Embed Size (px)
Citation preview

Seminar series 2
Protein structure validation

In 't verleden ligt het heden; in 't nu, wat worden zal.
The past: Linus Pauling‘Inventor’ of helix and strand.Inventor of Bioinformatics?!Worked on proteins.

The history of bioinformatics is proteins
The future of bioinformatics is proteins
Only the present is a bit confused……

Structure validation
Everything that can go wrong, will go wrong, especially with things as complicated as protein structures.

What is real?

What is real?
ATOM 1 N LEU 1 -15.159 11.595 27.068 1.00 18.46ATOM 2 CA LEU 1 -14.294 10.672 26.323 1.00 9.92ATOM 3 C LEU 1 -14.694 9.210 26.499 1.00 12.20ATOM 4 O LEU 1 -14.350 8.577 27.502 1.00 13.43ATOM 5 CB LEU 1 -12.829 10.836 26.772 1.00 13.48ATOM 6 CG LEU 1 -11.745 10.348 25.834 1.00 15.93ATOM 7 CD1 LEU 1 -11.895 11.027 24.495 1.00 13.12ATOM 8 CD2 LEU 1 -10.378 10.636 26.402 1.00 15.12

X-ray
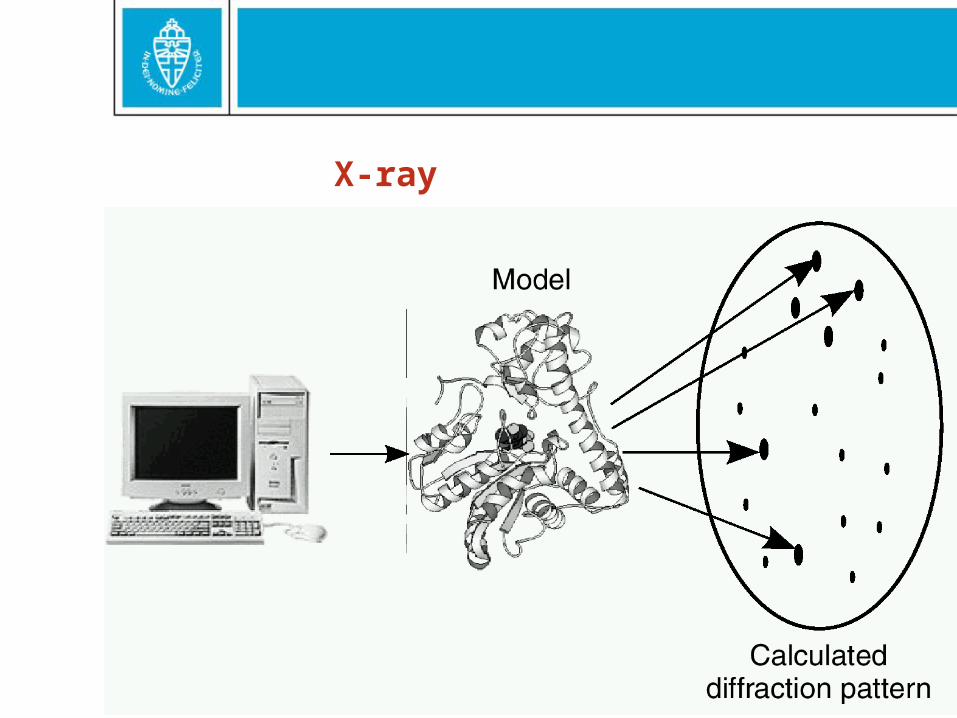
X-ray
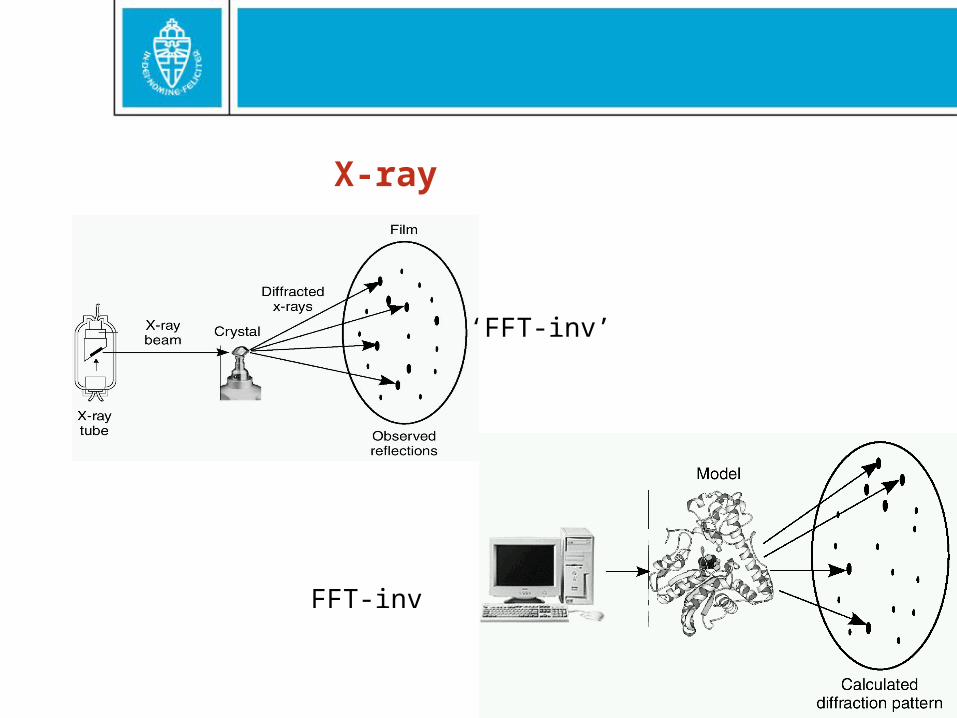
X-ray
‘FFT-inv’
FFT-inv

X-ray R-factor
Error = Σ w.(obs-calc)2
R-factor = Σ w.|obs-calc|
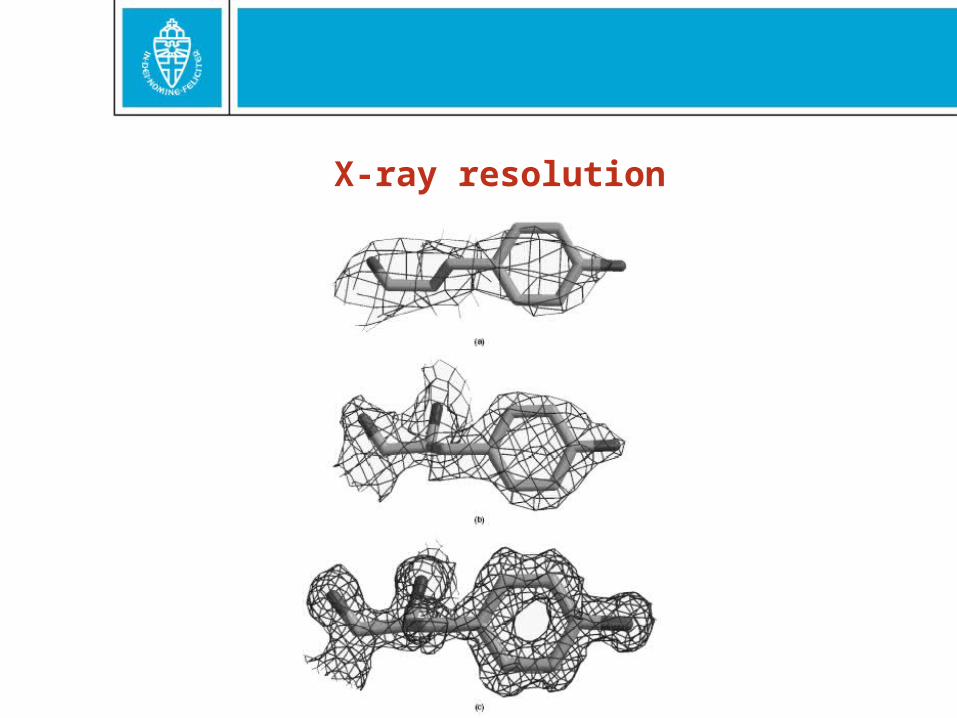
X-ray resolution

NMR data collection

NMR data
NMR data consists mainly of short inter-atomic distances between atoms. We call these NOEs.Most NOEs are between close neighbours in the sequence. Those hold little information.The ‘good’ NOEs are between atoms far away in the sequence. There are few of those, normally.NOEs are known with low precision. E.g. NOEs are binned 2.5-4.0, 4.0-5.5, and 5.5-7.0.

NMR Q-factor
Error = Σ NOE-violations + Energy term2

NMR versus X-ray
‘Error’ 1-2 Å 0.1-0.5 ÅMobility yes not reallyCrystal artefacts no yesMaterial needed 20 mg 1 mgCost of hardware 4 M Euro near infinite (share)Drug design no almost
Better combine and use the best of both worlds.

Why ?
Why does a sane (?) human being spend fourteen years to search for millions of errors in the PDB?

Because:
Everything we know about proteins comes from PDB files.
If a template is wrong the model will be wrong.
Errors become less dangerous when you know about them.

What do we check?
Administrative errors.Crystal-specific errors.NMR-specific errors.Really wrong things.Improbable things.Things worth looking at.Ad hoc things.

1FCC

Smile or cry?
A 5RXN 1.2 B 7GPB 2.9 C 1DLP 3.3 D 1BIW 2.5

X-ray specific

Further…4 The SCALE matrix gives a left handed axis system26 Scale matrix represents wrong crystal class4 Negated value in scale matrix11 Value in first row of scale matrix mistyped10 Value in second row of scale matrix mistyped6 Value in third row of scale matrix mistyped88 Determinant of MTRIX is incorrect195 Warning: New symmetry found62 Warning: MTRIX is not a pure rotation matrix165 Warning: Duplicate atoms encountered.57 Error: Threonine nomenclature problem324 Error: Weights outside the 0.0 -- 1.0 range709 Error: Weights outside the 0.0 -- 1.0 range520 Error: Decreasing residue numbers362 Error: Water clusters without contacts10973 Warning: Water molecules need moving

Further, further…1599 Error: B-factor over-refinement901 Error: Atoms too close to symmetry axes21090 Error: Abnormally short interatomic distances169 Note: No Van der Waals overlaps9100 Warning: Unusual bond lengths8214 Warning: Possible cell scaling problem18458 Warning: Unusual bond angles2515 Error: Ramachandran Z-score very low15408 Warning: Omega angles too tightly restrained4987 Error: Side chain planarity problems780 Warning: Inside/Outside residue distribution 12684 Warning: Backbone oxygen evaluation18612 Error: HIS, ASN, GLN side chain flips

Little things hurt big

How bad is bad?

Errors or discoveries?
Buried histidine.
Warning for buried histidine triggered biochemical follow -up and new mechanism for KH-module of Vigilin. (A. Pastore, 1VIG).

Contact Probability

Contact Probability

DACA

DACA

DACA

DACA

DACA

Contact probability box

Using contact probability
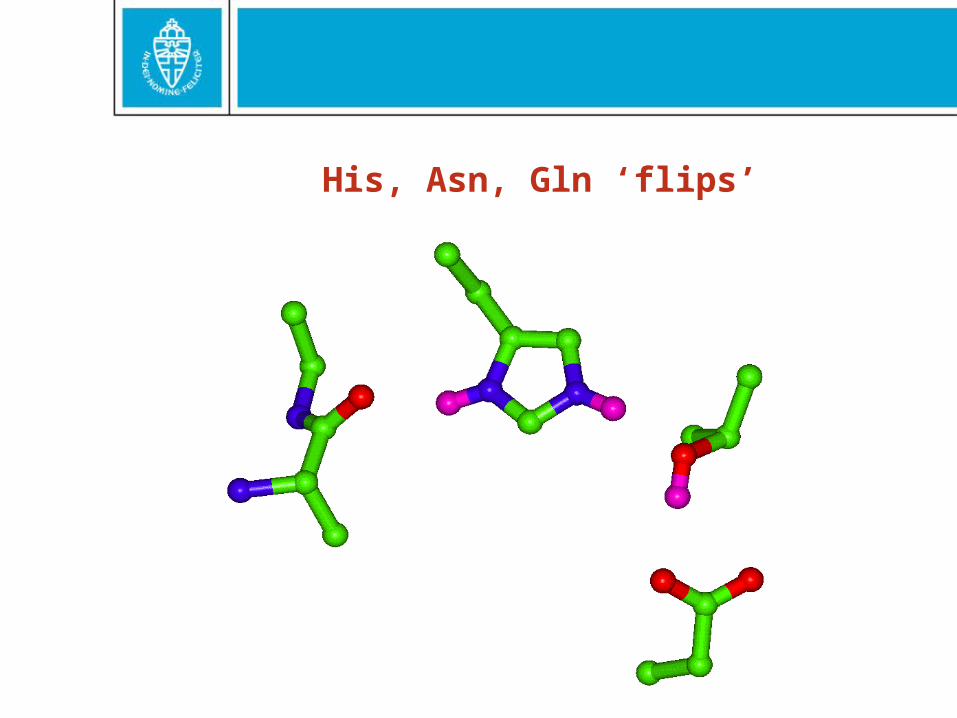
His, Asn, Gln ‘flips’

Where are the protons?

Hydrogen bond network

15% should be flipped

Your best check:

How difficult can it be?
1CBQ
2.2 A

How difficult can it be?
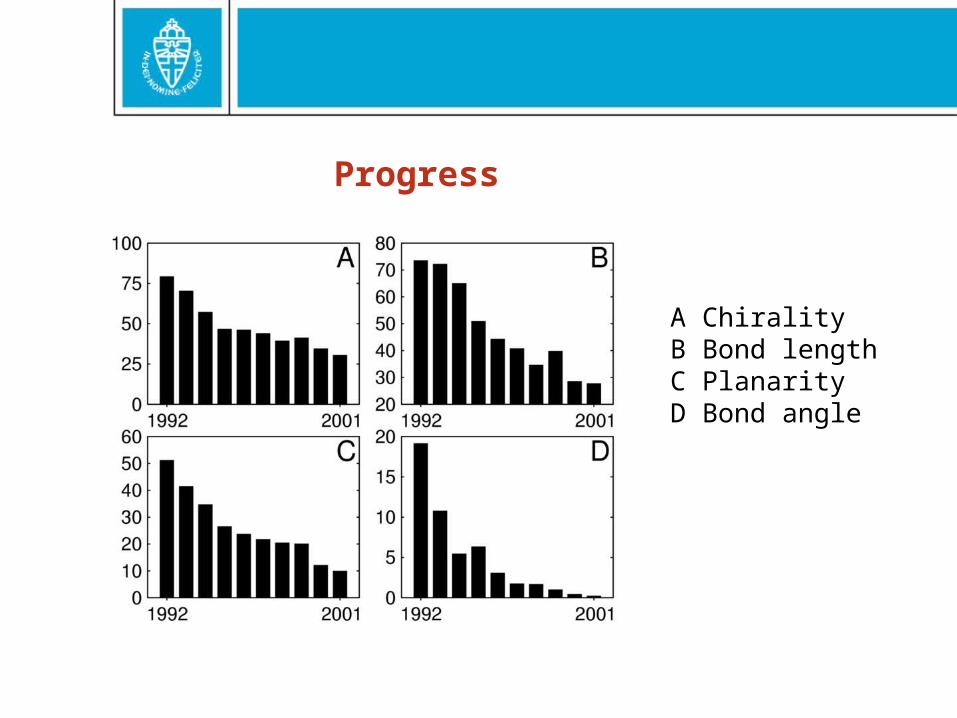
Progress
A ChiralityB Bond lengthC PlanarityD Bond angle

Progress
E Water islandF Bond angleG Atom on axisH Chain name

Progress
Chi-1 vs Chi 2
Ramachandran
Structures at 1.8 – 2.0 A

Conclusions
Everything that could go wrong has gone wrong.
Errors are on a ‘sliding scale’.
Error detection can detect a lot, but surely not everything (yet).
Acknowledgements:
Rob Hooft
Elmar Krieger Sander Nabuurs Chris SpronkRobbie JoostenMaarten Hekkelman





























