P) 3.2 Amol/L. B. p53 wild-type mammary glands were treated with ovarian hormones or RAs for 72 hours. Glands of experimental groupswere g-irradiated (black columns ), whereas control group glands were not (white columns ). Six hours after irradiation, all tissues were fixed and processedand paraffin embedded. p53 staining was done and p53-positive cells were counted. Estrogen + progesterone or RA can induce more p53 nuclearsequestration in response to irradiation (*, P < 0.05). Exposure time: 72 hours. Concentrations: 9-cis RA 1 Amol/L, all-trans RA 10 Amol/L, 4-HPR 20 Amol/L,estrogen 3.7 � 10�3 Amol/L, progesterone 3.2 Amol/L. C. Tissues were treated as described in A except that the concentrations of all-trans RA and 4-HPRwere lowered to 1 Amol/L. TUNEL assays were used to identify the apoptotic cells. The results were very similar with those observed in A (*, P < 0.05).D. The same was done as in B except that the concentrations of all-trans RA and 4-HPR were lowered to 1 Amol/L. p53 staining was done and p53-positivecells were counted. Estrogen + progesterone or 1 Amol/L RA also can induce more p53 nuclear sequestration in response to irradiation (*, P < 0.05).
Tu et al.
Mol Cancer Res 2005;3(8). August 2005
436

Retinoids signal through multiple pathways and seem to exert
protection at numerous levels. Pathways shared by the natural
and synthetic retinoid family seem to include an increased
expression and activation of TGF-h family members (23, 24).
Activation of p53 is another pathway by which retinoids may
exert their effects. Numerous examples have been cited which
indicate that retinoid exposure leads to the induction of p53
stability and transcriptional activities (25-28).
In this article, we addressed the question of whether natural
or synthetic retinoids can sensitize the ductal epithelial cells to
p53-dependent death induced by irradiation. We showed that
DNA damage–induced death by these retinoids is partially
dependent on p53 and TGF-h.
ResultsRetinoids Sensitize Ductal Epithelial Cells to Apoptosis inResponse to Radiation
Ionizing radiation is a complete mutagen and is a known
carcinogen of human breast and rodent mammary glands (29).
Here, we treated mouse mammary glands with g-radiation to
damage DNA and impart a response.
In Fig. 1A, terminal deoxyribonucleotidyl transferase–
mediated dUTP nick-end labeling (TUNEL) assays were used
to detect apoptotic cells. In response to DNA damage, virgin
BALB/c-p53+/+ mammary glands treated with ethanol failed to
induce a significant apoptotic response following irradiation
compared with unirradiated tissues (P > 0.05). However, when
the glands were treated with estrogen + progesterone or with all-
trans (10 Amol/L), 9-cis RA (1 Amol/L), or 4-HPR (20 Amol/L)
for 72 hours, the percentage of apoptotic cells in the irradiated
glands was increased significantly compared with the irradiated
ethanol-treated glands. There was, however, no difference
among effects of drug treatments (P > 0.05). Interestingly, in
the absence of irradiation, 9-cis RA caused more baseline death
than others, suggesting that it can cause DNA damage or stress
response which can mediate death. However, irradiation still
significantly further increased death. All the treatments caused a
sensitization to radiation-induced death that was statistically
significant (P < 0.05). This suggests that all three RAs can
sensitize ductal epithelial cells to respond to DNA damage.
To determine if p53 can respond to radiation in virgin tissue
exposed to retinoids, we examined the nuclear accumulation of
FIGURE 1 continued. E.Photos were taken of theglands with the treatmentsdescribed in A and B. Thedark brown stained cells wereTUNEL- or p53-positive cells;the green cells were negativecells counterstained withmethyl green.
Sensitivity to DNA Damage and Hormone-Based Strategies
Mol Cancer Res 2005;3(8). August 2005
437
p53 protein in response to radiation through immunohistochemistry.
In the unirradiated tissues, nuclear accumulation of p53 was
not observed. However, in the 9-cis RA–, all-trans RA–, and
4-HPR–treated groups, subsequent radiation caused significant
nuclear accumulation of the p53 protein (P < 0.05) compared
with the ethanol-treated group. No significant difference was
found when comparing the efficiencies of all-trans RA and
estrogen + progesterone or 9-cis RA and estrogen +
progesterone, but 4-HPR was found to be more effective than
estrogen + progesterone (P < 0.05). Because the pharmacologic
significances of high doses of retinoids (10 Amol/L all-trans
RA and 20 Amol/L 4-HPR) were not clear, we investigated the
effects of retinoids at lower doses (1 Amol/L all-trans RA and
4-HPR). In Fig. 1C and D, we can see the same effect of
retinoids at lower doses.
4-HPR and Estrogen + Progesterone Work AdditivelyBecause all three retinoids, as well as the ovarian hormones,
sensitize glands to cell death in response to DNA damage, we
were interested to determine whether overlapping or distinct
pathways were used to sensitize p53 and the cells to DNA
damage–induced cell death. If the pathways were distinct, then
we would expect to see an additive response. However, if they
were similar, we would expect to see no augmentation of the
response when both stimuli were presented together. We treated
virgin mammary glands with retinoids and ovarian hormones
for 72 hours, and then irradiated the tissue. Figure 2 shows that,
in response to radiation, estrogen plus progesterone sensitized
5.7% cells to apoptosis in ductal epithelial cells. All-trans RA
or 9-cis RA alone sensitized 7.1% and 7.6% cells to death,
respectively; however, the levels of apoptosis did not increase
when glands were treated with estrogen plus progesterone along
with all-trans and 9-cis RAs. In contrast, the response to
4-HPR with estrogen + progesterone seemed to be additive.
When we treated glands with estrogen + progesterone and
4-HPR, 9.4% cells were apoptotic, whereas only 6.9% cells
(statistically significant, P < 0.05) were apoptotic when glands
were treated with 4-HPR alone and only 5.7% cells (statistically
significant, P < 0.05) were apoptotic when glands were treated
with estrogen + progesterone. This suggests that 4-HPR
sensitizes cells to death through a distinct pathway from the
one used by the natural retinoids and ovarian hormones.
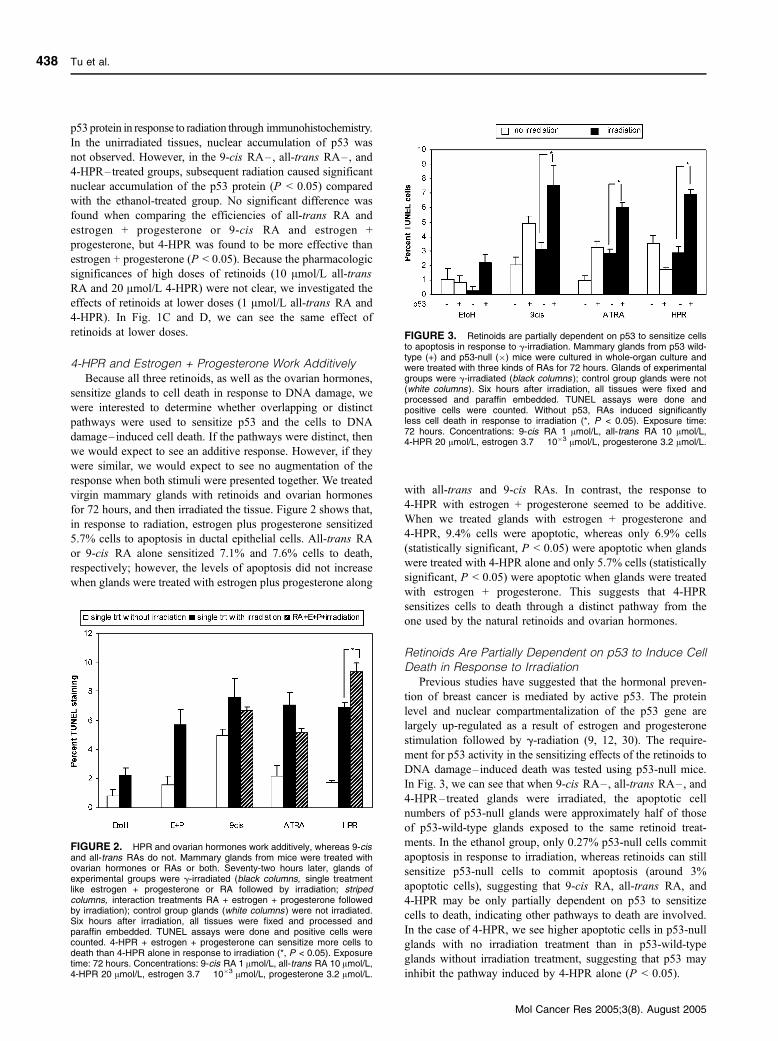
Retinoids Are Partially Dependent on p53 to Induce CellDeath in Response to Irradiation
Previous studies have suggested that the hormonal preven-
tion of breast cancer is mediated by active p53. The protein
level and nuclear compartmentalization of the p53 gene are
largely up-regulated as a result of estrogen and progesterone
stimulation followed by g-radiation (9, 12, 30). The require-
ment for p53 activity in the sensitizing effects of the retinoids to
DNA damage–induced death was tested using p53-null mice.
In Fig. 3, we can see that when 9-cis RA–, all-trans RA–, and
4-HPR–treated glands were irradiated, the apoptotic cell
numbers of p53-null glands were approximately half of those
of p53-wild-type glands exposed to the same retinoid treat-
ments. In the ethanol group, only 0.27% p53-null cells commit
apoptosis in response to irradiation, whereas retinoids can still
sensitize p53-null cells to commit apoptosis (around 3%
apoptotic cells), suggesting that 9-cis RA, all-trans RA, and
4-HPR may be only partially dependent on p53 to sensitize
cells to death, indicating other pathways to death are involved.
In the case of 4-HPR, we see higher apoptotic cells in p53-null
glands with no irradiation treatment than in p53-wild-type
glands without irradiation treatment, suggesting that p53 may
inhibit the pathway induced by 4-HPR alone (P < 0.05).
FIGURE 2. HPR and ovarian hormones work additively, whereas 9-cisand all-trans RAs do not. Mammary glands from mice were treated withovarian hormones or RAs or both. Seventy-two hours later, glands ofexperimental groups were g-irradiated (black columns, single treatmentlike estrogen + progesterone or RA followed by irradiation; stripedcolumns, interaction treatments RA + estrogen + progesterone followedby irradiation); control group glands (white columns ) were not irradiated.Six hours after irradiation, all tissues were fixed and processed andparaffin embedded. TUNEL assays were done and positive cells werecounted. 4-HPR + estrogen + progesterone can sensitize more cells todeath than 4-HPR alone in response to irradiation (*, P < 0.05). Exposuretime: 72 hours. Concentrations: 9-cis RA 1 Amol/L, all-trans RA 10 Amol/L,4-HPR 20 Amol/L, estrogen 3.7 � 10�3 Amol/L, progesterone 3.2 Amol/L.
FIGURE 3. Retinoids are partially dependent on p53 to sensitize cellsto apoptosis in response to g-irradiation. Mammary glands from p53 wild-type (+) and p53-null (�) mice were cultured in whole-organ culture andwere treated with three kinds of RAs for 72 hours. Glands of experimentalgroups were g-irradiated (black columns ); control group glands were not(white columns ). Six hours after irradiation, all tissues were fixed andprocessed and paraffin embedded. TUNEL assays were done andpositive cells were counted. Without p53, RAs induced significantlyless cell death in response to irradiation (*, P < 0.05). Exposure time:72 hours. Concentrations: 9-cis RA 1 Amol/L, all-trans RA 10 Amol/L,4-HPR 20 Amol/L, estrogen 3.7 � 10�3 Amol/L, progesterone 3.2 Amol/L.
Tu et al.
Mol Cancer Res 2005;3(8). August 2005
438
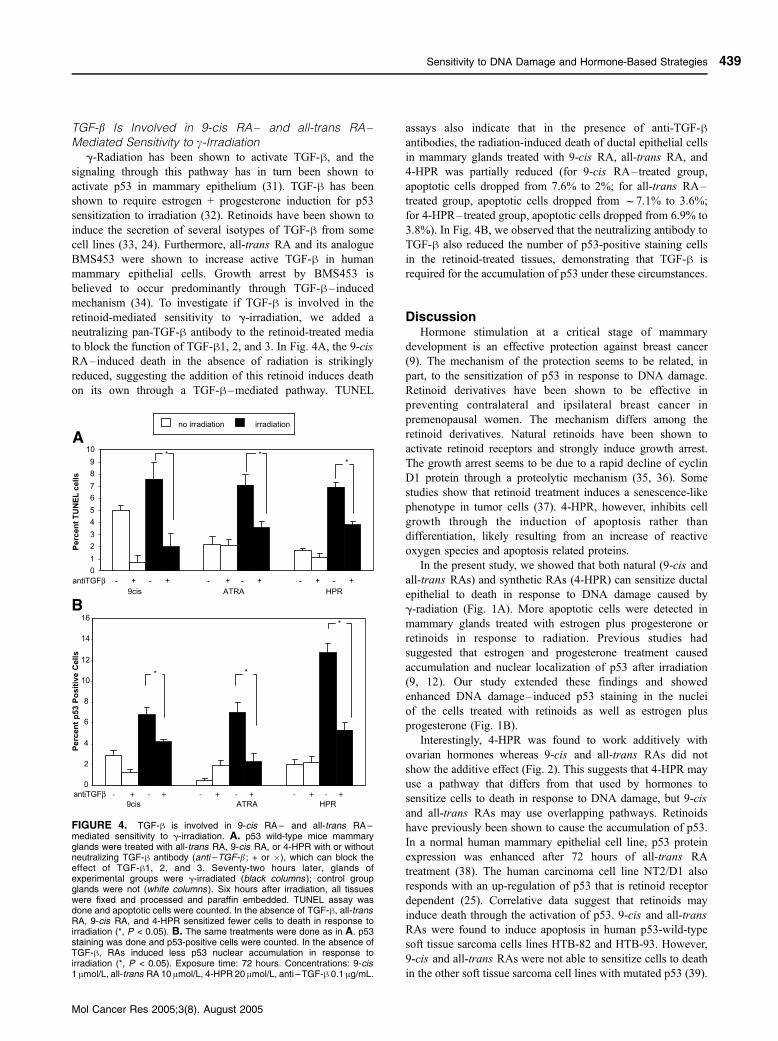
TGF-b Is Involved in 9-cis RA– and all-trans RA–Mediated Sensitivity to c-Irradiation
g-Radiation has been shown to activate TGF-h, and the
signaling through this pathway has in turn been shown to
activate p53 in mammary epithelium (31). TGF-h has been
shown to require estrogen + progesterone induction for p53
sensitization to irradiation (32). Retinoids have been shown to
induce the secretion of several isotypes of TGF-h from some
cell lines (33, 24). Furthermore, all-trans RA and its analogue
BMS453 were shown to increase active TGF-h in human
mammary epithelial cells. Growth arrest by BMS453 is
believed to occur predominantly through TGF-h–inducedmechanism (34). To investigate if TGF-h is involved in the
retinoid-mediated sensitivity to g-irradiation, we added a
neutralizing pan-TGF-h antibody to the retinoid-treated media
to block the function of TGF-h1, 2, and 3. In Fig. 4A, the 9-cis
RA–induced death in the absence of radiation is strikingly
reduced, suggesting the addition of this retinoid induces death
on its own through a TGF-h–mediated pathway. TUNEL
assays also indicate that in the presence of anti-TGF-hantibodies, the radiation-induced death of ductal epithelial cells
in mammary glands treated with 9-cis RA, all-trans RA, and
4-HPR was partially reduced (for 9-cis RA–treated group,
apoptotic cells dropped from 7.6% to 2%; for all-trans RA–
treated group, apoptotic cells dropped from f7.1% to 3.6%;
for 4-HPR–treated group, apoptotic cells dropped from 6.9% to
3.8%). In Fig. 4B, we observed that the neutralizing antibody to
TGF-h also reduced the number of p53-positive staining cells
in the retinoid-treated tissues, demonstrating that TGF-h is
required for the accumulation of p53 under these circumstances.
DiscussionHormone stimulation at a critical stage of mammary
development is an effective protection against breast cancer
(9). The mechanism of the protection seems to be related, in
part, to the sensitization of p53 in response to DNA damage.
Retinoid derivatives have been shown to be effective in
preventing contralateral and ipsilateral breast cancer in
premenopausal women. The mechanism differs among the
retinoid derivatives. Natural retinoids have been shown to
activate retinoid receptors and strongly induce growth arrest.
The growth arrest seems to be due to a rapid decline of cyclin
D1 protein through a proteolytic mechanism (35, 36). Some
studies show that retinoid treatment induces a senescence-like
phenotype in tumor cells (37). 4-HPR, however, inhibits cell
growth through the induction of apoptosis rather than
differentiation, likely resulting from an increase of reactive
oxygen species and apoptosis related proteins.
In the present study, we showed that both natural (9-cis and
all-trans RAs) and synthetic RAs (4-HPR) can sensitize ductal
epithelial to death in response to DNA damage caused by
g-radiation (Fig. 1A). More apoptotic cells were detected in
mammary glands treated with estrogen plus progesterone or
retinoids in response to radiation. Previous studies had
suggested that estrogen and progesterone treatment caused
accumulation and nuclear localization of p53 after irradiation
(9, 12). Our study extended these findings and showed
enhanced DNA damage–induced p53 staining in the nuclei
of the cells treated with retinoids as well as estrogen plus
progesterone (Fig. 1B).
Interestingly, 4-HPR was found to work additively with
ovarian hormones whereas 9-cis and all-trans RAs did not
show the additive effect (Fig. 2). This suggests that 4-HPR may
use a pathway that differs from that used by hormones to
sensitize cells to death in response to DNA damage, but 9-cis
and all-trans RAs may use overlapping pathways. Retinoids
have previously been shown to cause the accumulation of p53.
In a normal human mammary epithelial cell line, p53 protein
expression was enhanced after 72 hours of all-trans RA
treatment (38). The human carcinoma cell line NT2/D1 also
responds with an up-regulation of p53 that is retinoid receptor
dependent (25). Correlative data suggest that retinoids may
induce death through the activation of p53. 9-cis and all-trans
RAs were found to induce apoptosis in human p53-wild-type
soft tissue sarcoma cells lines HTB-82 and HTB-93. However,
9-cis and all-trans RAs were not able to sensitize cells to death
in the other soft tissue sarcoma cell lines with mutated p53 (39).
FIGURE 4. TGF-h is involved in 9-cis RA – and all-trans RA –mediated sensitivity to g-irradiation. A. p53 wild-type mice mammaryglands were treated with all-trans RA, 9-cis RA, or 4-HPR with or withoutneutralizing TGF-h antibody (anti –TGF-b ; + or �), which can block theeffect of TGF-h1, 2, and 3. Seventy-two hours later, glands ofexperimental groups were g-irradiated (black columns ); control groupglands were not (white columns ). Six hours after irradiation, all tissueswere fixed and processed and paraffin embedded. TUNEL assay wasdone and apoptotic cells were counted. In the absence of TGF-h, all-transRA, 9-cis RA, and 4-HPR sensitized fewer cells to death in response toirradiation (*, P < 0.05). B. The same treatments were done as in A. p53staining was done and p53-positive cells were counted. In the absence ofTGF-h, RAs induced less p53 nuclear accumulation in response toirradiation (*, P < 0.05). Exposure time: 72 hours. Concentrations: 9-cis1 Amol/L, all-trans RA 10 Amol/L, 4-HPR 20 Amol/L, anti –TGF-h 0.1 Ag/mL.
Sensitivity to DNA Damage and Hormone-Based Strategies
Mol Cancer Res 2005;3(8). August 2005
439

A study done with the subclones of the retinoid-sensitive
acute myeloblastic leukemia (OCI/AML-2) cell lines showed
9-cis RA had the most prominent effect of inducing apoptosis
and inhibiting cell growth compared with all-trans and 13-cis
RAs and that 9-cis RA was most effective at causing the
translocation of p53 from cytosol to nucleus (27). Because the
p53 pathway is involved in the protective response imparted
by hormones (14), and retinoids in cell lines have been shown
to induce p53 stability and transcriptional activity (38), we
further investigated the relationship between p53 and retinoids
in a whole-organ culture system. Mammary glands from wild-
type and p53 knockout mice were pretreated with retinoids
and then irradiated. The numbers of apoptotic cells were
compared with those of the p53-wild-type mammary glands
given the same treatments. From Fig. 3, we can see that
without p53, apoptosis was greatly reduced in response to
g-radiation. However, all three retinoids could still induce
death in f3% of ductal epithelial cells. This suggests that
9-cis RA, all-trans RA, and 4-HPR are able to sensitize cells
to irradiation-induced death through p53-dependent and
-independent pathways.
TGF-h inhibits cell proliferation and has been shown to be
associated with death in many kinds of epithelial cells. TGF-h1has been found to be rapidly activated in response to ionizing
radiation. Barcellos-Hoff et al. found that radiation-induced
apoptosis was a TGF-h1–dependent process in the mammary
epithelium. In addition, they found that glands from TGF-h1knockout animals or addition of TGF-h1 neutralizing anti-
bodies resulted in less p53 activation in response to g-radiation
(31). Previous studies in cell lines have indicated that retinoids
can also induce the activity of TGF-h (34, 40). All-trans RA
and its analogue BM453 were found to inhibit proliferation and
induce more total and active TGF-h in normal human
mammary epithelial cells (34). To investigate the involvement
of TGF-h in the cell death induced by RAs, we used
neutralizing TGF-h antibody to block the TGF-h functioning
to see if there were less apoptotic cells and p53-positive cells. In
Fig. 4A, we observed that with neutralizing TGF-h antibody,
the background death observed in response to 9-cis RA was
reduced, suggesting that 9-cis RA causes activation of TGF-hin the mammary gland and that this is responsible for the death
observed in the absence of radiation. Reduced cell death was
observed in response to radiation with all the retinoid
treatments. p53 staining on the same samples indicated that
the neutralizing TGF-h antibody decreased the p53-positive cell
numbers of 9-cis RA–, all-trans RA–, and 4-HPR–treated
tissues followed by irradiation. This suggests that all these
retinoids can stabilize or up-regulate p53 through TGF-h in the
gland and that this pathway is partially responsible for the
increased amount of death. Examples of other pathways which
might be involved in the induction of death include the up-
regulation of reactive oxygen species, retinoic acid receptor
h, and other apoptosis-related genes such as c-myc and c-jun
(36, 41).
In summary, our study showed that sensitization of the
mammary gland to radiation-induced responses may be a very
important pathway shared by numerous agents that act in
protecting the mammary gland. Retinoids, as well as ovarian
hormones, can sensitize p53 such that radiation will activate
TGF-h and result in the accumulation and activation of the p53
protein and death. Unlike ovarian hormones, retinoids may also
contain the ability to sensitize cells via other pathways,
particularly in the case of 4-HPR. The ability of 4-HPR to
enhance the death response in combination with other agents
suggests that it may use a different pathway or can affect cells
which are not responsive to the other agents. This is an
important observation because it indicates that 4-HPR may be
used in combination with other chemopreventive agents to
enhance protection of the mammary gland. This requirement for
both p53 and TGF-h in the death induced by 4-HPR suggests
that the activation of p53 by TGF-h is likely to be downstream
of the convergence of the retinoid pathway on the sensitization
of p53. Future studies will determine if ovarian hormones use
the retinoid pathway for the sensitization of p53 or if the
ovarian hormones and retinoids are acting on similar effectors.
In conclusion, we are able to show that multiple chemo-
preventive agents are able to sensitize p53 in the mammary
gland to death induced by radiation. This suggests that the
responsiveness of p53 is a critical protein target for chemo-
preventive strategies.
Materials and MethodsAnimals, Treatments, and Tissue Procedures
All procedures were conducted in accordance with the NIH
Guide for the Care and Use of Laboratory Animals. Virgin
BALB/c female mice, Trp53+/+ or Trp53�/�, 8 weeks of age,
were housed in plastic cages under a 12-hour dark/light cycle,
and were permitted free access to food and water. Fourth
inguinal mammary glands were removed from these mice and
were maintained in whole-organ culture for 72 hours. Then, the
glands were treated with 5 Gy of g-radiation using a 137Cs
irradiator. Six hours after g-radiation, tissues were processed
with gradient alcohol and paraffin embedded.
Whole-Organ CultureAge-matched 8-week-old virgin BALB/c-p53+/+ and BALB/
c-p53�/� mice were sacrificed with CO2 and fourth inguinal
mammary glands were isolated aseptically, cut in half
longitudinally, and then floated on siliconized lens paper in
60 mm tissue culture dishes containing 2 mL of medium (12).
Basic medium was made of serum-free DMEM/F12 buffer with
HEPES and NaHCO3, plus insulin (5 Ag/mL; Sigma, St. Louis,
MO), penicillin (100 units/mL), streptomycin (100 Ag/mL), and
gentamycin (10 Ag/mL; Life Technologies, Grand Island, NY).
Hormone-supplemented media contained 1 ng/mL (3.7 � 10�3
Amol/L) estradiol (Sigma) and 1 Ag/mL (3.2 Amol/L)
progesterone. RA-supplemented medium was prepared by
adding 1 Amol/L 9-cis RA, 20 Amol/L 4-HPR (Sigma), or
10 Amol/L all-trans RA (Sigma) to the basic medium,
respectively. The retinoid plus neutralizing TGF-h antibody–
supplemented medium was prepared by adding 0.1 Ag/mL
pan-specific TGF-h antibody (R&D systems, Inc., Minneapolis,
MN) to the RA-supplemented medium at respective concen-
trations. Negative control medium was prepared by adding a
similar percentage of ethanol to the basic medium. Whole-
organ cultures were maintained in an incubator supplied with
5% CO2 in air. Seventy-two hours later, whole-organ cultures
Tu et al.
Mol Cancer Res 2005;3(8). August 2005
440

were subjected to 5 Gy of g-radiation using a 137Cs irradiator,
whereas control whole-organ cultures were not irradiated. Six
hours after irradiation, tissues were fixed in 10% neutral-
buffered formalin and then embedded in paraffin.
ImmunohistochemistryTo detect p53, 4-Am-thick slides were baked at 37jC
overnight and were deparaffinized in xylene, rehydrated in
graded ethanols, rinsed in TBS, then subjected to microwave
antigen retrieval in 0.1 mol/L citrate buffer for 5 minutes with
two sets of 1-minute intervals. Slides were stained by a Dako
autostainer with Dako Envision Anti-rabbit kit (Dako Cytoma-
tion, Carpinteria, CA). The primary antibody used was rabbit
polyclonal CM5 anti-p53 antibody (1:200, Novacastra, New-
castle upon Tyne, United Kingdom). Slides were counterstained
with methyl green, dehydrated through 100% ethanol and
xylene, and coverslipped. Internal negative controls were
treated identically, omitting primary antibody labeling on the
tissues from age- and treatment-matched p53-wild-type mice. A
minimum of 3,600 mammary gland epithelial cells were
counted per treatment (1,200 cells per culture/triplicate cultures
per treatment). Differences in the percentage of cells with p53
staining with or without g-radiation were determined using a
test of two portions with a 95% confidence interval. Differences
of the effects among drug treatments were tested by ANOVA
method and Tukey’s multiple comparison procedure at 0.05
a level.
TUNEL AssayFour-micrometer-thick slides were baked at 37jC overnight
and were deparaffinized in xylene, rehydrated in graded
ethanol, rinsed in TBS, and incubated in 3% hydrogen peroxide
in methanol. Terminal deoxynucleotidyl transferase dUTP was
used to label the nick ends of fragmented DNA (FragEL DNA
Fragmentation Kit, Oncogene Research Products, Cambridge,
MA). Sections were stained with 3,3V-diaminobenzidine and
counterstained with methyl green. A minimum of 3,600
mammary gland epithelial cells was counted per treatment
(1,200 cells per culture/triplicate cultures per treatment).
Differences in the percentage of TUNEL-positive cells with
or without g-radiation were determined using a test of two
portions with a 95% confidence interval. Differences of the
effects among drug treatments were tested by ANOVA method
and Tukey’s multiple comparison procedure at 0.05 a level.
Acknowledgments
We thank Dr. Lou Roberts and Dr. Jenny Zhang for critical reading of themanuscript.
References1. Ellisen LW, Haber DA. Recent progress in the understanding and treatment ofbreast cancer. 2001, published online.
2. D’Cruz CM, Moody SE, Master SR, et al. Persistent parity-induced changes ingrowth factors, TGF-h3, and differentiation in the rodent mammary gland. MolEndocrinol 2002;16:2034 –51.
3. Russo IH, Russo J. Developmental stage of the rat mammary gland asdeterminant of its susceptibility to 7,12-dimethylben(a)anthracene. J Natl CancerInst 1978;61:1439–49.
4. Thordarson G, Jin E, Guzman RC, Swanson SM, Nandi S, Talamantes F.Refractoriness to mammary tumorigenesis in parous rats: is it caused by persistent
changes in the hormonal environment or permanent biochemical alterations in themammary epithelia? Carcinogenesis 1995;16:2847–53.
5. Guzman RC, Yang J, Rajkumar L, et al. Hormonal prevention of breast cancer:Mimicking the protective effect of pregnancy, Proc Natl Acad Sci U S A 1999;96:2520– 5.
6. Nandi S, Guzman R, Yang J. Hormones and mammary carcinogenesis inmice, rats, and humans: a unifying hypothesis. Proc Natl Acad Sci U S A 1995;92:3650–7.
7. Russo IH, Russo J. Mammary gland neoplasia in long-term rodent studies.Environ Health Perspect 1996;104:938–67.
8. Sivaraman L, Stephens LC, Markaverich BM, et al. Hormone inducedrefractoriness to mammary carcinogenesis in Wistar-Furth rats. Carcinogenesis1998;19:1573–81.
9. Medina D, Sivaraman L, Hilsenbeck SG, et al. Mechanisms of hormonalprevention of breast cancer. Ann N Y Acad Sci 2001;952:23 – 35.
10. Ginger MR, Gonzalez-Rimbau MF, Gay JP, et al. Persistent changes in geneexpression induced by estrogen and progesterone in the rat mammary gland. MolEndocrinol 2001;15:1993–2009.
11. Sivaraman L, Conneely OM, Medina D, O’Malley BW. p53 is a potentialmediator of pregnancy and hormone-induced resistance to mammary carcino-genesis. Proc Natl Acad Sci U S A 2001;98:12379–84.
12. Minter LM, Dickinson ES, Naber SP, et al. Epithelial cells cycling predictsp53 responsiveness to g-irradiation during post-natal mammary gland develop-ment. Development 2002;129:2997–3008.
13. Jerry J, Minter LM, Klaus AB, et al. Hormonal control of p53 andchemoprevention. Breast Cancer Res 2002;4:91 –4.
14. Medina D, Kittrell F, Shepard A, Contreras Alejandro, Rossen JM, Lydon J.Hormone dependence in premalignant mammary progression. Cancer Res 2003;63:1067–72.
15. Medina D, Kittrell FS. P53 function is required for hormone-mediatedprotection of mouse mammary tumorigenesis. Cancer Res 2003;63:6140–3.
16. Dragnev KH, Rigas JR, Dmitrovsky E. The retinoids and cancer preventionmechanisms. Oncologist 2000;5:361 –8.
17. Sacchi S, Russo D, Avvisati G, et al. All-trans retinoic acid in hematologicalmalignancies. Haematologica 1997;82:106–21.
18. Yang Q, Sakurai T, Kakudo K. Reviewed in: Retinoid, retinoic acid receptorh and breast cancer. Breast Cancer Res Treat 2002;76:167–73.
19. Verma AK. Retinoids in chemoprevention of cancer. J Biol Regul HomeostAgents 2003;17:92 –7.
20. Camacho LH. Clinical applications of retinoids in cancer medicine. J BiolRegul Homeost Agent 2003;17:98 –114.
21. Malone W, Perloff M, Crowell J, Sigman C, Higley H. Fenretinide: aprototype cancer prevention drug. Expert Opin Investig Drugs 2003;12:1829 –42.
22. Veronesi U, De Palo G, Marubini E, et al. Randomized trial of fenretinide toprevent second breast malignancy in women with early breast cancer. J NatlCancer Inst 1999 Nov 3;91:1847– 56.
23. Bonewald LF, Oreffo R, Lee C, Park-Snyder S, Twardzik D, Mundy G.Effects of retinol on activation of latent transforming growth factor-h by isolatedosteoclasts. Endocrinology 1997;138:657 –66.
24. Tokuyama H, Tokuyama Y. Retinoids enhance IgA production bylipopolysaccharide-stimulated murine spleen cells. Cell Immunol 1993 Sep;150:353 –63.
25. Curtin JC, Dragnev KH, Sekula D, Christie AJ, Dmitrovsky E,Spinella MJ. Retinoic acid activates p53 in human embryonal carcinomathrough retinoid receptor-dependent stimulation of p53 transactivation function.Oncogene 2001 May 3;20:2559 –69.
26. Tokumoto YM, Tang DG, Raff MC. Two molecularly distinct intracellularpathways to oligodendrocyte differentiation: role of a p53 family protein. EMBOJ 2001 Sep 17;20:5261 –8.
27. Koistinen P, Zheng A, Saily M, Siitonen T, Mantymaa P, Savolainen ER.Superior effect of 9-cis retinoic acid (RA) compared with all-trans RA and 13-cisRA on the inhibition of clonogenic cell growth and the induction of apoptosis inOCI/AML-2 subclones: is the p53 pathway involved? Br J Haematol 2002 Aug;118:401 –10.
28. Sun SY, Yue P, Wu GS, et al. Mechanisms of apoptosis induced by thesynthetic retinoid CD437 in human non-small cell lung carcinoma cells.Oncogene 1999 Apr 8;18:2357–65.
29. Barcellos-Hoff MH, Ravani SA. Irradiation mammary gland stromapromotes the expression of tumorigenic potential by unirradaited epithelial cells.Cancer Res 2000;60:1254– 60.
30. Kuperwasser C, Pinkas J, Hurlbut G. Cytoplasmic sequestration and
Sensitivity to DNA Damage and Hormone-Based Strategies
Mol Cancer Res 2005;3(8). August 2005
441

functional repression of p53 in the mammary epithelium is reversed by hormonaltreatment. Cancer Res 2000;60:2723 –9.
31. Ewan KB, Henshall-Powell RL, Ravani SA, et al. Transforming growthfactor-h1 mediates cellular response to DNA damage in situ . Cancer Res 2002;62:5627 –31.
32. Becker KA, Lu S, Dickinson ES, et al. Estrogen and progesterone regulateradiation-induced p53 activity in mammary epithelium through TGF-h-dependentpathways. Oncogene. Epub 2005 Jun 6.
33. Cosgaya JM, Perona R, Aranda A. Retinoic acid induces secretionof transforming growth factors by PC12 pheochromocytoma cells. Oncogene1997 Feb 6;14:579 –87.
34. Yang L, Jacek O, Reczek P, Brown P. The retinoic acid receptor antagonist,BM453, inhibits normal breast cell growth by inducing active TGFh and causingcell cycle arrest. Oncogene 2001;20:8025 –35.
35. Sherr CJ. G1 phase progression: cycling on cue. Cell 1994;79:551–5.
36. Nurse P. Ordering S phase andMphase in the cell cycle. Cell 1994;79:547–50.
37. Chang BD, Broude EV, Dokmanovic M, et al. A senescence-like phenotypedistinguishes tumor cells that undergo terminal proliferation arrest after exposureto anticancer agents. Cancer Res 1999;59:3761 –7.
38. Seewaldt VL, Dietze EC, Johnson BS, Steven JC, Parker MB. Retinoic acid-mediated G1-S-phase arrest of normal human mammary epithelial cells isindependent of the level of p53 protein expression. Cell Growth Differ 1999;10:49–59.
39. Brodowicz T, Wiltschke C, Grunt TW, et al. Inhibition of proliferation andinduction of apoptosis in soft tissue sarcoma cells by interferon-a and retinoids.J Cancer 1999;80:1350– 8.
40. Herbert BS, Sanders BG, Kline K. N -(4-hydroxyphenyl) retinamideactivation of transforming growth factor-h and induction of apoptosis in humanbreast cancer cells. Nutr Cancer 1999;34:121– 32.
41. Sun SY, Yue P, Lotan R. Induction of apoptosis by N -(4-Hydroxyphenol)retinamide and its association with reactive oxygen species, nuclear retinoic acidreceptors, and apoptosis-related genes in human prostate carcinoma cells. MolPharmacol 1999;55:403–10.
Tu et al.
Mol Cancer Res 2005;3(8). August 2005
442





























